

Abstract
The hydrophobic surfactant proteins, SP-B and SP-C, promote rapid adsorption by the surfactant lipids to the surface of the liquid that lines the alveolar air sacs of the lungs. To gain insights into the mechanisms of their function, we used X-ray diffuse scattering (XDS) and molecular dynamics (MD) simulations to determine the location of SP-B and SP-C within phospholipid bilayers. Initial samples contained the surfactant lipids from extracted calf surfactant with increasing doses of the proteins. XDS located protein density near the phospholipid headgroup and in the hydrocarbon core, presumed to be SP-B and SP-C, respectively. Measurements on dioleoylphosphatidylcholine (DOPC) with the proteins produced similar results. MD simulations of the proteins with DOPC provided molecular detail and allowed direct comparison of the experimental and simulated results. Simulations used conformations of SP-B based on other members of the saposin-like family, which form either open or closed V-shaped structures. For SP-C, the amino acid sequence suggests a partial α-helix. Simulations fit best with measurements of XDS for closed SP-B, which occurred at the membrane surface, and SP-C oriented along the hydrophobic interior. Our results provide the most definitive evidence yet concerning the location and orientation of the hydrophobic surfactant proteins.
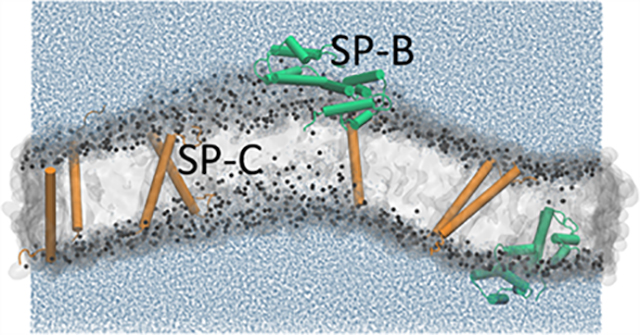
Graphical Abstract
INTRODUCTION
Pulmonary surfactant is a mixture of lipids and proteins secreted by the type II pneumocytes of the alveolar air sacs. The mixture acts as a surfactant, adsorbing to the surface of the liquid layer that lines the alveolus and forming a thin film that reduces surface tension.1 This function is essential for maintaining the integrity of the alveolus during normal breathing. Ventilation of premature neonates born before the lungs have adequate amounts of surfactant,2 and of adult animals depleted of surfactant by repeated lavage,3 shreds the thin barrier that separates alveolar air from capillary blood. The lungs develop pulmonary edema, which causes respiratory failure.
Two hydrophobic proteins, SP-B and SP-C, are required for the function of pulmonary surfactant. Over a relatively prolonged period, patients and genetically modified animals that lack SP-C develop pulmonary fibrosis.4,5 The effect of deficient SP-B is more immediate, equivalent to the injury that results from the absence of the complete surfactant mixture.6,7 Although SP-B and SP-C represent only ~1.5% (w:w) (~500:1 lipid:protein molar ratio) of pulmonary surfactant,8 their functional contribution is essential. Studies in vitro show that the proteins accelerate adsorption of the surfactant lipids to an air/water interface by orders of magnitude.9–11 SP-B is responsible for most of that effect.12
The mechanisms by which the proteins achieve their function remain unresolved. Structural information, which might provide functional insights, has been limited largely to secondary structure, based on the amino acid sequences and spectroscopic studies.1,13,14 SP-B belongs to the family of saposin-like proteins.15 These polypeptides form a series of amphipathic helices that adopt the antiparallel, cross-linked configuration of the saposin fold.16,17 Most of SP-C instead forms a hydrophobic helix.18 The locations of these proteins within the bilayer remain incompletely defined. The studies reported here used a combination of X-ray diffuse scattering (XDS) and molecular dynamics (MD) simulation to address that question.
METHODS
X-ray Diffuse Scattering (XDS).
Materials.
The hydrophobic constituents of calf surfactant (calf lung surfactant extract, CLSE), obtained by extracting19 phospholipid aggregates lavaged from freshly excised calf lungs,20 were provided by ONY, Inc. (Amherst, NY). Subfractions of the constituents in CLSE were obtained by using established methods8 based on gel permeation chromatography.21,22 These procedures yielded the isolated hydrophobic proteins (SPs) and the complete set of nonpolar and phospholipids (N&PL) without the proteins.8 Concentrations of the proteins and phospholipids were determined by colorimetric assays,23,24 using bovine serum albumin as the standard protein. Table 1 provides details concerning the known physical constants for bovine SP-B and SP-C and their ratio in the physiological mixture.
Table 1.
| protein | SP-B (dimer) | SP-C |
|---|---|---|
| molecular weight (Da) | 17,397 | 4042 |
| no. amino acids | 158 | 34 |
| net charge (e′) | +12 | +2 |
| weight ratio | 1 | 1 |
| molar ratio | 0.21 | 1 |
Dioleoylphosphatidylcholine (DOPC) was obtained from Avanti Polar Lipids (Alabaster, AL) and used without further characterization or purification. The following reagents were purchased: chloroform, trifluoroethanol, and methanol (Thermo Fischer Scientific, Pittsburgh, PA).
Sample Preparation.
Lipids and proteins were mixed in solutions of nonpolar solvents: N&PL:CLSE, 4 mg in 200 μL of chloroform:trifluoroethanol (TFE) (1:1 v:v); SP:DOPC, 4 mg in 200 μL of chloroform:methanol (3:1, v:v). The ratio of N&PL and CLSE was expressed as the mole fraction between 0 and 1.0 of phospholipid contributed by CLSE (XCLSE). Weight ratios were calculated by using an average molecular weight for the lipids in CLSE of 736 Da.27
The mixtures were plated onto silicon wafers (15 × 1.5 × 30 mm3) via the rock-and-roll method,28 in which rocking the silicon wafer continuously during solvent evaporation produces stacks of ~1800 well-aligned bilayers. Once immobile, further solvent was evacuated for at least 2 h. The sample was trimmed to a central strip 5 mm wide and parallel to the long edge of the wafer.28 A thick-walled, well-insulated X-ray chamber that maintained 100% humidity hydrated the films.29
Data Collection and Analysis.
XDS from oriented, fully hydrated samples were obtained on the G1 line at the Cornell High Energy Synchrotron Source (CHESS, Ithaca, NY) during two separate trips using wavelengths of 1.1775 and 1.0976 Å. Measurements with DOPC were performed at the experimentally convenient temperature of 30 °C, at which the membranes exist in the fluid phase. In mixtures of N&PL with CLSE below XCLSE of 0.9, the wide-angle X-ray pattern at 37 °C indicated a fluid/gel (Lα/Lβ) coexistence. The strong lamellar reflections from the gel phase dominated the low angle pattern from fluid phase structures and limited measurements of scattering that determine the form factor. Experiments at 40 °C for these samples with the surfactant lipids melted the gel phase and eliminated that problem.
The flat silicon wafer was rotated from −1.6° to 7° during the measurement to sample all scattered X-rays equally (30 s dezingered scans). The background was collected by setting the X-ray angle of incidence to −2.4°, where scattering from the sample did not contribute to the image. The analysis first subtracted backgrounds to remove extraneous scattering by air and Mylar. The images were then laterally symmetrized to increase the signal-to-noise ratio. In these experiments, when the samples became well hydrated, membrane fluctuations produced “lobes” of XDS30,31 (Figure 1). The analysis of these data begins with quantitation of the fluctuations by measuring the falloff of intensity in the lateral (qr) direction. The fitting procedure uses a nonlinear least-squares fit based on the free energy functional from liquid crystal theory
| (1) |
where N is the number of bilayers in the z (vertical) direction, Lr is the domain size in the r (horizontal) direction, Kc is the bending modulus, un is the vertical membrane displacement, and B is the compressibility modulus.32 This step determines the structure factor, S(q). S(q) is then fixed, and I(q), the intensity, is determined by using the NFIT program.31 In the next step, the scattering density profile (SDP) program33,34 performs the Lorentz and absorption corrections and calculates the form factor |F(qz)|:32
| (2) |
Fourier transformation of the form factor yields the electron density profile (EDP). Fitting the electron densities of the components to the profile (see the Results section) determines their locations. XDS can also be used with MD simulations. Agreement between the form factors generated by experimental measurement and the simulated structure supports the results of the simulation.
Figure 1.
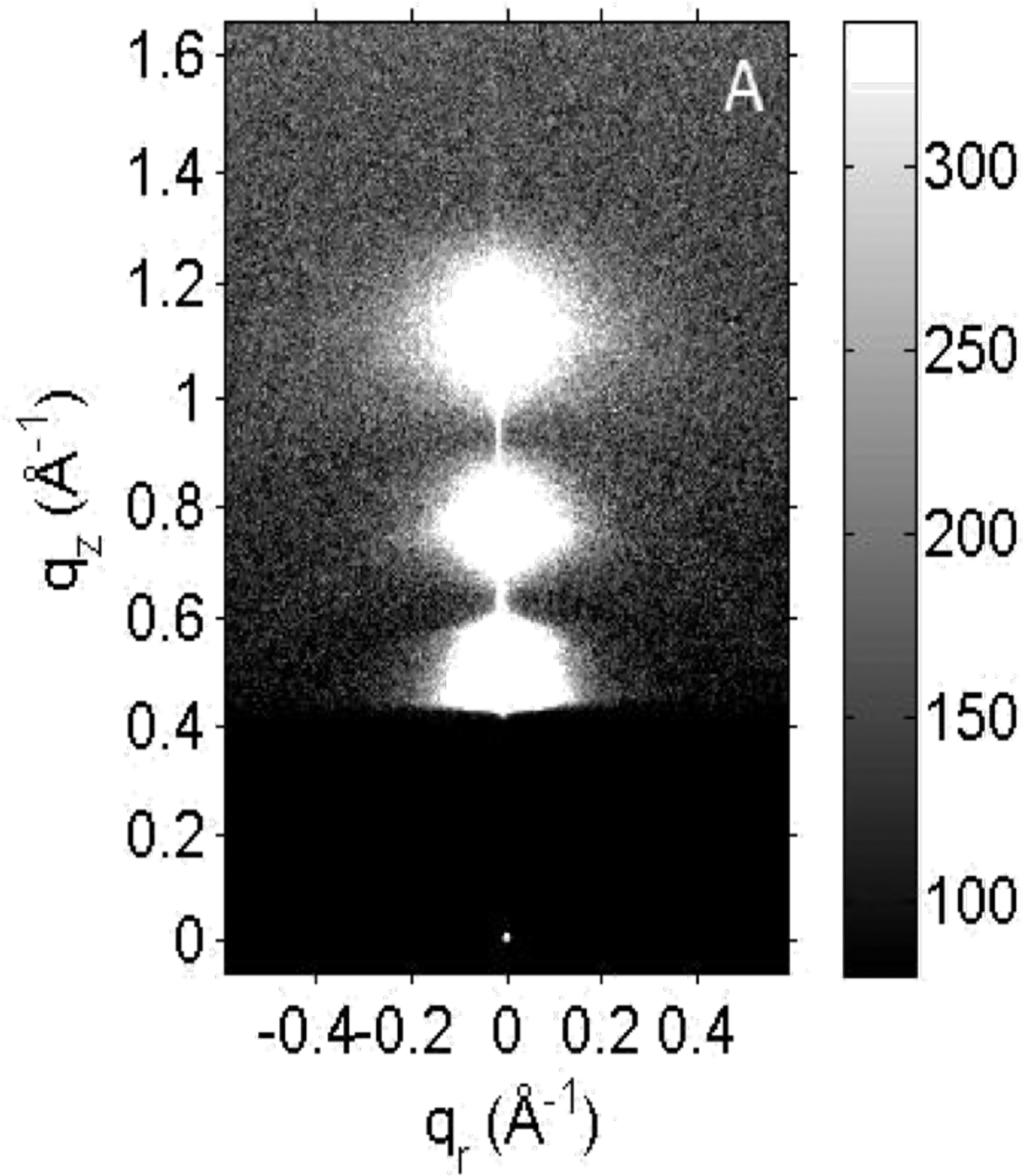
XDS pattern from XCLSE = 0.9 at 40 °C, with D-spacing = 81 Å. At this grayscale, lobes of white diffuse scattering obscure the diffracted lamellar orders. The X-ray beam appears as a small, white dot at qr, qz = 0 Å−1 through the semitransparent, dark rectangular beamstop.
We chose not to correct the qz positions in the form factors for membrane fluctuations,35 since a better agreement with MD simulations of the proteins in DOPC occurred without that correction. Because the SDP program is based on molecular volumes, the resulting EDPs are on an absolute scale (e/Å3). The modeled locations of the different components provided by the program yield structural parameters, such as DHH (peak head-to-head thickness), 2DC (hydrocarbon thickness), and AUC (area per unit cell), given by VC/DC, where VC is the volume of the unit cell. A detailed description of these analytical procedures has been published.36
Densitometry.
Calculation of the EDP on an absolute scale required the molecular volumes of the different components. We obtained the molecular volumes from densities of N&PL:CLSE mixtures at ~2% (w:w) in ~1.2 mL of water measured at 37 °C with an Anton-Paar DMA 5000M densitometer.37 The instrument uses a vibrating tube to detect shifts in the period of oscillation (τ) caused by changes in the density (ρ) of the solution according to the equation ρsample − ρair = κ(τsample − τair)2, where κ is an instrumental constant dependent on atmospheric pressure and room temperature. Samples of N&PL provided values for the lipids. With the N&PL:CLSE mixtures, molecular volume increased linearly with XCLSE, which provided a value for the mixed proteins. We assumed, based on the molar ratio and molecular weights of the two proteins, that SP-B and SP-C contributed equally to the averaged molecular volume. The measurements were repeated multiple times to obtain an average density.
MD Simulations.
We used a web-based graphical user interface for CHARMM38 to build membranes of SP:DOPC. The simulations used systems with dimensions between 7–22 nm laterally and 12–15 nm along the z direction. Simulations were performed using a NAPzT (constant number of atoms, constant area, constant pressure normal to the bilayer, constant temperature) ensemble with the open-source package GROMACS 2018.339,40 and the CHARMM36m force field.41–43 These simulations used a leapfrog algorithm to integrate the equations of motion with a time step of Δt = 0.002 ps. Periodic boundary conditions were applied along the x, y, and z axes. Electrostatic interactions were fully treated by using the particle mesh Ewald method.44,45 Lennard-Jones interactions were truncated at a cutoff distance (rc) of 1.2 nm and shifted from 1.0 nm to the cutoff distance, so that the interactions approached zero smoothly at rc. This procedure avoided any spurious artifacts at the cutoff distance. To minimize the effects of truncated interactions, we applied corrections to the pressure and energy.
These systems were simulated for 50 ns at temperature T = 303 K and pressure along the z direction of Pz = 1 bar (compressibility 4.5 × 10−5 bar−1) by using a Berendsen thermostat and barostat46 during equilibration. In the production periods (750 ns for small systems and 250 ns for large systems), we employed the v-rescaled thermostat coupled to a thermal reservoir with a coupling constant τ = 1.0 ps and a Parrinello–Rahman barostat with a coupling constant τ = 5.0 ps. All simulations used either 384 or 96 CPUs for large or small systems, respectively (see the Supporting Information for additional details).
Analysis of the atomic positions from the simulations used the program SIM-to-EXP.47 The program converts simulated data to an EDP and form factors for comparison with results obtained by XDS. The program also calculates the volume probability distributions for different components.48 Atoms were grouped into the headgroup, glycerol–carbonyl, chain region, methyl trough, and proteins.
RESULTS
Proteins with Surfactant Lipids.
N&PL with incremental amounts of CLSE showed the effect of increasing protein with the complete set of surfactant lipids. XDS from mixtures of CLSE and N&PL at full hydration had the typical pattern observed with hydrated, stacked fluid bilayers (Figure 1). The amplitudes of the scattering lobes and the points of crossover, where the form factor changes phase, varied minimally among the different samples (Figure 2). The amount of protein combined with the surfactant lipids had a relatively minor effect on the form factor.
Figure 2.
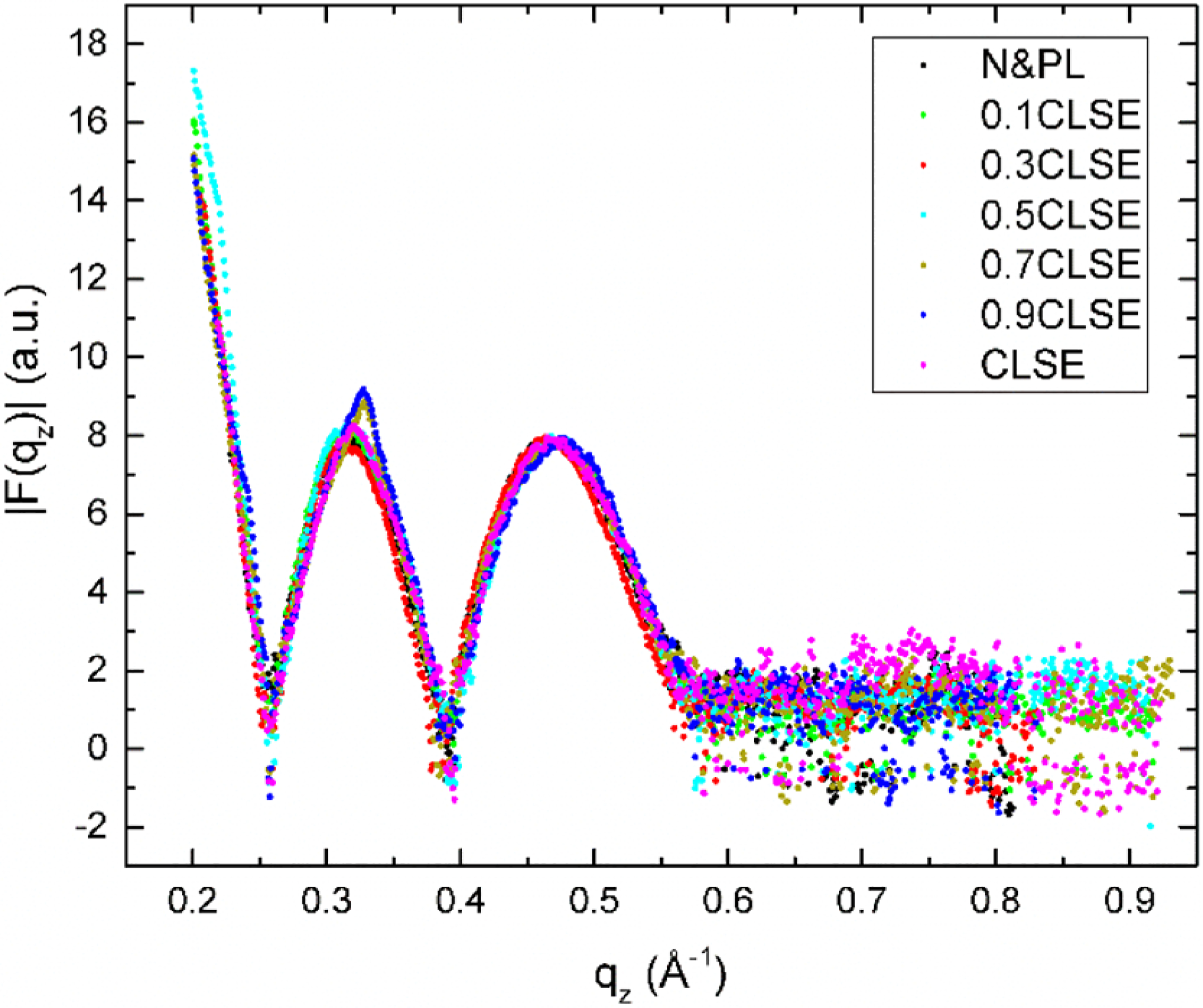
Form factors from XDS data for N&PL:CLSE mixtures obtained at 40 °C. D-spacings for these fully hydrated samples were ~80–120 Å.
The program SDP fit the experimental form factor to a model of the bilayer. The analysis provides the experimental EDPs, and the volume probabilities for the different component groups (Table 2 and Figure 3 for selected samples). The program fits the electron density from the protein exclusively in the headgroup (HG), exclusively in the hydrocarbon (CH), or with the electron density split between the two locations. The split density fit the experimental data best. Separate, distinct locations for the two proteins provided the simplest explanation for the split density. On the basis of their relative hydrophobicities, we speculated that SP-B contributed the density at the surface of the bilayer (Figure 3E,F,H,I, green shading) while SP-C occurred centrally (orange shading).
Table 2.
SDP Analysis of CLSE Mixtures at 40 °C
| XCLSE | protein/lipid mole ratio | volume (±3 Å3) | AL (±1 Å2) | DHH (±0.5 Å) | 2DC (±0.5 Å) | HG-protein (±1 Å)a | HC-protein (±1 Å)a |
|---|---|---|---|---|---|---|---|
| 0 | 0 | 1170 | 55.3 | 40.8 | 30.6 | N/A | N/A |
| 0.3 | 1:1330 | 1175 | 55.6 | 41.2 | 30.4 | 27.7 | 9.2 |
| 0.5 | 1:1108 | 1178 | 58.1 | 40.9 | 29.2 | 28.1 | 7.3 |
| 0.7 | 1:776 | 1182 | 57.0 | 40.5 | 29.9 | 24.6 | 5.8 |
| 1.0 | 1:554 | 1187 | 57.4 | 40.7 | 29.8 | 19.6 | 3.4 |
Distance from bilayer center (Å). HG- and HC-proteins refer to the proteins located in the headgroup and hydrocarbon regions, respectively.
Figure 3.
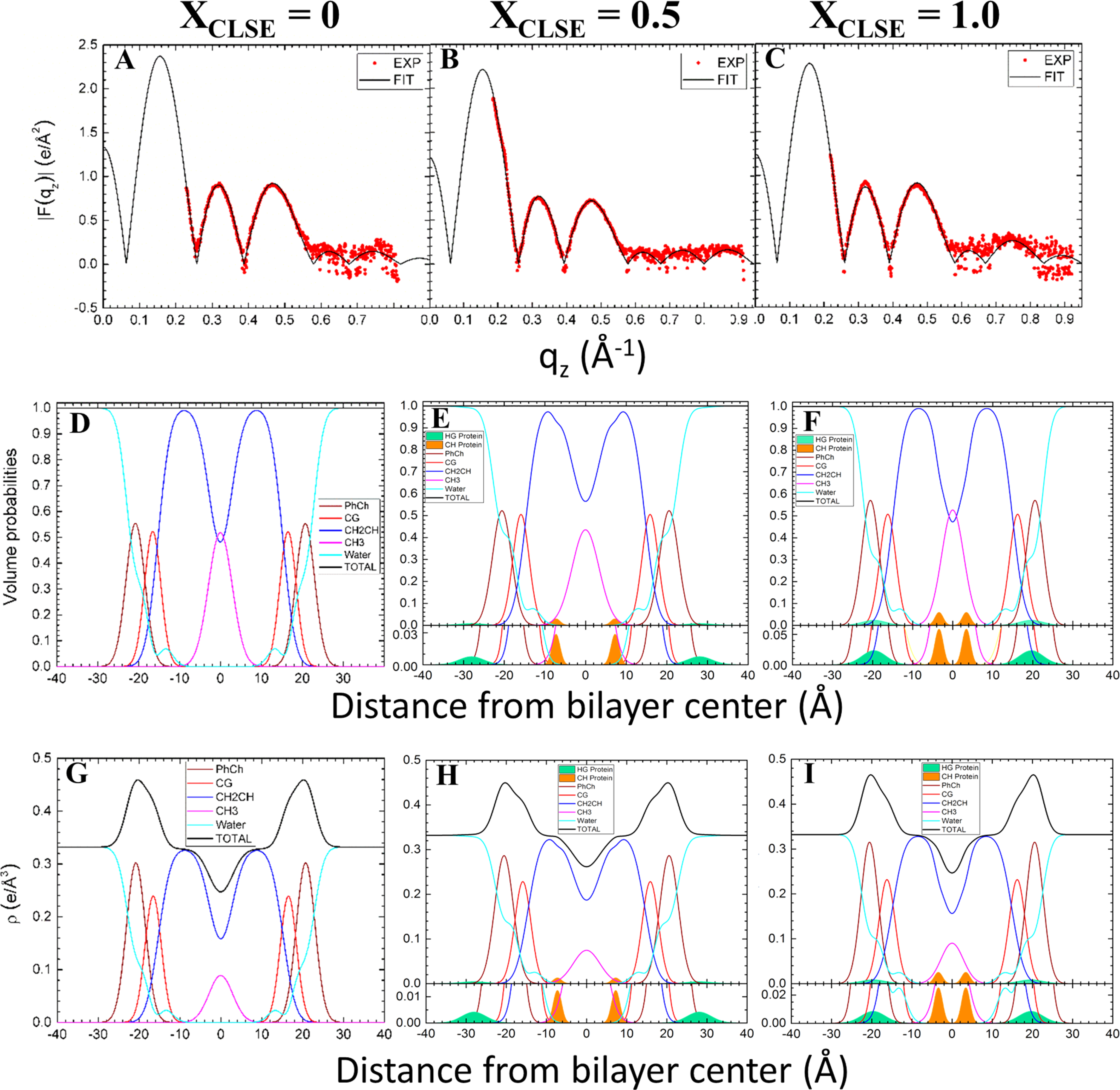
Analysis of XDS from N&PL and CLSE mixtures using the SDP modeling program. First row (panels A, B,C): form factors. Second row (panels D, E, F): volume probabilities. Third row (panels G, H, I): electron density profiles. Component groups: HG (headgroup) green protein; CH (hydrocarbon) orange protein; PhCh wine phosphocholine; CG red carbonyl glycerol; CH2CH blue methylene/methine; CH3 magenta methyl trough. In (E, F, H, and I), there is an inset at the bottom of each panel with an amplified Y-axis to show clearly the shapes and positions of the proteins’ distributions.
Varying the amounts of added protein shifted the location of each protein (Table 2 and Figure 3). With increasing XCLSE, both proteins moved further toward the center of the bilayer. The outer protein, presumed to be SP-B, which sat against the exterior of the bilayer at low XCLSE, moved into the headgroup. SP-C, which was located within the hydrocarbon core for all samples, moved closer to the bilayer’s midpoint. The dimensions of the bilayer and its hydrocarbon core changed minimally, but each protein shifted progressively inward (Table 2).
As expected, increased protein produced a larger volume per lipid (Table 2). Because the area per lipid, AL, is given by VC/DC, and DC remained relatively constant, the proteins also increased AL. Even the small amount of protein in CLSE (XCLSE = 1.0), with a protein:lipid molar ratio of 1:554, achieved this effect.
Proteins with DOPC.
In addition to the samples of N&PL:CLSE with the complete biological mixture of lipids, we also measured XDS from the single phospholipid DOPC with different amounts of the mixed proteins. Analysis with the program SDP again fit the data to a model of the membrane. The model again fit best with protein in two distinct positions (Figure 4). The outer protein, presumed to be SP-B, was located at the level of the headgroup. The presumed SP-C lay well within the hydrocarbon core. In contrast to the samples of N&PL:CLSE, the location of the proteins in DOPC moved slightly outward with increasing concentration (Figure 4). Greater amounts of the proteins, which shifted them toward the center of the mixed-lipid bilayer (Figure 3 and Table 2), had a minimal effect on their location in DOPC (Figure 4).
Figure 4.
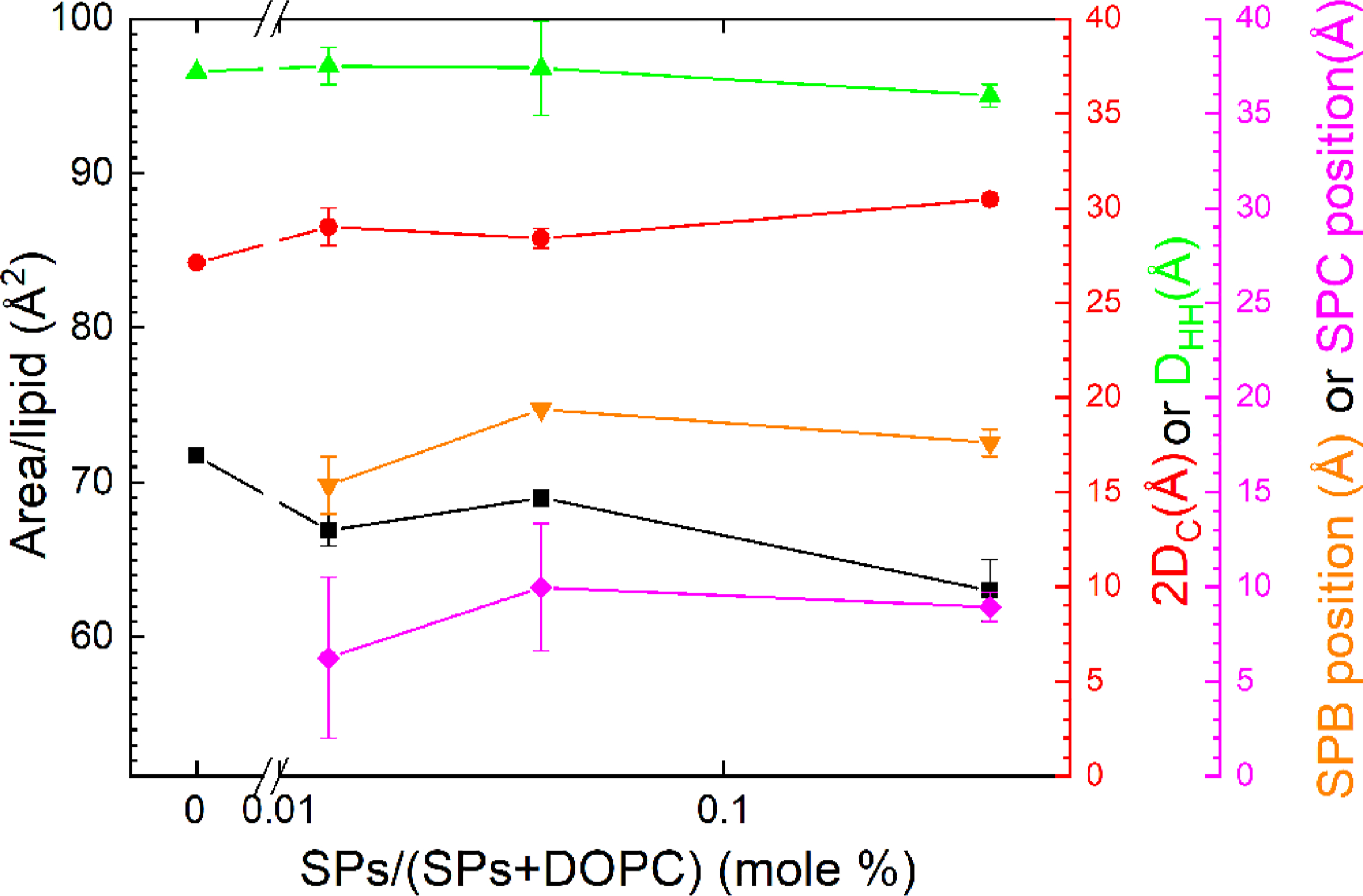
Experimental structural results from XDS data obtained at 30 °C. The highest concentration at 3.0 wt % corresponds to 1:250 SPs:DOPC molar ratio.
The compositional simplicity of the samples with DOPC facilitated MD simulations. That procedure provided information about the location and orientation of the proteins at the molecular level, beyond the detail available from XDS. The simulations also generated a model-independent form factor from its EDP for comparison with results from XDS.
The simulations initially considered systems large enough to include both SP-B and SP-C (Figure 5). These systems encountered out-of-plane fluctuations sufficiently large to prevent construction of a vertical EDP and assignment of components to specific vertical locations. Methods that have dealt with large undulations of other systems49 were unsuccessful here. The inability to construct a profile prevented comparison between simulated and experimental results.
Figure 5.
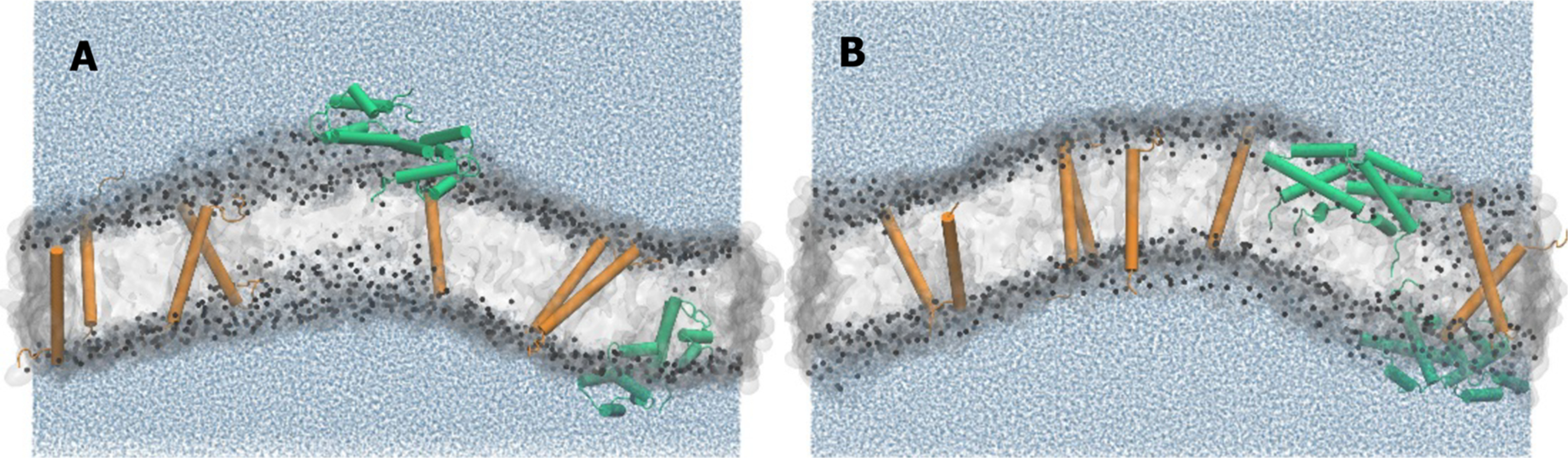
Snapshots from larger simulations containing both SP-B and SP-C. The protein:lipid molar ratios were ~1:130. Snapshots of the systems were taken at the end of the 250 ns simulation. SP-B is green and SP-C is orange in the cartoon representation. The cylinders represent α-helices. (A) Closed SP-B and transmembrane SP-C. (B) Open SP-B and transmembrane SP-C.
Simulations with smaller systems dampened the fluctuations. The periodic boundary condition constrained the location of the bilayer at the edge of the simulated box and restricted undulations. The limited size of these systems, however, accommodated only a single protein. In contrast to the experimental samples, the smaller simulated systems included only SP-B or SP-C rather than both proteins together. This obstacle prevented direct comparison of these simulated systems with the experimental samples containing both proteins.
To approximate results for a system with both proteins, we assumed that their locations were independent. The EDPs of such systems, if weighted appropriately, should be additive. The form factors, which relate directly to the EDP, should also be additive. A weighted average, based on the molar ratio of 1 SP-B:5 SP-C, of the form factors from the separate simulations with the individual peptides then provided simulated results with both proteins present. To correct for differences in the amplitudes of the experimental and simulated form factors, we multiplied the experimental amplitudes by a constant that achieved the best visual match with the averaged simulated curves, relying primarily on the fit between qz = 0.20–0.23 Å−1.
The simulations required the initial configurations of each protein. Membership of SP-B in the family of saposin-like proteins (Figure S1) provided initial configurations. The structures of some saposin-like proteins have been solved. Those peptides form 4–5 amphipathic helices arranged into two leaves.50 In different members of the family, the leaves adopt either an open, V-shaped configuration or a closed conformation (Figure S2). Our simulations therefore considered SP-B in either the open or closed configuration. The SP-B homodimer is unique among the saposin-like proteins in having a disulfide cross-link between the monomers. Prior experimental structures provided no guidance concerning that configuration. Attempts to dock the two monomers in a conformation that would facilitate formation of the inter-monomeric disulfide bond failed with both open and closed configurations. Our simulations therefore omitted that bridge (see the Supporting Information for details).
Prior data have suggested that SP-C has a simpler structure, forming a single helix that spans the lipid bilayer.13 Our results with XDS from the samples with CLSE, however, were also compatible with the protein located within the hydrophobic region, parallel to the plane of the bilayer. We therefore simulated SP-C embedded in the hydrocarbon core as well as in the transmembrane configuration.
DOPC alone, without the proteins, provided a simple control. Prior reports with slightly different methods have shown good agreement between the form factors produced by XDS and simulation.51 The results for the two approaches here similarly agreed (Figure S3). The AL in the simulations affected the level of agreement, with AL = 72 Å2 providing better results than 68 or 64 Å2 (data not shown). While neutron scattering obtains a smaller AL,33 XDS has consistently obtained AL = 72 Å2 for DOPC at 30 °C.52,53
The simulations used higher protein-to-lipid ratios than present in CLSE. The electron densities and volume probabilities of the proteins were therefore larger than for the samples with the surfactant lipids (Figure 3). The two initial configurations of each protein produced small differences in the structure of the DOPC bilayer. The grouped components of the lipids remained at roughly the same location (Figures 6–9). SP-B resided close to the phospholipid headgroup for both the open and closed configurations (Figures 6 and 7). The protein density for SP-C aligned parallel to the interface (Figure 8) was only slightly more bimodal than for the transmembrane configuration (Figure 9).
Figure 6.
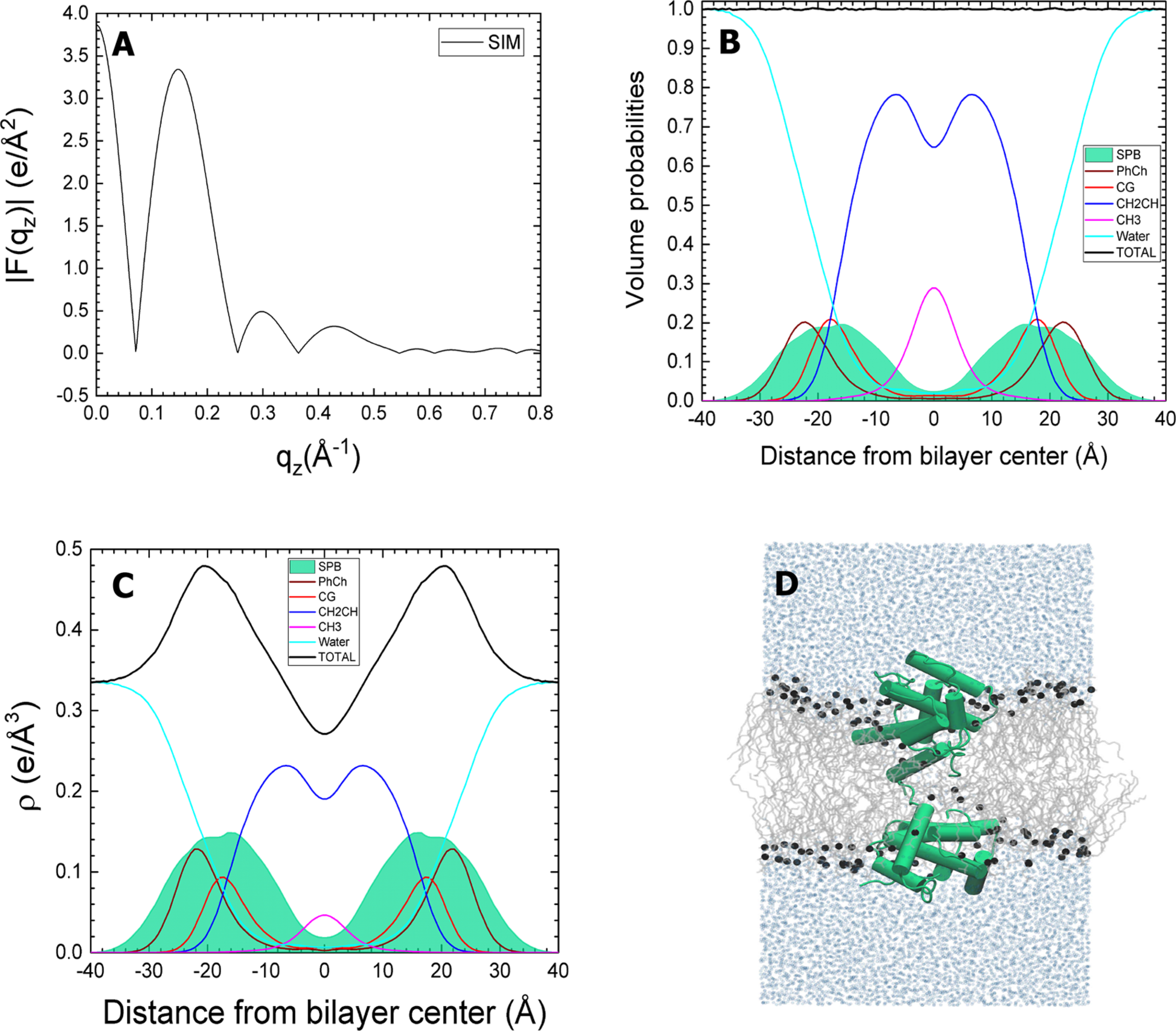
Closed SP-B in DOPC. The simulated SP-B:DOPC molar ratio is 1:40. (A) Form factors obtained from simulations. (B) Volume probabilities of components (as in caption to Figure 3). (C) Simulated electron density (ρ) profile for components. (D) Snapshot of the system taken near the end of the 750 ns simulation. The lipids are gray lines, phosphate atoms are black spheres, and water molecules are represented by light blue spheres. SP-B is shown in green.
Figure 9.
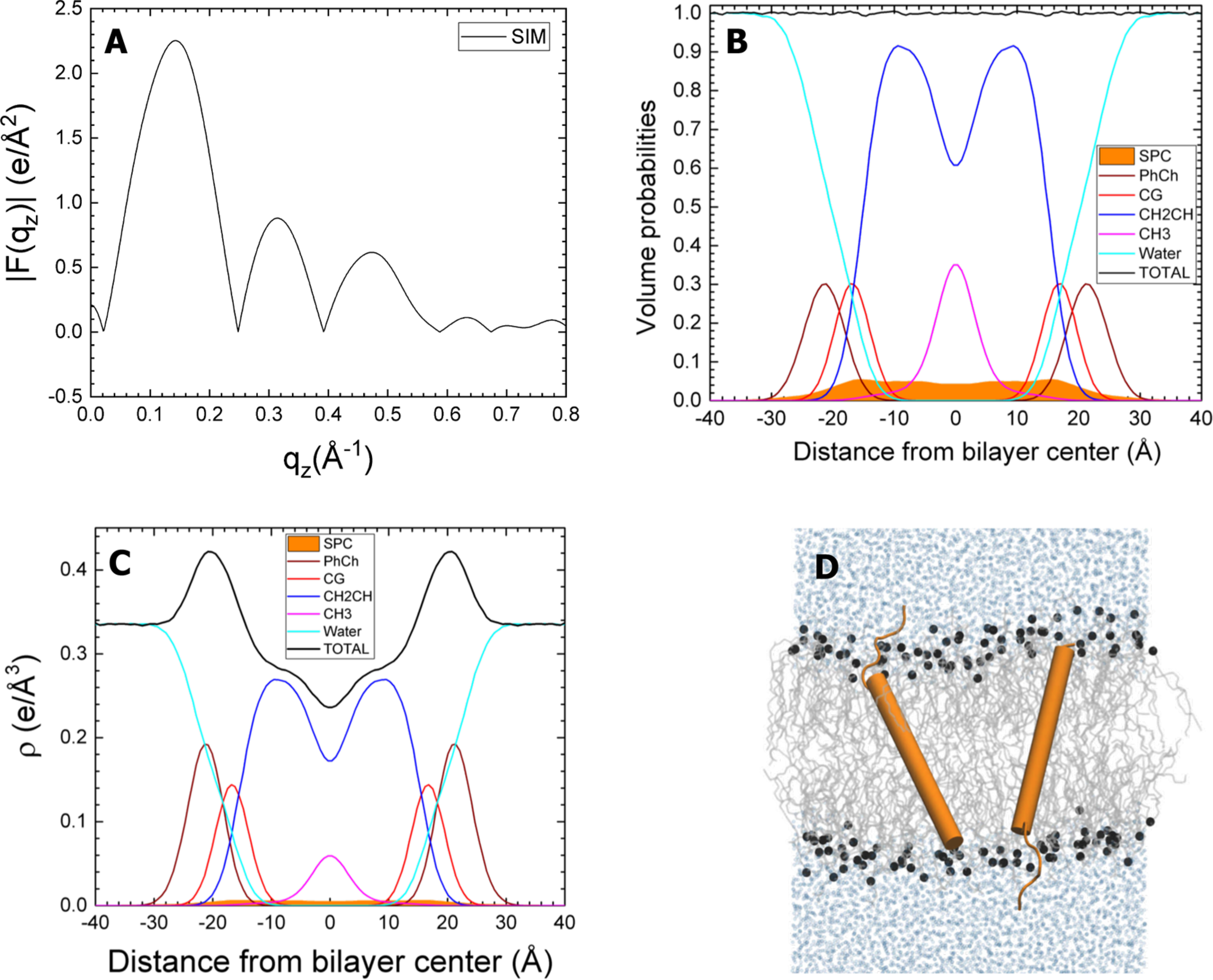
Simulated SP-C:DOPC, with transmembrane protein. The simulated SP-C:DOPC molar ratio is 1:80. Panels and component groups as in Figure 8.
Figure 7.
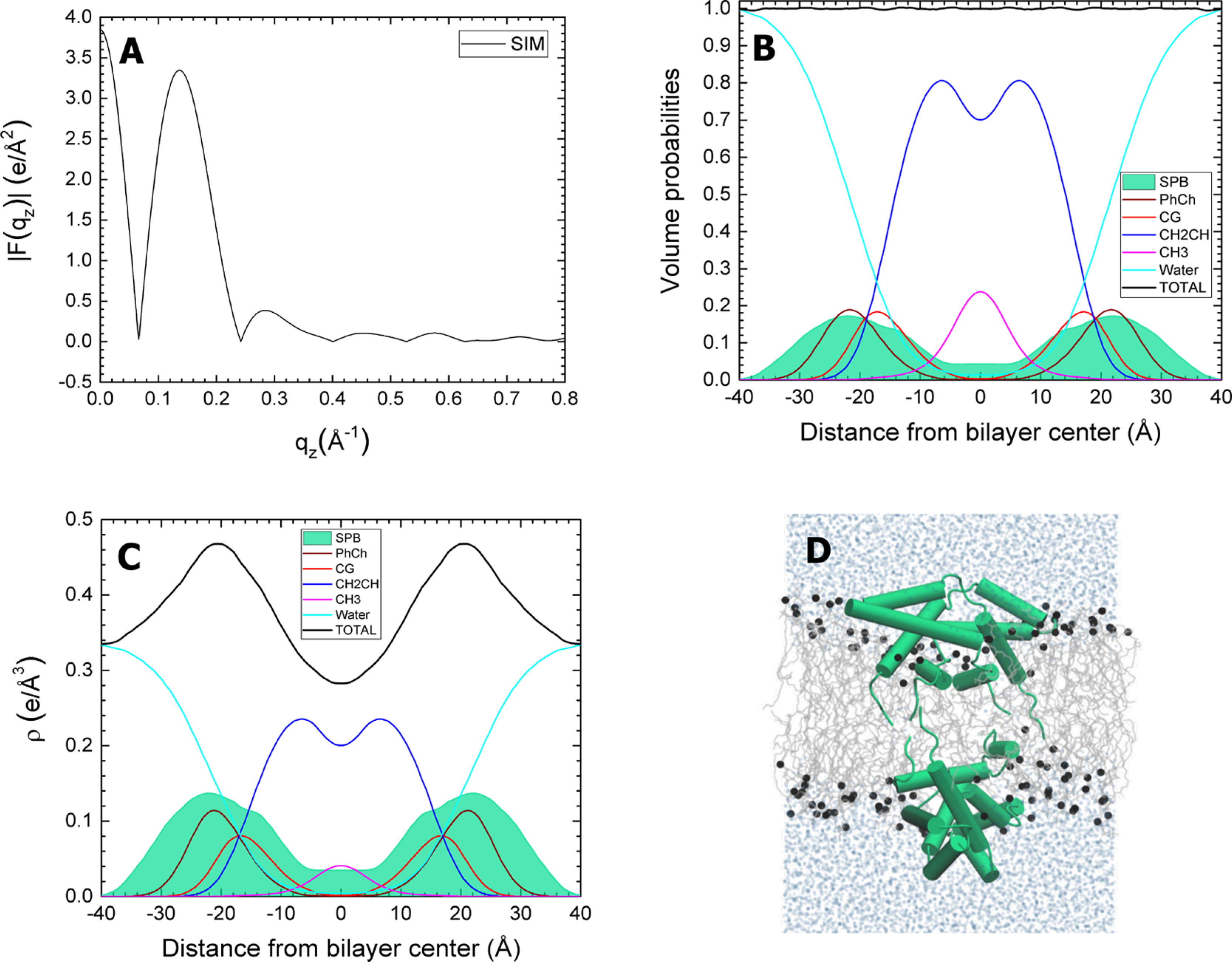
Open SP-B with DOPC. The simulated SP-B:DOPC molar ratio is 1:40. Panels and component groups as in Figure 6.
Figure 8.
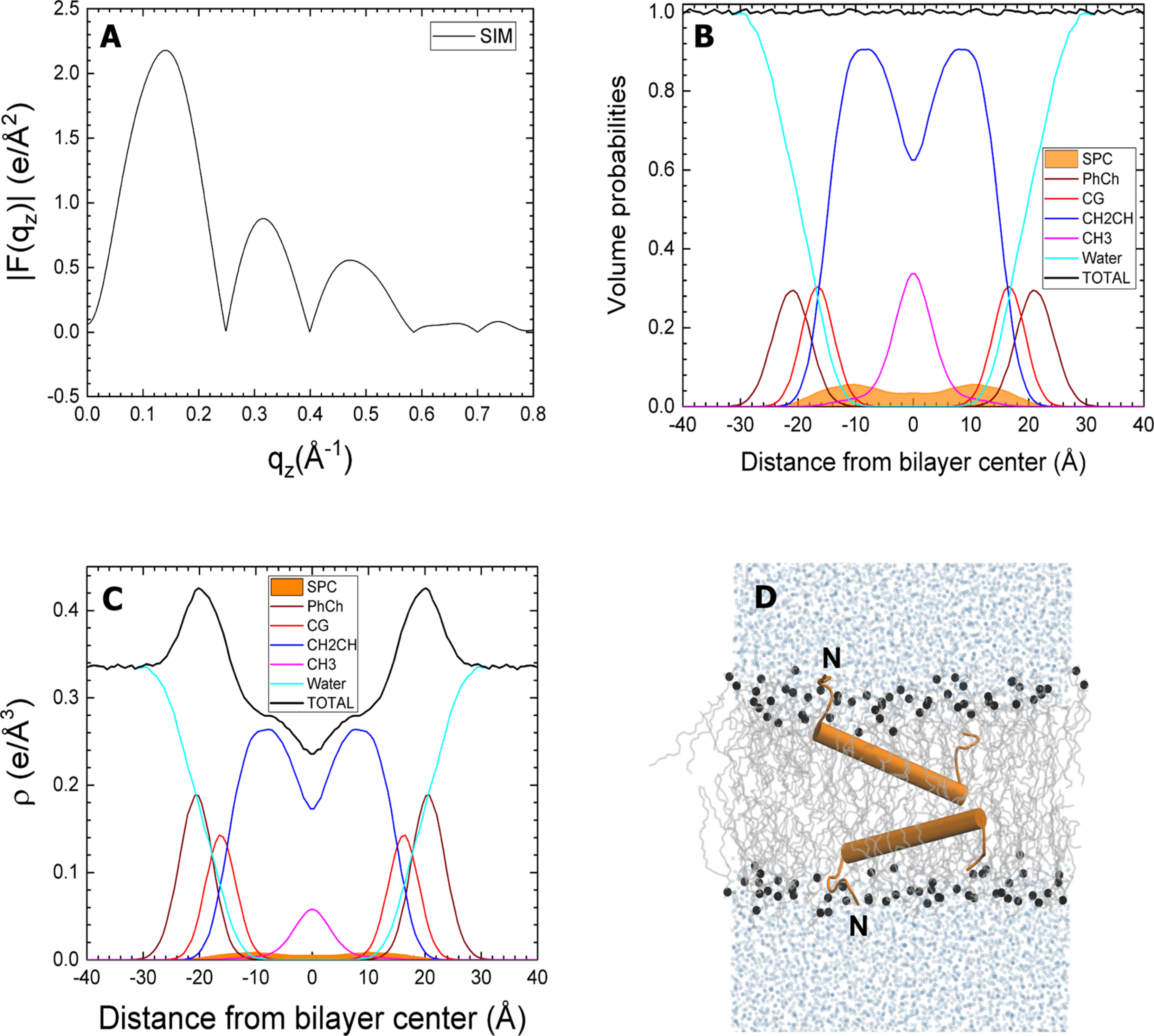
Simulated SP-C:DOPC, with protein in the hydrocarbon region. The simulated SP-C:DOPC molar ratio is 1:80. Panels and component groups as in Figure 6. SP-C is shown in orange.“N” indicates the amino terminus of the peptide. Although not shown here, the simulations included the two palmitoyl residues.
The averaged form factor consequently showed little variation with the different initial configurations of the proteins (Figure 10). These composite results, generated from the weighted average of the form factors for the simulations with each protein, generally agreed well with the experimental form factor from XDS. The crossover points for the experimental and simulated results were closest with closed SP-B and SP-C in the hydrocarbon region (Figure 10A). The differences were subtle, but sufficient to provide a slight preference for that combination. Perhaps the most important result was the similarity of the form factor for the weighted average of the separate systems, each with one individual protein, and the experimental results. This finding supported the assumption that the positions of the proteins are independent.
Figure 10.
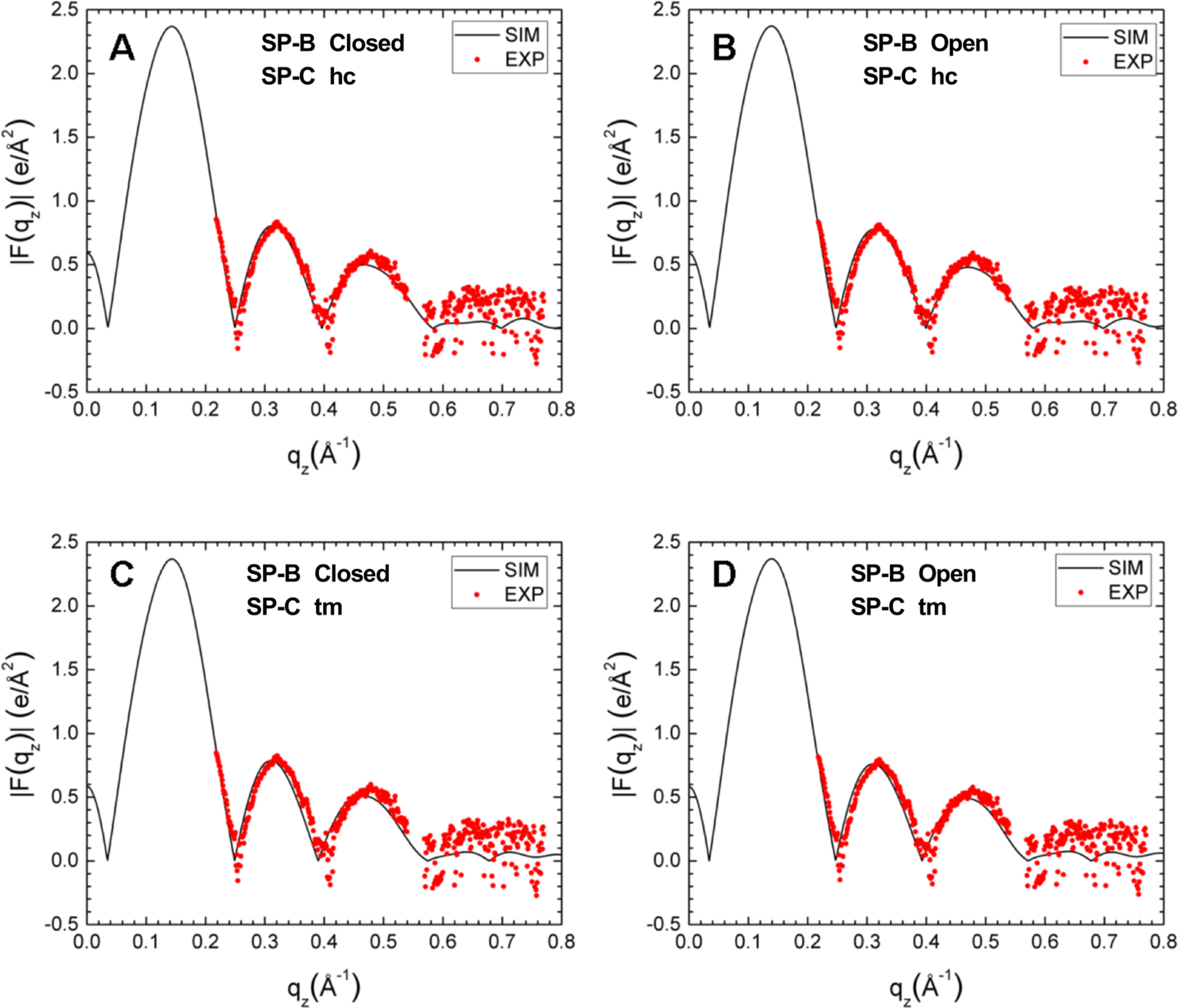
Simulated form factors (black traces), generated by weight-averaging simulations with individual proteins, compared to the experimental form factor obtained with 3.0 wt % protein (red symbols). Numbers below in parentheses quantitate the average difference in qz up to 0.55 Å−1 between experimental and simulated form factors. (A) Simulated closed SP-B and SP-C in hydrocarbon (hc) (0.055). (B) Simulated open SP-B and SP-C in hc (0.060). (C) Simulated closed SP-B and transmembrane (tm) SP-C (0.072). (D) Simulated open SP-B and tm SP-C (0.071).
DISCUSSION AND CONCLUSIONS
The studies reported here provide the most direct evidence yet concerning the location of the hydrophobic surfactant proteins within the bilayer. SP-B sits at the level of the headgroup, with some penetration into the hydrocarbon core. SP-C instead is located in the hydrophobic interior. These results agree with the preponderance of spectroscopic findings.13,14 Those studies generally located the proteins indirectly, based on interactions with and perturbation of components at different levels within the membrane. They leave open the possibility that the proteins could extend into other regions without causing detectable perturbations. Our studies determine the location of the proteins directly.
Our findings are self-consistent. XDS and MD simulation find the proteins in DOPC at comparable locations. In DOPC and the surfactant lipids, the proteins occupy equivalent positions. The proteins produce the same dramatic acceleration of adsorption with the two compositionally distinct lipids.9–11 The agreement between the different methods and the different systems supports the validity of our findings and their likely relevance to function.
Both the location of the proteins and their simulated behavior suggest restrictions on the possible orientations of the helices within the proteins. For SP-B, two distinct configurations would satisfy the amphipathic character of the helices. They could orient along the face of the bilayer, or they could form complexes that span the membrane. Helices in the perpendicular alignment could interact to form a porelike structure, with a central hydrophilic core, and hydrophobic helical surfaces facing the hydrocarbon core of the membrane.54 The location of SP-B by XDS in the headgroup region and the orientations found by simulations both argue that the helices lie along the face of the membrane.
Ideally the location of SP-B would provide insight into its function. The proteins promote fusion, both between two bilayers55 and between the adsorbing vesicle and the air/water interface or an interfacial film.10,56 Two models exist concerning the mechanisms by which the protein performs its function. The bridging model proposes that SP-B tethers the vesicle to the interface.13 A thermodynamic barrier, composed of hydration forces and entropic effects, limits the close approximation of two bilayers.57–60 That barrier involves the structure of adjacent solvent and the restriction of undulation in one bilayer by the presence of the other. Both contributions would be present for a bilayer approaching an air/water interface. A protein that binds the vesicle to the interface might reduce that barrier. Experimental evidence for the model is limited, but Monte Carlo61 and coarse-grained62 simulations support this possibility. The previously suggested superficial location of the protein in the bilayer, now confirmed here, has represented an attractive feature of the model.
The second model of adsorption emphasizes an initial continuous structure that would connect the adsorbing lipid bilayer with the nascent film. The proposed structure would consist of a stalk extending from the vesicle to the interface, directly comparable to the structure proposed as a key feature in the fusion of two bilayers.63 The stalk would represent a structure that could convey phospholipids from the vesicle to the interface without exposing the hydrophobic acyl groups to the aqueous environment. The thermodynamic cost of that exposure at physiological concentrations, multiple orders of magnitude above the critical micelle concentration, would be enormous. Several experimental findings support the possibility of a connecting stalk, and coarse-grained simulation demonstrates such a structure.62
The tight negative curvature of the two lipid leaflets that would constitute the stalk might require a significant energy of bending. SP-B could reduce that energy by making the lipids more flexible. To the best of our knowledge, measurements of how SP-B affects the modulus of bending remain unavailable.
Alternatively, the protein could shift the spontaneous curvature of the lipids toward the negative curvature of the stalk. Studies suggest that the hydrophobic surfactant proteins enhance negative curvature, both in lipids that become capable of forming inverse bicontinuous cubic phases64 and in the cylindrical monolayers of the inverse hexagonal phase.65 To increase negative curvature, SP-B should expand the hydrophobic face of the phospholipid monolayer more than the headgroup region. Our results here, particularly with the surfactant lipids, suggest the opposite. The location of SP-B predominantly in the region of the headgroup represents a significant challenge to the model in which the protein promotes adsorption by enhancing negative curvature.
Our results show that SP-C occurs in a different location, within the hydrocarbon core. The different positions of the two proteins is expected from their amino acid compositions. Based on the Wimley–White scale of hydrophobicity,66 the calculated free energy of transferring the proteins from a palmitoyloleoyl-phosphatidylcholine bilayer to the aqueous phase is −2.37 kcal/mol for SP-B and 0.44 kcal/mol for SP-C. SP-B should prefer a more hydrophilic environment, while SP-C favors immersion in hydrophobic regions. Our results also suggest that the proteins sort independently to their different locations. The good agreement between the results from the weighted average simulations and from experimental XDS supports the assumed independence of the positions for the two proteins.
Our data provide a slight preference for orientation of SP-C along the plane of the bilayer rather than across it. With DOPC and particularly the surfactant lipids, the volume probabilities obtained by XDS for the inner protein occur as two distributions, each centered within a single leaflet, rather than as a single distribution spanning the bilayer. The agreement between simulation and XDS is also slightly better for SP-C imbedded in the hydrocarbon core rather than in the transmembrane configuration.
That orientation of SP-C parallel to the face of the bilayer disagrees with the conclusions of prior studies using vibrational spectroscopy,67,68 which favored the transmembrane alignment. We speculate that the difference may reflect the lipids used, which may disguise the behavior of the protein. The primary sequence, LIPC*C*PVNIKRLLIVVVVVVLVVVVIV-GALLMGL,69 suggests that the roughly one-third of the protein at the amino terminal, with two positively charged residues (underlined; asterisk (*) indicates cysteine-linked palmitoyl residues), should favor the headgroup region. The remaining two-thirds of the sequence toward the carboxyl terminus is extraordinarily hydrophobic. That segment should form a helix that would lack a basis, other than the terminal carboxyl group, for interacting with the opposing hydrophilic surface of the bilayer. The orientation of the helix might then depend on the relative length of the helix and the span of the hydrophobic region of the bilayer. The helical content of the protein, found to be ~60%,68 would indicate participation of 21 amino acids. A single α-helix of that length would extend for 31.5 Å. The hydrophobic thickness for CLSE is 29.8 Å(Table 2). These distances suggest that SP-C must assume at least a small tilt angle (~19°), which could be larger depending on the configuration of the lipid chains. In a bilayer with a hydrophobic region of greater thickness, the orientation of the helix would be unconstrained. A thinner hydrocarbon core would restrict the helix to an orientation more parallel to the plane of the bilayer.
The studies with vibrational spectroscopy used phospholipids that form the gel phase either exclusively (dipalmitoylphosphatidylcholine:dipalmitoylphosphatidylglycerol (DPPC:DPPG);67 DPPG68) or predominantly (DPPC:egg PG, 8:2 mol:mol).68 Our studies instead provide results with lipids in the fluid phase. The thickness of the hydrocarbon region for gel phase bilayers (34.6 Å for DPPC)70 exceeds the length of the SP-C helix. The carboxy terminus would fail to reach the opposite face of the bilayer. The hydrophobic thickness in this case would provide no restriction on the helical orientation. For DOPC in the fluid phase, the hydrophobic thickness (~27 Å)52 should constrain the helical orientation to alignment more parallel to the face of the bilayer. We consider that our evidence is insufficient to establish the helical orientation of SP-C conclusively. We contend, however, that the evidence that the protein must align across the membrane is also inconclusive and that its orientation in the bilayer remains an open question.
Perhaps our most distinctive result seems likely to be functionally unimportant. With greater amounts of protein, both SP-B and SP-C move toward the center of bilayers formed by the surfactant lipids. This shift is most easily explained by electrostatic interactions. Greater amounts of the cationic proteins should reduce the net charge on a fixed level of anionic lipids. This change is absent with DOPC, which lacks an anionic headgroup. The proteins produce equivalent functional effects on the two sets of lipids.9–11 This progressive relocation therefore seems unlikely to represent a major factor in the mechanism by which the proteins promote adsorption.
All studies have their limitations. The relatively similar electron densities of protein and lipid limit the certainty with which XDS can determine their locations. The simulations in turn require the structure of the proteins as initial input. Unraveled membrane proteins do not fold to appropriate conformations over the course of these atomistic simulations. Although this issue seems unimportant for SP-C, the structure of SP-B is uncertain. The studies of other saposin-like proteins suggest two initial configurations. Our results fail to distinguish clearly between them.
One structural approximation for SP-B that seems likely to be unimportant is the absence in our simulations of the inter-monomeric disulfide bond. Mutation of the relevant cysteine to serine prevents formation of the disulfide linkage between the two monomers.71 Above a threshold concentration, which is considerably less than physiological levels, SP-B from mice with this mutation forms noncovalently linked dimers.72 The behavior in our simulations of the unlinked monomers seems likely to resemble the performance of the wild-type SP-B homodimer.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Hannah Smith for preparation of the oriented samples for the second set of measurements on samples of N&PL:CLSE and Alex Boscia for help with the densitometry. These studies were supported by the National Institutes of Health (HL060914, HL130130, and HL136734). Work in DPT’s group is supported by the Natural Sciences and Engineering Research Council (Canada), with additional support from the Canada Research Chair Program. Simulations were performed on Compute Canada resources, funded by the Canada Foundation for Innovation and partners.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.0c03665.
Additional materials and methods concerning MD simulations; structure of DOPC without proteins (PDF)
Contributor Information
Ryan W. Loney, Pulmonary and Critical Care Medicine, Oregon Health & Science University, Portland, Oregon 97239, United States
Sergio Panzuela, Centre for Molecular Simulation and Department of Biological Sciences, University of Calgary, Calgary, AB T2N 1N4, Canada; Department of Theoretical Physics and Condensed Matter, Universidad Autonoma de Madrid, E-28049́ Madrid, Spain.
Jespar Chen, Biological Physics Group, Department of Physics, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, United States.
Zimo Yang, Biological Physics Group, Department of Physics, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, United States.
Jonathan R. Fritz, Biological Physics Group, Department of Physics, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, United States
Zachary Dell, Biological Physics Group, Department of Physics, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, United States.
Valentina Corradi, Centre for Molecular Simulation and Department of Biological Sciences, University of Calgary, Calgary, AB T2N 1N4, Canada.
Kamlesh Kumar, Pulmonary and Critical Care Medicine, Oregon Health & Science University, Portland, Oregon 97239, United States.
D. Peter Tieleman, Centre for Molecular Simulation and Department of Biological Sciences, University of Calgary, Calgary, AB T2N 1N4, Canada.
Stephen B. Hall, Pulmonary and Critical Care Medicine, Oregon Health & Science University, Portland, Oregon 97239, United States.
Stephanie A. Tristram-Nagle, Biological Physics Group, Department of Physics, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, United States.
REFERENCES
- (1).Rugonyi S; Biswas SC; Hall SB The biophysical function of pulmonary surfactant. Respir. Physiol. Neurobiol 2008, 163, 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Robertson B Pathology and pathophysiology of neonatal surfactant deficiency (“respiratory distress syndrome,” “hyaline membrane disease”). In Pulmonary Surfactant, 1st ed.; Robertson B, Van Golde LMG, Batenburg JJ, Eds.; Elsevier Science Publishers: Amsterdam, 1984; pp 383–418. [Google Scholar]
- (3).Lachmann B; Robertson B; Vogel J In vivo lung lavage as an experimental model of the respiratory distress syndrome. Acta Anaesthesiol. Scand 1980, 24, 231–236. [DOI] [PubMed] [Google Scholar]
- (4).Nogee LM; Dunbar AE III; Wert SE; Askin F; Hamvas A; Whitsett JA A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med 2001, 344, 573–579. [DOI] [PubMed] [Google Scholar]
- (5).Nogee LM Alterations in SP-B and SP-C expression in neonatal lung disease. Annu. Rev. Physiol 2004, 66, 601–623. [DOI] [PubMed] [Google Scholar]
- (6).Nogee LM; Garnier G; Dietz HC; Singer L; Murphy AM; deMello DE; Colten HR A mutation in the surfactant protein B gene responsible for fatal neonatal respiratory disease in multiple kindreds. J. Clin. Invest 1994, 93, 1860–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Melton KR; Nesslein LL; Ikegami M; Tichelaar JW; Clark JC; Whitsett JA; Weaver TE SP-B deficiency causes respiratory failure in adult mice. Am. J. Physiol Lung Cell Mol. Physiol 2003, 285, L543–549. [DOI] [PubMed] [Google Scholar]
- (8).Hall SB; Wang Z; Notter RH Separation of subfractions of the hydrophobic components of calf lung surfactant. J. Lipid Res 1994, 35, 1386–1394. [PubMed] [Google Scholar]
- (9).Schram V; Hall SB Thermodynamic effects of the hydrophobic surfactant proteins on the early adsorption of pulmonary surfactant. Biophys. J 2001, 81, 1536–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Biswas SC; Rananavare SB; Hall SB Effects of gramicidin-A on the adsorption of phospholipids to the air-water interface. Biochim. Biophys. Acta, Biomembr 2005, 1717, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Loney RW; Anyan WR; Biswas SC; Rananavare SB; Hall SB The accelerated late adsorption of pulmonary surfactant. Langmuir 2011, 27, 4857–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang Z; Gurel O; Baatz JE; Notter RH Differential activity and lack of synergy of lung surfactant proteins SP-B and SP-C in interactions with phospholipids. J. Lipid Res 1996, 37, 1749–1760. [PubMed] [Google Scholar]
- (13).Johansson J; Curstedt T Molecular structures and interactions of pulmonary surfactant components. Eur. J. Biochem 1997, 244, 675–693. [DOI] [PubMed] [Google Scholar]
- (14).Hawgood S; Derrick M; Poulain F Structure and properties of surfactant protein B. Biochim. Biophys. Acta, Mol. Basis Dis 1998, 1408, 150–160. [DOI] [PubMed] [Google Scholar]
- (15).Patthy L Homology of the precursor of pulmonary surfactant-associated protein SP-B with prosaposin and sulfated glycoprotein 1. J. Biol. Chem 1991, 266, 6035–6037. [PubMed] [Google Scholar]
- (16).Andersson M; Curstedt T; Jörnvall H; Johansson J An amphipathic helical motif common to tumourolytic polypeptide NK-lysin and pulmonary surfactant polypeptide SP-B. FEBS Lett 1995, 362, 328–332. [DOI] [PubMed] [Google Scholar]
- (17).Olmeda B; García-Álvarez B; Gómez MJ; Martínez-Calle M; Cruz A; Pérez-Gil J A model for the structure and mechanism of action of pulmonary surfactant protein B. FASEB J 2015, 29, 4236–4247. [DOI] [PubMed] [Google Scholar]
- (18).Pérez-Gil J; Cruz A; Casals C Solubility of hydrophobic surfactant proteins in organic solvent/water mixtures. Structural studies on SP-B and SP-C in aqueous organic solvents and lipids. Biochim. Biophys. Acta, Lipids Lipid Metab 1993, 1168, 261–270. [DOI] [PubMed] [Google Scholar]
- (19).Bligh E; Dyer W A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol 1959, 37, 911–917. [DOI] [PubMed] [Google Scholar]
- (20).Notter RH; Finkelstein JN; Taubold RD Comparative adsorption of natural lung surfactant, extracted phospholipids, and artificial phospholipid mixtures to the air-water interface. Chem. Phys. Lipids 1983, 33, 67–80. [DOI] [PubMed] [Google Scholar]
- (21).Takahashi A; Fujiwara T Proteolipid in bovine lung surfactant: its role in surfactant function. Biochem. Biophys. Res. Commun 1986, 135, 527–532. [DOI] [PubMed] [Google Scholar]
- (22).Hawgood S; Benson BJ; Schilling J; Damm D; Clements JA; White RT Nucleotide and amino acid sequences of pulmonary surfactant protein SP 18 and evidence for cooperation between SP 18 and SP 28−36 in surfactant lipid adsorption. Proc. Natl. Acad. Sci. U. S. A 1987, 84, 66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ames BN Assay of inorganic phosphate, total phosphate and phosphatases. Methods Enzymol 1966, 8, 115–118. [Google Scholar]
- (24).Kaplan RS; Pedersen PL Determination of microgram quantities of protein in the presence of milligram levels of lipid with amido black 10B. Anal. Biochem 1985, 150, 97–104. [DOI] [PubMed] [Google Scholar]
- (25).Liu S; Zhao L; Manzanares D; Doherty-Kirby A; Zhang C; Possmayer F; Lajoie GA Characterization of bovine surfactant proteins B and C by electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom 2008, 22, 197–203. [DOI] [PubMed] [Google Scholar]
- (26).Kumar K; Chavarha M; Loney RW; Weiss TM; Rananavare SB; Hall SB The Lγ phase of pulmonary surfactant. Langmuir 2018, 34, 6601–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Markin CJ; Hall SB The anionic phospholipids of bovine pulmonary surfactant. Lipids 2020, In press. [DOI] [PubMed] [Google Scholar]
- (28).Tristram-Nagle SA Preparation of oriented, fully hydrated lipid samples for structure determination using X-ray scattering. Methods Mol. Biol 2007, 400, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kučerka N; Liu Y; Chu N; Petrache HI; Tristram-Nagle S; Nagle JF Structure of fully hydrated fluid phase DMPC and DLPC lipid bilayers using x-ray scattering from oriented multilamellar arrays and from unilamellar vesicles. Biophys. J 2005, 88, 2626–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lyatskaya Y; Liu Y; Tristram-Nagle S; Katsaras J; Nagle JF Method for obtaining structure and interactions from oriented lipid bilayers. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top 2000, 63, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Liu Y; Nagle JF Diffuse scattering provides material parameters and electron density profiles of biomembranes. Phys. Rev. E 2004, 69, 040901–1–040901–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhang R; Suter RM; Nagle JF Theory of the structure factor of lipid bilayers. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top 1994, 50, 5047–5060. [DOI] [PubMed] [Google Scholar]
- (33).Kučerka N; Nagle JF; Sachs JN; Feller SE; Pencer J; Jackson A; Katsaras J Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data. Biophys. J 2008, 95, 2356–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kučerka N; Perlmutter JD; Pan J; Tristram-Nagle S; Katsaras J; Sachs JN The effect of cholesterol on short- and long-chain monounsaturated lipid bilayers as determined by molecular dynamics simulations and X-ray scattering. Biophys. J 2008, 95, 2792–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Nagle JF; Tristram-Nagle S Structure of lipid bilayers. Biochim. Biophys. Acta, Rev. Biomembr 2000, 1469, 159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dupuy FG; Pagano I; Andenoro K; Peralta MF; Elhady Y; Heinrich F; Tristram-Nagle S Selective interaction of colistin with lipid model membranes. Biophys. J 2018, 114, 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hallinen KM; Tristram-Nagle S; Nagle JF Volumetric stability of lipid bilayers. Phys. Chem. Chem. Phys 2012, 14, 15452–15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Jo S; Kim T; Iyer VG; Im W CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- (39).Pronk S; Páll S; Schulz R; Larsson P; Bjelkmar P; Apostolov R; Shirts MR; Smith JC; Kasson PM; Van Der Spoel D; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Abraham MJ; Van der Spoel D; Lindahl E; Hess B GROMACS user manual version 2018.3. [Google Scholar]
- (41).Huang J; Mackerell AD CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; De Groot BL; Grubmüller H; MacKerell AD Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Darden T; York D; Pedersen L Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (45).Deserno M; Holm C How to mesh up Ewald sums. I. a theoretical and numerical comparison of various particle mesh routines. J. Chem. Phys 1998, 109, 7678–7693. [Google Scholar]
- (46).Berendsen HJC; Postma JPM; Van Gunsteren WF; Dinola A; Haak JR Molecular dynamics with coupling to an external bath. J. Chem. Phys 1984, 81, 3684–3690. [Google Scholar]
- (47).Kučerka N; Katsaras J; Nagle JF Comparing membrane simulations to scattering experiments: Introducing the SIMtoEXP software. J. Membr. Biol 2010, 235, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Petrache HI; Feller SE; Nagle JF Determination of component volumes of lipid bilayers from simulations. Biophys. J 1997, 72, 2237–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Braun AR; Brandt EG; Edholm O; Nagle JF; Sachs JN Determination of electron density profiles and area from simulations of undulating membranes. Biophys. J 2011, 100, 2112–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bruhn H A short guided tour through functional and structural features of saposin-like proteins. Biochem. J 2005, 389, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tristram-Nagle S; Nagle JF Lipid bilayers: thermodynamics, structure, fluctuations, and interactions. Chem. Phys. Lipids 2004, 127, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Tristram-Nagle S; Petrache HI; Nagle JF Structure and interactions of fully hydrated dioleoylphosphatidylcholine bilayers. Biophys. J 1998, 75, 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kučerka N; Tristram-Nagle S; Nagle JF Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol 2006, 208, 193–202. [DOI] [PubMed] [Google Scholar]
- (54).Eisenberg D Three-dimensional structure of membrane and surface proteins. Annu. Rev. Biochem 1984, 53, 595–623. [DOI] [PubMed] [Google Scholar]
- (55).Hawgood S; Efrati H; Schilling J; Benson BJ Chemical characterization of lung surfactant apoproteins: amino acid composition, N-terminal sequence and enzymic digestion. Biochem. Soc. Trans 1985, 13, 1092–1096. [DOI] [PubMed] [Google Scholar]
- (56).Oosterlaken-Dijksterhuis MA; Haagsman HP; van Golde LMG; Demel RA Interaction of lipid vesicles with monomolecular layers containing lung surfactant proteins SP-B or SP-C. Biochemistry 1991, 30, 8276–8281. [DOI] [PubMed] [Google Scholar]
- (57).Rand RP; Parsegian VA Hydration forces between phospholipid bilayers. Biochim. Biophys. Acta, Rev. Biomembr 1989, 988, 351–376. [Google Scholar]
- (58).Israelachvili JN; Wennerström H Entropic forces between amphiphilic surfaces in liquids. J. Phys. Chem 1992, 96, 520–531. [Google Scholar]
- (59).Leikin S; Parsegian VA; Rau DC; Rand RP Hydration forces. Annu. Rev. Phys. Chem 1993, 44, 369–395. [DOI] [PubMed] [Google Scholar]
- (60).McIntosh TJ; Simon SA Hydration and steric pressures between phospholipid bilayers. Annu. Rev. Biophys. Biomol. Struct 1994, 23, 27–51. [DOI] [PubMed] [Google Scholar]
- (61).Zaltash S; Palmblad M; Curstedt T; Johansson J; Persson B Pulmonary surfactant protein B: A structural model and a functional analogue. Biochim. Biophys. Acta, Biomembr 2000, 1466, 179–186. [DOI] [PubMed] [Google Scholar]
- (62).Baoukina S; Tieleman DP Lung surfactant protein SP-B promotes formation of bilayer reservoirs from monolayer and lipid transfer between the interface and subphase. Biophys. J 2011, 100, 1678–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Markin VS; Kozlov MM; Borovjagin VL On the theory of membrane fusion. The stalk mechanism. Gen. Physiol. Biophys 1984, 3, 361–377. [PubMed] [Google Scholar]
- (64).Chavarha M; Loney RW; Kumar K; Rananavare SB; Hall SB Differential effects of the hydrophobic surfactant proteins on the formation of inverse bicontinuous cubic phases. Langmuir 2012, 28, 16596–16604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Chavarha M; Loney RW; Rananavare SB; Hall SB Hydrophobic surfactant proteins strongly induce negative curvature. Biophys. J 2015, 109, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Wimley WC; White SH Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol 1996, 3, 842–848. [DOI] [PubMed] [Google Scholar]
- (67).Pastrana B; Mendelsohn R; Mautone AJ Fourier transform infrared studies of secondary structure and orientation of pulmonary surfactant SP-C and its effect on the dynamic surface properties of phospholipids. Biochemistry 1991, 30, 10058–10064. [DOI] [PubMed] [Google Scholar]
- (68).Vandenbussche G; Clercx A; Curstedt T; Johansson J; Jornvall H; Ruysschaert JM Structure and orientation of the surfactant-associated protein C in a lipid bilayer. Eur. J. Biochem 1992, 203, 201–209. [DOI] [PubMed] [Google Scholar]
- (69).Augusto LA; Li J; Synguelakis M; Johansson J; Chaby R Structural basis for interactions between lung surfactant protein C and bacterial lipopolysaccharide. J. Biol. Chem 2002, 277, 23484–23492. [DOI] [PubMed] [Google Scholar]
- (70).Nagle JF; Cognet P; Dupuy FG; Tristram-Nagle S Structure of gel phase DPPC determined by X-ray diffraction. Chem. Phys. Lipids 2019, 218, 168–177. [DOI] [PubMed] [Google Scholar]
- (71).Beck DC; Ikegami M; Na CL; Zaltash S; Johansson J; Whitsett JA; Weaver TE The role of homodimers in surfactant protein B function in vivo. J. Biol. Chem 2000, 275, 3365–3370. [DOI] [PubMed] [Google Scholar]
- (72).Zaltash S; Griffiths WJ; Beck D; Duan CX; Weaver TE; Johansson J Membrane activity of (Cys48Ser) lung surfactant protein B increases with dimerisation. Biol. Chem 2001, 382, 933–939. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.