

Abstract
Background
Prognostic markers for disease severity and identification of therapeutic targets in COVID‐19 are urgently needed. We have studied innate and adaptive immunity on protein and transcriptomic level in COVID‐19 patients with different disease severity at admission and longitudinally during hospitalization.
Methods
Peripheral blood mononuclear cells (PBMCs) were collected at three time points from 31 patients included in the Norwegian SARS‐CoV‐2 cohort study and analysed by flow cytometry and RNA sequencing. Patients were grouped as either mild/moderate (n = 14), severe (n = 11) or critical (n = 6) disease in accordance with WHO guidelines and compared with patients with SARS‐CoV‐2‐negative bacterial sepsis (n = 5) and healthy controls (n = 10).
Results
COVID‐19 severity was characterized by decreased interleukin 7 receptor alpha chain (CD127) expression in naïve CD4 and CD8 T cells. Activation (CD25 and HLA‐DR) and exhaustion (PD‐1) markers on T cells were increased compared with controls, but comparable between COVID‐19 severity groups. Non‐classical monocytes and monocytic HLA‐DR expression decreased whereas monocytic PD‐L1 and CD142 expression increased with COVID‐19 severity. RNA sequencing exhibited increased plasma B‐cell activity in critical COVID‐19 and yet predominantly reduced transcripts related to immune response pathways compared with milder disease.
Conclusion
Critical COVID‐19 seems to be characterized by an immune profile of activated and exhausted T cells and monocytes. This immune phenotype may influence the capacity to mount an efficient T‐cell immune response. Plasma B‐cell activity and calprotectin were higher in critical COVID‐19 while most transcripts related to immune functions were reduced, in particular affecting B cells. The potential of these cells as therapeutic targets in COVID‐19 should be further explored.
Keywords: B cells, COVID‐19, flow cytometry, monocytes, T cells, transcriptomics

Introduction
The outbreak of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has rapidly spread across the world and caused a global health emergency. Although patients usually present with mild disease, the condition may progress to pneumonia, ARDS and death [1, 2]. The clinical deterioration that often leads to hospitalization occurs typically one week after onset of symptoms and suggests that host immunity drives the propagation to severe coronavirus disease (COVID)‐19 [3].
The systemic cytokine profile in severe COVID‐19 resembles cytokine release syndromes, such as macrophage activation syndrome [4], with high plasma levels of pro‐inflammatory cytokines [2, 5]. Notably, pro‐inflammatory cytokine levels seem to be lower in SARS‐CoV‐2‐ than in sepsis‐induced ARDS [6]. COVID‐19 is also associated with lymphopenia, with a more pronounced depletion of CD8 T cells, and lymphocyte count is inversely correlated with severity [3, 7, 8, 9], an association also demonstrated in sepsis [10]. The lymphopenia in COVID‐19 may be more prolonged than in other viral infections [8]. Little is known about the cause of lymphopenia and whether T‐cell responses are helpful or harmful in COVID‐19 [8]. Interleukin 7 (IL‐7) known to restore lymphocyte counts and repertoire in lymphopenia [8] may be a potential target for therapeutics in COVID‐19.
Although studies report increased levels of T‐cell activation, dysfunctional T cells expressing inhibitory molecules such as PD‐1 and TIM‐3 are also increased in patients with severe and critical COVID‐19 [11, 12, 13, 14]. Interestingly, a recent publication shows that PD‐1‐expressing SARS‐CoV‐2‐specific CD8 T cells are in fact functional [15]. Thus, although most data indicate an activated yet exhausted and dysfunctional immune system in COVID‐19 [13], the function and characteristics of cell subsets involved in COVID‐19 immunity remain unclear.
Monocytes are potential drivers of hyperinflammation and ARDS in COVID‐19 [4, 16]. Alveolar‐residing monocytes and macrophages may be infected by SARS‐CoV‐2 through angiotensin‐converting enzyme (ACE‐2)‐dependent or independent pathways [16, 17, 18, 19, 20]. Grant et al. argue that the infection results in release of T‐cell chemoattractants creating a positive feedback loop that drives alveolar inflammation, possible facilitating dissemination of SARS‐CoV‐2 in the lungs [18]. Whether ACE‐2‐expression in circulating monocytes is associated with COVID‐19 severity remains unknown. Mononuclear phagocyte dysregulations associated with severe COVID‐19 have recently been documented [21]. Further, a relative and absolute decrease in classical (CD14++CD16‐) and non‐classical (CD14+CD16++) monocytes to the advantage of intermediate monocytes (CD14++CD16+) has been reported in severe COVID‐19 [21, 22, 23, 24]. Still, a conflicting study finds an increase in classical and non‐classical monocytes [16]. Thus, the distribution and relevance of the various monocyte lineages and subsets in COVID‐19 infection are not finally determined.
Understanding the mechanisms that drive detrimental pathophysiology is vital in order to find strategies for host‐directed therapies in COVID‐19 [25]. We therefore aimed to explore COVID‐19 immunity by characterizing T‐cell and monocyte subsets and their key markers of activation and exhaustion as well as transcriptome patterns of a broader variety of immune cells across the clinical disease spectrum in a Norwegian COVID‐19 cohort compared with patients with SARS‐CoV‐2‐negative bacterial sepsis and healthy subjects.
Materials and methods
Study design, setting and participants
Patients aged ≥ 18 years with confirmed COVID‐19 admitted to Oslo University Hospital, Norway, between 9 March and 12 June 2020 were included in a prospective observational study (Norwegian SARS‐CoV‐2 study; NCT04381819) [26, 27, 28]. Study participants not treated with experimental drugs, immunosuppressants or corticosteroids were included within four days (mean 1.6 days) of admission and followed with registration of clinical data and blood samples during hospitalization. SARS‐CoV‐2‐negative patients with community‐acquired bacterial sepsis were prospectively recruited from the medical intensive care unit (ICU) in the same time period. Gender‐, age‐ and ethnicity‐matched healthy controls without any known diseases were recruited from the general population. SARS‐CoV‐2 PCR tests were not performed; however, they had no symptoms or history of COVID‐19 or detectable plasma SARS‐CoV‐2 antibodies. Peripheral blood was obtained from COVID‐19 patients at inclusion, after one (6–9 days) and two (10–16 days) weeks and from sepsis patients at inclusion (≤4 days after admission). Peripheral blood was drawn into BD CPT™ Cell Preparation Tube containing sodium heparin as anti‐coagulant, and peripheral blood mononuclear cells (PBMC) were isolated according to the manufacturer´s instructions and stored at −150°C in freezing media containing 10% DMSO until analysis.
Diagnostic assessments and definitions
COVID‐19 was diagnosed in oro‐ and nasopharyngeal swabs using the cobas® SARS‐CoV‐2 nucleic acid test on a cobas 8800 system targeting the E‐ and ORF1 genes [29]. COVID‐19 severity was classified based on adapted WHO criteria [30]: mild/moderate disease as PaO2/FiO2‐ratio (P/F) ≥37 kPa (corresponding to SpO2 < 90% on room air); severe disease as P/F < 37 kPa; and critical disease as P/F < 10 kPa or in need of ICU treatment. For longitudinal analysis, patients were classified as (i) improved (Δ ≥ +5 kPa), (ii) stable (Δ −5 to +5 kPa P/F) or (iii) deteriorated (Δ ≤ −5 kPa) measured by change in P/F after one week. Sepsis was defined according to sepsis‐3 criteria, as suspected infection and Sequential Organ Failure Assessment (SOFA) score of ≥2 points [31].
Data collection
Medical history including comorbidities, clinical data, routine blood haematological and biochemical markers and radiological findings were collected according to a modified version of the International Severe Acute Respiratory and Emerging Infection Consortium (ISARIC isaric.tghn.org)/World Health Organization (WHO) Clinical Characterization Protocol (CCP) version 3.1 and entered into a secure web‐based REDCap database (Research Electronic Data Capture, Vanderbilt University, TN, hosted by University of Oxford, UK). Corresponding data for sepsis patients were recorded in a local database (MedInsight, Oslo, Norway).
Ethical considerations
Prior to inclusion, informed consent was collected from all patients or patient’s family members if the patient was not able to consent. The study was approved by the Regional Committee for Medical and Health Research Ethics in South‐Eastern Norway (reference numbers 106624 and 2019/306).
Flow cytometry
Thawed PBMCs rested for 2 h in a 37 °C incubator with 5% CO2. Cells were split into two suspensions, stained with a monocyte panel (1 × 106 cells) or T‐cell panel (1 × 106 cells) according to the manufacturer’s instructions (BD Biosciences). Overview of the antibodies is displayed in Tables S1 and S2. The gating strategies for monocytes and T cells are shown in Figures S1‐S3. Briefly, the three monocytes subsets were classified as classical (CD14++CD16‐), intermediate (CD14++CD16+) or non‐classical (CD14+CD16++) monocytes [32]. Fluorescence minus one (FMO) control for CD142, PD‐L1 and ACE‐2 was included. T‐cell maturity subsets were identified by their expression of CD4, CD8 and CD45RO and Tregs defined by CD3+CD4+CD25+CD127low. T‐cell activation and/or exhaustion were investigated by the expression of HLA‐DR, PD‐1 and IL‐7Rα. Cell acquisition was performed on FACS Canto II (BD Biosciences). Instrument calibration was performed according to the manufacturer’s instructions and compensation adjusted using antibody capture beads (CompBeads, BD Biosciences). Data analysis was performed using FlowJo (Tree Star Inc.), expressed as frequencies (%) or mean fluorescent intensity (MFI).
mRNA analysis
mRNA expression was analysed on PBMC pellets from a subset of the COVID‐19 cohort: critical (n = 3), severe (n = 5), mild/moderate (n = 7) COVID‐19 and healthy controls (n = 6). The cell pellet was immediately resuspended in RNAlater solution and extracted applying the Qiagen Allprep kit, according to the manufacturer’s instructions (Qiagen, Hilden, Germany), treated with DNase (Qiagen) and stored at −80°C until analysis. RNA concentrations and purity based on the 260/280 and the 260/230 ratio were assessed by spectrophotometer absorbance (NanoDrop ND‐1000; Thermo Fisher Scientific Inc., Waltham, MA). RNA was sequenced with 2 × 150 bp flow cell on the Illumina‐NovaSeq 6000 platform (OGC, Oxford, UK) at the Norwegian Sequencing Centre, Oslo, Norway. Contaminated adapters and reads with Phred quality scores below 30 in raw data were trimmed by fastp (version 0.20.1) [33]. Filtered reads were mapped to human transcript sequence (GRCh38.p13, Gencode release 34) [34] by Kallisto (version 0.46.1) with 200 bootstrap iterations [35].
Statistical analysis
Non‐parametric statistics were applied as follows: Mann–Whitney U‐test for independent, non‐paired groups, when comparing all COVID‐19 cases with healthy controls and septic with critical COVID‐19 patients. The Jonckheere–Terpstra trend test was used when comparing COVID‐19 severity groups. Wilcoxon signed‐rank test was used for paired longitudinal samples, whereas Mann–Whitney was used when comparing changes in expression from baseline to week 1 in the two respiratory function trajectory groups (P/F improved vs. P/F deteriorated/stable). Spearman correlation was applied where appropriate. A significance level of P < 0.05 was chosen. Due to the explorative nature of the study, the statistics calculated from the flow cytometry data were not corrected for multiple testing, as the findings in this study were considered as hypotheses generating rather than confirmatory. Statistical analysis was performed by SPSS statistical software (Macintosh version 26.0, IBM, Armonk, NY, USA).
The significantly differentially expressed transcripts obtained from the RNA sequencing were obtained by Sleuth (version 0.30.0) using the Wald test [36]. Transcript‐level information was summarized to gene level by tximport R package version 1.18 [37], and the differentially expressed genes were obtained by DESeq2 R package version 1.30.1 [38]. Data are presented as natural‐log scaled b‐value, which is analogous to the fold change. These data are corrected for inferential variance estimated from bootstraps. To plot the heatmap, the normalized estimated counts of significantly differentially expressed transcripts were obtained from bootstraps. They were further z‐score‐normalized before plotting. To estimate cell fractions with CIBERSORT, all the gene‐level normalized estimated counts were submitted to the online platform (cibersort.stanford.edu). The built‐in LM22 table was used as signature matrix [39]. For functional enrichment analysis, significantly differentially expressed transcripts (q ≤ 0.05) were uploaded to Metascape (metascape.org, database version 2020‐09‐16) [40]. Enriching was performed in Gene Ontology (biological process) [41, 42], Hallmark [43], Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome and PID pathways [44]. Terms and pathways with P ≤ 0.01, minimum count of 3, enrichment factor >1.5 were considered statistically significant.
Results
Clinical characteristics
A total of 31 COVID‐19 patients, five patients with bacterial sepsis and ten healthy controls were included in the study. Patient clinical and demographic characteristics are shown in Table 1. One critically ill COVID‐19 patient had a bacterial lower respiratory super‐infection. In the sepsis cohort, 4 of 5 patients had bacterial infections confirmed by positive culture (three positive blood cultures, one abscess culture) and 1 of 5 showed radiological and clinical findings consistent with bacterial pneumonia. A total of 12 COVID‐19 patients were sampled also after one week, hereof six with improved (ΔP/F + 13, 10 to 16 kPa (median, range)), three with stable (ΔP/F −3, −4 to −1 kPa) and three deteriorating respiratory function (ΔP/F −15, −23 to −8 kPa). The latter two groups were merged in further analysis.
Table 1.
Clinical characteristics of study participants
| Total COVID‐19 (n = 31) | Disease severity, COVID‐19 | Sepsis (n = 5) | |||
|---|---|---|---|---|---|
| Mild/Moderate (n = 14) | Severe (n = 11) | Critical (n = 6) | |||
| Age years, median (IQR) a | 57 (21) | 49 (30) | 57 (16) | 64 (16) | 51 (32) |
| Female (%) a | 11 (36) | 7 (50) | 3 (28) | 1 (17) | 2 (40) |
| Ethnicity, white (%) a | 22 (71) | 9 (64) | 10 (91) | 3 (50) | 5 (100) |
| Smoking (%) | |||||
| ‐ Former | 8 (26) | 2 (14) | 4 (36) | 4 (67) | 1 (20) |
| ‐ Current | 2 (7) | 2 (14) | 0 | 0 | 1 (20) |
| ARB/ACEi use (%) | 11 (36) | 4 (28) | 3 (28) | 4 (67) | 1 (20) |
| ICU LoS | 0 | 0 | 6 (0–14) | 4 (1–8) | |
| Mechanical ventilation days | 0 | 0 | 2 (0–10) | 0 | |
| Hospital LoS | 8 (1–26) | 5 (2–11) | 7 (1–10) | 20 (12–26) | 14 (9–18) |
| Comorbidities, n (%) | |||||
| Chronic heart disease | 7 (23) | 3 (21) | 0 | 4 (67) | 0 |
| Chronic renal disease | 3 (10) | 1 (7) | 0 | 2 (33) | 0 |
| Cancer | 1 (3) | 0 | 0 | 1 (17) | 1 (20) |
| Diabetes | 4 (13) | 1 (7) | 1 (9) | 1 (17) | 2 (40) |
| Hypertension | 14 (45) | 5 (36) | 4 (40) | 4 (67) | 1 (20) |
| Chronic lung disease | 8 (26) | 5 (36) | 3 (28) | 0 | 1 (20) |
| BMI, mean (range) | 28 (18–38) | 26 (18–36) | 29 (21–38) | 29 (26–32) | NA |
| Symptoms, n (%) | |||||
| Fever | 26 (84) | 11 (100) | 11 (100) | 4 (67) | NA |
| Cough | 28 (90) | 12 (86) | 11 (100) | 5 (83) | NA |
| Dyspnoea | 23 (74) | 8 (57) | 9 (82) | 6 (100) | NA |
| Fatigue | 28 (90) | 11 (79) | 12 (100) | 6 (100) | NA |
| Diarrhoea(−2) | 9 (29) | 6 (50) | 2 (18) | 1 (17) | NA |
| Days symptoms, mean (range) | 9 (0–17) | 8 (0–17) | 10 (3–17) | 8 (4–14) | NA |
| Routine clinical data at inclusion, mean (range) b | |||||
| Leukocytes ×109/L | 6.0 (2.6–12.0) | 4.9 (2.6–7.2) | 6.4 (3.5–12.0) | 7.9 (3.8–11.2) | 20.2 (11.2–46.7) |
| Lymphocytes ×109/L | 1.3 (0.4–2.1) | 1.5 (0.8–2.1) | 1.2 (0.5–1.7) | 1.1 (0.4–1.5) | 1.7 (0.5–2.7) |
| Neutrophils ×109/L | 4.2 (1.2–11.1) | 2.8 (1.2–5.3) | 4.8 (1.8–11.1) | 6.4 (1.9–9.4) | 17.3 (7.5–43.8) |
| Monocytes ×109/L | 0.5 (0.1–0.9) | 0.5 (0.1–0.9) | 0.4 (0.1–0.8) | 0.4 (0.2–0.5) | 1.4 (0.7–2.0) |
| NLR | 4.5 (0.6–23.0) | 2.1 (0.6–5.9) | 5.3 (1.3–22.2) | 8.4 (1.3–23.0) | 15.5 (2.8–36.4) |
| MLR | 0.4 (0.1–1.0) | 0.3 (0.1–0.6) | 0.4 (0.1–0.8) | 0.4 (0.2–1.0) | 0.9 (0.3–1.8) |
| Platelets ×109/L(−1) | 213 (120–427) | 216 (136–414) | 200 (127–419) | 230 (120–427) | 263 (98–510) |
| Procalcitonin μg/L(−2) | 0.22 (0.05–1.70) | 0.12 (0.05–0.60) | 0.13 (0.05–0.46) | 0.58 (0.16–1.70) | NA |
| CRP, mg/L | 66 (0–219) | 34 (0–191) | 73 (15–196) | 127 (45–219) | 146 (42–277) |
| Ferritin μg/L(−2) | 674 (7–2348) | 380 (7–992) | 789 (231–1408) | 1050 (354–2348) | NA |
| Fibrinogen g/L(−2) | 5.5 (2.6–8.7) | 4.7 (2.6–8.7) | 5.7 (4.1–7.2) | 6.5 (4.3–7.5) | NA |
| D‐dimer mg/L(−2) | 0.8 (0.2–2.9) | 0.5 (0.2–1.2) | 0.6 (0.2–1.1) | 1.6 (0.4–2.9) | NA |
| PaO2/FiO2 | 37.6 (8.5–58.2) | 48.6 (39.5–58.1) | 33.4 (21.7–48.8) | 19.5 (8.5–36.3) | 48.3 (30.7–61) |
Controls (n = 10) were 40% female, median age was 51.5 years, and 60% were white.
Clinical biochemical and haematological routine laboratory assays. ACEi, Angiotensin converter enzyme inhibitor; ARB, angiotensin receptor blocker; CRP, C‐reactive protein; IQR, interquartile range; LoS, Length of stay; MLR, Monocyte: lymphocyte ratio; NA, Not available; NLR, Neutrophil: lymphocyte ratio. Superscript in parentheses indicates number of missing values among COVID‐19 patients.
Absolute lymphocyte count decreased with increasing COVID‐19 severity (P = 0.041), while absolute neutrophil count (P = 0.002), neutrophil: lymphocyte ratio (NLR) (P = 0.003), procalcitonin (PCT) (P = 0.003) and C‐reactive protein (CRP) (P = 0.001) increased. Compared with patients with bacterial sepsis, critical COVID‐19 patients had lower white blood cell (WBC) counts (P = 0.009) and monocyte counts (P = 0.004) while lymphocyte and neutrophil counts, NLR and CRP were comparable.
T‐cell IL‐7Rα expression declines with increased COVID‐19 severity
We found no significant differences in the fractions of CD3, CD4 or CD8 T‐cell subsets across COVID‐19 severity groups (Figure S4A). Thus, we proceeded to explore T‐cell markers of differentiation/survival (CD127/IL‐7Rα), activation (HLA‐DR and CD25) and exhaustion (PD‐1). IL‐7Rα expression was generally higher in COVID‐19 patients compared with healthy controls (CD8 memory, P = 0.010; CD8 naïve, P = 0.037; CD4 memory, P = 0.011; Fig. 1a). An inverse association with COVID‐19 severity was observed for IL‐7Rα expression in these cell subsets, significant for naïve CD4 and CD8 T cells (Fig. 1a). IL‐7Rα expression in naïve CD4 T cells also correlated with P/F (r = 0.45, P = 0.021; Figure S5). Notably, both in sepsis patients and in critical COVID‐19 patients, IL‐7Rα expression was comparable to controls.
Fig. 1.
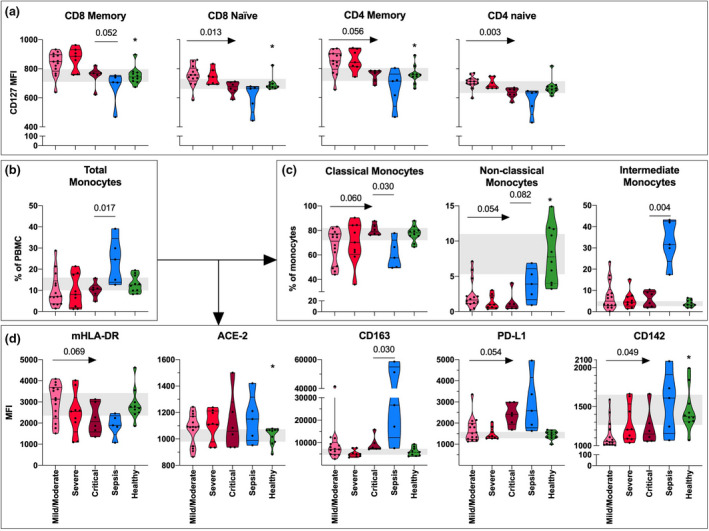
CD127/IL‐7Rα expression in T‐cell subsets and monocyte subsets and markers. (a) The expression (mean fluorescence intensity, MFI) of CD127/IL‐7Rα in memory (CD45RO+) and naïve (CD45RO‐) CD8 and CD4 T cells and Tregs in mild/moderate (n = 13), severe (n = 7) and critical (n = 6) COVID‐19, sepsis (n = 5) and healthy controls (n = 10) at baseline. (b) The frequency of total monocytes and (c) monocyte subsets and (d) the monocyte expression (MFI) of mHLA‐DR, ACE‐2, CD142, CD163 and PD‐L1 in total monocytes in mild/moderate (n = 14), severe (n = 9) and critical (n = 6) COVID‐19, sepsis (n = 5) and healthy controls (n = 10) at baseline. Arrow indicates significance calculated with Jonckheere–Terpstra trend test across COVID‐19 severity groups, and straight line indicates Mann–Whitney U‐test between sepsis and critical COVID‐19. * indicates significance comparing all COVID‐19 patients with healthy controls using Mann–Whitney U‐test. P‐values were considered significant when <0.05. Violin plots displaying median with quartiles. Grey area indicates the 95% CI of healthy controls.
Activation marker CD25 and exhaustion marker PD‐1 expression higher in critical COVID‐19 compared with bacterial sepsis
T‐cell expression of the activation marker HLA‐DR was similar across COVID‐19 severity groups and comparable to patients with bacterial sepsis (Figure S4B). Nevertheless, HLA‐DR expression in CD8 T cells was higher in COVID‐19 patients irrespective of severity compared with healthy controls (memory, P < 0.001, and naïve, P = 0.001) (Figure S4B). Likewise, there was no significant difference in CD25 and PD‐1 expression across COVID‐19 severity groups (Figure S4C/D), but in contrast to HLA‐DR these markers were increased in critical COVID‐19 compared with bacterial sepsis. Furthermore, PD‐1 expression and CD25 expression were higher in some T‐cell subsets in COVID‐19 patients compared with healthy controls (Figure S4D).
Non‐classical monocytes decline in critical COVID‐19 correlating to respiratory failure
Total monocyte frequencies were similar across COVID‐19 severity groups, but trends of increased fractions of classical monocytes and decreased fractions of non‐classical monocytes were observed with increased COVID‐19 severity (Fig. 1b and c). The percentage of non‐classical monocytes correlated with P/F, whereas classical monocytes were negatively correlated with P/F (P = 0.001) (Figure S5). Septic patients had higher fractions of total monocytes, non‐classical and intermediate monocytes but lower fraction of classical monocytes than critical COVID‐19 patients (Fig. 1b and c).
Monocyte markers of activation and suppression change with COVID‐19 severity
We analysed markers of function (monocytic (m)HLA‐DR, CD142, ACE‐2), differentiation (CD163) and suppressive activity (PD‐L1) in total monocytes (Fig. 1d). We observed a trend of decreased mHLA‐DR expression with COVID‐19 severity supported by a negative correlation with P/F (P = 0.004) (Figure S5). In contrast, there were no differences in ACE‐2 expression across COVID‐19 severity groups, but COVID‐19 patients independent of severity group had significantly higher levels of ACE‐2 compared with healthy controls (P = 0.034) (Fig. 1d). Of note, ACE‐2 expression was not associated with age (Figure S5) or treatment with an angiotensin blocker or ACE‐2 inhibitor (P = 0.982, Mann–Whitney). Both mHLA‐DR expression and ACE‐2 expression in critical COVID‐19 were comparable to levels in patients with sepsis (Fig. 1d). CD163 expression was comparable across COVID‐19 severity groups, but lower in critical COVID‐19 patients than sepsis patients (Fig. 1d). Further, we observed an increase in both PD‐L1 and TF (CD142) expression with increased COVID‐19 severity (Fig. 1d).
Treg PD‐1 and mHLA‐DR expression correlate with plasma inflammation markers
Ideally, COVID‐19 severity should be identified by biomarkers analysed in routine blood samples. The relationships between the immune cell subsets, clinical routine blood markers, age and COVID‐19 severity measured as P/F were investigated (Figs 2 and S5). The association between immune cell subsets and relevant clinical routine blood markers of COVID‐19 such as lymphocyte count, PCT, CRP, ferritin and lactate dehydrogenase (LDH) is displayed in Fig. 2. PD‐1 expression in Tregs correlated positively with all these markers except lymphocyte count, whereas mHLA‐DR showed an inverse correlation to CRP, ferritin and LDH. Finally, PD‐L1 expression on monocytes was correlated with CRP and the fraction of classical monocytes was negatively correlated with lymphocyte count (Figs 2 and S5). IL‐7Rα expression (in naïve CD8 T cells) was only correlated with D‐dimer (Figure S5).
Fig. 2.
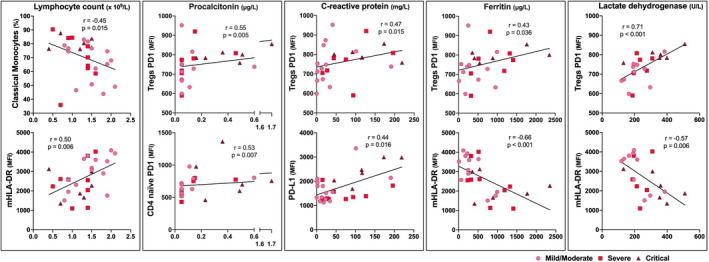
Baseline correlations between clinical blood markers and immune cell subsets. Correlations of five selected clinical routine blood inflammatory markers (x‐axis) relevant in COVID‐19 (absolute lymphocyte count, procalcitonin, C‐reactive protein, ferritin, lactate dehydrogenase) with the two immune cell subsets (y‐axis) demonstrating the strongest significant correlations (highest R value). Correlations were examined using Spearman's rank correlation coefficient. Pink dot indicates mild/moderate (n = 14), red square severe (n = 9) and purple triangle critical (n = 6) COVID‐19.
Monocyte PD‐L1 expression decrease upon respiratory improvement in COVID‐19
To investigate longitudinal dynamics in T cell and monocyte immunity in COVID‐19, we assessed the same cell subsets and markers in 12 patients whose respiratory function (P/F) improved (6/12) or deteriorated/remained stable (6/12) after one week of hospitalization. Expression of IL‐7Rα (CD127) tended to decline after one week in most T‐cell subsets in both P/F trajectory groups, significantly in memory CD8 T cells (Fig. 3a). In contrast, T‐cell expression of CD25, HLA‐DR and PD‐L1 did not change significantly during follow‐up (Figure S6B). The patterns were heterogeneous, but the fraction of intermediate monocytes increased at week 1 while an increase in non‐classical monocytes compared with baseline was observed first at week 2 (Fig. 3b). Overall, expression of mHLA‐DR, ACE‐2, CD163 and PD‐L‐1 on total monocytes did not significantly change during follow‐up, while CD142 expression increased in monocytes for most of the patients after one week, independent of clinical progression (Fig. 3c). Interestingly, monocyte expression of PD‐L1 decreased at week 1 in patients whose P/F improved compared with those whose P/F deteriorated or maintained stable (P = 0.015, Mann–Whitney).
Fig. 3.
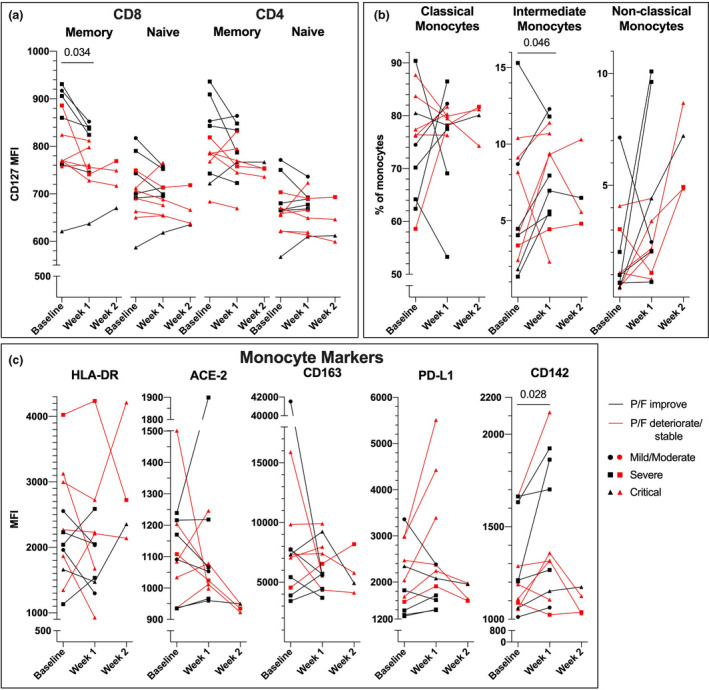
Dynamic changes in T cells and monocyte subsets during hospitalization in COVID‐19. (a) CD127/IL‐7Rα expression (mean fluorescence intensity; MFI) in T‐cell subsets, (b) monocyte subsets (%) and (c) expression (MFI) of monocyte markers (mHLA‐DR, ACE‐2, CD163, PD‐L1 and CD142) in COVID‐19 patients whose respiratory function (P/F) improved (n = 6, black line) or deteriorated/ remained stable (n = 6, red line) from baseline to week 1. Dot indicates mild/moderate (n = 1), square severe (n = 5) and triangle critical (n = 6) COVID‐19 severity; line represents mean value from each patient group. Significance calculated with Wilcoxon signed‐rank test including all patients from baseline to week 1 for all patients. No statistics were performed including data at week 2.
Differentially regulated transcriptomes in critical COVID‐19
Next, to gain in‐depth insight into the distribution of immune cells and engagement of pathways in COVID‐19, we performed RNA sequencing on PBMC in a subgroup of mild/moderate (n = 7), severe (n = 5) and critical (n = 3) COVID‐19 patients and healthy controls (n = 6) (Figs 4, 5, S7 and S8). When comparing mild/moderate to critical COVID‐19, we found 411 transcripts that were differentially expressed, of which 42 had higher whereas 369 had lower expression in critical COVID‐19 patients (Fig. 4a). The overlapping differentially expressed transcripts between mild/moderate vs critical and severe vs critical COVID‐19 patients are displayed in Fig. 4b. To assess the biological differences between mild/moderate and critical patients, we performed gene enrichment analysis. The three most differentially regulated pathways were ‘immune response‐activating signal transduction’ (GO0002757), ‘leucocyte activation involved in immune response’ (GO0002366) and ‘mRNA processing’ (GO0006397) (Figs 4c and S8B and D). In the GO pathway ‘immune response‐activating signal transduction’, only seven transcripts that were differentially expressed were higher in the critical compared with the mild/moderate COVID‐19 group, including pro‐inflammatory calprotectin (S100A8/9). There were lower levels of transcripts associated with regulatory functions (NFkB inhibitor zeta), apoptosis inhibitor genes such as BIRC and cell signalling (MAPK, NFkB, CREBBP, RAP1A) in critical compared with mild/moderate COVID‐19 patients (Fig. 4d and e).
Fig. 4.
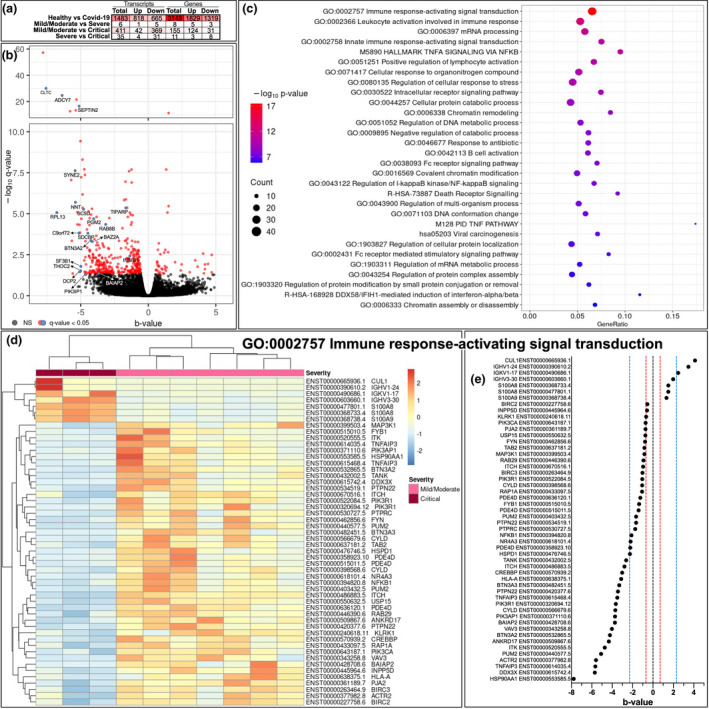
Comparison of the transcriptome in COVID‐19 severity groups. (a) Number of significantly (q < 0.05, Wald test) differentially expressed transcripts and genes comparing mild/moderate (n = 7), severe (n = 5) vs critical (n = 3) disease and healthy controls (n = 6). (b) Volcano plot displaying transcripts comparing mild/moderate and critical COVID‐19 patients. Each dot represents one transcript; coloured dots significantly differentially expressed transcripts. The blue dots represent overlapping differentially expressed transcripts between mild/moderate vs critical and severe vs critical COVID‐19, identified with their associated gene name. The y‐axis corresponds to the mean expression value of log 10 (q‐value), and the horizontal axis (x‐axis) displays the b‐value, which is analogous to fold change. (c) The 30 most enriched pathways (KEGG, Hallmark, Reactome and PID) and Gene Ontology (GO) terms from analysis within the 411 differentially expressed transcripts comparing mild/moderate and critical disease. Dot size reflects number of differentially expressed transcripts, and colour indicates P‐value (increasing from blue to red). X‐axis represents gene ratio, the fraction of differentially expressed transcripts in each pathway or GO term. (d) Heatmap displaying clustering of transcripts involved in the most enriched GO term from the enrichment analysis: ‘Immune response activating signal transduction’. (e) Comparing the magnitude of the significantly differentially expressed transcripts in the most enriched GO term across mild/moderate and critical severity groups. B‐value is analogous to fold change, and the red line indicates an approximate fold change of 2, while the blue line indicates an approximate fold change of 10.
Fig. 5.
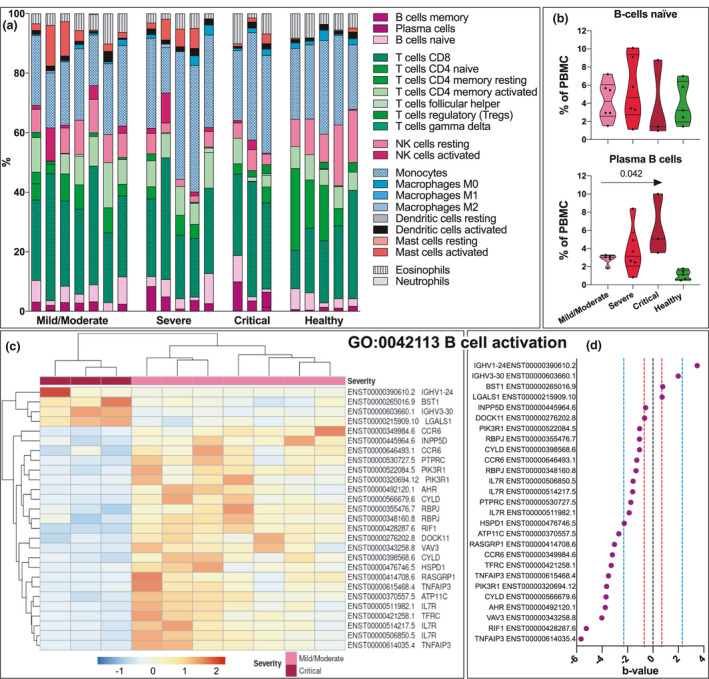
B‐cell transcriptomics in COVID‐19. (a) CIBERSORT analysis imputing different cell subsets present in PBMC from COVID‐19 patients, mild/moderate (n = 7), severe (n = 5), critical (n = 3) and healthy controls (n = 5). (b) Quantification of the estimated levels of naïve and plasma B cells in mild, severe and critical COVID‐19 and healthy controls. Arrow indicates significance calculated with Jonckheere–Terpstra trend test across COVID‐19 severity groups. (c) Heatmap displaying clustering of transcripts involved in the GO term ‘B cell activation’. (d) Comparing the magnitude of the significantly differentially expressed transcripts in the GO term B‐cell activation across mild/moderate and critical severity groups. B‐value is analogous to fold change, and the red line indicates an approximate fold change of 2, while the blue line indicates an approximate fold change of 10. Differentially expressed transcripts with q ≤ 0.05 in the Wald test were considered significant.
Plasma B‐cell levels and transcripts of function disturbances in critical COVID‐19
Using CIBERSORT analysis, we next evaluated the different cell populations based on gene expression pattern (Fig. 5a and b). The most striking differences were the estimated B‐cell portions in the subset of COVID‐19 samples (Fig. 5a). There was no difference in naïve B cells, but the analysis estimated an increase in plasma B cells with increased severity (Fig. 5b). Further, in the enrichment analysis, the ‘B cell activation’ (GO0042113) pathway was statistically different in mild/moderate compared with critically ill patients (Fig. 5b). A considerable number of transcripts related to B‐cell function were lower in critically ill patients (Fig. 5c and d), including three transcripts (one a nonsense‐mediated decay) coding for IL‐7Rα, suggesting a strongly reduced expression.
Discussion
We present cellular and gene analyses of immune profiles of cells from the innate and adaptive immune system from a cohort of patients with various severity of COVID‐19 and compared with septic patients. Our data show phenotypic alterations, which may indicate a dysfunctional adaptive and innate immune response in patients with severe and critical COVID‐19. IL‐7Rα expression decreased with COVID‐19 severity correlating to respiratory function. T‐cell exhaustion marker PD‐1 was higher in critical COVID‐19 than in septic patients with bacterial infections. In monocytes, the expression of PD‐L1 and ACE‐2 and the fraction of pro‐inflammatory classical monocytes increased, while mHLA‐DR expression and the fraction of anti‐inflammatory non‐classical monocytes decreased with COVID‐19 severity. Transcriptomic analyses revealed changes in pathways of both innate and adaptive immune responses. Pro‐inflammatory calprotectin and plasma B cells were increased while transcripts related to B‐cell function were reduced in critical COVID‐19.
We found a substantial number of differentially expressed transcripts in mild/moderate versus critical COVID‐19, indicating a possible shift in transcripts during disease progression. Surprisingly, there was a low amount of differentially expressed transcripts comparing mild/moderate to severe disease. In contrast, a large number of differentially expressed transcripts were found in mild vs severe COVID‐19 in a recent study [45]. However, stratification of the clinical groups will affect the results and the severe COVID‐19 group in the report from Aschenbrenner et al., corresponds to our critical COVID‐19 group and highlights the need for a common stratification of COVID‐19 severity as well as further in‐depth studies on the transcriptome. Interestingly, Calprotectin (S100A8/9), a potent alarmin and driver of innate immune response [46], was significantly higher in critical patients. Calprotectin has recently been suggested as a biomarker for COVID‐19 severity [24]. The findings of increased plasma B cells with potentially reduced B‐cell function in critical COVID‐19 indicate an important role for B cells in this disease. Recent reports support the findings of increased plasma B cells in critical COVID‐19 [47, 48]. While plasma B cells may produce neutralizing antibodies, they were also associated with severe disease and mortality [49]. Studies of B‐cell function and as a source of long‐term antibody production for protection are needed to determine their role in acute COVID‐19. Further, critical patients in general displayed lower levels of transcripts involved in regulation, apoptosis and cell signalling compared with mild/moderate disease.
To gain deeper insight into the immune responses, we have characterized several immune cells representative of both innate and adaptive immune responses. Our study showed that IL‐7Rα expression was significantly reduced with increased COVID‐19 severity, correlated with respiratory function measured as P/F and was comparable to levels found in septic patients. Further, transcripts of ILR7 were lower in critical COVID‐19. This may represent a dysfunctional immune response in septic patients suggestive of immune suppression [50]. Further, low levels of IL‐7Rα in people living with HIV lead to decreased T‐cell survival and proliferation [51]. We have previously shown that the plasma levels of soluble IL‐7 are increased in COVID‐19, but with no difference between the clinical severity groups [5]. Thus, we argue that cellular IL‐7Rα, rather than soluble IL‐7 levels, is predictive of COVID‐19 severity and as infection progresses T cells develop a unique exhausted phenotype characterized by loss of IL‐7Rα. IL‐7 therapy, shown to restore lymphocyte count and repertoire diversity in sepsis, could serve as a possible host‐directed therapy for advanced COVID‐19 [52]. Three ongoing randomized controlled clinical trials are exploring IL‐7 therapy for COVID‐19 (clinicaltrials.org) and our data support further investigation of IL‐7 as therapy.
While immunosuppression caused by cell exhaustion reflected in higher expression of PD‐1 and Tim‐3 has been shown to be associated with reduced viral clearance [12, 23, 53], a recent report shows that PD‐1 expressing SARS‐CoV‐2‐specific CD8 T cells seem to be functional [15]. We demonstrate an increased expression of PD‐1 on T cells in COVID‐19, but evenly distributed across the COVID‐19 severity groups, also questioning the degree of T‐cell exhaustion in critical COVID‐19 [26]. However, PD‐L1 on monocytes increased with COVID‐19 severity, and in deteriorating patients during the first week of follow‐up, whereas in patients whose P/F improved there was a decrease in the expression of PD‐L1. T‐cell depletion and exhaustion have been reported to underlay severe disease in SARS‐CoV‐infected mice [54] and possibly mediated through the PD‐1/PD‐L1 pathway [55]. Our findings may indicate that the continued increase in PD‐L1 may come as a result of increased disease severity and contribute to T‐cell suppression via the PD‐1/PD‐L1 signalling axis also reported by others [56]; however, the precise role of the PD‐1/PD‐L1 pathway and characteristics of T cells in COVID‐19 pathology should be explored in future studies.
Changes in monocyte function and phenotype may be related to severity of COVID‐19, since excessive monocyte/macrophage activation has been associated with hyperinflammation propagating disease severity in a subset of patients [2, 57, 58, 59]. We found that the frequencies of classical monocytes, an important contributor to the secretion of pro‐inflammatory cytokines, were increased in patients with severe disease and inversely correlated with P/F and lymphocyte count and thus may contribute to hyperinflammation and increased lung pathology [60, 61, 62]. In contrast, anti‐inflammatory non‐classical monocytes, known to patrol the vasculature [63], were reduced with increased COVID‐19 severity and correlated with P/F, suggesting a role in severe inflammatory lung damage [22, 24, 56, 62, 64]. Further, this subgroup seems to increase in later stages of disease, in accordance with Schulte‐Schrepping et al [56]. However, causality would need to be further explored in studies of pulmonary tissue samples. Compared with COVID‐19, the distribution of monocyte subset differs clearly from that seen in septic patients suggesting a viral‐specific phenotype.
Downregulation of mHLA‐DR is recognized as a marker for monocyte dysfunction in severe disease in general [3, 24] and for immunosuppression in sepsis [65]. We observed a decrease in mHLA‐DR expression with decreasing respiratory function, indicating less antigen‐presenting activity in more severe COVID‐19. Although volatile, mHLA‐DR expression overall persisted over time in COVID‐19 as previously shown [56]. Moreover, our data showing comparable levels in critical COVID‐19 and sepsis are in contrast to another study reporting higher expression of mHLA‐DR in COVID‐19 patients admitted to the ICU than in sepsis [65]. SARS‐CoV‐2 may enter the cells via ACE‐2 expressed on alveolar monocytes and macrophages to gain intracellular access [16, 18, 19] and this may contribute to morbidity in COVID‐19 [18, 66, 67]. We report higher levels of ACE‐2 expression on circulating monocytes in COVID‐19 patients compared with healthy individuals, suggesting that these cells may be susceptible for SARS‐CoV‐2 infection. The underlying mechanisms, however, need further investigation [17].
There are several limitations in our study. Our cohort is small, especially the groups with critical COVID‐19 and sepsis and RNA sequencing are performed on few patients. Likewise, only a fraction of the cohort was followed due to logistical constraints and discharge from the hospital. Methodologically, our flow cytometry analyses were limited to T cells and monocytes and phenotyping of Tregs and T‐cell subsets were crude without the use of markers such as FOXP3 or detailed maturity markers. Further, p‐values were not corrected for multiple testing on data from the flow cytometry analyses and should be interpreted thereafter. If applying a conservative Bonferroni adjustment for the number of tests (37), then only the correlations between LD and Tregs PD‐1 and Ferritin and mHLA‐DR remain significant. Still, our exploratory study presents several interesting findings that could lay grounds for further functional in vitro studies of larger cohorts addressing in‐depth cell properties to determine cell subset functionality in COVID‐19.
Conclusion
In our exploratory study of untreated COVID‐19 infection, we found that critical disease is characterized by activated and exhausted T cells with reduced IL‐7Rα expression and monocytes with decreased fractions of mHLA‐DR and the anti‐inflammatory non‐classical subset, whereas the fraction of the pro‐inflammatory classical monocytes and expression of PD‐L1, ACE‐2 and CD142 were increased. Further, in critical COVID‐19, pathways of both adaptive and innate immune responses are activated on a transcriptional level with increased transcripts of pro‐inflammatory mediators and increased numbers of plasma B cells that may have altered functionality. Our findings indicate an altered innate and adaptive immune phenotype in critical COVID‐19, contributing to the increasing knowledge on COVID‐19 pathogenesis. Nevertheless, further in‐depth studies of regulatory and functional mechanisms are required to identify targets for therapeutic intervention.
Funding
This study received funding from the Research Council of Norway (grant no 312780), South‐Eastern Norway Regional Health Authority (grant nos 2018084 and 39550), Oslo University Hospital, University of Oslo, and has received private donation from Vivaldi Invest A/S owned by Jon Stephenson von Tetzchner.
Conflict of interest
The authors declare no conflict of interest.
Author contribution
Erik Egeland Christensen: Conceptualization (equal); Data curation (equal); Formal analysis (supporting); Investigation (equal); Methodology (equal); Visualization (lead); Writing‐original draft (lead); Writing‐review & editing (equal). Marthe Jøntvedt Jørgensen: Conceptualization (equal); Data curation (equal); Formal analysis (lead); Investigation (equal); Methodology (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Kristin Grotle Nore: Conceptualization (supporting); Formal analysis (equal); Methodology (supporting); Writing‐original draft (supporting); Writing‐review & editing (supporting). Tuva B Dahl: Formal analysis (equal); Investigation (equal); Methodology (equal); Validation (equal); Writing‐original draft (supporting); Writing‐review & editing (supporting). Kuan Yang: Formal analysis (equal); Investigation (equal); Software (equal); Visualization (supporting); Writing‐review & editing (supporting). Trine Ranheim: Data curation (equal); Formal analysis (supporting); Investigation (supporting); Software (supporting). Camilla Huse: Data curation (supporting); Formal analysis (supporting); Investigation (supporting); Methodology (supporting); Writing‐review & editing (supporting). Andreas Lind: Formal analysis (supporting); Writing‐review & editing (supporting). Sarah Nur: Formal analysis (supporting); Methodology (supporting). Birgitte Stiksrud: Conceptualization (supporting); Data curation (supporting); Investigation (supporting); Methodology (supporting); Writing‐review & editing (supporting). Synne Jenum: Conceptualization (supporting); Data curation (supporting); Writing‐review & editing (supporting). Kristian Tonby: Conceptualization (supporting); Data curation (supporting); Writing‐review & editing (supporting). Jan Cato Holter: Conceptualization (supporting); Data curation (equal); Funding acquisition (supporting); Project administration (equal); Writing‐review & editing (supporting). Aleksander Rygh Holten: Conceptualization (supporting); Data curation (equal); Investigation (supporting); Writing‐review & editing (equal). Bente Halvorsen: Formal analysis (equal); Funding acquisition (equal); Methodology (equal); Supervision (supporting); Validation (equal); Writing‐review & editing (equal). Anne Ma Dyrhol‐Riise: Conceptualization (equal); Formal analysis (equal); Funding acquisition (equal); Investigation (equal); Project administration (equal); Resources (equal); Supervision (lead); Writing‐review & editing (equal).
Supporting information
Table S1. Monocyte antibody panel.
Table S2. T cell antibody panel.
Figure S1. T cell gating strategy.
Figure S2. Monocyte gating strategy.
Figure S3. Gating strategy of monocytes showed with FMO controls.
Figure S4. T cell subsets and activation and exhaustion markers in COVID‐19, sepsis and healthy controls.
Figure S5. Correlations between clinical parameters and exploratory immune markers in COVID‐19 (n = 31).
Figure S6. Longitudinal measurements of T cell subsets.
Figure S7. Comparison of the transcriptome in COVID‐19.
Figure S8. All differentially expressed transcripts and the second and third most enriched Gene Ontology terms comparing mild/moderate and critical COVID‐19 patients.
Acknowledgements
We thank the staff at the laboratory and clinical ward of the Departments of Infectious Diseases and Medical microbiology at Oslo University Hospital, Ullevål Hospital, for support, including the patient follow‐up, blood sampling and biobank processing.
Christensen EE, Jørgensen MJ, Nore KG, Dahl TB, Yang K, Ranheim T, et al. Critical COVID‐19 is associated with distinct leukocyte phenotypes and transcriptome patterns. J Intern Med 2021; 290: 677–692.
References
- 1. Fu L, Wang B, Yuan T, Chen X, Ao Y, Fitzpatrick T, et al. Clinical characteristics of coronavirus disease 2019 (COVID‐19) in China: A systematic review and meta‐analysis. J Infect 2020;80:656–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu YI, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giamarellos‐Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID‐19 patients with severe respiratory failure. Cell Host Microbe 2020;27:992–1000.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Merad M, Martin JC. Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol 2020;20:355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jørgensen MJ, Holter JC, Christensen EE, Schjalm C, Tonby K, Pischke SE, et al. Increased interleukin‐6 and macrophage chemoattractant protein‐1 are associated with Respiratory Failure In COVID‐19. Sci Rep 2020;10:21697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kox M, Waalders NJB, Kooistra EJ, Gerretsen J, Pickkers P. Cytokine levels in critically Ill patients with COVID‐19 and other conditions. JAMA 2020;324:1565–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tavakolpour S, Rakhshandehroo T, Wei EX, Rashidian M. Lymphopenia during the COVID‐19 infection: What it shows and what can be learned. Immunol Lett 2020;225:31–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Z, John Wherry E. T cell responses in patients with COVID‐19. Nat Rev Immunol 2020;20:529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mathew D, Giles JR, Baxter AE, et al. Deep immune profiling of COVID‐19 patients reveals distinct immunotypes with therapeutic implications. Science 2020;369: 1203.12–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drewry AM, Samra N, Skrupky LP, Fuller BM, Compton SM, Hotchkiss RS. Persistent lymphopenia after diagnosis of sepsis predicts mortality. Shock 2014;42:383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng HY, Zhang M, Yang CX, Zhang N, Wang X‐C, Yang X‐P, et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID‐19 patients. Cell Mol Immunol 2020;17:541–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID‐19). Front Immunol 2020;11:827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Biasi S, Meschiari M, Gibellini L, Bellinazzi C, Borella R, Fidanza L, et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID‐19 pneumonia. Nat Commun 2020;11:3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sekine T, Perez‐Potti A, Rivera‐Ballesteros O, Strålin K, Gorin JB, Olsson A, et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID‐19. Cell 2020;183:158–68.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rha MS, Jeong HW, Ko JH, Choi SJ, Seo I‐H, Lee JS, et al. PD‐1‐Expressing SARS‐CoV‐2‐Specific CD8(+) T cells are not exhausted, but functional in patients with COVID‐19. Immunity 2020;54:44–52.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang D, Guo R, Lei L, Liu H, Wang Y, Wang Y, et al. Frontline Science: COVID‐19 infection induces readily detectable morphologic and inflammation‐related phenotypic changes in peripheral blood monocytes. J Leukoc Biol 2021;109:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martinez FO, Combes TW, Orsenigo F, Gordon S. Monocyte activation in systemic Covid‐19 infection: Assay and rationale. EBioMedicine 2020;59:102964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grant RA, Morales‐Nebreda L, Markov NS, Swaminathan S, Querrey M, Guzman ER, et al. Circuits between infected macrophages and T cells in SARS‐CoV‐2 pneumonia. Nature 2021;590:635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jafarzadeh A, Chauhan P, Saha B, Jafarzadeh S, Nemati M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID‐19: Lessons from SARS and MERS, and potential therapeutic interventions. Life Sci 2020;257:118102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chu H, Chan JF, Wang Y, Yuen T‐T, Chai Y, Hou Y, et al. Comparative replication and immune activation profiles of SARS‐CoV‐2 and SARS‐CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID‐19. Clin Infect Dis 2020;71:1400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kvedaraite E, Hertwig L, Sinha I, Ponzetta A, Hed Myrberg I, Lourda M, et al. Major alterations in the mononuclear phagocyte landscape associated with COVID‐19 severity. Proc Natl Acad Sci U S A 2021;118:e2018587118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sánchez‐Cerrillo I, Landete P, Aldave B, Sánchez‐Alonso S, Sánchez‐Azofra A, Marcos‐Jiménez A, et al. COVID‐19 severity associates with pulmonary redistribution of CD1c+ DCs and inflammatory transitional and nonclassical monocytes. J Clin Invest 2020;130:6290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, et al. Pathogenic T‐cells and inflammatory monocytes incite inflammatory storms in severe COVID‐19 patients. National Sci Rev 2020;7:998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Silvin A, Chapuis N, Dunsmore G, Goubet A‐G, Dubuisson A, Derosa L, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID‐19. Cell 2020;182:1401–18.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ritchie AI, Singanayagam A. Immunosuppression for hyperinflammation in COVID‐19: a double‐edged sword? Lancet 2020;395:1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ueland T, Heggelund L, Lind A, Holten AR, Tonby K, Michelsen AE, et al. Elevated plasma sTIM‐3 levels in severe Covid‐19 patients. J Allergy Clin Immunol 2020;147:92–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holter JC, Pischke SE, de Boer E, Lind A, Jenum S, Holten AR, et al. Systemic complement activation is associated with respiratory failure in COVID‐19 hospitalized patients. Proc Natl Acad Sci U S A 2020;117:25018–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoel H, Heggelund L, Reikvam DH, Stiksrud B, Ueland T, Michelsen AE, et al. Elevated markers of gut leakage and inflammasome activation in COVID‐19 patients with cardiac involvement. J Intern Med 2020;289:523–31.[Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boutin CA, Grandjean‐Lapierre S, Gagnon S, Labbé A‐C, Charest H, Roger M, et al. Comparison of SARS‐CoV‐2 detection from combined nasopharyngeal/oropharyngeal swab samples by a laboratory‐developed real‐time RT‐PCR test and the Roche SARS‐CoV‐2 assay on a cobas 8800 instrument. J Clin Virol 2020;132:104615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. World Health Organization . Clinical Management of COVID‐19. Geneva, Switzerland: World Health Organization; 2020. [Google Scholar]
- 31. Singer M, Deutschman CS, Seymour CW, Shankar‐Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis‐3). JAMA 2016;315:801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ziegler‐Heitbrock L, Hofer TP. Toward a refined definition of monocyte subsets. Front Immunol 2013;4:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics 2018;34:i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 2019;47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bray NL, Pimentel H, Melsted P, Pachter L. Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol 2016;34:525–7. [DOI] [PubMed] [Google Scholar]
- 36. Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA‐seq incorporating quantification uncertainty. Nat Methods 2017;14:687–90. [DOI] [PubMed] [Google Scholar]
- 37. Soneson C, Love MI, Robinson MD. Differential analyses for RNA‐seq: transcript‐level estimates improve gene‐level inferences. F1000Research 2015;4:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun 2019;10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. The Gene Ontology Consortium . The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 2019;47:D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pratt D, Chen J, Welker D, Rivas R, Pillich R, Rynkov V, et al. NDEx, the network data exchange. Cell Syst 2015;1:302–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aschenbrenner AC, Mouktaroudi M, Krämer B, Oestreich M, Antonakos N, Nuesch‐Germano M, et al. Disease severity‐specific neutrophil signatures in blood transcriptomes stratify COVID‐19 patients. Genome Med 2021;13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pruenster M, Vogl T, Roth J, Sperandio M. S100A8/A9: From basic science to clinical application. Pharmacol Ther 2016;167:120–31. [DOI] [PubMed] [Google Scholar]
- 47. Bernardes JP, Mishra N, Tran F, Bahmer T, Best L, Blase JI, et al. Longitudinal multi‐omics analyses identify responses of megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID‐19. Immunity 2020;53:1296–314.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stephenson E, Reynolds G, Botting RA, Calero‐Nieto FJ, Morgan M, Tuong ZK, et al. The cellular immune response to COVID‐19 deciphered by single cell multi‐omics across three UK centres. medRxiv 2021: 2021. 01.13.21249725. [Google Scholar]
- 49. Woodruff MC, Ramonell RP, Nguyen DC, Cashman KS, Saini AS, Haddad NS, et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID‐19. Nat Immunol 2020;21:1506–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Asehnoune K, Roquilly A, Vourc'h M. Monitoring the adaptive immune system in septic shock patients: ready for prime time? Crit Care Med 2018;46:1867–9. [DOI] [PubMed] [Google Scholar]
- 51. Rethi B, Fluur C, Atlas A, Krzyzowska M, Mowafi F, Grützmeier S, et al. Loss of IL‐7Ralpha is associated with CD4 T‐cell depletion, high interleukin‐7 levels and CD28 down‐regulation in HIV infected patients. Aids 2005;19:2077–86. [DOI] [PubMed] [Google Scholar]
- 52. Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El Jammal T, et al. Should we stimulate or suppress immune responses in COVID‐19? Cytokine and anti‐cytokine interventions. Autoimmun Rev 2020;19:102567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fu B, Xu X, Wei H. Why tocilizumab could be an effective treatment for severe COVID‐19? J Transl Med 2020;18:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao J, Zhao J, Van Rooijen N, Perlman S. Evasion by stealth: inefficient immune activation underlies poor T cell response and severe disease in SARS‐CoV‐infected mice. PLoS Pathog 2009;5:e1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, et al. The diverse function of PD‐1/PD‐L pathway beyond cancer. Front Immunol 2019;10:2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schulte‐Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, et al. Severe COVID‐19 is marked by a dysregulated myeloid cell compartment. Cell 2020;182:1419–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet 2020;395:1033–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. Cytokine release syndrome in severe COVID‐19: interleukin‐6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J Antimicrob Agents 2020;55:105954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Russell B, Moss C, George G, Santaolalla A, Cope A, Papa S, et al. Associations between immune‐suppressive and stimulating drugs and novel COVID‐19‐a systematic review of current evidence. Ecancermedicalscience 2020;14:1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez‐Colón GJ, McKechnie JL, et al. A single‐cell atlas of the peripheral immune response in patients with severe COVID‐19. Nat Med 2020;26:1070–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single‐cell landscape of bronchoalveolar immune cells in patients with COVID‐19. Nat Med 2020;26:842–4. [DOI] [PubMed] [Google Scholar]
- 62. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID‐19 and influenza highlights the role of type I interferons in development of severe COVID‐19. Sci Immunol 2020;5:eabd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guilliams M, Mildner A, Yona S. Developmental and functional heterogeneity of monocytes. Immunity 2018;49:595–613. [DOI] [PubMed] [Google Scholar]
- 64. Gatti A, Radrizzani D, Viganò P, Mazzone A, Brando B. Decrease of non‐classical and intermediate monocyte subsets in severe acute SARS‐CoV‐2 infection. Cytometry A 2020;97:887–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kox M, Frenzel T, Schouten J, van de Veerdonk FL, Koenen H, Pickkers P. COVID‐19 patients exhibit less pronounced immune suppression compared with bacterial septic shock patients. Crit Care 2020;24:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Prebensen C, Myhre PL, Jonassen C, Rangberg A, Blomfeldt A, Svensson M, et al. SARS‐CoV‐2 RNA in plasma is associated with ICU admission and mortality in patients hospitalized with COVID‐19. Clin Infect Dis 2020; ciaa1338. 10.1093/cid/ciaa1338. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wichmann D, Sperhake JP, Lütgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy findings and venous thromboembolism in patients with COVID‐19: a prospective cohort study. Ann Intern Med 2020;173:268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Monocyte antibody panel.
Table S2. T cell antibody panel.
Figure S1. T cell gating strategy.
Figure S2. Monocyte gating strategy.
Figure S3. Gating strategy of monocytes showed with FMO controls.
Figure S4. T cell subsets and activation and exhaustion markers in COVID‐19, sepsis and healthy controls.
Figure S5. Correlations between clinical parameters and exploratory immune markers in COVID‐19 (n = 31).
Figure S6. Longitudinal measurements of T cell subsets.
Figure S7. Comparison of the transcriptome in COVID‐19.
Figure S8. All differentially expressed transcripts and the second and third most enriched Gene Ontology terms comparing mild/moderate and critical COVID‐19 patients.