

Abstract
Lead optimization, a critical step in early stage drug discovery, involves making chemical modifications to a small-molecule ligand to improve properties such as binding affinity. We recently developed DeepFrag, a deep-learning model capable of recommending such modifications. Though a powerful hypothesis-generating tool, DeepFrag is currently implemented in Python and so requires a certain degree of computational expertise. To encourage broader adoption, we have created the DeepFrag browser app, which provides a user-friendly graphical user interface that runs the DeepFrag model in users’ web browsers. The browser app does not require users to upload their molecular structures to a third-party server, nor does it require the separate installation of any third-party software. We are hopeful that the app will be a useful tool for both researchers and students. It can be accessed free of charge, without registration, at http://durrantlab.com/deepfrag. The source code is also available at http://git.durrantlab.com/jdurrant/deepfrag-app, released under the terms of the open-source Apache License, Version 2.0.
Introduction
The process of discovering and developing a new drug is both expensive and time-consuming. In the earliest steps, researchers seek to identify hit compounds that are active against a disease-implicated protein of interest. These hits must then undergo lead optimization, which involves adding or swapping chemical moieties with the goal of improving binding affinity or other chemical properties related to absorption, distribution, metabolism, excretion, and toxicity.1
Computer-aided drug discovery (CADD) can accelerate these early stage steps. For example, structure-based virtual screening (i.e., computer docking) can identify compounds that are promising candidate hits for subsequent experimental testing. Once a hit has been identified, a number of computational techniques can also further lead optimization, ranging from docking-based methods such as AutoGrow2−4 to more advanced, molecular dynamics (MD) “alchemical” methods5 such as thermodynamic integration,6 single-step perturbation,7 and free energy perturbation.8
We recently created a 3D convolutional neural network called DeepFrag9 that aims to further lead optimization. To train DeepFrag, we assembled a large set of crystal structures and systematically removed fragments from the cocrystallized ligands. We then asked DeepFrag to predict a molecular fingerprint (vector) describing the missing fragment. The predicted fingerprints most closely matched the corresponding missing fragments roughly 60% of the time when selecting from a reference library of ∼6,500 fragments. Remarkably, even when the network predicts the wrong fragment, the top predictions are often chemically similar and may well be more optimal. In prospective practice, DeepFrag can also be used to add novel fragments to an identified lead, in addition to swapping existing moieties.
To ensure usability, we took great care to document the DeepFrag Python source code and even created a Google Colab notebook so users can test the network without having to download or locally install any software, libraries, or dependencies,10 but even this approach limits accessibility to those who are experts in the field. While the negative impact of poor usability on software adoption among scientists should not be understated, it is particularly problematic in educational settings. Many students are unfamiliar with Python, and expecting students to download, install, and use a command-line program is often impractical.
To address these usability challenges, we have created the DeepFrag browser app. By “browser app”, we mean software that runs on users’ local computers, entirely in a web browser. Browser apps have some notable advantages over server apps, which run calculations on remote resources (“in the cloud”). For example, rather than require users to upload their proprietary data to a third-party server, browser apps download the software required to run the calculations locally in the browser’s secure sandboxed environment. Thanks to this de facto distributed approach, browser apps do not require an extensive and difficult-to-maintain remote computer infrastructure. Furthermore, calculations begin immediately on the user’s own computer, so there is no need to wait in lengthy queues for limited remote resources to become available.
The DeepFrag browser app will be a useful tool for the CADD research and educational communities. A working implementation can be accessed free of charge at http://durrantlab.com/deepfrag, without registration. Its source code is available at http://git.durrantlab.com/jdurrant/deepfrag-app, released under the terms of the Apache License, Version 2.0.
Results and Discussion
Input Parameters Tab
To run the DeepFrag browser app, users need only visit http://durrantlab.com/deepfrag, where they will encounter the “Input Parameters” tab illustrated in Figure 1, on the left. In the “Input Receptor and Ligand Files” subsection (Figure 1A), users can specify the protein receptor and ligand file for optimization in any of several popular formats. The contents of these files are loaded into the browser’s memory, but they are never transmitted/uploaded to any third-party server. Users who wish to simply test DeepFrag can instead click the “Use Example Files” button (not shown) to load a preprepared structure of H. sapiens peptidyl-prolyl cis–trans isomerase NIMA-interacting 1 (HsPin1p) bound to a small-molecule ligand (PDB 2XP9(11)).
Figure 1.

Input parameters tab (on the left) includes the (A) “Input Receptor and Ligand Files” and (B) “Molecular Viewer” subsections, as well as the (C) save/load and “Start DeepFrag” buttons. The Output tab (on the right) includes the (D) suggested-fragments table and (E) “Output Files” subsections. Some components are not shown to simplify the presentation.
The “Molecular Viewer” subsection (Figure 1B) contains a 3Dmol.js molecular viewer12 where the specified files are displayed. This subsection also includes two toggle buttons. The “Delete Atom” button allows users to remove ligand atoms from the structure by clicking on them. We included this optional feature anticipating that many users will wish to use DeepFrag to replace existing ligand moieties. The “Select Atom as Growing Point” toggle button allows users to indicate which ligand atom should serve as the growing point (i.e., connection point) that connects the predicted fragments to the parent ligand molecule. After users click the appropriate ligand atom, a yellow transparent sphere indicates the location of the growing point.
A slider allows the user to control the grid-ensemble size for ensemble (consensus) predictions (not shown in Figure 1). In the original DeepFrag publication,9 we evaluated the impact of sampling multiple random grid rotations for each protein/ligand input. A final ensemble fragment fingerprint was then calculated by averaging the predicted fingerprints associated with each rotated grid. This approach led to modest improvements in accuracy (∼1.5% TOP-1 accuracy in our tests when considering 32 rotations vs one9). To match the original DeepFrag implementation, the browser app performs the full 32 rotations by default. Users who wish to accelerate the in-browser calculation can optionally specify fewer rotations.
A checkbox allows users to control how the DeepFrag browser app performs grid rotations (not shown in Figure 1). In the original DeepFrag implementation,9 each of the ensemble-member grids is rotated randomly. To match the original implementation, the browser app also performs random rotations by default, but users can optionally instruct the browser app to (1) always rotate in 90° increments and (2) further consider grid reflections. These operations can be performed rapidly using the TensorFlow.js reverse and transpose functions, thus speeding the in-browser calculation. Given that the input structures are unlikely to have rotational or reflection symmetry along any of the primary axes, using 90° rotations and reflections should not introduce any bias into the predictions, but this optional, accelerated approach does differ from the thoroughly tested DeepFrag implementation described in our previous manuscript.9 We therefore suggest using the default browser-app settings to match the original implementation precisely.
Several buttons are present at the bottom of the Input Parameters tab (Figure 1C). The “Temporary Save” button saves the specified parameters (i.e., receptor/ligand files, growing point, etc.) to the browser’s session storage. These same parameters can be later restored using the “Load Saved Data” button. Otherwise, the user simply clicks the “Start DeepFrag” button to begin the DeepFrag run.
The DeepFrag browser app then generates tensor(s) from the input molecular structures and uses the trained model to predict appropriate molecular fragments. The prediction typically takes at most half a minute, even when running the DeepFrag app on a mobile phone.
Output Tab
DeepFrag displays the “Output” tab once the calculations are complete (Figure 1, illustrated on the right). The “Visualization” subsection again displays the specified receptor, ligand, and growing point for user convenience (not shown). Below the molecular visualization, a table shows the SMILES strings, molecular structures (generated using SmilesDrawer13), and DeepFrag scores of the top 20 predicted fragments, sorted from most to least promising (Figure 1D). To generate a 3D structure of a given “fused” parent/fragment composite molecule (e.g., for computer docking), users can simply click the corresponding fragment SMILES string to launch a separate molecule-preparation web app called Fuser (see the Git repository for details). As with the original DeepFrag implementation, fragment scores are calculated by considering the cosine similarity14 between the predicted fingerprint vector and the fingerprint vector of the corresponding fragment.
The “Output Files” subsection (Figure 1E) allows users to directly view DeepFrag output files. Users can also press the associated “Download” buttons to save the files to disk. These files include a more complete list of the predicted fragments (TSV format), the 3D coordinates of the selected growing point (JSON format), and the receptor and ligand files used for analysis (PDB format).
Compatibility
We have tested the DeepFrag browser app on the browser/operating-system combinations shown in Table 1. It works well on both desktop and mobile operating systems, as well as on all major browsers (e.g., Chrome, Edge, Firefox, and Safari).
Table 1. DeepFrag Browser and Operating-System Compatibility Tests.
| browser | operating system |
|---|---|
| Chrome 88.0.4324.87 | macOS 10.14.5 |
| Firefox 84.0 | macOS 10.14.5 |
| Safari 13.1.1 | macOS 10.14.5 |
| Chrome 87.0.4280.141 | Windows 10.0.19041 Home |
| Firefox 84.0.2 | Windows 10.0.19041 Home |
| Edge 87.0.664.75 | Windows 10.0.19041 Home |
| Chrome 87.0.4280.141 | Android 10 |
| Firefox 84.1.4 | Android 10 |
| Safari 14 | iPhone SE iOS 14.3 |
| Chromium 87.0.4280.141 | Ubuntu Linux 18.04.5 LTS |
| Firefox 84.0.2 | Ubuntu Linux 18.04.5 LTS |
Example of Use: HsPin1p
In our original manuscript describing the DeepFrag model,9 we provided several test cases showing how it can be used for lead optimization.9 To show that the original implementation and the browser app give comparable results, we here reproduce one of those tests, which focused on the cancer target HsPin1p15 bound to a phenyl-imidazole ligand (IC50: 8 μM; PDB 2XP9(11)). We chose this protein/ligand complex because neither the protein nor the ligand was included in the DeepFrag training or validation sets. DeepFrag is nondeterministic because it randomly rotates the voxel grids used as input; that is, the program by design gives slightly different results every time it is run. We thus do not expect the browser implementation to always suggest fragments that are identical to those reported previously; rather, we expect the fragments to be identical in many cases and at least similar otherwise (Figure 2).
Figure 2.
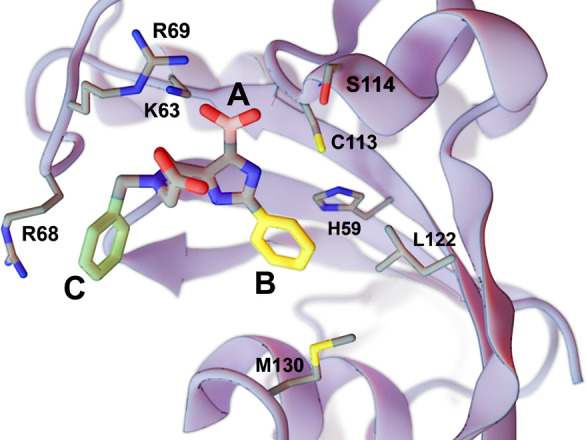
A crystal structure of HsPin1p bound to a phenyl-imidazole ligand (PDB 2XP9(11)). We reassessed a carboxyl fragment (A, in pink) and two phenyl fragments (B, in yellow; C, in green) using the DeepFrag browser app. Select labeled protein residues are shown in a sticks representation. The figure was rendered using BlendMol.16
We first used the DeepFrag browser app to remove carboxylate A (Figure 2, highlighted in pink) and to predict appropriate replacement moieties at the same position. Like the original DeepFrag implementation,9 the browser app also predicted the correct (known) carboxylate moiety. This choice is sensible given that the carboxylate enables electrostatic interactions with K63 and R69 and hydrogen bonds with C113 and S114.
We used the same approach to evaluate phenyl B (Figure 2, highlighted in yellow). The original DeepFrag implementation suggested a bicyclic replacement, *c1ccnc2c1C(=O)N(C)C2, at this position.9 When run from the browser app, this same fragment was ranked second, likely because it preserves π–π interactions with H59 while improving hydrophobic interactions with L122 and M130.9 The first browser-app-predicted fragment was also a bicyclic replacement, *c1ncnc2c1C(C)CC2O, that is similarly composed of a six-member aromatic ring fused to a five-member nonaromatic ring. Given these structural similarities, the top browser-app fragment may adopt a similar binding pose within the HsPin1p pocket.
Finally, we removed phenyl C (Figure 2, highlighted in green) and similarly used the browser app to predict appropriate replacements. The original DeepFrag implementation suggested methyl and ethyl replacements at this location, likely because they maintain potential hydrophobic interactions with the R68 side chain.9 The browser app suggested the same two fragments, though it preferred the ethyl replacement over the methyl.
This work demonstrates that the original DeepFrag implementation and the browser app make comparable fragment predictions, as expected given that the two implementations are functionally identical.
Example of Use: DNA Gyrase B (24 kDa Domain)
Having applied the DeepFrag browser app to an established test case (HsPin1p), we now provide a second, novel example that illustrates how DeepFrag can suggest fragment additions that improve binding affinity. We considered a recent fragment-based lead-optimization project undertaken by Ushiyama et al., which identified several E. coli DNA gyrase B (24 kDa domain) inhibitors, including one in the low-nanomolar range.17 Importantly, none of the crystal structures associated with the Ushiyama study (PDB IDs: 6KZV, 6KZX, 6KZZ, and 6L01(17)) were included in the original training, validation, or testing sets used to create the DeepFrag model. While some unassociated structures of E. coli DNA gyrase B were included in the training set, they were bound to ligands that are quite distinct (PDB IDs: 4DUH,186F86,196F94,19 and 6F8J(19)).
A number of the inhibitors that Ushiyama et al. identified share the same inhibitory 8-(methylamino)-2-oxo-N-phenyl-1,2-dihydroquinoline-3-carboxamide scaffold, which itself has an IC50 value of 0.24 μM per isothermal titration calorimetry.17 We applied DeepFrag to this scaffold to see if it could identify the same fragment additions that Ushiyama et al. selected and experimentally tested. There is no crystal structure of the scaffold itself bound to DNA gyrase B, but multiple crystal structures of bound analogues containing the scaffold are nearly superimposable. We therefore created a model of the receptor/scaffold complex from the 6KZZ structure17 by simply removing any ligand atoms that did not belong to the scaffold itself.
Ushiyama et al. tested a number of fragment additions at the phenyl para position. We used the DeepFrag browser app to evaluate this same position (Figure 3). The top DeepFrag-suggested addition was an acetic acid, *CC(=O)O, which Ushiyama et al. had also selected and tested. In their hands, this addition improved the IC50 value to 0.018 μM,17 likely because it enables electrostatic interactions with R136 and perhaps R76. The second DeepFrag-suggested fragment addition at the para position was a carboxyl group. This compound had also been tested and was found to have an improved IC50 value of 0.0017 μM.17
Figure 3.

An inhibitory scaffold bound to E. coli DNA gyrase B. The protein is shown as a blue ribbon, and key amino acids are shown as thin sticks (PDB ID 6KZZ(17)). The scaffold is shown as thick sticks, with the relevant atomic coordinates taken from the 6KZZ ligand.17 The benzene para and meta positions are marked with a dagger and a double dagger, respectively. A 2D depiction of the scaffold is overlaid, with its experimentally measured IC50 value (per Ushiyama et al.17). DeepFrag-suggested fragment additions at the para and ortho positions are ranked in the table below, with the associated IC50 values.
Ushiyama et al. also found that fragment additions at the ortho position improved IC50 values, albeit more modestly. The top DeepFrag-suggested addition at this position was a carboxyl group (Figure 3), an addition that Ushiyama et al. had also tested (IC50 0.21 μM).17 The second DeepFrag addition was an acetic acid, *CC(=O)O, which had an experimentally measured IC50 value of 0.16 μM.17
These examples illustrate that the DeepFrag browser app can suggest fragment additions similar to those a trained medicinal chemist might select and that those additions can in some cases dramatically improve binding affinity.
Conclusions
Our original DeepFrag model serves as a useful tool that aims to help trained medical chemists and structural biologists in their lead-optimization efforts, but as originally implemented, DeepFrag is a stand-alone Python program tailored primarily to expert computationalists. To enable use by a broader audience, we have implemented DeepFrag as a browser app. Researchers, educators, and students can easily experiment with DeepFrag optimization in their browsers, without ever having to upload possibly proprietary structures to a third-party server and without ever having to install any separate software.
The DeepFrag browser app will be a useful tool for the CADD research and education community. It is functionally identical to the original implementation and so yields comparable results, but the browser-app version additionally provides a user-friendly interface for setting up a DeepFrag run and for viewing predicted fragments. It is freely accessible at http://durrantlab.com/deepfrag. A copy of the source code can be obtained free of charge from http://git.durrantlab.com/jdurrant/deepfrag-app, released under the terms of the Apache License, Version 2.0.
Implementation
We relied on several web technologies to implement the original DeepFrag model as a browser app. Our implementation can be broadly divided into setup, calculation, and results. The workflow is illustrated in Figure 4 and described in detail below.
Figure 4.
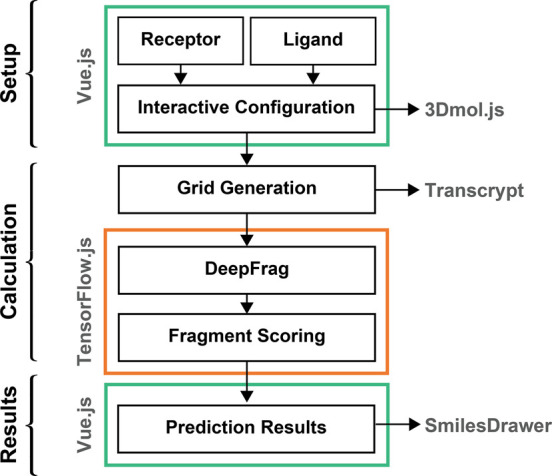
DeepFrag browser-app workflow can be broadly divided into components for setup, calculation, and displaying results. The setup and results include graphical user interfaces (GUIs) powered by Vue.js, and the calculations themselves depend on Transcrypt-compiled code and TensorFlow.js. 3Dmol.js12 and SmilesDrawer13 are useful JavaScript libraries for molecular visualization.
Setting up a DeepFrag Calculation
To simplify the process of setting up a DeepFrag calculation, we created a browser-based GUI so users can easily (1) load their receptor and ligand structures into the browser’s memory, (2) select the growing point, and (3) specify other DeepFrag parameters (Figure 4). To build the GUI, we used the same approach that we have used previously.20 In brief, the interface is written in the open-source Microsoft TypeScript programming language, which compiles to JavaScript and so can run in any modern web browser. It uses the open-source Vue.js framework (https://vuejs.org/) to provide reusable, consistently styled HTML-like components (e.g., buttons, input fields, etc.). Many of these components are derived from the open-source BootstrapVue library (https://bootstrap-vue.js.org/), which makes it easy to implement the color, size, and typography specifications of the Bootstrap4 framework (https://getbootstrap.com/). We also adapted our existing molecular-visualization Vue.js component20 for use in the DeepFrag app. This component leverages the 3Dmol.js JavaScript library,12 which displays molecular structures without requiring any separate installation or browser plugin.
To compile and assemble our TypeScript codebase and the third-party libraries described above, we used Webpack, an open-source module bundler (https://webpack.js.org/). This compilation process included Google’s Closure Compiler (https://developers.google.com/closure/compiler), which automatically optimizes TypeScript/JavaScript code for size and speed.
Running a DeepFrag Calculation
The DeepFrag model requires tensor grids as input. To convert molecular structures to grids in the browser, we first created a pure-Python implementation of the GPU-accelerated grid-generation code described in the original DeepFrag publication9 and transpiled it to JavaScript using the Transcrypt compiler (https://www.transcrypt.org/) (Figure 4).
To run the DeepFrag model itself in a browser environment, we used the Open Neural Network Exchange framework21 to convert our PyTorch model to the equivalent Tensorflow model.22 We then used TensorFlow.js (https://www.tensorflow.org/js) to run the model in the browser. Internally, the browser implementation is functionally identical to the stand-alone version.
Viewing DeepFrag Results
To view DeepFrag results (i.e., suggested fragments), we again created/reused the appropriate Vue.js components (Figure 4). Graphical depictions of the suggested fragments are generated from the output SMILES strings using the SmilesDrawer JavaScript library.13
Acknowledgments
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health [R01GM132353 to J.D.D.]. We would like to thank Dr. David R. Koes for helpful discussions. We also acknowledge the University of Pittsburgh’s Center for Research Computing for providing valuable computer resources.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.1c00103.
Names and SMILES strings of compounds mentioned in text (TXT)
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
Notes
Data and Software Availability. The DeepFrag browser app is freely accessible at http://durrantlab.com/deepfrag, without registration. The app source code is also available free of charge at http://git.durrantlab.com/jdurrant/deepfrag-app, released under the terms of the Apache License, Version 2.0. The browser app is based on the original DeepFrag Python program, also released under the terms of the Apache License, Version 2.0. This original implementation can be downloaded free of charge from http://durrantlab.com/deepfragmodel/. It was trained on data derived from the Binding MOAD,23 which is publicly accessible at https://bindingmoad.org/.
Supplementary Material
References
- Balani S. K.; Miwa G. T.; Gan L.-S.; Wu J.-T.; Lee F. W. Strategy of utilizing in vitro and in vivo ADME tools for lead optimization and drug candidate selection. Curr. Top. Med. Chem. 2005, 5, 1033–8. 10.2174/156802605774297038. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; Amaro R. E.; McCammon J. A. AutoGrow: A Novel Algorithm for Protein Inhibitor Design. Chem. Biol. Drug Des. 2009, 73, 168–178. 10.1111/j.1747-0285.2008.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; Lindert S.; McCammon J. A. AutoGrow 3.0: An Improved Algorithm for Chemically Tractable, Semi-Automated Protein Inhibitor Design. J. Mol. Graphics Modell. 2013, 44, 104–112. 10.1016/j.jmgm.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel J. O.; Durrant J. D. AutoGrow4: an open-source genetic algorithm for de novo drug design and lead optimization. Journal of Cheminformatics 2020, 12, 25. 10.1186/s13321-020-00429-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. Molecular dynamics simulations and drug discovery. BMC Biology 2011, 9, 71–79. 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adcock S. A.; McCammon J. A. Molecular dynamics: survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615. 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab F.; van Gunsteren W. F.; Zagrovic B. Computational study of the mechanism and the relative free energies of binding of anticholesteremic inhibitors to squalene-hopene cyclase. Biochemistry 2008, 47, 2945–2951. 10.1021/bi702067h. [DOI] [PubMed] [Google Scholar]
- Kim J. T.; Hamilton A. D.; Bailey C. M.; Domaoal R. A.; Wang L.; Anderson K. S.; Jorgensen W. L. FEP-guided selection of bicyclic heterocycles in lead optimization for non-nucleoside inhibitors of HIV-1 reverse transcriptase. J. Am. Chem. Soc. 2006, 128, 15372–15373. 10.1021/ja066472g. [DOI] [PubMed] [Google Scholar]
- Green H.; Koes D. R.; Durrant J. D.. DeepFrag: A Deep Convolutional Neural Network for Fragment-based Lead Optimization. bioRxiv, 2021. https://www.biorxiv.org/content/10.1101/2021.01.07.425790v1 (accessed 2021-05-14). [DOI] [PMC free article] [PubMed]
- Bisong E.Building Machine Learning and Deep Learning Models on Google Cloud Platform: A Comprehensive Guide for Beginners; Apress: Berkeley, CA, 2019; pp 59–64, 10.1007/978-1-4842-4470-8_7. [DOI] [Google Scholar]
- Potter A.; Oldfield V.; Nunns C.; Fromont C.; Ray S.; Northfield C. J.; Bryant C. J.; Scrace S. F.; Robinson D.; Matossova N.; Baker L.; Dokurno P.; Surgenor A. E.; Davis B.; Richardson C. M.; Murray J. B.; Moore J. D. Discovery of cell-active phenyl-imidazole Pin1 inhibitors by structure-guided fragment evolution. Bioorg. Med. Chem. Lett. 2010, 20, 6483–8. 10.1016/j.bmcl.2010.09.063. [DOI] [PubMed] [Google Scholar]
- Rego N.; Koes D. 3Dmol.js: molecular visualization with WebGL. Bioinformatics 2015, 31, 1322–4. 10.1093/bioinformatics/btu829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst D.; Reymond J.-L. SmilesDrawer: Parsing and Drawing SMILES-Encoded Molecular Structures Using Client-Side JavaScript. J. Chem. Inf. Model. 2018, 58, 1–7. 10.1021/acs.jcim.7b00425. [DOI] [PubMed] [Google Scholar]
- Xia P.; Zhang L.; Li F. Learning similarity with cosine similarity ensemble. Inf. Sci. 2015, 307, 39–52. 10.1016/j.ins.2015.02.024. [DOI] [Google Scholar]
- Nakatsu Y.; Yamamotoya T.; Ueda K.; Ono H.; Inoue M.-K.; Matsunaga Y.; Kushiyama A.; Sakoda H.; Fujishiro M.; Matsubara A.; Asano T. Prolyl isomerase Pin1 in metabolic reprogramming of cancer cells. Cancer Lett. 2020, 470, 106–114. 10.1016/j.canlet.2019.10.043. [DOI] [PubMed] [Google Scholar]
- Durrant J. D. BlendMol: Advanced Macromolecular Visualization in Blender. Bioinformatics 2019, 35, 2323–2325. 10.1093/bioinformatics/bty968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushiyama F.; Amada H.; Takeuchi T.; Tanaka-Yamamoto N.; Kanazawa H.; Nakano K.; Mima M.; Masuko A.; Takata I.; Hitaka K.; Iwamoto K.; Sugiyama H.; Ohtake N. Lead Identification of 8-(Methylamino)-2-oxo-1,2-dihydroquinoline Derivatives as DNA Gyrase Inhibitors: Hit-to-Lead Generation Involving Thermodynamic Evaluation. ACS Omega 2020, 5, 10145–10159. 10.1021/acsomega.0c00865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brvar M.; Perdih A.; Renko M.; Anderluh G.; Turk D.; Solmajer T. Structure-based discovery of substituted 4,5′-bithiazoles as novel DNA gyrase inhibitors. J. Med. Chem. 2012, 55, 6413–26. 10.1021/jm300395d. [DOI] [PubMed] [Google Scholar]
- Narramore S.; Stevenson C. E. M.; Maxwell A.; Lawson D. M.; Fishwick C. W. G. New insights into the binding mode of pyridine-3-carboxamide inhibitors of E. coli DNA gyrase. Bioorg. Med. Chem. 2019, 27, 3546–3550. 10.1016/j.bmc.2019.06.015. [DOI] [PubMed] [Google Scholar]
- Kochnev Y.; Hellemann E.; Cassidy K. C.; Durrant J. D. Webina: An Open-Source Library and Web App that Runs AutoDock Vina Entirely in the Web Browser. Bioinformatics 2020, 36, 4513–4515. 10.1093/bioinformatics/btaa579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J.; Lu F.; Zhang K., et al. ONNX: Open Neural Network Exchange; 2019. https://github.com/onnx/onnx (accessed 2021-05-14).
- Abadi M.; Agarwal A.; Barham P.; Brevdo E.; Chen Z.; Citro C.; Corrado G. S.; Davis A.; Dean J.; Devin M.. TensorFlow: Large-scale machine learning on heterogeneous systems; 2015. https://www.tensorflow.org/ (accessed 2021-05-14).; Software available from tensorflow.org.
- Hu L.; Benson M. L.; Smith R. D.; Lerner M. G.; Carlson H. A. Binding MOAD (Mother Of All Databases). Proteins: Struct., Funct., Genet. 2005, 60, 333–340. 10.1002/prot.20512. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.