

Abstract
The emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants that escape convalescent and vaccine-induced antibody responses has renewed focus on the development of broadly protective T-cell-based vaccines. Here, we apply structure-based network analysis and assessments of HLA class I peptide stability to define mutationally constrained CD8+ T cell epitopes across the SARS-CoV-2 proteome. Highly networked residues are conserved temporally among circulating variants and sarbecoviruses and disproportionately impair spike pseudotyped lentivirus infectivity when mutated. Evaluation of HLA class I stabilizing activity for 18 globally prevalent alleles identifies CD8+ T cell epitopes within highly networked regions with limited mutational frequencies in circulating SARS-CoV-2 variants and deep-sequenced primary isolates. Moreover, these epitopes elicit demonstrable CD8+ T cell reactivity in convalescent individuals but reduced recognition in recipients of mRNA-based vaccines. These data thereby elucidate key mutationally constrained regions and immunogenic epitopes in the SARS-CoV-2 proteome for a global T-cell-based vaccine against emerging variants and SARS-like coronaviruses.
Keywords: SARS-CoV-2, COVID-19, CD8+ T cells, vaccine, epitopes, variants, sarbecovirus, protection
Graphical abstract
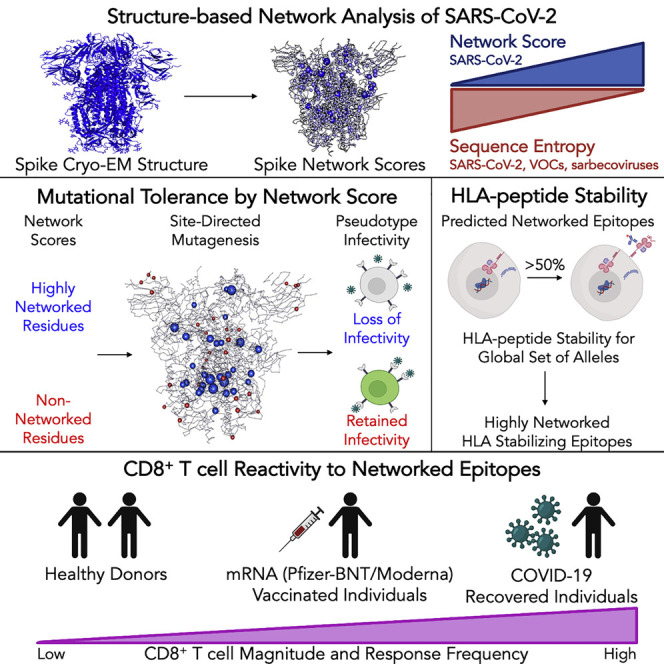
Structure-based network analyses identify regions in the SARS-CoV-2 proteome that are mutationally constrained and bear CD8+ T cell epitopes that are also conserved in emerging variants as well as other sarbecoviruses. These epitopes elicit stronger CD8+ T cell responses in convalescent individuals over mRNA vaccine recipients and provide a framework for a broad T-cell-based vaccine against coronaviruses.
Introduction
An effective vaccine for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been a major global health priority. While multiple vaccine candidates have been developed using a variety of platforms to deliver the viral spike protein and induce neutralizing antibodies (Baden et al., 2021; Folegatti et al., 2020; Keech et al., 2020; Polack et al., 2020; Sadoff et al., 2021), the emergence of the B.1.1.7 alpha (Tang et al., 2020), B.1.351 beta (Tegally et al., 2021), P.1 gamma (Voloch et al., 2020), and B.1.617.2 delta variants (Cherian et al., 2021) has raised substantial new concerns due to their increased transmissibility (Davies et al., 2021; Kumar et al., 2021) and ability to escape convalescent and vaccine-induced antibody responses (Garcia-Beltran et al., 2021; Hoffmann et al., 2021; Madhi et al., 2021; Wang et al., 2021; Wibmer et al., 2021). In addition, given that SARS-CoV-2 is the third coronavirus outbreak in the past 20 years (after SARS-CoV-1 and Middle East respiratory syndrome coronavirus [MERS-CoV]), significant additional concerns exist about a future pandemic due to the numerous SARS-like coronaviruses identified in the bat reservoir (Menachery et al., 2015, 2016). Thus, while existing vaccines are critical to curtail the ongoing pandemic, new vaccine candidates that can enhance protection against variants of concern (VOCs) and emerging coronaviruses are also urgently needed.
Neutralizing antibodies will certainly be a key component of a broadly protective vaccine, but the induction of virus-specific CD8+ T cells could greatly augment antibody-based protection. Individuals with mild coronavirus disease 2019 (COVID-19) disease have increased clonal expansion of CD8+ T cells in bronchoalveolar lavage fluid (Liao et al., 2020), robust CD8+ T cell reactivity to SARS-CoV-2 epitopes (Peng et al., 2020; Sekine et al., 2020), and rapid CD8+ T-cell-mediated viral clearance (Tan et al., 2021). In addition, patients with X-linked agammaglobulinemia who lack circulating B cells but have functional T lymphocytes develop only mild-to-moderate COVID-19 disease (Soresina et al., 2020), and antibody-mediated depletion of CD8+ T cells from convalescent macaques partially abrogates protective immunity (McMahan et al., 2021). From the perspective of a broad sarbecovirus vaccine, CD8+ T cells developed in response to SARS-CoV-1 infection exhibit long-lasting immune memory (Ng et al., 2016), and vaccine-induced CD8+ T cells protect against lethal SARS-CoV-1 challenge in mice (Channappanavar et al., 2014). Moreover, in contrast to spike-restricted neutralizing antibody responses, CD8+ T cells can target regions across the SARS-CoV-2 proteome and could therefore be directed at sites that are constrained from mutation in circulating VOCs and sarbecoviruses. This is becoming increasingly important given emerging evidence that SARS-CoV-2 can evade cellular immunity through mutation of HLA-class-I-restricted epitopes (Agerer et al., 2021).
Toward this goal, we leveraged an approach known as structure-based network analysis, which uses protein structure data and network theory to delineate topologically important residues based on their local connectivity, involvement in bridging interactions, and proximity to known protein ligands (Gaiha et al., 2019). We recently demonstrated that CD8+ T cell epitopes identified by this approach in the highly mutable human immunodeficiency virus (HIV) were mutationally constrained and preferentially targeted by individuals who successfully control HIV in the absence of therapy (Gaiha et al., 2019; McMichael and Carrington, 2019). Importantly, structure-based network analysis outperformed traditional sequence conservation metrics in its detection of mutation-intolerant residues across a spectrum of HIV and non-HIV proteins. While SARS-CoV-2 has a lower mutation rate than HIV, its rapid worldwide spread has provided ample opportunity for the emergence of viral escape variants, making the structure-based network approach particularly well suited given that the full extent of sequence variation continues to be defined.
Thus, in this study, we applied structure-based network analysis to high-quality protein structures for SARS-CoV-2 to define mutationally constrained (“highly networked”) residues. The mutational tolerance of highly networked residues was assessed through comparisons to viral sequence entropy data, site-directed mutagenesis and functional assessments of the SARS-CoV-2 spike protein, and correlations with deep mutational scanning of the receptor-binding domain (RBD). Using a recently established HLA class I peptide stability assay (Kaseke et al., 2021), we defined CD8+ T cell epitopes within highly networked regions for 18 globally prevalent HLA class I alleles. We then assessed the mutational resistance of these epitopes through analyses of SARS-CoV-2 primary isolates and their immunogenicity in individuals with convalescent disease and mRNA-based vaccine recipients. Collectively, these studies elucidate key regions within the SARS-CoV-2 proteome that are not only structurally constrained from mutation but also harbor a globally relevant set of CD8+ T cell epitopes to augment protection against SARS-CoV-2 variants and sarbecoviruses.
Results
Structure-based network analysis of SARS-CoV-2 identifies highly conserved residues across SARS-CoV-2 variants and sarbecoviruses
To identify mutationally constrained regions in the SARS-CoV-2 proteome, we applied structure-based network analysis (Gaiha et al., 2019) to define areas of topological structural importance. Based on available high-quality structural data, we were able to calculate amino acid network scores for open and closed spike protein conformations (Figure 1 A) and 14 additional proteins, which made up ∼44% of the viral proteome (Figures S1 A and S1B). Residue network scores were binned (<0, 0–2, 2–4, >4) and compared with sequence entropy values from SARS-CoV-2, sarbecoviruses (SARS-CoV-1/bat CoV), and MERS-CoV, which revealed a strong inverse relationship between network measures of topological importance and mutational frequencies (Figures 1B–1D). Network scores calculated using structural data for SARS-CoV-1 and MERS-CoV were also highly correlated with scores for SARS-CoV-2 (R = 0.78 and R = 0.67, respectively), indicating that highly networked residues are likely to be structurally conserved across lineage B and C betacoronaviruses (Figures S2 A and S2B).
Figure 1.

Structure-based network analysis of the SARS-CoV-2 proteome identifies amino acid residues conserved in lineage B and C coronaviruses
(A) Structure-based network analysis schematic for closed spike trimer (PDB: 6VXX) with amino acid residues (nodes) and non-covalent interactions (edges). Edge width indicates interaction strength and node size indicates relative network scores.
(B–D) Comparison of SARS-CoV-2 amino acid network scores (binned by network score: <0, 0–2, 2–4, and >4) with sequence entropy for SARS-CoV-2, sarbecoviruses (SARS-CoV-1/bat CoV), and MERS. (E) Alignment of SARS-CoV-2 network scores with sequence entropy values for SARS-CoV-2 in May 2020 and February 2021, sarbecoviruses (SARS-CoV-1/bat CoV) and MERS-CoV. Residues in blue indicate those with network scores >4. Network scores of residues mutated in the B.1.1.7 alpha (red triangles), B.1.351 beta (green triangles), P.1 gamma (yellow triangles), and B.1.617.2 delta variant (purple triangles) are depicted in gray. Yellow boxes indicate new areas of sequence variation in SARS-CoV-2 that emerged between May 2020 and February 2021. Statistical comparisons were made using Mann-Whitney U test. For comparisons of more than two groups, Kruskal-Wallis test with Dunn’s pos hoc analyses was used. Calculated p values were as follows: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Figure S1.

Structure-based network analysis of the SARS-CoV-2 proteome, related to Figure 1
(A) Network diagrams of SARS CoV-2 structural and accessory proteins and (B) NSPs. Node size indicates relative intra-protein network scores.
Figure S2.

Correlation of SARS-CoV-2 network scores with SARS-CoV-1 and MERS-CoV network scores, related to Figure 1
Scatterplots comparing SARS-CoV-2 network scores to (A) SARS-CoV-1 network scores and (B) MERS-CoV network scores. Correlations were calculated by Spearman’s rank correlation coefficient.
Alignment of SARS-CoV-2 residue network scores with sequence entropy values for SARS-CoV-2 (at two distinct time points), sarbecoviruses (CoV-1/bat CoV) and MERS-CoV also revealed numerous linear regions in which highly networked and conserved CD8+ T cell epitopes could be identified (Figure 1E). In addition, given that SARS-CoV-2 network scores were calculated at an early stage of the pandemic (May 2020), we aligned sequence entropy values at that time (45,603 sequences) to values obtained in February 2021 (661,816 sequences) and found that the vast majority of new sequence variation in SARS-CoV-2 has emerged in non-networked regions (Figure 1E; yellow boxes). Given the worldwide concern regarding new SARS-CoV-2 variants, we specifically evaluated residues mutated in the B.1.1.7 alpha, B.1.351 beta, P.1 gamma, and B.1.617 delta VOCs and observed that they had low network scores, with ∼82.1% having negative values and ∼96.4% having scores <1 (p = 0.0003 for comparison of VOC network scores to non-VOC) (Figure 1E; Table S1). This was similar to an analysis of network scores of spike escape variants identified by in vitro mutational scanning (Greaney et al., 2021) (Table S1). These data demonstrate that structure-based network analysis can predict regions of relative mutational constraint or freedom within SARS-CoV-2 and identify residues highly conserved across sarbecoviruses.
Mutation of highly networked SARS-CoV-2 spike residues impairs pseudotyped lentiviral infectivity and correlates with functional assessments of the spike RBD
To experimentally evaluate the relationship between SARS-CoV-2 network scores and mutational tolerance, we utilized a SARS-CoV-2 spike pseudotyped lentivirus assay (Crawford et al., 2020) (Figures S3 A–S3C) and engineered non-conservative point mutations for 10 pairs of sequence conserved spike residues that occupied either high (>2; blue) or low (<1; red) network score positions (Figures 2 A–2C and S4A; Table S2). We also engineered conservative point mutations for highly networked spike residues to further assess their mutational tolerance (Table S1). Pseudotyped lentiviruses with no spike protein (delta spike), wild-type (WT) spike protein, or mutant spike proteins were used to infect parental 293T cells or 293T cells expressing human ACE2 (293T-ACE2), and the level of infectivity was determined by ZsGreen expression following 3-day incubation (Figures S3A–S3C).
Figure S3.

Spike pseudotyped lentiviral infectivity assay, related to Figure 2
(A) Flow cytometry plots showing %ZsGreen-positive 293T and 293T-ACE2 cells after 60h incubation with ZsGreen backbone lentiviruses pseudotyped with no Spike protein (delta Spike; gray), wild-type (WT) Spike protein (green) or VSV-G (black) envelope protein. Composite pseudotyped lentiviral infectivity data of (B) 293T or (C) 293T-ACE2 cells at five-fold and two-fold dilutions of neat stock virus preparations.
Figure 2.

Mutation of highly networked residues in the viral spike protein impairs infectivity and RBD folding
(A) Location of networked (blue) and non-networked (red) residues in the closed (PDB: 6VXX) and open (PDB: 6VYB) conformations of the spike protein that were mutated in pseudotyped lentivirus.
(B and C) Comparison of network scores and Shannon entropy values between networked residues and non-networked residues selected for mutagenesis.
(D) Flow cytometry plots showing the percentage of ZsGreen-positive 293T-ACE2 cells after 60-h incubation with ZsGreen backbone lentiviruses pseudotyped with no spike protein (delta spike; gray), wild-type (WT) spike (green), VSV-G (black), or mutated spike proteins (dark blue, light blue, and red).
(E) Comparison of spike pseudotyped lentiviral infectivity of 293T-ACE2 cells after mutation of networked residues with non-conservative mutations (N, dark blue), networked residues with conservative mutations (C, light blue), and non-networked residues with non-conservative mutations (N, red). Data are means of technical triplicates from an experiment performed twice. Statistical analysis by one-way analysis of variance and Mann-Whitney U test.
(F) Scatterplot of full spike protein residue network scores and average effect of mutation on monomeric RBD folding. Residues in blue indicate those with high network scores but low effect on monomeric RBD folding (V362, A363, C391, V524, C525). Correlations were calculated by Spearman’s rank correlation coefficient.
(G) Location of highly networked residues with low effect on monomeric RBD folding (blue) within the RBD monomer (PDB: 6MOJ) and RBD-distal S1 domain (PDB: 6VXX).
(H) Percentage of ZsGreen-positive 293T-ACE2 cells after 60-h incubation with WT spike pseudotyped lentiviruses (green) and non-conservative (blue) or conservative (light blue) mutations to highly networked residues with low effect on monomeric RBD folding. Data are means of technical triplicates from an experiment performed twice.
(I) Scatterplot of SARS-CoV-2 RBD residue network scores and average effect of mutation on monomeric RBD folding stability. Residues in blue indicate those that previously had high network scores in the full spike protein but low scores in the RBD monomer.
(J) Scatterplot of Shannon entropy values of SARS-CoV-2 RBD residues and average effect of mutation on monomeric RBD folding.
Correlations were calculated by Spearman’s rank correlation coefficient. For comparisons of more than two groups, Kruskal-Wallis test with Dunn’s post hoc analyses were used. Calculated p values were as follows: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See also Figure S2 and Table S2.
Figure S4.

Shannon entropy values for residues mutated in SARS-CoV-2 spike protein and correlations of spike RBD values with functional mutagenesis data, related to Figure 2
(A) List of matched pairs of networked and non-networked residues in the SARS-CoV-2 Spike proteins targeted for mutagenesis. (B) Comparison of Shannon entropy values between networked residues and non-networked residues in the Sarbecovirus subgenus (SARS-CoV-1/Bat CoV), respectively. (C) Scatterplot of network score of RBD and average effect of mutation on monomeric RBD binding. Statistical comparisons were made using Mann-Whitney U test. (D) Scatterplot of SARS-CoV-1/Bat Shannon entropy values for the RBD and average effect of mutation on monomeric RBD folding. Correlations were calculated by Spearman’s rank correlation coefficient. Calculated P values were as follows: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Comparative assessment of pseudotyped lentiviruses harboring non-conservative mutations of spike residues with high or low network scores revealed highly statistically significant differences in 293T-ACE2 cell infectivity (Figures 2D and 2E). Moreover, conservative mutations of highly networked spike residues (Table S2) also led to substantial impairment of pseudotyped lentiviral infectivity (Figures 2D and 2E). Importantly, mutated spike residues with high or low network scores had no significant difference in sequence entropy for SARS-CoV-2 or sarbecoviruses (Figures 2C and S4 B), indicating that network score provides a level of resolution of mutational constraint beyond entropy, consistent with previous observations (Gaiha et al., 2019).
To further assess the mutational tolerance of highly networked residues, we utilized a published high-throughput mutagenesis dataset in which every residue within the SARS-CoV-2 spike RBD was mutated to all possible amino acid substitutions and assessed for its impact on protein folding stability and ACE2 binding (Starr et al., 2020). Correlation of full spike residue network scores with the average effect of residue mutation on RBD folding revealed a significant inverse correlation (R = −0.46, p = 9.5 × 10−11) (Figure 2F). Interestingly, there were five residues with high network scores (V362, A363, C391, V524, and C525) that did not have substantial impact on RBD protein folding when mutated (Figure 2F). We therefore evaluated the protein structure of the monomeric RBD (PDB: 6MOJ), which demonstrated that these residues were not within the RBD core or RBD-ACE2 binding surface (Figure 2G), likely explaining why they have little effect on either folding or ACE2 binding of the RBD monomer (Starr et al., 2020). However, evaluation of these residues in the full spike structure (PDB: 6VXX), which we utilized for our network score calculations, reveals that they are located at a critical hinge region between the RBD and distal S1 domain (Figure 2G) that mediates the conformational change between the open and closed states of the spike trimer (Gur et al., 2020; Meirson et al., 2020).
We therefore engineered conservative and non-conservative mutations for each of these five spike residues (Table S2) and found that they all had significant effects on pseudotyped lentiviral infectivity, with mutations of C391, V524, and C525 having the greatest effect (Figure 2H). We also generated network scores for the RBD monomer structure alone and observed a more robust inverse correlation with the effects of mutation on protein-folding stability (R = −0.67, p = 7.9 × 10−27) (Figure 2I) and ACE2 binding (R = −0.68, p = 6.1 × 10−25) (Figure S4C), indicating better agreement between the two methodologies when the same protein domain is used. Comparison of sequence entropy values from the SARS-CoV-2 RBD and the average effect of mutation on protein folding revealed a markedly lower magnitude correlation (R = 0.37, p = 3.4 × 10−7) (Figure 2J), which was also observed for comparisons with sarbecovirus entropy values (R = 0.38, p = 2.4 × 10−7) (Figure S4D). These data demonstrate that structure-based network analysis outperforms sequence conservation in its ability to identify mutationally constrained residues in the spike RBD and can also delineate residues that mediate ACE2 binding not detected by deep mutational scanning of the RBD monomer.
Identification of highly networked CD8+ T cell epitopes by HLA class I stabilization
To define CD8+ T cell epitopes within highly networked regions of SARS-CoV-2, we utilized a prioritization pipeline that integrates computational epitope prediction with experimental HLA class I stabilization (Figure 3 A). Epitope network scores were calculated (see STAR Methods, method details) for all possible 8, 9, 10, and 11 amino acid peptides for which structural data were available (16,604 possible CD8+ T cell epitopes). We subsequently down-selected on those peptides with an epitope network score >3.00 (2,235 epitopes), which was a similar cutoff used for protective epitopes identified in HIV (Gaiha et al., 2019). We then applied the NetMHCpan 4.1 epitope prediction algorithm (http://www.cbs.dtu.dk/services/NetMHCpan/) to define putative binders for 18 HLA class I alleles that provide >99% coverage of the global population (A∗0101, A∗0201, A∗0301, A∗2402, B∗0702, B∗0801, B∗1402, B∗1501, B∗2705, B∗3501, B∗3901, B∗4001, B∗4402, B∗5201, B∗5701, B∗5801, B∗8101, and Cw∗0701) (Sette and Sidney, 1999; Sidney et al., 2008). We recently demonstrated that HLA class I peptide stability plays a key role in mediating CD8+ T cell immunodominance hierarchies across the HIV proteome and outperformed predicted binding affinity (Kaseke et al., 2021). We therefore experimentally determined whether predicted highly networked SARS-CoV-2 epitopes (311 HLA-epitope pairs; Table S3) could bind and stabilize the 18 HLA alleles using an assay that leverages CRISPR-Cas9-edited transporter associated with antigen processing (TAP)-deficient mono-allelic HLA-class-I-expressing cell lines. Epitopes that achieved at least 50% relative HLA class I stabilization to an HLA-matched immunodominant HIV epitope were considered to be promising SARS-CoV-2 T cell immunogens given the observed in vivo CD8+ T cell targeting of HIV epitopes that reached this threshold (Streeck et al., 2009).
Figure 3.

Stabilization of HLA class I molecules by CD8+ T cell epitopes derived from highly networked regions
(A) Epitope prioritization pipeline for identification of highly networked CD8+ T cell epitopes in SARS-CoV-2. This image was made using BioRender.
(B) Representative concentration-based stabilization of surface HLA-A∗0301 following incubation with no peptide, immunodominant HIV HLA-A∗0301 epitope RK9 (100 μM), predicted highly networked SARS-CoV-2 epitopes for HLA-A∗0301 (100 μM), and B∗08-restricted HIV epitope FL8 (100 μM).
(C) Concentration-based HLA class I stabilization of predicted highly networked SARS-CoV-2 CD8+ T cell epitopes for HLA-A∗0301 (0.1-100 μM). The y axis depicts the anti-HLA MFI normalized to the highest value for each HLA class I allele (0-1). HIV HLA-A∗0301 RK9 epitope is indicated in red. SARS-CoV-2 epitopes with at least 50% relative HLA-A∗0301 stabilization in comparison to HIV RK9 are indicated in dark blue. The non-HLA-A∗03-restricted HIV epitope FL8 is depicted in light red. Data are means of technical duplicates from an experiment performed twice.
(D) Network-based depiction of A∗03 RK11 (NSP16; PDB ID: 6W4H, chain A) and A∗03 KR10 (spike; PDB: 6VXX).
(E) Sequence alignments of A∗03 RK11 and A∗03 KR10 with the corresponding sequence for SARS-CoV-2, including the emerging variants, bat CoV RaTG13, and all coronaviruses known to infect humans.
(F) Fractions of highly networked CD8+ T cell epitopes in SARS-CoV-2 with ≤1 amino acid mutation (blue), 2 mutations (green), 3 mutations (red), 4 mutations (orange), and 5 mutations (purple) in SARS-CoV-2 variants, bat CoV RaTG13, and all coronaviruses known to infect humans.
(G) Comparison of HLA class I peptide stabilization for SARS-CoV-2 ancestral epitopes and corresponding mutated epitopes in B.1.1.7 alpha (red; A∗02 VL9) and P.1 gamma (yellow; A∗01 SY10, A∗01 NY10, A∗01 NY11, and B∗35 SY10) at 100 μM peptide concentration. Statistical comparison was made using Wilcoxon matched-pairs test.
(H) Comparison of the fraction of HLA∗02-restricted highly networked (blue) and non-networked (red) epitope variants (Agerer et al., 2021) that achieve an allelic frequency >0.1 or >0.9. Statistical comparisons of epitope variant frequencies were made using Fisher’s exact test. Calculated p values were as follows: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
As a representative example, we incubated TAP-deficient HLA-A∗0301 mono-allelic cells with the immunodominant A∗0301-restricted HIV RLRPGGKKK epitope (RK9, Gag p17 20–28) and 15 highly networked SARS-CoV-2 peptides that were predicted to bind by NetMHCPan 4.1 and found five epitopes that successfully surface stabilized HLA-A∗0301 at a level >50% of HIV RK9 (Figure 3B). Evaluation of all 311 predicted highly networked epitopes for 18 HLA alleles at increasing peptide concentrations (0.1–100 μM) revealed that 109 epitopes reached >50% relative HLA class I stabilization, of which 56 were derived from SARS-CoV-2 non-structural proteins and 53 were derived from structural proteins and ORF3a (Figures 3C and S5 A; Table S3). Representative examples of HLA-stabilizing epitopes for HLA-A∗0301 include the RK11 epitope from NSP16 (ORF1a 6864-6874) and KR10 epitope from spike (310–319), both of which occupy topologically important positions in their respective viral proteins (Figure 3D). We also identified peptides that are frequently targeted during natural infection (e.g., LLYDANYFL, ORF3a 139–147) (Schulien et al., 2021) or can stabilize a number of HLA class I alleles (e.g., MIAQTYSAL, spike 869–877) (Figures S5B and S5C; Table S3) and induce T cell reactivity in distinct cohorts of recovered individuals (Peng et al., 2020).
Figure S5.

Concentration-based HLA class I-peptide stabilization of predicted SARS-CoV-2 CD8+ T cell epitopes, related to Figure 3
Concentration-based HLA class I stabilization of 311 predicted SARS-CoV-2 CD8+ T cell epitopes (0.1-100 μM) across 18 TAP-deficient mono-allelic HLA class I-expressing cell lines. The y axis depicts the anti-HLA MFI normalized to the known immunodominant HIV CD8+ T cell epitope (red) for each HLA class I allele. SARS-CoV-2 epitopes with > 50% relative HLA class I stabilization to the HIV immunodominant epitope indicated in dark blue and those with < 50% relative stabilization are indicated in light blue. (B) HLA class I-peptide stabilization of TAP-deficient B∗0702, B∗1402, B∗3901, B∗8101 and Cw∗0701 expressing cell lines following incubation with no peptide (gray), HLA-specific HIV immunodominant HIV epitope (10 μM, red) or Spike ML9 (10 μM, blue). (C) Comparison of normalized anti-HLA MFI for B∗0702, B∗1402, B∗3901, B∗8101 and Cw∗0701 following incubation with immunodominant HIV epitope or ML9 peptide at range of concentrations (0.1-100 μM). Statistical comparisons were made using Mann-Whitney U test.
Alignment of highly networked HLA-stabilizing epitopes with sequences of SARS-CoV-2 variants, bat CoV RaTG13, SARS-CoV-1, MERS-CoV and the common cold coronaviruses (HKU1, OC43, 229E, NL63) revealed that 65% of epitopes have ≤1 amino acid mutation, and >90% of epitopes have ≤2 amino acid mutations across sarbecoviruses (bat CoV, SARS-CoV-1), but substantially higher levels of sequence mismatch for non-lineage B betacoronaviruses (Figures 3E and 3F). Specific assessment of the mutations in the SARS-CoV-2 VOCs revealed that only three residues (spike S982A in B.1.1.7 alpha, nucleocapsid P80R in P.1 gamma, and spike H1101D in B.1.617.2 delta) were found to be mutated in the highly networked epitopes from structural and accessory proteins (Table S4), leading to exact sequence matching or <1 amino acid mutation for 100% of epitopes. We therefore assessed the impact of these mutations on HLA class I peptide stability and found no significant difference between ancestral sequence epitopes and five mutated epitopes in B.1.1.7 alpha and P.1 gamma (Figure 3G), indicating that highly networked CD8+ T cell epitopes would provide broad coverage of VOCs with maintained HLA class I presentation.
To determine whether highly networked epitopes have inherent mutational constraints that would mitigate against the emergence of viral escape variants, we utilized deep-sequencing data of 747 primary SARS-CoV-2 isolates that delineated the mutational frequencies of 26 HLA-A∗02-restricted epitopes (Agerer et al., 2021). Importantly, three of these epitopes were identified as being highly networked (ALNTLVKQL, spike 958–966; KLNDLCFTNV, spike 386–395; and VLNDILSRL, spike 976–984). Given that each viral isolate was sequenced to a similar depth and the prevalence of HLA-A∗02 was ∼30% in the affected population, we felt that this was a highly relevant dataset to compare the in vivo viral evolution of highly networked and non-networked epitopes. We therefore compared the frequencies of mutations at HLA anchor and TCR contact sites (position 2 through the terminal amino acid) that achieved an allelic frequency of >0.1 (i.e., tolerated mutations nearing fixation) and > 0.9 (i.e., achieved mutational fixation) (Agerer et al., 2021). This revealed a striking difference with 6.67% (2/30) of networked epitope variants having an allelic frequency >0.1 and 0% (0/30) having an allelic frequency >0.9, while 25.2% (66/262) of non-networked epitope variants had an allelic frequency >0.1 (p = 0.02) and 16.8% (44/262) achieved mutational fixation (>0.9; p = 0.01) (Figure 3H). Alternatively, while the networked epitopes represented 10.3% of the analyzed epitope sequences, they accounted for only 2.9% of all variant epitopes with allelic frequencies >0.1 and 0% of variants with allelic frequencies >0.9. These analyses suggest that highly networked epitopes have significant constraints on in vivo viral evolution in comparison to non-networked epitopes restricted by the same HLA allele.
Highly networked epitopes are recognized by CD8+ T cells induced by SARS-CoV-2 infection
To evaluate the immunogenicity of highly networked HLA-stabilizing SARS-CoV-2 epitopes, we assessed the CD8+ T cell responses within a cohort of 20 healthy donors (HDs) and 30 convalescent COVID-19 patients (Table 1 ). CD4-depleted peripheral blood mononuclear cells (PBMCs) were tested for reactivity to peptide pools of highly networked epitopes derived from non-structural proteins (NSPs; n = 56), structural and accessory proteins (SPs; n = 53), or a combination of non-structural and structural and accessory proteins (NSPs + SPs; n = 109) (Figure 4 A) using ex vivo interferon-γ (IFN-γ) enzyme-linked immunospot (ELISpot) assays (Figure 4B). Anti-CD3/CD28 antibodies and a pool of human cytomegalovirus (CMV), Epstein-Barr Virus (EBV), and influenza virus (CEF) peptides were used as positive controls, and DMSO was used as a negative control.
Table 1.
Characteristics of healthy donors and convalescent COVID-19 patients utilized for IFN-γ ELISpot assays
| Unexposed (n = 20) | COVID-19 (n = 30) | |
|---|---|---|
| Age (years) | 23–63 (median = 30, IQR = 16.5) | 20-63 (median = 36, IQR = 22.5) |
| Gender | ||
| Male (%) | 25% (5/20) | 23.3% (7/30) |
| Female (%) | 75% (15/20) | 76.6% (23/30) |
| Sample collection date (range) | January 2015 to January 2020 | April 2020 to August 2020 |
| Disease severity | ||
| Mild (%) | N/A | 70% (21/30) |
| Moderate (%) | N/A | 20% (6/30) |
| Severe (%) | N/A | 10% (3/30) |
| Symptoms | ||
| Cough | N/A | 43.3% (13/30) |
| Fever | N/A | 40% (12/30) |
| Anosmia | N/A | 23.3% (7/30) |
| Dyspnea | N/A | 23.3% (7/30) |
| Diarrhea | N/A | 0.07% (2/30) |
| Mylagias | N/A | 36.7% (11/30) |
| Days post-symptom resolution at collection | N/A | 7-92 (median = 30.5, IQR = 24.25) |
| Past medical history | ||
| Hypertension | N/A | 16.7% (5/30) |
| Hyperlipidemia | N/A | 0.07% (2/30) |
| Diabetes | N/A | 0.07% (2/30) |
| Asthma | N/A | 0.03% (1/30) |
Figure 4.

CD8+ T cells from convalescent COVID-19 individuals recognize highly networked epitopes derived from structural and accessory proteins
(A) Location of highly networked HLA-stabilizing CD8+ T cell epitopes in non-structural proteins (NSPs; green) and structural proteins (SPs; purple) across the SARS-CoV-2 proteome.
(B) Representative IFN-γ ELISpot data for two pairs of healthy donors (HDs) and COVID-19 patients following incubation with DMSO, anti-CD3/CD28 antibodies, the CEF peptide pool, the highly networked NSP peptide pool (n = 56), the highly networked SP peptide pool (n = 53), and the combined NSP + SP peptide pool (n = 109).
(C) Magnitude of IFN-γ+ CD8+ T cell responses to the CEF peptide pool in HDs (open, n = 20) and COVID-19 patients (filled, n = 30). Mild (filled circles, n = 21) and moderate-to-severe COVID-19 patients (filled diamonds, n = 9) are denoted.
(D) Magnitude of IFN-γ+ CD8+ T cell responses to the highly networked SARS-CoV-2 NSP epitope pool (green), the SP epitope pool (purple), and the combined NSP + SP epitope pool (blue) in HDs (open, n = 20) and COVID-19 patients (filled, n = 30). The number of positive responders relative to the total number of individuals analyzed is indicated.
(E) Magnitude of IFN-γ+ CD8+ T cell responses against the highly networked SARS-CoV-2 SP epitope pool (purple) in mild (n = 21) and moderate-to-severe COVID-19 patients (n = 9).
(F) Comparison of the magnitude of IFN-γ+ CD8+ T cell responses to SP and NSP + SP peptide pools in COVID-19 SP peptide pool responders (n = 15).
Statistical comparison was made using a Wilcoxon matched-pairs test. All other statistical comparisons were made using a Mann-Whitney U test. Calculated p values were as follows: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See also Table 1.
While CEF-specific CD8+ T cell responses were not significantly different between the two patient groups (Figure 4C), we observed significant differences in IFN-γ+ CD8+ T cell responses to highly networked HLA-stabilizing epitopes in the SP peptide pool (n = 53 epitopes) (1/20 HDs versus 15/30 COVID-19; p = 0.0003) and combined NSP + SP pool (n = 109 epitopes) (3/20 HDs versus 13/30 COVID-19; p = 0.001), but not the NSP pool alone (n = 56 epitopes) (2/20 HDs versus 8/30 COVID-19; p = 0.2627) (Figure 4D). This is consistent with prior reports that observed stronger SARS-CoV-2-specific CD8+ T cell responses to epitopes derived from higher-abundance SPs than from NSPs (Grifoni et al., 2020; Le Bert et al., 2020). In addition, we observed a higher average magnitude of IFN-γ+ CD8+ T cell response to the SP pool in convalescent COVID-19 patients with moderate-to-severe disease (n = 9) than in those with mild disease (n = 21) (Figure 4E), consistent with prior work (Peng et al., 2020). In patients who responded to the highly networked SP peptide pool, we also observed a decrease in CD8+ T cell reactivity of individual participants when incubated with the combination SP + NSP peptide pool (13/15 individuals) (Figure 4F). Importantly, these data demonstrate that highly networked epitopes from structural and accessory proteins that stabilize HLA molecules, which can be encompassed within 15 viral regions (Table 2 ), are immunogenic in natural infection and therefore viable candidates for a broadly protective T-cell-based vaccine.
Table 2.
Highly networked regions within SARS-CoV-2 structural and accessory proteins that contain stabilizing CD8+ T cell epitopes with global HLA coverage
| Amino acid sequence | Protein | Domain | AA coordinates |
|---|---|---|---|
| RGVYYPDKVFRSSV | spike | N-terminal S1 domain | 34–47 |
| KGIYQTSNFRVQPTESIVRF | spike | S1-RBD hinge domain | 310–329 |
| KLNDLCFTNVY | spike | RBD | 386–396 |
| FELLHAPATV | spike | RBD | 515–524 |
| TSNEVAVLYQDVNCTEV | spike | C-terminal S1 domain | 604–620 |
| TEILPVSMTKTSVDCTMY | spike | N-terminal S2 domain | 724–741 |
| PLLTDEMIAQYTSAL | spike | N-terminal S2 domain | 863–877 |
| YRFNGIGV | spike | N-terminal S2 domain | 904–911 |
| ALNTLVKQLSSNFGAISSVLNDILSRL | spike | S2 HR1 domain | 958–984 |
| KRVDFCGKGYHLMSFPQSAPHGVVF | spike | S2 CD domain | 1,038–1,062 |
| GVFVSNGTHW | spike | C-terminal S2 domains | 1,093–1,102 |
| NPLLYDANYFLCWHTNCYDYCIPYNSVTSSI | ORF3A | domains IV-VI | 137–167 |
| RLFARTRSMWSFNPETNILLNVPLHGTILTR PLLESELVIGAVILRGHLRIAGHHL | membrane | C-terminal domain | 101–156 |
| NSSPDDQIGYY | nucleocapsid | RNA-binding domain | 78–88 |
| RRGPEQTQGNFGDQELIRQGTDYKHWPQI AQFAPSASAFFGM | nucleocapsid | C-terminal dimerization domain | 277–318 |
Recipients of mRNA-based vaccines have reduced CD8+ T cell reactivity to highly networked epitopes
Given that mRNA-based vaccines for SARS-CoV-2 have been widely distributed, we sought to determine whether vaccine recipients have CD8+ T cell responses to highly networked epitopes from the viral spike protein. We therefore assessed the reactivity of CD8+ T cells from individuals who were at least 14 days post receipt of two doses of either the BNT162b2 (n = 13) or mRNA-1273 (n = 10) vaccines (Table S5). Stimulation of PBMCs with a full overlapping spike peptide pool yielded robust IFN-γ+ responses for recipients of both vaccines (18/23 vaccine recipients), as has previously been described (Jackson et al., 2020; Sahin et al., 2020) (Figure 5 A). However, spike peptide stimulation of PBMCs following depletion of CD4+ T cells led to markedly lower IFN-γ+ T cell magnitude and reactivity (9/23 responders) (Figures 5A and 5B), illustrating the preferential induction of spike-specific CD4+ T cells. Importantly, the magnitude of CEF-specific CD8+ T cell responses was maintained following CD4+ depletion (Figure 5C), indicating no significant loss in assay sensitivity for spike-specific CD8+ T cells. While ∼39% of mRNA-based vaccine recipients had detectable CD8+ T cell responses to the full spike peptide pool, an even smaller number had reactivity to a highly networked spike epitope pool (6/23 responders) (Figure 5D). Interestingly, three of the six vaccine recipients with responses to highly networked spike epitopes were individuals with prior SARS-CoV-2 infection (Table S5), further illustrating the immunogenicity of these epitopes during natural infection but the modest CD8+ T cell reactivity after mRNA-based vaccination.
Figure 5.

Recipients of mRNA-based vaccines have reduced CD8+ T cell reactivity to highly networked spike epitopes
(A) Representative IFN-γ ELISpot data for PBMCs from two mRNA-based vaccine recipients with and without CD4+ T cells following incubation with DMSO, anti-CD3/CD28 antibodies, the CEF peptide pool, the highly networked SP peptide pool (n = 53), the highly networked spike peptide pool (n = 28), and the full overlapping spike peptide pool.
(B) Comparison of the magnitude of IFN-γ+ T cell responses against full overlapping spike peptide pool before (open circles) and after CD4+ T cell depletion (filled circles) for 23 mRNA vaccine recipients. Blue circles represent individuals with prior SARS-CoV-2 infection. The number of positive responders relative to the total number of vaccinated individuals analyzed is depicted above each dataset.
(C) Comparison of the magnitude of IFN-γ+ T cell responses against the CEF peptide pool before (open circles) and after (filled circles) CD4+ T cell depletion.
(D) Comparison of the magnitude of IFN-γ+ CD8+ T cell responses reactive to full overlapping spike pool (gray) and highly networked SARS-CoV-2 spike epitope pool (red, n = 28 peptides).
(E) Representative CD8+ T cell responses after 6-day incubation of CFSE-loaded PBMCs with DMSO, anti-CD3/CD28 antibodies, the CEF peptide pool, the highly networked spike peptide pool, and the full overlapping spike peptide pool for three mRNA-based vaccine recipients, which include the two individuals shown in (A) and an additional vaccinated individual with prior COVID-19 infection.
(F) Comparison of the magnitude of proliferative CD8+ T cell responses (%CD8 CFSE low) following incubation with the CEF peptide pool (orange), the full overlapping spike peptide pool (gray), and the highly networked spike peptide pool (red). A positive response was defined as one with %CD8+ CFSE low cells at least 1.5× greater than background wells and greater than 0.2% CD8+ CFSE low cells in magnitude following background subtraction.
Statistical comparisons were made using a Wilcoxon matched-pairs test. Calculated p values were as follows: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See also Table S4.
To evaluate for the presence of functional memory CD8+ T cell responses to highly networked spike epitopes, we also performed a 6-day carboxyfluorescein succinimidyl ester (CFSE)-based proliferation assay. Similar to the IFN-γ ELISpot (Figure 5E), mRNA-based vaccine recipients had low and frequently undetectable proliferative CD8+ T cell responses to both full spike antigen (8/23 responders) and highly networked spike epitopes (4/23 responders) (Figure 5F). This suggests that mRNA-based vaccines induce reduced levels of CD8+ T cell reactivity against mutationally constrained regions of the spike protein, indicating an opportunity for additional vaccines that can elicit CD8+ T cell responses to highly networked epitopes.
Discussion
The development of immunogens that can induce CD8+ T cell responses to mutationally constrained epitopes could greatly augment current vaccines for SARS-CoV-2 given the emergence of variants that escape convalescent plasma and vaccine-induced antibody responses (Garcia-Beltran et al., 2021; Hoffmann et al., 2021; Madhi et al., 2021; Wang et al., 2021; Wibmer et al., 2021). Here, we combined structure-based network analysis and HLA class I peptide stability assessments to define a globally relevant set of CD8+ T cell epitopes across the SARS-CoV-2 proteome that are structurally constrained from mutation, conserved across VOCs and sarbecoviruses, and recognized by individuals who have recovered from COVID-19 disease. These results thereby provide a guide for the rational development of a global T-cell-based vaccine to counteract emerging SARS-CoV-2 variants and future SARS-like coronaviruses.
The ability of structure-based network analysis to define mutation intolerant residues in SARS-CoV-2 leverages prior work applying the approach to HIV (Gaiha et al., 2019). We confirmed our network-based predictions using a spike pseudotyped lentiviral infectivity assay (Crawford et al., 2020), and the strong agreement between our computational network scores and experimentally derived effects of mutation on spike RBD folding and ACE2 binding (Starr et al., 2020) further illustrated its predictive ability. Network score also outperformed sequence entropy in its identification of functionally important RBD residues. Moreover, as a complement to time- and resource-intensive approaches such as deep mutational scanning, structure-based network analysis was able to be rapidly deployed, allowing for the prompt prediction of mutationally constrained residues for ∼44% of the SARS-CoV-2 proteome.
Sequence analysis of SARS-CoV-2 demonstrated that most of the new sequence variation over the course of the pandemic has occurred in sites with low network scores. This suggests that viral regions identified by network analysis may have the potential to remain constrained from mutation even as the pandemic progresses. Moreover, while deep sequencing of primary SARS-CoV-2 isolates revealed subversion of CD8+ T cell surveillance through HLA-class-I-restricted epitope mutations (Agerer et al., 2021), we found that highly networked HLA-A∗02 epitopes exhibited significantly lower levels of mutational frequency than non-networked epitopes, indicating putative constraints on SARS-CoV-2 evasion from highly networked CD8+ T cell epitope responses.
The use of HLA class I peptide stability assays to define CD8+ T cell epitopes within highly networked regions builds on previous work linking epitope immunogenicity and immunodominance hierarchies to HLA class I stabilizing capacity (Harndahl et al., 2012; Rasmussen et al., 2016; Kaseke et al., 2021). The demonstrable CD8+ T cell reactivity to these epitopes in recovered individuals confirmed their immunogenicity and the agreement between the highly networked epitopes identified in this study and those found by orthogonal methodologies further illustrated the value of the HLA-peptide stability assays (Ferretti et al., 2020; Peng et al., 2020; Schulien et al., 2021). The higher magnitude responses against highly networked epitopes from higher abundance structural and accessory proteins was also consistent with prior reports examining CD8+ T cell targeting in SARS-CoV-1 (Li et al., 2008) and SARS-CoV-2 (Grifoni et al., 2020; Le Bert et al., 2020).
In addition to their immunogenicity, highly networked SARS-CoV-2 epitopes exhibited strong sequence homology with circulating variants and sarbecoviruses. The limited sequence homology between highly networked epitopes and common cold CoVs likely explains the absence of detectable CD8+ T cell responses in HDs but also underscores the benefit that a networked T cell vaccine could provide for uninfected individuals. Moreover, the observation that mRNA-based vaccine recipients had modest CD8+ T cell responses to highly networked spike epitopes suggests that there is also an opportunity to augment vaccine-induced immunity. Given that highly networked regions from structural and accessory proteins (which harbor 53 epitopes for 18 HLA class I alleles) can be encompassed within 315 amino acid residues (Table 2), the size of a focused immunogen would not preclude its incorporation into any number of vaccine delivery platforms. We therefore envision that this networked T cell immunogen could be delivered alongside a spike-based vaccine as a tandem molecule or as a separate co-delivered physical entity. This would ensure ample CD4+ T cell help to facilitate the induction of de novo responses to highly networked CD8+ T cell epitopes, which ideally would be highly proliferative with robust expression of cytotoxic effector molecules.
In summary, we integrated structure-based network analysis and HLA class I peptide stability assessments to define a global set of mutationally constrained CD8+ T cell epitopes. While variant spike vaccines may provide immunity for circulating VOCs, the induction of CD8+ T cell responses to highly networked epitopes could offer additional protection against newly emerging variants. A highly networked T cell vaccine would therefore be highly complementary to ongoing antibody-based vaccine efforts to provide the global population with broad immunity against continued SARS-CoV-2 evolution and future SARS-like coronaviruses.
Limitations of study
The present study assessed the in vivo mutational frequencies of highly networked and non-networked epitopes restricted by HLA-A∗02 using available sequence data (Agerer et al., 2021). Future studies evaluating epitopes restricted by additional HLA alleles will help further confirm the mutational constraints of highly networked epitopes. In addition, while the number of convalescent and HDs evaluated for CD8+ T cell responses to highly networked epitopes was limited, it was similar to previous studies characterizing cellular immunity to SARS-CoV-2 (Grifoni et al., 2020; Le Bert et al., 2020; Peng et al., 2020; Rodda et al., 2021). The present study only used IFN-γ ELISpot assays to evaluate CD8+ T cell reactivity within convalescent individuals, which may not fully detect CD8+ T cell responses or assess the full complement of CD8+ T cell functions. While the magnitude of IFN-γ responses that we observed in our study was comparable to what has been previously published for other convalescent COVID-19 cohorts (Le Bert et al., 2020; Peng et al., 2020), additional assays, such as the CD8+ T cell activation-induced marker assay (Grifoni et al., 2020), CFSE-based proliferation assay, or intracellular cytokine staining following peptide stimulation, could also be utilized.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-human HLA ABC (clone W6/32) labeled with APC fluorophore | Biolegend | Cat# 311410; RRID:AB_314879 |
| Mouse monoclonal anti-human CD3 (clone OKT3) | Biolegend | Cat# 317302; RRID:AB_571927 |
| Mouse monoclonal anti-human CD28 (clone CD28.2) | Biolegend | Cat# 302902; RRID:AB_314304 |
| Mouse monoclonal anti-human CD8 (clone SK1) labeled with APC fluorophore | Biolegend | Cat# 980904; RRID:AB_2616624 |
| Mouse monoclonal anti-human CD3 (clone SK7) labeled with PE-Cy7 fluorophore | Biolegend | Cat# 344816; RRID:AB_10640737 |
| LIVE/DEAD Violet Viability | Life Technologies | Cat# L34960 |
| CellTrace CFSE Cell Proliferation | Life Technologies | Cat# C34554 |
| Biological samples | ||
| PBMC Specimens: Healthy Donors | Ragon Institute | N/A |
| PBMC Specimens: COVID-19 Patients | MassCPR | N/A |
| PBMC Specimens: mRNA Vaccine Recipients | MGH | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV-2 Networked Epitope Peptide Pools | MGH Peptide Core | N/A |
| PepMix Full SARS-CoV-2 Spike Peptide Pool | JPT | PM-WCPV-S |
| CEF Extended Peptide Pool | Mabtech | Cat# 3618-1 |
| β2-Microglobulin | Sino Biological | Cat# 11976-H08H |
| Critical commercial assays | ||
| Human IFN-gamma ELISpot Basic Kit | MABTECH | Cat# 3420-2A |
| Human CD4 microbeads | Miltenyi Biotec | Cat# 130-045-101 |
| Experimental models: Cell lines | ||
| Human: 721.221 cells + Cas9 + HLA + sgRNA TAP | Kaseke et al., 2021 | N/A |
| Human: HEK293T cells | ATCC | N/A |
| Human: HEK293T-humanACE2 cells | A gift from Alex Balazs, Ragon Institute | N/A |
| Oligonucleotides | ||
| Primers for site-directed mutagenesis of HDM-SARS2-Spike-delta21, see Table S2 | This paper | Table S2 |
| Recombinant DNA | ||
| HDM-SARS2-Spike-delta21 | Crawford et al., 2020 | Addgene #155130 |
| pHAGE-CMV-Luc2-IRES-ZsGreen-W | BEI | NR-52516 |
| HDM-Hgpm2 | BEI | NR-52517 |
| HDM-tat1b | BEI | NR-52518 |
| pRC-CMV-Rev1b | BEI | NR-52519 |
| pHEF-VSVG | (Coleman et al., 2003) | Addgene #22501 |
| Software and algorithms | ||
| Structure-based Network Analysis Pipeline | Gaiha et al., 2019 | https://doi.org/10.5281/zenodo.2597484. |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Gaurav D. Gaiha (ggaiha@mgh.harvard.edu).
Materials availability
All requests for resources and reagents should be directed to and will be fulfilled by the lead contact. All reagents will be made available on request after completion of a Materials Transfer Agreement.
Data and code availability
All data supporting the findings of this study available within the paper and available from the lead contact upon request. The code used during this study to generate network scores is archived at Zenodo (https://zenodo.org/record/2597484). Viral sequence data of highly networked and non-networked epitope variants from primary SARS-CoV-2 isolates is available in the supplementary materials of the primary manuscript (Agerer et al., 2021).
Experimental model and subject details
Cell lines
The female cell line HEK293T were used for lentivirus production and ACE2-expressing HEK293T cells (a gift from A. Balazs, Ragon Institute) used for lentivirus infection were maintained in advanced DMEM (Sigma-Aldrich) supplemented with 10% FBS, 1X Penicillin-Streptomycin-L-Glutamine mixture (GIBCO), 1X non-essential amino acids (GIBCO), 1X sodium pyruvate (GIBCO), and 1X HEPES buffer (Corning) (D10). The human female B cell lines 721.221 were generated previously by γ-radiation of 721 cells and do not express HLA A and B alleles (Shimizu and DeMars, 1989). These cell lines were maintained in RPMI-1640 medium (Sigma-Aldrich) supplemented with 10% (v/v) FBS (Sigma-Aldrich) and 1X Penicillin-Streptomycin-L-Glutamine mixture (GIBCO). TAP-deficient mono-allelic HLA class I-expressing 721.221 cells were generated as described previously (please see companion manuscript) and maintained in 5ug/mL blasticidin (Invivogen), 0.5 ug/ml puromycin (Invivogen) and 1.5 mg/ml G418 (Invivogen).
Human subjects
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy human volunteers, SARS-CoV-2 infected patients and mRNA-based vaccine recipients by Ficoll gradient separation from ACD tubes. They were then cryopreserved and stored in liquid nitrogen prior to experimental use. The study was approved by the MGH Institutional Review Board. All subjects were between 18-70 years of age, provided informed consent and were confirmed to have a test positive for SARS-CoV-2 using PCR with reverse transcription from an upper respiratory tract (nose and throat) swab tested at an accredited laboratory and a subsequent negative PCR test following symptom resolution at least 7 days prior to sample acquisition. For infected individuals, the degree of disease severity was identified as mild, severe or critical infection, according to recommendations from the World Health Organization. Patients were classified as having mild symptoms if they did not require oxygen (that is, their oxygen saturation was 94% or greater on ambient air) and if their symptoms were managed at home. Moderate-to-severe infection was defined as one of the following conditions in a patient confirmed as having COVID-19: respiratory distress with a respiratory rate of > 30 breaths per minute; blood oxygen saturation of < 94%; or arterial oxygen partial pressure/FiO2 < 300 mmHg. All mRNA-based vaccine recipients received two doses of either the Pfizer-BioNTech BNT162b2 or Moderna mRNA-1273 vaccines and were at least 14 days post-administration of the second vaccine dose.
Method details
SARS-CoV-2 protein structures
For the analysis of the SARS-CoV-2 proteome, the following PDB files were utilized: NSP3 ADP ribose phosphatase domain (PDB: 6W02), NSP3 papain-like protease (PDB: 6W9C), NSP5 3CL protease (PDB: 6YB7), NSP7 (PDB: 6M7I, Chain C). NSP8 (PDB: 6M7I, Chain B, D), NSP9 (PDB: 6W4B), NSP10 (6W4H, Chain B), NSP12 RNA-dependent RNA polymerase (6M7I, Chain A), NSP15 (PDB: 6W01), NSP16 (PDB: 6W4H, Chain A), Spike closed conformation (PDB: 6VXX), Nucleocapsid RNA-binding domain (PDB: 6VYO), Nucleocapsid dimerization domain (PDB: 6WJI), ORF3a (PDB: 6XDC), ORF7a (PDB: 6W37), Spike open conformation (PDB: 6VYB), and Spike receptor binding domain (PDB: 6M0J). The membrane structure was downloaded from from DeepMind (https://deepmind.com/research/open-source/computational-predictions-of-protein-structures-associated-with-COVID-19) on April 8, 2020. MODELER (https://salilab.org/modeller/) was used to create homology models for the envelope protein using SARS-CoV-1 envelope (PDB: 5X29) as a template. Water molecules and solvents were removed from each PDB file prior to analysis.
SARS-CoV-1 protein structures
For the analysis of the SARS-CoV-1 proteome, the following PDB files were utilized: NSP3 ADP ribose phosphatase domain (PDB: 2FAV), NSP3 papain-like protease (PDB: 5Y3Q), NSP5 3CL protease (PDB: 1Q2W), NSP7 (PDB: 6NUR, Chain C). NSP8 (PDB: 6NUR, Chain B, D), NSP9 (PDB: 1QZ8), NSP10 (2XYQ, Chain B), NSP12 RNA-dependent RNA polymerase (6NUR, Chain A), NSP15 (PDB: 2H85), NSP16 (PDB: 2XYQ, Chain A), Spike (PDB: 5XLR), Nucleocapsid RNA-binding domain (PDB: 1SSK), and Nucleocapsid dimerization domain (PDB: 2GIB). Water molecules and solvents were removed from each PDB file prior to analysis.
MERS-CoV protein structures
For the analysis of the MERS proteome, the following PDB files were utilized: NSP3 ADP ribose phosphatase domain (PDB: 5HOL), NSP3 papain-like protease (PDB: 4RNA), NSP5 3CL protease (PDB: 4WME), NSP10 (5YN5, Chain B), NSP15 (PDB: 5YVD), NSP16 (PDB: 5YN5, Chain A), Spike (PDB: 5X59), Nucleocapsid RNA-binding domain (PDB: 4UD1), and Nucleocapsid dimerization domain (PDB: 6G13). Water molecules and solvents were removed from each PDB file prior to analysis.
Structure-based network analysis
Individuals network scores were calculated as described previously (Gaiha et al., 2019). For multimeric proteins, degree-based network values (second-order degree, ligand binding) in the protein’s highest oligomeric state were utilized prior to calculation of a normalized Z-score. For node edge betweenness metrics, the maximum normalized Z-score from monomer or multimeric conformation was incorporated into the final network score calculation. For analyses with multiple structures utilized to capture different conformational states for the same oligomeric structure (e.g., 6VXX and 6VYB, closed and open conformations), network Z-scores were averaged. All molecular assemblies were generated using the online server PDBePISA (https://www.ebi.ac.uk/pdbe/pisa/).
Protein network visualization
The positions of the alpha carbons of protein residues of interest were parsed from the corresponding PDB structure files using a script written in the Perl language version 5.18.2. Subsequent data processing and visualization were performed in the R language, version 3.6.3. Network scores for residues were mapped to alpha carbons in the corresponding structures by position. In cases where the PDB structures used to generate the network scores differed from the PDB structures used for visualization, and where individual residue substitutions had occurred, the alpha carbon position for the substituted residue in the structure PDB file was used. Protein network structures were created using R’s igraph package version 1.2.5. Using the alpha carbon positions as X, Y, and Z coordinates, nodes representing residues, were plotted in three dimensions using the R rgl library version 0.100.50, which implements OpenGL. Where indicated, residue nodes were colored and scaled by their corresponding network score, and inter-residue interaction strengths were represented as the thickness of edges between nodes. Two-dimensional network views of interest as defined by aspect, zoom and rotation, were selected manually, then extracted using the rgl par3d function and saved for later use. Views of interest were exported as PNG files.
Calculation of epitope network scores
Epitope network scores were calculated as described previously (Gaiha et al., 2019). Briefly, network scores from individual amino acid residues within and neighboring a CD8+ T cell epitope were combined and averaged based on their involvement as either HLA anchor, TCR contact or peptide processing residues. HLA anchor residues were defined based on previous delineations for each HLA allele (Marsh et al., 1999). Putative TCR contact residues were considered to be all remaining non-HLA anchor residues, excluding position 1, based on previously reported frequencies of TCR-peptide contacts (Calis et al., 2012). Flanking residues were defined as the five residues N-terminal and C-terminal to the epitope (ten in total). These three quantities were then summed to generate an overall composite network score for each CD8+ T cell epitope using the following formula:
where NS i is the ith network score for HLA anchor residues 1 through a, NS j is the jth network score for TCR contact residues 1 through b and NS k is the kth network score for peptide processing residues 1 through c. The normalized epitope network score (Table S3) was calculated by subtracting the lowest epitope network score from all epitope scores, such that all values were greater than or equal to zero. The normalized network score was utilized when comparing patient responses such that no CTL response would be assigned a negative value.
Reference genomes
For the analysis of the highly stabilizing epitopes across human coronaviruses, the following reference genomes were utilized: bat coronavirus RaTG13 (GenBank: MN996532.1), SARS-CoV1 (GenBank: AY274119.3), MERS (GenBank: JX869059.2), HCoV-OC43 (GenBank: AY391777.1), HCoV-HKU1 (GenBank: AY884001.1), HCoV-229E (GenBank: KY684760.1), and HCoV-NL63 (NCBI Reference Sequence: NC_005831.2).
Shannon entropy and conservation scoring
SARS CoV-2 (total downloaded sequences, 45,603 in May 2020; 661,816 sequences in February 2021), sarbecovirus (SARS-CoV-1/Bat; 4,416 sequences), and MERS (7,737 sequences) were downloaded from NCBI. Sarbecovirus and MERS sequences were downloaded on May 18, 2020, as were initial SARS-CoV-2 sequences. Additional SARS-CoV-2 sequences were downloaded on February 6, 2021. Using the protein sequence derived from SARS-CoV-2 PDB structures as a reference in each protein sequence alignment, amino acid frequencies at each amino acid position were tabulated. Shannon entropy, H(p), was calculated based on the following formula (Lund et al., 2005): where p a is the proportion of amino acid a at a given position and q a is the background frequency of amino acid a.
Generation of SARS-CoV-2 spike mutants
HDM-SARS2-Spike-delta21 was a gift from Jesse Bloom (Addgene plasmid # 155130; http://addgene.org/155130; RRID: Addgene_155130) and was modified to express one of several individual mutations using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) according to the manufacturer’s instructions. Back-to-back 5′ oligonucleotide primers were utilized to engineer individual mutants (Table S1) within the HDM-SARS2-Spike-delta21 plasmid. Confirmation of successful mutagenesis was accomplished by complete plasmid sequencing (MGH Sequencing Core). Full-length viral plasmids were propagated in Stellar competent cells (Takara Bio) and DNA plasmid stocks were prepared using a QiaPrep spin miniprep kit (QIAGEN).
Generation of SARS-CoV-2 spike pseudotyped lentivirus
SARS-CoV-2 Spike pseudotyped lentivirus was produced as previously described (Crawford et al., 2020). Briefly, HEK293T cells were transfected with 1 μg pHAGE-CMV-Luc2-IRES-ZsGreen-W (BEI), a lentiviral backbone plasmid expressing luciferase under a CMV promoter and an IRES followed by ZsGreen, 0.22 μg HDM-Hgpm2 (BEI), a lentiviral helper plasmid expressing HIV Gag-Pol under a CMV promoter, 0.22 μg HDM-tat1b (BEI), a lentiviral helper plasmid expressing HIV Tat under a CMV promoter, 0.22 μg pRC-CMV-Rev1b (BEI), a lentiviral helper plasmid expressing HIV Rev under a CMV promoter, and 0.34 μg of the plasmid encoding HDM-SARS2-Spike-delta21 using polyethylenimine (Polyplus) in serum-free Dulbecco’s Modified Eagle’s Medium (Sigma-Aldrich) supplemented with 25 mM HEPES buffer (Corning). Media was changed to D10 24h post-transfection. After 48h, pseudotyped lentivirus was harvested by filtering supernatant through a 0.45 μm low protein binding durapore membrane (Millipore). Frozen aliquots were stored at −80°C and viral concentrations were quantified using the colorimetric Reverse Transcriptase Assay (Sigma-Aldrich). All packaging plasmids were propagated in DH5α cells (NEB).
SARS-CoV-2 spike pseudotyped lentiviral infectivity assay
HEK293T and ACE2-expressing HEK293T cells were seeded at a density of 1.25 × 104 cells/well into a 96-well plate one day prior to infection with 60 μL wild-type or mutant Spike pseudotyped lentivirus diluted two-fold in D10 with 5 μg/mL Polybrene Transfection Reagent (Millipore). 24h following infection, an additional 140 μL of D10 was added and cells were cultured at 37°C and 5% CO2 for 48h. Cells were harvested, stained with viability dye, fixed in 2% paraformaldehyde and subsequently analyzed for ZsGreen expression via flow cytometry using a BD LSR II (BD Biosciences). Flow cytometric data were analyzed using FlowJo software (v10.1r5).
Peptide synthesis reagents
Fmoc-protected amino acids and synthesis resin, 2-Chlorotrityl chloride were purchased from Akaal Organics (Long Beach, CA). Dimethylformamide (DMF), N-methyl pyrrolidone (NMP), Acetonitrile and Methyl-tert. Butyl Ether (MTBE) were purchased from Fisher Bioreagents (Fair Lawn, NJ). 2-(6-Chloro-1-H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU) was purchased from AAPPTEC (Louisville, KY). Piperidine and Dichloromethane (DCM) were from EMD-Millipore (Billerica, MA). Diisopropylethylamine (DIEA), N-Methyl-morpholine (NMM), Triisoprpopyl-silane, 3,6-dioxa-1,8-octanedithiol (DODT) and trifluoroacetic acid (TFA) were purchased from Sigma–Aldrich.
Peptide synthesis and analysis
Peptides were synthesized on an automated robotic peptide synthesizer (AAPPTEC, Model 396 Omega) by using Fmoc solid-phase chemistry (Behrendt et al., 2016) on 2-chlorotrityl chloride resin (Barlos et al., 1991). The C-terminal amino acids were loaded using the respective Fmoc-Amino Acids in the presence of DIEA. Unreacted sites on the resin were blocked using methanol, DIEA and DCM (15:5:80 v/v). Subsequent amino acids were coupled using optimized (to generate peptides containing more than 90% of the desired full-length peptides) cycles consisting of Fmoc removal (deprotection) with 25% Piperidine in NMP followed by coupling of Fmoc-AAs using HCTU/NMM activation. Each deprotection or coupling was followed by several washes of the resin with DMF to remove excess reagents. After the peptides were assembled and the final Fmoc group removed, peptide resin was then washed with dimethylformamide, dichloromethane, and methanol three times each and air-dried. Peptides were cleaved from the solid support and deprotected using odor free cocktail (TFA/triisopropyl silane/water/DODT; 94/2.5/2.5/1.0 v/v) for 2.5h at room temperature (Teixeira et al., 2002). Peptides were precipitated using cold methyl tertiary butyl ether (MTBE). The precipitate was washed 2 times in MTBE, dissolved in a solvent (0.1% trifluoroacetic acid in 30%Acetonitrile/70%water) followed by freeze drying. Peptides were characterized by Ultra Performance Liquid Chromatography (UPLC) and Matrix Assisted Laser Desorption/Ionization Mass Spectrometry (MALDI-MS). All peptides were dissolved initially in 100% DMSO at a concentration of 40 mM, prior to dilution at the appropriate concentration in RPMI-1640 medium.
HLA class I-peptide concentration-based stability assay
For concentration-based HLA class I-peptide stability binding assays, 5x104 TAP-deficient mono-allelic HLA class I-expressing 721.221 cells were incubated with peptides in concentrations ranging from 0.1 to 100μM, and 3μg/mL of β2 m (Sino Biological, Wayne, PA, USA), in RPMI-1640 medium overnight at 26°C/5% CO2 for 18 hours. Controls without peptide, but the corresponding concentration of DMSO, were performed in parallel. Following overnight incubation, cells were incubated at 37°C/5% CO2 prior to staining for viability and HLA class I surface expression with HLA-ABC APC antibody (1:100), and subsequent analysis by flow cytometry.
Ex vivo ELISpot assay
IFN-γ ELISpot assays were performed according to the manufacturer’s instructions (Mabtech). PBMCs were first depleted of CD4+ T cells by CD4 depletion kit (Miltenyi Biotec). 500,000 CD4-depleted PBMCs per test were then incubated with SARS-CoV-2 peptide pools at a final concentration of 1 μg/ ml for 16–18h. CEF peptide pool (Mabtech; 1ug/mL), anti-CD3 (Clone OKT3, Biolegend, 1ug/mL) and anti-CD28 Ab (Clone CD28.2, Biolegend, 1ug/mL) were used as positive controls. To quantify antigen-specific responses, mean spots of the DMSO control wells were subtracted from the positive wells, and the results were expressed as spot-forming units (SFU) per 106 PBMCs. Responses were considered positive if the results were > 5 SFU/106 PBMCs following control subtraction. If negative DMSO control wells had > 30 SFU/106 PBMCs or if positive control wells (anti-CD3/anti-CD28 stimulation) were negative, the results were excluded from further analysis.
CD8+ T cell proliferation assay
PBMCs were suspended at 1 × 106/mL in PBS and incubated at 37°C for 20 min with 0.5 uM carboxyfluorescein succinimidyl ester (CFSE; Life Technologies). After the addition of serum and washes with PBS, cells were resuspended at 1 × 106/mL and plated into 96-well U-bottom plates (Corning) at 200 uL volumes. Peptide pools were added at a final concentration of 1 ug/mL. On day 6, cells were harvested, washed with PBS + 2% Fetal Bovine Serum, and stained with anti-CD3-PE-Cy7 (clone SK1; BioLegend), anti-CD8 APC (clone SK7; BioLegend), and LIVE/DEAD violet viability dye (Life Technologies). Cells were washed and fixed in 2% paraformaldehyde, prior to flow cytometric analysis on a BD LSR II (BD Biosciences). A positive response was defined as one with a percentage of CD3+ CD8+ CFSE low cells at least 1.5x greater than the highest of three negative-control wells and greater than 0.2% CD8+ CFSE low cells in magnitude following background subtraction.
Quantification and statistical analysis
The generation of dot plots, nonparametric statistical analysis, correction for multiple comparisons and non-parametric correlations (Spearman) were performed using the statistical programs in Graphpad Prism version 8.0. Differences between groups were evaluated using the non-parametric Mann Whitney U t test and Kruskal-Wallis test with Dunn’s post hoc analyses for correction of multiple comparisons, as indicated. Paired analyses were performed using the non-parametric Wilcoxon matched-pairs signed rank test. Comparisons of viral variants with allelic frequencies greater than a threshold value were performed using Fisher’s exact test.
Consortia
The members of the Massachusetts Consortium of Pathogen Readiness (MassCPR) Specimen Working Group are Betelihem A. Abayneh, Patrick Allen, Galit Allter, Diane Antille, Katrina Armstrong, Alejandro Balazs, Max Barbash, Siobhan Boyce, Joan Braley, Karen Branch, Katherine Broderick, George Daley, Ashley Ellman, Liz Fedirko, Keith Flaherty, Jeanne Flannery, Pamela Forde, Elise Gettings, David Golan, Amanda Griffin, Sheila Grimmel, Kathleen Grinke, Kathryn Hall, Meg Healey, Howard Heller, Deborah Henault, Grace Holland, Chantal Kayitesi, Evan C. Lam, Vlasta LaValle, Yuting Lu, Sara Luthern, Jordan Marchewska, Brittni Martino, Ilan Millstrom, Noah Miranda, Christian, Nambu, Susan Nelson, Marjorie Noone, Claire O’Callaghan, Christine Ommerborn, Lois Chris Pacheco, Nicole Phan, Falisha A Porto, Alexandra Reissis, Francis Ruzicka, Edward Ryan, Katheleen Selleck, Arlene Sharpe, Christianne Sharr, Sue Slaugenhaupt, Kimberly Smith Sheppard, Elizabeth Suschana, Vivine Wilson, Daniel Worrall. Those who processed samples are Alicja Piechocka-Trocha, Kristina Lefteri, Matt Osborn, Julia Bals, Yannic C. Bartsch, Nathalie Bonheur, Timothy M. Caradonna, Josh Chevalier, Fatema Chowdhury, Thomas J. Diefenbach, Kevin Einkauf, Jon Fallon, Jared Feldman, Kelsey K. Finn, Pilar Garcia-Broncano, Ciputra Adijaya Hartana, Blake M. Hauser, Chenyang Jiang, Paulina Kaplonek, Marshall Karpell, Eric C. Koscher, Xiaodong Lian, Hang Liu, Jinqing Liu, Ngoc L. Ly, Ashlin R. Michell, Yelizaveta Rassadkina, Kyra Seiger, Libera Sessa, Sally Shin, Nishant Singh, Weiwei Sun, Xiaoming Sun, Hannah J. Ticheli, Michael T. Waring, and Alex L. Zhu.
Acknowledgments
We thank Alejandro Balazs and Evan Lam for assistance with pseudotyped lentivirus infectivity assays; Maia Pavlovic, David Gregory, and Mark Poznansky for access to COVID-19 vaccinee specimens; and Shiv Pillai, Vinay Mahajan, and David Collins for their scientific advice. Access to convalescent patient samples was facilitated by the MassCPR. This study was supported by NIH grants P01 DK011794-51A1 (A.K.), R01AI149704 (B.D.W.), UM1AI144462 (G.D.G. and B.D.W.), and DP2AI154421 (G.D.G.) and a grant from the MassCPR (B.D.W. and G.D.G.). Additional support was provided by the Howard Hughes Medical Institute (B.D.W.); the Ragon Institute of MGH, MIT and Harvard (B.D.W. and G.D.G.); the Mark and Lisa Schwartz Foundation and Enid Schwartz (B.D.W.); and Sandy and Paul Edgerly. E.J.R. is supported by the Heed Ophthalmic Foundation. G.D.G. is supported by the Bill and Melinda Gates Foundation, a Burroughs Wellcome Career Award for Medical Scientists, and the Gilead HIV Research Scholars Program. This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research. The graphical abstract was prepared using BioRender.
Author contributions
Conceptualization, A.N., E.J.R., and G.D.G.; methodology, A.N., E.J.R., and G.D.G.; software, A.N. and E.J.R.; investigation, A.N., E.J.R., C.K., R.J.P., A.K., D.K., J.U., N.K.S., A.B., R.T.M., F.S., M.T.W., A.T., W.C.B., V.N., and G.D.G.; writing – original draft, A.N., E.J.R., and G.D.G.; writing – review & editing, A.N., E.J.R., C.K., R.J.P., A.K., D.K., J.U., N.K.S., R.T.M., A.T., V.N., M.C., B.D.W., and G.D.G.; funding acquisition, A.J.I., M.C., B.D.W., and G.D.G.; resources, A.K., M.C., B.D.W., and G.D.G.; supervision, M.T.W., A.J.I, M.C., B.D.W., and G.D.G.
Declaration of interests
E.J.R. and G.D.G. have filed patent application PCT/US2021/028245.
Inclusion and diversity
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
Published: June 30, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.06.029.
Supplemental information
Epitope mutations within SARS-CoV-2 variants of concern are bolded.
References
- Agerer B., Koblischke M., Gudipati V., Montaño-Gutierrez L.F., Smyth M., Popa A., Genger J.-W., Endler L., Florian D.M., Mühlgrabner V., et al. SARS-CoV-2 mutations in MHC-I-restricted epitopes evade CD8+ T cell responses. Sci. Immunol. 2021;6:eabg6461. doi: 10.1126/sciimmunol.abg6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden L.R., El Sahly H.M., Essink B., Kotloff K., Frey S., Novak R., Diemert D., Spector S.A., Rouphael N., Creech C.B., et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021;384:403–427. doi: 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlos K., Chatzi O., Gatos D., Stavropoulos G. 2-Chlorotrityl chloride resin. Studies on anchoring of Fmoc-amino acids and peptide cleavage. Int. J. Pept. Protein Res. 1991;37:513–520. [PubMed] [Google Scholar]
- Behrendt R., White P., Offer J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016;22:4–27. doi: 10.1002/psc.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calis J.J.A., de Boer R.J., Keşmir C. Degenerate T-cell recognition of peptides on MHC molecules creates large holes in the T-cell repertoire. PLoS Comput. Biol. 2012;8:e1002412. doi: 10.1371/journal.pcbi.1002412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R., Fett C., Zhao J., Meyerholz D.K., Perlman S. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J. Virol. 2014;88:11034–11044. doi: 10.1128/JVI.01505-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian S., Potdar V., Jadhav S., Yadav P., Gupta N., Das M., Rakshit P., Singh S., Abraham P., Panda S., et al. Convergent evolution of SARS-CoV-2 spike mutations, L452R, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. bioRxiv. 2021 doi: 10.1101/2021.04.22.440932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman J.E., Huentelman M.J., Kasparov S., Metcalfe B.L., Paton J.F.R., Katovich M.J., Semple-Rowland S.L., Raizada M.K. Efficient large-scale production and concentration of HIV-1-based lentiviral vectors for use in vivo. Physiol. Genomics. 2003;12:221–228. doi: 10.1152/physiolgenomics.00135.2002. [DOI] [PubMed] [Google Scholar]
- Crawford K.H.D., Eguia R., Dingens A.S., Loes A.N., Malone K.D., Wolf C.R., Chu H.Y., Tortorici M.A., Veesler D., Murphy M., et al. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses. 2020;12:513. doi: 10.3390/v12050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies N.G., Abbott S., Barnard R.C., Jarvis C.I., Kucharski A.J., Munday J.D., Pearson C.A.B., Russell T.W., Tully D.C., Washburne A.D., et al. CMMID COVID-19 Working Group. COVID-19 Genomics UK (COG-UK) Consortium Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021;372:eabg3055. doi: 10.1126/science.abg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti A.P., Kula T., Wang Y., Nguyen D.M.V., Weinheimer A., Dunlap G.S., Xu Q., Nabilsi N., Perullo C.R., Cristofaro A.W., et al. Unbiased Screens Show CD8+ T Cells of COVID-19 Patients Recognize Shared Epitopes in SARS-CoV-2 that Largely Reside outside the Spike Protein. Immunity. 2020;53:1095–1107.e3. doi: 10.1016/j.immuni.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folegatti P.M., Ewer K.J., Aley P.K., Angus B., Becker S., Belij-Rammerstorfer S., Bellamy D., Bibi S., Bittaye M., Clutterbuck E.A., et al. Oxford COVID Vaccine Trial Group Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet. 2020;396:467–478. doi: 10.1016/S0140-6736(20)31604-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiha G.D., Rossin E.J., Urbach J., Landeros C., Collins D.R., Nwonu C., Muzhingi I., Anahtar M.N., Waring O.M., Piechocka-Trocha A., et al. Structural topology defines protective CD8+ T cell epitopes in the HIV proteome. Science. 2019;364:480–484. doi: 10.1126/science.aav5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Beltran W.F., Lam E.C., St Denis K., Nitido A.D., Garcia Z.H., Hauser B.M., Feldman J., Pavlovic M.N., Gregory D.J., Poznansky M.C., et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell. 2021;184:2523. doi: 10.1016/j.cell.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney A.J., Starr T.N., Gilchuk P., Zost S.J., Binshtein E., Loes A.N., Hilton S.K., Huddleston J., Eguia R., Crawford K.H.D., et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe. 2021;29:44–57.e9. doi: 10.1016/j.chom.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S., et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur M., Taka E., Yilmaz S.Z., Kilinc C., Aktas U., Golcuk M. Conformational transition of SARS-CoV-2 spike glycoprotein between its closed and open states. J. Chem. Phys. 2020;153:075101. doi: 10.1063/5.0011141. [DOI] [PubMed] [Google Scholar]
- Harndahl M., Rasmussen M., Roder G., Dalgaard Pedersen I., Sørensen M., Nielsen M., Buus S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur. J. Immunol. 2012;42:1405–1416. doi: 10.1002/eji.201141774. [DOI] [PubMed] [Google Scholar]
- Hoffmann M., Hofmann-Winkler H., Krüger N., Kempf A., Nehlmeier I., Graichen L., Sidarovich A., Moldenhauer A.-S., Winkler M.S., Schulz S., et al. SARS-CoV-2 variant B.1.617 is resistant to Bamlanivimab and evades antibodies induced by infection and vaccination. bioRxiv. 2021 doi: 10.1101/2021.05.04.442663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson L.A., Anderson E.J., Rouphael N.G., Roberts P.C., Makhene M., Coler R.N., McCullough M.P., Chappell J.D., Denison M.R., Stevens L.J., et al. An mRNA vaccine against SARS-CoV-2—preliminary report. N. Engl. J. Med. 2020;383:1920–1931. doi: 10.1056/NEJMoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaseke C., Park R.J., Singh N.K., Koundajkian D., Bashirova A., Garcia Beltran W.F., Takou Mbah O.C., Ma J., Senjobe F. HLA class I-peptide stability mediates CD8+ T cell immunodominance hierarchies and facilitates HLA-associated immune control of HIV. Cell Rep. 2021;36 doi: 10.1016/j.celrep.2021.109378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keech C., Albert G., Cho I., Robertson A., Reed P., Neal S., Plested J.S., Zhu M., Cloney-Clark S., Zhou H., et al. Phase 1–2 Trial of a SARS-CoV-2 Recombinant Spike Protein Nanoparticle Vaccine. N. Engl. J. Med. 2020;383:2320–2332. doi: 10.1056/NEJMoa2026920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V., Singh J., Hasnain S.E., Sundar D. Possible link between higher transmissibility of B.1.617 and B.1.1.7 variants of SARS-CoV-2 and increased structural stability of its spike protein and hACE2 affinity. bioRxiv. 2021 doi: 10.1101/2021.04.29.441933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bert N., Tan A.T., Kunasegaran K., Tham C.Y.L., Hafezi M., Chia A., Chng M.H.Y., Lin M., Tan N., Linster M., et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584:457–462. doi: 10.1038/s41586-020-2550-z. [DOI] [PubMed] [Google Scholar]
- Li C.K.-F., Wu H., Yan H., Ma S., Wang L., Zhang M., Tang X., Temperton N.J., Weiss R.A., Brenchley J.M., et al. T cell responses to whole SARS coronavirus in humans. J. Immunol. 2008;181:5490–5500. doi: 10.4049/jimmunol.181.8.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Lund O., Nielsen M., Brunak S., Lundegaard C., Kesmir C. MIT Press; 2005. Immunological Bioinformatics. [Google Scholar]
- Madhi S.A., Baillie V., Cutland C.L., Voysey M., Koen A.L., Fairlie L., Padayachee S.D., Dheda K., Barnabas S.L., Bhorat Q.E., et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. N. Engl. J. Med. 2021;384:1885–1898. doi: 10.1056/NEJMoa2102214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh S.G.E., Parham P., Barber L.D. Elsevier; 1999. The HLA FactsBook. [Google Scholar]
- McMahan K., Yu J., Mercado N.B., Loos C., Tostanoski L.H., Chandrashekar A., Liu J., Peter L., Atyeo C., Zhu A., et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature. 2021;590:630–634. doi: 10.1038/s41586-020-03041-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMichael A.J., Carrington M. Topological perspective on HIV escape. Science. 2019;364:438–439. doi: 10.1126/science.aax4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirson T., Bomze D., Markel G. Structural basis of SARS-CoV-2 spike protein induced by ACE2. Bioinformatics. 2020;37:929–936. doi: 10.1093/bioinformatics/btaa744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D., Yount B.L., Jr., Debbink K., Agnihothram S., Gralinski L.E., Plante J.A., Graham R.L., Scobey T., Ge X.-Y., Donaldson E.F., et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015;21:1508–1513. doi: 10.1038/nm.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D., Yount B.L., Jr., Sims A.C., Debbink K., Agnihothram S.S., Gralinski L.E., Graham R.L., Scobey T., Plante J.A., Royal S.R., et al. SARS-like WIV1-CoV poised for human emergence. Proc. Natl. Acad. Sci. USA. 2016;113:3048–3053. doi: 10.1073/pnas.1517719113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng O.-W., Chia A., Tan A.T., Jadi R.S., Leong H.N., Bertoletti A., Tan Y.-J. Memory T cell responses targeting the SARS coronavirus persist up to 11 years post-infection. Vaccine. 2016;34:2008–2014. doi: 10.1016/j.vaccine.2016.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Pérez Marc G., Moreira E.D., Zerbini C., et al. C4591001 Clinical Trial Group Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M., Fenoy E., Harndahl M., Kristensen A.B., Nielsen I.K., Nielsen M., Buus S. Pan-Specific Prediction of Peptide-MHC Class I Complex Stability, a Correlate of T Cell Immunogenicity. J. Immunol. 2016;197:1517–1524. doi: 10.4049/jimmunol.1600582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodda L.B., Netland J., Shehata L., Pruner K.B., Morawski P.A., Thouvenel C.D., Takehara K.K., Eggenberger J., Hemann E.A., Waterman H.R., et al. Functional SARS-CoV-2-Specific Immune Memory Persists after Mild COVID-19. Cell. 2021;184:169–183.e17. doi: 10.1016/j.cell.2020.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoff J., Gray G., Vandebosch A., Cárdenas V., Shukarev G., Grinsztejn B., Goepfert P.A., Truyers C., Fennema H., Spiessens B., et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19. N. Engl. J. Med. 2021;384:2187–2201. doi: 10.1056/NEJMoa2101544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U., Muik A., Vogler I., Derhovanessian E., Kranz L.M., Vormehr M., Quandt J., Bidmon N., Ulges A., Baum A., et al. BNT162b2 induces SARS-CoV-2-neutralising antibodies and T cells in humans. medRxiv. 2020 doi: 10.1101/2020.12.09.20245175. [DOI] [Google Scholar]
- Schulien I., Kemming J., Oberhardt V., Wild K., Seidel L.M., Killmer S., Sagar, Daul F., Salvat Lago M., Decker A., et al. Characterization of pre-existing and induced SARS-CoV-2-specific CD8+ T cells. Nat. Med. 2021;27:78–85. doi: 10.1038/s41591-020-01143-2. [DOI] [PubMed] [Google Scholar]
- Sekine T., Perez-Potti A., Rivera-Ballesteros O., Strålin K., Gorin J.-B., Olsson A., Llewellyn-Lacey S., Kamal H., Bogdanovic G., Muschiol S., et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell. 2020;183:158–168.e14. doi: 10.1016/j.cell.2020.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette A., Sidney J. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics. 1999;50:201–212. doi: 10.1007/s002510050594. [DOI] [PubMed] [Google Scholar]
- Shimizu Y., DeMars R. Production of human cells expressing individual transferred HLA-A,-B,-C genes using an HLA-A,-B,-C null human cell line. J. Immunol. 1989;142:3320–3328. [PubMed] [Google Scholar]
- Sidney J., Peters B., Frahm N., Brander C., Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunol. 2008;9:1. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soresina A., Moratto D., Chiarini M., Paolillo C., Baresi G., Focà E., Bezzi M., Baronio B., Giacomelli M., Badolato R. Two X-linked agammaglobulinemia patients develop pneumonia as COVID-19 manifestation but recover. Pediatr. Allergy Immunol. 2020;31:565–569. doi: 10.1111/pai.13263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T.N., Greaney A.J., Hilton S.K., Crawford K.H.D., Navarro M.J., Bowen J.E., Tortorici M.A., Walls A.C., Veesler D., Bloom J.D. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. bioRxiv. 2020 doi: 10.1101/2020.06.17.157982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeck H., Jolin J.S., Qi Y., Yassine-Diab B., Johnson R.C., Kwon D.S., Addo M.M., Brumme C., Routy J.-P., Little S., et al. Human immunodeficiency virus type 1-specific CD8+ T-cell responses during primary infection are major determinants of the viral set point and loss of CD4+ T cells. J. Virol. 2009;83:7641–7648. doi: 10.1128/JVI.00182-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A.T., Linster M., Tan C.W., Le Bert N., Chia W.N., Kunasegaran K., Zhuang Y., Tham C.Y.L., Chia A., Smith G.J.D., et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021;34:108728. doi: 10.1016/j.celrep.2021.108728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.W., Tambyah P.A., Hui D.S. Emergence of a new SARS-CoV-2 variant in the UK. J. Infect. 2020;82:e27–e28. doi: 10.1016/j.jinf.2020.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H., Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J., Doolabh D., Pillay S., San E.J., Msomi N., et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature. 2021;592:438–443. doi: 10.1038/s41586-021-03402-9. [DOI] [PubMed] [Google Scholar]
- Teixeira A., Benckhuijsen W.E., de Koning P.E., Valentijn A.R.P.M., Drijfhout J.W. The use of DODT as a non-malodorous scavenger in Fmoc-based peptide synthesis. Protein Pept. Lett. 2002;9:379–385. doi: 10.2174/0929866023408481. [DOI] [PubMed] [Google Scholar]
- Voloch C.M., Ronaldo da Silva F., de Almeida L.G.P., Cardoso C.C., Brustolini O.J., Gerber A.L., de C Guimarães A.P., Mariani D., da Costa R.M., Ferreira O.C., et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. medRxiv. 2020 doi: 10.1101/2020.12.23.20248598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Schmidt F., Weisblum Y., Muecksch F., Barnes C.O., Finkin S., Schaefer-Babajew D., Cipolla M., Gaebler C., Lieberman J.A., et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature. 2021;592:616–622. doi: 10.1038/s41586-021-03324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibmer C.K., Ayres F., Hermanus T., Madzivhandila M., Kgagudi P., Oosthuysen B., Lambson B.E., de Oliveira T., Vermeulen M., van der Berg K., et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021;27:622–625. doi: 10.1038/s41591-021-01285-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Epitope mutations within SARS-CoV-2 variants of concern are bolded.
Data Availability Statement
All data supporting the findings of this study available within the paper and available from the lead contact upon request. The code used during this study to generate network scores is archived at Zenodo (https://zenodo.org/record/2597484). Viral sequence data of highly networked and non-networked epitope variants from primary SARS-CoV-2 isolates is available in the supplementary materials of the primary manuscript (Agerer et al., 2021).