

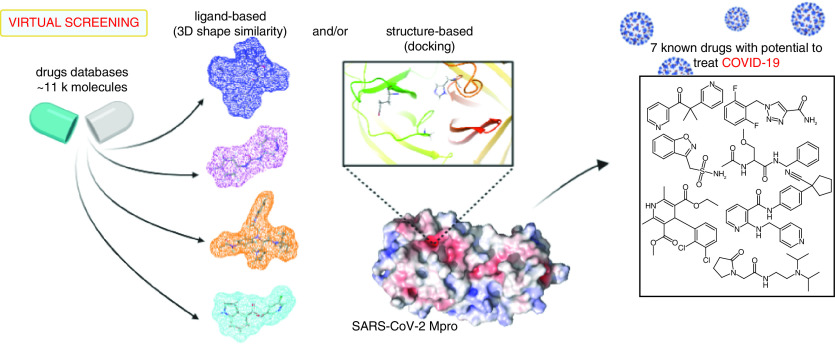
Abstract
Background: The new coronavirus pandemic has had a significant impact worldwide, and therapeutic treatment for this viral infection is being strongly pursued. Efforts have been undertaken by medicinal chemists to discover molecules or known drugs that may be effective in COVID-19 treatment – in particular, targeting the main protease (Mpro) of the virus. Materials & methods: We have employed an innovative strategy – application of ligand- and structure-based virtual screening – using a special compilation of an approved and diverse set of SARS-CoV-2 crystallographic complexes that was recently published. Results and conclusion: We identified seven drugs with different original indications that might act as potential Mpro inhibitors and may be preferable to other drugs that have been repurposed. These drugs will be experimentally tested to confirm their potential Mpro inhibition and thus their effectiveness against COVID-19.
Keywords: : COVID-19, drug repositioning, ligand-based drug discovery, main-protease (Mpro), SARS-CoV-2, structure-based drug discovery
Graphical abstract
In December 2019, in Wuhan, China, cases of pneumonia associated with severe acute respiratory syndrome were reported, which would later be diagnosed as a new variety of corona virus – SARS-CoV-2. On 11 February 2020, the WHO named it corona virus disease 2019 (COVID-19) and in March of the same year a pandemic was declared. Sadly, to date, there is no specific treatment against the disease and research in the field is ongoing [1–3].
Molecularly, the virus infection mechanism in host cells starts after the virus surface protein, known as Spike protein, binds to the receptor on the surface of human cells, ACE-2. This connection generates a conformational change in the viral structure, allowing the fusion between the virus and the host membrane and, thus, permitting the pathogen to release its genetic material in the infected organism [4]. Two proteases play a fundamental role in the infection process of SARS-CoV-2, with the sequencing of its genome, these proteases were identified as being, respectively, PLpro and 3CLpro - the latter also known as main protease, Mpro [5].
SARS-CoV 3C-like protease (SARS-CoV 3CLpro), as a part of the replicase polyproteins, cleaves a functional polypeptide and consequently leads to the maturation of SARS-CoV. Because of its functional importance in the SARS-CoV replication cycle, SARS-CoV 3CLpro is considered a potential target to develop novel anti-SARS drugs [6].
Because COVID-19 has a fast transmission speed, several research groups have been working together and disseminating their results quickly. The rapid dissemination of proteomics results, for example, has allowed researchers in the field of drug development to use these data to propose structures capable of stopping the progression of COVID-19 [7,8].
Several structures of the Mpro protease are already available in the protein structure repository Protein Data Bank (PDB) [9]. From these data, it is possible to use virtual screening (VS) techniques, a fast and efficient methodology for development of new drugs that can be linked to a database of drugs that have already been approved by regulatory agencies. In this way, it is possible to shorten the development time for new drugs, allowing these compounds that have known activity against a certain biological target to be studied against another biological target and go through the process known as repositioning of drugs [10–15].
In the present work, we sought to identify known drugs (approved by regulatory agencies around the world) with the potential to interact and inhibit the SARS-CoV-2 Mpro (i.e., by applying the so-called drug repositioning or drug repurposing strategy). This could be done by means of employing computational chemistry techniques [15] to identify potential drug candidates for COVID-19 treatment. We used an innovative strategy to scan an in-house compilation of known approved drugs, specifically by application of hybrid ligand- and structure-based VS approaches. The idea is to speed up the workflow toward in vitro tests, making it possible to foresee data concerning the biological activity of known drugs [5,11] and, in this way, possibly helping combat the pandemic. Therefore, we determined seven drugs, acting by inhibitory mechanism of enzyme Mpro, as potential candidates for COVID-19 treatment. Furthermore, the binding mode toward Mpro of these seven drugs was compared with known antivirals previously repurposed for COVID-19 (lopinavir, ritonavir and darunavir), which indeed inhibit the enzyme and showed promising in vitro results but have not demonstrated improvement of disease treatment in subsequent clinical studies. Next, these seven drugs will be tested in vitro to confirm their inhibition potential toward Mpro and thus their effectiveness against the new coronavirus.
Materials & methods
Drugs library preparation
There is not a single official database of drugs approved by the US FDA; moreover, those available with this classification have differing numbers of compounds. Hence, we compiled a pool of compounds originating from drug databases approved by different agencies or organizations, comprising approximately 8,000 molecules. Our drug library was compiled into three main datasets, Drugbank, the FDA and ZincWorld; these were gathered from the following databases: DrugBank (Release Version 5.1.5) [16], Zinc15 (subset: world not fda) [17], Selleckchem subset FDA-approved Drug Library (https://www.selleckchem.com/), Enamine subset approved Drugs Collection (https://www.enaminestore.com/platedsets/fda-approved) e-Drug3D [18] and the BindingDB subset of FDA approved drugs [19].
Preparation of the drug molecules contained in these databases was carried out by removing duplicate/redundant molecules using OpenBabel [20], considering InChI (IUPAC InchI identifier), adding hydrogens, setting partial charges and adjusting bond orders with Discovery Studio [21] and saving the corresponding output files in mol2 extension format. Compounds were also processed with FILTER [22] using no properties filtering, but its default functions of canonicalization, pKa normalization and salt were removed. In sequence, drug databases were processed using OMEGA [23,24] for conformer generation, with the flipper flag turned on (for specification of unassigned stereocenters), adjustment of the strain energy (above the energy of global minimum conformer) below 9.0 kcal/mol and a root mean square deviation (RMSD) of 0.6 Å as a cutoff for conformer identity. For ROCS [25,26], as the ligand-based VS (LBVS) approach, up to 300 conformers generated per molecule; for docking, as structure-based VS (SBVS) approach, only one 3D conformer was generated per molecule.
Targets & ligands: selection & preparation
Selection of crystallographic structures to be used as targets in these structure-based studies, as well as corresponding complex ligands in ligand-based studies, was carried out by inspecting more than 90 available structures in the PDB, considering those of Mpro of the ‘old coronavirus' (SARS-CoV) and those available for the new coronavirus (SARS-CoV-2). Initially, we avoided apo structures, MPro structures in complex with ligands bound to different sites or those with covalent bond within the active site of the enzyme. Finally, we selected the following protein–ligand complexes: PDB ID 6W63 (‘Structure of COVID-19 main protease bound to potent broad-spectrum non-covalent inhibitor X77,’ at 2.10-Å resolution [27]); PDB ID 5R83 (‘PanDDA analysis group deposition – crystal structure of SARS-CoV-2 Mpro in complex with Z44592329,’ at 1.58-Å resolution [28]); and PDB ID 4MDS (‘Discovery of N-(benzo[1,2,3]triazol-1-yl)-N-(benzyl)acetamido)phenyl) carboxamides as severe acute respiratory syndrome coronavirus SARS-CoV 3CLpro inhibitors – bound to ML300,’ at 1.60-Å resolution [29]).
In addition, we selected one of the most potent noncovalent inhibitors of SARS-CoV MPro, as reported by Ghosh et al. [30], with IC50 value of 30 nM and here named C10 (5-chloropyridin-3-yl 1H-indole-4-carboxylate). This ligand has been also used as query/reference in our ligand-based approaches.
MPro protein structures (retrieved from complexes of PDB IDs 6W63, 5R83 and 4MDS) were prepared before docking runs by keeping only the corresponding primary chain; removing water, ligands, metals and cofactors; and adding hydrogens using the Protein Preparation Wizard tool implemented in Maestro software [31,32].
It is worth noting that four of the ligands were here used in ligand-based methodologies. As previously cited, three were retrieved from corresponding complexes PDB IDs 6W63, 5R83 and 4MDS, respectively X77 (N-(4-tert-butylphenyl)-N-[(1R)-2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl]-1H-imidazole-4-carboxamide), Z44592329 (N-phenyl-N′-pyridin-3-ylurea), ML300 (N-[4-(acetylamino)phenyl]-2-(1H-benzotriazol-1-yl)-N-[(1R)-2-[(2-methylbutan-2-yl)amino]-1-(1-methyl-1H-pyrrol-2-yl)-2-oxoethyl]acetamide) and the fourth named here C10, as mentioned earlier. Crystallographic poses (conformation and orientation) from the three complexes ligands were kept unchanged. For C10, we performed a docking run using GOLD [33,34] in which the chosen pose was obtained with best score and in consensus within 10 best scoring poses; thus, this pose was used as query/template/reference in subsequent ligand-based strategies.
Validation of docking procedures on SBVS & queries performance on LBVS
SBVS (with docking) strategies have been validated by two procedures: redocking and receiver operating characteristic (ROC) curve analysis. Redocking was performed using GOLD default settings and the previously prepared protein from each of the three selected PDB mentioned earlier. Previous to redocking simulations, simple preparation of compounds X77, Z44592329 and ML300 was carried out by same ‘standard’ procedure adopted in this work (i.e., that mentioned for preparation of the drug databases).
ROC curve analysis has been applied to the docking strategies used here as a second validation procedure. This docking validation was performed using GOLD software for each one of the three selected and prepared Mpro structures retrieved from PDB. The purpose of this validation procedure is to assess the ability of such docking methodology to recover known active compounds from a dataset of actives contaminated with ‘decoys.’ One dataset containing approximately 40 actives was used for each of the three inhibitor–enzyme complexes selected in this work. To do this, we randomly selected forty actives for each target from a small library that was built with ~140 known Mpro inhibitors of SARS-CoV and SARS-CoV-2, compiled from literature [27–30,35–44], considering either reported data concerning their IC50 values or existence of respective PDB files (see Supplementary Table 1). Decoy generation was executed from the webserver Database of Useful Decoys Enhanced (DUD-E) [45], which furnished 50 decoys per active compound. Each dataset (40 actives and 2000 decoys) was then submitted as input in the GOLD docking software, using the exact same settings as those further used in GOLD runs of VS studies. ROC curves were built using the Screening Explorer webserver (http://stats.drugdesign.fr/) [46] and subsequently evaluated by analysis of the corresponding area under the curve (AUC) values.
Validation of LBVS was also performed by ROC curve analysis, that is, by analyzing the performance of each selected ‘query’ in the shape similarity comparison screened by ROCS. Selected queries for this purpose were the three ligands X77, Z44592329 and ML300 in their crystallographic poses (conformation), as well as the best scored docking pose of C10, as previously mentioned. For each ligand, we used the same corresponding datasets (as stated in the previous paragraph) compiled from literature, consisting of 40 actives, plus 50 decoys per active compound (generated by DUD-E), and these were used as input database to run vROCS [47]. Results were ranked and evaluated according to the Tanimoto Combo (shape and color) similarity indices, obtained for each one of the corresponding four queries, and thus used to plot ROC curves (using vROCS and the Screening Explorer webserver as well), which were then evaluated according the AUC values obtained.
Structure- & ligand-based VS
We performed seven independent VS: three only using structure-based methodology (docking) and four using mixed ligand- and structure-based methodologies (ROCS + docking) (see Table 1).
Table 1. Number of compounds prioritized and filtered in each step/filter performed in virtual screening (VS) workflows.
| Main methodology | Reference PDB/query | VS filter | Drugbank | FDA | Zincworld |
|---|---|---|---|---|---|
| Structure-based VS | 6W63 | – | 1089 | 4061 | 3475 |
| Docking | 100 | 100 | 100 | ||
| DEREK (Tox) | 41 | 45 | 39 | ||
| Visual inspection | |||||
| 5R83 | – | 1089 | 4061 | 3475 | |
| Docking | 100 | 100 | 100 | ||
| DEREK (Tox) | 49 | 48 | 41 | ||
| Visual inspection | |||||
| 4MDS | – | 1089 | 4061 | 3475 | |
| Docking | 100 | 100 | 100 | ||
| DEREK (Tox) | 50 | 41 | 42 | ||
| Visual inspection | |||||
| Ligand- and structure-based VS | X77 (6W63.pdb) | – | 1089 | 4061 | 3475 |
| ROCS + EON | 100 | 100 | 100 | ||
| DEREK (Tox) | 33 | 49 | 34 | ||
| Docking + Visual inspection | |||||
| Z44592329 (5R83.pdb) | – | 1089 | 4061 | 3475 | |
| ROCS + EON | 100 | 100 | 100 | ||
| DEREK (Tox) | 46 | 53 | 50 | ||
| Docking + visual inspection | |||||
| ML300 (4MDS.pdb) | – | 1089 | 4061 | 3475 | |
| ROCS + EON | 100 | 100 | 100 | ||
| DEREK (Tox) | 44 | 50 | 40 | ||
| Docking + visual inspection | |||||
| C10 | – | 1089 | 4061 | 3475 | |
| ROCS + EON | 100 | 100 | 100 | ||
| DEREK (Tox) | 34 | 45 | 41 | ||
| Docking + visual inspection | |||||
Structure-based strategies (i.e., mainly conducted by docking) were done using GOLD software [33,34]. For this, we used individual drug databases previously prepared as well as the three selected Mpro enzymes. Hence, we selected the best 100 compounds scored by ChemPLP fitness function, that were subsequently inputted into DEREK to assess their toxicity profiles [48,49]. From these analyses, we filtered compounds that afforded alerts as ‘certain’ and ‘plausible,’ according to Lhasa expert system predictions. Survivors compounds were then inspected by docking about probable interactions within active sites of corresponding enzymes so that we only kept molecules that showed consensus poses as well as interactions especially regarding those with His41, and Cys145 [50] and Glu 166 observed in the crystallographic complexes [28,29,51].
Furthermore, we applied mixed VS workflows by means of ligand-based shape and electrostatic potential similarities, using ROCS [25] and EON [52], respectively. For this, we used all the drug databases noted earlier individually, as well as the four ligand queries. We then retrieved the best 100 scored compounds regarding their Tanimoto similarity indices (ROCS_TanimotoCombo and EON_ET_Combo), obtained for each query. These compounds were then assessed with regard their toxicity profiles using DEREK [48,49], filtering out compounds that afforded alerts as ‘certain’ and ‘plausible.’ Next, survivor compounds were submitted to docking (SBVS) using GOLD to inspect their interactions within the active sites of the corresponding enzymes, especially regarding those with His41 and Cys145, as well as Glu166 observed in the crystallographic complexes.
Finally, the predicted binding modes for the selected compounds were compared with those of the drugs lopinavir, ritonavir, darunavir and boceprevir, which were approved by the FDA for the SARS-CoV-2 treatment.
Results & discussion
Mpro-inhibitor structures
The decision to use the PDB ID 4MDS complex [29] (of SARS-CoV Mpro), in this work, is due to the existence of few SARS-Cov-2 MPro structures deposited in the PDB in complex with ligands showing high potency or binding affinity. Reliability of using this structure was confirmed by the high sequential similarity and identity shared between the enzymes of the two viruses (sequence identity of 96.1 and 99.02% sequence similarity, with only 12 mutations, all of which are outside the region of interest – the catalytic site) [50,53].
The high similarity shared by Mpro of SARS-CoV and Mpro of SARS-CoV-2, together with the fact of that they present practically identical catalytic sites, allows the use of both available co-crystals to elucidate interactions between the enzyme active site and SARS-CoV-2 inhibitors [53–55].
Surrounding both Mpro catalytic sites, there is only one difference/mutation, represented by a serine in SARS-CoV-2 and by an alanine in SARS-CoV; nevertheless, their sidechains point to the outer region of the binding site [53]. Moreover, recent studies have shown that both binding sites possess similar substract specificities, suggesting that drugs developed for SARS-CoV might also be efficient in inhibiting the SARS-CoV-2 enzyme [56].
Furthermore, superposition of the PDB ID 4MDS enzyme structure (MPro SARS-CoV) with the PDB ID 5R83 (Mpro SARS-CoV-2) revealed that the region of the site is composed mostly of the same amino acid residues (and rotamers), with the exception of different rotamers observed for the Asn142 and Gln189 residues of the two structures (Figure 1).
Figure 1. Superposition of catalytic/active sites of enzymes structures PDB IDs 4MDS (SARS-CoV Mpro) and 5R83 (SARS-CoV-2 Mpro).

The Ribbons diagram reveals the same amino acid residues (in stick representations) as well as rotamers constituting the corresponding active sites, except for Asn142 and Gln189.
The PDB ID 4MDS (of SARS-CoV) enzyme structure has a cavity slightly larger than the corresponding SARS-CoV-2 cavity, where a main chain movement due to the larger and more branched inhibitor is observed compared with that observed in the PDB ID 5R83 structure. The difference in cavity sizes between the MPro structures of the two viruses is, in fact, only due to the size of the corresponding complexed inhibitors. This hypothesis is confirmed by the PDB ID 6W63 (SARS-CoV-2 Mpro) structure, which presents an inhibitor equally large and branched compared with the PDB ID 4MDS inhibitor structure of SARS-CoV Mpro.
Another important aspect that motivated us to use the PDB ID 4MDS structure was the presence of an inhibitor chemical and that it is structurally different from the inhibitor present in the PDB ID 6W63 structure, even though the cavities are very similar and correspondent in size and shape. The virus-1 enzyme inhibitor is more branched than the SARS-CoV-2 enzyme and similar to some of the main protease inhibitors on the market.
Furthermore, our comparative analyzes here performed with both the Mpro structures of SARS-CoV and SARS-CoV-2 contribute to the clarification of their structural similarity, corroborating with studies that were previously carried out with this objective [24,50,51] and thus allowing the use of previous results from the literature (since 2003) on relevant structural characteristics of compounds to act as Mpro SARS-CoV-2 inhibitors as well.
Validation of the docking & shape-queries performances
Regarding to the docking validation procedures, a standard redocking protocol was carried out. Redocking was performed using the GOLD software to verify the ability of the docking methodology used here to reproduce the crystallographic poses (in terms of conformation and orientation – or ‘pose’) of the original ligands located at the three proteins binding sites. Results are shown in Figure 2A–C, where a suitable convergence of the crystallographic poses for X77, Z44592329, and ML300 (respectively of the PDB IDs 6W63, 5R83 and 4MDS) can be seen. In fact, the top-ranked poses of each ligand, obtained using GOLD, have RMSD values of 0.6, 0.9 and 1.7 Å, respectively, in agreement with that expected for a redocking validation procedure [57,58].
Figure 2. Redocking validation of structures employed.
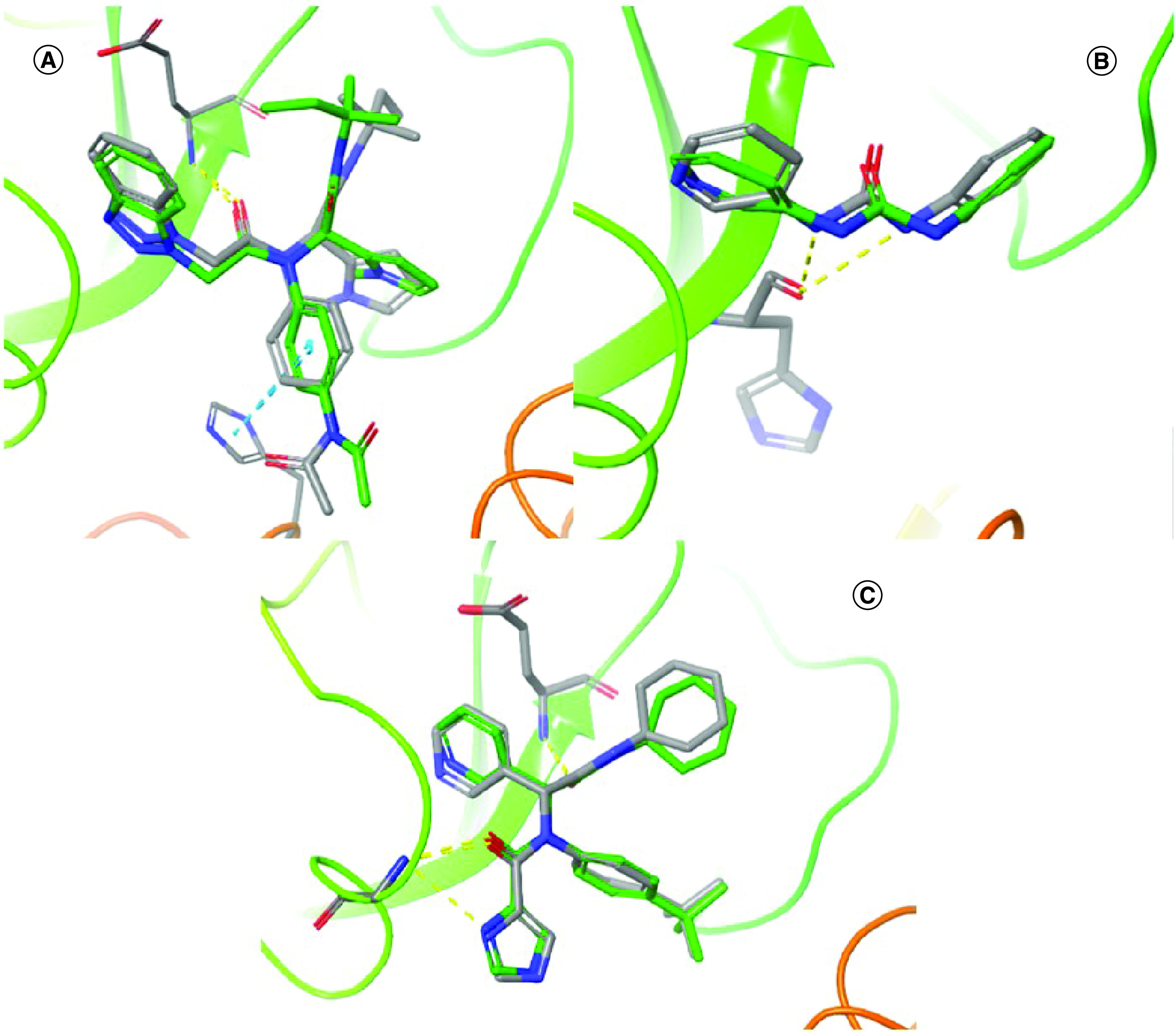
Superpositions showing the agreement between the crystallographic poses (with stick representations and carbon atoms in green) of (A) X77, (B) Z44592329 and (C) ML300 with the top-ranked ones obtained (with stick representations, in color by atom). Hydrogen bonds (dashed lines, in yellow) and pi–pi stacking (dashed lines, in blue) are also visualized.
A second docking validation procedure was also employed, as described earlier, to assess the ability of this docking methodology and recover known active compounds from a dataset containing a large number of DUD-E-generated decoys.
It is worth noting that we randomly compiled one individual dataset of 40 actives for each validation with each PDB structure. In fact, the three crystallographic Mpro inhibitors (two of SARS-CoV-2 and one of SARS-CoV) have different molecular weights, volumes and shapes, inducing and requiring an apparent flexibility of the active site of such enzyme to occupy and interact with residues of interest. Moreover, the small library compiled from the literature (see Supplementary Table 1), from where we retrieved 40 actives to rationalize the validation datasets, includes inhibitors of both the SARS-CoV-2 and the SARS-CoV Mpro structures and reasonable structure diversity. Thus, due to the structural similarity of the enzymes and inhibitors of the two SARS-CoV Mpro crystallographic structures used here, the random selection of actives in the validation datasets proved to be efficient.
For the GOLD docking results obtained for each enzyme and each selected dataset, compounds were ranked using the ChemPLP score function and the corresponding binaries codes (1 to active, 0 to decoy) used to plot ROC curves, correlating sensitivity (y-axis) against 1-specificity (x-axis). In general, sensitivity means the ‘true positive,’ whereas 1-specificity signifies a ‘false positive’ [59]. In this way, compounds were classified regarding true–false positive–negative ratios and represented in terms of sensitivity and specificity to build the ROC curves. Figure 3 demonstrates the resulting curves and respective AUC values, which were 0.645, 0.732 and 0.655 for the PDB IDs 6W63, 5R83 and 4MDS, respectively.
Figure 3. Receiver operating characteristic (ROC) curves obtained for validation of docking and shape-similarity methodologies.
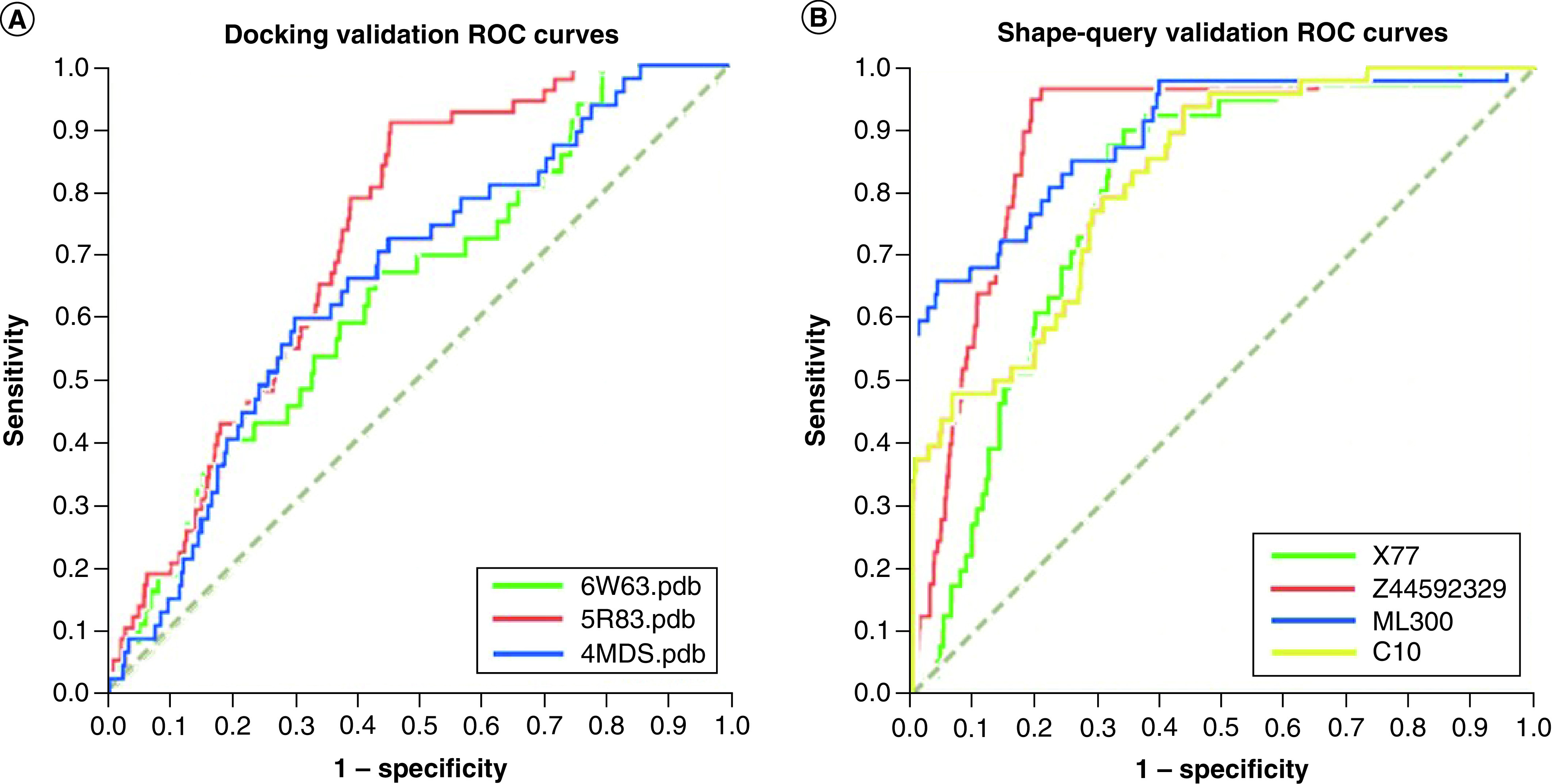
(A) validation of docking protocols and respective enzyme structures (PDB IDs 6W63, 5R83 and 4MDS). (B) ROCS validation of shape-query similarity for inhibitors X77, Z44592329, ML300 and C10.
We also validated our ROCS queries before applying them to large databases in VS prospections by means of analyses of ROC curves generated by vROCS. When using the ligand X77 we obtained an AUC value of 0.801, for Z44592329 an AUC value of 0.839, for ML300 an AUC value of 0.895, and for C10 an AUC value of 0.824, as depicted in Figure 3.
Docking AUC results that were not close to the unit could be attributed to trivial differences observed between the active compounds and the decoys, to active compounds belonging to limited chemical classes or to their possessing similar physicochemical properties. However, because these limitations should rely principally on the selection of the compounds dataset, which was indeed constituted of variable chemical classes (see Supplementary Table 1), we believe it represents a satisfactory result because it is relatively far from a 0.5 AUC random value.
Finally, both the structure-based (by docking, with GOLD) and ligand-based (by shape similarity, with ROCS) methodologies employed were successful in each validation procedure according to their ROC curve analyses. They showed sufficiently good performance to ensure reliable use of the corresponding protocols and queries in prospective VS studies.
VS workflows
Considering the previously described methodological strategies (see Materials & methods), including database preparation and validation of methodologies, we carried out the VS workflows. The databases were used separately so that results were obtained for each database and for each of three proteins (three SBVS workflows) and four ligands (four LBVS + SBVS workflows), for a total of seven VS workflow results for each database. Thus, we could analyze consensus between results in each VS workflow as a way to select final potential hits.
By comparing results obtained with workflows consisting of only SBVS and of both LBVS + SBVS, the LBVS + SBVS strategies were shown to be more consistent because results obtained using LBVS workflows showed drugs as potential hits with a higher number of consensus docking poses (higher homogeneity between them) and also the indication that these compacts can interact with residues Glu166 and His41 in their corresponding final visual inspection. Use of these two methodologies together has shown significant improvement in results obtained in research from our group [60,61], allowing elimination of extensive or bulky ligands in the initial stages of the screening protocols so that only molecules with similar characteristics to the queries of interest proceed to later stages of each workflow.
Importantly, the docking algorithm used to perform conformer generation was the same as that used in all the docking simulations, and 10 docking runs (poses) were conducted for each ligand. Hence, drugs with potential to be repurposed for Mpro inhibition that presented a homogeneity among the corresponding sets of docked poses were selected (some ligands presented unanimity for 10 poses in this analysis; for examples, see metyrapone and zonisamide in Table 2).
Table 2. Potential repurposing drugs retrieved from drug databases and selected by virtual screening (VS) workflows and references, as well as number of corresponding consensus poses observed in the docking final inspection.
| Drug | Database | VS workflow | Reference/query | Consensus poses | Ligand/pocket coverage† |
|---|---|---|---|---|---|
| Metyrapone | DrugBank | LBVS | 4MDS | 10 | 96.77/23.56 – P1 |
| DrugBank | LBVS | 5R83 | 9 | 100.00/38.22 – P2 | |
| FDA | LBVS | 6W63 | 5 | 58.07/21.35 – P2 64.52/62.77 – P6 |
|
| Rufinamide | DrugBank | LBVS | 5R83 | 8 | 100.00/35.25 – P2 |
| FDA | LBVS | 5R83 | 7 | 100.00/35.83 – P2 | |
| FDA | LBVS | 6W63 | 3 | 88.00/36.60 – P2 20.00/12.85 – P6 |
|
| Zonisamide | DrugBank | LBVS | 6W63 | 10 | 100.00/36.50 – P2 27.27/7.45 -– P6 |
| FDA | LBVS | 6W63 | 10 | 95.46/35.55 – P2 27.27/7.64 – P6 |
|
| Lacosamide | FDA | LBVS | 6W63 | 9 | 47.22/25.75 – P2 63.89/74.53 – P6 |
| Apatinibe | FDA | LBVS | 4MDS | 8 | 83.02/33.75 – P1 |
| Pramiracetam | DrugBank | LBVS | 6W63 | 8 | 65.96/37.98 – P2 42.55/71.72 – P6 |
| Felodipine | FDA | LBVS | 4MDS | 7 | 100.00/35.50 – P1 |
Ligand/pocket coverage values predicted using DoGSiteScorer [62,63] (more details in Supplementary Table 2 & Supplementary Figures 1–3).
Moreover, these seven selected drugs maintained key interactions within the active site of enzymes in a similar manner as observed in the crystallographic complex structures, particularly with regard to the hydrogen bond with Glu166. Furthermore, in the majority of poses, the key interaction with His41 was established, which was also observed in the crystallographic complex PDB ID 4MDS; when this is not present, as in the case of the drug felodipine, it was substituted in one interaction with Gly143.
Table 2 shows the potential drugs to be repurposed as Mpro inhibitors, retrieved from corresponding databases and obtained from respective VS workflows. These same compounds are also illustrated in Figure 4, showing their 3D and 2D representations and their potential as Mpro inhibitor candidates.
Figure 4. The 3D (stick) and 2D (line) representations of seven drugs as repurposing candidates for Sars-Cov-2 Mpro (in Ribbons diagram) inhibitors and future COVID-19 treatment.
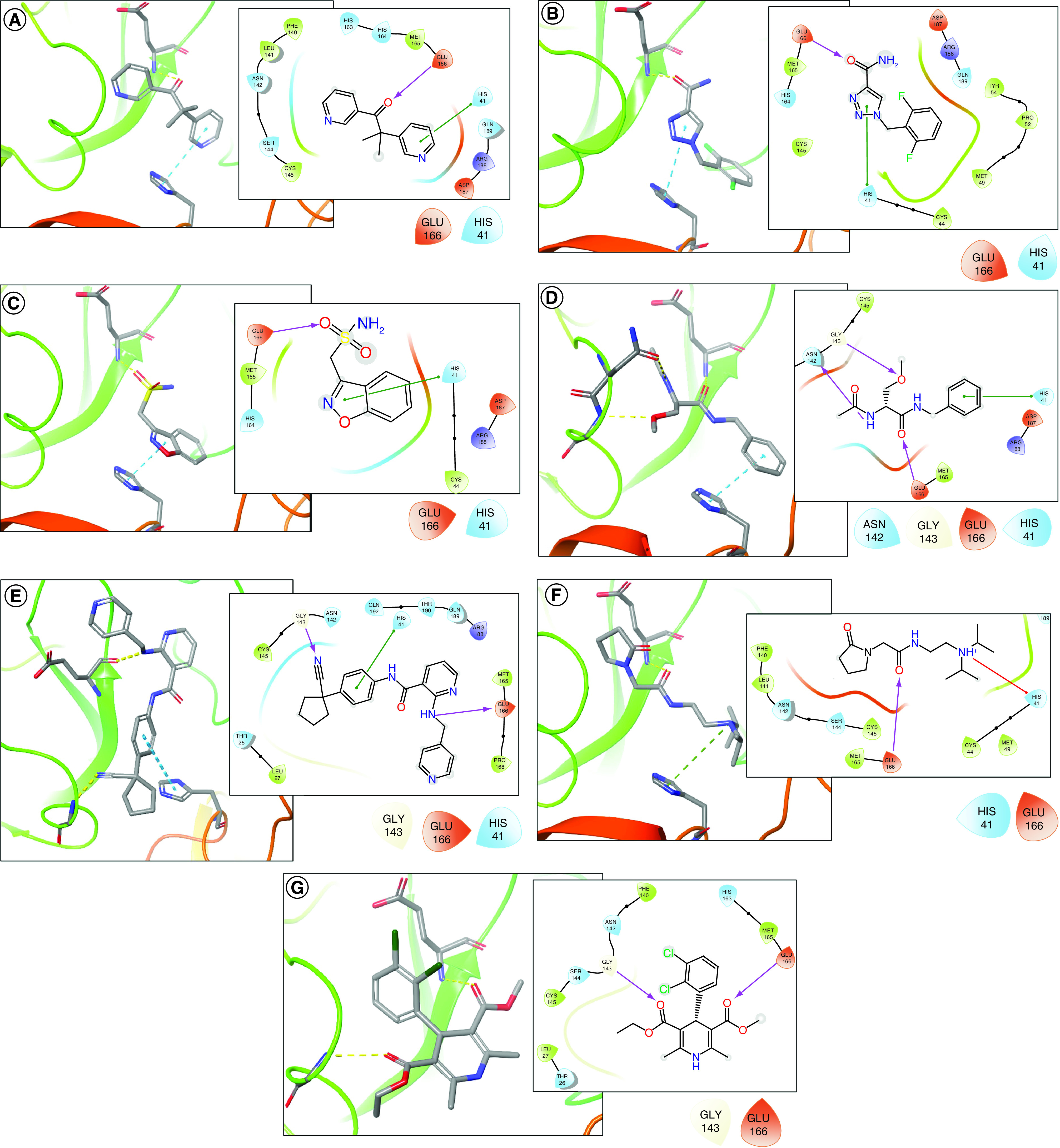
(A) Metyrapone, (B) rufinamide, (C) zonisamide, (D) lacosamide, (E) apatinibe, and (F) pramiracetame and (G) felodipine.
The first three drugs shown in Table 2 – metyrapone, rufinamide and zonisamide – had the most consistent results. Metyrapone, which has been used for the diagnosis of adrenal insufficiency and treatment of Cushing syndrome, was recovered in three simulations, each with a different query, demonstrating the strong tendency of this drug to interact effectively with the Mpro enzyme, even with small conformational changes in it. The anticonvulsant rufinamide was also recovered in three simulations. The adjuvant zonisamide used in the treatment of epilepsy also showed interesting results, achieving consensus in all the docking poses in two simulation routes in two drug databases.
The other four drugs selected, despite having been recovered by only one of the simulations performed, presented a high number of docking poses in consensus. Lacosamide, which is used as an adjuvant in epilepsy treatment, presented a consensus of nine poses in the simulation using the FDA database and the reference structure PDB ID 6W63. Apatinib and Pramiracetam, used to treat gastric cancer and as a CNS stimulant, respectively, demosntrated consensus of eight poses among their corresponding results. Finally, the calcium channel blocker Felodipine, originally to control hypertension, remains a potential drug to treat of COVID-19; this drug showed a consensus of seven poses using the FDA database and the structure of the PDB ID 4MDS inhibitor as a reference.
Results of docking simulations allowed us to infer that proximity between Glu166 and Hys41 residues at the catalytic site of Mpro favored the selection of compounds bearing groups that are able to act as hydrogen bond acceptors next to other groups capable of transferring electrons by pi–pi or cation–pi interactions. In this way, interactions presented by the majority of drugs in this study show oxygen with electron pairs near an aromatic ring.
In addition, we evaluated the likelihood that these seven drug hits indeed interact with the binding pocket from each SARS-CoV-2 Mpro used in docking studies. For this, we employed the software DoGSiteScorer [62,63], which is a tool for pocket detection and protein druggability assessment. Preliminarily, we detected the most significant pockets in each Mpro protein (PDB IDs 4MDS, 5R83 and 6W63) and found that the respective active sites (where corresponding original crystallographic ligands were bound) presented favorable values of clusters' number, chemical descriptors and druggability scores (see Supplementary Table 2).
Furthermore, as shown in the last column of Table 2, when retrieving the best docking poses for each of the seven drug hits and evaluating which pocket should preferably host these ligands, all of them presented unique and highest scores toward potential binding in the active sites (see also Supplementary Figures 1–3). Worth noting that the main active site (with highest number of clusters) predicted using DoGSiteScorer are represented by P1 for PDB ID 4MDS, P2 for 5R83, and P2 + P6 for 6W63. Thus, this evaluation should strongly indicate that top poses prefer the active site over other sites present in the surface of SARS-CoV-2 Mpro.
The presence of a hydrogen bond acceptor group is represented by a carbonyl in metyrapone (Figure 4A); an amide oxygen in rufinamide, lacosamide and pramiracetam (Figure 4B, D & F, respectively); a sulfonamide oxygen in zonisamide (Figure 4C); and an ester in felodipine (Figure 4G). In the case of apatinibe (Figure 4e), the drug presents a secondary amine in place of carbonyl that is able to interact with amide oxygen of Glu166 by means of donating a hydrogen bond. All these groups present distances ranging from 2.0 to 2.5 Å from the amide nitrogen of Glu166 – or from the corresponding oxygen for apatinibe – which therefore should have strong hydrogen bonding (represented by yellow dotted lines in 3D images and by purple arrows in 2D images).
Moreover, selected drugs present an aromatic ring or a protonated amine next to the aforementioned oxygen that forms a hydrogen bond with Glu166. Both groups are capable of interacting by electron transfer with imidazole from Hys41 (represented by blue dotted lines in 3D and green lines in 2D images), with the exception of felodipine, which does not interact with this residue. There is a pyrimidine ring in metyrapone, a triazole in rufinamide, benzioxazole in zonisamide and benzene in lacosamide and apatinibe; all of these are distanced at a maximum of 4.4 Å when face-to-face or 5.5 Å when edge-to-face with Hys41 imidazole. These groups and distances allow the occurrence of pi–pi interactions with residue when drugs bear aromatic rings or cation–pi interactions when they bear a protonated amine.
Other interactions are yet to be observed, such as lacosamide, which other than previously mentioned interactions, is still able to establish a hydrogen bond between its ether acceptor group and the side chain nitrogen from Gly143 and also donate a hydrogen bond from its amide nitrogen to carbonyl of Asn142.
To enhance the quality of selection of these seven compounds, we compared their binding interactions with the corresponding ones from known drugs that have been reported as promising repurposing candidates for COVID-19. We used lopinavir, ritonavir and darunavir, three antivirals known to act as protease inhibitors that prevent viral replication of HIV and also have been shown to inhibit SARS-CoV-2 Mpro in vitro [64]. Nonetheless, the use of these drugs in clinical studies to treat patients with COVID-19 have not improved the clinical picture of hospitalized patients or patients with severe cases of the disease [65–69].
In addition, these three drugs did not show a high affinity for Mpro inhibition when evaluating the corresponding docking results, obtained by applying the same protocol and conditions performed in the present work (Figure 5).
Figure 5. Representations (atoms in stick) of docking results obtained for lopinavir, ritonavir and darunavir with SARS-CoV-2 Mpro.
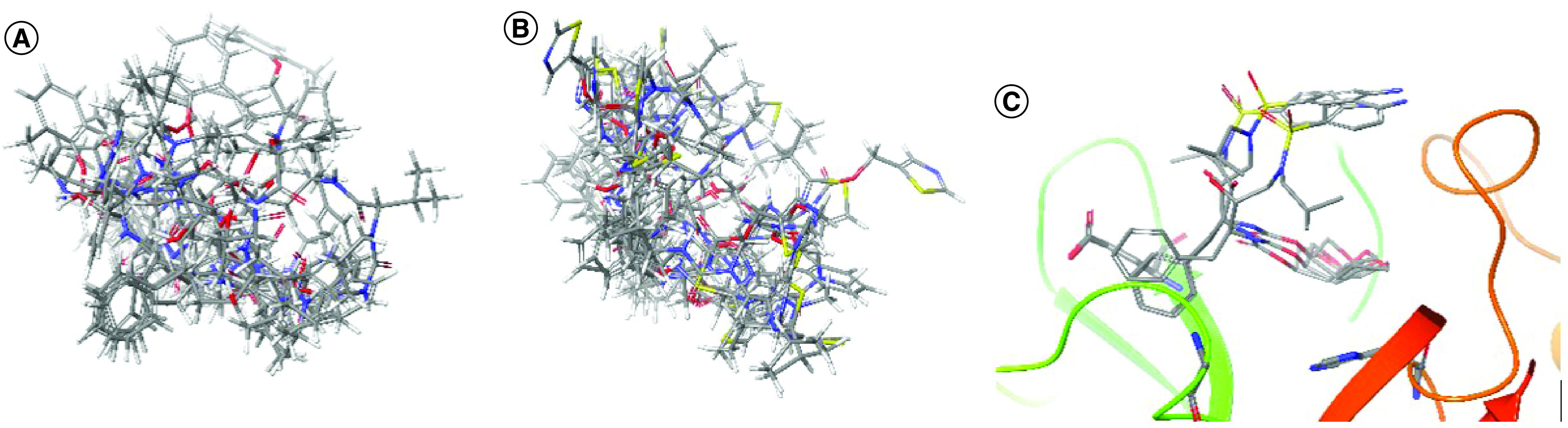
Absence of consensus (among 10 docking poses generated using GOLD) for (A) lopinavir and (B) ritonavir. (C) The superposition of the docking poses reveals no interaction with the main residues at the Mpro (in Ribbons diagram) catalytic site, where only three poses could be obtained in consensus.
Lopinavir shows in vitro data concerning the inhibition of viral replication, with an EC50 value of 5.73 μM, CC50 of 74.44 μM and SI of 12.99 [70]. However, in our docking studies, it had no consensus results within 10 poses generated using GOLD (Figure 5A), and none of the poses had simultaneous key interactions with more than one of the main residues (Glu166, His41 and Gly143) present at the enzyme catalytic site. The same behavior was observed for ritonavir (Figure 5B), which is usually used in clinical practice with lopinavir–ritonavir and had an EC50 value of 8.63 μM, CC50 of 74.11 μM and SI of 8.59 [70].
Darunavir, which had EC50value of 46.41 μM, CC50 >81 μM and SI >1.75 [70], presented consensus in three of 10 poses generated by docking simulation, with variation of the positions only the sulfonamide and isobutyl groups (oriented toward the entrance of the active site), but it did not show key interactions with the main residues of the catalytic site (Figure 5C). This drug also did not have satisfactory clinical data and has not been approved as a possible COVID-19 treatment [68,69].
These drugs have presented significant and promising results concerning the inhibition of COVID-19 viral replication; however, this was not noted for in vivo or clinical studies. This could be due to the low bioavailability of free drug (not bound to plasma proteins) – that is, in their corresponding pharmacological active forms [69]. Hence, we expect that the seven drugs repurposed here will have an even greater affinity toward inhibition of SARS-CoV-2 Mpro and overcome the drawbacks related to clinical and in vivo studies, given the results we hope to obtain with appropriate assays that will be performed in the future.
Conclusion
In view of the urgent need for a safe and reliable COVID-19 treatment, drug repositioning studies prove to be a quick and important alternative for discovery of drugs that are already approved or regulated and could also act against the new coronavirus. The use of this approach eliminates a long and important step during drug development – pharmacokinetics evaluation – which is responsible for a large number of failures in drug development.
Thus, we have developed and described two systematic VS workflows that present excellent validation results, with the aim of repositioning drugs to act as potential inhibitors of the SARS-CoV-2 Mpro. More specifically, seven workflows were conducted using LBVS approaches, and we ultimately selected seven promising drugs with strong indications of acting against the coronavirus.
Following the same docking protocols adopted in the selection of the seven drugs with repurposing potential, we compared the methodology application of known repurposed drugs – lopinavir, ritonavir and darunavir. These three drugs are described in the literature as potential and expressive inhibitors of the enzyme under study; however, they did not provide clinical improvement in patients hospitalized with COVID-19, which may be due to their low bioavailability in the human body. Furthermore, analysis of the main docking interactions for the seven compounds that we selected have shown more consensual results, pointing to a greater affinity for Mpro than lopinavir, ritonavir and darunavir, which have not shown an equal consensus of poses in docking simulations.
These drugs will be followed by in vitro inhibition tests with the SARS-CoV-2 Mpro and further in vitro assays to reduce viral load in cells infected with the new coronavirus and prove their potential.
Future perspective
The worldwide impact of the COVID-19 pandemic has pushed the development of novel strategies to contain this viral spread. In addition to known vaccination plans that countries have been adopting, it would be worthwhile to determine efficient therapeutic strategies to circumvent viral mortality in serious cases. Thus, drug repurposing is a strategy that could be successfully applied in our work, along with the use of hybrid virtual screening. Hence, we identified 7 known drugs that provide interesting insights for SARS-CoV-2 Mpro inhibition and binding interactions, that shall, after corresponding assays are developed, help this field of drug discovery to evolve.
Summary points.
Hybrid ligand- and structure-based virtual screening
Drug repurposing using a compilation of approximately 8,000 approved drugs
A variety of recently published SARS-CoV-2 crystallographic complexes
Robust validation of both 3D shape-similarity and docking methodologies
Consensus analysis of docking poses from ligand based virtual screening
Seven known drugs studied as potential Mpro inhibitors
Comparison with known drugs repurposed for SARS-CoV-2, such as lopinavir and ritonavir
Further experimental assays will confirm whether these seven drugs have the potential to treat COVID-19
Supplementary Material
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.future-science.com/doi/suppl/10.4155/fmc-2021-0025
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest
- 1.Young BE, Ong SWX, Kalimuddin S et al. Epidemiologic features and clinical course of patients infected with SARS-CoV-2 in Singapore. JAMA. 1–7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet. Infect. Dis. 3099(20), 19–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pang J, Gao S, Sun Z, Yang G. Discovery of small molecule PLpro inhibitor against COVID-19 using structure-based virtual screening, molecular dynamics simulation, and molecular mechanics/Generalized Born surface area (MM/GBSA) calculation. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pacific J. Allergy Immunol. (2020). [DOI] [PubMed] [Google Scholar]
- 5.Arya R, Das A, Prashar V, Kumar M. Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs.. chemrxiv.org. (2020). [Google Scholar]
- 6.Tsai KC, Chen SY, Liang PH et al. Discovery of a novel family of SARS-CoV protease inhibitors by virtual screening and 3D-QSAR studies. J. Med. Chem. 49(12), 3485–3495 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Srinivasan S, Cui H, Gao Z et al. Structural genomics of SARS-CoV-2 indicates evolutionary conserved functional regions of viral proteins. Viruses. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ul Qamar MT, Alqahtani SM, Alamri MA, Chen L-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 10(4), 313–319 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berman HM, Westbrook J, Feng Z et al. The Protein Data Bank. Nucleic Acids Res. 28(1), 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palos I, Lara-Ramirez EE, Lopez-Cedillo JC et al. Repositioning FDA drugs as potential cruzain inhibitors from Trypanosoma cruzi: virtual screening, in vitro and in vivo studies. Molecules. 22(6), (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reddy AS, Pati SP, Kumar PP et al. Virtual screening in drug discovery – a computational perspective. Curr. Protein Pept. Sci. 8(4), 329–51 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Zhou Y, Hou Y, Shen J et al. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 6(1), (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hakmi M, Bouricha EM, Kandoussi I et al. Repurposing of known anti-virals as potential inhibitors for SARS-CoV-2 main protease using molecular docking analysis. 16(4), 301–306 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alves VM, Bobrowski T, Melo-Filho CC et al. QSAR Modeling of SARS-CoV M pro inhibitors identifies sufugolix, cenicriviroc, proglumetacin, and other drugs as candidates for repurposing against SARS-CoV-2. Mol. Inform. 40(1), 2000113 (2021). [DOI] [PubMed] [Google Scholar]; • Good example of Computer-Aided Drug Design (CADD) and Quantitative Structure-Activity Relationship (QSAR) studies performed in the same context.
- 15.Wu C, Liu Y, Yang Y et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Good example of structure-based virtual screening in same context.
- 16.Wishart DS. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 34(90001), D668–D672 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sterling T, Irwin JJ. ZINC 15 – ligand discovery for everyone. J. Chem. Inf. Model. 55(11), 2324–2337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Douguet D. Data sets representative of the structures and experimental properties of FDA-approved drugs. ACS Med. Chem. Lett. 9(3), 204–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu T, Lin Y, Wen X et al. BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 35(suppl. 1), D198–D201 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Boyle NM, Banck M, James CA et al. Open Babel: an open chemical toolbox. J. Cheminform. 3(33), (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.BIOVIA. Dassault Systèmes – Discovery Studio Visualizer. (2017). https://discover.3ds.com/discovery-studio-visualizer-download
- 22.OpenEye. Scientific Software. (2020). www.eyesopen.com
- 23.OpenEye. Scientific Software. OMEGA 4.0.0.4. (2020). www.eyesopen.com [Google Scholar]
- 24.Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 50(4), 572–584 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.OpenEye Scientific Software. ROCS 3.3.1.2. (2018).
- 26.Hawkins PCD, Skillman AGG, Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 50(1), 74–82 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Mesecar AD. Structure of COVID-19 main protease bound to potent broad-spectrum non-covalent inhibitor X77 - 6W63. (2020). [Google Scholar]
- 28.Fearon D, Powell AJ, Douangamath A et al. PDB ID:5R83 PanDDA analysis group deposition – crystal structure of SARS-CoV-2 main protease in complex with Z44592329. (2020). [Google Scholar]
- 29.Turlington M, Chun A, Tomar S et al. Discovery of N-(benzo[1,2,3]triazol-1-yl)-N-(benzyl)acetamido)phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding. Bioorganic Med. Chem. Lett. 23(22), 6172–6177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh AK, Gong G, Grum-Tokars V et al. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorganic Med. Chem. Lett. 18(20), 5684–5688 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madhavi Sastry G, Adzhigirey M, Day T et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 27(3), 221–234 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Maestro Schrödinger L. Schrödinger release 2020–4. (2020). https://www.schrodinger.com/products/maestro
- 33.Verdonk ML, Cole JC, Hartshorn MJ et al. Improved protein–ligand docking using GOLD. Proteins. 52(January), 609–623 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Jones G, Willett P, Glen RC et al. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748 (1997). [DOI] [PubMed] [Google Scholar]
- 35.Shie JJ, Fang JM, Kuo CJ et al. Discovery of potent anilide inhibitors against the severe acute respiratory syndrome 3CL protease. J. Med. Chem. 48(13), 4469–4473 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Jin Z, Du X, Xu Y et al. Structure of Mpro fromSARS-CoV-2 and discovery of its inhibitors. Nature 582(7811), 289– 293 (2020). [DOI] [PubMed] [Google Scholar]; • Representative example; probably one of the first structure (SARS-CoV-2 crystallographic complex) deposited on PDB in 2020.
- 37.Zhang L, Lin D, Kusov Y et al. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. 63(9), 4562–4578 J. Med. Chem. (2020). [DOI] [PubMed] [Google Scholar]
- 38.Jeon S, Ko M, Lee J et al. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 64(7), e00819–e00820 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; • From a previous screen of 3000 approved drugs against SARS-CoV, 50 were selected to perform antiviral activity assays with SARS-CoV-2, finding potential in 24.
- 39.Zhang L, Lin D, Sun X et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 368(6489), 409–412 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galasiti Kankanamalage AC, Kim Y, Damalanka VC et al. Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element. Eur. J. Med. Chem. 150, 334–346 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blanchard JE, Elowe NH, Huitema C et al. High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem. Biol. 11(10), 1445–1453 2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacobs J, Grum-Tokars V, Zhou Y et al. Discovery, synthesis, and structure-based optimization of a series of N-(tert-Butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL. J. Med. Chem. 56(2), 534–546 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramajayam R, Tan KP, Liu HG, Liang PH. Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorganic Med. Chem. 18(22), 7849–7854 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J, Pettersson HI, Huitema C et al. Design, synthesis, and evaluation of inhibitors for severe acute respiratory syndrome 3C-like protease based on phthalhydrazide ketones or heteroaromatic esters. J. Med. Chem. 50 (8), 1850–1864 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of Useful Decoys, Enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 55(14), 6582–6594 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Empereur-Mot C, Zagury JF, Montes M. Screening explorer – an interactive tool for the analysis of screening results. J. Chem. Inf. Model. 56(12), 2281–2286 (2016). [DOI] [PubMed] [Google Scholar]
- 47.OpenEye Scientific Software. vROCS 3.3.2.2. (2020).
- 48.Marchant CA, Briggs KA, Long A. In silico tools for sharing data and knowledge on toxicity and metabolism: Derek for Windows, Meteor, and Vitic. Toxicol. Mech. Methods. 18(2–3), 177–187 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Barber C, Hanser T, Judson P, Williams R. Distinguishing between expert and statistical systems for application under ICH M7. Regul. Toxicol. Pharmacol. 84, 124–130 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Ton AT, Gentile F, Hsing M et al. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol. Inform. 39(8), 1–7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mesecar AD. (CSGID) C for SG of ID.PDB ID: 6W63 structure of COVID-19 main protease bound to potent broad-spectrum non-covalent inhibitor X77. (2020). [Google Scholar]
- 52.OpenEye Scientific Software. EON 2.3.1.2. (2018). www.eyesopen.com
- 53.Shitrit A, Zaidman D, Kalid O et al. Conserved interactions required for inhibition of the main protease of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Sci. Rep. 10(1), 1–11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Describes the similarity between the SARS-CoV and SARS-CoV-2 structures.
- 54.Hall DC, Ji H-F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis. 35, 101646 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hui DS, Azhar E I, Madani TA et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health – the latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 91, 264–266 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rut W, Groborz K, Zhang L et al. Substrate specificity profiling of SARS-CoV-2 main protease enables design of activity-based probes for patient-sample imaging. bioRxiv. (2020) (Epub ahead of print). [Google Scholar]
- 57.Cole JC, Murray CW, Nissink JWM et al. Comparing protein-ligand docking programs is difficult. Proteins Struct. Funct. Bioinforma. 60(3), 325–332 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Hevener KE, Zhao W, Ball DM et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 49(2), 444– 460 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Triballeau N, Acher F, Brabet I et al. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 48(7), 2534–2547 (2005). [DOI] [PubMed] [Google Scholar]
- 60.de Almeida JR, Figueiro M, Almeida WP et al. Discovery of novel dual acetylcholinesterase inhibitors with antifibrillogenic activity related to Alzheimer's disease. Future Med. Chem. 10(9), 1037–1053 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Federico LB, Silva GM, de Fraga Dias A et al. Identification of novel αβ-tubulin modulators with antiproliferative activity directed to cancer therapy using ligand and structure-based virtual screening. Int. J. Biol. Macromol. 165, 3040–3050 (2020). [DOI] [PubMed] [Google Scholar]; • Corroborates the robustness of the protocol used.
- 62.Volkamer A, Kuhn D, Grombacher T et al. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 52(2), 360–372 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Volkamer A, Griewel A, Grombacher T, Rarey M. Analyzing the topology of active sites: on the prediction of pockets and subpockets. J. Chem. Inf. Model. 50(11), 2041–2052 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Bolarin JA, Oluwatoyosi MA, Orege JI et al. Therapeutic drugs for SARS-CoV-2 treatment: current state and perspective. Int. Immunopharmacol. 90, 107228 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Loshak D. No benefit for lopinavir–ritonavir in severe COVID-19. JAMA - J. Am. Med. Assoc. 323(20), 1999 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Cao B, Wang Y, Wen D et al. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med. 382(19), 1787–1799 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giacomelli A, Pagani G, Ridolfo AL et al. Early administration of lopinavir/ritonavir plus hydroxychloroquine does not alter the clinical course of SARS-CoV-2 infection: a retrospective cohort study. J. Med. Virol. 93(3), 1421–1427 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Meyer S, Bojkova D, Cinatl J et al. Lack of antiviral activity of darunavir against SARS-CoV-2. Int. J. Infect. Dis. 97, 7–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smolders EJ, te Brake LH, Burger DM. SARS-CoV-2 and HIV protease inhibitors: why lopinavir/ritonavir will not work for COVID-19 infection. Antivir. Ther. (May), 2–4 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Yamamoto N, Matsuyama S, Hoshino T, Yamamoto6 N. Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro.. bioRxiv. (2020) (Epub ahead of print). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.