

Abstract
SARS coronavirus 2 is neutralized by proteins that block receptor‐binding sites on spikes that project from the viral envelope. In particular, substantial research investment has advanced monoclonal antibody therapies to the clinic where they have shown partial efficacy in reducing viral burden and hospitalization. An alternative is to use the host entry receptor, angiotensin‐converting enzyme 2 (ACE2), as a soluble decoy that broadly blocks SARS‐associated coronaviruses with limited potential for viral escape. Here, we summarize efforts to engineer higher affinity variants of soluble ACE2 that rival the potency of affinity‐matured antibodies. Strategies have also been used to increase the valency of ACE2 decoys for avid spike interactions and to improve pharmacokinetics via IgG fusions. Finally, the intrinsic catalytic activity of ACE2 for the turnover of the vasoconstrictor angiotensin II may directly address COVID‐19 symptoms and protect against lung and cardiovascular injury, conferring dual mechanisms of action unachievable by monoclonal antibodies. Soluble ACE2 derivatives therefore have the potential to be next generation therapeutics for addressing the immediate needs of the current pandemic and possible future outbreaks.
Keywords: ACE2, avidity, COVID‐19, decoy receptor, protein engineering, SARS coronavirus 2
1. INTRODUCTION
Not long following its emergence in China in late 2019, SARS coronavirus 2 (SARS‐CoV‐2) became an unprecedented public health emergency for our generation. The absence of immunity, superspreader events 1 and presymptomatic 2 and asymptomatic 3 , 4 transmission have all combined to favor this respiratory virus' global spread. The virus is devastating to the elderly and other vulnerable groups with certain predisposing conditions, and elicits surprisingly heterogenous disease symptoms collectively known as COVID‐19, with the most common being fever, dry cough, hypoxemia and pneumonia, 5 , 6 , 7 , 8 but also unusual neurological symptoms 9 and coagulopathy. 10 It remains unclear which aspects of disease are the result of disseminated virus infection of multiple tissues vs dysregulation of signaling pathways, including cytokine storms 11 , 12 and aberrant angiotensin and kinin peptide processing. 13 , 14 , 15
Virus entry is mediated by interactions between the viral spike, a trimeric complex of protein S, and angiotensin‐converting enzyme 2 (ACE2) on a host cell membrane. 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 S is proteolytically processed as two subunits, S1 and S2, that remain noncovalently associated until ACE2 is bound by a receptor‐binding domain (RBD) in the S1 subunit, triggering conformational changes that include S1 shedding, exposure of a fusion peptide in S2 and fusion of the viral envelope with the host membrane. 21 , 24 , 25 , 26 , 27 Antibodies targeting multiple epitopes on S, but especially the RBD, can block ACE2 engagement or prevent membrane fusion from occurring, and numerous monoclonal antibody therapies are now in preclinical and clinical development. 28 , 29 , 30 , 31 , 32 , 33 However, coronaviruses have moderate to high‐mutation rates 34 , 35 and there is a perceived risk that drug resistant SARS‐CoV‐2 variants might begin circulating as antibody therapies are more widely used. New SARS‐CoV‐2 variants with increased transmissibility and partial immune escape have now emerged, 36 , 37 , 38 , 39 and the virus will continue to mutate as it becomes endemic and population immunity builds. Indeed, Emergency Use Authorization from the U.S. Food and Drug Adnministration for anti‐SARS‐CoV‐2 antibody bamlanivimab as a monotherapy has been withdrawn due to decreased efficacy against new virus variants (FDA revocation letter, April 16, 2021). The risk of the virus acquiring resistance to monoclonal antibody therapies is mitigated by combining noncompeting antibodies in cocktails. 40
An alternative strategy is to use ACE2 itself as a soluble decoy receptor that competes for receptor‐binding sites on S 22 , 41 , 42 , 43 (Figure 1). ACE2 is an 805 amino acid (a.a.) protein that comprises a protease domain (a.a. 19‐615), a collectrin‐like dimerization domain (a.a. 616‐729), and a single‐span transmembrane domain (a.a. 741‐765). 16 It is widely expressed in vascular endothelium throughout the body and is notably found at high levels in the epithelia of the lung and gastrointestinal tract, 44 which are both important sites of infection and symptoms. 5 , 6 , 45 Neuropilins, transmembrane proteins with promiscuous interactions for growth factors and growth factor receptors, bind the C‐terminus of the processed S1 subunit to further facilitate cell entry, and neuropilin expression likely influences SARS‐CoV‐2 tissue tropism. 46 , 47 The major attraction of using an entry receptor like ACE2 as a soluble decoy is that, in principle, the virus has limited mutational mechanisms for escape without simultaneously losing affinity for the native, membrane anchored form. 48 Soluble decoy receptors are used clinically for a variety of indications, although none are yet approved drugs for viruses. 49 Wild type (WT), soluble ACE2 (sACE2) is an investigational drug for acute respiratory distress that has been rapidly repurposed as a SARS‐CoV‐2 antiviral 41 and was recently evaluated in a phase 2 COVID‐19 clinical trial with promising results in severely ill patients (ClinicalTrials.gov Study NCT04335136; Apeiron Biologics media release 12 March 2021). This drug candidate has become the starting point for multiple engineering efforts to solve key issues surrounding pharmacokinetics, affinity, and avidity for the creation of next generation ACE2 derivatives with superior efficacy. These efforts are reviewed here.
FIGURE 1.
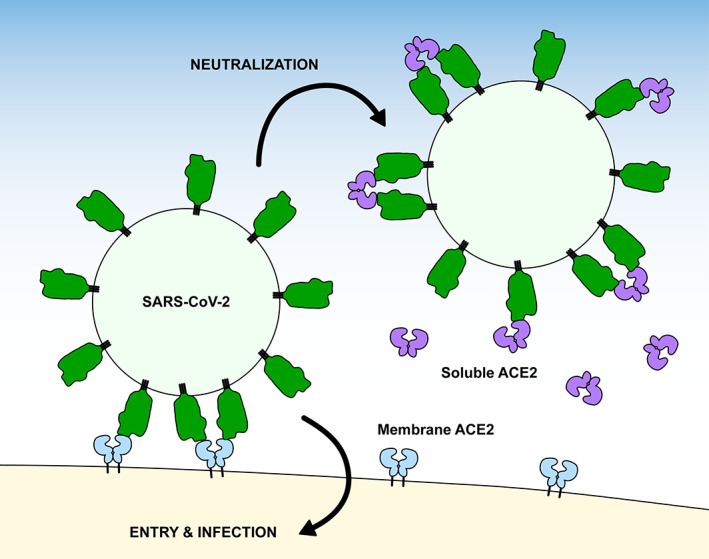
Soluble ACE2 neutralizes SARS‐CoV‐2 infection. Soluble ACE2 (violet) competes with native, membrane‐anchored ACE2 receptors (blue) for binding sites on viral spikes (green). ACE2, angiotensin‐converting enzyme 2; SARS‐CoV‐2, SARS coronavirus 2
2. MUTAGENESIS OF ACE2 FOR ENHANCED AFFINITY
Soluble decoy receptors should ideally bind the SARS‐CoV‐2 spike tighter than, and out‐compete, the WT ACE2 receptor. Tight affinity is also necessary for effective virus neutralization at typical doses for biologic drugs. The importance of affinity is further emphasized when soluble decoys are considered against the therapeutic alternative of affinity‐matured monoclonal antibodies, which bind S of SARS‐CoV‐2 with tight nanomolar to picomolar K d 28 , 29 , 30 , 33 ; by comparison, WT sACE2 has only moderate affinity (K d ≈ 20 nM). 16 , 50 To this end, mutagenic approaches, either targeted or through selection of diverse libraries of variants, provide a means for engineering decoy receptors with enhanced affinity.
In the first report 51 that mutations within ACE2 can indeed be found that increase S affinity, Chan et al. performed deep mutagenesis on full length ACE2. 50 , 52 A library of ACE2 variants was created that encoded all possible single amino acid substitutions at 117 sites, covering both the regions that interface with the RBD as well as the ACE2 active site. The library was then selected by fluorescence activated cell sorting (FACS) for ACE2 surface expression in human cells and tight binding to the RBD of SARS‐CoV‐2 protein S (Figure 2). ACE2 residues buried at the interface with S tend to be more conserved, while ACE2 residues at the interface periphery or within the active site were mutationally tolerant. Mutations of ACE2 residues N90 and T92, which form an N‐glycosylation motif, were universally enriched for high binding affinity (with the exception of T92S), indicating that N90 glycosylation hinders binding, at least in this experiment's cell line. Other mutations were also found dispersed across multiple sites that increase S binding and some of these were rapidly screened as combinations of mutations in soluble ACE2 conjugated to superfolder GFP (sfGFP). 53 Expression medium containing sACE2‐sfGFP, without purification, can be incubated directly with cells expressing S and bound sACE2‐sfGFP detected by flow cytometry. Ultimately, variants of sACE2 carrying four or three mutations (Figure 2C), dubbed sACE2.v2 and sACE2.v2.4, were engineered for an optimal balance of high affinity, expression, and yield. Impressively, monomeric sACE2.v2, which spans ACE2 residues 19‐615 that form the extracellular protease domain, outcompeted WT sACE2 fused to dimeric Fc of IgG1 and also competed effectively with anti‐RBD Ig derived from COVID‐19 positive patient sera. Soluble ACE2 forms a natural dimer (which we refer to as sACE22) if the construct is expanded up to ACE2 residues 730‐740, which includes the dimerization/collectrin‐like domain immediately C‐terminal of the protease domain. Soluble ACE22 has tighter monovalent affinity for the SARS‐CoV‐2 RBD than the protease domain alone, perhaps due to structural stabilization. 50 , 54 , 55 The dimer also has increased apparent affinity and neutralization of authentic virus through avidity, demonstrating that each protease domain in sACE22 can bridge two S proteins. The leading variant from these efforts, dimeric sACE22.v2.4, had a picomolar K d for the SARS‐CoV‐2 RBD and achieved 50% relative infection inhibition of authentic virus at subnanomolar concentrations. Surprisingly, it showed similar neutralization efficacy against SARS‐CoV‐1, a related betacoronavirus responsible for a smaller epidemic in 2003, despite no consideration of binding to this virus during mutagenesis. This implied potential efficacy against other zoonotic coronaviruses (reviewed in Reference 56) that utilize ACE2 and may spill over to humans in the future. This was confirmed by demonstrating sACE22.v2.4 tightly binds diverse RBD sequences from SARS‐related bat betacoronaviruses that use ACE2 as an entry receptor. 48 Furthermore, an in vitro selection of mutations within the RBD from SARS‐CoV‐2 failed to find S variants that lose affinity for the engineered sACE22.v2.4 decoy but retain binding to the WT host ACE2 receptor, thus confirming the hypothesis that mechanisms for viruses to escape soluble decoy receptors are limited. 48
FIGURE 2.
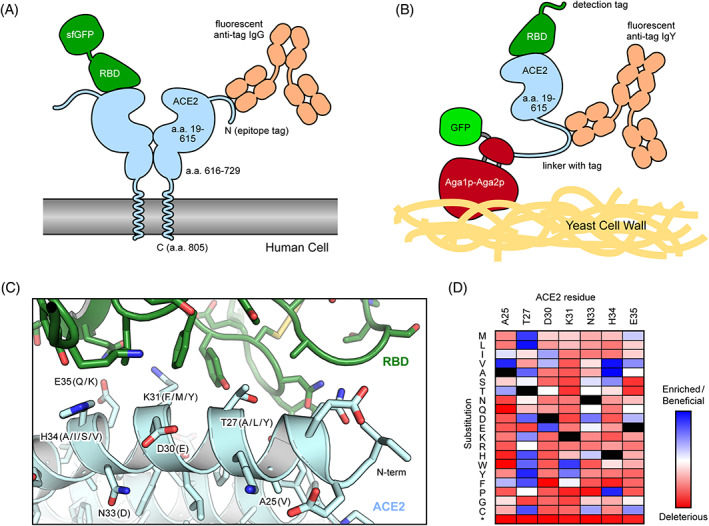
Selection strategies for enhancing the affinity of ACE2 for the RBD of SARS‐CoV‐2. A, In human cell selections of ACE2 variants, surface expression of full‐length ACE2 with N‐terminal epitope tags was detected via fluorescent anti‐tag antibodies. Cells expressing ACE2 clones with high binding to RBD‐sfGFP were collected by FACS. B, In FACS selections of yeast, the protease domain of ACE2 was expressed as a fusion to Aga2p for display on the yeast surface. Displayed protein was detected via a C‐terminal fusion to GFP or immunostaining of an epitope tag in the connecting linker. Bound RBD was labeled via biotin or an epitope tag for fluorescence detection. C, Structure (PDB 6M17) of RBD (green) bound to ACE2 (pale blue). Common mutations in high‐affinity engineered decoys are indicated in parentheses. D, Mutational landscape showing how single amino acid substitutions in ACE2 increase (blue) or decrease (red) binding to RBD. Data are from Chan et al. 50 ACE2, angiotensin‐converting enzyme 2; FACS, fluorescence activated cell sorting; RBD, receptor‐binding domain; SARS‐CoV‐2, SARS coronavirus 2; sfGFP, superfolder GFP
In the aforementioned study, single mutations in ACE2 identified by deep mutagenesis were combined based on rational human intuition. This is a major limitation, as it is not clear whether the best suite of mutations was chosen in the final optimized variant sACE22.v2.4. Human bias can be removed by in vitro selection of combinatorial mutant libraries and other studies have done exactly this (described below). 54 , 57 Another solution is to use computational modeling to predict which mutations can be combined without compromising protein stability or binding free energy. Computational modeling, informed by the deep mutational scan, guided the design of a modified ACE2 derivative with eight mutations (one predicted to decrease catalytic activity) that bound S of SARS‐CoV‐2 with over 100‐fold tighter affinity. 58 Six of the mutations were among the most enriched in the deep mutational scan and it is worth noting that the works described in this review frequently converge on very similar mutations to a small number of sites (Figure 2C).
In a similar deep mutagenesis experiment, Heinzelman and Romero investigated the influence on S protein binding of single‐nucleotide variants (SNVs) in the ACE2 protease domain, evaluating nearly 4000 amino acid substitutions. 59 Mutations were made using error‐prone PCR, a rapid and reliable way of generating library diversity but which fails to capture multiple nucleotide changes within a single codon, unless error rates are excessively high. The library of the ACE2 protease domain was screened by yeast surface display (Figure 2B) and while most mutations decreased binding (it is, after all, easy to “break” a protein through random mutagenesis), approximately 4% of mutations were found to increase RBD binding. While mutations in the active site tended to have little effect (with the caveat that the background ACE2 sequence used for library generation already had catalytic activity knocked out), the authors found that residues in the chloride‐binding site of ACE2, which regulates its peptidase activity 60 and is over 40 Å from the RBD interface, do affect binding. In addition, a separate distal cluster of hydrophobic residues (L236, F588, L591, and L595) was also identified as a critical site affecting binding despite being over 30 Å from the RBD interface. By interrogating SNVs, the authors were able to statistically evaluate the clinical and epidemiological relevance of ACE2 allelic variants in the human population, and they estimated that roughly 1 in 10 000 people may have SNVs that increase spike binding and roughly 4 in 1000 have SNVs that decrease spike binding. However, these predictions will need to be confirmed by genetics studies of patient cohorts, especially since mutations in ACE2 with altered affinity for S may also have other effects that impact infection in vivo, such as expression changes in human tissues. Overall, this work highlights the importance of considering long‐range effects of mutations distal from the functional binding site.
The deep mutational scans of ACE2 were based on a single round of sequence diversification and selection, whereas others have employed multiple rounds with mutations being combined to direct the in vitro evolution of ACE2 toward exceptionally tight affinity for SARS‐CoV‐2 S. As mentioned above, this also removes human bias in finding optimum combinations of mutations with high affinity. Higuchi et al focused mutagenesis to ACE2 residues 18‐102 and 272‐409 in the protease domain, respectively denoted as PD1 and PD2, that form the RBD interface. 57 Binding was assayed using the method previously developed for deep mutagenesis, 50 with human cells expressing the ACE2 library incubated with RBD‐sfGFP and sorted by FACS. Mutations within the “PD1” region were selected first, followed by mutagenesis within the “PD2” region and additional rounds of selection, although additional mutations within the PD2 region did not substantially improve affinity. Molecular interactions with the RBD are mostly localized to the ACE2 N‐terminus 16 encompassed in their PD1 library. The ACE2 variant identified with highest affinity, termed 3N39 with seven substitutions (Figure 2C), bound RBD with picomolar K d. There is a risk during in vitro affinity maturation that a decoy receptor will be “over‐engineered” or “over‐fitted” for its target, such that tight binding is achieved at the expense of other important attributes such as protein stability or breadth against diverse virus variants. In this particular case, and despite having a much higher mutational load than sACE2.v2.4, 3N39‐IgG1 Fc fusions were soluble with minimal aggregation by size exclusion chromatography and resistance did not emerge when authentic SARS‐CoV‐2 was passaged in the protein's presence. The engineered 3N39‐Fc fusion outperformed the equivalent WT ACE2‐Fc fusion for virus neutralization in vitro and reduced virus burden with excellent amelioration of lung pathology and inflammation in hamsters when administered 2 hours post SARS‐CoV‐2 challenge.
The Kortemme, Hobman, and Wells groups went even further in using multiple rounds of engineering, diversification, and selection to drive down the dissociation constant to the picomolar range. 54 Computational alanine scanning identified ACE2 residues H34, Q42, and K353 as hot spots that contribute disproportionately to the binding energy and these residues (with their neighbors) were the focus for computational mutagenesis. Computationally designed ACE2 variants were purified as Fc fusions and binding to S was improved 3‐ to 11‐fold. Four of these ACE2 variants served as “parents” that were then matured through yeast surface display, resulting in a further 14‐fold improvement in apparent K d. Deep mutagenesis data from Chan et al 50 was then considered, from which new ACE2 variants were designed with mutations that were not alanine scan‐based hotspots and included sites outside the RBD interface that likely stabilize conformation. The final ACE2 variants had exceptionally tight affinity, with the lowest K d variants carrying 4 to 8 substitutions (Figure 2C). It was also independently confirmed that additional increases in apparent affinity can be achieved through inclusion of the ACE2 collectrin‐like dimerization domain for avid binding and protein stabilization. 50 , 54 , 55 In this study, deliberate action was taken to inactivate ACE2 enzymatic activity. This has historically been done by mutating two critical Zn2+‐coordinating residues (H374N and H378N), 61 but because these substitutions resulted in undesirable destabilization in the variant background, an alternative mutation was adopted: H345L within the substrate‐binding cavity. It has since been noted that a common assay for measuring ACE2 catalytic activity with a fluorogenic substrate mimic does not necessarily reflect proteolysis of the relevant in vivo substrate (angiotensin II) and therefore some caution must be used in interpreting measurements of catalysis reported by different groups. 62 ACE2 mutants of residue H345 in particular may retain proteolytic activity toward angiotensin II. While the rationale for this mutation is the elimination of vasodilatory effects due to excess ACE2 peptidase activity that may cause hypotension, it is worth considering the potential benefits of keeping enzymatic activity (discussed below).
Pangolin (Pangolin‐CoV‐2020) and bat (isolate RaTG13) coronaviruses, in addition to human SARS‐CoV‐1 and ‐2, can initiate membrane fusion using not only human ACE2 but many other animal orthologs. 55 This informed the substitution of human ACE2‐D30 for glutamate, 55 which is found in ACE2 of other species and is able to better reach K417 of the RBD for salt bridge formation. 16 , 17 In addition, inclusion of the dimerization domain together with the protease domain further facilitated the engineering of a sACE2‐Ig fusion protein effective against multiple betacoronavirus strains. The discovery of the affinity‐enhancing D30E mutation and benefits of including the dimerization domain overlap with independent work by others. 50 , 54 Another contributing factor was increased avidity (to be discussed further below) due to design of a tetrameric sACE2‐Ig configuration. 55 The concept of borrowing residues from the ACE2 proteins of other species that bind zoonotic coronaviruses with naturally higher affinity than the human ortholog has been taken even further. 63 Residues in human ACE2 were systematically substituted with their counterparts in high‐affinity bat ACE2 orthologs, providing a set of five mutations that could be combined to yield an sACE22‐Fc derivative with increased efficacy for pseudovirus neutralization. Overall, it is apparent from all these studies that a truly effective therapeutic approach utilizing decoy ACE2 receptors will likely incorporate a multifaceted strategy of affinity enrichment, avidity, and Fc fusions.
One final aspect to touch upon regarding affinity enhancement through mutagenesis is the connection between binding affinity and ACE2 enzymatic activity. For example, the sACE22.v2.4 variant has reduced catalytic activity even though its three amino acid substitutions are not within the active site, 50 thus demonstrating potentially unappreciated coupling between S affinity and ACE2 catalysis. Others have observed that some mutations that knock out ACE2 activity can increase S binding, 64 although many mutations in the active site have no effect on S affinity. 50 , 59 Bioinformatics and mutagenesis based on comparisons of ACE2 between species reveals epistatic relationships between residues at the virus binding surface and the active site. 65 Furthermore, we again highlight the key finding by Heinzelman and Romero that mutations to a chloride‐binding motif of ACE2, which is 40 Å from the RBD interface and is directly involved in regulating peptidase activity, can affect spike affinity, possibly due to conformational effects. 59 The binding of trimeric SARS‐CoV‐2 spike protein to WT ACE2 also increases its peptidase activity 3‐ to 10‐fold, with the SARS‐CoV‐2 RBD inducing bending of the N‐terminal protease subdomain toward the C‐terminal protease subdomain “…reminiscent of a closing dam.” 66 Although the precise mechanisms remain unclear, there are nonetheless multiple clues incriminating ACE2 conformation and dynamics on both catalytic activity and S affinity. We believe from a therapeutic perspective (to be discussed further below), maintaining catalytic activity is preferable due to the protective effects of ACE2 products on the pulmonary and cardiovascular systems.
3. ACE2 FUSIONS WITH IMMUNOGLOBULIN FC
Despite a recent clinical trial of WT sACE22 in Europe 67 and the studies reviewed above demonstrating the realization of picomolar affinity decoy receptors, it is likely that low sACE2 serum stability will limit its successful application as a therapeutic. The serum half‐life of dimeric sACE22 in vivo is on the order of hours, 68 , 69 thereby requiring frequent dosing to maintain sACE22 in circulation. As a consequence, it is questionable whether sACE22 can be safely administered in an outpatient setting. Serum stability of biologic drugs is greatly increased by fusion of the therapeutic protein with the Fc region of IgG. 70 Serum proteins are turned over by constant internalization and lysosomal degradation, especially by endothelial cells. Internalized IgG engages the neonatal Fc receptor (FcRn) within endosomal compartments to direct its trafficking back to the plasma membrane, thereby returning the protein to the serum (Figure 3A). The previously highlighted works describing the discovery of high‐affinity sACE2 variants 50 , 54 , 55 , 57 have already extended their findings to fusions with IgG1 Fc and we believe that sACE2‐Fc fusions will be the standard going forward for the production of high‐affinity decoy receptors with excellent pharmacokinetics.
FIGURE 3.
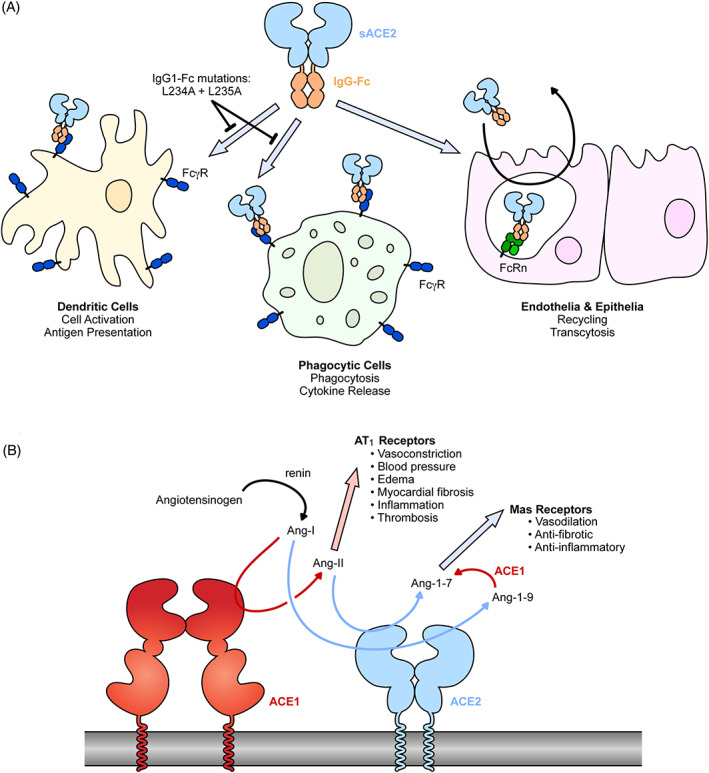
Common modifications to soluble ACE2: fusions with IgG‐Fc and elimination of catalytic activity. A, The Fc region of IgG can be fused to the extracellular domains of ACE2. The Fc moiety is recognized by inflammatory FcγRs on various cell types for immune effector functions. IgG‐Fc also binds FcRn within endosomal compartments following pinocytosis by endothelial and epithelial cells to mediate recycling or transcytosis, impacting biodistribution and serum stability. B, The conversion of angiotensin peptide hormones by renin, ACE1 and ACE2. ACE2, angiotensin‐converting enzyme 2
Recombinant human sACE22 has been developed over nearly two decades as a therapeutic for lung inflammation and acute respiratory distress syndrome (ARDS). Its administration has been shown to be protective against cardiovascular 71 and pulmonary pathologies and to treat hypoxemia in multiple animal models, 72 , 73 , 74 , 75 , 76 and it has acceptable safety without causing episodes of hypotension in healthy and mechanically ventilated human subjects. 69 , 77 However, while sACE22 demonstrated the expected in vivo biochemical activity in ARDS patients, there was no statistically significant improvement in clinical outcomes in a phase 2 trial. The clinicians commented that “…lower plasma concentrations (of sACE22) over a longer duration may be more effective as a result of more sustained production of” ACE2 products. 77 These findings motivated the first published report of a sACE22‐Fc fusion tested in vivo, in which the serum half‐life of murine sACE22 was extended to over a week in mice with substantially higher maximum serum activity. 76 Recombinant sACE22‐Fc mitigated hypertension following injection of the vasopressor (and ACE2 substrate) angiotensin II for up to 7 days, whereas unfused sACE22 lost function in serum within hours. The Fc fusion protein reduced blood pressure and cardiac and renal fibrosis in a transgenic mouse model for hypertension and albuminuria in which renin is massively overexpressed. 76 This work demonstrated serum longevity of sACE22 fused to IgG1‐Fc as well as the potent physiological effects of ACE2 catalytic activity.
More recently with the emergence of SARS‐CoV‐2, multiple groups have explored IgG1‐Fc fusions of sACE2 or sACE22, consistently finding no adverse toxicity in small animals. 42 , 57 , 62 , 64 Importantly, an engineered sACE2‐Fc derivative had identical pharmacokinetics to the WT equivalent, showing that mutations to enhance SARS‐CoV‐2 affinity did not alter in vivo clearance. 57 However, there are large differences in reported serum half‐lives ranging from minutes when levels of the ACE2 moiety are measured, 57 vs days when levels of the IgG1‐Fc moiety are detected instead. 42 , 64 Only a single study shows long serum half‐life of both moieties by use of a sandwich ELISA with an anti‐ACE2 capture antibody and anti‐IgG1 detection antibody. 62 We believe these discrepancies point toward cleavage of the connecting linker at the fusion site, liberating short‐lived sACE2 and long‐lived IgG1‐Fc fragments. While the serum half‐life of unfused human sACE22 is 8.5 hours in mice, 78 this is after intraperitoneal administration in which resorption of macromolecules into the blood is delayed by hours; 79 , 80 direct intravenous administration almost certainly gives a much shorter serum half‐life. Studies may therefore differ substantially in their reported pharmacokinetics depending on the linker sequence and the methods for serum analysis; this is an important area for future research that has been neglected. Despite uncertainties in the pharmacokinetic profiles, multiple groups have now shown that various fusions of WT or engineered sACE2 with IgG fragments can effectively reduce virus burden and lung pathology in animal models of infection, whether administered prophylactically or therapeutically. 57 , 64 , 81 , 82 , 83 It remains to be determined whether engineered derivatives and Fc fusions of human sACE2 will be immunogenic in humans, and if so, whether anti‐drug antibodies will have any negative impact on short term treatment of COVID‐19 patients. The answers to these questions will likely remain unknown until clinical trials are conducted.
The Fc region of IgG subclasses engage multiple Fc receptors to effect function, including not only FcRn to mediate serum stability and uptake into the lungs, but also a suite of inflammatory Fc γ receptors (FcγR). 84 These receptors are expressed on a variety of cell types, especially in the immune system, and are responsible for effector functions such as antibody‐dependent cellular cytotoxicity, cytokine release, antigen presentation, and co‐stimulation (Figure 3A). Until different Fc fusion proteins are properly assessed in controlled in vivo experiments, it is unclear whether these effector functions will aid in clearance and recovery from SARS‐CoV‐2 infection. Iwanaga et al have hypothesized that interactions of a sACE22‐Fc biologic with inflammatory FcγRs may exacerbate inflammation and cytokine storms in COVID‐19 patients. 64 Consequently, two leucine residues in the IgG1‐Fc moiety were mutated to alanines (L234A and L235A). These so‐called LALA mutations disrupt FcγR interactions while leaving FcRn binding intact for high serum stability. The LALA variant of sACE22‐Fc had long serum half‐life measured by IgG1 ELISA, was distributed to lung tissues, and retained virus neutralization efficacy. Similarly, sACE22 has been fused with the Fc region of IgG4, which naturally has reduced interactions with inflammatory FcγRs. 85 Finally, a fusion of the ACE2 protease domain with a dimerization motif and an albumin binding domain (serum albumin interacts with FcRn like immunoglobulins but lacks other immune effector functions) increases protein longevity in vivo and reduces SARS‐CoV‐2 replication and pathology in a human ACE2 transgenic mouse model. 82 However, Fc‐mediated effector functions of monoclonal antibodies are important for optimal protection against SARS‐CoV‐2 infection in vivo 86 and it may be that efforts toward silencing effector functions in sACE2 fusion proteins will negatively impact their in vivo efficacy.
For some antibodies, binding to viral epitopes can increase infection and pathogenicity, known as antibody‐dependent enhancement (ADE). This has been a major obstacle in the development of vaccines to dengue and respiratory syncytial viruses, but also occurs in many other viral infections due to various mechanisms. 87 , 88 , 89 , 90 , 91 For example, antibodies can promote uptake of viruses by phagocytic cells through Fc receptor interactions, or antibodies can generate highly inflammatory antibody‐virus aggregates. Concerningly, ADE was observed in animals vaccinated against SARS‐CoV‐1 and MERS‐CoV, 89 , 92 , 93 but at this point in time ADE has not prevented the rapid development of safe vaccines for COVID‐19. 94 , 95 , 96 Only a subset of monoclonal antibodies with distinct mechanisms for binding S increase SARS‐CoV‐2 infection in tissue culture (in both FcγR‐dependent and independent ways), 97 , 98 and even so at least some of these antibodies still protect against disease in animals, emphasizing how non‐neutralizing antibodies can mediate virus clearance in the host. 97 Importantly, enhanced disease has not occurred in animal studies where sACE2‐IgG fusions have proven to be effective at reducing SARS‐CoV‐2 burden and disease pathology. 57 , 64 , 81 , 83 Human macrophages are also not infected by SARS‐CoV‐2 in the presence of convalescent plasma, suggesting macrophages are not permissive for productive infection even when allowing for FcγR‐mediated uptake. 99 A very recent study demonstrated that secreted and recombinant sACE2 may enhance SARS‐CoV‐2 infection of cell lines that express low levels of ACE2, 100 but the evidence was exclusively from in vitro experiments where the normal pathways for virus attachment may have been by‐passed. Certainly, in humans, sACE22 has proven to be safe and effective at reducing viral load and COVID‐19 symptoms (ClinicalTrials.gov Study NCT04335136; Apeiron Biologics media release 12 March 2021). Therefore, while enhanced disease remains of potential concern, evidence at this point in time suggests it will not limit the efficacy of IgG‐fused soluble decoy receptors to treat or prevent COVID‐19.
4. CATALYTICALLY ACTIVE OR INACTIVE SACE2?
Most reports describing the engineering of sACE2 to enhance its affinity through targeted substitutions, or to improve pharmacokinetics through Fc fusions, have deliberately eliminated ACE2 catalytic activity. 42 , 54 , 58 , 64 , 85 The assumption is that administration of a catalytically active decoy receptor will adversely interact with physiology to have unacceptable toxicity. Soluble decoy receptors for other viruses have also been engineered to eliminate the normal biological activity for safety and efficacy reasons. 101 However, there are strong arguments that ACE2 catalytic activity might be beneficial for treating COVID‐19 symptoms.
In the renin‐angiotensin system (RAS), a set of proteases regulates the production of angiotensin peptide hormones 102 , 103 (Figure 3B). Renin is released by the kidneys when renal blood flow is low and it catalyzes the conversion of angiotensinogen in the blood to angiotensin I (Ang‐I). Ang‐I is in turn a substrate for angiotensin‐converting enzyme (ACE or ACE1), which cleaves the peptide to produce the vasoconstrictor Ang‐II that has a wide range of effects through the angiotensin II type 1 receptor (AT1R). Ang‐II promotes vasoconstriction and elevated blood pressure and volume, and under conditions where Ang‐II is high there can be increased inflammation, myocardial fibrosis, and thrombosis. Ang‐II is proteolyzed by ACE2 to produce Ang‐1‐7, 104 , 105 , 106 , 107 which has vasodilatory properties and is cardioprotective. 108 Both ACE1 and ACE2 are active in lung tissue (and elsewhere) and also catalyze the conversion of kinin peptides, the consequences of which are still speculative in the context of COVID‐19. 15 , 109 Genetic studies and administration of sACE2 in animal models have demonstrated that tilting the RAS system in favor of Ang‐1‐7 has numerous benefits for the treatment of lung injury and inflammation. 72 , 73 , 74 Indeed, it has been proposed that COVID‐19 symptoms may in part manifest from RAS dysregulation, 41 , 103 especially due to loss of ACE2 activity as the protein is shed from the plasma membrane following cell infection. 110 , 111 , 112 , 113 In two investigations for biomarkers associated with COVID‐19, Ang‐II levels were elevated in hospitalized COVID‐19 patients and correlated with both viral load and severity of hypoxemic respiratory failure. 13 , 114 However, a third study found no such correlation 115 and any causality between decreased ACE2 activity and disease symptoms remains inconclusive. Nonetheless, catalytically active sACE2 offers two potential mechanisms of action. First, it blocks receptor‐binding sites on SARS‐CoV‐2 spikes to neutralize infection. Second, it promotes the degradation of Ang‐II and production of Ang‐1‐7 to directly address COVID‐19 symptoms. A clinical trial testing WT sACE22 in critically ill COVID‐19 patients was recently completed (ClinicalTrials.gov Study NCT04335136; Apeiron Biologics media release 12 March 2021) and suggests, along with a preliminary case report, 67 that both mechanisms of action are at play. This provides sACE2 a tremendous advantage over monoclonal antibodies, which are safe and effective at reducing viral load 116 but do not directly address symptoms in sick patients. However, at this time no direct comparison has been made in animal models of COVID‐19 to evaluate catalytically active vs inactive sACE2, alongside engineered variants with altered S affinity. The hypothesis that sACE2 will have dual mechanisms of action therefore remains unproven.
Different mutations have been introduced into sACE2 to eliminate catalytic activity, the most common being H374N and H378N to prevent coordination of an essential Zn2+ ion. 61 Others have used alternative mutations within the ACE2 active site, 54 , 64 although it has recently been shown that a common fluorescence assay to assess ACE2 enzymatic activity does not accurately represent proteolysis of Ang‐II, suggesting caution when considering claims surrounding mutational effects on catalysis. 62 A number of ACE2 variants with reduced catalytic activity bind slightly tighter to SARS‐CoV‐2 S, suggesting possible conformational coupling between the catalytic and S binding sites.
5. ACE2 AND AVIDITY
The apparent affinity of ACE2 for S incorporates both affinity (site specific binding strength) and avidity (the total sum of all binding interactions). ACE2 is a natural dimer and dimeric sACE22 is orders of magnitude more effective at neutralizing authentic virus than the monomeric sACE2 protease domain. 50 The three subunits in a trimeric S spike are conformationally heterogenous, and the RBDs may be exposed for ACE2 interactions in an “up”/“open” conformation or hidden in a “down”/“closed” conformation. 18 , 117 The dominant conformations of prefusion S trimers are either all RBDs down or one RBD in the up state, yet there are minor populations with additional RBDs up and accessible for ACE2 interactions. 118 , 119 , 120 There is limited evidence that dimeric ACE22 can bridge two S subunits in a single trimeric spike (ie, intra‐spike avidity) 121 and atomic structures suggest the spatial orientation of natural dimeric ACE22 is incompatible with binding two RBDs in a single S trimer 16 , 122 (Figure 4). This is not to say that more than one ACE22 dimer cannot bind a trimeric spike, and indeed structures have been determined with all three RBDs of trimeric S in the up state and bound to three ACE2 protease domains. 122 Rather, a single ACE22 dimer in which the orientations of the protease domains are governed by the collectrin‐like dimerization domains cannot simultaneously engage two subunits in a single S trimer, unless spike flexibility is greater than is captured in current high‐resolution structures. Avidity effects are therefore most likely due to dimeric ACE22 cross‐linking two separate viral spikes, which is supported by cryo‐electron tomography showing moderate spike density on the viral envelope suitable for bridging interactions. 118 , 119 , 120 We speculate that clustering of dimeric ACE22 via bridging interactions with S trimers at the virus‐cell membrane synapse might even aid endocytosis and infection. However, protein engineers are not limited to the geometric architecture of natural ACE22 dimers and have at their disposal tools for creating higher multimeric forms that may sterically complement exposed RBDs on native S trimers (Figure 5).
FIGURE 4.
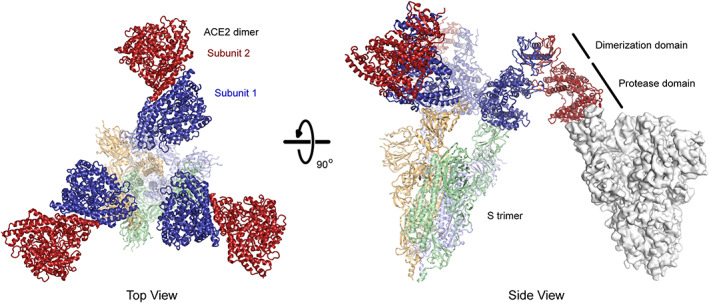
The predicted basis for avid binding of natural ACE22 dimers is inter‐spike bridging. Three copies of ACE22 dimerized via the collectrin‐like domain (subunits in dark red and blue; PDB 6M17) are overlaid with the structure of trimeric SARS‐CoV‐2 S (subunits are pale shades; PDB 7KMS) with all three RBDs in the up conformation and bound to ACE2 protease domains. Note that in each ACE22 dimer, subunit 1 (blue) is bound to a RBD while subunit 2 (red) is directed away from the spike axis where it is accessible for a bridging interaction to another spike (an example is shown as a gray surface at right with a single RBD in the up conformation; PDB 7KNB). Artificial oligomers of sACE2 missing the natural collectrin‐like dimerization domain are not limited to this geometry and might bind with intra‐spike avidity. ACE2, angiotensin‐converting enzyme 2; RBD, receptor‐binding domain; SARS‐CoV‐2, SARS coronavirus 2
FIGURE 5.
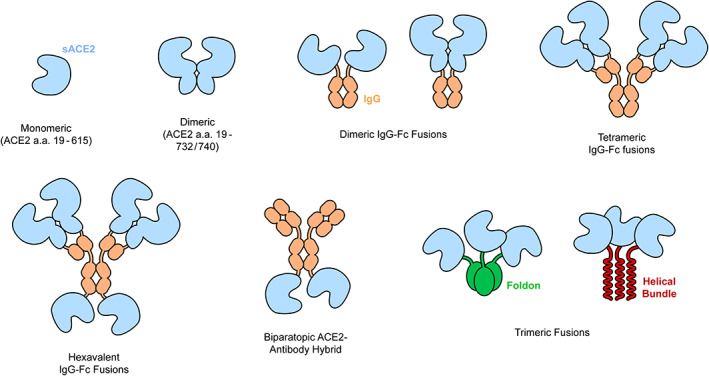
Strategies to increase sACE2•S avidity. S of SARS‐CoV‐2 binds the monomeric ACE2 protease domain (a.a. 19‐615). A longer version of sACE2 (a.a. 19‐732/740) forms a stable dimer that avidly binds virus. Higher levels of multimerization can be accomplished by various N‐ or C‐terminal fusions to IgG heavy and light chains. Trimeric sACE2 proteins have also been constructed to complement trimeric S spikes. Finally, ACE2 has been fused to an anti‐SARS‐CoV‐2 monoclonal to create an avid biparatopic antibody hybrid. ACE2, angiotensin‐converting enzyme 2; SARS‐CoV‐2, SARS coronavirus 2
The creation of sACE2‐Fc fusions automatically forms dimeric ACE2 constructs that are superior to monomeric sACE2, 54 , 63 , 121 although we do not foresee any obvious avidity improvements of Fc‐mediated dimers over natural dimeric sACE22. By adjusting the fusion site between sACE2 and IgG1 from the hinge to the CH1 and LH domains instead, thereby replacing the variable domains with sACE2, an assembly with four ACE2 chains is created. 55 This can be further expanded by fusing additional ACE2 moieties to the C‐terminus of the H chains to create a hexavalent sACE2‐IgG construct, although this arrangement compromised yield. 55 As expected, avid binding to spikes increases as the oligomeric assemblies get larger.
Trimeric sACE2 that spatially aligns three ACE2 protease domains for interactions with S in which all RBDs adopt an up conformation will have very high avidity and has been explored by two groups. 123 , 124 The ACE2 protease domain has been fused to trimerization motifs: the C‐terminal foldon domain of T4 fibritin or a three‐helix bundle. The sACE2 trimers can simultaneously bind multiple RBDs in a spike with low picomolar apparent K d. The sACE2‐foldon trimer was found to have slightly better inhibitory activity against pseudotyped virus. Affinity‐enhancing mutations have also been combined with trimeric sACE2‐foldon. 124
Lastly, avidity has been increased by fusing an anti‐S antibody to sACE2 to create a hybrid, biparatopic ACE2‐IgG fusion. 125 While this protein still contains two ACE2 moieties like other sACE2‐IgG fusions, it is distinct in that avidity is also increased by combining bivalent ACE2 binding with a neutralizing antibody that recognizes a non‐competing epitope on the spike N‐terminal domain. The antibody was selected by yeast display from SARS‐CoV‐2 survivors. The biparatopic ACE2‐IgG hybrid has greatly improved neutralization potency against pseudotyped and authentic virus. Whether this fusion construct compromises serum stability remains to be determined.
6. VESICLE‐BASED ACE2 NANOPARTICLE DECOYS
While this review has focused on soluble ACE2 receptors and their associated Ig‐fusions, work has also been done utilizing membrane‐anchored ACE2. These decoys are based on cell‐derived vesicles or fabricated nanoparticles derived from cells expressing membrane‐embedded ACE2. 126 , 127 , 128 Extracellular vesicles produced from cells over‐expressing ACE2 together with TMPRSS2—which is required for membrane fusion following ACE2/S binding 21 —were more effective at neutralizing pseudotyped viruses than purified sACE2, perhaps because of inbuilt avidity from high‐density receptor display. 127 TMPRSS2 was required for optimum activity and it is possible bound viruses fused with the vesicles, effectively creating irreversible neutralization. Vesicles have been further functionalized by the use of membranes from monocytes that naturally express cytokine receptors, such that both virus and cytokines are sequestered on the nanoparticle surface. 128 Inhibited cytokine signaling might ameliorate potentially life‐threatening inflammatory responses associated with severe COVID‐19. Cytokine neutralization activity was confirmed in vivo by suppressed inflammation in a lipopolysaccharide‐induced lung injury model in mice. While vesicle‐based nanoparticles are scientifically exciting and show efficacy for virus and cytokine neutralization, they will almost certainly suffer a tortuous regulatory pathway to the clinic due to composition heterogeneity and safety concerns.
7. ACE2 MIMETICS
As an alternative to using the natural receptor as a soluble decoy, miniproteins that mimic the SARS‐CoV‐2 binding epitope on ACE2 have been computationally designed and optimized through rounds of deep mutagenesis and selection. 129 , 130 These miniproteins have helical structure and share attributes of the ACE2 N‐terminal helix that forms key interactions with S. The miniproteins bind S with low nanomolar or picomolar K ds and can neutralize authentic SARS‐CoV‐2 infection both in vitro and in vivo. Their key advantage is that miniproteins can be manufactured at scale from bacteria, whereas sACE2 and Fc fusion proteins require mammalian cell expression platforms. There is also precedence for hyperstable miniproteins being non‐immunogenic. 131 However, small proteins tend to have short serum half‐lives and while this can be remedied by fusion to IgG1‐Fc, this negates their primary advantage of bacterial expression. Most likely, miniproteins will need to be administered directly to the nasal passages or lungs to compensate for poor serum stability, as was done in animal experiments. 130 This is not necessarily a disadvantage and may contrast favorably in clinical practice with convalescent plasma and monoclonal antibodies that are infused intravenously. The major disadvantage of these miniproteins is that, while they might mimic some aspects of ACE2 binding in the sense that they use helical segments at the interface, they are not true receptors and lack the breadth of ACE2 derivatives. To begin with, the designed miniproteins poorly bind S of SARS‐CoV‐1 and are not pan‐specific inhibitors. A deep mutational scan of the SARS‐CoV‐2 RBD also provided hints of possible escape mutations, even if not common, 130 and at least one of the designed miniproteins with exceptionally tight affinity to S from the original Wuhan SARS‐CoV‐2 isolate has a near total loss of affinity to newer virus variants in circulation. 132 Computational calculations predict that the miniproteins in general will have reduced affinity to highly transmissible SARS‐CoV‐2 variants carrying a N501Y mutation in S; 133 this mutation markedly increases affinity between ACE2 and the RBD. 48 We propose that at least some miniproteins are “over‐fitted” for the receptor‐binding surface of a single S sequence, such that they are at risk of reduced potency as S mutates. There may be important lessons here for the engineering of ACE2 mutants and it is likely there is a balance between achieving ever higher affinity at the expense of breadth as ACE2 derivatives increasingly look different from WT receptors.
8. CONCLUSION
Emergency Use Authorization from the U.S. Food and Drug Administration has now been granted for the first monoclonal antibody therapies for COVID‐19, with others under development. It has been conclusively demonstrated that soluble ACE2 decoys can be engineered to rival the affinities of monoclonal antibodies and, provided IgG fusions confer desirable pharmacokinetics in humans, there is no reason to expect soluble ACE2 decoys cannot be as effective as monoclonals in reducing viral load in patients. Anti‐SARS‐CoV‐2 monoclonals have received a preferential regulatory environment for clinical advancement due to an in‐depth understanding of antibody biochemistry, pharmacokinetics, and general toxicology. Decoy receptors will always come with additional risks related to immunogenicity and safety, and the outstanding question for the community is whether they provide truly unique benefits that outweigh those risks. The advantages of soluble ACE2 are its breadth against SARS‐associated coronaviruses and its potential for alleviating COVID‐19 symptoms through intrinsic enzymatic activity. We believe these advantages justify continued efforts toward advancing these proteins to the clinic.
CONFLICT OF INTEREST
The University of Illinois has filed a provisional patent for engineered decoy receptors with Erik Procko as an inventor. Erik Procko is a co‐founder of Orthogonal Biologics, Inc.
ACKNOWLEDGMENTS
Erik Procko was supported by NIH award R01AI129719.
Jing W, Procko E. ACE2‐based decoy receptors for SARS coronavirus 2. Proteins. 2021;89(9):1065–1078. 10.1002/prot.26140
Funding information National Institute of Allergy and Infectious Diseases, Grant/Award Number: R01AI129719
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Adam DC, Wu P, Wong JY, et al. Clustering and superspreading potential of SARS‐CoV‐2 infections in Hong Kong. Nat Med. 2020;26:1714‐1719. [DOI] [PubMed] [Google Scholar]
- 2. He X, Lau EHY, Wu P, et al. Temporal dynamics in viral shedding and transmissibility of COVID‐19. Nat Med. 2020;26:672‐675. [DOI] [PubMed] [Google Scholar]
- 3. Bai Y, Yao L, Wei T, et al. Presumed asymptomatic carrier transmission of COVID‐19. JAMA. 2020;323:1406‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Long QX, Tang XJ, Shi QL, et al. Clinical and immunological assessment of asymptomatic SARS‐CoV‐2 infections. Nat Med. 2020;26:1200‐1204. [DOI] [PubMed] [Google Scholar]
- 5. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zheng Z, Peng F, Xu B, et al. Risk factors of critical & mortal COVID‐19 cases: a systematic literature review and meta‐analysis. J Infect. 2020;81:e16‐e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang W, Tang J, Wei F. Updated understanding of the outbreak of 2019 novel coronavirus (2019‐nCoV) in Wuhan, China. J Med Virol. 2020;92:441‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ellul MA, Benjamin L, Singh B, et al. Neurological associations of COVID‐19. Lancet Neurol. 2020;19:767‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Becker RC. COVID‐19 update: Covid‐19‐associated coagulopathy. J Thromb Thrombolysis. 2020;50:54‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soy M, Keser G, Atagündüz P, Tabak F, Atagündüz I, Kayhan S. Cytokine storm in COVID‐19: pathogenesis and overview of anti‐inflammatory agents used in treatment. Clin Rheumatol. 2020;39:2085‐2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song P, Li W, Xie J, Hou Y, You C. Cytokine storm induced by SARS‐CoV‐2. Clin Chim Acta. 2020;509:280‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Y, Yang Y, Zhang C, et al. Clinical and biochemical indexes from 2019‐nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63:364‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo J, Huang Z, Lin L, Lv J. Coronavirus disease 2019 (COVID‐19) and cardiovascular disease: a viewpoint on the potential influence of angiotensin‐converting enzyme inhibitors/angiotensin receptor blockers on onset and severity of severe acute respiratory syndrome coronavirus 2 Infec. J Am Heart Assoc. 2020;9:e016219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roche JA, Roche R. A hypothesized role for dysregulated bradykinin signaling in COVID‐19 respiratory complications. FASEB J. 2020;34:7265‐7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science. 2020;367:1444‐1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lan J, Ge J, Yu J, et al. Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature. 2020;581:215‐220. [DOI] [PubMed] [Google Scholar]
- 18. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 2020;367:1260‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade‐Long structural studies of SARS coronavirus. J Virol. 2020;94:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271‐280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li W, Moore MJ, Vasilieva N, et al. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS‐CoV‐2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5:562‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tortorici MA, Veesler D. Chapter four ‐ structural insights into coronavirus entry. Adv Virus Res. 2019;105:93‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 2020;181:281‐292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mille JK, Whittaker GR. Host cell entry of Middle East respiratory syndrome coronavirus after two‐step, furin‐mediated activation of the spike protein. Proc Natl Acad Sci U S A. 2014;111:15214‐15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walls AC, Tortorici MA, Snijder J, et al. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad Sci U S A. 2017;114:11157‐11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hansen J, Baum A, Pascal KE, et al. Studies in humanized mice and convalescent humans yield a SARS‐CoV‐2 antibody cocktail. Science. 2020;369:1010‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brouwer PJM, Caniels TG, van der Straten K, et al. Potent neutralizing antibodies from COVID‐19 patients define multiple targets of vulnerability. Science. 2020;369:643‐650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wec AZ, Wrapp D, Herbert AS, et al. Broad neutralization of SARS‐related viruses by human monoclonal antibodies. Science. 2020;369:731‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang C, Li W, Drabek D, et al. A human monoclonal antibody blocking SARS‐CoV‐2 infection. Nat Commun. 2020;11:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu Y, Wang F, Shen C, et al. A noncompeting pair of human neutralizing antibodies block COVID‐19 virus binding to its receptor ACE2. Science. 2020;368:1274‐1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rogers TF, Zhao F, Huang D, et al. Isolation of potent SARS‐CoV‐2 neutralizing antibodies and protection from disease in a small animal model. Science. 2020;369:956‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pyrc K, Dijkman R, Deng L, et al. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J Mol Biol. 2006;364:964‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24:490‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Volz E, Mishra S, Chand M, et al. Transmission of SARS‐CoV‐2 lineage B.1.1.7 in England: insights from linking epidemiological and genetic data. medRxiv. 2021;2020:20249034. [Google Scholar]
- 37. Faria NR, Claro IM, Candido D, Moyses Franco LA, Andrade PS, Coletti TM, Silva CAM, Sales FC, Manuli ER, Aguiar RS, et al. Virological. Genomic characterisation of an emergent SARS‐CoV‐2 lineage in Manaus: preliminary findings. https://virological.org/t/genomic‐characterisation‐of‐an‐emergent‐sars‐cov‐2‐lineage‐in‐manaus‐preliminary‐findings/586. Accessed February 12, 2021.
- 38. Tegally H, Wilkinson E, Giovanetti M, et al. Emergence and rapid spread of a new severe acute respiratory syndrome‐related coronavirus 2 (SARS‐CoV‐2) lineage with multiple spike mutations in South Africa. medRxiv. 2020;2020:20248640. [Google Scholar]
- 39. Wibmer CK, Ayres F, Hermanus T, et al. SARS‐CoV‐2 501Y.V2 escapes neutralization by south African COVID‐19 donor plasma. Nat Med. 2021;27:622‐625. [DOI] [PubMed] [Google Scholar]
- 40. Baum A, Fulton BO, Wloga E, et al. Antibody cocktail to SARS‐CoV‐2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369:1014‐1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Monteil V, Kwon H, Prado P, et al. Inhibition of SARS‐CoV‐2 infections in engineered human tissues using clinical‐grade soluble human ACE2. Cell. 2020;181:905‐913.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lei C, Qian K, Li T, et al. Neutralization of SARS‐CoV‐2 spike pseudotyped virus by recombinant ACE2‐Ig. Nat Commun. 2020;11:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hofmann H, Geier M, Marzi A, et al. Susceptibility to SARS coronavirus S protein‐driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem Biophys Res Commun. 2004;319:1216‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin L, Jiang X, Zhang Z, et al. Gastrointestinal symptoms of 95 cases with SARS‐CoV‐2 infection. Gut. 2020;69:997‐1001. [DOI] [PubMed] [Google Scholar]
- 46. Cantuti‐Castelvetri L, Ojha R, Pedro LD, et al. Neuropilin‐1 facilitates SARS‐CoV‐2 cell entry and infectivity. Science. 2020;370:856‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Daly JL, Simonetti B, Klein K, et al. Neuropilin‐1 is a host factor for SARS‐CoV‐2 infection. Science. 2020;370:861‐865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chan KK, Tan TJC, Narayanan KK, Procko E. An engineered decoy receptor for SARS‐CoV‐2 broadly binds protein S sequence variants. Sci Adv. 2021;7:eabf1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Usmani SS, Bedi G, Samuel JS, et al. THPdb: database of FDA‐approved peptide and protein therapeutics. PLoS One. 2017;12:e0181748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chan KK, Dorosky D, Sharma P, et al. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science. 2020;369:1261‐1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Procko E. The sequence of human ACE2 is suboptimal for binding the S spike protein of SARS coronavirus 2. bioRxiv. 2020;2020:994236. [Google Scholar]
- 52. Procko E. Deep mutagenesis in the study of COVID‐19: a technical overview for the proteomics community. Expert Rev Proteomics. 2020;17(9):633‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79‐88. [DOI] [PubMed] [Google Scholar]
- 54. Glasgow A, Glasgow J, Limonta D, et al. Engineered ACE2 receptor traps potently neutralize SARS‐CoV‐2. Proc Natl Acad Sci U S A. 2020;117:28046‐28055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li Y, Wang H, Tang X, et al. SARS‐CoV‐2 and three related coronaviruses utilize multiple ACE2 Orthologs and are potently blocked by an improved ACE2‐Ig. J Virol. 2020;94:e01283‐e01220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS‐CoV‐2. Nat Rev Microbiol. 2021;19:155‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Higuchi Y, Suzuki T, Arimori T, et al. High affinity modified ACE2 receptors protect from SARS‐CoV‐2 infection in hamsters. bioRxiv. 2020;2020:299891. [Google Scholar]
- 58. Cohen‐Dvashi H, Weinstein J, Katz M, et al. Coronacept – a potent immunoadhesin against SARS‐CoV‐2. bioRxiv. 2020;2020:247940. [Google Scholar]
- 59. Heinzelman P, Romero PA. Discovery of human ACE2 variants with altered recognition by the SARS‐CoV‐2 spike protein. bioRxiv. 2020;2020:301861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rushworth CA, Guy JL, Turner AJ. Residues affecting the chloride regulation and substrate selectivity of the angiotensin‐converting enzymes (ACE and ACE2) identified by site‐directed mutagenesis. FEBS J. 2008;275:6033‐6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Moore MJ, Dorfman T, Li W, et al. Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin‐converting enzyme 2. J Virol. 2004;78:10628‐10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu P, Xie X, Gao L, Jin J. Designed variants of ACE2‐fc that decouple anti‐SARS‐CoV‐2 activities from unwanted cardiovascular effects. Int J Biol Macromol. 2020;165:1626‐1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mou H, Quinlan BD, Peng H, et al. Mutations derived from horseshoe bat ACE2 orthologs enhance ACE2‐fc neutralization of SARS‐CoV‐2. PLoS Pathog. 2021;17:e1009501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Iwanaga N, Cooper L, Rong L, et al. Novel ACE2‐IgG1 fusions with improved in vitro and in vivo activity against SARS‐CoV2. bioRxiv. 2020;2020:152157. [Google Scholar]
- 65. Castiglione GM, Zhou L, Xu Z, Neiman Z, Hung C‐F, Duh EJ. The ancient cardioprotective mechanisms of ACE2 bestow SARS‐CoV‐2 with a wide host range. bioRxiv. 2021;2021:425115. [Google Scholar]
- 66. Lu J, Sun PD. High affinity binding of SARS‐cov‐2 spike protein enhances ACE2 carboxypeptidase activity. J Biol Chem. 2020;295:18579‐18588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zoufaly A, Poglitsch M, Aberle JH, et al. Human recombinant soluble ACE2 in severe COVID‐19. Lancet Respir Med. 2020;8:1154‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wysocki J, Ye M, Rodriguez E, et al. Targeting the degradation of angiotensin II with recombinant angiotensin‐converting enzyme 2: prevention of angiotensin II‐dependent hypertension. Hypertension. 2010;55:90‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Haschke M, Schuster M, Poglitsch M, et al. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin‐converting enzyme 2 in healthy human subjects. Clin Pharmacokinet. 2013;52:783‐792. [DOI] [PubMed] [Google Scholar]
- 70. Strohl WR. Fusion proteins for half‐life extension of biologics as a strategy to make biobetters. BioDrugs. 2015;29:215‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Crackower MA, Sarao R, Oliveira‐dos‐Santos AJ, Da Costa J, Zhang L. Angiotensin‐converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822‐828. [DOI] [PubMed] [Google Scholar]
- 72. Imai Y, Kuba K, Rao S, et al. Angiotensin‐converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zou Z, Yan Y, Shu Y, et al. Angiotensin‐converting enzyme 2 protects from lethal avian influenza a H5N1 infections. Nat Commun. 2014;5:3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Treml B, Neu N, Kleinsasser A, et al. Recombinant angiotensin‐converting enzyme 2 improves pulmonary blood flow and oxygenation in lipopolysaccharide‐induced lung injury in piglets. Crit Care Med. 2010;38:596‐601. [DOI] [PubMed] [Google Scholar]
- 75. Johnson JA, West J, Maynard KB, Hemnes AR. ACE2 improves right ventricular function in a pressure overload model. PLoS One. 2011;6:e20828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu P, Wysocki J, Souma T, et al. Novel ACE2‐fc chimeric fusion provides long‐lasting hypertension control and organ protection in mouse models of systemic renin angiotensin system activation. Kidney Int. 2018;94:114‐125. [DOI] [PubMed] [Google Scholar]
- 77. Khan A, Benthin C, Zeno B, et al. A pilot clinical trial of recombinant human angiotensin‐converting enzyme 2 in acute respiratory distress syndrome. Crit Care. 2017;21:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jan W, Minghao Y, Eva R, et al. Targeting the degradation of angiotensin II with recombinant angiotensin‐converting enzyme 2. Hypertension. 2010;55:90‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Al Shoyaib A, Archie SR, Karamyan VT. Intraperitoneal route of drug administration: should it be used in experimental animal studies? Pharm Res. 2019;37:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Regoeczi E, Zaimi O, Chindemi PA, Charlwood PA. Absorption of plasma proteins from peritoneal cavity of normal rats. Am J Physiol Metab. 1989;256:E447‐E452. [DOI] [PubMed] [Google Scholar]
- 81. Tada T, Fan C, Chen JS, et al. An ACE2 microbody containing a single immunoglobulin fc domain is a potent inhibitor of SARS‐CoV‐2. Cell Rep. 2020;33:108528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hassler L, Wysocki J, Gelarden I, et al. A novel soluble ACE2 protein totally protects from lethal disease caused by SARS‐CoV‐2 infection. bioRxiv. 2021;2021:435191. [Google Scholar]
- 83. Sims JJ, Greig JA, Michalson KT, et al. Intranasal gene therapy to prevent infection by SARS‐CoV‐2 variants. bioRxiv. 2021;2021:439149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half‐life. Front Immunol. 2019;10:1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Svilenov HL, Sacherl J, Reiter A, et al. Efficient inhibition of SARS‐CoV‐2 strains by a novel ACE2‐IgG4‐fc fusion protein with a stabilized hinge region. bioRxiv. 2020;2020:413443. [Google Scholar]
- 86. Schäfer A, Muecksch F, Lorenzi JCC, et al. Antibody potency, effector function, and combinations in protection and therapy for SARS‐CoV‐2 infection in vivo. J Exp Med. 2020;218:e20201993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Katzelnick LC, Gresh L, Halloran ME, et al. Antibody‐dependent enhancement of severe dengue disease in humans. Science. 2017;358:929‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tirado SMC, Yoon K‐J. Antibody‐dependent enhancement of virus infection and disease. Viral Immunol. 2003;16:69‐86. [DOI] [PubMed] [Google Scholar]
- 89. Su S, Du L, Jiang S. Learning from the past: development of safe and effective COVID‐19 vaccines. Nat Rev Microbiol. 2020;19(3):211‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kim HW, Canchola JG, Brandt CD, et al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol. 1969;89:422‐434. [DOI] [PubMed] [Google Scholar]
- 91. Lee WS, Wheatley AK, Kent SJ, DeKosky BJ. Antibody‐dependent enhancement and SARS‐CoV‐2 vaccines and therapies. Nat Microbiol. 2020;5:1185‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wan Y, Shang J, Sun S, et al. Molecular mechanism for antibody‐dependent enhancement of coronavirus entry. J Virol. 2020;94:e02015‐e02019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wang Q, Zhang L, Kuwahara K, et al. Immunodominant SARS coronavirus epitopes in humans elicited both enhancing and neutralizing effects on infection in non‐human primates. ACS Infect Dis. 2016;2:361‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Baden LR, El Sahly HM, Essink B, et al. Efficacy and safety of the mRNA‐1273 SARS‐CoV‐2 vaccine. N Engl J Med. 2020;384:403‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA Covid‐19 vaccine. N Engl J Med. 2020;383:2603‐2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Voysey M, Clemens SAC, Madhi SA, et al. Safety and efficacy of the ChAdOx1 nCoV‐19 vaccine (AZD1222) against SARS‐CoV‐2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet. 2021;397:99‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Li D, Edwards RJ, Manne K, et al. The functions of SARS‐CoV‐2 neutralizing and infection‐enhancing antibodies in vitro and in mice and nonhuman primates. bioRxiv. 2021;2020:424729. [Google Scholar]
- 98. Wu F, Yan R, Liu M, et al. Antibody‐dependent enhancement (ADE) of SARS‐CoV‐2 infection in recovered COVID‐19 patients: studies based on cellular and structural biology analysis. medRxiv. 2020;2020:20209114. [Google Scholar]
- 99. García‐Nicolás O, V'kovski P, Zettl F, Zimmer G, Thiel V, Summerfield A. No evidence for human monocyte‐derived macrophage infection and antibody‐mediated enhancement of SARS‐CoV‐2 infection [internet]. Front Cell Infect Microbiol. 2021;11:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yeung ML, Teng JLL, Jia L, et al. Soluble ACE2‐mediated cell entry of SARS‐CoV‐2 via interaction with proteins related to the renin‐angiotensin system. Cell. 2021;184:2212‐2228.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Park J, Gill KS, Aghajani AA, et al. Engineered receptors for human cytomegalovirus that are orthogonal to normal human biology. PLoS Pathog. 2020;16:e1008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kuba K, Imai Y, Ohto‐Nakanishi T, Penninger JM. Trilogy of ACE2: a peptidase in the renin‐angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol Ther. 2010;128:119‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Gheblawi M, Wang K, Viveiros A, et al. Angiotensin‐converting enzyme 2: SARS‐CoV‐2 receptor and regulator of the renin‐angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res. 2020;126:1456‐1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin‐converting enzyme. Cloning and functional expression as a captopril‐insensitive carboxypeptidase. J Biol Chem. 2000;275:33238‐33243. [DOI] [PubMed] [Google Scholar]
- 105. Turner AJ, Tipnis SR, Guy JL, Rice G, Hooper NM. ACEH/ACE2 is a novel mammalian metallocarboxypeptidase and a homologue of angiotensin‐converting enzyme insensitive to ACE inhibitors. Can J Physiol Pharmacol. 2002;80:346‐353. [DOI] [PubMed] [Google Scholar]
- 106. Vickers C, Hales P, Kaushik V, et al. Hydrolysis of biological peptides by human angiotensin‐converting enzyme‐related carboxypeptidase. J Biol Chem. 2002;277:14838‐14843. [DOI] [PubMed] [Google Scholar]
- 107. Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1‐9. Circ Res. 2000;87:E1‐E9. [DOI] [PubMed] [Google Scholar]
- 108. Dasgupta C, Zhang L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discov Today. 2011;16:22‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Garvin MR, Alvarez C, Miller JI, et al. A mechanistic model and therapeutic interventions for covid‐19 involving a ras‐mediated bradykinin storm. Elife. 2020;9:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Heurich A, Hofmann‐Winkler H, Gierer S, Liepold T, Jahn O, Pöhlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88:1293‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Haga S, Yamamoto N, Nakai‐Murakami C, et al. Modulation of TNF‐α‐converting enzyme by the spike protein of SARS‐CoV and ACE2 induces TNF‐α production and facilitates viral entry. Proc Natl Acad Sci. 2008;105:7809‐7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kuba K, Imai Y, Rao S, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat Med. 2005;11:875‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wallentin L, Lindbäck J, Eriksson N, et al. Angiotensin‐converting enzyme 2 (ACE2) levels in relation to risk factors for COVID‐19 in two large cohorts of patients with atrial fibrillation. Eur Heart J. 2020;41:4037‐4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wu Z, Hu R, Zhang C, Ren W, Yu A, Zhou X. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID‐19 patients. Crit Care. 2020;24:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Rieder M, Wirth L, Pollmeier L, et al. Serum ACE‐2, angiotensin II, and aldosterone levels are unchanged in patients with COVID‐19. Am J Hypertens. 2020. ;34(3):278–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Chen P, Nirula A, Heller B, et al. SARS‐CoV‐2 neutralizing antibody LY‐CoV555 in outpatients with Covid‐19. N Engl J Med. 2021;384:229‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Huang Y, Yang C, Feng XX, Xu W, Wen LS. Structural and functional properties of SARS‐CoV‐2 spike protein: potential antivirus drug development for COVID‐19. Acta Pharmacol Sin. 2020;41:1141‐1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ke Z, Oton J, Qu K, et al. Structures and distributions of SARS‐CoV‐2 spike proteins on intact virions. Nature. 2020;588:498‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Turoňová B, Sikora M, Schürmann C, et al. In situ structural analysis of SARS‐CoV‐2 spike reveals flexibility mediated by three hinges. Science. 2020;370:203‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yao H, Song Y, Chen Y, et al. Molecular architecture of the SARS‐CoV‐2 virus. Cell. 2020;183:730‐738.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lui I, Zhou XX, Lim SA, et al. Trimeric SARS‐CoV‐2 spike interacts with dimeric ACE2 with limited intra‐spike avidity. bioRxiv. 2020;2020:109157. [Google Scholar]
- 122. Zhou T, Tsybovsky Y, Gorman J, et al. Cryo‐EM structures of SARS‐CoV‐2 spike without and with ACE2 reveal a pH‐dependent switch to mediate endosomal positioning of receptor‐binding domains. Cell Host Microb. 2020;28:867‐879.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Guo L, Bi W, Wang X, et al. Engineered trimeric ACE2 binds viral spike protein and locks it in “three‐up” conformation to potently inhibit SARS‐CoV‐2 infection. Cell Res. 2021;31:98‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Xiao T, Lu J, Zhang J, et al. A trimeric human angiotensin‐converting enzyme 2 as an anti‐SARS‐CoV‐2 agent. Nat Struct Mol Biol. 2021;28:202‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Miao X, Luo Y, Huang X, et al. A novel biparatopic hybrid antibody‐ACE2 fusion that blocks SARS‐CoV‐2 infection: implications for therapy. MAbs. 2020;12:1804241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wang C, Wang S, Chen Y, et al. Membrane nanoparticles Derived from ACE2‐rich cells block SARS‐CoV‐2 infection. ACS Nano. 2021;15:6340‐6351. [DOI] [PubMed] [Google Scholar]
- 127. Cocozza F, Névo N, Piovesana E, et al. Extracellular vesicles containing ACE2 efficiently prevent infection by SARS‐CoV‐2 spike protein‐containing virus. J Extracell Vesicles. 2020;10:e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rao L, Xia S, Xu W, et al. Decoy nanoparticles protect against COVID‐19 by concurrently adsorbing viruses and inflammatory cytokines. Proc Natl Acad Sci U S A. 2020;117:27141‐27147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Cao L, Goreshnik I, Coventry B, et al. De novo design of picomolar SARS‐CoV‐2 miniprotein inhibitors. Science. 2020;370:426‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Linsky TW, Vergara R, Codina N, et al. De novo design of potent and resilient hACE2 decoys to neutralize SARS‐CoV‐2. Science. 2020;370:1208‐1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Chevalier A, Silva D‐A, Rocklin GJ, et al. Massively parallel de novo protein design for targeted therapeutics. Nature. 2017;550:74‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Javanmardi K, Chou C‐W, Terrace CI, et al. Rapid characterization of spike variants via mammalian cell surface display. bioRxiv. 2021;2021:437622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Williams AH, Zhan C‐G. Fast prediction of binding affinities of the SARS‐CoV‐2 spike protein mutant N501Y (UKvariant) with ACE2 and miniprotein drug candidates. J Phys Chem B. 2021;125:4330‐4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.