

Abstract

Efforts directed at curtailing the bioavailability of intracellular iron could lead to the development of broad-spectrum anticancer drugs given the metal’s role in cancer proliferation and metastasis. Human ribonucleotide reductase (RNR), the key enzyme responsible for synthesizing the building blocks of DNA replication and repair, depends on Fe binding at its R2 subunit to activate the catalytic R1 subunit. This work explores an intracellular iron chelator transmetalative approach to inhibit RNR using the titanium(IV) chemical transferrin mimetic (cTfm) compounds Ti(HBED) and Ti(Deferasirox)2. Whole-cell EPR studies reveal that the compounds can effectively attenuate RNR activity though seemingly causing different changes to the labile iron pool that may account for differences in their potency against cells. Studies of Ti(IV) interactions with the adenosine nucleotide family at pH 7.4 reveal strong metal binding and extensive phosphate hydrolysis, which suggest the capacity of the metal to disturb the nucleotide substrate pool of the RNR enzyme. By decreasing intracellular Fe bioavailability and altering the nucleotide substrate pool, the Ti cTfm compounds could inhibit the activity of the R1 and R2 subunits of RNR. The compounds arrest the cell cycle in the S phase, indicating suppressed DNA replication, and induce apoptotic cell death. Cotreatment cell viability studies with cisplatin and Ti(Deferasirox)2 reveal a promising synergism between the compounds that is likely owed to their distinct but complementary effect on DNA replication.
Keywords: Titanium(IV) chemical transferrin mimetic complexes, intracellular iron(III) transmetalation, ribonucleotide reductase inhibition, metal-based anticancer strategy, titanium(IV) binding of nucleotides
Introduction
In the drive to develop anticancer drugs with greater potency and cell specificity, efforts have shifted to targeting molecular mechanisms and biological species responsible for cancer cell proliferation and metastasis.1 One particular effort focusing on the relationship between iron (Fe) and cancer has been driven by the particular skill sets of bioinorganic and coordination chemists in collaboration with cancer biologists. Fe, the most important and abundant essential transition metal in the human body, is required for many cellular processes, including oxygen and electron transport, DNA synthesis, cell cycle regulation, and in the functioning of many proteins.2,3 As such, cells strictly regulate the uptake, storage, and distribution of Fe to maintain its homeostasis to ensure proper cell development and replication.3 Just as it is vital to healthy cells, Fe is critical to the survival of cancer cells. Cancer cells have a higher requirement for Fe than normal cells resulting in several key Fe-based cancer hallmarks including the overexpression of the transferrin receptor 1(TfR1) to increase endocytotic delivery of Fe(III) by human serum transferrin (sTf) into the cells,3−5 increased expression of intracellular Fe–proteins, and decreased expression of the Fe export protein ferroportin1 (Fpn1) to minimize Fe departure.6 The net effect is greater Fe use and decreased storage in cancer cells relative to healthy cells to meet the higher metabolic demand of cancer cells and to facilitate cellular proliferation due to the role of Fe-dependent ribonucleotide reductase (RNR) in DNA synthesis7,8 as well as facilitate metastasis via Fe-based matrix metalloproteinases.9 When exceeding normal levels in the body, Fe can even lead to the onset of certain cancers because of Fenton-based overproduction of reactive oxygen species (ROS).10
Due to the extensive role of Fe in cancer progression, several interdisciplinary anticancer strategies have been designed to decrease the bioavailability of iron in cancer cells. Certain strategies operate by blocking iron uptake into the cells with extracellular Fe(III) chelators11 or agents that serve as inhibitors of different components of the sTf endocytotic pathway.12 These particular strategies are limited by not being selective to cancer cells and thus would decrease the general availability of Fe to the body. Other approaches involve depleting Fe bioavailability within cells using Fe(II) and Fe(III) intracellular chelators13 or consist of inhibition of iron-dependent enzymes.7,14 These strategies are also not selective to cancer cells, but selectivity could be improved with a drug delivery system. In this work, we explore an iron chelator transmetalative approach to inhibit the human RNR enzyme as an anticancer strategy. This approach (Scheme 1) consists of using an iron chelator to exchange a non-Fe metal for Fe intracellularly to decrease Fe bioavailability and exploit the chemical properties of the released non-Fe metal to synergistically inhibit RNR and then induce cell death. RNR is composed of two dimeric subunits, R1 (α2), which contains the active site that reduces nucleotide diphosphates to produce their deoxy forms used for DNA replication and repair, and R2 (β2), which contains a di-iron cofactor responsible for activating the enzyme by generating a tyrosyl radical. This radical travels from the R2 subunit to the active site in R1 enabling the radical-based reduction process. Currently, there are several classes of RNR inhibitors designed for anticancer application: R1 inhibitors, R2 inhibitors, R1–R2 interaction inhibitors, and translational inhibitors.15−18 Our approach centers on coupling the cytotoxic properties of iron chelators in targeting the intracellular labile iron pool (LIP)19 with the chemical properties of titanium(IV) (Ti(IV)) to mimic Fe(III) and its biomolecular binding and strongly bind to phosphates.20−22 Nucleotide binding of Ti(IV) is of particular interest.21 Ti(IV) is a biocompatible metal ion with generally low toxicity to the human body.23 Its hydrolysis thermodynamic product, titanium dioxide, is also biocompatible and is believed to be readily cleared from the body despite poor solubility.23−25 With the judicious choice of ligands, Ti(IV) coordination compounds can be highly cytotoxic.26−29 As such, small-molecular-weight compounds of Ti(IV) bound by Fe(III) chelators are designed to operate exclusively in the intracellular environment by transmetalating with the LIP30 to convert Fe(III) into inert metal incapable of activating the RNR enzyme and triggering cellular attack by Ti(IV). To facilitate this strategy, a class of Fe(III) chelators termed chemical transferrin mimetics (cTfms) (Figure 1a) is used, because the chelators, in mimicking the Fe(III) binding modality of serum Tf as opposed to the Ti(IV) modality,20,31 have a thermodynamic preference for Fe(III) coordination versus Ti(IV). Fe(III)-induced transmetalation has been explored with the cTfm ligands N,N′-di(o-hydroxybenzyl)-ethylenediamine-N,N′-diacetic acid (HBED) and deferasirox (Def also known as DFX) (Figure 1a). HBED is a hexadentate chelator (log K[FeHBED]− = 39.01),32 which was previously explored as a drug for the treatment of iron overload diseases33 and for regulating iron toxicity in traumatic brain injury.34 Def is an FDA-approved, orally administered tridentate iron chelator (log K[Fe(Def)2]− = 38.6).35 It is used for treating iron overload diseases36 and exhibits promise for chemotherapy application37−40 The Ti(IV) complexes of HBED ([TiO(HBED)]−) and Def ([Ti(Def)2]2–) demonstrate a potent cytotoxic effect.41,42
Scheme 1.
Figure 1.

Metal ion coordination to the chemical transferrin mimetic (cTfm) chelators. (a) Physiologically relevant (pH 7.4) Fe(III) coordination by human serum transferrin (sTf) and the cTfm chelators HBED and Def. (b) ORTEP diagram of the neutral compound Ti(Def)2 (Ti[C42H26N6O8]) with ellipsoids set at 50% probability (hydrogen atoms were omitted for clarity). (c) The pH-dependent speciation of the Ti(IV) HBED at a 1:1 metal/ligand ratio and micromolar concentrations. (d) The pH-dependent speciation of the Ti(IV) deferasirox at a 1:2 metal/ligand ratio and micromolar concentrations. The complete speciation diagrams can be found in Figure S1.
By profiling the mechanistic details of the cytotoxic behavior of the Ti(IV) cTfm compounds, insight is obtained about how they alter the bioavailability of the LIP and attenuate the activity of the Fe-dependent RNR enzyme and subsequently attenuate DNA replication. We unravel aspects of the elusive biochemistry of Ti(IV) by identifying how in binding nucleotides it can disturb the RNR nucleotide substrate pool. We also provide evidence for the [Ti(Def)2]2– compound exhibiting synergistic cytotoxicity in combination with cisplatin. The study presents a promising strategy for possible dual-RNR R1 and R2 inhibition for an anticancer application that centers on manipulating physiological metal coordination chemistry and speciation for therapeutic effect.
Results
Characterization of the Ti(Def)2 Complexation
In this study, we sought to better understand the structure and corresponding solution behavior/speciation of the Ti(IV) cTfm compounds for deeper insight into their mechanism of action. Previously, we published the synthesis and spectroscopic characterization of the neutral Ti(Def)2 (Ti[C42H26N6O8]) compound.42 Herein, we report the crystallization and single-crystal X-ray diffraction analysis of this compound. Single crystals of this compound were obtained from its saturated DMF solution (see Supporting Information). Orange-colored plate-like crystals developed within a few hours of storage at 4 °C. The X-ray structure of neutral Ti(Def)2 (Figure 1b) reveals that Ti(IV) is coordinated in a meridional fashion by the Def ligands with a low strain that results in twisting of the phenol rings.43 To the best of our knowledge, our Ti(Def)2 crystal structure represents Ti(IV) as the second metal to be shown by X-ray diffraction to be bound by Def. The first metal was vanadium(V) in a dinuclear structure, in which the V(V) ions are hexacoordinated in a 1:1 metal/ligand stoichiometry.44 Our structure is the first monometallic bisDef structure. A summary of the crystal data, structure solution, and refinement are listed in Table S1 with select bond lengths and angles in Table S2.
A ligand competition experiment was performed using UV–vis spectroscopy to quantify the formation constant of the [Ti(Def)2]2– complex at pH 7.4. In this study, separate 1 mL aqueous solutions of 26.7 μM [Ti(citrate)3]8– ([Ti(Cit)3]8–), a biorelevant labile source of Ti(IV), were reacted with varying concentrations of Def (0 to 417 μM) in a background that included 7 mM citrate to ensure that [Ti(Cit)3]8– is the sole Ti(IV) citrate species present at pH 7.4,45 0.1 M KCl to maintain an ionic strength of 0.1 M, and 5% DMF (v/v) to maintain Def solubility.45 Citrate serves as an intracellular metal chelator, forming part of the LIP speciation. All solutions were left to equilibrate for 24 h in the dark. The absorbance of the solutions was then scanned in the wavelength range of 300 to 600 nm. In this range, only the [Ti(Def)2]2– complex absorbs with an absorbance maximum of 365 nm and extinction coefficient of 14 900 M–1cm–1 due to a ligand to metal charge transfer (LMCT) band.42 The formation constant of the [Ti(Def)2]2– complex was determined by fitting the saturation curve signal at 365 nm (Figure S2 is a rearrangement of this data). By accounting for the constant citrate and proton concentrations and all competing equilibria in solution (Supporting Information), the [Ti(Def)2]2– formation constant was determined to be log K = 38.8 ± 0.1.
Physiologically Relevant Speciation of Ti(IV) Complexation by the cTfm Ligands HBED and Def
Having determined the [Ti(Def)2]2– formation constant and having previously performed spectropotentiometric studies of Ti(IV) in interaction with both HBED and Def, we are now able to provide a complete physiologically relevant pH-dependent speciation of the Ti(IV) complexes of HBED and Def as it pertains to the role of the chelators as cTfm ligands.42,46 At a 1:1 metal/ligand ratio and micromolar concentrations in 0.1 M ionic strength (0.1 M KCl) and 2.6% DMF (v/v), HBED coordinates Ti(IV) in mononuclear complexation in the pH 3 to 10 range. At pH 3, HBED binds Ti(IV) in a hexadentate modality comparable to its coordination of Fe(III) (Figures 1c and S1).46 This neutral Ti(HBED) compound is highly stable though poorly water-soluble. Its solubility is enhanced with a cosolvent like ethanol, DMF, or DMSO. Its formation constant (log K = 34.07)47 is lower than that of [FeHBED]− (log K = 39.01).32 While HBED hexadentate coordination remains prevalent for Fe(III) at pH 7.4 up to pH 11,32 the same is not true for Ti(IV) (Figures 1c and S1). In the pH 6 to 10.0 range, partial ligand dissociation of the phenolate and possibly carboxylate arms dominates due to Ti(IV) hydrolysis (Figures 1c and S1).46 These higher pH species can be solubilized in water at μM levels. The collective speciation results attest to the generally higher affinity of HBED for Fe(III) versus Ti(IV) and validate HBED as a cTfm ligand.41
At a 1:2 metal/ligand ratio and micromolar concentrations in 0.1 M ionic strength (0.1 M KCl) and 30% DMSO (v/v), Def coordinates Ti(IV) in a virtually consistent mononuclear complexation in the pH 4 to 8 range (Figures 1d and S1).42 Throughout this pH range, two ligand molecules bind the metal in tridentate fashion. The only change in speciation is due to deprotonation of the benzoic acid group on the two molecules. At pH 4, both benzoic acids are fully protonated yielding the neutral Ti(Def)2 complex, which is extremely water-insoluble. Like neutral Ti(HBED), the solubility of Ti(Def)2 is enhanced with a cosolvent such as ethanol, DMF, or DMSO. Above pH 7.0, both benzoic acid moieties are deprotonated, and the dianionic species [Ti(Def)2]2– is dominant. This species can be solubilized in water at high μM levels. The Ti(IV) Def speciation profile is comparable to that of Fe(III).43 Based on the ligand competition experiment performed in this work, the [Ti(Def)2]2– formation constant (log K = 38.8 ± 0.1) was determined to be virtually identical to that of [Fe(Def)2]3– (38.6).35 Given the relatively high iron content (∼μM) in cells,19 Fe(III) is expected to outcompete Ti(IV) for Def binding as demonstrated previously,42 validating it as a cTfm ligand.
The speciation studies reveal that the HBED and Def complexes of Ti(IV) are extremely stable in aqueous solution particularly under the physiologically relevant pH range of 5 to 8.42,46,47 Their stability was also examined in the context of their interaction with serum biomolecules that can induce the dissociation of other Ti(IV) compounds such as citrate, transferrin, and albumin. These studies demonstrated that these Ti(IV) cTfm compounds are extremely robust in the presence of physiological levels of these small molecules and proteins,31,42 which suggests that they would remain intact as they enter into the cellular environment.
Cell Studies Provide Valuable Insight into the Effect of the Ti(IV) cTfm Complexes and Metal-Free Ligands on Intracellular Iron, R2 RNR Activity, and Cellular Proliferation
Having obtained a clear picture of the behavior of the Ti(IV) cTfm compounds in physiological relevant solution, they were then examined in terms of their antiproliferative/cytotoxic effect on human cells. The neutrally charged forms of Ti(HBED) and Ti(Def)2 compounds were submitted to the National Cancer Institute (NCI) 60 human tumor cell line anticancer drug screen (Table S3). This screen is performed by diluting stock solutions of the compounds in cell media (∼pH 7), and the Ti(IV) compounds are expected to speciate accordingly. At the one-dose test of 10 μM administered for 48 h, [Ti(Def)2]2– demonstrates stronger antiproliferative potency than [TiO(H+–HBED)]− against the different cell line types. To gain mechanistic insight into the cellular effect of the compounds, we decided to focus on Jurkat leukemia cells as a case study because of the stronger antiproliferative behavior of the Ti(IV) cTfm complexes against leukemia cells versus other cell types. Furthermore, Jurkat cells express the RNR enzyme at sufficiently high levels to facilitate the use of whole-cell electron paramagnetic resonance (EPR) to monitor the active state of the enzyme’s Fe-dependent R2 subunit. The complexes and the metal-free ligands are antiproliferative against the Jurkat cell line (Table S4 and Figure S3). In agreement with the NCI screen, [Ti(Def)2]2– demonstrated greater potency than [TiO(H+–HBED)]− (a 24 h IC50 of 9.1 ± 1.5 μM vs 24.7 ± 1.2 μM). A possible difference in the general potency of the compounds may be due to a difference in lipophilicity, which would affect their relative uptake into cells. These compounds are expected to enter cells via a passive diffusion pathway given that a membrane receptor or transporter is not known to recognize them.41,42,46 The partition coefficients (log DpH 7.4) of [TiO(H+–HBED)]− and [Ti(Def)2]2– were measured at pH 7.4 using the shake-flask method.48 The [TiO(H+–HBED)]− value of −0.34 falls in the range of a solute that would be expected to have membrane permeability problems, whereas the [Ti(Def)2]2– value of 0.23 falls in an optimal range for membrane permeability. Future studies on the cell uptake pathways of these compounds are warranted to determine if lipophilicity differences influence the antiproliferative potency of the compounds.
Another possibility for the difference in compound potency could be how they alter the bioavailability of the intracellular LIP following transmetalation and subsequently inhibit the RNR enzyme. To this end, whole-cell EPR experiments were performed following the method of Tomat et al.49,50 to monitor the effect of 50 μM of [Ti(Def)2]2–, [TiO(H+–HBED)]−, and the metal-free ligands on the intracellular LIP within 2.54 × 106 Jurkat cells as gauged, in part, by inhibition of the R2 subunit of the enzyme. This subunit contains a di-iron cofactor, which after binding two Fe(II) ions and oxidizing them to the +3 state with the assistance of O2, results in subunit activation by generating a tyrosyl radical. This radical travels 35 Å by a long-range proton-coupled electron transfer to the active site cysteine generating a thiyl radical, which initiates nucleotide reduction.51 The tyrosyl radical is detected at g ≈ 2 by EPR. Following treatment of the Jurkat cells with [Ti(Def)2]2–, [TiO(H+–HBED)]−, and the metal-free ligands, the cells were washed with buffer, and a relative quantitation of the tyrosyl radical signal was measured by double integration of the baseline-corrected spectra. In comparison with untreated cells at pH 7.4, the tyrosyl signal was reduced in cells after a 3 h treatment with HBED2–, [TiO(H+–HBED)]−, Def–, and [Ti(Def)2]2– by 56, 83, 75, and 62%, respectively (Figure 2a). Based on comparable EPR studies with the tridentate iron chelator triapine and its metalated complexes (Fe(III), Ga(III), Cu(II), and Zn(II)),52−54 there are several possible contributions to the decreased tyrosyl radical signal induced by the Ti(IV) cTf complexes. First, the intracellular bioavailability of Fe is reduced by cTfm chelation resulting in decreased activation of apo (metal-free)-enzyme. Second, the Fe cTfm complexes that form could exhibit redox activity similar to the ferrous compound of triapine, [Fe(II)(triapine)2], which is capable of reducing the tyrosyl radical. Third, the Ti(IV) cTfm complexes may trigger the release of Fe from the cofactor site, which then destabilizes the tyrosyl radical. At present, none of these possibilities can be ruled out until direct work examining the effect of the Ti(IV) cTfm complexes on the enzyme using recombinant protein or the protein extracted from cells is conducted.
Figure 2.
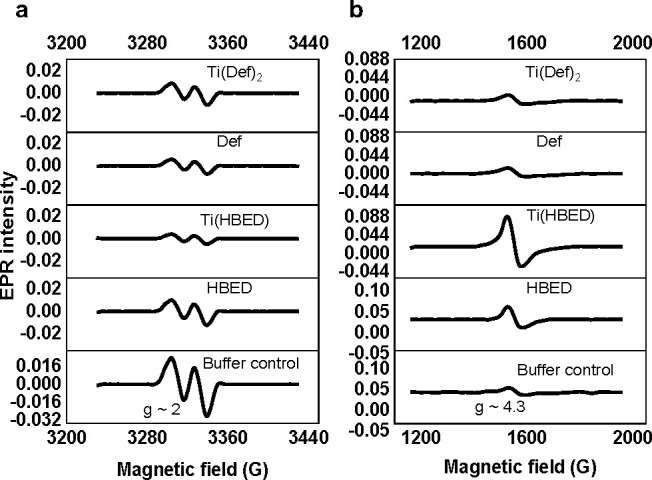
Whole-cell EPR spectra for the 3 h treatment of Jurkat cells with 50 μM [Ti(Def)2]2–, Def–, [TiO(H+–HBED)]1–, and HBED2– and also the buffer control. The EPR signal corresponding to (a) the tyrosyl radical of the RNR enzyme (g ≈ 2) and to the (b) intracellular high-spin Fe(III) (S = 5/2; g ≈ 4.3). Experimental conditions: microwave frequency, 9.338 GHz; microwave power, 2 mW; magnetic field modulation amplitude, 0.5 mT for the tyrosyl radical and 1 mT for high-spin Fe(III) (S = 5/2), temperature: 20 K. All EPR spectra were baseline-corrected.
The high-spin S = 5/2 Fe(III) EPR signal (g ≈ 4) was also monitored in the Jurkat whole-cell experiment. The data demonstrate a surprising difference in this signal caused by [TiO(H+–HBED)]− and HBED2– and by [Ti(Def)2]2– and Def–, which if they react with the LIP can be expected to form the [Fe(HBED)]− and [Fe(Def)2]3– species at pH 7.4, respectively. This is true also for the metal-free cTfm chelators whether they directly bind Fe(II), because the Fe(II) complexes can be rapidly oxidized intracellularly to the Fe(III) complex.43,55 The EPR spectra collected for [Fe(HBED)]− and [Fe(Def)2]3– reference standards at pH 7.4 reveal that both complexes consist of high-spin Fe(III) species (Figure S4). It was expected that all cTfm compounds would increase the cellular high-spin Fe(III) signal, as less labile Fe(III) would be available in the cytosol and would shift the cellular LIP to increase Fe(III) levels to re-establish equilibrium. In comparison to untreated Jurkat cells, [TiO(H+–HBED)]− and HBED2– do increase the high-spin Fe(III) signal; however, [Ti(Def)2]2– and Def– appear to have no effect (Figure 2b). These results indicate that Def– and [Ti(Def)2]2– either do not react with the LIP, which is very unlikely, or if they do, they form redox-active Fe(III) complexes (within the time frame in which the experiment was performed) that can rapidly redox cycle and thus do not increase the high-spin Fe(III) signal.
Previous mechanistic studies on Fe(III) transmetalation of [TiO(H+–HBED)]− and [Ti(Def)2]2– at pH 7.4 using [Fe(Cit)2]5– as a model for labile Fe(III)56 sheds light on the differing high-spin S = 5/2 Fe(III) EPR signal profiles. The complex [Ti(Def)2]2– is able to form a relatively long-lived Fe intermediate, [Fe(Cit)(Def)]4–, whereas [TiO(H+–HBED)]− does not.41,42 To examine the redox activity of this intermediate, an in situ approximation of it was prepared by reacting [Fe(Cit)2]5– and Def– at pH 7.4 in a 1:1 ratio. The in situ intermediate was examined by electrospray ionization mass spectrometry (ESI-MS) (prepared in salt-free 5 mM NH4HCO3 buffer) and by EPR (prepared under the same conditions as the other Fe(III) complexes). The ESI-MS data suggest that, at minimum, there is a mixture of the [Fe(Def)2]3– and [Fe(Cit)(Def)]4– species (Figure S5). The EPR data reveal a high-spin Fe(III) signal (S = 5/2) that is distinct from that of [Fe(Cit)2]5– at pH 7.4 but with features similar to [Fe(Def)2]3– at pH 7.4 (Figure S4). Cyclic voltammetry was used to measure or estimate the reduction potential of all relevant metal complexes in the Fe(III) transmetalation reactions at pH 7.4 (Table 1 and Figure S6). [Ti(Def)2]2–, [Fe(Def)2]3–, and [TiO(H+–HBED)]− have negative reduction potential values (E1/2) for the 1e– reduction of the metal ions. These values are outside the biological redox window of the intracellular environment.57 The value for [Fe(HBED)]− is on the edge of this window. The voltammogram for the in situ prepared [Fe(Cit)(Def)]4– solution shows reduction processes that are similar to the ones observed in the data for [Fe(Cit)2]5– (two irreversible reduction events for two different Fe(III) citrate species) and for [Fe(Def)2]3–, which confirms that there is a mixture of species present in the sample (Figure S6). Collectively, the data suggest that the generation of species like [Fe(Cit)(Def)]4– in the reaction pathway of Def– and [Ti(Def)2]2– with the LIP may be redox-active and could possibly redox cycle. If such species form in the time frame in which the whole-cell EPR data were collected for Jurkat cells treated with Def– and [Ti(Def)2]2–, then they would account for why no increase in the high-spin S = 5/2 Fe(III) signal was observed.
Table 1. Reduction Potential Values (E1/2) vs Normal Hydrogen Electrode (NHE) for Metal Complexes Involved in the Fe(III) Transmetalation of [Ti(Def)2]2– and [TiO(H+–HBED)]− at pH 7.4a.
| compound | E1/2 (V) |
|---|---|
| [Ti(Def)2]2– | –0.77 |
| [TiO(H+–HBED)]− | –0.60 |
| [Fe(Def)2]3– | –0.59 |
| [Fe(HBED)]− | –0.28 |
| [Fe(Cit)2]5– | –0.38* and 0.16* |
| [Fe(Cit)(Def)]4– | –0.75*, −0.41*, 0.19* |
* represents peak potential values of the cathodic peak (Ec) due to a mixture of species.
Regardless of the exact molecular details of how the cTfm complexes interact with the LIP, by inhibiting the RNR enzyme they are expected to alter the cell cycle process and significantly decrease cell survival. Cell cycle studies at pH 7.4 were performed using flow cytometry, where the DNA was quantified in cells treated with the compounds (at concentrations to avoid extensive cell death) and stained using propidium iodide (PI) (Figure 3a). All the compounds resulted in time-dependent S-phase cell cycle arrest, indicative of the cells being unable to duplicate DNA. An apoptosis study was performed using flow cytometry by staining the compound treated and untreated Jurkat cells (for 24 and 72 h) with the dyes annexin-V-conjugated Alexa Fluor 488 and PI. In the study, 20 μM cisplatin and 50 μM [Ti(Cit)3]8– were used as positive and negative controls, respectively (Figure 3b).42,58 The data indicate that all of the Ti(IV) cTfm compounds and metal-free ligands induce early and late apoptosis, consistent with their behavior observed in human lung cells.42
Figure 3.

Effect of compounds (written without charge states for simplicity) on Jurkat cells. (a) Cell cycle analyses using flow cytometry at 3, 10, and 24 h of treatment with 29 μM Def, 18.8 μM [Ti(Def)2]2–, and 26.3 μM HBED– and [TiO(H+–HBED)]−. (b) Time-dependent apoptosis assay performed using flow cytometry after treating the cells with 20 μM cisplatin, 50 μM [Ti(Cit)3]8–, 19 μM [Ti(Def)2]2–, 27 μM [TiO(H+–HBED)]−, 29 μM Def–, 27 μM HBED2–, and buffer control for 24 and 72 h and then staining with annexin V–Alexa Fluor 488/propidium iodide (PI). * p-value ≤ 0.05 vs the corresponding control group.
cTfm Complexes Are Not Cytotoxic to Iron-Rich Erythrocytes
In their current form, the Ti(IV) cTfm compounds are not selective in their antiproliferative/cytotoxic effect toward cancer cells versus noncancer cells, although slightly greater potency has been observed against cancer cells.41,42 Nonetheless, we sought to further explore their general toxicity profile by examining their effect on erythrocytes. Erythrocytes, the most common type of blood cells, are a major source of Fe within blood due to housing high levels of hemoglobin (Hb). When Hb is released from the erythrocytes because of hemolysis, the free Hb becomes involved in specific diseases such as chronic vascular disease, inflammation, and thrombosis.59 Drug-induced hemolysis is a frequent hematological associated adverse drug reaction. Erythrocytes were incubated with [TiO(H+–HBED)]−, [Ti(Def)2]2–, and the metal-free ligands at pH 7.4. Even at 100–200 μM concentrations, the compounds are unable to induce significant hemolysis as determined by the low release of hemoglobin relative to positive control samples lysed with Triton X-100 (Figure S7).
Transmetalation reactions between 12.5 μM Hb (in the form of Fe(III)–hemoglobin; methemoglobin (MHb)) and excess amounts of cTfm compounds were also investigated at pH 7.4 for 96 h. At 24 h time intervals, MHb was monitored at its Soret band of 405 nm and between 500 and 650 nm, the visible absorbance due to the Fe(III).60 When accounting for background absorbances of the compounds, no changes in these signals were observed in any of the reactions, indicative of the protein being unperturbed by the compounds (Figure S8). The same is expected to be true for Fe(II)–hemoglobin, the main protein form, given the higher affinity of the cTfm ligands for Fe(III) than for Fe(II) (log K[Fe(II)HBED]2– = 22.7 and log K [Fe(II)(Def)2]4– = 14).32,43,61
Exploring the Potential Synergism between [Ti(Def)2]2– and Cisplatin
While the Ti(IV) cTfm compounds demonstrate promise as anticancer agents, we wanted some preliminary insight into their potential to work in combination with another drug that can inhibit DNA replication. Due to the greater potency of [Ti(Def)2]2– vs [TiO(H+–HBED)]− against Jurkat cells and other cancer cell lines screened, the compound was examined in a cotreatment cell viability study with the Pt(II) drug cisplatin. The objective was to examine a possible synergism in their antiproliferative/cytotoxic behaviors based on their distinct but potentially complementary effects on DNA replication. For cisplatin and other Pt(II)- or even Pt(IV)-containing anticancer compounds, DNA is considered the primary cellular target. In the reducing intracellular environment, Pt would exist in the +2 charge state, and in this form, it predominantly binds to the N7 position of the guanine bases of DNA, possibly in the monoadduct form or producing inter/intrastrand cross-links.62,63 Coordination to the DNA bases can severely distort the molecular structure and, in extreme cases, induce strand breaks.62,63
Jurkat cells were subjected to a 48 h cell viability dose response screening against [Ti(Def)2]2– and cisplatin from 0.1 to 10 μM concentration (Figure S9) and then to a 48 h cotreatment with 1:1 and 1:2 [Ti(Def)2]2– to cisplatin mole equivalent ratios over the same concentration range but as a combined concentration of both compounds (Figure S9). The behavior of the combinations was analyzed by the traditional isobolographic method.64,65 An isobologram is constructed by plotting the two IC50 values for each individual drug alone along the x- and y-axes of the coordinate system. The two points are connected by a straight line called the addition line.66 The addition line is accompanied by the error range presented by dashed lines, as derived from the error values of the individual IC50 values. The IC50 values for the drugs used in the combination are then determined and plotted on the same graph. Points lying on the line of addition correspond to the simple addition of the combination. Points lying above the line of addition indicate that the behavior of the two drugs in combination is antagonistic, and thus, their effect is less than expected from their individual responses. Points under the line of addition correspond to synergism and indicate that the activity of the combination is greater than that expected from the activities of the two drugs given separately. An isobologram analysis of the cotreatment (Figure 4, Table S4, and Figure S9) shows that at the 1:1 ratio the compounds behave modestly synergistically (IC50 combination index (CI) = 0.953; Supporting Information), and at the 1:2 ratio, they behave strongly synergistically (IC50 CI = 0.730).
Figure 4.

Isobolographic analysis of the antiproliferative activity of the 1:1 and 1:2 [Ti(Def)2]2–/cisplatin combinations against Jurkat cells for 48 h.
Ti(IV) Inhibition of DNA Replication
An important component of the Ti(IV) cTfm drug design that we sought to explore in this work was the contribution of the Ti(IV) to the antiproliferative/cytotoxic effect, particularly as it pertains to the metal ion’s phosphate chemistry. Ti(IV) binding to DNA has long been thought to be the foundation for its cytotoxicity based on studies with the former anticancer drug candidate titanocene dichloride. This compound operates as a prodrug, because it is extremely ligand-exchange-labile in aqueous solution, rapidly dissociating all of its ligands to release Ti(IV) to sTf at pH 7.4, which is believed to be transported via TfR-mediated endocytosis into cells.31,67,68 Following endosome acidification, adenosine triphosphate (ATP) can effectively remove Ti(IV) from sTf and transport it into the cytosol, ultimately directing it to the nucleus to target DNA.69 Ti(IV) in ion form, perhaps as the hydrolyzed titanyl ion (TiO2+), can coordinate to the phosphate moieties of the DNA backbone (Figure S10a).20,21,70 A study performed by directly reacting titanocene dichloride with a DNA fragment has shown that the titanocenyl moiety may bind intact to the phosphate moieties at pH 5.3, but at pH 7, one of the cyclopentadienyl rings dissociates (Figure S10a).70 However, this study does not account for the above-mentioned relevant physiological interactions with the compound. Titanocene Y, a structurally more stable compound, is believed to retain the cyclopentadienyl rings bound to the Ti(IV), while the metal ion coordinates to the DNA phospho groups (Figure S10a).71 Regardless of how Ti(IV) from titanocenyl compounds coordinates to DNA, its binding structurally distorts DNA and can prevent binding of topoisomerase, which impedes the onset of replication.71
While [TiO(H+–HBED)]− and [Ti(Def)2]2– entry into cells is different from titanocene dichloride by being independent of sTf,42 a study was performed to determine whether Ti(IV) dissociated from [TiO(H+–HBED)]− and [Ti(Def)2]2– following intracellular transmetalation possibly localizes within the DNA of Jurkat cells. After the cells were treated at concentrations of the cTfm compounds that would induce apoptosis, a DNA fragment was isolated over time courses of 3, 10, and 24 h following treatment and run on an agarose gel (Figure S11). The mobility of the DNA was unaffected compared to control samples without Ti(IV). Inductively coupled plasma-optical emission spectrometry (ICP-OES) confirmed that no Ti(IV) content was found in the DNA fragment (Supporting Information).
Given that [TiO(H+–HBED)]− and [Ti(Def)2]2– have the capacity to transmetalate with the intracellular LIP, Ti(IV) release could facilitate its interaction with nucleotides, which could hinder DNA replication. Nucleotide binding of Ti(IV) has been previously explored using titanocene dichloride as the source of Ti(IV).21,67,69 A Ti(IV) ATP compound was synthesized by reacting Ti(IV) with ATP at a 1:6 mol equivalent ratio in water. A white compound, which precipitated from the highly acidic solution (pH 1.34), was characterized by C, H, N elemental analysis and a Ti(IV) colorimetric analysis yielding the molecular formula for a multinuclear neutral compound Ti6O7(C10H14N5O13P3)5(H2O)34 (Ti6O7(ATP)5(H2O)34), with a weight of 3617.55 g/mol. The elemental composition of C, H, N, O, P, and Ti was confirmed by energy dispersive X-ray spectroscopy–scanning electron microscopy (EDS–SEM), and an amorphous morphology was observed by powder X-ray diffraction (Figure S12). Fourier transform infrared (FTIR) analysis of the compound compared to ATP alone reveals phosphate coordination of Ti(IV) (Figure S13; Supporting Information). A full structural characterization of the solid-state form of the compound was not achieved, but nonetheless, the elemental formulation suggests a ligand/Ti (L/Ti) ratio of less than 1 with possible oxide (and even aqua) bridges linking the metal ions due to the extremely hydrolytic nature of Ti(IV).20
The Ti6O7(ATP)5(H2O)34 compound is not soluble under acidic conditions but is soluble at pH 7.4, although it appears to form a precipitate after some time in solution. Interestingly, the 31P NMR spectrum of a pH 7.4 solution of the Ti6O7(ATP)5(H2O)34 compound (Figures 5 and S14) at 24 h (to achieve equilibration) shows that Ti(IV) not only binds to ATP, it is also able to induce the hydrolysis of the phosphates producing ADP, AMP, and inorganic phosphate. A similar behavior was observed after 1 h in solution. To help identify the various species in the spectrum for Ti6O7(ATP)5(H2O)34, 31P NMR spectra (Figure S15) were collected of 5 mM ATP, ADP, and AMP at pH 7.4 in the presence and absence of 1 mM [Ti(Cit)3]8– after 24 h in solution. As indicated by the changes in the chemical shifts of the phosphate groups of ATP, ADP, and AMP, all three compounds appear to be Ti(IV)-bound (Table 2). Although 31P NMR data alone is not sufficient to fully determine the speciation that results from these reactions, the changes in the chemical shifts suggest that Ti(IV) coordinates to the β and γ phosphate groups of ATP, the β phosphate of ADP, and the sole phosphate group of AMP (Figure S10b). The coordination of these particular groups is proposed, because the changes of the 31P chemical shifts of these groups is most significant in the presence of Ti(IV) (Table 2). Due to the excess of the nucleotides (L/Ti ≥ 3), the six coordination sites of Ti(IV) are proposed to be saturated by the nucleotides (Figure S10b). Surprisingly, there is no indication of metal-free phosphorus signal, even though an excess amount of the nucleotides was used with respect to Ti(IV), and there was also 3 equiv of citrate present in solution. This observation suggests both high-affinity binding of the phosphate groups and rapid exchange between free and Ti(IV)-bound nucleotides. More broadly, this observation demonstrates how Ti(IV) could affect the nucleotide pool at pH 7.4 even when a much larger excess of nucleotides is expected relatively to Ti(IV) uptake into cells. The 31P NMR spectrum of ATP in the presence of [Ti(Cit)3]8– sample does demonstrate that some amount of ADP is generated at a ratio of ∼5.3:1 ATP/ADP as determined by averaging peak integration.
Figure 5.

31P NMR spectra for (a) Ti6O7(ATP)5(H2O)34 and (b) ATP at pH 7.4 and 5 mM concentration based on ATP content. Also refer to Table 2 as well as Figures S14 (a zoom-in of this figure) and S15.
Table 2. Summary of 31P NMR data at pH 7.4 for Ti6O7(ATP)5(H2O)34 and for AMP, ADP, and ATP in the Presence and Absence of [Ti(Cit)3]8–a.
| sample | chemical shift (δ, ppm) |
|---|---|
| AMP (Figure S15g) | 3.35 (s) |
| Ti(IV) + AMP (Figure S15f) | 3.13 (s) |
| ADP (Figure S15e) | –6.80 (d, I = 1.00), −11.21 (d, I = 1.01) |
| Ti(IV) + ADP (Figure S15d) | –7.69 (d, I = 1.00), −11.26 (d, I = 1.02) |
| ATP (Figure 5b) | –7.59 (s(wide), I = 1.00), −11.60 (d, I = 1.11), −22.49 (t, I = 1.02) |
| Ti(IV) + ATP (Figure S15b) | –7.37 (d, I = 5.37), −11.22 (d, ADP species, I = 1.00), −11.55 (d, I = 5.56), −22.28 (t, I = 5.04) |
| Ti6O7(ATP)5(H2O)34 (Figure 5a) | 3.30 (s, free AMP, I = 2.02) |
| 1.995 (s, free phosphate, I = 1.00) | |
| –6.56 (d, ATP species, I = 0.867) | |
| –6.85 (d, ADP species, I = 0.997) | |
| –11.21 (d, ADP species, I = 0.996) | |
| –11.55 (d, ATP species, I = 0.99) | |
| –22.20 (t, ATP species, I = 0.7441) |
The pH 7.4 solution behavior of the nucleotides when in excess of Ti(IV) (L/Ti ≥ 3) is in contrast to that exhibited by Ti6O7(ATP)5(H2O)34. The 31P NMR spectrum of Ti6O7(ATP)5(H2O)34 (Figure 5) reveals that phosphate hydrolyzed products predominate with AMP, ADP, and ATP (whether Ti(IV)-free or -bound) present at a ratio of 2.31:1.14:1.0, respectively (Table 2). A full comprehension of the speciation of this compound is not entirely possible, but the likely explanation for the dramatically different solution behavior is that the hydrolyzed Ti(IV) moieties in this compound, which are believed to form due to the absence of ligand saturation of its coordination sites, facilitates Ti(IV)-induced hydrolysis of the phosphate groups at pH 7.4 (Figure S10 b). The phosphorus integration for free inorganic phosphate is lower than what mass balance would suggest, but this may be owed to the formation of hydrolyzed Ti(phosphate) species, which are quite insoluble72 and would not be detectable in this solution-based 31P NMR study. There is no indication of diphosphate or even triphosphate formation. Excess presence of nucleotides combined with the presence of citrate as an additional metal binder could inhibit Ti(IV)-induced phosphate hydrolysis. Nucleotide binding of Ti(IV) and hydrolysis of their phosphate groups could provide a distinct route to inhibit the R1 subunit of RNR by chemically modifying the nucleotide substrates. The rates of Ti(IV)-induced phosphate hydrolysis of nucleotides at pH 7.4 have to be determined, but this work demonstrates that within the hours to days time scale of the antiproliferative/cytotoxic effects of the Ti(IV) cTfm compounds, Ti(IV) released intracellularly should be able to interact with the nucleotide pool. Whether the behavior of phosphate hydrolysis can be quickly corrected by the actions by kinases requires further exploration.
Discussion
Previous in vitro kinetic studies have shown that in the presence of a labile Fe(III) source,41,42 the Ti(IV) cTfm compounds undergo Fe(III) transmetalation on a physiologically relevant time scale, which supports the feasibility of this mechanism occurring intracellularly and effecting iron-dependent processes that enable cell survival and replication. Supplementing cells with Fe makes them more resilient to treatment with these compounds, highlighting the importance of Fe to their activity.41,42 As part of their toxicological profile, we have determined that the compounds are unable to perturb Fe(III)-bound sTf, the main Fe(III) species in blood serum,42 nor perturb the Fe reservoir of erythrocytes. These results speak to the high stability of the Ti(IV) cTfm compounds and to their reactivity being triggered by the cellular LIP.
By using Jurkat cells as a case study, we were able to examine the utility of iron transmetalation to inhibit the RNR enzyme and to reveal mechanistic details of the antiproliferative/cytotoxic behavior of the Ti(IV) cTfm complexes, [TiO(H+–HBED)]− and [Ti(Def)2]2– (Scheme 2). The whole-cell EPR studies on inhibition of the tyrosyl radical signal of R2 RNR present a few different possibilities as to how the Ti(IV) cTfm complexes might inhibit the RNR enzyme, specifically the R2 subunit. Through their transmetalation with Fe(III) from the LIP, the complexes attenuate the bioavailability of cellular Fe, and this decrease could directly impact activation of apoRNR enzyme. Clearly, this form of the enzyme would not constitute a large population of the enzyme, and this mechanistic route should then be able to inhibit other intracellular Fe-dependent enzymes and proteins, like the Fe–S cluster-containing ones. Studies with the iron chelator triapine have shown that perturbing the LIP and scavenging some of the Fe does not lead to a general inhibition of all Fe-dependent proteins.54 For instance, the enzyme aconitase was not inhibited.54 Rather, triapine forms a redox-active ferrous complex, [Fe(triapine)2], with the LIP that serves as a specific Fe-bound R2 RNR inhibitor capable of directly reducing the tyrosyl radical.53,54 Whether [TiO(H+–HBED)]− or [Ti(Def)2]2– can facilitate this same specific Fe-bound R2 RNR inhibition remains to be examined. Given the potential redox-active Fe heteroleptic intermediates that [Ti(Def)2]2– may form by transmetalation with the LIP, there is the possibility for it to exhibit a comparable specific R2 inhibitory role. This behavior might account for the generally higher antiproliferative potency of [Ti(Def)2]2– against different cell lines. The whole-cell EPR tyrosyl radical signal inhibition could also be explained by the Ti(IV) cTfm complexes inducing Fe release from the R2 di-iron cofactor site possibly by cTfm dissociation and then chelation of the Fe or even by transmetalation. Either route would subsequently destabilize the tyrosyl radical. To determine the feasibility of Fe cofactor dissociation, studies will have to be performed with the enzyme itself.
Scheme 2. Proposed Mechanism of Action for Ti(IV) cTfm as Combined R1 and R2 RNR Inhibitors.

(a) Decreasing the LIP via transmetalation would prevent Fe binding of the R2 site and enzyme activation. (b) Ti(IV) modification of the nucleotide pool would inhibit substrate binding to the catalytic site of the R1 subunit. Created with BioRender.com.
During cell division, human RNR performs a central role in regulating deoxyribonucleotide diphosphate (dNDP) levels for genomic maintenance.7,8 In mammalian cells, the dNDP pool reaches its peak during the S-phase to fulfill the requirement of nucleotides for DNA replication.73 Cell cycle studies showed that all of the cTfm compounds resulted in time-dependent S-phase arrest, indicative of the cells being unable to duplicate DNA. This finding is consistent with RNR inhibition by the compounds.
An intriguing discovery of this work is the potential for Ti(IV) to impede DNA replication by way of inhibiting RNR. A true understanding of the antiproliferative mechanism of the action of Ti(IV) has always been elusive, because earlier understandings of Ti(IV) activity were based on titanocene dichloride, which demonstrates very poor solution stability and is prone to hydrolysis-induced precipitation. STf binding of Ti(IV) dissociated from the compound regulates its blood transport and attenuates its cytotoxic potential.31 The Ti(IV) then localizes on nuclear DNA, but this interaction and the corresponding onset of apoptosis is observed in vitro at very high concentrations of titanocene dichloride,74 which correlates with its very high IC50 values observed for many cell lines and likely its ultimate failure in clinical trials.74 Ti(IV) from [TiO(H+–HBED)]− and [Ti(Def)2]2–, however, does not localize in the DNA of Jurkat cells at concentrations near their IC50 values. Reactions with ATP, ADP, and AMP at pH 7.4 show the strong capacity of Ti(IV) to be bound by the phosphate groups of the nucleotides and at near equal levels as the nucleotides to induce phosphate hydrolysis. Ti(IV) was previously thought capable of hydrolyzing nucleotides only under acidic conditions.21 If this behavior is possible within the cellular environment, then the Ti(IV) cTfm complexes have another route by which to inhibit the RNR enzyme (Scheme 2): either by cleaving the phosphates of the nucleotide substrates and making them unable to bind to the RNR active site (which could be corrected by certain kinases) or by binding to them so that they are not recognized as RNR substrates, a behavior observed for gallium(III).75 The 31P NMR study performed in this work suggests that Ti(IV) is able to interact with nucleotides even beyond its coordination site capacity given that there appears to be a rapid ligand exchange between bound and free nucleotides when the L/Ti ratio was 5. Further studies of this broader nucleotide effect are warranted by evaluating the 31P NMR spectra of higher L/Ti ratios. While it is not yet clear how Ti(IV) released into the cellular milieu following transmetalation will speciate, if Ti(IV) can bind and chemically modify nucleotide diphosphates then it may be able to decrease the RNR substrate pool.
Given the distinct but complementary targeting of DNA by the Ti(IV) cTfm complexes and cisplatin and other Pt-containg anticancer drugs, a Jurkat cotreatment study was performed to determine their ability to work in synergism. Cisplatin is commonly used in combination with other drugs to treat a variety of different cancer types including, but not limited to ovarian, testicular, and bladder cancers.76−80 The combination of two or more drugs can help to achieve an enhanced desired effect but with a reduced dosage of each drug, which may consequently reduce the side effects of the drugs and the chances for resistance development.81,82 Exploring the 1:1 and 2:1 cisplatin/Ti(Def)2 ratios of the compounds against Jurkat cells led to favorable synergistic and potent cytotoxic effects, suggesting that their combinatorial treatment should be screened more broadly against other cancer cell lines to consider future in vivo characterization.
Conclusion
Altogether, this work presents the Ti(IV) cTfm complexes as exhibiting the potential to serve as dual R1 and R2 RNR inhibitors (Scheme 2). Further studies are necessary to improve the potency of this family of compounds, to examine cotreatment with other lead drugs to determine synergistic cytotoxicity and to undertake mechanistic studies with the enzyme itself to evaluate the specificity of the Ti(IV) cTfm compounds for inhibiting the enzyme. In a broader context, this work reveals the power of using a transmetalative approach to exploit the distinct chemistry and cellular targets of cytotoxic metals in combination with the Fe debilitating power of key iron chelators to produce a multimodal broad-spectrum anticancer drug.
Experimental Section
Chemicals and Materials
N′-Di(o-hydroxybenzyl)ethylenediamine-N,N′-diacetic acid (HBED) was purchased from Strem Chemicals. 4-[(3Z,5E)-3,5-Bis(6-oxocyclohexa-2,4-dien-1-ylidene)-1,2,4-triazolidin-1-yl] benzoic acid (Def) was purchased from Focus Synthesis LCC (San Diego, CA). K2[Ti(citrate)3]·2H2O (Ti(IV) tricitrate) was synthesized following a previously published literature protocol.1 Fe(C6H5O7) (Fe(III) monocitrate) was purchased from Sigma, and FeCl3 was purchased from Alfa Aesar. Trisodium citrate was purchased from Acros Organics. Adenosine 5′-triphosphate disodium salt trihydrate (ATP), adenosine 5′-diphosphate disodium salt trihydrate (ADP), and adenosine 5′-monophosphate disodium salt trihydrate (AMP) were purchased from Fisher Scientific. Cisplatin was purchased Abcam. Ti atomic standard solution (1000 μg/mL) was purchased from Fluka Analytical. Human apo serum transferrin (T1147) and human methemoglobin (H7379) were purchased from Sigma. DNAzol was purchased from Molecular Research Center, Inc., and gelRed nucleic acid stain was purchased from (Biotium, Hayward, USA). Jurkat cells clone E6-1 was obtained from ATCC (ATCC TIB-152) authenticated with the certificate of analysis. RPMI-1640 media was purchased from Sigma and supplemented with fetal bovine serum (FBS; HyClone) and antibiotic solution prepared with 11 mg/mL of streptomycin and 7 mg/mL of penicillin (Calbiochem, EMD Biosciences Inc.). Mycoplasm removal agent was used in the preparation of cell stocks (Biorad). Single-donor human red blood cells resuspended in Alsever’s solution were obtained from Innovative Research (Novi, MI) authenticated with the certificate of analysis. Annexin V–Alexa Fluor 488 conjugate was purchased from Invitrogen (Thermo Fisher Scientific). 3-(4,5Ddimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma. All other chemicals were of high purity and used as received. All aqueous solutions were prepared with autoclaved (121 °C and 18 psi) high-quality nanopure water (18.2 MΩ·cm resistivity) at 25 °C, PURELAB flex system (ELGA Corporation).
Instrumentation
All UV–vis analyses were performed with either a Cary 300 UV–vis or NanoDrop 2000 (both from Agilent Technologies). The pH adjustments were done using a Thermo Scientific Orion Star A211 and an Orion 9157BNMD electrode, calibrated with standard buffer solutions at pH 4, 7, and 10. The electrode was calibrated before each measurement in units of mV. Continuous wave (CW)-EPR spectra were recorded at the University of Arizona, EPR Facility on an X-band EPR spectrometer Elexsys E500 (Bruker) equipped with the ESR900 flow cryostat (Oxford instruments). For cyclic voltammetric analysis, a VMP3 multichannel potentiostat (Biologic Science Instrument, USA) equipped with EC-Lab software and a three-electrode system was employed. Cell viability was determined using the MTT assay and the T20 automated cell counter (Bio-Rad). Cell cycle analysis was done using flow cytometry (C6 Accuri, BD Sciences). The agarose gel was imaged using a Sapphire Biomolecular Imager (Azure Biosystems, Dublin, CA). X-ray diffraction was performed using a Rigaku XtalLAB SuperNova single-microfocus Cu Kα radiation (λ = 1.5417 Å) source equipped with a HyPix3000 X-ray detector in transmission mode operating at 50 kV and 1 mA within the CrystallizPRO software ver. 1.171.39.43c. FTIR absorbance spectra were collected by using a Nicolet iS50 FTIR Spectrometer (ThermoFisher Scientific, WI). The energy dispersive X-ray spectrum (EDS) was obtained in a scanning electron microscope, a JEOL JSM-5800LV with 20 kV operating voltage. The ICP-OES was carried out using PerkinElmer Optima 8000 optical emission (PerkinElmer, Inc.). 31P nuclear magnetic resonance (31P NMR) recordings were carried out on a Bruker Ascend Aeon 700 NMR spectrometer with a 10 mm direct Z123952_0041 multinuclear probe. 96-Well plates were analyzed with an Infinite M200 PROTecan Microplate Reader.
Sample Solution Preparation
The neutral Ti(IV) compounds Ti(HBED) and Ti(Deferasirox)2 (Ti(Def)2) were synthesized by following our previously published protocol.42,47 The compounds were dissolved at 40 mM in DMF and then diluted to desired concentrations in 1× PBS (pH 7.4) for all the experiments except EPR studies (HEPES buffer). All final solutions contained no more than 1% DMF (v/v) unless specified otherwise. The Fe(III) compounds [Fe(Def)2]3– and [Fe(HBED)]− were prepared in situ by mixing 1 equiv of FeCl3 (dissolved in 10 mM HCl) with 2 equiv of Def (dissolved in DMF) and 1 equiv of HBED (dissolved in water), respectively, and then adjusting the pH to 7.4 with NaOH.35,41,43 The in situ preparation of a Fe(III) dicitrate ([Fe(Cit)2]5–) solution involved adding 1 mole equiv of aqueous sodium citrate to an aqueous solution of Fe(III) monocitrate, dissolved while heating. An in situ solution of [Fe(citrate)(Def)]4– ([Fe(Cit)(Def)]4–) was prepared by mixing an aqueous solution of [Fe(Cit)2]5– and Def (in DMF) in a 1:1 mol ratio and adjusting to pH 7.4. The in situ preparation of [Fe(Def)2]3– and [Fe(HBED)]− was validated by their characteristic LMCT bands: [Fe(Def)2]3–: λmax = 420 nm, ε = 5720 M–1 cm–1 and λ = 477 nm, ε = 4050 M–1 cm–1; [Fe(HBED)]−: λmax = 484 nm, ε = 3970 M–1 cm–1) using UV–vis spectroscopy.35,41,43 ([Fe(Cit)2]5–) is a mixture of [Fe(Cit)2]5– and other Fe(III) citrate species at pH 7.4.56,83 The exact nature of the speciation of all in situ prepared compounds was confirmed by modeling with the software “Species”.84 [Fe(Cit)(Def)]4– is characterized as described in the manuscript.
Characterization of Ti(Deferasirox)2 (Ti(Def)2) (Ti(C21H13N3O4)2) by Single-Crystal X-ray Diffraction
A neutral form of Ti(Def)2 was prepared following our previously published protocol.42 The compound was dissolved in DMF to saturation by vortexing until a clear solution was obtained. The solution was then covered with a poked parafilm and stored in the refrigerator at 4 °C. Orange-colored plate-like crystals developed within a few hours. A suitable single crystal was mounted on a MiTeGen MicroLoop. Structural elucidation was performed using a Rigaku XtalLAB SuperNova single-microfocus Cu Kα radiation (λ = 1.5417 Å) source equipped with a HyPix3000 X-ray detector in transmission mode operating at 50 kV and 1 mA within the CrystAlis Pro software ver. 1.171.39.43c.85 The data collection was carried out at 100 K using an Oxford Cryosystems Cryostream 800 cooler. The crystal structure was solved by direct methods using the program ShelXT86 and refined by full-matrix least-squares on F2 using ShelXL87 within the Olex2 (v1.2-ac3) software.88 Gaussian absorption correction was applied for all data. Crystal data: C42H26N6O8Ti, mol. wt. = 790.59 g/mol, orange plate, 0.133 × 0.08 × 0.049 mm, triclinic, P1̅, a = 12.2524(2) Å, b = 14.8895(3) Å, c = 15.0625(3) Å, α = 111.308(2)°, β = 104.390(2)°, γ = 99.092(2)°, V = 2383.98(9), Z = 2, ρcalc = 1.101 g·cm–3. The X-ray diffraction data were collected at T = 100.01(10) K, with Cu Kα radiation, μ = 1.943 mm–1, 48 025 reflections measured (6.628 ≤ 2θ ≤ 137.756°), 8773 unique (Rint = 0.0354, Rsigma = 0.0262), which were used in all calculations. The final R1 was 0.0547 (I > 2σ(I)), and wR2 was 0.1901 for all data. All non-hydrogen atoms were anisotropically refined. All the H atoms were placed in their calculated positions and then refined using the riding model with atom–H lengths of 0.95 Å (C–H) and 0.84 Å (O–H). Isotropic displacement parameters for these atoms were set to 1.2 (C–H) or 1.5 (O–H) times the Ueq of the parent atom. Idealized tetrahedral OH was refined as rotating groups. Large solvent-accessible voids in the crystal structure of Ti(Def)2 are occupied by interstitial solvent molecules, whose crystallographic disorder could not be modeled satisfactorily. Consequently, the diffraction data set of Ti(Def)2 was modified by the “solvent mask” feature of the OLEX2 program before the final refinement. The Ti(Def)2 structure is comparable to that of Ti(BHPT)2, which we previously published.42 The BHPT ligand possesses the same core bisphenoltriazole metal binding moieties as Def but lacks the benzoic acid substituent. As previously noted by Steinhauser et al., meridional coordination of this ligand is the only possible arrangement, but an entirely planar configuration of the three rings would result in significant stress.43 Therefore a low-strain meridional coordination results in twisting of the phenol rings in both the Ti(BHPT)2 and Ti(Def)2 structures. The average Ti–N bond lengths of Ti(Def)2 are 2.145 Å, just as they are for Ti(BHPT)2. The average Ti–O bond lengths of Ti(Def)2 are 1.887 Å, similar to those for Ti(BHPT)2 (1.888 Å). The presence of the benzoic acid substituent of Def causes a slight increase in the distortion of the bisphenoltriazole coordination. This can be observed in the N–Ti–N bond angle (173.54°) of Ti(Def)2 deviating from linearity more so than that of Ti(BHPT)2(177.92°). The average O–Ti–O bond angles (163.27°) of Ti(Def)2 are only slightly different from those of Ti(BHPT)2 (162.68°). A summary of the crystal data, structure solution, and refinement are listed in Table S1 with selected bond lengths and angles in Table S2. CCDC 2016873 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK; fax: + 44 1223 336033.
Jurkat Cell Culturing
Jurkat cells are nonadherent cells, and they were cultured in an RPMI-1640 media (supplemented with 10% FBS (v/v) and 1% antibiotic solution (v/v) of penicillin/streptomycin) in non-tissue-culture-treated 25 mL flasks according to the known protocol provided by the supplier ATCC. The cell passaging was done at 70% confluency by taking 3 mL of cells and diluting to 25 mL of fresh media. The cells were incubated at 37 °C in 5% CO2 atmosphere.
Whole Jurkat Cell EPR Analysis
Jurkat cells with a cell count of 2.54 × 106 cells/mL were treated with 50 μM of each compound HBED2–, Def–, [TiO(H+–HBED)]−, and [Ti(Def)2]2– with 0.05% v/v of DMSO. In contrast, the control group was treated with HEPES buffer (15 mM, pH 7.5, 5% glycerol (v/v)) and 0.05% v/v of DMSO. The cells were incubated for 3 h. After the corresponding incubation time, the cells were centrifuged for 10 min at 1020 rpm. The supernatant was discarded, and the cell pellet was resuspended in 2 mL of 1× PBS. The samples were centrifuged again for 10 min at 1020 rpm, and the supernatant was discarded. The cell pellet was resuspended in 0.5 mL of the HEPES buffer, and the solution was transferred to the EPR tubes. The tubes were centrifuged for 10 min, and the excess supernatant was carefully removed. The EPR tubes were frozen at −50 °C and then stored in liquid nitrogen until they could be measured by EPR. EPR analysis was done using conditions: microwave frequency, 9.338 GHz; microwave power, 2 mW; magnetic field modulation amplitude, 0.5 mT for the whole-cell tyrosyl radical detection and 1 mT for whole-cell high-spin Fe(III) (S = 5/2) detection; temperature, 20 K. Measurements were done in triplicate.
Acknowledgments
We acknowledge Dr. Carlos R. Cabrera’s researchers Dr. Nadja E. Solis-Marcano and Dr. Myreisa Morales Cruz for their assistance with CV. We thank Dr. Ritika Gautam and Dr. Yamixa Delgado for their insight into experiments and the UPR RP Materials Characterization Center for technical support with SEM-EDS. A.D.T. and K.G. were supported by the NIH 5SC1CA190504 grant (which also funded S.A.L.-R.), the UPR RP FIPI (which also funded R.K.S.), PBDT grants, and the NIH INBRE 5P20GM103475 grant. S.C.P.O. was also funded by the INBRE award and the PR LSAMP 1400868 grant. A.M.V.S. was supported by the NIH MARC 5T34GM007821-38 grant. J.A.B.R., I.R., and L.H.M. were supported by the NIH RISE 5R25GM061151-17 grant. N.A. was supported by the ACS Project SEED program. M.K. was funded by the NSF REU 1560278 grant. A.V.W. was funded by the NIH 1R01HL090933-01A2 grant. E.T. was funded by the NIH GM127646 grant. The X-ray work was collected with instrumentation provided by the NSF MRI grant CHE-1626103.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00078.
Author Contributions
K.G. and A.D.T. designed the research; K.G., S.C.P.O, J.A.B.R., I.R., S.A.L.R., A.M.V.S., E.A.A., L.H.M., R.K.S., N.A., M.K., A.V.W., A.V.A., E.T., and A.D.T. performed research and analyzed data; K.G. and A.D.T. wrote the manuscript with commentary provided by E.A.A., E.T., I.R., and J.A.B.R. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Weinberg R.The biology of cancer, 2nd ed.; Garland Science: New York, 2013; p 960. [Google Scholar]
- Cairo G.; Bernuzzi F.; Recalcati S. A precious metal: Iron, an essential nutrient for all cells. Genes Nutr. 2006, 1 (1), 25–39. 10.1007/BF02829934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane D. J. R.; Merlot A. M.; Huang M. L. H.; Bae D. H.; Jansson P. J.; Sahni S.; Kalinowski D. S.; Richardson D. R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta, Mol. Cell Res. 2015, 1853 (5), 1130–1144. 10.1016/j.bbamcr.2015.01.021. [DOI] [PubMed] [Google Scholar]
- Sutherland R.; Delia D.; Schneider C.; Newman R.; Kemshead J.; Greaves M. Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proc. Natl. Acad. Sci. U. S. A. 1981, 78 (7), 4515. 10.1073/pnas.78.7.4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott C. C.; Ryu M.-S. Special delivery: distributing iron in the cytosol of mammalian cells. Front. Pharmacol. 2014, 5, 173. 10.3389/fphar.2014.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinnix Z. K.; Miller L. D.; Wang W.; D’Agostino R.; Kute T.; Willingham M. C.; Hatcher H.; Tesfay L.; Sui G.; Di X.; Torti S. V.; Torti F. M. Ferroportin and Iron Regulation in Breast Cancer Progression and Prognosis. Sci. Transl. Med. 2010, 2 (43), 43ra56. 10.1126/scitranslmed.3001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannargudi M. B.; Deb S. Clinical pharmacology and clinical trials of ribonucleotide reductase inhibitors: is it a viable cancer therapy?. J. Cancer Res. Clin. Oncol. 2017, 143 (8), 1499–1529. 10.1007/s00432-017-2457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene B. L.; Kang G.; Cui C.; Bennati M.; Nocera D. G.; Drennan C. L.; Stubbe J. Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem. 2020, 89, 45–75. 10.1146/annurev-biochem-013118-111843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal D.; Goodison S.; Nicholson B.; Tarin D.; Urquidi V. Expression of matrix metalloproteinase 8 (MMP-8) and tyrosinase-related protein-1 (TYRP-1) correlates with the absence of metastasis in an isogenic human breast cancer model. Differentiation 2003, 71 (2), 114–25. 10.1046/j.1432-0436.2003.710202.x. [DOI] [PubMed] [Google Scholar]
- Toyokuni S. Iron-induced carcinogenesis: The role of redox regulation. Free Radical Biol. Med. 1996, 20 (4), 553–566. 10.1016/0891-5849(95)02111-6. [DOI] [PubMed] [Google Scholar]
- Kicic A.; Chua A. C. G.; Baker E. Desferrithiocin is a more potent antineoplastic agent than desferrioxamine. Br. J. Pharmacol. 2002, 135 (6), 1393–1402. 10.1038/sj.bjp.0704507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taetle R.; Honeysett J. M.; Bergeron R. Combination iron depletion therapy. J. Natl. Cancer Inst. 1989, 81 (16), 1229–35. 10.1093/jnci/81.16.1229. [DOI] [PubMed] [Google Scholar]
- Gaur K.; Vázquez-Salgado A. M.; Duran-Camacho G.; Dominguez-Martinez I.; Benjamín-Rivera J. A.; Fernández-Vega L.; Carmona Sarabia L.; Cruz García A.; Pérez-Deliz F.; Méndez Román J. A.; Vega-Cartagena M.; Loza-Rosas S. A.; Rodriguez Acevedo X.; Tinoco A. D. Iron and Copper Intracellular Chelation as an Anticancer Drug Strategy. Inorganics 2018, 6 (4), 126. 10.3390/inorganics6040126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J. M.; Zhou B. S.; Di Bilio A. J.; Zhu L. J.; Wang T. L.; Qi C.; Shih J.; Yen Y. A ferrous-Triapine complex mediates formation of reactive oxygen species that inactivate human ribonucleotide reductase. Mol. Cancer Ther. 2006, 5 (3), 586–592. 10.1158/1535-7163.MCT-05-0384. [DOI] [PubMed] [Google Scholar]
- Wijerathna S. R.; Ahmad M. F.; Xu H.; Fairman J. W.; Zhang A.; Kaushal P. S.; Wan Q.; Kiser J.; Dealwis C. G. Targeting the Large Subunit of Human Ribonucleotide Reductase for Cancer Chemotherapy. Pharmaceuticals 2011, 4 (10), 1328–1354. 10.3390/ph4101328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J.; Liu X.; Zhu L.; Yen Y. Targeting ribonucleotide reductase for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 1423–1437. 10.1517/14728222.2013.840293. [DOI] [PubMed] [Google Scholar]
- Torrents E. Ribonucleotide reductases: essential enzymes for bacterial life. Front. Cell. Infect. Microbiol. 2014, 4, 52. 10.3389/fcimb.2014.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Liu G.; Huang M. Ribonucleotide reductase metallocofactor: assembly, maintenance and inhibition. Front. Biol. 2014, 9 (2), 104–113. 10.1007/s11515-014-1302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantchik Z. I. Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45. 10.3389/fphar.2014.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena M.; Loza Rosas S.; Gaur K.; Sharma S.; Perez Otero S. C.; Tinoco A. D. Exploring titanium(IV) chemical proximity to iron(III) to elucidate a function for Ti(IV) in the human body. Coord. Chem. Rev. 2018, 363, 109–125. 10.1016/j.ccr.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M. L.; Guo Z. J.; Sadler P. J. Titanium(IV) targets phosphoesters on nucleotides: implications for the mechanism of action of the anticancer drug titanocene dichloride. JBIC, J. Biol. Inorg. Chem. 2001, 6 (7), 698–707. 10.1007/s007750100248. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Low T. Y.; Hennrich M. L.; van der Toorn H.; Schwend T.; Zou H.; Mohammed S.; Heck A. J. Enhancing the identification of phosphopeptides from putative basophilic kinase substrates using Ti (IV) based IMAC enrichment. Mol. Cell. Proteomics 2011, 10 (10), M110.006452. 10.1074/mcp.M110.006452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loza-Rosas S. A.; Saxena M.; Delgado Y.; Gaur K.; Pandrala M.; Tinoco A. D. A ubiquitous metal, difficult to track: towards an understanding of the regulation of titanium(iv) in humans. Metallomics 2017, 9 (4), 346–356. 10.1039/C6MT00223D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner K. M.; Valentine A. M. Bioinorganic Chemistry of Titanium. Chem. Rev. 2012, 112 (3), 1863–1881. 10.1021/cr1002886. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Sharma R. K.; Gaur K.; Cátala Torres J. F.; Loza-Rosas S. A.; Torres A.; Saxena M.; Julin M.; Tinoco A. D. Fueling a Hot Debate on the Application of TiO2 Nanoparticles in Sunscreen. Materials 2019, 12 (14), 2317. 10.3390/ma12142317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tshuva E. Y.; Peri D. Modern cytotoxic titanium(IV) complexes; Insights on the enigmatic involvement of hydrolysis. Coord. Chem. Rev. 2009, 253 (15–16), 2098–2115. 10.1016/j.ccr.2008.11.015. [DOI] [Google Scholar]
- Tshuva E. Y.; Ashenhurst J. A. Cytotoxic Titanium(IV) Complexes: Renaissance. Eur. J. Inorg. Chem. 2009, 2009 (15), 2203–2218. 10.1002/ejic.200900198. [DOI] [Google Scholar]
- Cini M.; Bradshaw T. D.; Woodward S. Using titanium complexes to defeat cancer: the view from the shoulders of titans. Chem. Soc. Rev. 2017, 46 (4), 1040–1051. 10.1039/C6CS00860G. [DOI] [PubMed] [Google Scholar]
- Fernández-Vega L.; Ruiz Silva V. A.; Domínguez-González T. M.; Claudio-Betancourt S.; Toro-Maldonado R. E.; Capre Maso L. C.; Sanabria Ortiz K.; Pérez-Verdejo J. A.; Román González J.; Rosado-Fraticelli G. T.; Pagán Meléndez F.; Betancourt Santiago F. M.; Rivera-Rivera D. A.; Martínez Navarro C.; Bruno Chardón A. C.; Vera A. O.; Tinoco A. D. Evaluating Ligand Modifications of the Titanocene and Auranofin Moieties for the Development of More Potent Anticancer Drugs. Inorganics 2020, 8 (2), 10. 10.3390/inorganics8020010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hider R. C.; Kong X. L. Iron speciation in the cytosol: an overview. Dalton Trans. 2013, 42 (9), 3220–3229. 10.1039/C2DT32149A. [DOI] [PubMed] [Google Scholar]
- Tinoco A. D.; Saxena M.; Sharma S.; Noinaj N.; Delgado Y.; Quiñones González E. P. Q.; Conklin S. E.; Zambrana N.; Loza-Rosas S. A.; Parks T. B. Unusual synergism of transferrin and citrate in the regulation of Ti(IV) speciation, transport, and toxicity. J. Am. Chem. Soc. 2016, 138 (17), 5659–5665. 10.1021/jacs.6b01966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma R.; Motekaitis R. J.; Martell A. E. Stability of metal-ion complexes of N,N’-bis(2-hydroxybenzyl)ethylenediamine-N,N’-diacetic acid. Inorg. Chim. Acta 1994, 224 (1–2), 151–155. 10.1016/0020-1693(94)04012-5. [DOI] [Google Scholar]
- Bergeron R. J.; Wiegand J.; Brittenham G. M. HBED ligand: preclinical studies of a potential alternative to deferoxamine for treatment of chronic iron overload and acute iron poisoning. Blood 2002, 99 (8), 3019–3026. 10.1182/blood.V99.8.3019. [DOI] [PubMed] [Google Scholar]
- Khalaf S.; Ahmad A. S.; Chamara K.; Dore S. Unique Properties Associated with the Brain Penetrant Iron Chelator HBED Reveal Remarkable Beneficial Effects after Brain Trauma. J. Neurotrauma 2019, 36 (1), 43–53. 10.1089/neu.2017.5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz U.; Hegetschweiler K.; Acklin P.; Faller B.; Lattmann R.; Schnebli H. P. 4-[3,5-Bis(2-hydroxyphenyl)-1,2,4-triazol-1-yl]- benzoic Acid: A Novel Efficient and Selective Iron(III) Complexing Agent. Angew. Chem., Int. Ed. 1999, 38 (17), 2568–2570. . [DOI] [PubMed] [Google Scholar]
- Taher A. T.; Porter J.; Viprakasit V.; Kattamis A.; Chuncharunee S.; Sutcharitchan P.; Siritanaratkul N.; Galanello R.; Karakas Z.; Lawniczek T.; Ros J.; Zhang Y. Y.; Habr D.; Cappellini M. D. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood 2012, 120 (5), 970–977. 10.1182/blood-2012-02-412692. [DOI] [PubMed] [Google Scholar]
- Bedford M. R.; Ford S. J.; Horniblow R. D.; Iqbal T. H.; Tselepis C. Iron Chelation in the Treatment of Cancer: A New Role for Deferasirox?. J. Clin. Pharmacol. 2013, 53 (9), 885–891. 10.1002/jcph.113. [DOI] [PubMed] [Google Scholar]
- Ibrahim O.; O’Sullivan J. Iron chelators in cancer therapy. BioMetals 2020, 33 (4–5), 201–215. 10.1007/s10534-020-00243-3. [DOI] [PubMed] [Google Scholar]
- Moghadam S. M. M.; Alibolandi M.; Babaei M.; Mosafer J.; Saljooghi A. S.; Ramezani M. Fabrication of deferasirox-decorated aptamer-targeted superparamagnetic iron oxide nanoparticles (SPION) as a therapeutic and magnetic resonance imaging agent in cancer therapy. JBIC, J. Biol. Inorg. Chem. 2021, 26 (1), 29–41. 10.1007/s00775-020-01834-8. [DOI] [PubMed] [Google Scholar]
- U.S. National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/home (accessed 2021-04-06).
- Parks T. B.; Cruz Y. M.; Tinoco A. D. Applying the Fe(III) Binding Property of a Chemical Transferrin Mimetic to Ti(IV) Anticancer Drug Design. Inorg. Chem. 2014, 53 (3), 1743–1749. 10.1021/ic4028749. [DOI] [PubMed] [Google Scholar]
- Loza-Rosas S. A.; Vázquez-Salgado A. M.; Rivero K. I.; Negrón L. J.; Delgado Y.; Benjamin-Rivera J. A.; Vazquez-Maldonado A. L.; Parks T. B.; Munet-Colon C.; Tinoco A. D. Expanding the therapeutic potential of the iron chelator deferasirox in the development of aqueous stable Ti(IV) anticancer complexes. Inorg. Chem. 2017, 56, 7788–7802. 10.1021/acs.inorgchem.7b00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauser S.; Heinz U.; Bartholomä M.; Weyhermüller T.; Nick H.; Hegetschweiler K. Complex formation of ICL670 and related ligands with Fe(III) and Fe(II). Eur. J. Inorg. Chem. 2004, 2004 (21), 4177–4192. 10.1002/ejic.200400363. [DOI] [Google Scholar]
- Stucky S.; Koch N. J.; Heinz U.; Hegetschweiler K. 3,5-Bis(2-hydroxyphenyl)-1H-1,2,4-triazole based ligands — protonation and metal complex formation. Chem. Pap. 2008, 62 (4), 388–397. 10.2478/s11696-008-0043-x. [DOI] [Google Scholar]
- Collins J. M.; Uppal R.; Incarvito C. D.; Valentine A. M. Titanium(IV) Citrate Speciation and Structure under Environmentally and Biologically Relevant Conditions. Inorg. Chem. 2005, 44 (10), 3431–3440. 10.1021/ic048158y. [DOI] [PubMed] [Google Scholar]
- Tinoco A. D.; Thomas H. R.; Incarvito C. D.; Saghatelian A.; Valentine A. M. Cytotoxicity of a Ti(IV) compound is independent of serum proteins. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (13), 5016–5021. 10.1073/pnas.1119303109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco A. D.; Incarvito C. D.; Valentine A. M. Calorimetric, Spectroscopic, and Model Studies Provide Insight into the Transport of Ti(IV) by Human Serum Transferrin. J. Am. Chem. Soc. 2007, 129 (11), 3444–3454. 10.1021/ja068149j. [DOI] [PubMed] [Google Scholar]
- DesMarteau D.; Lu C. Syntheses and lipophilicity measurement of N α/ N-terminus-1,1-dihydroperfluoroalkylated α-amino acids and small peptides. J. Fluorine Chem. 2007, 128, 1326–1334. 10.1016/j.jfluchem.2007.07.003. [DOI] [Google Scholar]
- Akam E. A.; Chang T. M.; Astashkin A. V.; Tomat E. Intracellular reduction/activation of a disulfide switch in thiosemicarbazone iron chelators. Metallomics 2014, 6 (10), 1905–1912. 10.1039/C4MT00153B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam R.; Akam E. A.; Astashkin A. V.; Loughrey J. J.; Tomat E. Sirtuin inhibitor sirtinol is an intracellular iron chelator. Chem. Commun. (Cambridge, U. K.) 2015, 51 (24), 5104–5107. 10.1039/C5CC00829H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolberg M.; Strand K. R.; Graff P.; Andersson K. K. Structure, function, and mechanism of ribonucleotide reductases. Biochim. Biophys. Acta, Proteins Proteomics 2004, 1699 (1–2), 1–34. 10.1016/S1570-9639(04)00054-8. [DOI] [PubMed] [Google Scholar]
- Kowol C. R.; Trondl R.; Heffeter P.; Arion V. B.; Jakupec M. A.; Roller A.; Galanski M.; Berger W.; Keppler B. K. Impact of metal coordination on cytotoxicity of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (triapine) and novel insights into terminal dimethylation. J. Med. Chem. 2009, 52 (16), 5032–43. 10.1021/jm900528d. [DOI] [PubMed] [Google Scholar]
- Popovic-Bijelic A.; Kowol C. R.; Lind M. E. S.; Luo J. H.; Himo F.; Enyedy E. A.; Arion V. B.; Graslund A. Ribonucleotide reductase inhibition by metal complexes of Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone): A combined experimental and theoretical study. J. Inorg. Biochem. 2011, 105 (11), 1422–1431. 10.1016/j.jinorgbio.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye Y.; Long M. J. C.; Stubbe J. Mechanistic Studies of Semicarbazone Triapine Targeting Human Ribonucleotide Reductase in Vitro and in Mammalian Cells: Tyrosyl radical quenching not involving reactive oxygen species. J. Biol. Chem. 2012, 287 (42), 35768–35778. 10.1074/jbc.M112.396911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eplattenier F. L.; Murase I.; Martell A. E. New Multidentate Ligands. VI. Chelating Tendencies of N,N’-Di(2-hydroxybenzyl)ethylenediamine-N,N’-diacetic Acid. J. Am. Chem. Soc. 1967, 89 (4), 837–843. 10.1021/ja00980a019. [DOI] [Google Scholar]
- Silva A. M. N.; Kong X.; Parkin M. C.; Cammack R.; Hider R. C. Iron(III) citrate speciation in aqueous solution. Dalton Trans. 2009, (40), 8616–8625. 10.1039/b910970f. [DOI] [PubMed] [Google Scholar]
- Go Y.-M.; Jones D. P. The Redox Proteome. J. Biol. Chem. 2013, 288 (37), 26512–26520. 10.1074/jbc.R113.464131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T. T.; Strozynski M.; Thiede B. Quantitative phosphoproteome analysis of cisplatin-induced apoptosis in Jurkat T cells. Proteomics 2017, 17 (11), 1600470. 10.1002/pmic.201600470. [DOI] [PubMed] [Google Scholar]
- Schaer D. J.; Buehler P. W.; Alayash A. I.; Belcher J. D.; Vercellotti G. M. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121 (8), 1276–1284. 10.1182/blood-2012-11-451229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F.; Alayash A. I. Determination of extinction coefficients of human hemoglobin in various redox states. Anal. Biochem. 2017, 521, 11–19. 10.1016/j.ab.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell A.; Motekaitis R.; Chen D.; Hancock R.; McManus D. Selection of new Fe(III)/Fe(II) chelating agents as catalysts for the oxidation of hydrogen sulfide to sulfur by air. Can. J. Chem. 1996, 74, 1872–1879. 10.1139/v96-210. [DOI] [Google Scholar]
- Bertini I.; Gray H. B.; Stiefel E. I.; Valentine J. S.. Biological Inorganic Chemistry: Structure and Reactivity. University Science Book, 2006; p 95–135. [Google Scholar]
- Dabrowiak J. C.Metals in Medicine, 2nd ed.; Wiley, 2017; p 91–146. [Google Scholar]
- Chou T.-C.; Talalay P. Quantitative Analysis of Dose-Effect Relationships: The Combined effects of Multiple Drugs or Enzyme Inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Tallarida R. J.; Porreca F.; Cowan A. Staistical Analysis of Drug-Drug and Site-Site Interactions with Isobolograms. Life Sci. 1989, 45, 947–961. 10.1016/0024-3205(89)90148-3. [DOI] [PubMed] [Google Scholar]
- Gessner P. K. Isobolographic analysis of interactions: an update on applications and utility. Toxicology 1995, 105, 161–179. 10.1016/0300-483X(95)03210-7. [DOI] [PubMed] [Google Scholar]
- Guo M. L.; Sun H. Z.; McArdle H. J.; Gambling L.; Sadler P. J. Ti(IV) uptake and release by human serum transferrin and recognition of Ti(IV) transferrin by cancer cells: Understanding the mechanism of action of the anticancer drug titanocene dichloride. Biochemistry 2000, 39 (33), 10023–10033. 10.1021/bi000798z. [DOI] [PubMed] [Google Scholar]
- Tinoco A. D.; Eames E. V.; Valentine A. M. Reconsideration of serum Ti(IV) transport: Albumin and transferrin trafficking of Ti(IV) and its complexes. J. Am. Chem. Soc. 2008, 130 (7), 2262–2270. 10.1021/ja076364+. [DOI] [PubMed] [Google Scholar]
- Guo M. L.; Sadler P. J. Competitive binding of the anticancer drug titanocene dichloride to N,N-ethylenebis(o-hydroxyphenylglycine) and adenosine triphosphate: a model for Ti-IV uptake and release by transferrin. Dalton Trans. 2000, (1), 7–9. 10.1039/a908759a. [DOI] [Google Scholar]
- McLaughlin M. L.; Cronan J. M.; Schaller T. R.; Snelling R. D. DNA-metal binding by antitumor-active metallocene dichlorides from inductively coupled plasma spectroscopy analysis: titanocene dichloride forms DNA-Cp2Ti or DNA-CpTi adducts depending on pH. J. Am. Chem. Soc. 1990, 112 (24), 8949–8952. 10.1021/ja00180a046. [DOI] [Google Scholar]
- Erxleben A.; Claffey J.; Tacke M. Binding and hydrolysis studies of antitumoural titanocene dichloride and Titanocene Y with phosphate diesters. J. Inorg. Biochem. 2010, 104 (4), 390–6. 10.1016/j.jinorgbio.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Trublet M.Titanium(IV) Phosphates: The Next Generation of Wastewater Sorbents. Doctoral thesis, Luleå University of Technology: Luleå, Sweden, 2018. [Google Scholar]
- Rampazzo C.; Miazzi C.; Franzolin E.; Pontarin G.; Ferraro P.; Frangini M.; Reichard P.; Bianchi V. Regulation by degradation, a cellular defense against deoxyribonucleotide pool imbalances. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2010, 703 (1), 2–10. 10.1016/j.mrgentox.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Christodoulou C. V.; Eliopoulos A. G.; Young L. S.; Hodgkins L.; Ferry D. R.; Kerr D. J. Anti-proliferative activity and mechanism of action of titanocene dichloride. Br. J. Cancer 1998, 77 (12), 2088–2097. 10.1038/bjc.1998.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan J.; Antholine W. E.; Chitambar C. R. Effect of gallium on the tyrosyl radical of the iron-dependent M2 subunit of ribonucleotide reductase. Biochem. Pharmacol. 1992, 44 (12), 2403–2408. 10.1016/0006-2952(92)90686-D. [DOI] [PubMed] [Google Scholar]
- Agarwal R.; Kaye S. B. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3 (7), 502–16. 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- Boulikas T.; Vougiouka M. Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs. Oncol. Rep. 2004, 11, 559–595. 10.3892/or.11.3.559. [DOI] [PubMed] [Google Scholar]
- Armstrong D. K.; Bundy B.; Wenzel L.; Huang H. Q.; Baergen R.; Lele S.; Copeland L. J.; Walker J. L.; Burger R. A. Intraperitoneal Cisplatin and Paclitaxel in Ovarian Cancer. N. Engl. J. Med. 2006, 354, 34–43. 10.1056/NEJMoa052985. [DOI] [PubMed] [Google Scholar]
- von der Maase H.; Hansen S. W.; Roberts J. T.; Dogliotti L.; Oliver T.; Moore M. J.; Bodrogi I.; Albers P.; Knuth A.; Lippert C. M.; Kerbrat P.; Sanchez Rovira P.; Wersall P.; Cleall S. P.; Roychowdhury D. F.; Tomlin I.; Visseren-Grul C. M.; Conte P. F. Gemcitabine and Cisplatin Versus Methotrexate, Vinblastine, Doxorubicin, and Cisplatin in Advanced or Metastatic Bladder Cancer: Results of a Large, Randomized, Multinational, Multicenter, Phase III Study. J. Clin. Oncol. 2000, 18, 3068–3077. 10.1200/JCO.2000.18.17.3068. [DOI] [PubMed] [Google Scholar]
- Numahata K.; Hoshi S.; Hoshi K.; Yasuno N.; Kubo M.; Sasagawa I.; Ohta S. Remarkable response to neoadjuvant therapy with methotrexate, vinblastine, adriamycin, and Cisplatin for undifferentiated bladder carcinoma: a case report and literature review. Case Rep. Oncol. 2014, 7 (3), 746–50. 10.1159/000369004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S. M.; Saravolatz L. D. Monotherapy versus combination therapy. Med. Clin. North Am. 2006, 90 (6), 1183–95. 10.1016/j.mcna.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Toews M. L.; Bylund D. B. Pharmacologic principles for combination therapy. Proc. Am. Thorac. Soc. 2005, 2 (4), 282–9. (discussion 290–291) 10.1513/pats.200504-037SR. [DOI] [PubMed] [Google Scholar]
- Vukosav P.; Mlakar M.; Tomišić V. Revision of iron(III)-citrate speciation in aqueous solution. Voltammetric and spectrophotometric studies. Anal. Chim. Acta 2012, 745, 85–91. 10.1016/j.aca.2012.07.036. [DOI] [PubMed] [Google Scholar]
- Sukhno I. V.; Buzko V. Y.; Pettit L. D.. Software for the quantitative treatment of equilibria in solution. https://www.acadsoft.co.uk/aq_solutions.htm (accessed 1 December 2020).
- CrysAlis PRO, CrysAlis CCD, and CrysAlis RED; Rigaku Oxford Diffraction: Oxfordshire, England, 2016.
- Sheldrick G. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71 (1), 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71 (1), 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42 (2), 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.