

Abstract
Extant fold‐switching proteins remodel their secondary structures and change their functions in response to environmental stimuli. These shapeshifting proteins regulate biological processes and are associated with a number of diseases, including tuberculosis, cancer, Alzheimer's, and autoimmune disorders. Thus, predictive methods are needed to identify more fold‐switching proteins, especially since all naturally occurring instances have been discovered by chance. In response to this need, two high‐throughput predictive methods have recently been developed. Here we test them on ORF9b, a newly discovered fold switcher and potential therapeutic target from the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2). Promisingly, both methods correctly indicate that ORF9b switches folds. We then tested the same two methods on ORF9b1, the ORF9b homolog from SARS‐CoV‐1. Again, both methods predict that ORF9b1 switches folds, a finding consistent with experimental binding studies. Together, these results (a) demonstrate that protein fold switching can be predicted using high‐throughput computational approaches and (b) suggest that fold switching might be a general characteristic of ORF9b homologs.
Keywords: fold‐switching proteins, metamorphic proteins, protein folding, SARS‐CoV‐2
1. INTRODUCTION
Fold‐switching proteins remodel their secondary structures and change their functions in response to environmental stimuli. 1 These proteins challenge the long‐held paradigm that the amino acid sequence of a globular protein encodes its unique stable structure. 2 Furthermore, fold switchers occur in all kingdoms of life, perform over 30 different functions, and are triggered by nearly a dozen stimuli. 1 Additionally, the structural transitions of some fold switchers regulate biological processes, such as the expression of bacterial virulence genes 3 and the circadian rhythm of cyanobacteria. 4
Given the growing amount of evidence suggesting that fold switchers play important regulatory roles, 5 it is not surprising that a number of them are associated with different human diseases. For example, PimA, which undergoes an α‐helix <−> β‐strand transition, initiates the biosynthetic pathway of virulence factors produced by M. tuberculosis. 6 Human lymphotactin (a.k.a. XCL1) iso‐energetically populates two β‐sheet conformations with completely different hydrogen bonding patterns 7 , 8 and is associated with autoimmune disorders. 9 Furthermore, human Chloride Intracellular Channel 1 (CLIC1) remodels its secondary structure and changes its function from a glutathione reductase 10 to a chloride channel 11 that balances intracellular chloride levels when cells undergo oxidative stress due to cancer 12 or Alzheimer's. 13
The biological relevance and increasing number of identified fold switchers have motivated the development of computational methods that predict more. The need for accurate predictive methods is especially acute because, to date, all naturally occurring fold switchers have been discovered by chance. Two years ago, we reported that discrepancies between predicted and experimentally determined protein structures can indicate fold switching. 14 More recently, we developed a sequence‐based method that predicts fold switchers with high levels of statistical significance. 15
Here, both predictive approaches are tested on the newly discovered fold switcher, ORF9b 16 (Figure 1). This protein is from the genome of the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2), the cause of the current global pandemic. When expressed, ORF9b binds the human mitochondrial protein Tom70, an outer membrane protein that acts as a host‐dependency factor for SARS‐CoV‐2. 16 In other words, viral titers in Caco‐2 cells with TOMM70 (the gene that encodes Tom70) are significantly higher than in cells without. ORF9b‐Tom70 binding has been proposed to have one of two cellular effects 16 : (a) modulating interferon and apoptosis signaling or (b) decreasing mitochondrial import efficiency, leading to mitophagy.
FIGURE 1.
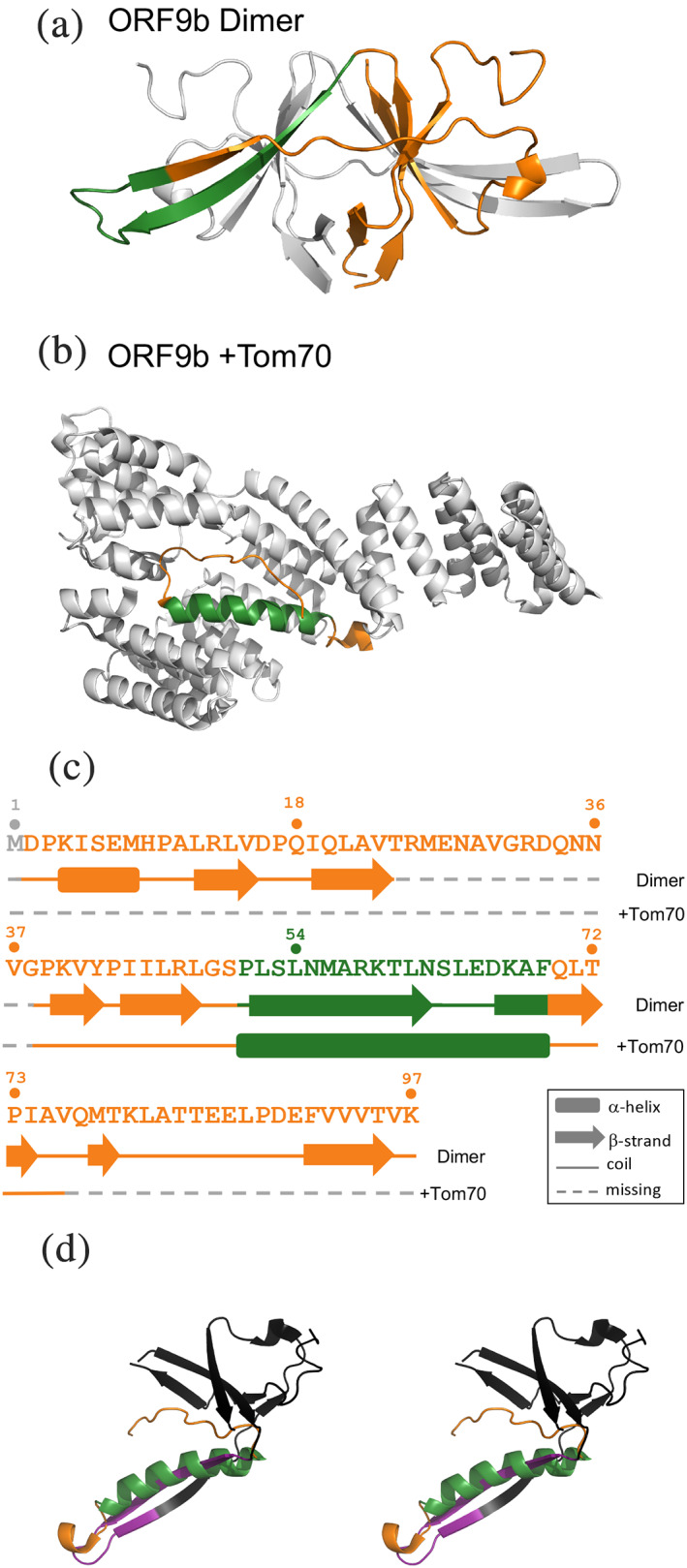
The SARS‐CoV‐2 protein, ORF9b, switches folds. (a). ORF9b forms a homodimer (PDB ID: 6Z4U); chain A is colored orange; its fold‐switching region is colored dark green; chain B is gray to illustrate how its domains are swapped. (b). The structure of ORF9b (dark green and orange) in complex with human TOM70 (gray) is shown below (PDB ID: 7KDT). (c). The sequence of ORF9b assumes different secondary structures in different contexts: an all‐β homodimer (upper secondary structure diagram) and an α‐helix when bound to Tom70 (lower secondary structure diagram). Residue numbers are shown directly above the sequence. Regions of ORF9b that assume a β‐hairpin in the homodimer and an α‐helix in complex with human Tom70 are shown in dark green; the rest of ORF9b is shown in orange, except for regions of missing electron density (dashed lines shown in gray). (d). Stereo view of both ORF9b conformations superimposed. Only one unit of the homodimer is shown; its fold‐switching region is purple; the rest is black. The α‐helical form is colored as in previous panels: dark green for fold switching, orange for the rest. All ribbon diagrams were made using PyMOL 22
Isolated ORF9b folds into a β‐sheet topology that forms a domain‐swapped dimer, part of which transforms into a long α‐helix when bound to Tom70 (Figure 1). Interestingly, ORF9b has two infection‐driven phosphorylation sites (S50 and S53), 17 which contact Tom70 directly but are solvent‐exposed in the ORF9b dimer. Thus, it has been hypothesized that phosphorylation weakens ORF9b's interactions with Tom70, causing its homodimeric β‐sheet fold to become more energetically favorable. 16
Since ORF9b undergoes a large α‐helix <−> β‐sheet transition, it is a suitable target for both the sequence‐ and structure‐based predictive methods reported previously. 15 , 18 Consistent with experimental observations, both methods indicate that ORF9b switches folds. These methods were then tested on the SARS‐CoV‐1 ORF9b homolog, hereafter called ORF9b1, which is also binds Tom70 in situ 16 but has not been shown to switch folds. Again, both methods predict that ORF9b1 switches folds. Together, these results (a) corroborate previous work demonstrating that low‐resolution, high‐throughput methods can predict fold switching and (b) suggest that fold switching might be a general characteristic of ORF9b homologs.
2. RESULTS
2.1. Structure‐based predictions suggest that SARS‐CoV‐2 ORF9b switches folds
Previous work has shown that inconsistencies between experimentally determined secondary structures and homology‐based secondary structure predictions often indicate protein fold switching. 14 These predictions are homology‐based because the algorithms that generate them (in this case PSIPRED, 19 SPIDER2, 20 and JPred4 21 ) leverage conservation patterns from homologous sequences to infer secondary structure.
Secondary structure predictions of full‐length ORF9b were calculated using PSIPRED, 19 SPIDER2, 20 and JPred4 21 (Figure 2) and compared with the experimentally determined structures. Interestingly, all three secondary structure predictors suggest helical propensities in the region of ORF9b that forms an α‐helix when it binds Tom70 (this has been reported previously for JPred4, 16 but not the other secondary structure predictors). Homology‐based predictions of this region uniformly disagree with the experimentally determined secondary structure of the ORF9b dimer, which forms a domain‐swapped β‐hairpin (Figure 1, green). By contrast, these predictions suggest more helical content than was observed in the cryo‐EM structure of the ORF9b‐Tom70 complex. Accordingly, low prediction accuracies (Methods) were observed from all three secondary structure predictors. Specifically, prediction accuracies ranged from 48–59% when referenced against the full‐length experimentally determined ORF9b dimer and 51–62% when compared with the cryo‐EM‐resolved region of ORF9b that binds Tom70 (Figure 2). These accuracies fall well below previously benchmarked predictor accuracies, all three of which exceed 80%. 19 , 20 , 21 Additionally, these accuracies are lower than the mean/median secondary structure prediction accuracies for fold‐switching regions of proteins reported previously, which range from 67–68%/68–71%, respectively. 14 Thus, in line with previous findings, 1 , 14 these inaccurate secondary structure predictions are consistent with the experimental observation that ORF9b switches folds.
FIGURE 2.
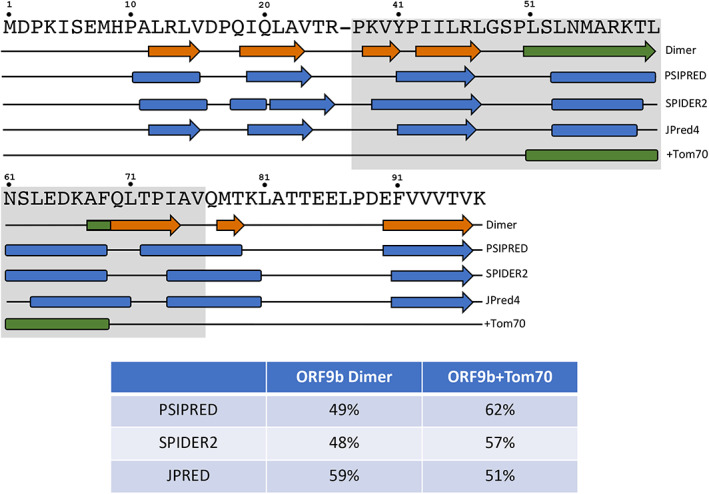
Three state‐of‐the art algorithms inaccurately predict the secondary structures of both experimentally determined forms of ORF9b. Names of the three algorithms lie to the right of their corresponding predictions (three middle secondary structure diagrams, all colored blue). Coils are represented by black lines; α‐helices/β‐strands are represented by rounded rectangles/arrows. Predictions of α‐helices and β‐strands that span ≥2 contiguous residues are shown. Experimentally determined secondary structures of the ORF9b Dimer/ORF9b + Tom70 are shown above/below the predictions and are colored as in Figure 1: green secondary structures switch folds; all other secondary structures are shown in orange. The dash in ORF9b's sequence (above secondary structure diagrams) represents an area of missing electron density in both structures; the gray background corresponds to the region of the ORF9b structure that could be resolved by cryo‐EM when in complex with Tom70. The table reports secondary structure prediction accuracies of each algorithm referenced against each experimentally determined structure
2.2. Sequence‐based predictions suggest that SARS‐CoV‐2 ORF9b switches folds
Given that the structure‐based method correctly inferred that ORF9b switches folds, the next step was to determine whether ORF9b fold switching could be inferred from its sequence alone. It has been shown previously that JPred4 predicts fold switching from sequence more robustly than other secondary structure predictors, 18 and α‐helix <−> β‐strand prediction discrepancies from JPred4 between whole protein sequences and excised sequence fragments can be a robust indicator of fold switching. 15 Such a fragment (residues 59–80) was identified in ORF9b (Figure 3). Within the context of its parent protein, this fragment is predicted to form N‐ and C‐terminal α‐helices, whereas the excised fragment is predicted to assume an α‐helix at the N‐terminus and β‐strands towards the C‐terminus.
FIGURE 3.
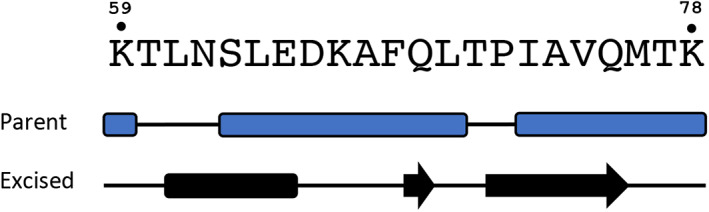
JPred4 predicts significantly different secondary structures for a fold‐switching ORF9b sequence fragment depending on its context. When within its parent sequence, the fragment is predicted to be α‐helical (above), but when excised from its parent, it is predicted to be a mix of α‐helix and β‐strand (below). The sequence is shown above both predictions with residue numbers shown above for reference. Coils are represented by black lines; α‐helices/β‐strands are represented by rounded rectangles/arrows
Based on previously reported thresholds, 15 these prediction discrepancies between parent and excised sequence fragments are significant, consistent with the observation that ORF9b switches folds. Specifically, both the total predicted secondary structure content (50%) and the 75% discrepancy between α‐helix <−> β‐strand predictions exceed the minimum parameters 15 (secondary structure content ≥35% and α‐helix <−> β‐strand prediction discrepancies ≥50%). Although the region with α‐helix <−> β‐strand discrepancy begins 5‐residues C‐terminal to the region experimentally observed to switch from α‐helix <−> β‐strand, we note that the β‐strand predictions of the excised fragment overlap with some regions of the ORF9b dimer that also fold into β‐strands. Furthermore, as reported previously, 15 , 18 this method uses the differences in predicted α‐helix and β‐strand to infer fold switching. Predictions need not adhere exactly to a solved protein structure to make this inference. Indeed, as shown in the previous section and in previous work, 14 there is generally poor correspondence between the predicted and experimentally determined secondary structures of fold‐switching proteins. Furthermore, slightly off‐register α‐helix <−> β‐strand prediction discrepancies have been observed in other fold switchers such as RfaH and Ovalbumin, but such discrepancies are very rare in proteins expected not to switch folds. 15
2.3. Both predictive methods suggest that ORF9b's homolog from SARS‐CoV‐1 also switches folds
Since both of our predictive methods indicate that ORF9b switches folds, they were used to assess whether its homolog from SARS‐CoV‐1 (hereafter called ORF9b1) might switch also. ORF9b1 is the only ORF9b homolog with a solved crystal structure in the PDB. Its experimentally determined structure assumes the same dimeric fold as ORF9b (RMSD = 0.94 Å using PyMOL 22 ), and its sequence is 69% identical to ORF9b's (Figure 4a).
FIGURE 4.
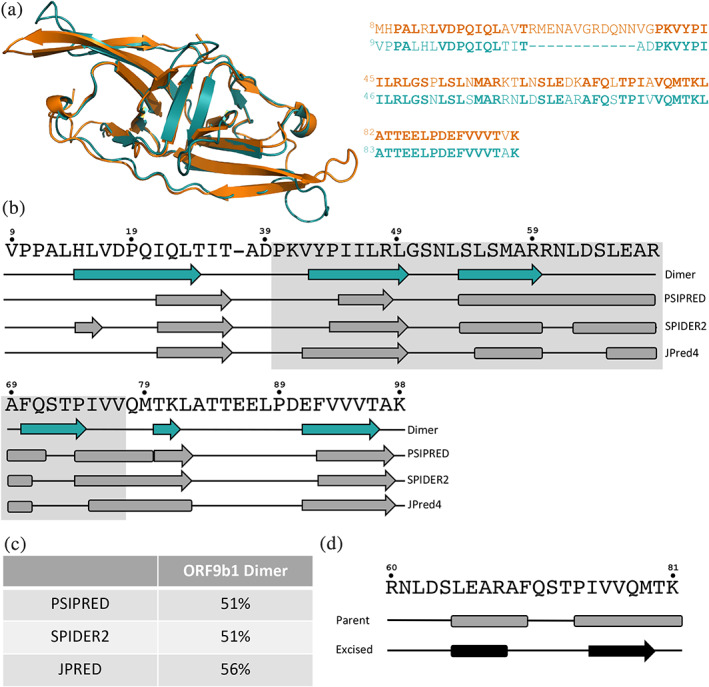
Both predictive methods suggest that SARS‐CoV‐1 ORF9b (ORF9b1) switches folds. (a) Both ORF9b1 (teal, pdb ID: 2CME) and ORF9b (orange, pdb ID: 6Z4U) 22 ), and their sequences are 69% identical (bold residues are identical; sequences are colored to correspond with their respective protein structures). (b). The experimentally determined secondary structure of ORF9b1 (teal secondary structure diagram) was predicted inaccurately by three state‐of‐the‐art algorithms. Names of the three algorithms lie to the right of their corresponding predictions (gray). Predictions of α‐helices and β‐strands that span ≥2 contiguous residues are shown. The region with the gray background corresponds to the region of the ORF9b‐Tom70 that could be resolved by cryo‐EM. Residue numbers are shown above the sequence. (c) Secondary structure prediction accuracies (Methods) from each algorithm with reference to the solved structure are shown in the Table. (d) JPred4 predicts significantly different secondary structures for an ORF9b1 sequence fragment when it is within its parent sequence (above) and when it is excised from its parent (below). This sequence aligns exactly with the sequence fragment of ORF9b shown in Figure 3. The sequence of this fragment is shown above both predictions with residue numbers shown above. In both secondary structure diagrams, coils are represented by black lines; α‐helices/β‐strands are represented by rounded rectangles/arrows
As with ORF9b, both predictive approaches suggest that ORF9b1 switches folds (Figure 4b‐d). Specifically, all three secondary structure predictors suggest that the region of ORF9b1 analogous to the ORF9b region that interacts with human Tom70 is helical, even though it folds into a β‐hairpin in the experimentally determined ORF9b1 structure (Figure 4a,b). The prediction that this region can form an α‐helix is plausible given that ORF9b1 (a) coimmunoprecipitates with Tom70 in both HEK293T and A549 cells and (b) colocalizes with Tom70 in HeLaM cells. 16 Additionally, predictions from all three algorithms had lower‐than‐expected accuracies, 19 , 20 , 21 ranging from 51–59% (Figure 4c), similar to the prediction accuracies the ORF9b dimer. Furthermore, JPred4 predictions of the ORF9b1 sequence fragment analogous to the ORF9b sequence in Figure 3 differ significantly depending on whether the fragment is excised or contexualized within its parent sequence (Figure 4d): 41% predicted secondary structure content, 56% α <−> β discrepancies, both of which, again, exceed the significance thresholds for fold switching reported previously. 15
3. DISCUSSION
Extant fold‐switching proteins remodel their secondary structures and change their functions in response to environmental stimuli. 1 These shapeshifting proteins regulate biological processes 5 and are associated with a number of human diseases. 6 , 9 , 12 In December 2020, ORF9b, a protein from the SARS‐CoV‐2 genome and a possible therapeutic target for coronaviruses, 16 was reported to switch between a homodimeric β‐sheet fold and an α‐helix that binds human Tom70. 16
We tested two recently developed predictive methods on ORF9b to assess whether they could identify it as a fold switcher. Both methods–one structure‐based, 14 the other sequence‐based 15 –were successful. The structure‐based method identified the region experimentally observed to assume α‐helix in one structure and β‐strand in the other, while the sequence‐based prediction began within five residues of this fold‐switching region. These results suggest that structure‐based predictions might identify the precise locations of fold‐switching regions more accurately than sequence‐based predictions. Nevertheless, the previously reported robustness of the sequence‐based method 15 suggests that it is an adequate binary classifier for fold switchers/single‐fold proteins. Interestingly, sequence‐based predictions identify the part of the domain‐swapped β‐strands of ORF9b and ORF9b1 as helices in both whole‐sequence and excised sequence fragments. This finding suggests that this region of the sequence might have intrinsic helical propensities that are energetically outweighed by favorable interactions with neighboring β‐sheets, a possibility consistent with other studies suggesting that protein secondary structure formation can be context‐dependent. 23 , 24
Our predictions suggest that ORF9b1, the ORF9b homolog from SARS‐CoV‐1, also switches folds. This finding is consistent with a couple lines of experimental evidence demonstrating that ORF9b1, like ORF9b, binds Tom70. 16 Based on the ORF9b‐Tom70 structure, it is not obvious how ORF9b1 could bind to Tom70 without assuming an α‐helical fold, even though its only experimentally determined structure suggests that it folds into a β‐sheet topology. The sequences of ORF9b1 and ORF9b are 69% identical. Reconstructed ancestors with similar levels of sequence identity to the metamorphic protein XCL1 have also been shown to switch folds, 7 demonstrating that fold switching can be conserved among protein homologs, though this is not always the case. 25
The folds of ORF9b differ from those of the fold switchers used to develop our predictive methods. 14 , 15 Since these methods correctly infer that ORF9b switches folds, they have the potential to identify other proteins that undergo novel fold‐switching transitions. Given that these methods are high‐throughput, they can be tested on numerous sequences and structures. We are optimistic that they will reveal new fold switchers in future work.
4. METHODS
4.1. Secondary structure predictions of ORF9b and ORF9b1
Secondary structure predictions of ORF9b (PDB 26 IDs: 6Z4U, chain A and 7KTD, chain B) and ORF9b1 (PDB ID 2CME, chain A) sequences (both parent and fragments) were determined using the JPred4 (http://www.compbio.dundee.ac.uk/jpred/) and PSIPRED webservers (http://bioinf.cs.ucl.ac.uk/psipred/). SPIDER2 predictions were determined from position‐specific scoring matrices generated locally by running three rounds of PSI‐BLAST 27 on the UniRef90 28 database from 7/2014 with up to 10,000 alignments with a maximum e‐value of 0.05. For consistency, each residue from all three predictors were assigned one of three secondary structure annotations: “H” for α‐helix, “E” for extended β‐strand, and “C” for coil.
4.2. Secondary structure prediction accuracy calculations
Experimentally determined secondary structures of ORF9b and ORF9b1 were calculated locally using DSSP 29 and simplified to the same three‐state classification system as the secondary structure predictors: “H” for α‐helix, “E” for extended β‐strand, and “C” for all other DSSP annotations. Additionally, chain breaks were annotated as “‐“. Sequences were aligned using the pairwise2.align.localxs function from Biopython 30 with a gap‐forming score of −1 and gap‐elongation score of −0.5. Predicted/calculated secondary structures were then re‐registered according to the Biopython sequence alignments. Secondary structure prediction accuracies were calculated using the Q total (or Q 3 ) metric, 31 in which experimentally determined and predicted secondary structures are compared one‐by‐one, residue‐by‐residue and normalized by the length of the sequences compared. Chain breaks were excluded from both scoring and normalization.
AUTHOR CONTRIBUTIONS
Lauren L. Porter: Conceptualization; formal analysis; funding acquisition; investigation; methodology; visualization; writing‐original draft; writing‐review & editing.
ACKNOWLEDGEMENTS
The author thanks Loren Looger and Marius Clore for helpful discussion. This work utilized the computational resources of the NIH HPS Biowulf cluster (http://hpc.nih.gov). This work was supported by the Intramural Research Program of the National Library of Medicine, National Institutes of Health.
Porter LL. Predictable fold switching by the SARS‐CoV‐2 protein ORF9b . Protein Science. 2021;30:1723–1729. 10.1002/pro.4097
Funding information National Institutes of Health; National Library of Medicine
REFERENCES
- 1. Porter LL, Looger LL. Extant fold‐switching proteins are widespread. Proc Natl Acad Sci U S A. 2018;115:5968–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. [DOI] [PubMed] [Google Scholar]
- 3. Kang JY, Mooney RA, Nedialkov Y, et al. Structural basis for transcript elongation control by nusg family universal regulators. Cell. 2018;173:1650–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang YG, Cohen SE, Phong C, et al. Circadian rhythms. A protein fold switch joins the circadian oscillator to clock output in cyanobacteria. Science. 2015;349:324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim AK, Porter LL. Functional and regulatory roles of fold‐switching proteins. Structure. 2021;29:6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giganti D, Albesa‐Jove D, Urresti S, et al. Secondary structure reshuffling modulates glycosyltransferase function at the membrane. Nat Chem Biol. 2015;11:16–18. [DOI] [PubMed] [Google Scholar]
- 7. Dishman AF, Tyler RC, Fox JC, et al. Evolution of fold switching in a metamorphic protein. Science. 2021;371:86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tuinstra RL, Peterson FC, Kutlesa S, Elgin ES, Kron MA, Volkman BF. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc Natl Acad Sci U S A. 2008;105:5057–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lei Y, Takahama Y. Xcl1 and xcr1 in the immune system. Microbes Infect. 2012;14:262–267. [DOI] [PubMed] [Google Scholar]
- 10. Al Khamici H, Brown LJ, Hossain KR, et al. Members of the chloride intracellular ion channel protein family demonstrate glutaredoxin‐like enzymatic activity. PLoS One. 2015;10:e115699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Littler DR, Harrop SJ, Fairlie WD, et al. The intracellular chloride ion channel protein clic1 undergoes a redox‐controlled structural transition. J Biol Chem. 2004;279:9298–9305. [DOI] [PubMed] [Google Scholar]
- 12. Li BP, Mao YT, Wang Z, et al. Clic1 promotes the progression of gastric cancer by regulating the mapk/akt pathways. Cell Physiol Biochem. 2018;46:907–924. [DOI] [PubMed] [Google Scholar]
- 13. Milton RH, Abeti R, Averaimo S, et al. Clic1 function is required for beta‐amyloid‐induced generation of reactive oxygen species by microglia. J Neurosci. 2008;28:11488–11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mishra S, Looger LL, Porter LL. Inaccurate secondary structure predictions often indicate protein fold switching. Protein Sci. 2019;28:1487–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mishra S, Looger LL, Porter LL. A sequence‐based method for predicting extant fold switchers that undergo alpha‐helix <−> beta‐strand transitions. bioRxiv. 2021. 10.1101/2021.01.14.426714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gordon DE, Hiatt J, Bouhaddou M, et al. Comparative host‐coronavirus protein interaction networks reveal pan‐viral disease mechanisms. Science. 2020;370:eabe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bouhaddou M, Memon D, Meyer B, et al. The global phosphorylation landscape of sars‐cov‐2 infection. Cell. 2020;182:685–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim AK, Looger LL, Porter LL. A high‐throughput predictive method for sequence‐similar fold switchers. Biopolymers. 2021;e23416. 10.1002/bip.23416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buchan DWA, Jones DT. The psipred protein analysis workbench: 20 years on. Nucleic Acids Res. 2019;47:W402–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heffernan R, Dehzangi A, Lyons J, et al. Highly accurate sequence‐based prediction of half‐sphere exposures of amino acid residues in proteins. Bioinformatics. 2016;32:843–849. [DOI] [PubMed] [Google Scholar]
- 21. Drozdetskiy A, Cole C, Procter J, Barton GJ. Jpred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015;43:W389–W394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
- 23. Minor DL Jr, Kim PS. Context‐dependent secondary structure formation of a designed protein sequence. Nature. 1996;380:730–734. [DOI] [PubMed] [Google Scholar]
- 24. Porter LL, He Y, Chen Y, Orban J, Bryan PN. Subdomain interactions foster the design of two protein pairs with approximately 80% sequence identity but different folds. Biophys J. 2015;108:154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Littler DR, Brown LJ, Breit SN, Perrakis A, Curmi PM. Structure of human clic3 at 2 a resolution. Proteins. 2010;78:1594–1600. [DOI] [PubMed] [Google Scholar]
- 26. Berman HM, Battistuz T, Bhat TN, et al. The protein data Bank. Acta Cryst D. 2002;58:899–907. [DOI] [PubMed] [Google Scholar]
- 27. Altschul SF, Madden TL, Schaffer AA, et al. Gapped blast and psi‐blast: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. UniProt C. The universal protein resource (uniprot) in 2010. Nucleic Acids Res. 2010;38:D142–D148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers. 1983;22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 30. Cock PJ, Antao T, Chang JT, et al. Biopython: Freely available python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rost B, Sander C. Prediction of protein secondary structure at better than 70% accuracy. J Mol Biol. 1993;232:584–599. [DOI] [PubMed] [Google Scholar]