

Abstract
Introduction:
Pulmonary invasive mucinous adenocarcinomas (IMA) commonly present with spatially separate lung lesions. Clonal relationship between such lesions, particularly those involving contralateral lobes, is not well established. Here, we used comparative genomic profiling to address this question.
Methods:
Patients with genomic analysis performed on 2 IMA located in different lung regions were identified. Molecular assays included DNA-based next-generation sequencing (NGS) for 410–468 genes (MSK-IMPACT), RNA-based NGS for 62 genes (MSK-Fusion), or non-NGS assays.
Results:
Comparative genomic profiling was performed on 2 separate IMAs in 24 patients, of whom 19 had contralateral lesions. Tumors from all but one patient shared matching driver alterations, including KRAS (n=19), NRG1 (n=2), ERBB2 (n=1) or BRAF (n=1). In addition, in patients with paired tumors profiled by NGS (n=12), shared driver alterations were accompanied by up to 4 (average 2.6) other identical mutations, further supporting the clonal relationship between the tumors. Only in a single patient separate IMAs harbored entirely non-overlapping mutation profiles, supporting clonally unrelated, distinct primary tumors. Notably, in a subset of patients (n=3), molecular testing confirmed a clonal relationship between the original resected IMAs and subsequent contralateral IMA presenting after an extremely long latency (8.1–11.7 years).
Conclusions:
Comparative molecular profiling supports that nearly all separate pulmonary IMA lesions represent intrapulmonary spread arising from a single tumor and documents a subset with a remarkably protracted course of intrapulmonary progression. This study reinforces the unique biology and clinical behavior of IMAs while further highlighting the value of genomic testing for clarifying the clonal relationship between multiple lung carcinomas.
Keywords: Invasive mucinous adenocarcinoma, KRAS, lung, NSCLC, comparative molecular profiling, molecular diagnostics
Introduction
Pulmonary invasive mucinous adenocarcinoma (IMA) – formerly mucinous bronchioalveolar carcinoma – is a highly distinct subtype of lung adenocarcinoma, representing approximately 3–5% of all lung adenocarcinomas1–3. Histologically, the tumor cells are characterized by columnar morphology with abundant mucinous or eosinophilic cytoplasm and small basally located nuclei. These tumors frequently have abundant extracellular mucin and tend to grow along the alveolar walls in a so-called lepidic pattern4. Genomically, IMAs are characterized by activating KRAS mutations in the majority of cases and frequent NRG1 fusions in the remainder5–9. Clinically, IMAs commonly arise in never or light smokers. While these tumors may present as solitary lesions, they have a tendency for multifocality which may involve different lobes10–13. In addition, some patients have a diffuse, consolidative presentation resembling lobar or multilobar pneumonia14–19. In contrast to non-mucinous carcinomas, nodal and distant metastases are rare5.
Biologically, lung carcinomas with spatially separate lesions comprise two distinct processes: one is separate, clonally unrelated primary tumors and the other is intrapulmonary spread of a single tumor, also referred to as intrapulmonary metastasis (IPM). This distinction is critical as it directly impacts tumor staging, prognosis, and treatment decisions. Specifically, separate primary tumors are staged individually, whereas IPMs are staged as T3, T4, or M1a for separate lesions involving same lobe, ipsilateral different lobe, or contralateral lobe, respectively20. Recent studies have highlighted the clinical utility of genomic profiling, particularly next-generation sequencing (NGS), for establishing clonal relationships between lung carcinomas21–25. However, these genomic efforts have been primarily focused on lung adenocarcinomas with non-mucinous histology, which represent the predominant type of lung carcinomas. In a recent study from our group, comprehensive NGS was applied to assess clonal relationships among a large set of lung adenocarcinomas; however, IMAs were entirely excluded given their distinctive clinicopathological features and their unique propensity to form additional lung lesions25.
It is generally presumed that IMAs with consolidative, pneumonic lesions represent intra-pulmonary spread of a single tumor9. However, this has not been confirmed by molecular methods, especially for lesions involving different lung lobes. In addition, clonal relationship between IMAs presenting as discrete, oligo-nodular lesions has not been studied in detail. Here, we present the results of comparative genomic analysis performed to clarify the clonal relationship of IMAs with spatially separate lesions.
Materials and Methods
Study Design
Patients for whom genomic profiling was performed on 2 IMAs during a 14-year period (2007–2020) were identified. The study was approved by the institutional review board at Memorial Sloan Kettering Cancer Center (MSKCC).
Histologic, Radiographic, and Clinical Review
The diagnosis of IMA was confirmed by two pathologists (SY, NR) using the 2015 World Health Organization (WHO) classification4. Clinicopathologic and radiographic data were used to exclude metastasis from non-pulmonary mucinous tumors. Computed tomography (CT) of the chest was reviewed, and the lesions were classified as follows: (i) nodular lesion - a rounded or solid opacity with a clear, well-defined periphery; and (ii) pneumonic lesion - a diffuse consolidation that is involving at least the majority of a single lobe26,27. Metachronous tumors from same patient were enumerated by the chronological order in which they were sampled (e.g., T1 and T2). Clinical information and outcomes data were obtained by review of the electronic medical records.
Genetic Testing
Tumor specimens were analyzed as part of prospective clinical care using a variety of clinically-validated molecular assays in the Molecular Diagnostics Service clinical laboratory as follows: (i) Sanger sequencing of KRAS codons 12 and 13, (ii) mutation screening for 92 mutations in 8 genes, by Matrix-Assisted Laser Desorption/Ionization-Time Of Flight (MALDI-TOF) mass spectrometry (MS) (Sequenom, San Diego, CA) (Supplemental Table 1)28,29, and/or (iii) targeted DNA and RNA-based next-generation sequencing (NGS) using MSK-IMPACT30 and MSK-Fusion31, respectively. In brief, MSK-IMPACT is a hybridization capture-based NGS assay that sequences the entire exons and select introns of 410–468 genes (Supplemental Table 2). MSK-Fusion is an RNA-based NGS assay that utilizes Anchored Multiplex PCR technology for targeted sequencing of 62 genes (Supplemental Table 3), which is performed on all tumors lacking a mitogenic driver by MSK-IMPACT. For cases that were negative for drivers by non-NGS assays, a high-sensitivity locked nucleic acid (LNA)-PCR sequencing was subsequently performed for enhanced detection of KRAS mutations, as previously described32,33. DNA and RNA extractions were performed using standard operating procedures on formalin-fixed paraffin-embedded tissue on tumor specimens with matched blood normal control for MSK-IMPACT.
Clonality Assessment
Somatic mutations and structural variants from paired tumors were compared to determine their clonal relationship as previously described25. In brief, pairs with 2 or more shared alterations by NGS were considered definitely clonal, whereas pairs with entirely non-overlapping, unique mutational profiles by NGS, including distinct driver alterations, were classified non-clonal (separate primaries). Tumors sharing only 1 hotspot driver alteration were classified as likely clonal (see Discussion). As described previously25, manual review of the sequence reads was performed for all tumor pairs to confirm the presence (in clonal pairs) or absence (in non-clonal pairs) of shared mutations at low variant allele frequencies (VAF) (i.e., <5% VAF). In addition, the cases were reviewed manually for silent coding mutations and intronic mutations, and those with high quality read support were included for comparative analysis (Supplemental Table 4).
For tumors classified as clonal, we determined the odds of a full set of shared mutations occurring by chance. This probability of chance co-occurrence was calculated by using IMA-specific mutation prevalence derived from an internal database of IMAs that were sequenced using MSK-IMPACT (n=136)8,34.
Results
Patient and tumor characteristics
We identified 24 patients in whom 2 IMAs, located in different lung regions, were analyzed molecularly. The patient and tumor characteristics are summarized in Table 1. Among the 24 tumor pairs, 11 presented synchronously and 13 metachronously. Nineteen tumor pairs were contralateral, 3 involved an ipsilateral different lobe and 2 were located in the same lobe (the latter were all anatomically separate, discrete nodules). Radiologically, tumor pairs presented as discrete nodules (n=18), pneumonic infiltrate plus an isolated contralateral nodule (n=3) or contralateral pneumonic infiltrates (n=3).
Table 1:
Summary of patient and tumor characteristics
| Patient characteristics | N=24 patients |
|---|---|
| Age (years) | |
| Mean | 64 |
| Range | 30–81 |
| Gender | |
| Female | 15 (63%) |
| Male | 9 (37%) |
| Pack years | |
| Mean | 13 |
| Range | 0–55 |
| Tumor pair characteristics | N=24 pairs |
| Timeline of presentation | |
| Synchronous | 11 |
| Metachronous | 13 |
| Radiologic presentation of tumor pairs | |
| Both nodules | 18 (75%) |
| Contralateral | 13 |
| Ipsilateral different lobe | 3 |
| Ipsilateral same lobe | 2 |
| Pneumonic infiltrate and contralateral nodule | 3 (13%) |
| Both pneumonic infiltrates (contralateral) | 3 (13%) |
| Specimen type of tumor pairs | |
| Both resections | 9 (38%) |
| Resection + biopsy | 9 (38%) |
| Both biopsies | 6 (25%) |
Molecular testing methods
The cohort included a total of 48 individual tumors, of which 29 (60%) were tested by MSK-IMPACT followed by MSK-Fusion if no driver alteration was identified (Tables 2 and 3). The remaining 19 (40%) cases were analyzed using non-NGS assays, including Sanger sequencing for KRAS mutations (n=6) or MALDI-TOF MS for hotspot mutations in major NSCLC drivers (n=13). With this approach, 13 tumor pairs from individual patients (54%) had both tumors sequenced using NGS, 8 pairs (33%) had both tumors profiled using non-NGS assays, and 3 pairs (13%) were tested using a combination of NGS and non-NGS assays.
Table 2:
Clinicopathologic and molecular features of individual tumors
| Pt | Age (yrs) | Sex | Tumor | Presentation and latency (yrs) | Radiology | Size (cm) | Molecular method | MAPK driver | Additional shared alterations (for NGS) | Chance co-occurrence |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 61 | F | T1 | Metachronous 1.1 (C) | Nodule | 1.8 | MALDI-TOF MS# | KRAS G12D | N/A | 0.055 |
| T2 | Nodule | 1.7 | IMPACT | KRAS G12D | ||||||
| 2 | 60 | M | T1 | Metachronous 2.2 (C) | Nodule | 2 | MALDI-TOF MS# | KRAS G12D | N/A | 0.055 |
| T2 | Nodule | 2.5 | IMPACT | KRAS G12D | ||||||
| 3 | 30 | M | T1 | Metachronous 11.7 (C) | Nodule | 3.2 | IMPACT | KRAS G12D | CDKN2A H83Y, TP53 R248Q | 1.5E-09 |
| T2 | Nodule | 2.8 | IMPACT | KRAS G12D | ||||||
| 4 | 61 | F | T1 | Synchronous (C) | Nodule | 3.2 | IMPACT | KRAS G12D | ARID1A M1361Cfs*120 | 3.0E-06 |
| T2 | Nodule | 2 | IMPACT | KRAS G12D | ||||||
| 5 | 77 | M | T1 | Synchronous (C) | Nodule | 0.8 | Sanger | KRAS G12D | N/A | 0.055 |
| T2 | Nodule | 3 | Sanger | KRAS G12D | ||||||
| 6 | 74 | F | T1 | Synchronous (C) | Pneumonic | 6.8 | MALDI-TOF MS | KRAS G12D | N/A | 0.055 |
| T2 | Pneumonic | 8.4 | MALDI-TOF MS | KRAS G12D | ||||||
| 7 | 46 | M | T1 | Synchronous (C) | Nodule | 3.6 | MALDI-TOF MS | KRAS G12D | N/A | 0.055 |
| T2 | Pneumonic | 7.2 | MALDI-TOF MS | KRAS G12D | ||||||
| 8* | 69 | F | T1 | Synchronous (S) | Nodule | 3.2 | IMPACT | KRAS G12D | N/A | N/A |
| T2 | Nodule | 1.6 | IMPACT | KRAS G12A | ||||||
| 9 | 66 | F | T1 | Synchronous (C) | Pneumonic | 5.2 | IMPACT | KRAS G12V | SOX9 H169Pfs*83, RPTOR V971I, RAF1 K367Nfs*8, CDKN2A R80Pfs*65 |
4.7E-19 |
| T2 | Pneumonic | 5.8 | IMPACT | KRAS G12V | ||||||
| 10 | 70 | F | T1 | Synchronous (C) | Nodule | 1.3 | IMPACT | KRAS G12V | SMARCB1 R377C, SMAD4 N107Kfs*3, FYN G275S | 8.7E-15 |
| T2 | Nodule | 1.2 | IMPACT | KRAS G12V | ||||||
| 11 | 66 | F | T1 | Metachronous 2 (ID) | Nodule | 3 | IMPACT | KRAS G12V | TP53 R175H, NOTCH3 A1044T, KMT2C N3388S | 3.5E-14 |
| T2 | Nodule | 1.2 | IMPACT | KRAS G12V | ||||||
| 12 | 66 | M | T1 | Synchronous (C) | Pneumonic | 5.8 | MALDI-TOF MS | KRAS G12V | N/A | 0.055 |
| T2 | Nodule | 3.4 | MALDI-TOF MS | KRAS G12V | ||||||
| 13 | 65 | F | T1 | Metachronous 8.1 (C) | Nodule | 1.4 | Sanger | KRAS G12V | N/A | 0.055 |
| T2 | Nodule | 0.7 | Sanger | KRAS G12V | ||||||
| 14 | 69 | F | T1 | Metachronous 8.6 (C) | Nodule | 3 | MALDI-TOF MS | KRAS G12V | N/A | 0.055 |
| T2 | Nodule | 2 | MALDI-TOF MS# | KRAS G12V | ||||||
| 15 | 61 | M | T1 | Metachronous 9.1 (S) | Nodule | 1.2 | MALDI-TOF MS | KRAS G12V | N/A | 0.055 |
| T2 | Nodule | 0.8 | IMPACT | KRAS G12V | ||||||
| 16 | 75 | M | T1 | Metachronous 1 (C) | Nodule | 3.4 | IMPACT | KRAS G12C | ARID1A G276Efs* 87, BMPR1A P465A, KMT2A I1132V, NKX2–1 Q234L |
1.2E-19 |
| T2 | Nodule | 4.7 | IMPACT | KRAS G12C | ||||||
| 17 | 68 | F | T1 | Metachronous 1.4 (ID) | Nodule | 5.6 | IMPACT | KRAS G12C | SDHB S144N | 7.5E-07 |
| T2 | Nodule | 1.5 | IMPACT | KRAS G12C | ||||||
| 18 | 56 | F | T1 | Metachronous 2.9 (C) | Nodule | 4.5 | MALDI-TOF MS | KRAS G12C | N/A | 0.014 |
| T2 | Nodule | 1.1 | MALDI-TOF MS | KRAS G12C | ||||||
| 19 | 64 | F | T1 | Metachronous 3.6 (C) | Nodule | 2 | Sanger# | KRAS G12A | N/A | 0.0026 |
| T2 | Nodule | 1.1 | Sanger | KRAS G12A | ||||||
| 20 | 59 | F | T1 | Synchronous (C) | Nodule | 1 | IMPACT | KRAS G12R | PRKD1 A137T, ERBB4 T731M | 2.5E-12 |
| T2 | Nodule | 1.6 | IMPACT | KRAS G12R | ||||||
| 21 | 81 | M | T1 | Metachronous 1.1 (C) | Pneumonic | 15.5 | IMPACT, Fusion | CD74-NRG1 | CTNNB1 N387K, SMARCA4 D362N, STK11 c.374+220_c.863–1del, NOTCH3-CNTNAP4 |
4.2E-21 |
| T2 | Nodule | 1.9 | IMPACT, Fusion | CD74-NRG1 | ||||||
| 22 | 74 | M | T1 | Metachronous 2.4 (ID) | Nodule | 7.8 | IMPACT, Fusion | F11R-NRG1 | MAP3K1 c.2179+27A>G | 2.9E-09 |
| T2 | Nodule | 1.2 | IMPACT, Fusion | F11R-NRG1 | ||||||
| 23 | 73 | F | T1 | Synchronous (C) | Pneumonic | 9.8 | IMPACT | BRAF K483E | STK11 Q37*, NKX2–1 K214_Y215delinsN, RBM10 X630_splice | 8.5E-18 |
| T2 | Pneumonic | 9.3 | IMPACT | BRAF K483E | ||||||
| 24 | 48 | F | T1 | Synchronous (C) | Nodule | 3.2 | IMPACT | ERBB2 V658_V659insR | SMAD4 D355Y, RBM10 R251*, NBN V210I | 8.5E-18 |
| T2 | Nodule | 0.8 | IMPACT | ERBB2 V658_V659insR |
Pt=Patient; F=Female; M=Male; Yrs=Years; N/A=Not applicable (refers to comparisons where one or both tumors were tested by non-NGS methods, and therefore status of non-driver genes is not known); C=Contralateral; ID=Ipsilateral different lobe; S=Same lobe; IMPACT=MSK-IMPACT; FUSION=MSK-Fusion;
Initial testing resulted in a false-negative result; the mutation was detected only after using a locked nucleic acid-PCR sequencing.
Indicates the sole patient with clonally-unrelated tumors
Table 3:
Summary of molecular results
| Molecular testing methods | |
|---|---|
| Individual tumors | N=48 tumors |
| NGS assays (MSK-IMPACT, MSK-Fusion) | 29 (60%) |
| Non-NGS assays | 19 (40%) |
| Tumor pairs | N=24 pairs |
| Both tumors tested by NGS assays | 13 (54%) |
| Both tumors tested by non-NGS assays | 8 (33%) |
| NGS and non-NGS assays | 3 (13%) |
| Genetic alterations and clonality assessment | |
| Mitogenic driver alteration per patient | N=24 tumor pairs |
| Matching KRAS mutations | 19 (79%) |
| Matching NRG1 fusions | 2 (8%) |
| Matching BRAF mutations | 1 (4%) |
| Matching ERBB2 insertions | 1 (4%) |
| Each tumor with different KRAS mutation | 1 (4%) |
| Number of additional shared alterations in pairs with matching drivers by NGS | N=12 pairs |
| Range | 1–4 |
| Mean | 2.6 |
| Clonal relationship* | N=24 pairs |
| Definitely clonal | 12 (50%) |
| Likely clonal | 11 (46%) |
| Definitely non-clonal (separate primaries) | 1 (4%) |
| Probability of chance co-occurrence of shared alterations for pairs interpreted as clonally-related | N=23 pairs |
| Pairs with ≥2 shared alterations (definitely clonal) | N=12 |
| Median | 2.2 × 10−14 |
| Range | 4.2 × 10−21 − 3.0 ×10−6 |
| Pairs with a shared driver alteration only (likely clonal) | N=11 |
| Median | 0.055 |
| Range | 0.0026 – 0.055 |
As detailed in the Methods and Results, “definitely clonal” defined as sharing ≥2 mutations, “likely clonal” defined as sharing a single hotspot mutation, and “non-clonal” defined as having distinct driver mutations and lacking any shared mutations.
Landscape of somatic alterations and tumor pair clonality assessment
Molecular testing of 48 individual tumors revealed canonical mitogenic driver alterations and other pathogenic variants predicted to activate the MAPK pathway in all cases (Figure 1). To assess tumor clonal relationships, we compared the genetic profiles of tumors in individual patients. This revealed that with single exception, all tumors in individual patients shared identical driver alterations: KRAS mutations (n=19), NRG1 fusions (n=2), BRAF p.K483E mutation (n=1) and ERBB2 in-frame insertion p.V658_V659insR (n=1). Of 12 tumor pairs with shared drivers analyzed by NGS, all pairs harbored between 1 to 4 (average 2.6) additional shared mutations. Full mutational data are provided in Supplemental Table 4.
Figure 1: Summary of genomic findings.

The top rows show the patient number and the individual tumor designations as T1 and T2. #Initial testing by Sanger sequencing or MALDI-TOF MS resulted in a false-negative result; the mutation was detected only after re-testing with a locked nucleic acid-PCR sequencing. *Additional shared alterations other than the shared pathogenic MAPK alterations as provided in the panel above.
In 4 samples (each from a different patient), initial testing with MALDI-TOF MS (n=3) or Sanger sequencing (n=1) was negative for mutations, while the other tumors in these patients were positive for a KRAS mutation. However, subsequent re-testing using high-sensitivity LNA-PCR revealed pathogenic KRAS mutations in all negative samples. On histologic re-review, tumor cell content was low to borderline (<20% of all nucleated cells) due the abundance of extra-cellular mucin and admixed inflammation.
Based on the approach outlined in the Methods, IMAs with ≥2 shared somatic alterations were classified as definitely clonal (n=12; 50%), tumors with a single shared alteration as likely clonal (n=11; 46%) and a single tumor pair with entirely unique profiles in each tumor lacking any overlapping alterations as non-clonal (4%).
Next, for pairs classified as clonal, we measured the level of support for the clonality classifications based on probability of chance co-occurrence of a full set of shared alterations using IMA-specific prevalence of each alteration (see Methods; Figure 1 and Supplementary Table 5). For tumors with ≥2 shared alterations (n=12; all for paired NGS assays), the probability of chance co-occurrence of a full set of alterations was virtually nil (median: 2.2 × 10−14, range: 4.2 × 10−21 – 3.0 × 10−6). For tumors with only a single shared alteration (n=11; all non-NGS assays), the probability of chance co-occurrence ranged from 0.0026 – 0.055 (0.055 representing a 1 in 18 chance).
In the sole patient with distinct driver mutations, one tumor harbored a KRAS p.G12D and the other, a KRAS p.G12A. In addition, one tumor harbored an additional unique mutation (PDCD1 c.76+114G>A) and the other harbored 2 additional unique mutations (NKX2–1 p.Q350Hfs*88 and IGF1R p.R753Q). Manual review of sequencing reads did not identify any overlap in mutations even at a sub-threshold level (<5% VAF). This patient was a 69-year-old never-smoker with synchronous IMAs (3.2 cm and 1.6 cm) presenting as 2 discrete nodules in the same lobe. There was no evidence of underlying interstitial lung disease or known cancer predisposition syndrome.
Representative radiologic and histologic findings in the analyzed tumor pairs are illustrated in Figure 2. Although the clonally unrelated pair showed some histologic differences (Figure 2E), morphologic heterogeneity was also seen in clonally related tumors (see Figure 2C).
Figure 2: Radiologic, histologic, and genomic findings for select tumor pairs that were analyzed using next-generation sequencing.
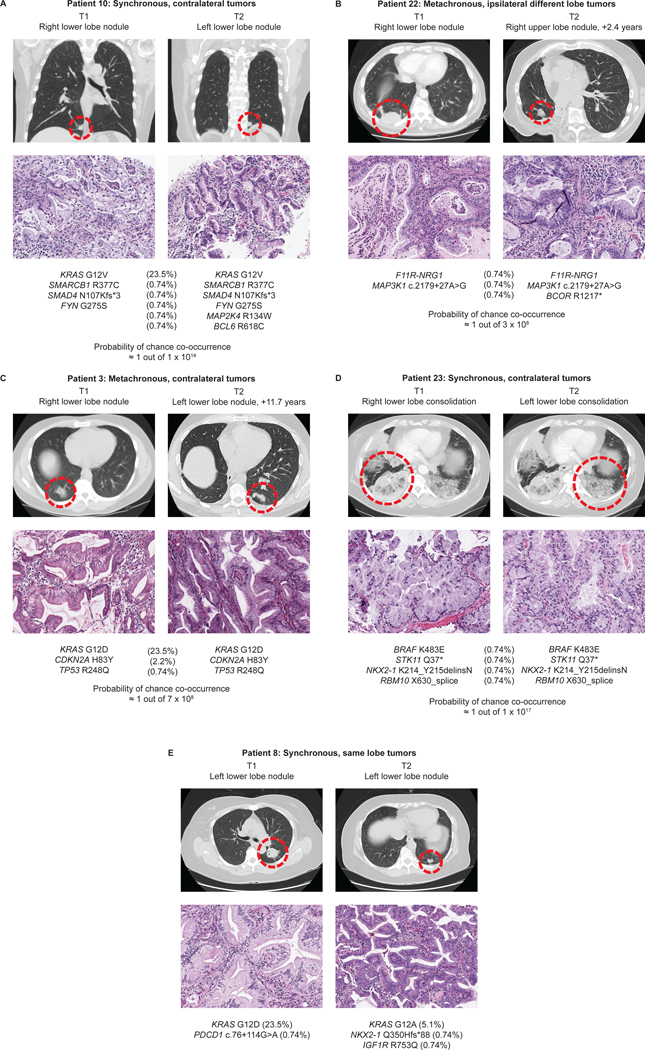
A-D: Tumor pairs classified as clonally related. E: The sole patient in this study with tumor pair classified as clonally unrelated (separate primary IMAs). Histologic sections (20x objective) and the molecular results are provided below. The IMA-specific prevalence is provided for each genetic alteration in parenthesis. Probability of chance co-occurrence is provided for pairs with shared genetic alterations (A-D).
Patient presentation and progression: The description of a subset of clonally related IMAs with an extremely protracted course of intrapulmonary spread.
Next, we reviewed the initial radiologic presentation and subsequent progression of patients with clonally related IMAs (n=23). Figure 3A and Figure 3B summarize the chronological relationship of the index lesions that were genotyped as well as other pulmonary lesions in patient with metachronous and synchronous presentation, respectively. Clinical course of the patients was highly heterogenous, with the presence of pneumonic infiltrates being significantly associated with poor overall survival (Log-rank test, p=0.0007; Figure 3C). Despite confirmed intrapulmonary spread, none of the patients had nodal or distant metastases on presentation or during the available follow-up.
Figure 3: Radiologic presentation and the timeline of progression of clonally-related IMAs.

Progression timeline for patients with metachronous tumors (A) and synchronous tumors (B), and (C) overall survival based on the presence of pneumonic infiltrates. Each row represents individual patients and the course of their disease. The horizontal axis represents time in years. Shading legend: red death of disease (DD), gray death of other causes (DOC), blue no evidence of disease (NED) and yellow alive with disease (AWD). T1 and T2 refer to index lesions that underwent molecular analysis. Superscript values represent additional lesions identified radiologically.
Notably, among the patients with clonally related metachronous lesions, we identified a subgroup with an unusually long time course of intrapulmonary progression. This included 3 patients (P3, P13, P14) in whom resection of the initial IMA was followed by the development of an isolated contralateral IMA 8.1 – 11.7 years later. In one additional patient (P15), an isolated IMA nodule developed after 9.1 years in same lobe away from the resection margin of the prior wedge resection. Clinicopathologically, all subsequent tumors in these 4 patients were favored to represent unrelated new primary tumors given the long time interval. However, molecular profiling supported their clonal relationship. For the patient with an 11.7-year latency between the tumors (P3), paired NGS revealed shared KRAS G12D mutation plus 2 additional shared identical mutations, providing definite support for clonal recurrence (Figure 2C). While the three remaining tumor pairs did not have paired NGS testing on both tumors, they were found to share a KRAS G12V, suggesting that these are clonal (see Discussion). Even after resection of the recurrent lesions, all 4 patients have remained free of disease during up to 4.8 years of follow-up. In another patient (P19), isolated recurrent IMA developed 3.6 years after resection of the initial IMA, and the patient remained disease-free for 8.4 years of follow-up after resection of that lesion. Altogether, the 5 aforementioned patients were thus alive with no evidence of disease 11.9 to 16.4 years after initial IMA resection despite developing molecularly-supported, limited intrapulmonary recurrence.
Discussion
This is the first comprehensive molecular study to date to examine clonal relationships between spatially separate IMA lesions. By performing comparative genomic analysis, we found that in all but one of the 24 patients in this study, these lesions represented intrapulmonary spread of a single tumor despite the contralateral location in the majority of lesions. In addition, we describe remarkable instances of extremely protracted clinical course of molecularly-supported intrapulmonary progression in a subset of patients.
The first major finding in this study is the confirmation that pneumonic IMAs involving different lobes, including contralateral sites, represent intrapulmonary spread of a single tumor. While single lobe involvement by pneumonic IMA is generally presumed to represent the spread of a single tumor, the clonal nature of pneumonic IMAs involving different lobes is a novel observation. The presumption of the clonal nature of pneumonic IMAs in a single lobe has been supported by the histologic hallmark of these tumors to form loosely-interconnected skipping foci surrounding the main tumor, which are thought to arise through aerogenous spread of tumor cells, recently described as spread through air spaces (STAS)14–17. It can be hypothesized that multi-lobar disease, including contralateral spread of IMA, is propagated via floating mucin through major airways and across the carina, in line with the clinical symptom of bronchorrhea in some patients with IMA18,19.
The second major finding relates to IMAs presenting as discrete, isolated synchronous or metachronous nodules, which comprised the majority of cases in this study. Here we demonstrate that such lesions, with only single exception, also represent intrapulmonary spread of the same tumor. Intrapulmonary spread manifesting as a single additional nodule or oligo-nodular disease, in some cases involving contralateral lobes, is not a well-recognized pattern of IMA progression. Here, using molecular methods, we confirm that this pattern in most cases represents a limited, localized form of IMA intrapulmonary spread.
Notably, here we document a remarkable subset of IMA patients (n=4) in whom intrapulmonary spread manifested after an exceptionally long latency (8.1 to 11.7 years). In these patients, initial IMA resection was followed by development of a single new IMA lesion in a different location. For all these patients, a new primary tumor was strongly favored based on clinicoradiologic grounds, given the (i) extremely long latency between tumors, (ii) solitary nature of the second nodule, and (iii) contralateral location in 3 of these patients. In sharp contrast to the clinicopathologic impression, genomic profiling supported tumors being clonal in all cases. We speculate that these remarkable cases represent IMAs with slow growth rate and limited aerogenous tumor spread, leading to the protracted formation of isolated additional nodules. All tumors with such progression harbored KRAS mutations and the initial tumors were fully resected nodules; however, specific genomic or clinicopathologic predictors of such slow-progressive disease are not clear from the available data. To our knowledge, this is the first documentation using molecular techniques of instances of ultra-late IMA recurrence. Importantly, this finding suggests that patients with IMAs may benefit from extended clinical surveillance for recurrence after surgical resection.
In this study, we identified a single outlier case of a patient with two unrelated primary IMAs that presented as synchronous separate nodules in the same lobe. These tumors had entirely non-overlapping mutational profiles with different KRAS drivers (G12D vs. G12A) and additional mutations that were unique to each tumor. Given that the prevalence of IMA is low (~5% of all lung adenocarcinomas), the odds of 2 independent IMAs arising in a single patient by chance is low but not nil. It is known that underlying fibrosing interstitial lung disease (ILD) may predispose to formation of multiple IMAs35,36. However, no background ILD was identified pathologically or on imaging studies for this patient. Yet, the possibility that this patient may have an occult risk factor or subclinical predisposition for developing multiple unrelated IMAs cannot be excluded. Overall, although our data show that nearly all cases of multiple IMAs represent intrapulmonary spread of a single tumor, there are exceptions, and these cases can be clarified only by comparative genomic profiling.
Overall, the molecular landscape of IMAs with spatially separate lesions in our series is consistent with findings from prior studies on the distribution of driver alterations in IMAs in general, which includes KRAS mutations in the major of cases and NRG1 fusions in a minority5–9. We also found activating BRAF mutation p.K483E37 and an in-frame insertion in the ERBB2 transmembrane domain (p.V658_V659insR) that are likely oncogenic8,38,39. Our results indicate that intrapulmonary spread can occur in the context of various driver alterations.
Our study highlights several specific technical challenges for comparative molecular analysis of IMAs. First is related to the low tumor cellularity that is frequently encountered in IMAs. This is typically due to the abundance of mucin, stromal cells, and admixed inflammation in the tumor microenvironment. Thus, IMAs are particularly prone to false-negative results with lower-sensitivity assays such as Sanger sequencing and other multiplexed small gene panels. In our study, false-negative results by initial non-NGS assay led to a false impression of distinct genomic profiles in 4 patients. Yet, on re-testing with higher-sensitivity LNA-PCR sequencing, KRAS mutations were identified in all samples that were negative by lower-sensitivity methods.
Another challenge relates to the use of single gene assays or small hotspot panels. Given the high prevalence of KRAS G12D mutations in IMAs (23.5%)8, the odds of a patient with 2 unrelated, separate primary IMAs harboring KRAS G12D entirely by chance is 5.5% (or 1 in 18). Thus, in our series, analysis by non-NGS assays provided classification only as “likely” clonal for shared prevalent KRAS mutations, whereas paired analysis by broad panel NGS detected multiple shared mutations in all clonally related cases, thus providing a definitive confirmation of clonal relationship.
Despite the clear advantages of NGS for comparative tumor profiling, a unique consideration for IMAs is their overall low prevalence among lung adenocarcinomas (3–5%). Thus, the pre-test probability of second IMA representing a related tumor rather than a new primary is high. Given this consideration and in the context of our findings showing that additional IMA lesions are in almost all cases clonal, we propose that in the setting of multiple IMAs, shared single driver alteration may represent sufficient evidence to support tumor clonality. We note that this contrasts sharply with multiple non-mucinous carcinomas in smokers, which in our recent study had a high chance of incidentally sharing the same KRAS (usually G12C) driver mutation25.
This represents the first comprehensive study to date in which genomic approaches were used to establish clonal relationship in a large series of IMAs with separate pulmonary lesions. Although a number of studies have applied clinical NGS and other sequencing methods to determine clonal relationship between multiple lung cancers21–25, IMAs have been either excluded by study design25, or only a few cases were profiled given their low prevalence40,41. Notably, one prior study has suggested independent origin of different areas of pneumonic IMAs based on discordant TP53 mutations (1/1 patient)42, and another found discrepant results from KRAS Sanger sequencing (1/2 patients)41. Here, we used an extended panel of >400 genes, assays with high analytical sensitivity and a strict definition of clonal relationship to show that multiple IMAs are usually clonally related. Interestingly, among the 12 patients that were classified as definitely clonal by comparative NGS, 4 patients had tumor pairs where one tumor (e.g., T1 or T2) harbored at least one private, non-driver mutation that was not present in the other tumor. The presence of these private mutations does not contradict our assessment of clonality, but rather reflects the concept of branched tumor evolution in which clonal “truncal” mutations are accompanied by divergent “branch” mutations8,43.
Review of the overall presentation and progression in IMA patients with molecularly confirmed intrapulmonary spread in our series highlighted several unique features of this disease. First, we confirm that even in patients with confirmed intrapulmonary spread, IMAs rarely develop nodal or distant metastases, consistent with prior reports6,11. Second, we observed a wide spectrum of disease presentations and clinical outcomes, ranging from extensive to limited lung involvement, including the aforementioned subset with a remarkably slow course of pulmonary spread. Overall, the prognosis of patients with IMAs has been a controversial subject with studies showing a wide range of clinical behaviors5,11,44–46. While our study was not designed to investigate prognostic factors in IMA, we noted that among this cohort of patients with confirmed intra-pulmonary spread, the presence of pneumonic infiltrates was associated with poor outcome, as also suggested in prior studies10,12,26,47.
Our results may help optimize the clinical management of patients with IMAs, who may be candidates for molecular targeted therapies, including investigational agents against tumors with NRG1, KRAS, and ERBB2 alterations8,48. While these alterations were detected in some cases, patients in our cohort did not receive targeted therapies given the relative recency of these investigational treatments. Despite this, our findings offer important insights for future therapy selection and suggest that for patients with unresectable multifocal IMAs, driver alteration at a single site is in most cases representative of other lesions; however, confirmatory testing of an additional site could be considered in candidates for systemic targeted therapies.
Although intrapulmonary “metastasis” is currently the standard terminology to describe intrapulmonary spread of lung carcinomas20, metastatic disease generally implies lymphatic or hematogenous dissemination. Given that for IMAs aerogenous spread is likely the predominant mechanism of dissemination though the lung, “intrapulmonary spread” may be more appropriate terminology, particularly given the apparent indolent nature of limited intrapulmonary progression in some patients.
In conclusion, this study reinforces the concept that IMA represents a unique variant of lung adenocarcinoma unified by a distinct molecular profile and a propensity for intrapulmonary progression. While speculated in the past, our study, for the first time, provides a comprehensive molecular evidence supporting that intrapulmonary spread represents the predominant mechanism of pneumonic interlobar spread and formation of additional isolated nodules in IMAs, including tumors that were suspected to represent separate primaries on clinical grounds. In addition, our data expand on the utility of broad NGS-based testing for clonality assessment of multiple lung carcinomas.
Supplementary Material
Acknowledgements
This work was supported by the National Cancer Institute Cancer Center (core grant P30- CA008748).
Funding: This work was supported by the National Cancer Institute Cancer Center (core grant P30-CA008748).
Footnotes
The authors have no relevant conflicts of interest pertaining to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kadota K, Yeh YC, D’Angelo SP, et al. Associations between mutations and histologic patterns of mucin in lung adenocarcinoma: Invasive mucinous pattern and extracellular mucin are associated with KRAS mutation. Am J Surg Pathol. 2014;38(8):1118–1127. doi: 10.1097/PAS.0000000000000246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Russell PA, Wainer Z, Wright GM, Daniels M, Conron M, Williams RA. Does lung adenocarcinoma subtype predict patient survival?: A clinicopathologic study based on the new international association for the study of lung cancer/American thoracic society/European respiratory society international multidisciplinary lung adenocarcinoma classification. J Thorac Oncol. 2011;6(9):1496–1504. doi: 10.1097/JTO.0b013e318221f701 [DOI] [PubMed] [Google Scholar]
- 3.Rekhtman N, Ang DC, Riely GJ, Ladanyi M, Moreira AL. KRAS mutations are associated with solid growth pattern and tumor-infiltrating leukocytes in lung adenocarcinoma. Mod Pathol. 2013;26(10):1307–1319. doi: 10.1038/modpathol.2013.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. Fourth Edition. Volume 7. International Agency for Research on Cancer; 2015. [DOI] [PubMed] [Google Scholar]
- 5.Shim HS, Mari-Kenudson, Zheng Z, et al. Unique genetic and survival characteristics of invasive mucinous adenocarcinoma of the lung. J Thorac Oncol. 2015;10(8):1156–1162. doi: 10.1097/JTO.0000000000000579 [DOI] [PubMed] [Google Scholar]
- 6.Kadota K, Yeh YC, D’Angelo SP, et al. Associations between mutations and histologic patterns of mucin in lung adenocarcinoma: Invasive mucinous pattern and extracellular mucin are associated with KRAS mutation. Am J Surg Pathol. 2014;38(8):1118–1127. doi: 10.1097/PAS.0000000000000246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez-Cuesta L, Plenker D, Osada H, et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov. 2014;4(4):415–422. doi: 10.1158/2159-8290.CD-13-0633 [DOI] [PubMed] [Google Scholar]
- 8.Chang J, Arcila M, Montecalvo J, et al. Next-Generation Sequencing and Clinicopathologic Analysis of 101 Pulmonary Invasive Mucinous Adenocarcinomas Focusing on Comparison Among Molecular Subtypes. In: Modern Pathology. Vol 31.; 2018:723–763. doi: 10.1038/modpathol.2018.22 [DOI] [PubMed] [Google Scholar]
- 9.Cha YJ, Shim HS. Biology of invasive mucinous adenocarcinoma of the lung. Transl Lung Cancer Res. 2017;6(5):508–512. doi: 10.21037/tlcr.2017.06.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casali C, Rossi G, Marchioni A, et al. A single institution-based retrospective study of surgically treated bronchioloalveolar adenocarcinoma of the lung: clinicopathologic analysis, molecular features, and possible pitfalls in routine practice. J Thorac Oncol. 2010;5(6):830–836. doi: 10.1097/jto.0b013e3181d60ff5 [DOI] [PubMed] [Google Scholar]
- 11.Lee HY, Cha MJ, Lee KS, et al. Prognosis in resected invasive mucinous adenocarcinomas of the lung: Related factors and comparison with resected nonmucinous adenocarcinomas. J Thorac Oncol. 2016;11(7):1064–1073. doi: 10.1016/j.jtho.2016.03.011 [DOI] [PubMed] [Google Scholar]
- 12.Watanabe H, Saito H, Yokose T, et al. Relation between thin-section computed tomography and clinical findings of mucinous adenocarcinoma. Ann Thorac Surg. 2015;99(3):975–981. doi: 10.1016/j.athoracsur.2014.10.065 [DOI] [PubMed] [Google Scholar]
- 13.Nie K, Nie W, Zhang YX, Yu H. Comparing clinicopathological features and prognosis of primary pulmonary invasive mucinous adenocarcinoma based on computed tomography findings. Cancer Imaging. 2019;19(1). doi: 10.1186/s40644-019-0236-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohori NP, Yousem SA, Griffin J, et al. Comparison of extracellular matrix antigens in subtypes of bronchioloalveolar carcinoma and conventional pulmonary adenocarcinoma: An immunohistochemical study. Am J Surg Pathol. 1992;16(7):675–686. doi: 10.1097/00000478-199207000-00006 [DOI] [PubMed] [Google Scholar]
- 15.Gaeta M, Blandino A, Pergolizzi S, et al. Patterns of recurrence of bronchioloalveolar cell carcinoma after surgical resection: a radiological, histological, and immunohistochemical study. Lung Cancer. 2003;42(3):319–326. doi: 10.1016/s0169-5002(03)00362-3 [DOI] [PubMed] [Google Scholar]
- 16.Nguyen E, Hakimi M, Chavoshan B, Stringer W, French SW. Lepidic predominant adenocarcinoma with aerogenous spread of mucin in a young patient - A case report. Exp Mol Pathol. 2014;96(3):400–404. doi: 10.1016/j.yexmp.2014.04.005 [DOI] [PubMed] [Google Scholar]
- 17.Gaikwad A, Souza CA, Inacio JR, et al. Aerogenous Metastases: A Potential Game Changer in the Diagnosis and Management of Primary Lung Adenocarcinoma. Am J Roentgenol. 2014;203(6):W570–W582. doi: 10.2214/AJR.13.12088 [DOI] [PubMed] [Google Scholar]
- 18.Takao M, Takagi T, Suzuki H, et al. Resection of mucinous lung adenocarcinoma presenting with intractable bronchorrhea. J Thorac Oncol. 2010;5(4):576–578. doi: 10.1097/JTO.0b013e3181d3ccdf [DOI] [PubMed] [Google Scholar]
- 19.Popat N, Raghavan N, McIvor RA. Severe bronchorrhea in a patient with bronchioloalveolar carcinoma. Chest. 2012;141(2):513–514. doi: 10.1378/chest.11-0956 [DOI] [PubMed] [Google Scholar]
- 20.Amin M, Edge S, Greene F, et al. AJCC Cancer Staging Manual. 8th ed. Springer International Publishing: American Joint Commission on Cancer; 2017. [Google Scholar]
- 21.Patel SB, Kadi W, Walts AE, et al. Next-Generation Sequencing: A Novel Approach to Distinguish Multifocal Primary Lung Adenocarcinomas from Intrapulmonary Metastases. J Mol Diagnostics. 2017;19(6):870–880. doi: 10.1016/j.jmoldx.2017.07.006 [DOI] [PubMed] [Google Scholar]
- 22.Roepman P, ten Heuvel A, Scheidel KC, et al. Added Value of 50-Gene Panel Sequencing to Distinguish Multiple Primary Lung Cancers from Pulmonary Metastases: A Systematic Investigation. J Mol Diagnostics. 2018;20(4):436–445. doi: 10.1016/j.jmoldx.2018.02.007 [DOI] [PubMed] [Google Scholar]
- 23.Wu C, Zhao C, Yang Y, et al. High discrepancy of driver mutations in patients with NSCLC and synchronous multiple lung ground-glass nodules. J Thorac Oncol. 2015;10(5):778–783. doi: 10.1097/JTO.0000000000000487 [DOI] [PubMed] [Google Scholar]
- 24.Mansuet-Lupo A, Barritault M, Alifano M, et al. Proposal for a Combined Histomolecular Algorithm to Distinguish Multiple Primary Adenocarcinomas from Intrapulmonary Metastasis in Patients with Multiple Lung Tumors. J Thorac Oncol. 2019;14(5):844–856. doi: 10.1016/j.jtho.2019.01.017 [DOI] [PubMed] [Google Scholar]
- 25.Chang JC, Alex D, Bott M, et al. Comprehensive next-generation sequencing unambiguously distinguishes separate primary lung carcinomas from intrapulmonary metastases: Comparison with standard histopathologic approach. Clin Cancer Res. 2019;25(23):7113–7125. doi: 10.1158/1078-0432.CCR-19-1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nie K, Nie W, Zhang YX, Yu H. Comparing clinicopathological features and prognosis of primary pulmonary invasive mucinous adenocarcinoma based on computed tomography findings. Cancer Imaging. 2019;19(1):47. doi: 10.1186/s40644-019-0236-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, Remy J. Fleischner Society: Glossary of terms for thoracic imaging. Radiology. 2008;246(3):697–722. doi: 10.1148/radiol.2462070712 [DOI] [PubMed] [Google Scholar]
- 28.Chitale D, Gong Y, Taylor BS, et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene. 2009;28(31):2773–2783. doi: 10.1038/onc.2009.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rekhtman N, Tafe LJ, Chaft JE, et al. Distinct profile of driver mutations and clinical features in immunomarker-defined subsets of pulmonary large-cell carcinoma. Mod Pathol. 2013;26(4):511–522. doi: 10.1038/modpathol.2012.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng DT, Mitchell TN, Zehir A, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagnostics. 2015;17(3):251–264. doi: 10.1016/j.jmoldx.2014.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benayed R, Offin M, Mullaney K, et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin Cancer Res. 2019;25(15):4712–4722. doi: 10.1158/1078-0432.CCR-19-0225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arcila M, Lau C, Nafa K, Ladanyi M. Detection of KRAS and BRAF mutations in colorectal carcinoma: Roles for high-sensitivity locked nucleic acid-PCR sequencing and broad-spectrum mass spectrometry genotyping. J Mol Diagnostics. 2011;13(1):64–73. doi: 10.1016/j.jmoldx.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nafa K, Hameed M, Arcila ME. Locked nucleic acid probes (LNA) for enhanced detection of low-level, clinically significant mutations. In: Methods in Molecular Biology. Vol 1392. Humana Press Inc.; 2016:71–82. doi: 10.1007/978-1-4939-3360-0_8 [DOI] [PubMed] [Google Scholar]
- 34.Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7(6):596–609. doi: 10.1158/2159-8290.CD-16-1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honda T, Sakashita H, Masai K, et al. Deleterious Pulmonary Surfactant System Gene Mutations in Lung Adenocarcinomas Associated With Usual Interstitial Pneumonia. JCO Precis Oncol. 2018;(2):1–24. doi: 10.1200/po.17.00301 [DOI] [PubMed] [Google Scholar]
- 36.Masai K, Tsuta K, Motoi N, et al. Clinicopathological, Immunohistochemical, and Genetic Features of Primary Lung Adenocarcinoma Occurring in the Setting of Usual Interstitial Pneumonia Pattern. J Thorac Oncol. 2016;11(12):2141–2149. doi: 10.1016/j.jtho.2016.07.034 [DOI] [PubMed] [Google Scholar]
- 37.Dela Cruz FS, Diolaiti D, Turk AT, et al. A case study of an integrative genomic and experimental therapeutic approach for rare tumors: Identification of vulnerabilities in a pediatric poorly differentiated carcinoma. Genome Med. 2016;8(1):116. doi: 10.1186/s13073-016-0366-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ou SHI, Schrock AB, Bocharov E V., et al. HER2 Transmembrane Domain (TMD) Mutations (V659/G660) That Stabilize Homo- and Heterodimerization Are Rare Oncogenic Drivers in Lung Adenocarcinoma That Respond to Afatinib. J Thorac Oncol. 2017;12(3):446–457. doi: 10.1016/j.jtho.2016.11.2224 [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto H, Toyooka S, Ninomiya T, et al. Therapeutic Potential of Afatinib for Cancers with ERBB2 ( HER2 ) Transmembrane Domain Mutations G660D and V659E. Oncologist. 2018;23(2):150–154. doi: 10.1634/theoncologist.2017-0345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy SJ, Harris FR, Kosari F, et al. Using Genomics to Differentiate Multiple Primaries From Metastatic Lung Cancer. J Thorac Oncol. 2019;14(9):1567–1582. doi: 10.1016/j.jtho.2019.05.008 [DOI] [PubMed] [Google Scholar]
- 41.Pagan CA, Shu CA, Crapanzano JP, et al. Synchronous Pulmonary Adenocarcinomas. Am J Clin Pathol. March 2020. doi: 10.1093/ajcp/aqaa023 [DOI] [PubMed] [Google Scholar]
- 42.Nanki N, Fujita J, Hojo S, et al. Evaluation of the clonality of multilobar bronchioloalveolar carcinoma of the lung: Case report. Am J Clin Oncol Cancer Clin Trials. 2002;25(3):291–295. doi: 10.1097/00000421-200206000-00019 [DOI] [PubMed] [Google Scholar]
- 43.Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N Engl J Med. 2017;376(22):2109–2121. doi: 10.1056/nejmoa1616288 [DOI] [PubMed] [Google Scholar]
- 44.Yoshizawa A, Motoi N, Riely GJ, et al. Impact of proposed IASLC/ATS/ERS classification of lung adenocarcinoma: Prognostic subgroups and implications for further revision of staging based on analysis of 514 stage i cases. Mod Pathol. 2011;24(5):653–664. doi: 10.1038/modpathol.2010.232 [DOI] [PubMed] [Google Scholar]
- 45.Warth A, Muley T, Meister M, et al. The novel histologic International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification system of lung adenocarcinoma is a stage-independent predictor of survival. J Clin Oncol. 2012;30(13):1438–1446. doi: 10.1200/JCO.2011.37.2185 [DOI] [PubMed] [Google Scholar]
- 46.Russell PA, Wainer Z, Wright GM, Daniels M, Conron M, Williams RA. Does lung adenocarcinoma subtype predict patient survival?: A clinicopathologic study based on the new international association for the study of lung cancer/American thoracic society/European respiratory society international multidisciplinary lung adenocarcinoma classification. J Thorac Oncol. 2011;6(9):1496–1504. doi: 10.1097/JTO.0b013e318221f701 [DOI] [PubMed] [Google Scholar]
- 47.Shimizu K, Okita R, Saisho S, Maeda A, Nojima Y, Nakata M. Clinicopathological and immunohistochemical features of lung invasive mucinous adenocarcinoma based on computed tomography findings. Onco Targets Ther. 2017;10:153–163. doi: 10.2147/OTT.S121059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drilon A, Somwar R, Mangatt BP, et al. Response to ERBB3-directed targeted therapy in NRG1 -rearranged cancers. Cancer Discov. 2018;8(6):686–695. doi: 10.1158/2159-8290.CD-17-1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.