

Abstract

Research in natural products, the genetically encoded small molecules produced by organisms in an idiosyncratic fashion, deals with molecular structure, biosynthesis, and biological activity. Bioinformatics analyses of microbial genomes can successfully reveal the genetic instructions, biosynthetic gene clusters, that produce many natural products. Genes to molecule predictions made on biosynthetic gene clusters have revealed many important new structures. There is no comparable method for genes to biological activity predictions. To address this missing pathway, we developed a machine learning bioinformatics method for predicting a natural product’s antibiotic activity directly from the sequence of its biosynthetic gene cluster. We trained commonly used machine learning classifiers to predict antibacterial or antifungal activity based on features of known natural product biosynthetic gene clusters. We have identified classifiers that can attain accuracies as high as 80% and that have enabled the identification of biosynthetic enzymes and their corresponding molecular features that are associated with antibiotic activity.
Introduction
Natural products from bacteria, fungi, and plants have long been a rich source of useful molecules. Some 23.5% of FDA-approved drugs are small molecule natural products or chemically modified analogs of natural products, and an additional 22.5% are synthetic drugs designed to hit the same target as a natural product.1 High-throughput screening and bioinformatics analysis have increased efficiencies in steps on the discovery to drug pipeline, but identifying desired functions such as antibiotic activity require production, purification, and assaying, which collectively form a major bottleneck.2 The historic order, assaying, production, and purification, has the same collective bottleneck but with the added disadvantage of ultimately identifying a previously discovered molecule.3,4 Several different mass spectrometry or NMR-based methods can be used to increase efficiencies in some steps, but these in themselves do not obviate the need to have molecules to test.5−8 Overall, the process of going from activity in an extract of a bacterial culture to the discovery of a novel active molecule remains a time-consuming roadblock in using natural products as a source of antibiotics and other therapeutic agents.
One step that can be performed efficiently at scale is identifying promising biosynthetic gene clusters (BGC) in the growing number of available bacterial genomes. However, the “promising” rankings that emerge have been based on genes to structure connections. Existing genome mining tools, which include antiSMASH,9 PRISM,10 Deep-BGC,11 SMURF,12 as well as others that are geared toward identifying specific classes of natural products such as PKminer,13 2metDB,14 RiPP Miner,15 BAGEL,16 RODEO,17 and NeuRiPP,18 can be used to identify BGCs in genomes and compare them to BGCs for known natural products. There are over 147,000 BGC sequences that have been identified by antiSMASH alone,19,20 and prioritization based on structural novelty, which has resulted in many new molecules being discovered, has not really addressed the issue of finding molecules with useful functions. There are numerous architecturally fascinating molecules being published that lack any identified function. If the activity of a natural product could be predicted from its BGC, searches could be prioritized to increase focus only on those most likely to produce a natural product with the activity of interest.
Typically, it is not possible to predict the activity of natural products from a BGC. The presence of known resistance markers or duplication of an essential gene can indicate that a BGC produces an inhibitor for the duplicated gene.21 Existing tools to prioritize BGCs with resistance markers include the Resistance Gene Identifier (RGI)22 and the Antibiotic Resistant Target Seeker (ARTS).23 However, these methods limit discovery to natural products with antibacterial activity and, except in the case of resistance through gene duplication, are less likely to identify natural products that act through novel mechanisms of action. A recently reported genome mining tool, Deep-BGC, uses machine learning to predict antibacterial, cytotoxic, inhibitor, and antifungal activity, but due to a small training set size (370), it does so with only low accuracy.11 The activity predictions made by Deep-BGC were based only on protein family (PFAM) domains and no other types of genetic feature that could be predictive of activity. PRISM 4, another recently reported method for predicting natural product activity from the sequence of BGCs, first predicted the structure of the natural product from the BGC and then used chemical fingerprints to predict its activity.24 There is still an unmet need for a more general method of predicting a natural product’s biological activity from its BGC sequence using both biosynthetic genes and other genetic features such as resistance markers.
We here report a bioinformatics-based machine learning method to predict natural product activities from BGC sequences. To do this, we broke BGCs down into features that describe the type of genes and biosynthetic capabilities present in the cluster using automatic annotations such as PFAM domains and the Resistance Gene Identifier (RGI). We then trained machine learning models to predict the likelihood that a natural product will have activity such as antibacterial or antifungal with a library assembled from the MiBIG database and the literature. The resulting classifiers predict activity with accuracies up to 80%. In theory, this tool could also be trained to predict any other bioactivity if a high quality training dataset was available.
Results and Discussion
Generating a Training Data Set
In order to develop a machine learning model that can predict natural product bioactivity, we assembled a dataset of known BGCs paired with the activity of their products, represented BGCs as vectors based on the number of times various gene annotations appeared in the cluster, and trained several different binary classifiers on the resulting dataset (Figure 1A). To assemble a training dataset, we used bacterial BGCs listed in the Minimum Information about a Biosynthetic Gene Custer (MIBiG) database (version 1.4).25 For each metabolite, we searched the literature to determine what was known about its bioactivity. Specifically, we searched to determine if a metabolite had recorded antibacterial, antifungal, cytotoxic, antitumor, or other activities. We only included compounds that had a documented activity or function or that had been shown to lack antibacterial, antifungal, or antitumor activity in our dataset. We recorded activity as a binary yes/no value and due to the difficulties in comparing the level of activity measured by different assays and did not require a specific level of activity. We assumed that a natural product had only the activities that it was reported to have and not any additional activities that were not reported. It is likely that this assumption will be incorrect in some cases because not every natural product has been exhaustively tested for the activities for which we developed classifiers. This incomplete information will likely contribute to error in predictions. If a BGC was documented to produce multiple products, we counted it as active if one or more of the produced compounds was active. A full list of natural products used in the training set is available in Table S1.
Figure 1.

Machine learning method for predicting natural product activities. (A) Schematic illustrating our machine method for predicting natural product bioactivity. First, we generated a dataset of known BGCs and the activity of the natural product they produce, and we then represented the BGCs as a vector denoting the number of times different types of genes occurred in the cluster. Finally, we trained machine learning models to predict bioactivity. (B) Illustration of how example annotations provide information about the structure of erythromycin, e.g., how many polyketide units and sugars are in the molecule, as well as the approximate level of reduction of the polyketide backbone. Gene cluster rendered using Geneious Prime 2020.0.3 (https://www.geneious.com).
Using existing bioinformatics techniques, we identified the number of times different families of proteins occurred in the biosynthetic gene cluster. We used the annotations produced by antiSMASH 4.1,26 protein family (PFAM) classifications, smCOG, CDs motifs, and predictions of monomers for polyketides and nonribosomal peptides. To include more information about the biosynthetic machinery in each BGC, we further broke down some of the PFAM domains that were most associated with activity into sub-PFAM domains using sequence similarity networks (SSNs), which allow for the clustering of protein sequences into groups with high sequence similarity and often similar function.27,28 These annotations provide information about the types of functional groups or substructures present in the molecule, such as sugars, amines, and halogens. They can also provide information about the general level of oxidation or reduction of the molecule, especially in the case of polyketides where modules require specific domains to reduce the growing polyketide chain (Figure 1B). We used the Resistance Gene Identifier (RGI) 3.2.129 to identify genes that had similarity to known resistance genes, which we expect to be predictive of antibacterial activity. In total, this analysis resulted in 1809 features for 1003 BGCs.
Training and Optimizing Machine Learning Models
We trained and optimized binary classifiers on six different binary classification problems: (1) antibacterial, (2) anti-Gram-positive, (3) anti-Gram-negative, (4) antifungal, antitumor, or cytotoxic (antifungal/antitumor/cytotoxic), (5) antifungal, and (6) antitumor or cytotoxic. We used binary classifiers rather than multilabel classification because many molecules have multiple activities and therefore belong to multiple classes. We chose to make the combined antifungal/antitumor/cytotoxic classifier, by considering a compound active if it has one or more of these activities, since they all indicate activity against eukaryotic cells. We used classifiers available in the Python scikit-learn library to perform classifications. We used random forest with extra-randomized trees, support vector machine (SVM), and logistic regression with regularization because these classifiers enable interpretation of which features are important for predictions. Parameters for each classifier were optimized to maximize average accuracy in a 10-fold cross-validation, a process in which over 10 trials, a different one-tenth of the data is held out from training and used to evaluate classifier accuracy (Figure S1 and Table S2).
The balanced accuracy metric in scikit-learn was used to compare the performance of the optimized classifiers. The balanced accuracy metric is a more accurate reflection of classifier performance in the case of imbalanced datasets, where one class dominates the dataset. Our dataset is very imbalanced for the antifungal (20% of the dataset) and anti-Gram-negative (17% of the dataset) classification problems. We compared each classifier to the performance of a classifier trained on a scrambled version of the features, which represents random guessing. All of the scrambled data classifiers had balanced accuracies of approximately 50%; this is the expected result because binary classifiers can trivially achieve 50% balanced accuracy by always guessing the same label, regardless of how balanced the class labels are (see proof in the Supporting Information). All classifiers, except for the antifungal logistic regression classifier, perform significantly better (p < 0.001 for antifungal classification, p < 0.0001 for all other binary classifiers) than their randomized counterpart on 10-fold cross-validation (Figure 2). Performance was generally independent of the classification method (Figure 2). Classifiers for the antibacterial, anti-Gram-positive, and antifungal/antitumor/cytotoxic classification problems were all highly accurate, with balanced accuracies ranging from 74 to 80% (Figure 2). The accuracy for classifying natural products as cytotoxic or antitumor was slightly less accurate (ranging from 69 to 73%). The anti-Gram-negative and antifungal classifiers are the least accurate (with accuracies between 57 and 64% for antifungal and between 66 and 70% for anti-Gram-negative). We used cross-validation to limit the degree that overfitting influenced our estimated accuracies. However, we did use the entire training set to optimize parameters for the classifiers, so it is likely that there is still some degree of overfitting.
Figure 2.
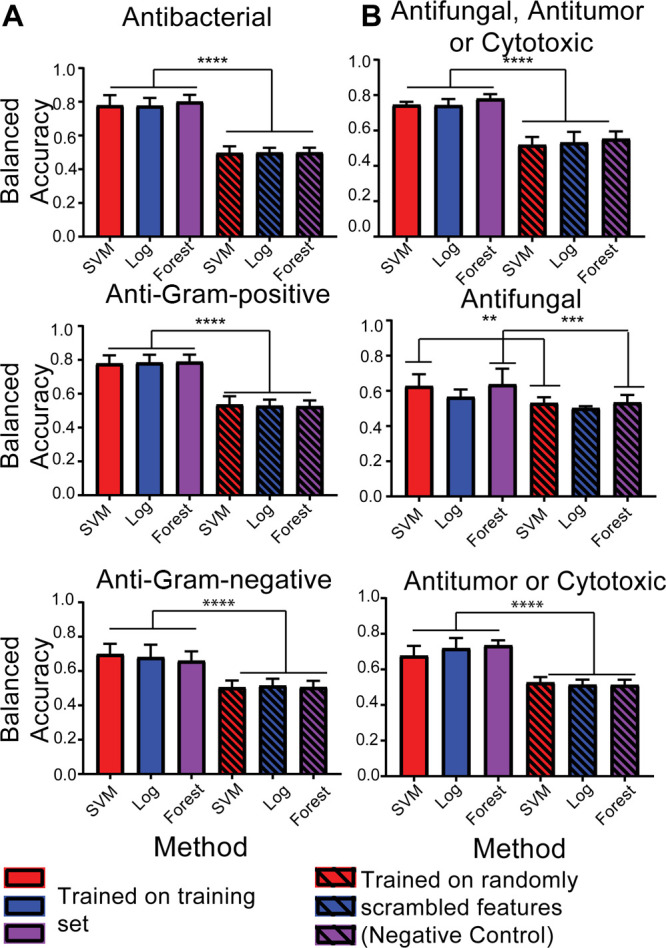
Balanced accuracy of classifiers. Balanced accuracy, or the average of the true positive rate and true negative rate, in a 10-fold cross-validation for (A) antibacterial classifiers and (B) antifungal/antitumor classifiers. Solid bars represent classifiers trained on training dataset; hashed bars are trained on randomly scrambled. Significance was determined using a one-way ANOVA.
In addition to balanced accuracy, we also generated receiver operator characteristic (ROC) curves and precision–recall curves for each classifier. ROC curves plot the false positive rate of a classifier vs the true positive rate, with a higher area under the curve (AROC), indicating that the classifier has a better true to false positive ratio. Our classifiers perform well with AROCs ranging between 0.57 and 0.79 (Figure S2). Precision–recall curves (Figure S3) plot the recall (x axis), or true positives/(true positives + false negatives), vs precision (y axis), or true positives/(true positives + false positives), and are a better way to gauge the accuracy of classifiers applied to unbalanced datasets. A good classifier will have a higher area under the curve, not sacrificing precision for a higher recall. By both the ROC and precision–recall curve metrics, the antibacterial, anti-Gram-positive, and antifungal/antitumor/cytotoxic classifiers perform very well while the anti-Gram-negative and antifungal classifiers do not perform as well. One possible reason for the lower accuracies of the anti-Gram-negative and antifungal classifiers is that natural products with anti-Gram-negative or antifungal activity make up a smaller portion of the dataset, 17% and 20%, respectively. The assumption that if a natural product was never recorded to have an activity then it does not have that activity likely contributes to error in all our classifiers but may especially affect the antifungal classifier because many compounds in the dataset were only tested for antitumor activity and not antifungal activity. Due to historical focus on the discovery of natural products with the activities for which we developed classifiers along with the use of bioactivity guided fractionation, our dataset is likely biased toward active natural products, especially antibacterial molecules. Therefore, it is likely that the probabilities calculated by our method are overestimated. Future work will focus on assessing the degree of this overestimate by experimentally validating the classifiers’ predictions.
Our classifiers performed better than those in Deep-BGC11 for the same activity classification, likely because of our larger training sets and inclusion of additional features such as resistance markers and sub-PFAM domains. Deep-BGC used only a random forest classifier, which is more difficult to interpret in terms of which features are associated with activity than the logistic regression classifier we used to interpret how our classifiers make predictions. Random forests have an importance score that can identify which features are the most important for classification, but the importance score does not specify whether the feature is associated with the presence or absence of activity, which the coefficients of a logistic regression do. Our analysis of the coefficient scores and the inclusion of sub-PFAM domains as features made it possible for us to identify molecular features associated with activity and to understand how our classifiers make their predictions. It is more difficult to compare the accuracy of our classifiers to PRISM 4 because they only reported the ROC curve, but comparison of these curves shows that our antibacterial and antitumor classifiers performed better than PRISM 4,24 while the antifungal classifiers performed approximately as well as PRISM 4. Our improved performance could be due to inclusion of non-biosynthetic features such as resistance markers and transporters or due to more thorough optimization of classifier hyperparameters.
Our classifiers were trained on activity alone and do not make any predictions about the mechanism of action, although some information about mechanism of action is contained in the resistance gene features and PFAM domains. Therefore, it is possible that our classifiers could identify natural products with previously undiscovered mechanisms of action. A recently reported deep learning method for predicting antibiotic activity based on molecular structure successfully used a similar mechanism-blind approach.30 Future experimental work will focus on determining mechanism of action for any active molecules identified by our classifiers in order to test this hypothesis.
The precision–recall curves for our classifiers provide insight into how our classifiers could be used to prioritize BGCs for study. For example, if a researcher is using heterologous expression of a variety of BGC classes, a relatively time-consuming approach, they would want to screen only a few BGCs and would want a few false positives (high precision). Even for the antifungal classifier, the precision–recall curve shows that it is possible to achieve a high precision of 78% (2 in 10 of the BGCs screened would be expected to be a false positive) with a recall of 33% (Figure S3). If the BGCs in question were amenable to high throughput cloning and heterologous expression, then the researcher would be willing to sacrifice some precision to obtain a higher recall. In the case of the antifungal classifier, the precision–recall curve shows that it is possible to achieve a recall of 90% and still have a precision of 30%. In practice, these accuracies might be slightly overestimated due to the similarity of the training and validation data and because the historical use of bioactivity guided fractionation likely inflates the number of active compounds in our training set. Specifically, we expect that the classifiers will be more accurate for BGCs that share features with those in our training set and will therefore be most accurate for BGCs for the major classes of natural products (nonribosomal and ribosomal peptides, polyketides, terpenes, alkaloids, and saccharides). If a BGC for a novel natural product class is discovered, it is possible that our classifiers could still make accurate predictions if it shares tailoring enzymes, transporters, or resistance markers with BGCs in the training set but researchers should treat these predictions with caution. Future work will determine the TPR and FPRs for the classifiers on BGCs for different levels of novelty relative to the training data set.
Different Features Play a Role in Different Prediction Problems
We used the trained logistic regression classifiers to determine which features are most important for each classification problem. Using the coefficients from the logistic regression, we ranked features by their importance to the prediction. Features with a large positive coefficient are highly associated with the activity, while those with a large negative coefficient are strongly associated with not having activity. Because we used elastic net regularization, most of the coefficients are zero or close to zero, indicating that they are not important for making predictions (Figure S4). We chose to analyze only the 50 features with the highest coefficients and 50 with the lowest coefficients. Figure 3 shows pie charts with classification of the top and bottom 50 features for each classification problem; Table S3 lists the specific features.
Figure 3.

PFAM domains associated with activity. Pie charts showing the 50 protein family domains most or least associated with activity, as measured by their coefficients in the logistic regression classifier. A full list of these PFAM domains is available in Table S3.
The types of features associated with activity vary considerably between the different types of activities for which we developed classifiers. For example, for all antibacterial classifiers, many transporter genes were highly associated with activity, accounting for 16–36% of the top 50 features. Transporters are less important for predicting antifungal or antitumor activity, accounting for only 6–14% of the top 50 features (Figure 3). This is probably due to the importance of transporters as a resistance mechanism; a dedicated transporter may be required to provide bacteria resistance to antibacterial secondary metabolites, while antifungal molecules could possibly be transported by a general transporter. Some transporter genes also appear in the bottom 50 features for each classifier, suggesting that only certain transporters are associated with those activities. For example, in the case of the antibacterial classifier, multiple PFAM domains that are part of ABC-transporters are highly associated with antibacterial activity. Conversely, some major facilitator transporters are in the top 50 genes and some are in the bottom 50 (Table S3). More work will be required to understand why certain transporters are associated with antibacterial compounds and others are not. Genes associated with resistance that were not transporters were generally not highly correlated with antibacterial activity. Initially, we were surprised by this result but resistance mechanisms such as drug modification, target modification, or target duplication are all specific to a class of molecules or mechanism of action. Transporters can provide resistance to many molecule classes that act through different mechanisms and are therefore more useful as a general predictor of antibacterial activity.
There were also many biosynthetic genes associated with activity. Seven aminotransferase genes were in the top 50 genes associated with anti-Gram-negative activity (none were in the top 50 for Gram-positive). This is consistent with a known Gram-negative permeability rule, that molecules containing an amine are more likely to accumulate in Gram-negative bacteria.31 Genes associated with RiPP biosynthesis, especially lantipeptides, are strongly associated with antibacterial activity in general and with anti-Gram-positive activity but not with anti-Gram-negative activity or other activities (Figure 3). Lantipeptides often have activity against Gram-positive bacteria but rarely have activity against Gram-negative bacteria,32 fungi,33 or tumor cells. Genes for synthesizing the other major classes of natural products are present in both the top and bottom 50 genes, and it is difficult to draw any conclusions about how these correlations relate to structure–activity relationships. This is unsurprising because many of these genes, such as those responsible for NRPS and PKS synthesis, are responsible for the synthesis of diverse molecules with a variety of functions.
PFAM Sub-Families Give Insight into Molecular Features Associated with Function
While analyzing how PFAM, antiSMASH, and resistance marker features correlated with activity was useful to draw some general conclusions about what types of biosynthetic genes and natural product classes are associated with various activities, they failed to provide much insight into how specific chemical features might contribute to activity. Therefore, we made Sequence Similarity Networks (SSNs) for some of the PFAMs most associated with activity to break them down into sub-PFAMs. SSNs, which group proteins based on pairwise sequence similarity, have been shown to cluster proteins with similar enzymatic activities and substrates.27,28 We colored each SSN based on the activity of the natural product produced by the cluster the domain was from and prioritized SSNs where there were one or more clusters dominated by a single activity. We first examined the SSN network generated for the aminotransferase class I and II PFAM domain (accession number PF00155) because it contained multiple clusters that were associated with different activities (Figure 4A). We then examined the literature to determine the enzymatic activity of each activity-associated cluster. We identified three sub-PFAM domains that were both associated with activity and had a single enzymatic activity (Figure 4A,B).
Figure 4.

Aminotransferase class I and II SSN. (A) Each node in the sequence similarity network represents a domain from a BGC. Nodes are colored based on the activity of the natural product produced by the biosynthetic gene cluster, pink denotes antibacterial activity, blue antifungal, antitumor, or cytotoxic activity, red both activities, green neither activity, and black unknown activity. Clusters are numbered based on the number of sequences assigned to them. For clarity, clusters with three or fewer members are not shown. (B) Cluster 3 is involved in the biosynthesis of phenylglycine and its analogs. Cluster 6 installs an amine on the sugar mycosamine, and cluster 8 is involved in the synthesis of various sugars found in aminoglycoside antibiotics. (C) Examples of nonribosomal peptide antibiotics that contain at least one phenylglycine analog, highlighted in red.
The sixth largest cluster associated with antifungal activity produced the sugar mycosamine.34 BGCs containing this mycosamine-producing sub-PFAM domain are all polyene macrolides with similar structures (Figure S5). The eighth largest cluster is associated with antibacterial activity, and the sub-PFAM domains in this cluster all transfer amines to sugars in aminoglycoside antibiotics, e.g., kanosamine, part of the antibiotic kanamycin (Figure 4B).35 Therefore clusters 6 and 8 both produce substructures of a single class of natural product. In the case of the polyene macrolides, the mycosamine sugar has been shown to be essential for activity, likely through its interactions with sterols in the fungal membrane.36 This is encouraging as it shows that the SSN approach can rediscover classes of active molecules and identify the biosynthetic machinery that installs the groups that are essential for activity.
Unlike clusters 6 and 8, cluster 3, whose members produce l-phenylglycine and its hydroxylated analogs, hydroxyphenylglycine (Hpg) and dihydroxyphenylglycine (Dpg), was not confined to one class of molecule.37,38 All the BGCs in our database containing phenylglycine transaminase produced natural products with antibacterial activity, but they produced molecules with very divergent structures (Figure 4C). In line with this observation, these natural products also do not all act through the same mechanism of action. For example, glycopeptide antibiotics such as vancomycin inhibit cell wall synthesis by binding to the d-Ala-d-Ala terminal of growing peptide chains in the cell wall, preventing cell wall remodeling,39 while nocardicin A is a β-lactam that inhibits cell wall synthesis by binding to penicillin binding proteins,40 and pristinamycin inhibits protein synthesis by binding to the ribosome.41 This suggests that, unlike mycosamine in polyenes, phenylglycine supports activity in a general way rather than through a specific mechanism or binding interaction. One possible explanation for phenylglycine’s association with activity is that unlike proteinogenic amino acids, phenylglycine does not have a β-carbon, so it has fewer rotatable bonds than proteinogenic aromatic amino acids like phenylalanine.38 Therefore, phenylglycine-containing peptides should be more rigid than they would be if they contained a proteinogenic amino acid instead, reducing the entropic cost for binding to a target. There is evidence that the structural rigidity of glycopeptide antibiotics are thought to be especially important for their activity.37,42 Alanine scanning studies have shown that two Hpg residues in the antibiotic feglymycin and four Hpg residues in ramoplanin are important for activity.43,44
However, it is not clear why phenylglycine appears to be associated only with antibacterial activity and no other activities. It is possible that the reason for this is evolutionary, that once a bacteria’s competitor evolved resistance to an ancient phenylglycine-containing antibiotic, evolutionary pressure maintained the antibacterial function of the molecule but altered it to work through an alternative mechanism. Interestingly, many of the phenylglycine-containing antibiotics, including glycopeptide antibiotics, nocardicin, ramoplanin, and feglymycin, act through inhibiting cell wall formation through different targets.39,40,44,45 Additional study will be required to determine if phenylglycine transaminases are useful as a handle for antibacterial discovery and to determine the mechanism by which phenylglycine and its analogs support activity.
We examined 46 other SSNs for trends similar those observed for the aminotransferase class I and II PFAM domain. We found that five SSNs had at least one cluster associated with activity and, as determined by literature search, had an enzymatic activity distinct from other enzymes not in the cluster. The functional groups produced by these enzymes are summarized in Figure 5, and the SSNs are shown in Figures S6–S9. While five of these functional groups, including the previously mentioned mycosamine and aminoglycoside sugars, were restricted to one class of active molecule, another 10 of these were not specific to one natural product scaffold (Figure 5). The sub-PFAM domains that produce these functional groups could be useful handles for discovering novel bioactive natural product scaffolds. It is likely that most of these functional groups are associated with activity because they change general properties of the molecule to improve its activity. For example, we identified one sub-PFAM domain that N-methylates the backbone or side chain of non-ribosomal peptides. It is known that N-methylation of peptides can improve their cell permeability in mammalian cells, increase protease resistance, and stabilize the conformation of cyclic peptides.46−48 It is likely that many of the other functional groups identified through this analysis also support activity by making the molecule more permeable to the target organism’s membrane or improve binding to target proteins. Bacteria may have evolved these sub-PFAM domains to improve the activity of their natural products analogous to how medicinal chemists use specific chemical modifications to improve the drug-like properties of a molecule. These activity-associated domains could both be used to identify new BGCs that produce active molecules and to engineer known natural products to improve their activity.
Figure 5.

Summary of molecular features associated with activity. List of molecular features associated with (A) antibacterial, (B) antifungal, antitumor, or cytotoxic, and (C) antibacterial, antifungal, antitumor, or cytotoxic activities identified through analysis of SSNs of PFAM domains from BGCs. Yellow boxes indicate features that were not associated with a single molecular scaffold or mechanism of action. These sub-PFAM domains could be useful handles for the discovery of bioactive compounds with novel scaffolds.
Classifiers Perform Well on Holdout Set BGCs with Some Similarity to BGCs in the Training Set
To assess how well our classifiers will perform on novel BGCs, we applied it to a holdout set of BGCs that were not used during the development and optimization of the classifiers. A holdout set is useful for assessing the algorithm because it ensures that the optimization process did not produce a classifier that works only on the training set and will not work as well on other data. To generate a holdout set, we used BGCs that were added to the MiBIG database when it was upgraded from version 1.4 to version 2.0.49 Our holdout set consisted of 258 BGCs for which we could find information about the activity of the associated natural product(s) (Table S5).
To assess the accuracy of the classifiers on the holdout set, we split the holdout set into subsets based on how similar the BGCs were to BGCs in the training set. To do this, we used the “knownclusterblast” feature of antiSMASH,26 which scores how similar BGCs are to known clusters. We split the holdout set into six subsets: clusters not recognized by antiSMASH and therefore lacking a knownclusterblast score (ND) and maximum percentage of genes that show similarity to genes in a single training set cluster of 0, (0–25], (25–50], (50–75], and (75–100] (Figure 6 and Figures S10–S12). The balanced accuracy increased with the similarity of all genes for almost all classifiers. This means that while the classifiers cannot accurately predict the activity for a natural product when its BGC has no similarity to any training set BGC, as the similarity increases, so does the accuracy of the prediction. All classifiers, except the antifungal SVM classifier, perform well on the 75–100 holdout set with balanced accuracies ranging from 67 to 98% (Figure 6 and Figures S10–S12). Most classifiers also perform well on the 50–75 holdout set, with accuracies as high as 85%. This indicates that if a novel cluster has a knownclusterblast similarity score of at least 50 with a BGC in the training set, then the predictions made by the classifiers are fairly accurate. We expect that the addition of more BGCs to the training set would increase its diversity and therefore improve the accuracy of predictions made by the classifiers.
Figure 6.

Performance of classifiers on holdout set. (A) Balanced accuracy of random forest antibacterial, antifungal/antitumor/cytotoxic, and antifungal classifiers applied to subsets of the holdout set, split based on the maximum known cluster BLAST similarity score to BGCs in the training set. ND indicates the subset where antiSMASH did not identify the cluster and therefore did not determine a known cluster BLAST score. (B) Comparison of aurantinin B50 and bacillaene51 structures and BGCs; genes that were identified as similar by known cluster BLAST are colored in the same color. (C) Comparison of aurantimycin A53 and polyoxypeptin54 structures and BGCs; genes that were identified as similar by known cluster BLAST are colored in the same color.
The molecules produced by BGCs with similarity scores higher than 50 can still be quite different. One example from the holdout set that demonstrates this is the aurantinin cluster.50 The closest match to aurantinin in the training set is the bacillaene cluster,51 with a similarity score of 57%. Despite the high similarity, aurantinin and bacillaene have quite different molecular structures (Figure 6B). Aurantinin has multiple cycles and is glycosylated,50 while bacillaene is linear, not glycosylated, and contains peptides in addition to the main polyketide backbone.51 Despite their different structures, aurantinin and bacillaene both have antibacterial activity.50,52 The classifiers were able to successfully predict that aurantinin has antibacterial activity (60% average probability). We also looked for examples in the holdout set that were very similar structurally to a molecule from the training set but that had different activities. We found that aurantimycin53 is similar both in structure and BGC sequence (67% similarity score) to polyoxypeptin54 (Figure 6C). Despite their similarity, aurantimycin is documented to have both antibacterial and antitumor activity,55 both of which were correctly predicted by the classifier, with probabilities of 58% for antibacterial activity and 85% for antifungal, antitumor, or cytotoxic activity. In the literature we reviewed, polyoxypeptin was only documented to have antitumor activity.56 Together, these examples suggest that the classifiers are not merely relying on the most similar cluster to make their predictions and are able to distinguish between similar BGCs that produce molecules with different activities. Therefore, we expect that the model will perform well on novel BGCs if they have some degree of similarity to BGCs in the training set and that the similarity required for accurate predictions does not mean that the molecules produced by the BGCs will themselves be similar.
Conclusions
We developed a bioinformatics method for predicting biological activities, antibacterial, antifungal, or antitumor, from biosynthetic gene clusters. The ability to predict natural product activity from BGC sequences alone will make it possible to prioritize clusters that are most likely to produce molecules with a desired activity, minimizing the number of times the bottlenecks of natural product discovery need to be encountered to discover a novel active compound. Our approach used machine learning to generate classifiers, which have balanced accuracies of at least 57% and up to 79%. Classifiers’ performance depends on training sets, and they work best for problems where there are many positive and negative examples in the dataset, which is why we focused on antibacterial activity. The method does not perform as well when molecules with the desired activity sparsely populate the dataset. In our analysis, this feature of machine learning is most pronounced for the antifungal classifiers, for which our method has the lowest accuracy. It is likely that increasing the size of the dataset will further improve the accuracy of all classifiers, especially the antifungal and anti-Gram-negative classifiers. By examining which features of BGCs are associated with activity, we gained new insights into which molecular features of natural products are associated with their activity. Some of these observations were consistent with previously discovered structure–activity relationships, for example, that molecules with an amine are more likely to accumulate in Gram-negative bacteria31 or that mycosamine is essential for the activity of antifungal polyenes.36 This concordance demonstrates that trained classifiers can be used to identify structural features associated with activity. Future work will be focused on validating our machine learning approach as a tool to accelerate functional natural product discovery and on investigating the connection between molecular features identified by our classifiers and activity.
Methods
Assembly of Training Dataset
We assembled a training dataset from BGCs available from the MiBIG (version 1.4) database.25 We then searched the literature for each natural product to determine if it had documented antibacterial, antifungal, antitumor, or cytotoxic activity. If a natural product was documented to not have a given activity, we recorded it as not having the activity in the dataset. Unfortunately, we found that reports rarely note the absence of activity. Therefore, we also assumed that a molecule did not have a given activity if there were no reports stating that it had that activity and if it was described as having a different activity (e.g., activity against a different organism, siderophore activity, ionophore activity, antioxidant activity, etc.). If the molecule had no recorded activity or function and no assays specifically showing its inactivity, we assumed that it was not tested for activity and we excluded it from our dataset.
Selection of Features
We ran antiSMASH 4.126 on the BGCs in the dataset and wrote a Python script to extract PFAM, CDS motif, smCOG, and polyketide and non-ribosomal peptide monomer prediction annotations of BGCs from an antiSMASH output file. We also ran RGI 3.2.129 on all clusters and wrote a script to extract resistance marker features. Features were only included if they appeared at least five times in the dataset. Further details on settings used to run antiSMASH and RGI as well as more details on the scripts are available in the Supporting Information. Scripts are available as a Jupyter notebook here: https://github.com/allie-walker/Natural-product-function. Since our initial development of the classification algorithm, new versions of antiSMASH and RGI became available. The command line version of our prediction algorithm, also available on GitHub, accepts input files from antiSMASH 5 and RGI 5 as well as the versions used in this paper.
Sequence Similarity Networks of PFAM Domains
To add additional information about the biosynthetic capabilities of the BGCs to the features, we broke down PFAM domains into sub-PFAM domains using the SSN algorithm on the EFI-EST webserver.28 We made SSNs for the top 30 PFAM domains associated with antibacterial or antifungal/antitumor/cytotoxic activity as measured by χ2 value, excluding any PFAM domains that appeared in fewer than 35 training set BGCs. We applied the EFI-EST tool to the sequences of selected PFAM domains that appeared in our training set BGCs. We chose alignment scores for clustering cutoffs that would result in distinct clusters in the SSN (listed in Table S4). We designated each cluster that contained more than five sequences and occurred in at least three BGCs a sub-PFAM domain and included it as a feature for training. SSN networks shown in Figure 4 and Figures S6–S9 were visualized using Cytoscape.57
Optimization and Training of Classifiers
We used modules from the scikit-learn Python library58 to perform all machine learning. Before training regression and SVM classifiers, we scaled all features using a min max scaler (scikit-learn MinMaxScaler). To assess which parameters produced the best results, we measured the average accuracy for each parameter set over a 10-fold cross-validation, where one-tenth of the data was held out from training in each trial and then used to assess accuracy. For the logistic regression classifier, we used the Stochastic Gradient Decent Classifier module (SGDClassifier) from scikit-learn with loss=’log’ and reg=’elasticnet’. We then tested all combinations of α = 0.5, 0.3, 0.2, 0.1, 10–2, 10–3, 10–4, 10–5, and 10–6 and l1 ratios = 0.5, 0.2, 0.1, 0.05, 10–2, 10–3, and 10–4 (Figure S1A and Table S2). For the SVM classifier, we used the SVC module from scikit-learn. We tested the accuracy of the SVM with linear and rbf kernels and tested the C (l2 regularization parameter) values = 100, 10, 1, 0.5, 0.1, and 0.01. For the rbf kernel, we also tested kernel coefficients, γ = 0.001, 0.01, 0.1, 1, and 10 (Figure S1B and Table S2). To train random forest classifiers, we used the ExtraTreesClassifier module from scikit-learn with bootstrap = True, max_features=’auto’, and criterion=’gini’. We then tested all combinations of n_estimators = 1, 5, 10, 15, 20, 25, 50, and 100 and max_depth = 10, 20, 50, 100, 1000, and None (Figure S1C). We then chose the combination of parameters with the highest average accuracy for each classification problem for all subsequent analysis (Table S2).
Calculating Accuracy Metrics for Trained Classifiers
Methods from the scikit-learn metrics module were used to analyze the accuracy of our classifiers. The methods we used were balanced_accuracy_score, roc_curve, roc_auc_score, and precision_recall_curve. For the balanced accuracy and AROC scores, we took the average of scores from a 10-fold cross-validation. For plots showing ROC and precision recall curves, we displayed a curve from one trial of the cross-validation. Statistical significance of improvement over random guessing was determined in PRISM using and one-way ANOVA test.
Acknowledgments
This work was funded by NIH Grant R01AT009874 (J.C.) and NIH post-doctoral fellowship, F32GM128267 (A.S.W.). We thank Prof. Laurent Lessard for helpful discussions on machine learning techniques, Friederike Biermann for testing the code associated with this manuscript, and Harvard Medical School Research Computing for computational resources.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c01304.
Additional information on the computational methods used in this study along with the supporting figures and tables referenced in the text (PDF)
The authors declare no competing financial interest.
Notes
All input files and code required to reproduce the results in this paper and make predictions on novel BGCs are available on our GitHub page: https://github.com/allie-walker/Natural-product-function, and all third-party software we used to generate features of BGCs are also freely available at https://antismash.secondarymetabolites.org/#!/start, https://card.mcmaster.ca/analyze/rgi, and https://efi.igb.illinois.edu/efi-est/.
Supplementary Material
References
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- Kingston D. G. Modern natural products drug discovery and its relevance to biodiversity conservation. J. Nat. Prod. 2011, 74, 496–511. 10.1021/np100550t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genilloud O. Actinomycetes: still a source of novel antibiotics. Nat. Prod. Rep. 2017, 34, 1203–1232. 10.1039/C7NP00026J. [DOI] [PubMed] [Google Scholar]
- Kong D. X.; Guo M. Y.; Xiao Z. H.; Chen L. L.; Zhang H. Y. Historical variation of structural novelty in a natural product library. Chem. Biodiversity 2011, 8, 1968–1977. 10.1002/cbdv.201100156. [DOI] [PubMed] [Google Scholar]
- Mohimani H.; Gurevich A.; Shlemov A.; Mikheenko A.; Korobeynikov A.; Cao L.; Shcherbin E.; Nothias L. F.; Dorrestein P. C.; Pevzner P. A. Dereplication of microbial metabolites through database search of mass spectra. Nat. Commun. 2018, 9, 4035. 10.1038/s41467-018-06082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieckmann R.; Graeber I.; Kaesler I.; Szewzyk U.; von Döhren H. Rapid screening and dereplication of bacterial isolates from marine sponges of the sula ridge by intact-cell-MALDI-TOF mass spectrometry (ICM-MS). Appl. Microbiol. Biotechnol. 2005, 67, 539–548. 10.1007/s00253-004-1812-2. [DOI] [PubMed] [Google Scholar]
- Bradshaw J.; Butina D.; Dunn A. J.; Green R. H.; Hajek M.; Jones M. M.; Lindon J. C.; Sidebottom P. J. A rapid and facile method for the dereplication of purified natural products. J. Nat. Prod. 2001, 64, 1541–1544. 10.1021/np010284g. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Idelbayev Y.; Roberts N.; Tao Y.; Nannapaneni Y.; Duggan B. M.; Min J.; Lin E. C.; Gerwick E. C.; Cottrell G. W.; Gerwick W. H. Small Molecule Accurate Recognition Technology (SMART) to Enhance Natural Products Research. Sci. Rep. 2017, 7, 14243. 10.1038/s41598-017-13923-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K.; Shaw S.; Steinke K.; Villebro R.; Ziemert N.; Lee S. Y.; Medema M. H.; Weber T. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. 10.1093/nar/gkz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinnider M. A.; Merwin N. J.; Johnston C. W.; Magarvey N. A. PRISM 3: expanded prediction of natural product chemical structures from microbial genomes. Nucleic Acids Res. 2017, 45, W49–W54. 10.1093/nar/gkx320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan G. D.; Prihoda D.; Palicka A.; Soukup J.; Klempir O.; Rampula L.; Durcak J.; Wurst M.; Kotowski J.; Chang D.; Wang R.; Piizzi G.; Temesi G.; Hazuda D. J.; Woelk C. H.; Bitton D. A. A deep learning genome-mining strategy for biosynthetic gene cluster prediction. Nucleic Acids Res. 2019, 47, e110. 10.1093/nar/gkz654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaldi N.; Seifuddin F. T.; Turner G.; Haft D.; Nierman W. C.; Wolfe K. H.; Fedorova N. D. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Biol. 2010, 47, 736–741. 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Yi G. S. PKMiner: a database for exploring type II polyketide synthases. BMC Microbiol. 2012, 12, 169. 10.1186/1471-2180-12-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann B. O.; Ravel J. Chapter 8. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009, 458, 181–217. 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- Agrawal P.; Khater S.; Gupta M.; Sain N.; Mohanty D. RiPPMiner: a bioinformatics resource for deciphering chemical structures of RiPPs based on prediction of cleavage and cross-links. Nucleic Acids Res. 2017, 45, W80–W88. 10.1093/nar/gkx408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel A. J.; de Jong A.; Song C.; Viel J. H.; Kok J.; Kuipers O. P. BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. 10.1093/nar/gky383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietz J. I.; Schwalen C. J.; Patel P. S.; Maxson T.; Blair P. M.; Tai H. C.; Zakai U. I.; Mitchell D. A. A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat. Chem. Biol. 2017, 13, 470–478. 10.1038/nchembio.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Los Santos E. L. C. NeuRiPP: Neural network identification of RiPP precursor peptides. Sci. Rep. 2019, 9, 13406. 10.1038/s41598-019-49764-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K.; Pascal Andreu V.; de Los Santos E. L. C.; Del Carratore F.; Lee S. Y.; Medema M. H.; Weber T. The antiSMASH database version 2: a comprehensive resource on secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2019, 47, D625–D630. 10.1093/nar/gky1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K.; Shaw S.; Kautsar S. A.; Medema M. H.; Weber T. The antiSMASH database version 3: increased taxonomic coverage and new query features for modular enzymes. Nucleic Acids Res. 2021, 49, D639–D643. 10.1093/nar/gkaa978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Li J.; Millán-Aguiñaga N.; Zhang J. J.; O’Neill E. C.; Ugalde J. A.; Jensen P. R.; Mantovani S. M.; Moore B. S. Identification of Thiotetronic Acid Antibiotic Biosynthetic Pathways by Target-directed Genome Mining. ACS Chem. Biol. 2015, 10, 2841–2849. 10.1021/acschembio.5b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcock B. P.; Raphenya A. R.; Lau T. T. Y.; Tsang K. K.; Bouchard M.; Edalatmand A.; Huynh W.; Nguyen A. V.; Cheng A. A.; Liu S.; Min S. Y.; Miroshnichenko A.; Tran H. K.; Werfalli R. E.; Nasir J. A.; Oloni M.; Speicher D. J.; Florescu A.; Singh B.; Faltyn M.; Hernandez-Koutoucheva A.; Sharma A. N.; Bordeleau E.; Pawlowski A. C.; Zubyk H. L.; Dooley D.; Griffiths E.; Maguire F.; Winsor G. L.; Beiko R. G.; Brinkman F. S. L.; Hsiao W. W. L.; Domselaar G. V.; McArthur A. G. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. 10.1093/nar/gkz935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanjary M.; Kronmiller B.; Adamek M.; Blin K.; Weber T.; Huson D.; Philmus B.; Ziemert N. The Antibiotic Resistant Target Seeker (ARTS), an exploration engine for antibiotic cluster prioritization and novel drug target discovery. Nucleic Acids Res. 2017, 45, W42–W48. 10.1093/nar/gkx360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinnider M. A.; Johnston C. W.; Gunabalasingam M.; Merwin N. J.; Kieliszek A. M.; MacLellan R. J.; Li H.; Ranieri M. R. M.; Webster A. L. H.; Cao M. P. T.; Pfeifle A.; Spencer N.; To Q. H.; Wallace D. P.; Dejong C. A.; Magarvey N. A. Comprehensive prediction of secondary metabolite structure and biological activity from microbial genome sequences. Nat. Commun. 2020, 11, 6058. 10.1038/s41467-020-19986-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema M. H.; Kottmann R.; Yilmaz P.; Cummings M.; Biggins J. B.; Blin K.; de Bruijn I.; Chooi Y. H.; Claesen J.; Coates R. C.; Cruz-Morales P.; Duddela S.; Dusterhus S.; Edwards D. J.; Fewer D. P.; Garg N.; Geiger C.; Gomez-Escribano J. P.; Greule A.; Hadjithomas M.; Haines A. S.; Helfrich E. J.; Hillwig M. L.; Ishida K.; Jones A. C.; Jones C. S.; Jungmann K.; Kegler C.; Kim H. U.; Kotter P.; Krug D.; Masschelein J.; Melnik A. V.; Mantovani S. M.; Monroe E. A.; Moore M.; Moss N.; Nutzmann H. W.; Pan G.; Pati A.; Petras D.; Reen F. J.; Rosconi F.; Rui Z.; Tian Z.; Tobias N. J.; Tsunematsu Y.; Wiemann P.; Wyckoff E.; Yan X.; Yim G.; Yu F.; Xie Y.; Aigle B.; Apel A. K.; Balibar C. J.; Balskus E. P.; Barona-Gómez F.; Bechthold A.; Bode H. B.; Borriss R.; Brady S. F.; Brakhage A. A.; Caffrey P.; Cheng Y. Q.; Clardy J.; Cox R. J.; De Mot R.; Donadio S.; Donia M. S.; van der Donk W. A.; Dorrestein P. C.; Doyle S.; Driessen A. J.; Ehling-Schulz M.; Entian K. D.; Fischbach M. A.; Gerwick L.; Gerwick W. H.; Gross H.; Gust B.; Hertweck C.; Höfte M.; Jensen S. E.; Ju J.; Katz L.; Kaysser L.; Klassen J. L.; Keller N. P.; Kormanec J.; Kuipers O. P.; Kuzuyama T.; Kyrpides N. C.; Kwon H. J.; Lautru S.; Lavigne R.; Lee C. Y.; Linquan B.; Liu X.; Liu W.; Luzhetskyy A.; Mahmud T.; Mast Y.; Mendez C.; Metsä-Ketelä M.; Micklefield J.; Mitchell D. A.; Moore B. S.; Moreira L. M.; Müller R.; Neilan B. A.; Nett M.; Nielsen J.; O’Gara F.; Oikawa H.; Osbourn A.; Osburne M. S.; Ostash B.; Payne S. M.; Pernodet J. L.; Petricek M.; Piel J.; Ploux O.; Raaijmakers J. M.; Salas J. A.; Schmitt E. K.; Scott B.; Seipke R. F.; Shen B.; Sherman D. H.; Sivonen K.; Smanski M. J.; Sosio M.; Stegmann E.; Sussmuth R. D.; Tahlan K.; Thomas C. M.; Tang Y.; Truman A. W.; Viaud M.; Walton J. D.; Walsh C. T.; Weber T.; van Wezel G. P.; Wilkinson B.; Willey J. M.; Wohlleben W.; Wright G. D.; Ziemert N.; Zhang C.; Zotchev S. B.; Breitling R.; Takano E.; Glöckner F. O. Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. 10.1038/nchembio.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K.; Wolf T.; Chevrette M. G.; Lu X.; Schwalen C. J.; Kautsar S. A.; Suarez Duran H. G.; de Los Santos E. L. C.; Kim H. U.; Nave M.; Dickschat J. S.; Mitchell D. A.; Shelest E.; Breitling R.; Takano E.; Lee S. Y.; Weber T.; Medema M. H. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017, 45, W36–W41. 10.1093/nar/gkx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson H. J.; Morris J. H.; Ferrin T. E.; Babbitt P. C. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS One 2009, 4, e4345 10.1371/journal.pone.0004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlt J. A.; Bouvier J. T.; Davidson D. B.; Imker H. J.; Sadkhin B.; Slater D. R.; Whalen K. L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854, 1019–1037. 10.1016/j.bbapap.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia B.; Raphenya A. R.; Alcock B.; Waglechner N.; Guo P.; Tsang K. K.; Lago B. A.; Dave B. M.; Pereira S.; Sharma A. N.; Doshi S.; Courtot M.; Lo R.; Williams L. E.; Frye J. G.; Elsayegh T.; Sardar D.; Westman E. L.; Pawlowski A. C.; Johnson T. A.; Brinkman F. S.; Wright G. D.; McArthur A. G. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes J. M.; Yang K.; Swanson K.; Jin W.; Cubillos-Ruiz A.; Donghia N. M.; MacNair C. R.; French S.; Carfrae L. A.; Bloom-Ackermann Z.; Tran V. M.; Chiappino-Pepe A.; Badran A. H.; Andrews I. W.; Chory E. J.; Church G. M.; Brown E. D.; Jaakkola T. S.; Barzilay R.; Collins J. J. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 181, 475–483. 10.1016/j.cell.2020.04.001. [DOI] [PubMed] [Google Scholar]
- Richter M. F.; Drown B. S.; Riley A. P.; Garcia A.; Shirai T.; Svec R. L.; Hergenrother P. J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field D.; Cotter P. D.; Hill C.; Ross R. P. Bioengineering Lantibiotics for Therapeutic Success. Front. Microbiol. 2015, 6, 1363. 10.3389/fmicb.2015.01363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr K. I.; Volz C.; Jansen R.; Wray V.; Hoffmann J.; Bernecker S.; Wink J.; Gerth K.; Stadler M.; Müller R. Pinensins: the first antifungal lantibiotics. Angew. Chem., Int. Ed. 2015, 54, 11254–11258. 10.1002/anie.201500927. [DOI] [PubMed] [Google Scholar]
- Nedal A.; Sletta H.; Brautaset T.; Borgos S. E. F.; Sekurova O. N.; Ellingsen T. E.; Zotchev S. B. Analysis of the mycosamine biosynthesis and attachment genes in the nystatin biosynthetic gene cluster of Streptomyces noursei ATCC 11455. Appl. Environ. Microbiol. 2007, 73, 7400–7407. 10.1128/AEM.01122-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo F.; Kitayama Y.; Miyanaga A.; Hirayama A.; Eguchi T. Biochemical and Structural Analysis of a Dehydrogenase, KanD2, and an Aminotransferase, KanS2, That Are Responsible for the Construction of the Kanosamine Moiety in Kanamycin Biosynthesis. Biochemistry 2020, 59, 1470–1473. 10.1021/acs.biochem.0c00204. [DOI] [PubMed] [Google Scholar]
- Palacios D. S.; Dailey I.; Siebert D. M.; Wilcock B. C.; Burke M. D. Synthesis-enabled functional group deletions reveal key underpinnings of amphotericin B ion channel and antifungal activities. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6733–6738. 10.1073/pnas.1015023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard B. K.; Thomas M. G.; Walsh C. T. Biosynthesis of L-p-hydroxyphenylglycine, a non-proteinogenic amino acid constituent of peptide antibiotics. Chem. Biol. 2000, 7, 931–942. 10.1016/S1074-5521(00)00043-0. [DOI] [PubMed] [Google Scholar]
- Al Toma R. S.; Brieke C.; Cryle M. J.; Süssmuth R. D. Structural aspects of phenylglycines, their biosynthesis and occurrence in peptide natural products. Nat. Prod. Rep. 2015, 32, 1207–1235. 10.1039/C5NP00025D. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M.; Jones P. G.; Kennard O.; Williams D. H.; Smith G. A. Structure of vancomycin and its complex with acetyl-D-alanyl-D-alanine. Nature 1978, 271, 223–225. 10.1038/271223a0. [DOI] [PubMed] [Google Scholar]
- Berenguer J.; De Pedro M. A.; Vazquez D. V. Interaction of nocardicin A with the penicillin-binding proteins of Escherichia coli in intact cells and in purified cell envelopes. Eur. J. Biochem. 1982, 126, 155–159. 10.1111/j.1432-1033.1982.tb06760.x. [DOI] [PubMed] [Google Scholar]
- Tenson T.; Lovmar M.; Ehrenberg M. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J. Mol. Biol. 2003, 330, 1005–1014. 10.1016/S0022-2836(03)00662-4. [DOI] [PubMed] [Google Scholar]
- Fowler B. S.; Laemmerhold K. M.; Miller S. J. Catalytic site-selective thiocarbonylations and deoxygenations of vancomycin reveal hydroxyl-dependent conformational effects. J. Am. Chem. Soc. 2012, 134, 9755–9761. 10.1021/ja302692j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänchen A.; Rausch S.; Landmann B.; Toti L.; Nusser A.; Süssmuth R. D. Alanine scan of the peptide antibiotic feglymycin: assessment of amino acid side chains contributing to antimicrobial activity. ChemBioChem 2013, 14, 625–632. 10.1002/cbic.201300032. [DOI] [PubMed] [Google Scholar]
- Nam J.; Shin D.; Rew Y.; Boger D. L. Alanine scan of [L-Dap(2)]ramoplanin A2 aglycon: assessment of the importance of each residue. J. Am. Chem. Soc. 2007, 129, 8747–8755. 10.1021/ja068573k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch S.; Hänchen A.; Denisiuk A.; Löhken M.; Schneider T.; Süssmuth R. D. Feglymycin is an inhibitor of the enzymes MurA and MurC of the peptidoglycan biosynthesis pathway. ChemBioChem 2011, 12, 1171–1173. 10.1002/cbic.201100120. [DOI] [PubMed] [Google Scholar]
- Bockus A. T.; Schwochert J. A.; Pye C. R.; Townsend C. E.; Sok V.; Bednarek M. A.; Lokey R. S. Going Out on a Limb: Delineating The Effects of beta-Branching, N-Methylation, and Side Chain Size on the Passive Permeability, Solubility, and Flexibility of Sanguinamide A Analogues. J. Med. Chem. 2015, 58, 7409–7418. 10.1021/acs.jmedchem.5b00919. [DOI] [PubMed] [Google Scholar]
- Fiacco S. V.; Roberts R. W. N-Methyl scanning mutagenesis generates protease-resistant G protein ligands with improved affinity and selectivity. ChemBioChem 2008, 9, 2200–2203. 10.1002/cbic.200800208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee J.; Gilon C.; Hoffman A.; Kessler H. N-methylation of peptides: a new perspective in medicinal chemistry. Acc. Chem. Res. 2008, 41, 1331–1342. 10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]
- Kautsar S. A.; Blin K.; Shaw S.; Navarro-Muñoz J. C.; Terlouw B. R.; van der Hooft J. J. J.; van Santen J. A.; Tracanna V.; Suarez Duran H. G.; Pascal Andreu V.; Selem-Mojica N.; Alanjary M.; Robinson S. L.; Lund G.; Epstein S. C.; Sisto A. C.; Charkoudian L. K.; Collemare J.; Linington R. G.; Weber T.; Medema M. H. MIBiG 2.0: a repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020, 48, D454–D458. 10.1093/nar/gkz882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Zhu X.; Cao M.; Wang C.; Zhang C.; Lu Z.; Lu F. Genomics-Inspired Discovery of Three Antibacterial Active Metabolites, Aurantinins B, C, and D from Compost-Associated Bacillus subtilis fmb60. J. Agric. Food Chem. 2016, 64, 8811–8820. 10.1021/acs.jafc.6b04455. [DOI] [PubMed] [Google Scholar]
- Butcher R. A.; Schroeder F. C.; Fischbach M. A.; Straight P. D.; Kolter R.; Walsh C. T.; Clardy J. The identification of bacillaene, the product of the PksX megacomplex in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 1506–1509. 10.1073/pnas.0610503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P. S.; Huang S.; Fisher S.; Pirnik D.; Aklonis C.; Dean L.; Meyers E.; Fernandes P.; Mayerl F. Bacillaene, a novel inhibitor of procaryotic protein synthesis produced by Bacillus subtilis: production, taxonomy, isolation, physico-chemical characterization and biological activity. J. Antibiot. 1995, 48, 997–1003. 10.7164/antibiotics.48.997. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Wang L.; Wan D.; Qi J.; Gong R.; Deng Z.; Chen W. Characterization of the aurantimycin biosynthetic gene cluster and enhancing its production by manipulating two pathway-specific activators in Streptomyces aurantiacus JA 4570. Microb. Cell Fact. 2016, 15, 160. 10.1186/s12934-016-0559-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y.; Wang Y.; Huang T.; Tao M.; Deng Z.; Lin S. Identification and characterization of the biosynthetic gene cluster of polyoxypeptin A, a potent apoptosis inducer. BMC Microbiol. 2014, 14, 30. 10.1186/1471-2180-14-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräfe U.; Schlegel R.; Ritzau M.; Ihn W.; Dornberger K.; Stengel C.; Fleck W. F.; Gutsche W.; Härtl A.; Paulus E. F. Aurantimycins, new depsipeptide antibiotics from Streptomyces aurantiacus IMET 43917. Production, isolation, structure elucidation, and biological activity. J. Antibiot. 1995, 48, 119–125. 10.7164/antibiotics.48.119. [DOI] [PubMed] [Google Scholar]
- Umezawa K.; Nakazawa K.; Ikeda Y.; Naganawa H.; Kondo S. Polyoxypeptins A and B Produced by Streptomyces: Apoptosis-Inducing Cyclic Depsipeptides Containing the Novel Amino Acid (2S,3R)-3-Hydroxy-3-methylproline. J. Org. Chem. 1999, 64, 3034–3038. 10.1021/jo981512n. [DOI] [PubMed] [Google Scholar]
- Shannon P.; Markiel A.; Ozier O.; Baliga N. S.; Wang J. T.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedergosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V.; Vanderplas J.; Passos A.; Cournapeau D.; Brucher M.; Perrot M.; Duchesnay E. Scikit-learn: Machine Learning in Python. J. Mach. Learn. 2011, 12, 2825–2830. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.