

SUMMARY
Here we report that MYC rescues early human reprogramming cells from a proliferation pause induced by OCT3/4, SOX2 and KLF4 (OSK). We identified ESRG, as a marker of early reprogramming cells, which turned on as early as day 3 after OSK induction. On day 4, ESRG positive (+) cells converted to a TRA-1–60 (+) intermediate state toward induced pluripotent stem cells. The early ESRG (+) or TRA-1–60 (+) cells showed a proliferation pause due to increased p16 INK4A, cyclin-dependent kinase inhibitor p21, and decreased endogenous MYC caused by OSK. Exogenous MYC did not enhance the appearance of initial reprogramming cells, but reactivated their proliferation and improved reprogramming efficiency. MYC increased expression of LIN41, which potently suppressed p21 post-transcriptionally. MYC suppressed p16 INK4A. These changes inactivated retinoblastoma protein (RB) and reactivated proliferation. The RB-regulated proliferation pause does not occur in immortalized fibroblasts, leading to high reprogramming efficiency even without exogenous MYC.
Keywords: reprogramming, pluripotency, induced pluripotent stem cell, proliferation, senescence, immortalization, MYC, LIN41, post-transcriptional regulation
INTRODUCTION
Enforced expression of the specific transcription factors (i.e., OCT3/4, SOX2, KLF4 and MYC (abbreviated as OSKM)) reprograms the somatic cell fate to pluripotency (Takahashi et al., 2007; Takahashi and Yamanaka, 2006). These factors in concert induce dynamic changes in gene expression, such as the suppression of somatic cell genes and activation of pluripotency-associated genes (Buganim et al., 2013). Initially, all the four factors were thought to be essential for reprogramming. However, exogenous MYC is dispensable, but greatly enhances the efficiency of reprogramming by OSK (Nakagawa et al., 2008; Wernig et al., 2008). Many factors promote OSK-mediate reprogramming, but MYC remains one of the strongest activators of induced pluripotent stem cell (iPSC) generation.
MYC was one of the first proto-oncogenes identified in human more than 30 years ago (Hayward et al., 1981; Vennstrom et al., 1982). Over-expression of MYC is a hallmark associated to up to the 70% of all human cancers (Ciriello et al., 2013; Dang, 2012; Gabay et al., 2014). It belongs to a basic helix-loop-helix leucine-zipper (bHLH-LZ) family of transcription factors and requires a partner protein MAX to bind the canonical E-box elements (CACGTG) or other non-canonical variants (CANNTG) (Blackwell et al., 1990; Blackwood and Eisenman, 1991; Lin et al., 2012). MYC primarily functions as a transcription activator through interaction with other activators, such as histone acetyltransferases and Mediator (Vervoorts et al., 2003). However, MYC can also function as a transcriptional suppressor by interacting with other partners, such as MIZ1 and Polycomb Repressive Complex (PRC) 2 (Haupt et al., 1991; Peukert et al., 1997; van Lohuizen et al., 1991). In addition, MYC may function as a universal amplifier of expressed genes (Lin et al., 2012; Wolf et al., 2015).
MYC is involved in various biological processes. It stimulates the cell cycle by promoting DNA synthesis (Dominguez-Sola and Gautier, 2014). It alters metabolism from oxidative phosphorylation to glycolysis, the latter of which is active in pluripotent stem cells and cancer cells (Cliff et al., 2017; Dang, 2013, 2015; Folmes et al., 2013). MYC also activates non-coding RNAs, including microRNAs (miRNAs), such as miR17–92 expression in mouse embryonic stem cells (ESCs) (Lin et al., 2009; Smith et al., 2010). MYC regulates, and in many cases suppresses, developmental genes, such as GATA6 and HOX genes (Chappell and Dalton, 2013). In addition, MYC affects signaling pathways. For example, MYC suppresses the ERK/MAPK pathway through DUSP2/7, whereas activates Wnt pathway through PRC2 (Chappell et al., 2013; Fagnocchi et al., 2016). Moreover, MYC can induce global epigenomic changes that affect binding of other transcription factors and nuclear architecture (Kieffer-Kwon et al., 2017; Kress et al., 2015).
MYC facilitates early stages of reprogramming, but the molecular mechanism remains controversial (Polo et al., 2012; Soufi et al., 2012; Sridharan et al., 2009). It may enhance early steps of reprogramming by repressing fibroblast-specific gene expression and upregulating the metabolic program of the embryonic state (Sridharan et al., 2009). They also showed that exogenous MYC is required only for the initial five days after OSK induction to promote iPSC generation. Alternatively, MYC may facilitate binding of OSK to their target genes in the first 48 hours of reprogramming (Soufi et al., 2012). Another study showed that MYC, together with KLF4, elicited the first wave of changes during reprogramming, including induction of proliferation, metabolic changes, and loss of genetic program of fibroblasts (Polo et al., 2012). Thus, how MYC promote early stages of reprogramming remains to be determined (Knoepfler, 2008).
In the current study, we utilized three unprecedented opportunities to clarify the role of MYC during reprogramming. Firstly, we identified a specific marker of reprogramming that is activated as early as 3 days after OSKM introduction. This allowed us to examine the role of MYC in the early stages during human iPSC generation. Since only a small fraction of cells that receive OSK(M) can start reprogramming, data would be misleading without purification by the specific marker. Secondly, we generated teratoma-derived fibroblasts (TdFs) that reprogram as efficiently by OSK alone as with OSKM. Exogenous MYC did not further enhance iPSC generation in this line. To our knowledge, this is the only cell line identified for which reprogramming is not enhanced by exogenous MYC. Therefore, TdFs provide a unique tool to help determine the role of MYC. Third, as we previously described, the heterochronic developmental factor LIN41/TRIM71, like MYC, enhances reprogramming when added to OSK (Worringer et al., 2014). Here, we provide a clear molecular mechanism by which LIN41 enhances reprogramming and demonstrate that this function is closely related to MYC as it is a critical downstream effector for MYC-enhancement of reprogramming. Collectively, our data show that MYC promotes human iPSC generation primarily by facilitating the escape of an OSK-induced proliferation blockade in early reprogramming cells by repressing RB, rather than enhancing OSK binding, suppressing fibroblast-specific genes, or inducing metabolic changes.
RESULTS
ESRG Is an Early Marker of Human Cellular Reprogramming
To understand what happens in the initial phase of reprogramming, we tried to identify markers that detect reprogramming cells before TRA-1–60. We analyzed the global gene expression of five human somatic cell types (i.e., dermal fibroblasts (HDFs, mesoderm), adipose-derived mesenchymal stem cells (HAdMSCs, mesoderm), astrocytes (HAs, ectoderm), bronchial epithelial cells (HBECs, endoderm) and prostate epithelial cells (HPrECs, endoderm)) and their TRA-1–60 (+) reprogramming progenies on day 7 after introduction of OSKM (fold-change (FC)>5.0, false discovery rate (FDR)<0.05) using gene expression arrays (Figure 1A). We identified 194 upregulated (Table S1) and 74 downregulated (Table S2) genes that were common in all the five somatic cells. Known early reprogramming markers (e.g., L1TD1, SALL4 and NANOG) and other pluripotency-associated genes (e.g., TGDF1, DNMT3L and ESRG) were among the upregulated genes (Buganim et al., 2012; O’Malley et al., 2013; Takahashi et al., 2014).
Figure 1: Identifying the Early Marker of Reprogramming.
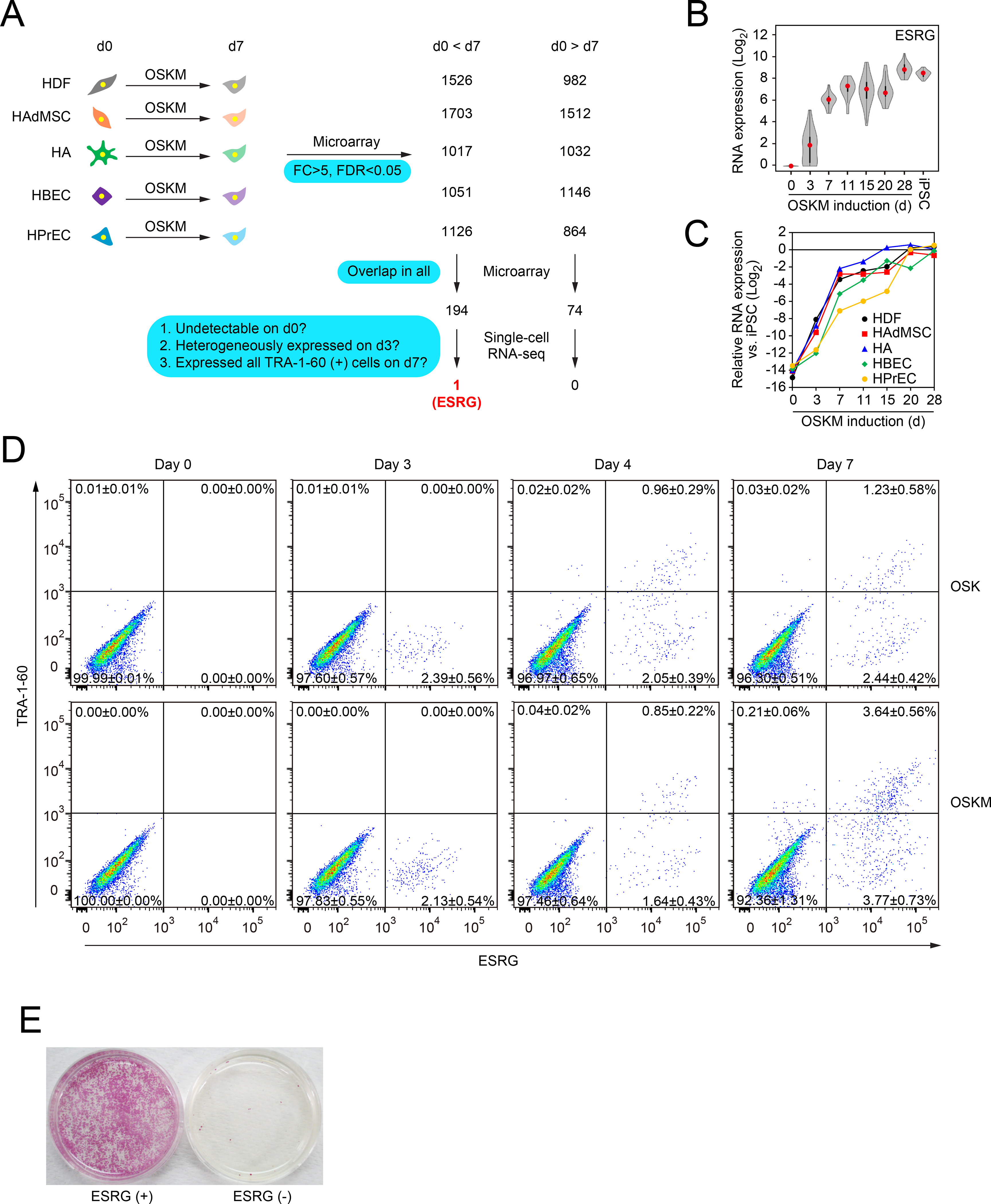
A, Extraction of commonly changed genes and narrowing down the candidate of reprogramming marker. We used microarrays to compare global gene expression of somatic cells (HDFs, HAdMSCs, HAs, HBECs and HPrECs) on day 0 (d0) and their TRA-1–60 (+) progenies on day 7 (d7) post-transduction of OSKM. n=3. By comparing differentially expressed genes between parental cells and TRA-1–60 (+) cells among five cell types, we obtained the indicated common genes. These commonly changed genes were evaluated by single-cell RNA-seq to narrow down the candidates. Blue boxes show the criteria of analyses. See also Tables S1 and S2.
B, Expression of ESRG during reprogramming. Shown are the results of expressing ESRG in parental HDFs (d0), OSKM-expressing cells on day 3 (d3), TRA-1–60 (+) cells at the indicated time points (d7–28), and iPSCs analyzed by single-cell RNA-seq. Twenty-four single cells were analyzed for each time point. Red dots indicate median values. Gray hour-glass shapes represent the distribution of RPKM + 1 value. See also Figure S1.
C, Expression of ESRG during reprogramming. Shown is the expression of ESRG in parental cells (d0), OSKM-expressing cells on day 3 (d3), TRA-1–60 (+) cells at the indicated time points (d7–28) derived from HDFs, HAdMSCs, HAs, HBECs and HPrECs analyzed by microarray. n=3.
D, TRA-1–60 (+) cells emerged only from ESRG (+) cells. Shown are representative histograms of ESRG-Clover (x-axis) and TRA-1–60 (y-axis) expression in ESRG-Clover fibroblasts (day 0) and those on days 3, 5 and 7 post-transduction by OSK or OSKM. n=3. See also Figure S1
E, ESRG (+) cells are origins of iPSCs. We introduced OSKM into ESRG-Clover fibroblasts by retroviral transduction and purified ESRG (+) and (−) cells by cell sorting. Sorted cells (100,000) were plated onto MMC-treated SNL feeder layers. A terminal stain was performed with red alkaline phosphatase on day 24. n=3.
We expected that some of the 194 commonly upregulated genes function as early markers of reprogramming. We rationalized that good markers should fulfill the following criteria: (1) undetectable expression in somatic cells, (2) heterogeneous expression in cells on day 3, and (3) expression in all TRA-1–60 (+) cells on day 7 (Figure 1A). We used single-cell RNA sequencing (RNA-seq) to examine parental HDFs, OSKM-expressing cells on day 3, TRA-1–60 (+) cells on days 7–28, and iPSCs. This analysis revealed that only one gene, ESRG encoding a human endogenous retrovirus type-H (HERV-H)-driven long non-coding RNA (lncRNA), fulfilled all the three criteria (Figures 1A and 1B) (Li et al., 2013). We then examined the expression of ESRG during iPSC generation from the five types of somatic cells and confirmed similar expression patters (Figure 1C). These data show that ESRG functions as an early marker of reprogramming toward human iPSCs regardless of starting somatic cell types.
To better monitor ESRG (+) cells, we generated an iPSC line carrying an ESRG-Clover allele by CRISPR/Cas9-mediated homologous recombination (Figures S1A–S1E). The resulting reporter clone revealed that undifferentiated iPSCs specifically and uniformly expressed Clover fluorescent proteins driven by the endogenous ESRG promoter (Figures S1F–S1H). We transduced OSK or OSKM into fibroblasts derived from ESRG-Clover iPSCs. Before transduction, fibroblasts expressed neither ESRG nor TRA-1–60 (Figures 1D, S1F–S1H). On day 3, we detected 2.39±0.56% and 2.13±0.54% of ESRG-Clover (+) cells after transduction of OSK and OSKM, respectively (Figure 1D). On day 3, we did not detect TRA-1–60 (+) cells, suggesting that ESRG is activated earlier than TRA-1–60 during reprogramming. On day 4, a small number (0.96±0.29% by OSK and 0.85±0.22% by OSKM) of ESRG (+)/TRA-1–60 (+) cells emerged (Figure 1D). On the other hand, ESRG (+) proportion on day 4 did not significantly increase from that on day 3 (2.39% vs 3.01% by OSK and 2.49% vs 2.13% by OSKM) (Figure 1D). In addition, we used cell sorting to purify ESRG (+) cells on day 3. After re-plating, ESRG (+) cells produced many iPSC colonies, whereas ESRG (−) cells resulted in very few (Figure 1E). These data demonstrate that TRA-1–60 (+) intermediate reprogramming cells and iPSCs emerged only from ESRG (+) cells.
Seven days after OSKM induction, the proportion of ESRG (+)/TRA-1–60 (+) cells and ESRG (+)/TRA-1–60 (−) cells rose to 3.64±0.56% and 3.77±0.73%, respectively (Figure 1D). Thus, the ESRG population on day 7 increased threefold from that of day 4. Notably, ESRG (−)/TRA-1–60 (+) cells were practically zero by day 7 (Figure 1D), and OSK induced ESRG (+) cells on day 3 and TRA-1–60 (+) cells on day 4 as comparable to OSKM (Figure 1D). However, without exogenous MYC, the proportion of ESRG (+)/TRA-1–60 (+) cells and ESRG (+)/TRA-1–60 (−) cells remained low on day 7 (1.23±0.58% and 2.44±0.52%, respectively). These data suggest that MYC does not facilitate initial emergence of ESRG and TRA-1–60 (+) cells, but rather promotes expansion of early reprogramming cells between day 4 and day 7.
Early Reprogramming Cells Do Not Proliferate
To understand how MYC promotes expansion of early reprogramming cells, we examined gene expression at a single-cell level 3 days after transduction of OSKM into HDFs. By gene ontology (GO) analysis, genes of which expression levels showed reverse-correlation to ESRG expression were significantly enriched with cell cycle–related terms (Figure 2A and Table S3). The Gene Set Enrichment Analysis (GSEA) indicated that the expression levels of 612 out of 1076 cell cycle–related genes showed reverse correlation to ESRG expression (p=1.95e-10 by Fisher’s exact test, Figure 2B and Table S3). Based on these results, we next examined proliferation profile of early reprogramming cells by means of incorporation of 5-bromo-2-deoxyuridine (BrdU). Three days after OSKM induction, BrdU (+) cells were readily detected in ESRG (−) cells, but not in ESRG (+) cells (Figure 2C). This is also the case with TRA-1–60 (+) cells and TRA-1–60 (−) cells on day 4 (Figure 2D). On the other hand, on day 7, BrdU (+) cells significantly increased in TRA-1–60 (+) cells, whereas decreased in TRA-1–60 (−) cells (Figure 2D). Comparable results were observed in TRA-1–60 (+) cells on days 4 and 7 after OSKM induction into other types of somatic cells, including HAdMSCs, HAs, HBECs and HPrECs (Figure 2E). These data demonstrated that ESRG (+) cells do not proliferate when they emerge at day 3 and convert to TRA-1–60 (+) cells at day 4 after OSKM induction, regardless of the origin of the somatic cells. By day 7 after OSKM transduction, however, early reprogramming cells initiate proliferation.
Figure 2: Cell-Cycle Progression Is Dispensable for Initiating Reprogramming.
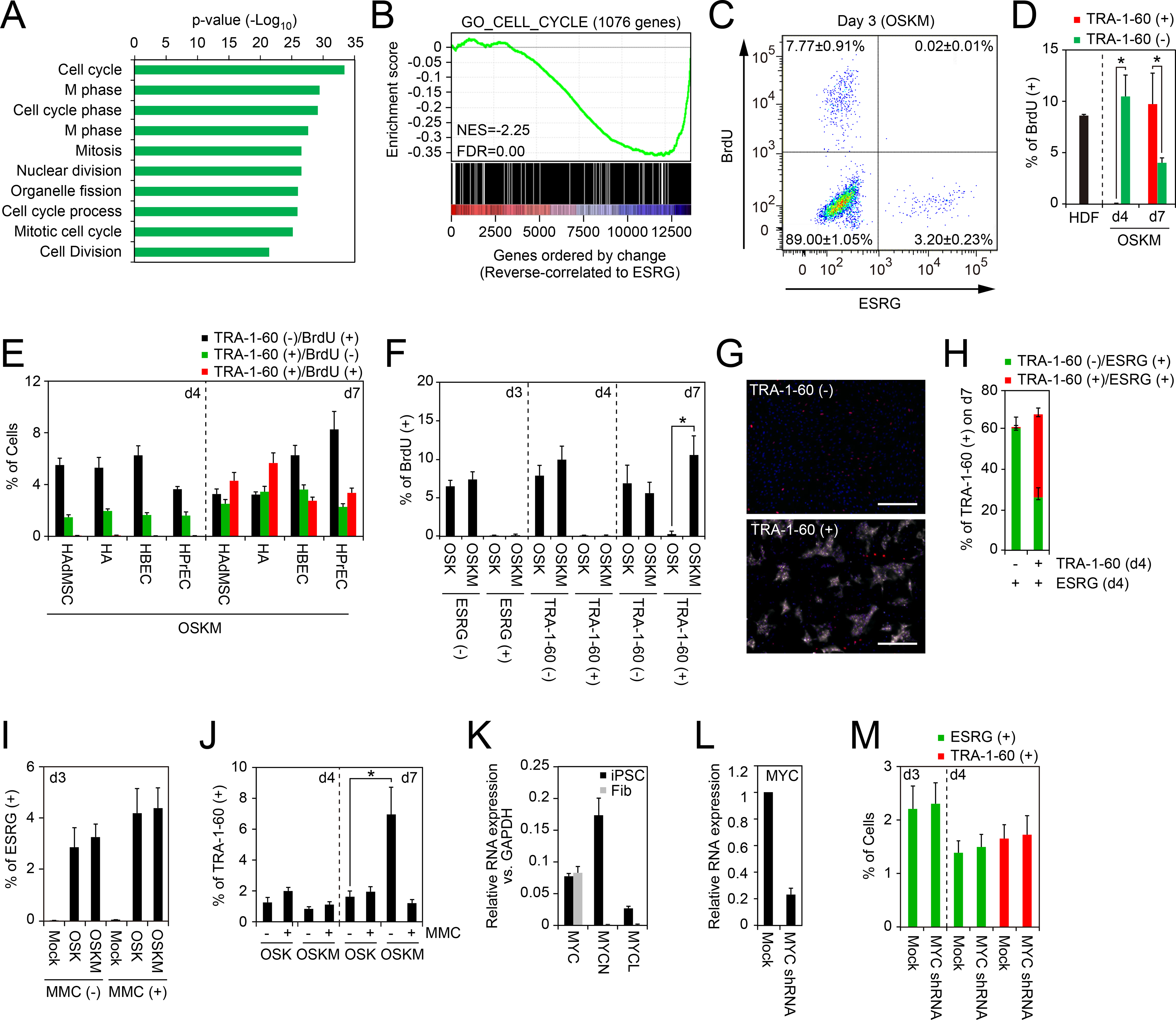
A, Gene ontology analyses of genes varied according to ESRG. Top 200 genes reverse-correlated to ESRG were classified by the gene ontology analysis. The p-values of top 10 enriched terms are shown. See also Table S3.
B, Gene Set Enrichment Analysis showing enrichment of cell-cycle-related genes in the set of genes that show reverse correlation with ESRG expression. X-axis shows genes ranked by Pearson correlation coefficient for ESRG expression. Left, positively correlated genes; right, negatively correlated genes. See also Table S3.
C, Proliferation of ESRG (+) cells. Shown is a representative histogram of the BrdU incorporation of OSKM-transduced ESRG-Clover fibroblasts on day 3. n=3.
D, Proliferation of newly converted TRA-1–60 (+) cells from HDFs. Shown are the percentages of the BrdU incorporation of parental HDFs (d0), TRA-1–60 (+) or (−) cells on days 4 (d4) or 7 (d7). *p<0.05 by unpaired t-test (n=3).
E, Proliferation of newly converted TRA-1–60 (+) cells from other cell types. Shown are the percentages of TRA-1–60 (+)/BrdU (−) (black), TRA-1–60 (+)/BrdU (+) (red) or TRA-1–60 (−)/BrdU (+) cells on days 4 (d4) or 7 (d7). n=3.
F, MYC enhances the proliferation of TRA-1–60 (+) cells. Shown are the percentages of BrdU (+) incorporation in ESRG (+) cells on day 3 (d3) and TRA-1–60 (+) cells on days 4 (d4) or 7 (d7) induced by OSK or OSKM. *p<0.05 by unpaired t-test (n=3).
G, MYC does not enhance the continuity of TRA-1–60 conversion. Shown are representative immunocytochemistry images of the cells on day 7 derived from sorted TRA-1–60 (+) or (−) cells in ESRG (+) population on day 4 post-transduction of OSKM stained with TRA-1–60 (white) and OCT3/4 (red) antibodies. Nuclei were visualized by Hoechst 33342 staining. Bars indicate 100 μm.
H, TRA-1–60 (−) cells on day 4 did not convert to TRA-1–60 (+) fate. Shown are quantitative results of ESRG (+)/TRA-1–60 (+) cells (red) and ESRG (+)/TRA-1–60 (−) cells (green) in the cells shown in Figure 2G.
I, Percentages of ESRG (+) cells with or without mitomycin C treatment. Shown are the percentages of ESRG (+) cells from MMC-treated (+) or non-treated (−) ESRG-Clover fibroblasts on day 3 post-transduction of OSKM. n=3. See also Figure S2.
J, MYC does not affect first-emergence of TRA-1–60 (+) cells. The percentage of TRA-1–60 (+) cells from MMC-treated (+) or non-treated (−) HDFs on days 4 and 7 post-transduction of OSK or OSKM. n=3. See also Figure S2.
K, Expression of MYC family genes in iPSCs and fibroblasts. Shown are relative expression of c-MYC, MYCN and MYCL compared to GAPDH expression in iPSC and fibroblasts (Fib) analyzed by qRT-PCR. n=3.
L, Knockdown of c-MYC in fibroblasts. Shown are the expression of c-MYC in fibroblasts which were introduced with empty vector (Mock) or c-MYC shRNA analyzed by qRT-PCR. n=3.
M, Endogenous MYC does not affect the initiation of reprogramming. Shown are the percentages of ESRG (+) cells (green) on days 3 and 4 and TRA-1–60 (+) cells on day 4 induced by OSK with or without MYC shRNA. n=3.
MYC Promotes Proliferation in Reprogramming Cells
We then determined if proliferation in early reprogramming cells depended on exogenous MYC. We induced OSK or OSKM and examined the proportion of TRA-1–60 (+) cells on days 4 and 7. Notably, the proportions of new TRA-1–60 (+) cells induced by OSK or OSKM on day 4 were comparable (Figure 1D). In contrast, on day 7, we observed increases in the proportion of TRA-1–60 (+) cells with OSKM, but not with OSK (Figure 1D). This tendency correlated well with BrdU incorporation into TRA-1–60 (+) cells (Figure 2F). ESRG (+) cells and TRA-1–60 (+) cells did not incorporate BrdU, except that TRA-1–60 (+) cells on day 7 after OSKM induction, indicating that MYC contributed to the re-establishment of proliferation. To confirm whether MYC promotes proliferation of TRA-1–60 (+) cells rather than conversion to a TRA-1–60 (+) fate, we separated ESRG (+)/TRA-1–60 (+) or ESRG (+)/TRA-1–60 (−) cells on day 4 of reprogramming and analyzed the existence of TRA-1–60 (+) cells on day 7. As a result, ESRG (+)/TRA-1–60 (−) cells on day 4 produced few TRA-1–60 (+) cells by day 7 (Figures 2G and 2H), suggesting that MYC did not promote de novo conversion to TRA-1–60 (+) fate.
Initiation of ESRG (+) and TRA-1–60 (+) Fate Depends on Neither Proliferation nor MYC
The finding that early reprogramming cells stop proliferating prompted us to examine whether proliferation is required for emergence of ESRG (+) and TRA-1–60 (+) cells. To this end, we treated ESRG-Clover fibroblasts and HDFs with mitomycin C (MMC) to arrest their proliferation 1 day after OSK(M) transduction. Counting of treated cells over 7 days showed that MMC treatment resulted in negligible proliferation, without detectable BrdU labeling (Figures S2A and S2B). However, ESRG (+) cells were readily detected even from MMC-treated fibroblasts on day 3 post-transduction of OSK or OSKM (Figure 2I). Interestingly, the proportion of ESRG (+) cells in MMC-treated cells was comparable to that of untreated fibroblasts (Figure 2I). This was also the case with the proportions of TRA-1–60 (+) cells on day 4 (Figure 2J). On day 7, the proportion of TRA-1–60 (+) cells did not increase from day 4 in MMC-treated cells, whereas it increased significantly in OSKM-transduced MMC-untreated cells (Figure 2J). Comparable results were observed with aphidicolin, a reversible inhibitor of eukaryotic nuclear DNA replication and cell-cycle progression (Figures S2C and S2D).
Next, we examined whether endogenous MYC was required for early reprogramming. Among the three MYC family members, including MYC (also known as c-MYC), MYCN and MYCL, we found that only MYC was expressed in fibroblasts (Figure 2K). Thus, we knocked down endogenous MYC using short hairpin RNA (shRNA) in OSK-induced reprogramming (Figure 2L). However, suppression of endogenous MYC altered the emergence of neither ESRG (+) cells on day 3 nor TRA-1–60 (+) cells on day 4 (Figure 2M).
RB Activity Is Crucial for Proliferation Pause in Early Reprogramming Cells
To address mechanisms of the proliferation pause in early reprogramming cells, we analyzed single-cell RNA-seq data to determine if genes that correlated with ESRG expression associated with specific functional pathway(s) (Table S4). We found that the retinoblastoma protein (RB)/E2F pathway was highly associated with genes that reversely correlated with ESRG expression (Figures 3A and S3). Between 6717 genes that reversely correlated to ESRG expression and 4405 genes of which promoter regions were occupied by E2F1, 2339 genes overlapped (p<2.2e-16 by Fisher’s exact test, Figure 3A). Consistently, western blot analyses demonstrated that phosphorylated-RB, an inactive form of RB, increased as reprogramming progressed (Figure 3B). We also found that ESRG (+) cells on day 3 post-transduction of OSK or OSKM highly expressed CDKN1A and INK4A (Figures 3C and S4A). TP53 mRNA, which encodes p53 tumor suppressor, was also induced by OSK or OSKM in ESRG (+) cells (Figures 3C and S4A). The increase of p53, at least in part, contributed to the increase of CDKN1A mRNA (el-Deiry et al., 1993). In addition, we found that OSK suppressed the expression of the endogenous MYC (Figure 3D). All of these changes should contribute to activation of RB protein (Munoz-Espin and Serrano, 2014), sequestration of E2F and decrease of proliferation.
Figure 3: RB Activity Must Be Attenuated for TRA-1–60 (+) Cell Proliferation.
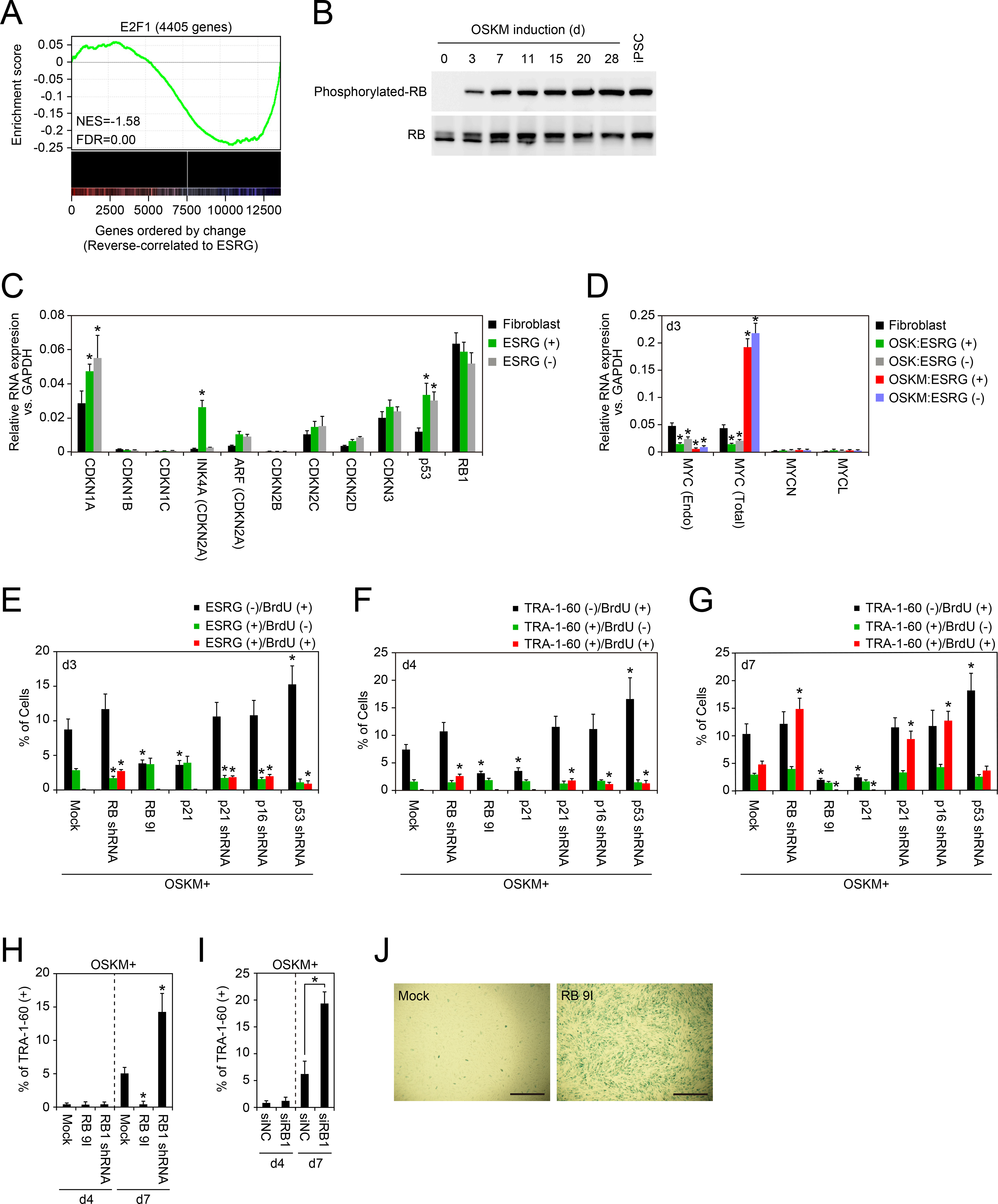
A, Gene Set Enrichment Analysis plot showing enrichment of genes with binding of E2F1 at promoters (±500 bp from transcription start site) in set of genes that show reverse correlation with ESRG expression. X-axis shows genes ranked by Pearson correlation coefficient for ESRG expression. Left, positively correlated genes; right, negatively correlated genes. See also Figure S3 and Table S3 and S4.
B, Phosphorylation statuses of RB protein during reprogramming. Phosphorylated (upper) and total (lower) RB proteins during reprogramming from HDFs to iPSCs induced by OSKM were detected by western blotting.
C, Expression of CDK inhibitors in the early stages of reprogramming. Shown are relative expressions of genes encoding CDK inhibitor proteins in ESRG-Clover fibroblast, ESRG (+) or (−) cells on day 3 post-transduction of OSKM analyzed by qRT-PCR. *p<0.05 vs. fibroblasts by unpaired t-test (n=3). See also Figure S4.
D, OSK suppresses endogenous MYC expression. Shown is the relative expression of endogenous MYC, total MYC, MYCN and MYCL, compared to those of GAPDH in ESRG (+) or (−) cells induced by OSK or OSKM on day 3. *p<0.05 vs. fibroblasts by unpaired t-test (n=3).
E, RB pathway affects proliferation of ESRG (+) cells. Shown are the percentages of ESRG (−)/BrdU (+) cells (black), ESRG (+)/BrdU (−) cells (green) and ESRG (+)/BrdU (+) cells (red) on day 3 post-transduction of OSKM along with each indicated factor to ESRG-Clover fibroblasts. * p<0.05 vs. Mock by unpaired t-test (n=3). See also Figure S4.
F, RB pathway affects proliferation of new TRA-1–60 (+) cells. Shown are the percentages of TRA-1–60 (−)/BrdU (+) cells (black), TRA-1–60 (+)/BrdU (−) cells (green) and TRA-1–60 (+)/BrdU (+) cells (red) on day 4 post-transduction of OSKM along with each indicated factor to HDFs. *p<0.05 vs. Mock by unpaired t-test (n=3). See also Figure S4.
G, RB pathway affects proliferation of TRA-1–60 (+) cells. Shown are the percentages of TRA-1–60 (−)/BrdU (+) cells (black), TRA-1–60 (+)/BrdU (−) cells (green) and TRA-1–60 (+)/BrdU (+) cells (red) on day 7 post-transduction of OSKM along with each indicated factor to HDFs. *p<0.05 vs. Mock by unpaired t-test (n=3). See also Figure S4.
H, Effects of RB on early stage of reprogramming. The graph shows the relative proportion of TRA-1–60 (+) cells on 7 days after transduction of OSKM along with empty vector (Mock), RB 9I or RB1 shRNA to HDFs. RB inactivation enhances the proportion of TRA-1–60 (+) cells. *p<0.05 vs. Mock by Dunnett’s test (n=3). See also Figure S4.
I, Inactivation of RB facilitates expansion of early TRA-1–60 (+) cells. Shown are the effects of RB1 knockdown by siRNA transfection on OSKM-induced TRA-1–60 (+) cell proportion on days 4 (d4) and 7 (d7). *p<0.05 by unpaired t-test (n=3). See also Figure S4.
J, RB activity is associated with senescence in early phase of reprogramming. Representative images of HDFs on 7 days after transduction of OSKM along with empty vector (Mock) or RB 9I with staining for senescence associated β-gal. Bars indicate 200 μm.
To study roles of RB1, p16, p21 and p53 in early reprogramming cells, we suppressed their expression by specific shRNAs. We found that inhibition of each these tumor suppressor genes induced DNA synthesis in ESRG (+) cells on day 3 and TRA-1–60 (+) cells on day 4 as judged by BrdU incorporation (Figures 3E, 3F, S4B and S4C). Also, they facilitated DNA synthesis of TRA-1–60 (+) cells on day 7 (Figures 3G and S4C). These data showed that the three tumor suppressor gene products, p16, p21 and p53, cooperatively activate RB and suppress proliferation. The increased DNA synthesis by RB1 shRNA did not immediately increase the proportion of TRA-1–60 (+) cells on day 4, but it did significantly increase on day 7 after OSK or OSKM transduction (Figures 3H and S4E). We observed comparable results with transfection of siRNA against RB1 (Figures 3I and S4F).
On the other hand, forced expression of constitutive active mutant of RB (RB-9I) or p21 strongly suppressed the proliferation of TRA-1–60 (+) cells on day 7 after OSKM induction (Figures 3E–3G). As a consequence, activation of the RB pathway significantly reduced the proportion of TRA-1–60 (+) cells on day 7 induction of OSKM, but not of OSK (Figures 3G, 3H, S4D and S4E). After transduction of RB-9I along with OSKM, many cells became positive for senescence-associated β-gal (Figure 3J). Importantly, they did not affect the emergence of ESRG (+) cells on day 3 and TRA-1–60 (+) cells on day 4 (Figures 3E, 3F, S4B and S4C). These data suggest that RB does not interfere the emergence of early reprogramming cells, but does inhibit their proliferation and induce senescence.
MYC Activates LIN41, Which Promotes RB Inactivation
To understand the mechanisms of how exogenous MYC attenuates RB activity in early reprogramming cells, we selected 13 pluripotency-associated genes and transcription factors genes (Figure 4A) from the list of genes that we found were commonly upregulated during early reprogramming from five different somatic cells (Figure 1A and Table S1). We examined whether forced expression of these 13 genes increased the proportion of TRA-1–60 (+) cells. As a result, we found that one of them, LIN41 (also known as TRIM71), significantly increased TRA-1–60 (+) cells on day 7 after OSKM transduction (Figure 4A). Although we previously showed that LIN41 enhanced human iPSC generation, its molecular mechanism(s) remains to be determined (Worringer et al., 2014).
Figure 4: LIN41 Enhances Early Reprogramming Process with RB Inactivation.
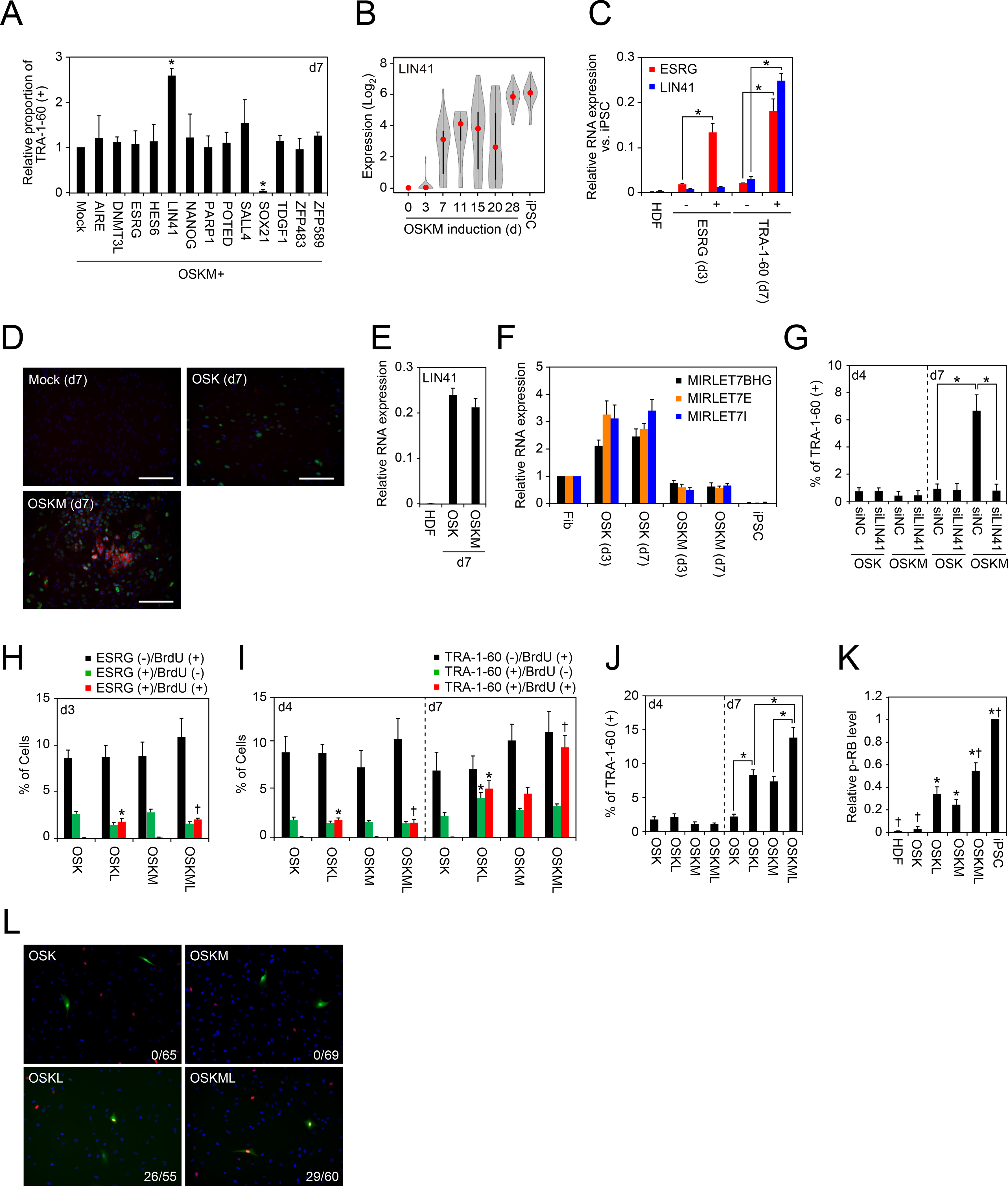
A, LIN41 enhances the proportion of TRA-1–60 (+) cells from HDFs. Shown are relative proportions of TRA-1–60 (+) cells on 7 days after transduction of OSKM along with each of indicated factor into HDFs. *p<0.05 by Dunnett’s test (n=3).
B, LIN41 is activated in TRA-1–60 (+) cells during reprogramming. Shown is the expression of LIN41 during reprogramming from HDFs to iPSCs analyzed by single-cell RNA-seq. Twenty-four single cells were analyzed for each sample. Red dots indicate median values. Gray hour-glass shapes represent the distribution of RPKM + 1 value.
C, LIN41 is silenced in ESRG (+) cells. Shown is the expression of ESRG and LIN41 in HDFs, ESRG (+) or (−) cells on day 3, and TRA-1–60 (+) or (−) cells on day 7 analyzed by qRT-PCR. *p<0.05 by unpaired t-test (n=3).
D, OSKM induces LIN41 protein expression in early stage of reprogramming. Panels show the immunocytochemistry of HDFs transduced with empty vector (Mock, upper left), OSK (upper right) or OSKM (lower left) with LIN41 (red) and OCT3/4 (green) antibodies. Nuclei were visualized by Hoechst 33342 staining. Bars indicate 100 μm.
E, MYC does not affect LIN41 mRNA expression. Shown is the relative expression of LIN41 transcript in TRA-1–60 (+) cells induced by OSK or OSKM on day 7, compared to Mock. n=3.
F, MYC suppresses OSK-induced let-7 expression. Shown is the relative expression of MIRLET7BHG, MIRLET7E and MIRLET7I pre-miRNAs in ESRG (+) cells on day 3 and TRA-1–60 (+) cells on day 7 induced by OSK or OSKM, compared to those in fibroblasts (Fib). *p<0.05 vs. fibroblast by unpaired t-test (n=3).
G, LIN41 is required for efficient reprogramming in early phase. Shown are the effects of LIN41 knockdown by siRNA transfection on OSK or OSKM-induced TRA-1–60 (+) cell proportion on days 4 (d4) and 7 (d7). *p<0.05 by unpaired t-test (n=3).
H, LIN41 affects proliferation of ESRG (+) cells. Shown are the percentages of ESRG (−)/BrdU (+) cells (black), ESRG (+)/BrdU (−) cells (green) and ESRG (+)/BrdU (+) cells (red) on day 3 post-transduction of OSK or OSKM along with empty vector (Mock) or LIN41 to ESRG-Clover fibroblasts. *p<0.05 vs. OSK and †p<0.05 vs. OSKM by unpaired t-test (n=3). OSKM data are identical to Mock in Figure 3E.
I, LIN41 affects proliferation of TRA-1–60 (+) cells. Shown are the percentages of TRA-1–60 (−)/BrdU (+) cells (black), TRA-1–60 (+)/BrdU (−) cells (green) and TRA-1–60 (+)/BrdU (+) cells (red) on days 4 and 7 post-transduction of OSK or OSKM along with empty vector (Mock) or LIN41 to HDFs. *p<0.05 vs. OSK and †p<0.05 vs. OSKM by unpaired t-test (n=3). OSKM data are identical to Mock in Figure 3F and 3G.
J, LIN41 facilitates expansion of early TRA-1–60 (+) cells. Shown are the effects of LIN41 overexpression on OSK or OSKM-induced TRA-1–60 (+) cell proportion on days 4 (d4) and 7 (d7). *p<0.05 by unpaired t-test (n=3).
K, LIN41 increases phosphorylation states of RB. Shown are the relative intensities of phosphorylated-RB in HDFs, iPSCs and HDFs expressing OSK, OSKL, OSKM or OSKML on day 4 by western blotting. *p<0.05 vs. OSK and †p<0.05 vs. OSKM by t-test (n=3). See also Figure S5.
L, Status of phosphorylated-RB in ESRG (+) cells. Shown are representative images of ESRG (+) cells (green) after 3 days of OSK, OSKL, OSKM or OSKML transduction stained with phosphorylated-RB (red) antibody. Nuclei (blue) were visualized by Hoechst 33342 staining. Indicated number at lower right corner of each panel is phosphorylated-RB (+)/ESRG (+) cells.
Single-cell RNA-seq revealed that endogenous LIN41 expression was detected only in TRA-1–60 (+) cells (Figure 4B). Compared to the expression pattern of ESRG (Figure 1B), the expression of LIN41 on day 3 after OSKM transduction was lower. To confirm this, we examined the expression of LIN41 and ESRG in ESRG-Clover (+) cells on day 3 and TRA-1–60 (+) cells on day 7 after OSKM transduction (Figure 4C). We found that LIN 41 expression remained low on day 3, but increased on day 7. Immunocytochemistry detected endogenous LIN41 proteins 7 days after transduction with OSKM but not OSK (Figure 4D). On the other hand, both OSK and OSKM increased LIN41 mRNA to comparable levels on day 7 (Figure 4E). We also found that let-7 family miRNAs, which target LIN41 mRNA to inhibit translation, were suppressed by MYC (Figure 4F) (Chang et al., 2008). These data showed that OSK induces LIN41 mRNA, but without MYC, LIN41 protein cannot be produced due to let-7.
Knockdown of endogenous LIN41 did not change the proportion of new TRA-1–60 (+) cells on day 4 but canceled MYC-induced expansion of TRA-1–60 (+) cells on day 7 (Figure 4G). In contrast, LIN41 knockdown did not affect the proportion of TRA-1–60 (+) cells induced by OSK on days 4 and 7 (Figure 4G), which was reasonable since LIN41 protein was not expressed in these early reprogramming cells after OSK transduction. These data suggest that induction of LIN41 is responsible for the expansion of TRA-1–60 (+) cells by MYC.
We then examined the effect of exogenous LIN41 on early reprogramming cells. When transduced together with OSK or OSKM, LIN41 induced proliferation of ESRG (+) cells on day 3 (Figure 4H) and TRA-1–60 (+) cells on day 4 (Figure 4I), as judged by BrdU incorporation. This is in great contrast to MYC, which did not induce DNA synthesis on day 3 and 4 when co-transduced with OSK. On the other hand, on day 7, both LIN41 and MYC promoted cell proliferation (Figure 4I) and increased the proportion (Figures 4J) of TRA-1–60 (+) cells. These data suggest that activation of LIN41 protein expression induced by MYC promotes the expansion of early TRA-1–60 (+) cells.
Next, to confirm whether LIN41 contributes to inactivation of RB activity, we checked the phosphorylation statuses of RB in HDFs transduced with OSK and OSKM with or without LIN41. We found that OSKM but not OSK caused the increase of phosphorylated-RB in HDFs albeit at a low efficiency (Figures 4K and S5). Noteworthy, exogenous LIN41 with either OSK or OSKM significantly increased phosphorylated-RB (Figures 4K and S5). Furthermore, immunohistochemistry detected many ESRG (+) cells on day 3 were positive for phosphorylated-RB when exogenous LIN41 was added to OSK or OSKM (Figure 4L). In sharp contrast, we did not detect phosphorylated-RB in ESRG (+) cells without LIN41. Thus, LIN41 promotes phosphorylation of RB in early reprogramming cells.
LIN41 Suppresses p21 Protein Expression
To better understand how LIN41 contributes to RB inactivation, we searched for LIN41’s target RNAs by immunoprecipitation of endogenous LIN41 from human ESCs. As a result, we found that CDKN1A mRNA encoding p21 was the most highly enriched among LIN41-bound mRNAs (Figure 5A and Table S5). Other RB-related genes, including RB1 and INK4A, were not found as LIN41’s targets. In TRA-1–60 (+) cells on day 7, exogenous LIN41 with OSK or OSKM significantly lowered the p21 protein level (Figure 5B). Notably, MYC induced LIN41 protein expression and lowered p21 protein levels, albeit with lower efficiency than exogenous LIN41 (Figure 5B).
Figure 5: LIN41 Post-Transcriptionally Suppresses p21.
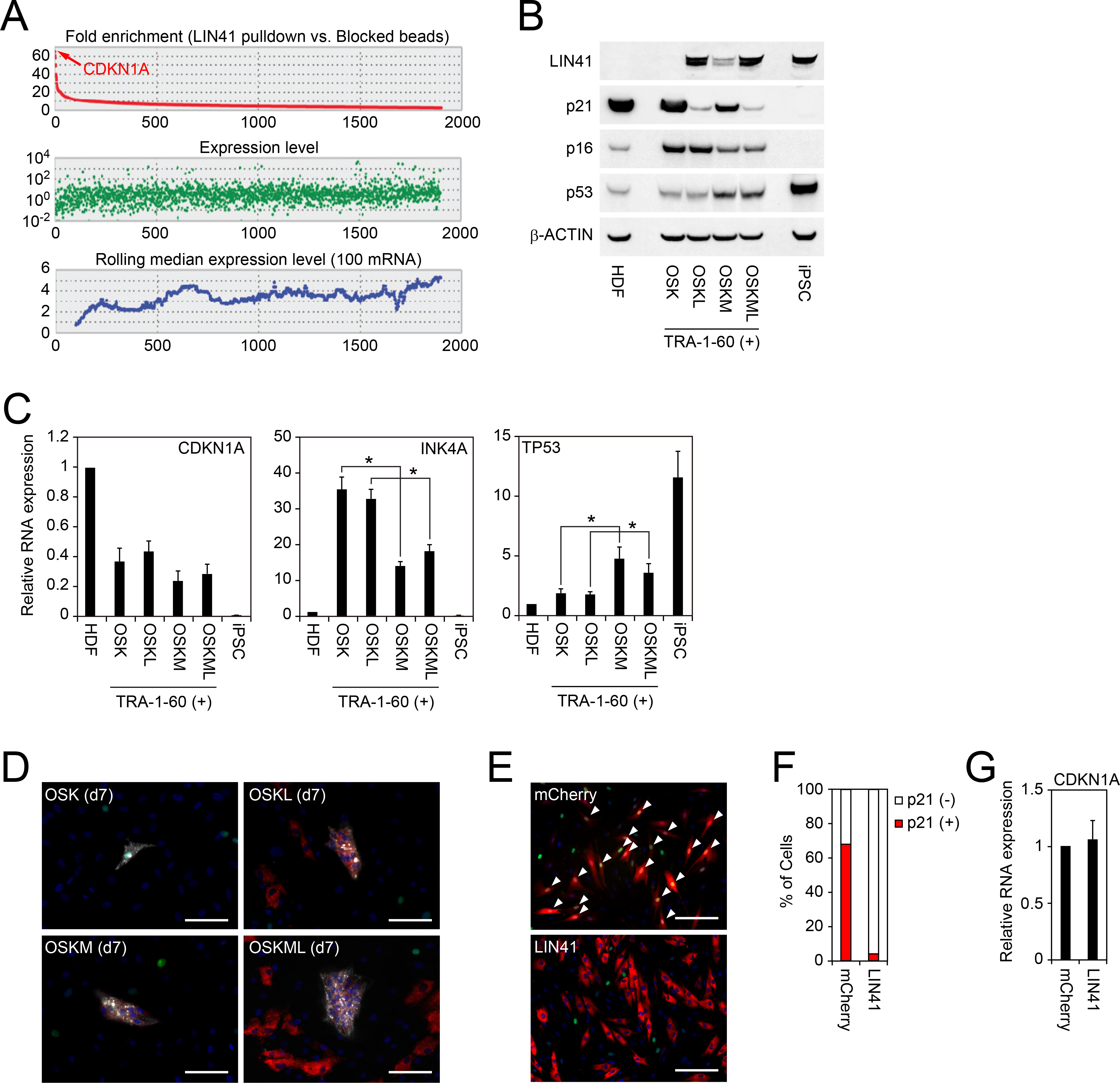
A, LIN41-interacting RNAs. Upper panel shows fold enrichment of precipitated RNAs by LIN41 pulldown compared to those by beads blocked with LIN41 peptides. Also, RNA expression levels (middle) and rolling median expression levels (lower) are shown. n=3. See also Table S5.
B, Expression of LIN41, p21 and p16 proteins in TRA-1–60 (+) cells on day 7. Western blot showing expression of LIN41, p21, p16, p53 and β-ACTIN proteins in HDFs, TRA-1–60 (+) on day 7 post-transduction of OSK, OSKL, OSKM or OSKML, and iPSCs.
C, Expression of CDKN1A, INK4A and TP53 mRNAs in TRA-1–60 (+) cells on day 7 analyzed by qRT-PCR. *p<0.05 by unpaired t-test (n=3).
D, Shown are representative immunocytochemistry images of TRA-1–60 (+) cells on day 7 post-transduction of OSK, OSKL, OSKM or OSKML stained with TRA-1–60 (white), LIN41 (red) and p21 (green) antibodies. Nuclei were visualized using Hoechst 33342 (blue). Bars indicate 25 μm.
E, LIN41 suppresses p21 protein expression in reprogramming intermediates. Shown are representative images of immunocytochemistry of p21 (green) in HDFs expressing mCherry (red in left) or LIN41 (red in right). Nuclei were visualized using Hoechst 33342 (blue). Bars indicate 100 μm. White arrowheads indicate the cells which express both transgene and p21.
F, LIN41 suppresses the expression of p21 protein. The graph shows the quantitative results of Figure 5E. n=50.
G, LIN41 does not alter the expression of CDKN1A mRNA. Shown are relative expression of CDKN1A in HDFs expressing mCherry or LIN41. n=3.
We hypothesized that LIN41, which is an RNA-binding protein, regulated p21 protein levels rather than transcription in early stage of reprogramming. In accordance with this hypothesis, LIN41 did not affect CDKN1A mRNA expression (Figure 5C). Immunocytochemistry of cultures on day 7 post-transduction of various combination of reprogramming factors, such as OSK, OSKL, OSKM and OSKML, showed a few in which LIN41 and p21 were merged (Figure 5D). In addition, immunohistochemistry with anti-p21 antibody showed that forced expression of LIN41 alone in HDFs was sufficient to eliminate endogenous p21 proteins, although CDKN1A mRNA level was not changed by LIN41 (Figures 5E–5G). These data suggest that CDKN1A mRNA is a direct target of LIN41 in human cells rather than an ESC-specific miRNA-dependent mechanism (Chang et al., 2012).
MYC Suppresses p16, Another Mechanism of RB Inactivation
In addition to LIN41-mediated p21 suppression, we found that MYC downregulated p16 expression, which was induced by OSK (Figure 5B). In contrast to the post-transcriptional regulation of p21 encoded by CDKN1A, MYC downregulated the RNA levels of INK4A in early reprogrammed cells (Figure 5C). LIN41 affected neither p16 protein nor INK4A mRNA levels (Figures 5B and 5C). These data suggest MYC suppressed RB activity via dual inhibition of p16 and p21 by different modes of actions.
Immortalization Bypasses the Proliferation Pause
We found that a fibroblast line established from an iPSC-derived teratoma (teratoma-derived fibroblasts: TdFs) is different from HDFs in that the efficiency of iPSC generation by OSK alone was significantly higher (Figure 6A). MYC did not further increase OSK-mediated iPSC generation from TdFs (Figure 6B). The MYC-independency of TdFs gave us an unprecedented opportunity to elucidate roles of MYC in promoting OSK-mediated reprogramming from non-immortalized cells.
Figure 6: Immortalization Bypasses the Proliferation Pause.
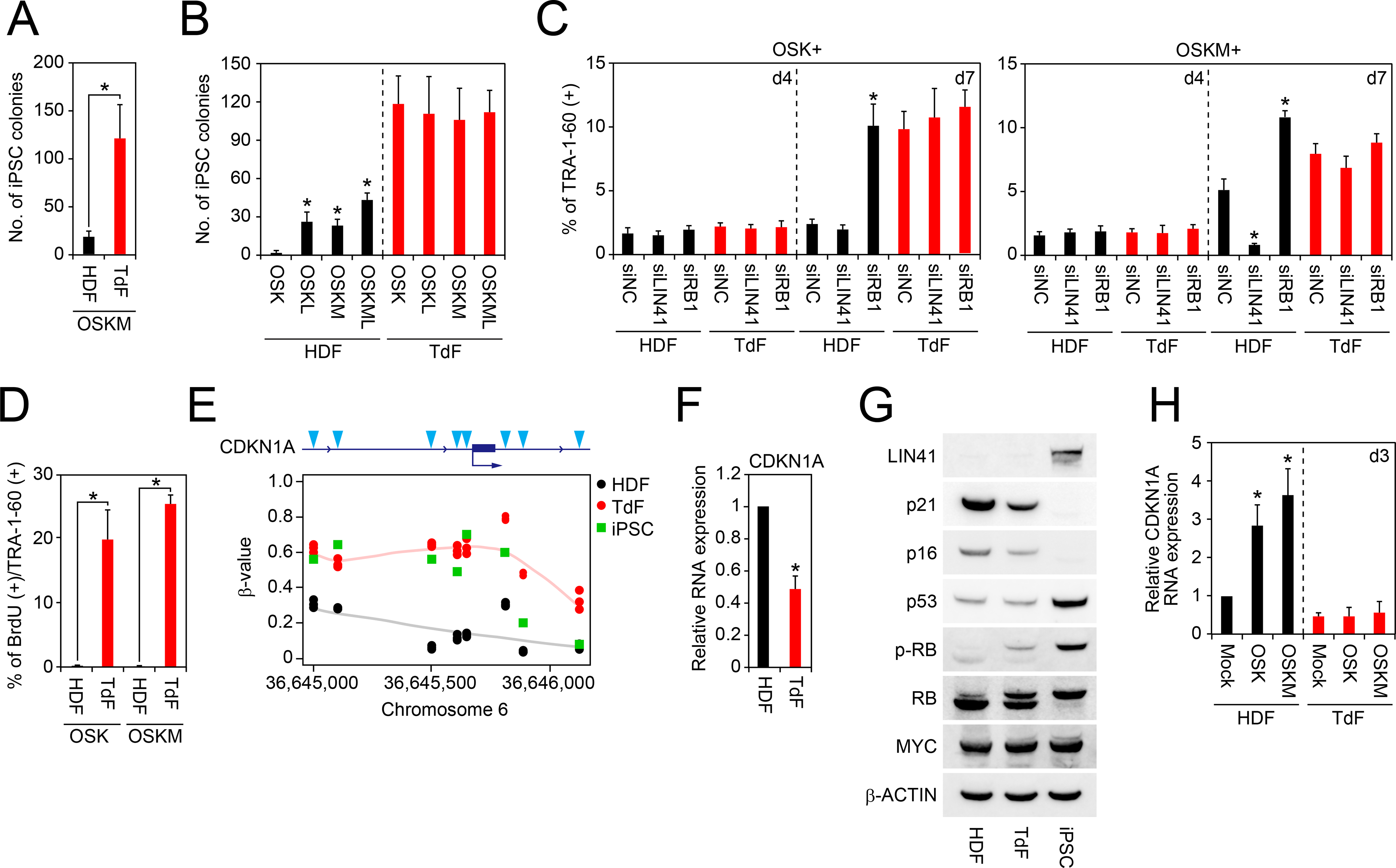
A, The number of iPSC colonies from HDFs and TdFs. Shown are number of iPSC colonies on day 24 derived from 5 × 104 cells of OSKM-transduced HDFs and TdFs. *p<0.05 by unpaired t-test (n=3).
B, Effects of exogenous MYC and LIN41 on iPSC generation from HDFs or TdFs. Shown are the number of iPSC colonies from HDFs or TdFs on day 24 post-transduction of OSK, OSKL, OSKM or OSKML. *p<0.05 by Dunnett’s test (n=3).
C, Effects of LIN41 or RB1 knockdown on the TRA-1–60 (+) cell proportion from HDFs or TdFs. Shown are the effects of LIN41 or RB1 knockdown by siRNA transfection on OSK or OSKM-induced TRA-1–60 (+) cell proportion from HDFs or TdFs on days 4 and 7. *p<0.05 by Dunnett’s test (n=3).
D, BrdU incorporation of newly converted TRA-1–60 (+) cells from TdFs or HDFs. Shown are the percentage of the BrdU incorporation of TRA-1–60 (+) cells from HDFs or TdFs on day 4 post-transduction of OSK or OSKM. *p<0.05 by unpaired t-test (n=3).
E, DNA methylation status at upstream region of CDKN1A gene. Shown are β-value of DNA methylation in each CpG site (blue arrowheads) at the upstream region of CDKN1A gene in HDFs (black), TdFs (red) and iPSCs (green) analyzed by Infinium. n=3.
F, Expression of CDKN1A in HDFs and TdFs. Shown is the relative expression of CDKN1A in HDFs and TdFs analyzed by qRT-PCR. *p<0.05 vs. HDFs by unpaired t-test (n=3).
G, Expression of cell-cycle-related proteins in HDFs, TdFs and iPSCs. Western blot showing expression of LIN41, p21, p16, p53, MYC and β-ACTIN proteins in HDFs, TdFs and iPSCs.
H, No induction of CDKN1A by OSKM in TdFs. Shown is the relative expression of CDKN1A mRNA in HDFs (black) or TdFs (red) on day 3 post-transduction of empty vector (Mock), OSK or OSKM. *p<0.05 by Dunnett’s test (n=3).
We found that, even in TdFs, the proportion of TRA-1–60 (+) cells at day 4 was low (~2%) and comparable to that of HDFs (Figure 6C). This showed that the initial appearance of reprogramming cells is not responsible for the high reprogramming efficiency of TdFs. However, ~20% of TRA-1–60 (+) cells from TdFs showed BrdU incorporation on day 4 after OSK or OSKM (Figure 6D). This resulted in a significant increase of TdFs-derived TRA1–60 (+) cells to ~10% on day 7 (Figure 6C). Of note, inhibiting endogenous LIN41 or RB1 did not affect the proportion of TdFs-derived TRA-1–60 (+) cells on day 7 (Figure 6C). These data showed that TdFs do not require MYC to overcome an RB-mediated proliferation pause in early reprogramming cells.
To clarify why TdFs bypass the proliferation pause, we analyzed DNA methylation and gene expression in HDFs and TdFs. CpG sites at the upstream region of CDKN1A gene in TdFs and in iPSCs were hyper-methylated, compared to those in HDFs (Figure 6E). In parallel, expression of CDKN1A mRNA and p21 protein in TdFs was significantly lower than that in HDFs (Figures 6F and 6G). Furthermore, OSK or OSKM transduction significantly increased CDKN1A expression on day 3 in HDFs, but not in TdFs (Figure 6H). We also detected lower p16 and higher phosphorylated-RB expression in TdFs than in HDFs (Figure 6G). On the other hand, c-MYC and p53 protein expression in TdFs was comparable to those in HDFs (Figure 6G). In addition, TdFs were resistant to premature senescence induced by forced-expression of constitutive active mutant of HRas (HRasV12, mutated Gly12 to Val) or RB 9I (Figures S6A and S6B). These data demonstrated that constitutive inactivation of p21, p16, and RB in TdFs contributes to MYC independency.
We next used TdFs to examine other MYC functions that had been reported as mechanisms to promote reprogramming, including changing metabolisms (Folmes et al., 2013), facilitating OSK binding to their targets (Soufi et al., 2012), and suppressing fibroblast-specific genes (Li et al., 2010; Nakagawa et al., 2010). We cultured HDF, TdFs, and iPSCs in culture media containing 2-deoxy-D-glucose (2-DG), a potent blocker of glycolytic metabolism (Woodward and Hudson, 1954). In this condition, iPSCs did not survive since they largely depend on glycolytic metabolism (Figure S6C). In sharp contrast, both HDFs and TdFs proliferated normally. In addition, we examined expression levels of oxidative phosphorylation markers (e.g., ACO1, IDH2, MDH1, and SDHD), and glycolysis markers (e.g., ENO3, PYGM, SLC16A1, and SLC2A4) (Varum et al., 2011) by qRT-PCR. We found that TdFs and HDFs share similar expression patterns, indicative of oxidative metabolism (Figure S6D). Thus, it is unlikely that metabolic effects of MYC play a major role in promoting iPSC generation.
We next examined whether OSK binding sites are more accessible in TdFs than in HDFs. To this end, we analyzed DNA methylation signatures of CpG islands of OSK binding sites (Figure S6E). We found no evidence that these sites were open, as characterized by lower DNA methylation. Indeed, TdFs showed slightly, but significantly higher DNA methylation for all the three transcription factors, including OCT3/4, SOX2, and KLF4. In addition, the expression levels of OSK-binding genes were comparable in TdFs and HDFs (Figure S6F). Thus increase in OSK biding is not responsible for the high reprogramming efficiency of TdFs.
Finally, we examined the expression levels fibroblast-specific genes. We selected 2449 genes of which expression levels were higher at least 5 fold in HDFs than in iPSCs. RNA seq analyses showed that the expression levels of these genes were significantly lower in TdFs than in HDFs, but still much higher than in iPSCs (Figure S6G). This result suggest that the suppression of fibroblast-specific genes may also contribute to the high reprogramming efficiency of TdFs.
DISCUSSION
MYC affects several important biological processes through DNA binding and subsequent transcriptional activation or inhibition of a disperse set of target genes. To define MYC’s role in reprogramming, previous reports compared OSK to OSKM reprogramming and found several MYC-dependent correlations. These include mainly enhancement/alteration of OSK binding, promotion of ESC-like metabolic changes, and enhancement of proliferation (Chappell and Dalton, 2013; Soufi et al., 2012; Sridharan et al., 2009). Each of these varied observed changes may or may not be of play a major role to the outcome of reprogramming (enhanced colony formation), and we are particularly cognizant of the leap from correlation to causation in this complicated system. To specifically address the significance of a particular function of a multifunctional protein, we need to isolate that particular function and determine its importance. Mutating MYC to isolate a specific biological process (chromatin accessibility vs. metabolism vs. proliferation) is not feasible: the varied biological pathways all depend on the same transcription factor/DNA-interaction. In this work, we identified two unexpected methods for replacing one particular aspect of MYC—enhancement of proliferation. This enabled us to weigh the contribution of MYC’s contribution to proliferation in reprogramming. Both of these methods will be discussed in detail below.
In addition to the strategies for replacing MYC, identification of ESRG as an early maker of reprogramming proved useful as it allowed us to precisely define cellular and molecular events during first few days after OSK or OSKM induction. Our data demonstrated that OSK elicited two important molecular events in human cells: initiation of reprogramming process and RB-mediated proliferation pause or blockade. MYC does not affect the first event, but promotes iPSC generation via escaping the proliferation pause.
The first molecular event that OSK elicits is to initiate the reprogramming process, including activating early markers, such as ESRG and TRA-1–60. We and others reported that activation of endogenous retroviruses is crucial for iPSC generation in human cells. ESRG is a lncRNA driven by long terminal repeat (LTR) 7 of a HERV-H group endogenous retrovirus (Li et al., 2013; Lu et al., 2014; Ohnuki et al., 2014; Wang et al., 2014). We found that the LTR7 of ESRG contains the binding site of KLF4, suggesting that OSK directly activates its expression (Ohnuki et al., 2014). To our surprise, MYC did not promote this initial process of reprogramming. This is in contrast to a previous report that MYC enhanced the binding of OSK to its targets. Furthermore, we found that even endogenous MYC seemed dispensable for the appearance of ESRG (+) cells. Thus MYC does not play a crucial role in this very early event during human iPSC generation.
The other molecular event that OSK elicits is the proliferation pause in early reprograming cells. Very few BrdU (+) cells were detected in ESRG (+) cells on day 3 and TRA-1–60 (+) cells on day 4 after induction of OSK. We found that the OSK-mediated proliferation pause was attributable to RB activation. We also found that OSK induced expression of p16 and p53. OSK also induced transient increase of p21 mRNA on day 3. Suppression of these genes by shRNAs induced proliferation. Thus, cooperation of these multiple tumor suppressor gene products is responsible for RB activation and the proliferation pause in early reprogramming cells. Previously, evidence of an oncogene-induced senescence-like process was detected via transcriptional changes and described as reprogramming induced senescence (Banito et al., 2009; Utikal et al., 2009). In this paper, we refer to the period from day 3 to day 7 as a proliferation pause or blockade, but have no objection to alternatively calling the OSK-initiated, p21/p16-driven cessation of proliferation “reprogramming induced senescence.” We think the two processes are likely one and the same. With regards to this process, this work demonstrates the utility of ESRG to define the precise timing as well as demonstrates mechanisms by which reprogramming cells are able to overcome this blockade.
MYC provided an escape from the proliferation pause elicited by OSK in early reprogramming cells. An important mechanism is let-7/LIN41/p21 axis. OSK increased transcripts of let-7 family members, but addition of MYC suppressed this increase. We found LIN41 mRNA expression was induced by OSK in early reprogramming cells. However, LIN41 protein level remained low due to the increased expression of let-7 that inhibits translation of LIN41 mRNA (Lin et al., 2007; Slack et al., 2000). MYC, by suppressing let-7 expression, increases LIN41 protein. We also found that CDKN1A mRNA encoding p21 is the most enriched mRNA binding target in LIN41 pull down from human ESCs (Figure 5A). Furthermore, LIN41 expression in fibroblasts dramatically represses p21 translation (Figure 5E). Thus, MYC decreased p21 protein expression, leading to inactivation of RB and initiation of proliferation.
Though less dramatic than the effects on the let-7/LIN41/p21 axis, we additionally found that MYC decreased mRNA and protein levels of p16. When MYC alone is overexpressed, it induces p16 expression (Guney et al., 2006), but when it is co-expressed with RAS, MYC suppresses RAS-induced senescence (Hydbring et al., 2010). We suspect that a similar mechanism operates during reprogramming. In contrast to p16, MYC increased mRNA expression of p53, which should have an inhibitory influence on reprogramming. We suppose that the suppression of p21 and p16 is sufficient to overcome negative effects of increased p53.
Since a proliferation pause was commonly observed in early reprogramming cells derived from five types of somatic cells, it might be a required step for reprogramming. However, teratoma-derived fibroblasts (TdFs) reprogram without going through a proliferation pause. Furthermore, addition of exogenous LIN41 with OSK allows even somatic cells to avoid the onset of the proliferation pause. Therefore, a proliferation pause is not required in reprogramming, but rather is a barrier to reprogramming that is common to normal somatic cells (five out of five lines) when induced with OSK.
TdFs gave us a unique opportunity to delineate the roles of MYC in promoting reprogramming. TdFs were highly efficient in iPSC generation, and MYC did not further enhance reprogramming. This suggests that the main role(s) of MYC is already achieved in TdFs. We found an RB-mediated proliferation pause does not occur in TdFs even without exogenous MYC. OSK and OSKM reprogramming efficiency was equal (Figure 6B). In sharp contrast, we did not observe metabolic changes in favor of reprogramming in TdFs (Figure S6C). Our data also suggested that the binding of OSK to their targets was comparable between TdFs and HDFs (Figures S6E and S6F). We did observe a significant down-regulation of fibroblast-specific genes in TdFs compared to HDFs, suggesting that this intermediate level of expression may contribute to high reprogramming efficiency of TdFs (Figure S6G). However, given that TdFs have an intermediate level of HDF markers, further downregulation of many fibroblast-related genes and upregulation of many ES-related genes are necessary to become iPSCs. If the previously proposed chromatin accessibility role of MYC significantly impacted reprogramming efficiency, we would expect that TdFs would show a partial improvement upon addition of MYC. Additionally, we would expect that the emergence of early reprogramming cells should be increased if the down-regulation of fibroblast genetic program offered an easier route to reprogramming. Neither of these predictions (depending on a significant role for chromatin remodeling by MYC in reprogramming) is true in TdFs. Since proliferation pause-resistant TdFs do not benefit from MYC whatsoever, we conclude that the main contribution of MYC in proliferation pause-responsive somatic cells is to overcome proliferation pause, rather than to improve reprogramming through other means, for instance, chromatin accessibility.
We previously showed that LIN41, a cytoplasmic protein and a let-7 miRNA translationally repressed target, dramatically enhanced iPSC generation (Worringer et al., 2014). We identified EGR1, a transcription factor involved in differentiation, as a target of LIN41. We showed EGR1 protein was upregulated by LIN41 knockdown and LIN41 overexpression suppressed iPSC generation. Based on these data, we hypothesized that positive action of LIN41 in reprogramming is attributable to suppression of EGR1. However, our current results clearly show that p21 is the most crucial target of LIN41 in promoting iPSC generation. Importantly, LIN41 enhances the reprogramming of OSK to the same extent as MYC (Figure 6B, see right side HDF panel). This is not coincidental as both LIN41 and MYC provide similar outcomes with regard to proliferation pause. OSKL reprogrammed cells entirely subvert the proliferation pause, by early and tight translational suppression of p21. OSKM entered the proliferation pause, but eventually escaped after down-regulation of let-7 miRNA, followed by de-repression of endogenous LIN41 protein and subsequently p21 translational silencing. Again, LIN41 likely replaced only one specific biological function out of the various proposed mechanisms of MYC, but achieved all of MYC’s reprogramming enhancement. We therefore conclude that the significant contribution of MYC is that shared with LIN41—the ability to overcome proliferation pause.
We believe that the metabolic effects described as contingent upon MYC could well be viewed as secondary to the continuation of reprogramming beyond escape from proliferation pause (the primary effect). In other words, when OSK is compared to OSKM, one is comparing blocked cells to cells that have escaped proliferation pause and continue to reprogram and thus are continuing to gain ESC-like attributes (e.g., ESC-like metabolism). Nevertheless, whether or not a metabolic contribution is provided by MYC through transcriptional activity on metabolic genes or as a consequence of the continued reprogramming provided by escape from proliferation blockade, we demonstrated that an equivalent outcome is obtained by two methods that likely replace only MYC’s effect on proliferation. Therefore, we favor a model emphasizing MYC’s contribution as escaping proliferation pause.
In conclusion, our data showed that the main function of MYC during human iPSC generation is to override an RB-mediated proliferation pause in early reprogramming cells. MYC achieves this suppressing let-7 miRNA and derepressing the newly emerging endogenous LIN41 transcript. This leads to LIN41 protein-dependent translational repression of p21. Exogenous LIN41 fulfills the role of endogenous LIN41 and therefore MYC. In TdFs, the proliferation pause does not operate during reprogramming, resulting in MYC-independent high efficiency of iPSC generation. The emergence of early reprogramming cells, however, is very inefficient even with MYC or in TdFs. Therefore, the RB pathway is responsible for the proliferation pause, but not the low efficiency of the appearance of early reprogramming cells. Understanding the cause of the low conversion efficiency to the state demarcated by ESRG positivity, an early prerequisite for (but not guarantee of inevitable reprogramming), remains an important future task.
EXPERIMENTAL PROCEDURES
Further details and outline of resources used in this work can be found in Supplemental Experimental Procedures.
Quantification and Statistical Analysis |
Data are presented as mean ± standard deviation. Sample number (n) indicates the number of biological replicates in each experiment. The number of experimental repeats are indicated in figure legends. To determine statistical significance, we used Dunnett’s test for one-to-many comparison, and unpaired t-test for the comparisons between two groups using the Excel 2013 (Microsoft) and the Kaleida graph software (HULINKS) unless otherwise noted. Statistical significance in main figures was set at p < 0.05 indicated by asterisk.
DATA AVAILABILITY
Gene expression microarray, Infinium and deep sequencing results are accessible in the Gene Expression Omnibus database of the National Center for Biotechnology Information website (accession number: GSE89455 and GSE90015). Raw images are accessible in the Mendeley website (https://data.mendeley.com/datasets/wh59xb6sz6/draft?a=7db44554–6446-4c66-babe-d2080c43f0a1).
Supplementary Material
Table S3: Reverse-correlated gene ranking to ESRG (related to Figure 2A)
The list shows genes ranked as reverse-correlating to ESRG expression on day 3 post-transduction of OSKM by single cell RNA-seq.
Table S4: The over-representation of gene sets in set of genes that show reverse correlation with ESRG expression (related to Figures 3A and S3)
Curated canonical pathways (C2) in MSigDB (version 6.0) were selected as the gene sets. RB/E2F related gene sets are described in red letters within the table.
ACKNOWLEDGMENTS
We would like to thank A. Hotta, T. Kitamura, H. Niwa, K. Okita, H. Suemori and J. Wang for providing materials, B. Conklin, M. Ohnuki and K. Tomoda for discussions, and S. Arai, T. Hookway, Y. Inoue, K. Kaneko, T. Kato, J. Kuwahara, M. Lancero, Y. Nomiya, Y. Sawamura, I. Teramoto, M. Umekage and the Gladstone Core Facilities of Stem Cell, Genomics, Flow Cytometry for technical assistance. We are also grateful to K. Essex, R. Fujiwara, H. Imagawa, R. Kato, E. Minamitani, Y. Miyake, S. Takeshima and Y. Uematsu for administrative support, and G. Howard for editorial assistance. This work was supported by Grants-in-Aid for Scientific Research from the Japanese Society for the Promotion of Science (JSPS) and from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT); a grant from the Funding Program for World-Leading Innovative Research and Development in Science and Technology (First Program) of the JSPS; a grant from Core Center for iPS Cell Research, Research Center Network for Realization of Regenerative Medicine, MEXT; a grant from Japan Foundation for Applied Enzymology; and the iPS Cell Research Fund. The study was also supported by funding from Mr. Hiroshi Mikitani, Mr. Marc Benioff, the L.K. Whittier Foundation, the Roddenberry Foundation, the Gladstone Institutes, and NHLBI/NIH (U01-HL100406, U01-HL098179). The Gladstone Institutes received support from a National Center for Research Resources Grant RR18928–01.
Footnotes
DECLARATION OF INTERESTS
K.S. is employed by I Peace, Inc. K. Tanabe is a founder of I Peace, Inc. S.Y. is a scientific advisor (without salary) of iPS Academia Japan. K. Takahashi is on the scientific advisory board of I Peace, Inc. without salary.
REFERENCES
- Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, Pinho S, Silva JC, Azuara V, Walsh M, et al. (2009). Senescence impairs successful reprogramming to pluripotent stem cells. Genes & development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, and Weintraub H (1990). Sequence-specific DNA binding by the c-Myc protein. Science (New York, NY) 250, 1149–1151. [DOI] [PubMed] [Google Scholar]
- Blackwood EM, and Eisenman RN (1991). Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science (New York, NY) 251, 1211–1217. [DOI] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, and Jaenisch R (2012). Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 150, 1209–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, and Jaenisch R (2013). Mechanisms and models of somatic cell reprogramming. Nat Rev Genet 14, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HM, Martinez NJ, Thornton JE, Hagan JP, Nguyen KD, and Gregory RI (2012). Trim71 cooperates with microRNAs to repress Cdkn1a expression and promote embryonic stem cell proliferation. Nature communications 3, 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, and Mendell JT (2008). Widespread microRNA repression by Myc contributes to tumorigenesis. Nature genetics 40, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell J, and Dalton S (2013). Roles for MYC in the establishment and maintenance of pluripotency. Cold Spring Harbor perspectives in medicine 3, a014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell J, Sun Y, Singh A, and Dalton S (2013). MYC/MAX control ERK signaling and pluripotency by regulation of dual-specificity phosphatases 2 and 7. Genes & development 27, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, and Sander C (2013). Emerging landscape of oncogenic signatures across human cancers. Nature genetics 45, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliff TS, Wu T, Boward BR, Yin A, Yin H, Glushka JN, Prestegaard JH, and Dalton S (2017). MYC Controls Human Pluripotent Stem Cell Fate Decisions through Regulation of Metabolic Flux. Cell stem cell 21, 502–516.e509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2013). MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harbor perspectives in medicine 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2015). Web of the extended Myc network captures metabolism for tumorigenesis. Cancer cell 27, 160–162. [DOI] [PubMed] [Google Scholar]
- Dominguez-Sola D, and Gautier J (2014). MYC and the control of DNA replication. Cold Spring Harbor perspectives in medicine 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, and Vogelstein B (1993). WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Fagnocchi L, Cherubini A, Hatsuda H, Fasciani A, Mazzoleni S, Poli V, Berno V, Rossi RL, Reinbold R, Endele M, et al. (2016). A Myc-driven self-reinforcing regulatory network maintains mouse embryonic stem cell identity. Nature communications 7, 11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CD, Martinez-Fernandez A, Faustino RS, Yamada S, Perez-Terzic C, Nelson TJ, and Terzic A (2013). Nuclear reprogramming with c-Myc potentiates glycolytic capacity of derived induced pluripotent stem cells. Journal of cardiovascular translational research 6, 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay M, Li Y, and Felsher DW (2014). MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harbor perspectives in medicine 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guney I, Wu S, and Sedivy JM (2006). Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16(INK4a). Proceedings of the National Academy of Sciences of the United States of America 103, 3645–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Alexander WS, Barri G, Klinken SP, and Adams JM (1991). Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell 65, 753–763. [DOI] [PubMed] [Google Scholar]
- Hayward WS, Neel BG, and Astrin SM (1981). Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature 290, 475–480. [DOI] [PubMed] [Google Scholar]
- Hydbring P, Bahram F, Su Y, Tronnersjo S, Hogstrand K, von der Lehr N, Sharifi HR, Lilischkis R, Hein N, Wu S, et al. (2010). Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proceedings of the National Academy of Sciences of the United States of America 107, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer-Kwon KR, Nimura K, Rao SSP, Xu J, Jung S, Pekowska A, Dose M, Stevens E, Mathe E, Dong P, et al. (2017). Myc Regulates Chromatin Decompaction and Nuclear Architecture during B Cell Activation. Molecular cell 67, 566–578.e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS (2008). Why myc? An unexpected ingredient in the stem cell cocktail. Cell stem cell 2, 18–21. [DOI] [PubMed] [Google Scholar]
- Kress TR, Sabo A, and Amati B (2015). MYC: connecting selective transcriptional control to global RNA production. Nature reviews Cancer 15, 593–607. [DOI] [PubMed] [Google Scholar]
- Li G, Ren C, Shi J, Huang W, Liu H, Feng X, Liu W, Zhu B, Zhang C, Wang L, et al. (2013). Identification, expression and subcellular localization of ESRG. Biochemical and biophysical research communications 435, 160–164. [DOI] [PubMed] [Google Scholar]
- Li R, Liang J, Ni S, Zhou T, Qing X, Li H, He W, Chen J, Li F, Zhuang Q, et al. (2010). A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell stem cell 7, 51–63. [DOI] [PubMed] [Google Scholar]
- Lin CH, Jackson AL, Guo J, Linsley PS, and Eisenman RN (2009). Myc-regulated microRNAs attenuate embryonic stem cell differentiation. The EMBO journal 28, 3157–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young RA (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Hsieh LC, Kuo MW, Yu J, Kuo HH, Lo WL, Lin RJ, Yu AL, and Li WH (2007). Human TRIM71 and its nematode homologue are targets of let-7 microRNA and its zebrafish orthologue is essential for development. Molecular biology and evolution 24, 2525–2534. [DOI] [PubMed] [Google Scholar]
- Lu X, Sachs F, Ramsay L, Jacques PE, Goke J, Bourque G, and Ng HH (2014). The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat Struct Mol Biol 21, 423–425. [DOI] [PubMed] [Google Scholar]
- Munoz-Espin D, and Serrano M (2014). Cellular senescence: from physiology to pathology. Nature reviews Molecular cell biology 15, 482–496. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, and Yamanaka S (2008). Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nature biotechnology 26, 101–106. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Takizawa N, Narita M, Ichisaka T, and Yamanaka S (2010). Promotion of direct reprogramming by transformation-deficient Myc. Proceedings of the National Academy of Sciences of the United States of America 107, 14152–14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley J, Skylaki S, Iwabuchi KA, Chantzoura E, Ruetz T, Johnsson A, Tomlinson SR, Linnarsson S, and Kaji K (2013). High-resolution analysis with novel cell-surface markers identifies routes to iPS cells. Nature 499, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuki M, Tanabe K, Sutou K, Teramoto I, Sawamura Y, Narita M, Nakamura M, Tokunaga Y, Watanabe A, Yamanaka S, et al. (2014). Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peukert K, Staller P, Schneider A, Carmichael G, Hanel F, and Eilers M (1997). An alternative pathway for gene regulation by Myc. The EMBO journal 16, 5672–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo JM, Anderssen E, Walsh RM, Schwarz BA, Nefzger CM, Lim SM, Borkent M, Apostolou E, Alaei S, Cloutier J, et al. (2012). A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 151, 1617–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, and Ruvkun G (2000). The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Molecular cell 5, 659–669. [DOI] [PubMed] [Google Scholar]
- Smith KN, Singh AM, and Dalton S (2010). Myc represses primitive endoderm differentiation in pluripotent stem cells. Cell stem cell 7, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Donahue G, and Zaret KS (2012). Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan R, Tchieu J, Mason MJ, Yachechko R, Kuoy E, Horvath S, Zhou Q, and Plath K (2009). Role of the murine reprogramming factors in the induction of pluripotency. Cell 136, 364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Sasaki A, Yamamoto M, Nakamura M, Sutou K, Osafune K, and Yamanaka S (2014). Induction of pluripotency in human somatic cells via a transient state resembling primitive streak-like mesendoderm. Nature communications 5, 3678. [DOI] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, Khalil A, Rheinwald JG, and Hochedlinger K (2009). Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 460, 1145–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, and Berns A (1991). Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 65, 737–752. [DOI] [PubMed] [Google Scholar]
- Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley C.A.t., Ramalho-Santos J, Van Houten B, and Schatten G (2011). Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PloS one 6, e20914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennstrom B, Sheiness D, Zabielski J, and Bishop JM (1982). Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. Journal of virology 42, 773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, Austen M, and Luscher B (2003). Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO reports 4, 484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Xie G, Singh M, Ghanbarian AT, Rasko T, Szvetnik A, Cai H, Besser D, Prigione A, Fuchs NV, et al. (2014). Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature. [DOI] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Cassady JP, and Jaenisch R (2008). c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell stem cell 2, 10–12. [DOI] [PubMed] [Google Scholar]
- Wolf E, Lin CY, Eilers M, and Levens DL (2015). Taming of the beast: shaping Myc-dependent amplification. Trends in cell biology 25, 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward GE, and Hudson MT (1954). The effect of 2-desoxy-D-glucose on glycolysis and respiration of tumor and normal tissues. Cancer research 14, 599–605. [PubMed] [Google Scholar]
- Worringer KA, Rand TA, Hayashi Y, Sami S, Takahashi K, Tanabe K, Narita M, Srivastava D, and Yamanaka S (2014). The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell stem cell 14, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3: Reverse-correlated gene ranking to ESRG (related to Figure 2A)
The list shows genes ranked as reverse-correlating to ESRG expression on day 3 post-transduction of OSKM by single cell RNA-seq.
Table S4: The over-representation of gene sets in set of genes that show reverse correlation with ESRG expression (related to Figures 3A and S3)
Curated canonical pathways (C2) in MSigDB (version 6.0) were selected as the gene sets. RB/E2F related gene sets are described in red letters within the table.
Data Availability Statement
Gene expression microarray, Infinium and deep sequencing results are accessible in the Gene Expression Omnibus database of the National Center for Biotechnology Information website (accession number: GSE89455 and GSE90015). Raw images are accessible in the Mendeley website (https://data.mendeley.com/datasets/wh59xb6sz6/draft?a=7db44554–6446-4c66-babe-d2080c43f0a1).