

Abstract
Rationale:
The mechanisms underlying atrial fibrillation (AF), the most common clinical arrhythmia, are poorly understood. Nucleoplasmic Ca2+ regulates gene-expression, but the nature and significance of nuclear Ca2+-changes in AF are largely unknown.
Objective:
To elucidate mechanisms by which AF alters atrial cardiomyocyte (CM) nuclear Ca2+ ([Ca2+]Nuc) and Ca2+/calmodulin-dependent protein kinase-II (CaMKII)-related signaling.
Methods And Results:
Atrial CMs were isolated from control and AF-dogs (kept in AF by atrial tachypacing [600 bpm × 1 week]). [Ca2+]Nuc and cytosolic [Ca2+] ([Ca2+]Cyto) were recorded via confocal microscopy. Diastolic [Ca2+]Nuc was greater than [Ca2+]Cyto under control conditions, while resting [Ca2+]Nuc was similar to [Ca2+]Cyto; both diastolic and resting [Ca2+]Nuc increased with AF. Inositol-trisphosphate-receptor (IP3R) stimulation produced larger [Ca2+]Nuc increases in AF versus control CMs, and IP3R-blockade suppressed the AF-related [Ca2+]Nuc-differences. AF upregulated nuclear protein-expression of IP3R-type 1 (IP3R1) and of phosphorylated CaMKII (immunohistochemistry and immunoblot), while decreasing the nuclear/cytosolic expression-ratio for histone deacetylase type-4 (HDAC4). Isolated atrial CMs tachypaced at 3 Hz for 24 hours mimicked AF-type [Ca2+]Nuc changes and L-type calcium current (ICaL) decreases versus 1-Hz-paced CMs; these changes were prevented by IP3R knockdown with short-interfering RNA directed against IP3R1. Nuclear/cytosolic HDAC4 expression-ratio was decreased by 3-Hz pacing, while nuclear CaMKII and HDAC4 phosphorylation were increased. Either CaMKII-inhibition (by autocamtide-2-related peptide) or IP3R-knockdown prevented the CaMKII-hyperphosphorylation and nuclear-to-cytosolic HDAC4 shift caused by 3-Hz pacing. In human atrial CMs from AF patients, nuclear IP3R1-expression was significantly increased, with decreased nuclear/non-nuclear HDAC4 ratio. MicroRNA-26a was predicted to target ITPR1 (confirmed by Luciferase assay) and was downregulated in AF atrial CMs; microRNA-26a silencing reproduced AF-induced IP3R1 upregulation and nuclear diastolic Ca2+-loading.
Conclusion:
AF increases atrial CM nucleoplasmic Ca2+-handling by IP3R1-upregulation involving miR-26a, leading to enhanced IP3R1-CaMKII-HDAC4 signaling and ICaL-downregulation.
Keywords: Nuclear calcium, arrhythmia (mechanisms), heart disease, inositol trisphosphate receptors, Arrhythmias, Atrial Fibrillation, Calcium Cycling/Excitation-Contraction Coupling, Ion Channels/Membrane Transport, Pathophysiology
Graphical Abstarct
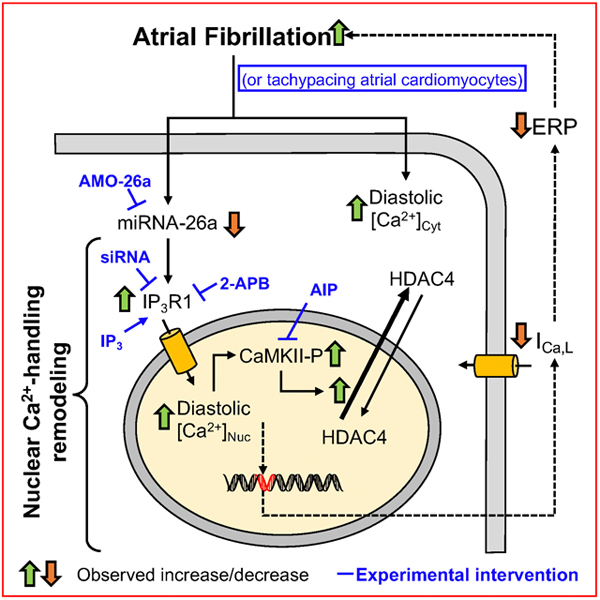
INTRODUCTION
Atrial fibrillation (AF) is a highly prevalent clinical arrhythmia, with an increasing age-dependent incidence and association with enhanced morbidity and mortality.1 AF is well-recognized to produce atrial cardiomyocyte (CM) Ca2+-handling abnormalities that participate in arrhythmogenesis and the atrial contractile dysfunction associated with increased stroke risk.2,3 AF induces important changes in atrial ion-channel expression, structure and electrical function that make AF more likely to be induced and maintained, changes commonly referred to as “atrial remodeling”.4 Changes in cellular Ca2+-content and distribution contribute to activating the signaling pathways involved in atrial remodeling.5,6
Atrial-cardiomyocyte gene expression is importantly modified in various animal models of AF and in AF-patients.7,8 Nuclear envelope (NE) Ca2+-signaling mechanisms can mediate changes in gene-transcription associated with cardiac remodelling, in response to stimuli like endothelin-induced inositol 1,4,5-trisphosphate (IP3) production, with subsequent NE IP3-receptor (IP3R)-activation, NE Ca2+-release and nuclear Ca2+/calmodulin protein-kinase type-II (CaMKII) activation that promotes nuclear export of class II histone deacetylase (HDAC) that modifies gene transcription.9,10 Nuclear Ca2+-handling is remodeled in cardiac hypertrophy and failure, and is presumed to be involved in the associated transcriptional changes.11 There is no information in the literature about whether atrial CM nuclear Ca2+-handling is altered in AF and, if so, what the underlying mechanisms and consequences might be. This study was designed to address the following specific hypotheses: 1) nuclear Ca2+-handling is altered in AF; 2) the observed changes are due to discrete alterations in Ca2+-handling protein expression and function; 3) AF-related altered nuclear Ca2+-handling drives nuclear-restricted changes in key intracellular signaling, including CaMKII-phosphorylation and HDAC translocation.
METHODS
This section briefly summarizes key methods; for methodological details, see Detailed Methods Supplement.
Data Availability.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animal Model.
Animal-care procedures were approved by the Animal Research Ethics Committee of the Montreal Heart Institute (protocol 2018-47-12) and followed Canadian Council on Animal Care Guidelines. A total of 86 adult mongrel dogs of both sexes were studied, divided into control and AF groups. Animals were arbitrarily assigned to treatment groups in sequence, attempting to balance control and intervention animals within similar time windows to avoid any time-dependent bias. AF was maintained by pacing the right atrium (RA) at 600 bpm×1 week.12,13 On day 7, dogs were anesthetized and ventilated. RA effective refractory periods (ERPs) were measured and mean induced AF-duration was determined. No major adverse events occurred, and all dogs were included in the final analyses.
Canine Atrial CM Isolation, Culture and in Vitro Pacing.
Atrial CMs were isolated with previously-described methods.14 CMs were plated at low density onto laminin-coated coverslips. Isolated CMs were subjected to 24-hr in vitro pacing at 1 Hz (P1, sinus rate) or 3 Hz (P3, in vitro model of AF 5).
Confocal Ca2+ Imaging.
The Ca2+-indicators Fluo-4 AM and Fluo-5N AM were used15–17 for two-dimensional confocal Ca2+-imaging. Images were acquired in line-scan mode (3 or 6 ms scan−1; pixel size 0.103μm). CMs were field stimulated with 2 platinum electrodes.
In Situ Calibration of Fluo-4 Fluorescence.
Calibration solutions were prepared as described by Ljubojevic et al.17 Saponin-permeabilized CMs were used for calibration. Minimal Fluo-4 fluorescence (Fmin) was measured during exposure to Ca2+-free calibration solution and maximal Fluo-4 fluorescence (Fmax) during exposure to a calibration solution containing a saturating free [Ca2+] of 1 mM.
Preparation of Nuclear-enriched Fractions.
Nuclear and non-nuclear fractions were isolated based on a previously-described approach.18 Canine atrial CMs were gently suspended in 3 volumes of hypotonic buffer per volume of CMs, and incubated on ice for 20 minutes. 50 μl 10% NP- 40 was added to 1 ml of suspended CMs after incubation and CMs were vortexed for 10 seconds at the highest setting. The homogenate was centrifuged for 5 minutes at 125×g and 4°C. The supernatant containing the non-nuclear and nuclear fractions was centrifuged for 10 minutes at 1910×g and 4°C to separate the nuclear (pellet) from non-nuclear (supernatant) fractions. The supernatant (non-nuclear fraction) was collected into a new tube, frozen in liquid-N2 and stored at −80°C for subsequent analysis. The pellet (nuclear fraction) was washed 3 times with Storage Buffer (250-mM sucrose, 20-mM KCl, 1-mM MgCl2, 1-mM dithiothreyitol, 50-mM HEPES, pH 7.0, containing one tablet of Protease Inhibitor Cocktail per 50 ml), frozen in liquid-N2 and stored at −80°C for subsequent analysis.
Nuclear and non-nuclear fractions from human atrial CMs were isolated from right atrial (RA) appendages obtained from sinus rhythm (Ctl) and >6 month (chronic) AF (cAF) patients, as previously described.19 Experimental protocols were approved by ethical review board of University Hospital Essen (no. 12-5268-BO) and were conducted in accordance with the Declaration of Helsinki. Each patient provided written informed consent. The antibodies used for immunoblotting of human samples are listed in the Major Resources Table.
Immunostaining and Immunoblot.
Atrial CMs were plated on 25*25 mm coverslips and then fixed for 20 minutes with 4% paraformaldehyde in phosphate-buffered saline (PBS). The CMs were then incubated with a blocking/permeabilizing buffer for 1 hour and incubated overnight at 4°C with the primary antibody (details in Major Resources Table) diluted in PBS. After washing (3×5 minutes) with PBS, CMs were incubated in the dark at room temperature for 1 hour, with Alexa-conjugated secondary antibodies diluted in PBS. Images were acquired with an Olympus FluoView FV1000 confocal laser-scanning microscope equipped with a 40×/1.3 oil immersion objective. Lysis buffer was prepared by adding one tablet of Protease Inhibitor Cocktail to 10 ml buffer. The nuclear fraction was then lysed by adding to the pellet a volume of lysis buffer three times the volume of the pellet. The non-nuclear and nuclear fractions were kept at –80°C for subsequent assay. Membranes were blocked for 1 hour at room temperature and probed with primary antibodies overnight at 4°C. After extensive washing, membranes were further incubated with secondary antibodies conjugated to horseradish peroxidase and immunoreactive bands detected with enhanced chemiluminescence (ECL).
Inositol-trisphosphate Receptor (ITPR) Gene Knockdown with siRNA.
Freshly-isolated canine atrial CMs were plated on laminin-coated glass cover slips (25*25 mm). Atrial CMs were transfected with inositol-trisphosphate receptor type-1 (ITPR1) and type-2 (ITPR2) gene siRNA (100 nM for each), or with the negative control (medium: Lipofectamine® RNAiMAX). SiRNAs and Lipofectamine® RNAiMAX were separately mixed with Opti-MEM®I Reduced Serum Medium for 5 min. The two mixtures were then combined and incubated for 5 minutes at room temperature. The Lipofectamine® RNAiMAX/siRNA mixture was added to the CMs, which were then incubated at 37°C for 24 hours. CMs were paced at 1 or 3 Hz for 24 hours followed by Ca2+-transient measurement, L-type calcium current (ICaL) recording or were collected for protein/mRNA assay.
Identification and Study of miRNAs Targeting ITPR1.
We used MirTarget V3, PicTar, TargetScan7.2, and the PubMed database to identify miRNAs predicted to target ITPR1. Only one miRNA, miR-26, fulfilled all of the criteria used (see Methods Supplement). Freshly-isolated CMs were transfected with miR-26a or miR-NC (20 nM for Luciferase studies; 100 nM for others), or 10 nM AMO-26a. After 24 hours of transfection, cells were used for Ca2+-transient recording or and kept at −80 °C for immunoblot and RT-qPCR studies.
Ca2+-current Recording.
The whole-cell tight-seal patch-clamp to record currents in voltage-clamp mode at 37±2°C. For details, see Methods Supplement.
Data Analysis.
Custom-made software (available at https://github.com/FengXiongCA/Nucleus-Cytosolic-Ca) was used to analyse Ca2+-transients. Image J was used to analyse immunostaining images. GraphPad Prism 8.0, Origin 5.0 and SAS release 9.4 (SAS Institute Inc., Cary, NC, USA) were used for data analysis. Wherever multiple CMs were measured per dog, numbers are given as n/N, where n indicates number of CMs studied from N dogs.
Mixed effect models were used for all analyses unless specified otherwise. For analyses involving the fixed effect condition (AF vs. CTL), the homogeneity of variance between conditions (AF vs. CTL) was verified using the Brown and Forsythe’s test. Some of the analyses were multilevel; involving (1) the individual dog and (2) cells within each dog. For these analyses, the random effect within the model was the intercept to take into account correlation of multiple cells within dogs. Other analyses involved fixed effects as repeated factor(s) such as pacing frequency (1 vs 3 Hz), knockdown-probe versus control, endothelin-1 (ET-1) exposure (before vs after), exposure to 2APB+IP3 (before versus after), exposure to IP3 (before versus after), 2APB (2APB, 2APB+ET-1, CTL), tetracaine (CTL+Tetracaine, CTL+Tetracaine+IP3), 2APB-concentration (5-μM, 25-μM, 50-μM, CTL), AIP (without vs with) and basic cycle-length (150, 200, 250, 300, 350 ms).
For repeated measure models, the unstructured covariance structure to model the within-dog errors was used. For multilevel repeated models, the random effects included in the model were the intercept, cell and the repeated factor to take into account correlation within dogs, within cells*dogs and within repeated factor*dogs. In addition, the multilevel linear model was used to measure the association between nucleus and cytosol.
All variables with a non-normal distribution were transformed using a natural logarithm to produce normally-distributed data for analyses.
Data from human atrial samples, in which each patient contributed a single data point, were compared with unpaired Student’s t-test or Mann-Whitney U-test for normally and non-normally distributed data, respectively.
Results are shown as scatterplots wherever possible and if not, as violin plots in almost all cases. Group data in scatterplots are presented as geometric mean±standard error and violin plot data show median and interquartile range; any exceptions to these general rules are specified in the figure legends. A 2-tailed P<0.05 was considered statistically significant.
RESULTS
In Vivo Experiments.
All AF-dogs showed atrial remodeling, with significantly reduced AERPs (Online Figure IA). RA filling pressures were not significantly changed in AF-dogs (Online Figure IB); left-ventricular end-diastolic pressure was modestly but significantly increased (Online Figure IC). The mean duration of pacing-induced AF was significantly increased in AF-dogs (Online Figure ID).
Calibration of Ca2+ Fluo-4 Fluorescence Signals in Canine Atrial CMs.
Fluo-4 has different Ca2+-binding affinities and fluorescent properties in different intracellular compartments,15–17 and Fluo-4 fluorescence signals had to be transformed into calibrated [Ca2+] separately for nucleoplasmic vs cytosolic compartments. Online Figure IIA shows original 2-dimensional confocal-microscopy images of Fluo-4 fluorescence during the calibration protocol at different [Ca2+] concentrations. Fluo-4 fluorescent signals from nucleoplasmic and cytosolic compartments with seven different [Ca2+] concentrations from 0 to 1000 μM were obtained from CTL (n/N=47/14 CMs/dogs) and AF (n/N=31/5) CMs (Online Figure IIB-D). The Fluo-4 fluorescence versus [Ca2+] curves were fitted with the Hill equation: F={(Fmax−Fmin)/(1+(Kd/[Ca2+])n)}+Fmin (Online Figure IIB, C). The obtained calibration curves were used to calculate the Kd (Online Figure IID). Fluo-4 showed slightly different Ca2+-binding affinities between the nucleoplasmic and cytosolic compartments in CTL and AF.
AF-induced Nuclear Structure Remodeling.
Canine atrial CMs were loaded with the low-affinity Ca2+-indicator Fluo-5N to reveal nuclear structure in vivo. Fluo-5N staining suggested nuclear enlargement and loss of invaginations in AF-dog CMs (Online Figure IIIA). Immunostaining of fixed CMs for the nuclear protein lamin (Online Figure IIIA) confirmed decreased NE-invagination density, along with increased nuclear length and perimeter in AF (Online Figure IIIB, C).
AF-induced [Ca2+]Nuc Changes.
Figure 1A shows original line-scan images of Fluo-4 signals recorded from intact CTL and AF CMs. Field-stimulation at 1 Hz elicited synchronized Ca2+-transients in the cytosol and nucleus. Figure 1B shows the calibrated Ca2+-signal as a function of time at individual pixels in the nucleus and cytosol. Diastolic [Ca2+]Nuc (end-cycle value at the onset of each activation at 1 Hz) was higher than [Ca2+ ]Cyto in CTL and increased further in AF. The greater diastolic [Ca2+] in the nucleus of AF-dogs could be due to a larger absolute Ca2+-load or to slowed kinetics, which do not allow [Ca2+]Nuc to return to baseline in a single cycle. To address this issue, we followed the Ca2+-transient to steady state after a series of pulses at 1 Hz. Figure 1C shows simultaneously-recorded cytosolic and nuclear Ca2+-transients pulsed at 1 Hz for 1 minute followed by a 10-second pause. The nuclear Ca2+-transient decayed more slowly than cytosolic, taking 2.9±0.2 s to reach the baseline value of 134±12 nM. The cytosolic Ca2+-transient took 1.2±0.12 s to reach the baseline value of 128±13 nM. These results suggest that the larger diastolic [Ca2+] in the nucleus during repeated activation (Figure 1B) is due to the slower decay kinetics of the nuclear Ca2+-transient rather than to subcellular differences in baseline [Ca2+]. The corresponding data in AF-cells show that the [Ca2+] at steady state in AF-cells was once again similar in cytosol and nucleus; however, the nuclear [Ca2+] averaged 55% higher in AF versus CTL (P<0.001), indicating a true (rather than purely kinetic) increase in AF. Corresponding kinetic data are shown in Online Figure IV. These results indicate that [Ca2+]Nuc activation and decay kinetics were slower than cytosolic.
Figure 1.

A. Line-scan imaging of cytosolic and nucleoplasmic Ca2+-transients (CaTs) in CTL and AF atrial cardiomyocytes at 1 Hz. B. Left, Original recordings of [Ca2+]Nuc and [Ca2+]Cyto transients in 1-Hz stimulated atrial cardiomyocytes. Right, Mean±SEM diastolic [Ca2+] C. Left, Original recordings of [Ca2+]Nuc and [Ca2+]Cyto transients for the last of a series of pulses at 1 Hz followed by a 10-s pause. Right, resting [Ca2+]. D. CaT amplitude; E. time to peak; F. time from peak [Ca2+] to 50% decline [DT50] of nucleoplasmic and cytosolic CaTs from CTL (black) and AF (red) atrial cardiomyocytes; G. Mean±SEM time-averaged [Ca2+] over one cycle for control (CTL) and atrial-fibrillation (AF) dog atrial cardiomyocytes. P-values in black reflect CTL vs. AF, P-values in blue italics reflect nucleus vs. cytosol based on multilevel mixed effects models (n/N=CMs/dogs). Group data are shown as violin plots with median (solid line) ± interquartile range (dotted lines).
Nucleoplasmic Ca2+-transient amplitude decreased significantly with AF; the simultaneously measured cytosolic transient amplitude decreased slightly but not significantly (Figure 1D). The kinetics of the nuclear Ca2+-transients during repeated pulsing were significantly slower than cytosolic: both time-to-peak and time to 50% signal-decay (DT50) were significantly greater in AF (Figure 1E, F). We calculated the time-averaged [Ca2+] per cycle as an index of the average [Ca2+] sensed by Ca2+-dependent signals that may work on a slower kinetic scale than contraction-relaxation. These values were greatly increased in AF-cells, e.g. by 65% in the nucleus (Figure 1G).
In order to relate these changes to the total SR [Ca2+]-load, we measured this by integrating Na+, Ca2+-exchange current evoked by a caffeine-induced SR Ca2+-release, according to standard methods.19 The results (Online Figure V) indicate that AF increased net SR [Ca2+]-content.
A rapid puff of caffeine (10 mM) reversibly suppressed Fluo-5N fluorescence both in the NE and in its tubular structures (Online Figure VI). This result confirms the ability of the NE to release and take up Ca2+ in our cells.
Expression of Ca2+-Handling Proteins.
To study the nuclear expression of Ca2+-handling proteins like IP3R, SERCA2a and RyR2, we performed immunostaining and immunoblots. IP3R1-staining was seen throughout the cell, but appeared stronger over the nuclear envelope (NE; Figure 2A). For IP3Rs and other NE-localized proteins, we specifically quantified NE-staining on immunochemistry with the use of a mask to delineate the NE. IP3R1-immunostaining in both the NE and non-nuclear compartments increased with AF; IP3R2-staining also appeared to increase with AF but to a lesser extent, and not significantly in the NE. SERCA2a-expression exhibited a striated organization, with some concentration in the NE (Online Figure VIIA). SERCA2a-expression was not significantly affected by AF (Online Figure VIIB). RyR2 also showed a striated pattern (Online Figure VIIIA). RyR2 staining decreased with AF (Online Figure VIIIB). The AF-related expression-changes were further assessed by Western Blot analysis of purified nuclear and non-nuclear fractions. Consistent with immunostaining results, IP3R expression increased with AF, particularly for IP3R1 (Figure 2B), whereas SERCA2a expression did not change (Online Figure VIIC, D) and RyR2 decreased (Online Figure VIIIC, D). Expression of phosphorylated RyR2 also decreased with AF.
Figure 2.

A. Original 2-dimensional images and violin plots with median (solid line) ± interquartile range (dotted lines) fluorescence intensity from control (CTL) and atrial fibrillation (AF) dog atrial cardiomyocytes (CMs) after immunostaining for NPC (green), IP3R1 and IP3R2 (red). B. Mean±SEM IP3R1 and IP3R2 immunoblot band intensities/GAPDH or histone H3 band intensities from CTL and AF atrial CM nuclear and non-nuclear fractions. C. Line-scan imaging of cytosolic and nucleoplasmic CaTs in one permeabilized CTL atrial CM before and after 20-μM IP3. D. Effect of IP3 and IP3+2APB (50 μM) on mean±SEM CTL atrial CM diastolic [Ca2+]Nuc and [Ca2+]Cyto. E. Line-scan imaging of cytosolic and nucleoplasmic CaTs in permeabilized AF atrial CM before and after IP3. F. Effect of IP3 and IP3+2APB (50 μM) on mean±SEM AF atrial CM diastolic [Ca2+]Nuc and [Ca2+]Cyto. In panels A and B, P-values in black reflect CTL vs. AF based on multilevel mixed effects models (A) or regular mixed effects models (B). In panels D and F, P-values in black reflect CTL/AF vs. CTL+IP3/AF+IP3, whereas P-values in blue italics reflect [Ca2+]nuc vs. [Ca2+]cyto based on repeated measures multilevel mixed effects models (n/N=CMs/dogs).
Role of IP3R-system Remodeling.
Endothelin-1 Effect on [Ca2+] in Atrial CMs.
Intracrine IP3-signaling is an important regulator of nucleoplasmic [Ca2+] and mediates endothelin-dependent changes.20 Online Figure IXA shows recordings of Ca2+-transients before and after the application of endothelin (ET-1, 100 nM) in CTL and AF atrial CMs. During 1-Hz field stimulation, ET-1 increased diastolic [Ca2+]Nuc and [Ca2+ ]Cyto in both CTL (by 67%, 52% respectively) and AF (by 82%, 63% ) atrial CMs (Online Figure IXB), and significantly increased nuclear Ca2+-transient amplitude only in AF-cells (Online Figure IXC). ET-1 did not alter time to peak or DT50 (Online Figures IXD, E). Atrial CMs pretreated with the IP3R antagonist 2-amino-thoxydiphenyl borate (2APB, 50 μM) failed to show a response of the Ca2+-transient to 100-nM ET-1 (Online Figure IXF, G), suggesting that the ET-1 effect is mediated by IP3Rs.
IP3-dependent Ca2+-signals in Permeabilized Atrial CMs.
In further experiments, we investigated the IP3 effect directly in saponin-permeabilized atrial CMs. Figure 2C shows 2-dimensional confocal line-scan images of a permeabilized CTL atrial CM before and after IP3-application. IP3 (20 μM) increased both nucleoplasmic and cytosolic [Ca2+] in control CMs (Figure 2D). In AF-cells, IP3 effects on nucleoplasmic and cytosolic [Ca2+] appeared to be enhanced (Figure 2E, F; Online Figure XA, B), with nucleoplasmic [Ca2+] increasing to a level significantly greater than cytosolic with IP3 (Figure 2F). The IP3R-blocker 2APB prevented the effect of IP3 on nuclear and cytosolic [Ca2+] in permeabilized CMs (Figure 2D, F), and eliminated the nuclear-cytosolic difference in AF-cells, consistent with an important role of IP3R-mediated Ca2+-transport in the nucleus. The RyR2-blocker tetracaine (0.7 mM) failed to block the effect of IP3 on [Ca2+]Nuc and [Ca2+]cyto (Online Figure XC, D).
Effects of 2APB on Nucleoplasmic Ca2+ Regulation.
The AF-related increases in nuclear IP3R-expression and IP3R-mediated [Ca2+]Nuc responses to ET-1 and IP3 point to a potentially-important role for IP3R-upregulation in the [Ca2+]Nuc-changes associated with AF. To investigate further the potential role of IP3R-mediated Ca2+-entry in the absence of an IP3R-agonist, we initially evaluated the effect of 2APB on Ca2+-transients in field-stimulated atrial-CMs. Figure 3A shows original recordings from a CTL CM, with clear 2APB-induced changes in the nuclear Ca2+-transient but little apparent effect on the cytosolic signal. Mean data in Figure 3B show statistically significant decreases only in [Ca2+]Nuc, at 2APB concentrations ≥25 μM. In AF-CMs large 2APB-effects are seen on [Ca2+]Nuc without clear effects on [Ca2+]Cyto (Figure 3C). Indeed, 2APB produced substantial and statistically-significant decreases in mean diastolic [Ca2+]Nuc, but not [Ca2+]Cyto (Figure 3D). Whereas in CTL cells the nuclear-cytosolic diastolic-Ca2+ differences continued to be statistically significant after 2APB (Figure 2B), in AF-cells 2APB eliminated the statistical significance of nuclear-cytosolic diastolic-Ca2+ differences, pointing to a particularly important contribution of IP3R-mediated Ca2+ transport in the nucleus of AF-cells. The amplitudes of [Ca2+]Nuc and [Ca2+]Cyto transients were not significantly affected by 2APB, similar to the kinetics (Online Figure XI).
Figure 3.

Effects of 2-APB on nucleoplasmic and cytosolic Ca2+-transients (CaTs). A. Original recordings of [Ca2+]Nuc and [Ca2+]Cyto in one control (CTL) dog atrial cardiomyocyte (CM) before and after application of 50 μM 2-APB. B. Violin plots with median (solid line) ± interquartile range (dotted lines) values of diastolic [Ca2+] and CaT amplitude before and after 5, 25, 50 μM 2-APB in CTL CMs. C. Original recordings of [Ca2+]Nuc and [Ca2+]Cyto in one atrial-fibrillation (AF) dog atrial CM before and after application of 50 μM 2-APB. D. Mean±SEM values of diastolic [Ca2+] and CaT-amplitude before and after 5, 25, 50 μM 2-APB in AF CMs. P-values in black reflect pre-2APB vs. 2APB, P-values in blue italics reflect nucleus vs. cytosol based on repeated measures multilevel mixed effects models (n/N=CMs/dogs).
Effects of IP3R-Knockdown on [Ca2+]Nuc.
While the effects of 2APB suggest an important role of IP3Rs in AF-related [Ca2+]Nuc changes, the drug has many potential effects other than blocking IP3Rs, for example inhibiting store-operated Ca2+-fluxes.21 To further test the role of nuclear IP3Rs in AF, we knocked down IP3Rs with siRNA. Canine atrial CMs appear to predominantly express ITPR1 and ITPR2 (Online Figure XIIA). Twenty-four-hour exposure to ITPR1and ITPR2 siRNA selectively suppressed ITPR1 and ITPR2 expression respectively, with the combination having a particularly strong effect (Online Figure XIIB, C). Online Figure XIIIA shows confocal line-scan images of permeabilized atrial CMs before and after the application of IP3, without or with siRNA-mediated knockdown of ITPR1and ITPR2. ITPR (1+2) knockdown strongly suppressed the IP3 effect (Online Figure XIIIB).
To evaluate the effect of ITPR knockdown in an AF-model, we turned to in vitro tachypaced cultured canine atrial CMs, which we previously showed to mimic in vivo AF-induced cellular electrophysiological remodeling.5 We incubated 1-Hz (P1) and 3-Hz (P3) paced CMs with siRNA targeting IP3R1 and/or IP3R2. Online Figure XIIIC shows confocal line-scan images of calibrated intracellular Ca2+ recorded during field-stimulation at 1 Hz from CMs that had been subjected to 3-Hz pacing for 24 hours, with and without ITPR knockdown. Online Figure XIIID shows [Ca2+]-time curves from small sectors in nuclear and cytosolic regions. [Ca2+]Nuc was greatly increased in P3-CMs, with changes qualitatively similar to those seen in CMs from AF-dogs (e.g., compare to Figure 3A and C). ITPR (1+2) knockdown predominantly affected [Ca2+]Nuc, with a particularly large effect on P3 CMs and no appreciable effect on [Ca2+]Cyto (see mean data in Online Figure XIIIE). These results strongly support a central role for IP3Rs in [Ca2+]Nuc changes with AF.
To dissect which IP3R-types play a role in regulating atrial CM [Ca2+]Nuc, we incubated P1 and P3 CMs with siRNA selectively targeting ITPR1 or ITPR2. ITPR1-knockdown had large effects on diastolic [Ca2+]Nuc (Figure 4A), similar to those of combined ITPR (1+2) knockdown, but did not change Ca2+-kinetics (Online Figure XIV). On the other hand, ITPR2-knockdown produced no significant changes (Figure 4B). These findings suggest that IP3R1 plays a predominant role in regulating the nucleoplasmic [Ca2+] in canine atrial CMs.
Figure 4.

A. Line-scan images and Ca2+-transients (CaTs) from single pixels at the regions indicated, along with median (solid line) ± interquartile range (dotted lines) violin plots for diastolic [Ca2+] and CaT-amplitude in cytosol and nucleus of 1-Hz (P1) and 3-Hz (P3) conditioned cardiomyocytes (CMs) with or without ITPR1 knockdown (KD), recorded at 1 Hz. B. Line-scan images and CaTs from single pixels at the regions indicated, along with mean±SEM violin plots for diastolic [Ca2+] and CaT-amplitude of cytosolic and nucleoplasmic CaTs in P1 and P3 CMs with or without ITPR2 KD, recorded at 1Hz. P-values in black reflect P1 vs P3, P-values in blue italics reflect with vs. without ITPR1/ITPR2 KD based on multilevel mixed effects models (n/N=CMs/dogs).
Modulation of CaMKII-HDAC Signaling by AF-related Nuclear Ca2+-handling Remodeling.
Ca2+/calmodulin-dependent protein kinase IIδ (CaMKIIδ) is the dominant isoform of CaMKII in the heart.22 CaMKIIδ phosphorylation of HDAC4 causes translocation to the cytoplasm, allowing transcription of a range of genes involved in cardiac hypertrophy and remodeling.22,23 We therefore tested how AF-related nuclear Ca2+-handling remodeling influences CaMKIIδ-HDAC4 signaling. Figure 5A shows confocal-microscopy images of atrial CMs from CTL and AF-dogs, immunostained for total CaMKIIδ (T-CaMKIIδ), Thr287-CaMKIIδ (p-CaMKIIδ), and HDAC4. Each image is accompanied by an NPC-stained (green) image to indicate the location of the nuclear envelope. AF did not significantly change T-CaMKIIδ protein-expression. Conversely, p-CaMKIIδ increased with AF in both nuclear and non-nuclear regions. Consequently, AF increased the CaMKIIδ phosphorylation-ratio, particularly in the nucleus. In addition, AF decreased the HDAC4[nuc]/HDAC4[cyto] ratio, suggesting HDAC4 export (Figure 5B). Immunoblot showed a selective AF-induced T-CaMKIIδ increase in non-nuclear extracts and a prominent p-CaMKIIδ increase in the nuclear extracts, resulting in an increased p-CaMKIIδ/T-CaMKIIδ fractional phosphorylation ratio only in the nucleus (Figure 5C, D).
Figure 5.

A. Original 2-dimensional images from control (CTL) and atrial-fibrillation (AF) atrial cardiomyocytes (CMs) after immunostaining for NPC (green), T-CaMKIIδ, p-CaMKIIδ (Thr287) and HDAC4 (red). B. T-CaMKIIδ, p-CaMKIIδ and HDAC4 immunofluorescence quantification. C. Immunoblots for T-CaMKIIδ, p-CaMKII and HDAC4 from CTL and AF atrial CM nuclear (Nuc) and non-nuclear (Non-Nuc) fractions. D. Mean±SEM T-CaMKIIδ, p-CaMKIIδ and HDAC4 protein-expression levels. P-values in black reflect CTL vs. AF, P-values in blue reflect Nuc vs. Non-Nuc based on multilevel mixed effects models (B) or regular mixed effects models (D). E. Overall and individual-dog means for IP3R1, T-CaMKIIδ, p-CaMKIIδ and HDAC4 immunofluorescence intensity from P1 CMs and P3 CMs with or without ITPR1 knockdown (KD). P-values in black reflect P1 vs. P3, P-values in blue italics reflect with vs. without ITPR1 KD based on multilevel mixed effects models, using data from all CMs from each dog (as non-independent data points), with dog as the independent unit of analysis.
In additional studies, canine atrial CMs were subjected to 24-hr in vitro pacing at 1 Hz or 3 Hz. Examples of IP3R1, CaMKIIδ and HDAC immunofluorescence are shown for paced CMs with and without ITPR1 knockdown in Online Figures XV-XVIII. Figure 5E shows quantification of mean per-dog data for cellular immunofluorescence of IP3R1, T-CaMKIIδ, p-CaMKIIδ, the p-CaMKIIδ/T-CaMKIIδ ratio and the nuclear/cytosolic HDAC4-ratio in 1-Hz and 3-Hz paced CMs, with and without ITPR1-knockdown. IP3R1-expression increased significantly with AF and was effectively reduced by IP3R1 knockdown. Suppression of IP3R1-expression greatly attenuated the p-CaMKIIδ and p-CaMKIIδ/T-CaMKIIδ ratio changes caused by 3-Hz pacing, particularly in the nucleus, and eliminated the decrease in nuclear/non-nuclear HDAC4-ratio seen with 3-Hz pacing. These results are consistent with the notion that CaMKII hyperphosphorylates HDAC4 to cause its export.
To further assess the role of CaMKIIδ in AF-related HDAC export, experiments were performed with in vitro paced CMs exposed to autocamtide CaMKII inhibitory peptide (AIP) or its vehicle in the culture medium. CaMKII-inhibition suppressed tachypacing-induced nuclear CaMKIIδ hyperphosphorylation and HDAC4 extranuclear translocation (Figure 6).
Figure 6.

A. Original 2-dimensional images from 1-Hz (P1) and 3-Hz (P3) conditioned and P3+AIP atrial cardiomyocytes (CMs) after immunostaining for NPC, T-CaMKIIδ, p-CaMKIIδ (Thr287) and HDAC4. B. Mean ± SEM of p-CaMKIIδ/T-CaMKIIδ and HDAC4[nuc] /HDAC4[cyto] ratios, with and without the cell-permeable CaMKII-inhibitor AIP. P-values in black reflect P1 vs. P3, P-values in blue reflect Control vs. AIP based on multilevel mixed effects models (n/N=CMs/dogs). C. Immunoblots and quantification of HDAC4 in subcellular fractions and nuclear/non-nuclear ratio from human atrial samples. P-values reflect control (CTL) vs. chronic AF (cAF) based on Mann Whitney test. Histone H3 and GAPDH were loading controls.
Remodeling of Nuclear CaMKII-HDAC4 Signaling in Atrial CMs of cAF Patients.
The results of Western-blot assays on human atrial CM nuclear and non-nuclear fractions are shown in Figures 6C and 7. Non-nuclear fractions expressed GAPDH but not histone H3; the converse was true of the nuclear fractions (Figure 7A). Nuclear IP3R1 was upregulated in cAF patients (Figure 7B); no IP3R1-signal could be detected in the non-nuclear fraction. Nuclear SERCA2a-expression was not altered in cAF-patient CM-nuclei, whereas SERCA2a was downregulated in the non-nuclear fraction (Online Figure VIIC). T-CaMKIIδ was upregulated in the nuclear fraction and showed a tendency to upregulation in the non-nuclear fraction. p-CaMKIIδ was upregulated in both nuclear and non-nuclear fractions (Online Figure VIID). Consistent with the immunofluoresence results in canine atrial CMs, the nuclear/non-nuclear ratio of HDAC4 was significantly reduced in AF-patients, compatible with enhanced nuclear export (Figure 6C).
Figure 7.

A. Western blot showing separation of nuclear and non-nuclear fractions from human atrial cardiomyocytes. B. Western blots and quantification of nuclear IP3R1 (Ctl=sinus-rhythm control samples; cAF=chronic AF patient samples). No IP3R1-signal could be detected in the non-nuclear fraction. C. Western blots and quantification of SERCA2a in subcellular fractions. D. Western blots and quantification of total CaMKIIδ and p-CaMKIIδ (Thr287) in subcellular fractions. P-values reflect Ctl vs. cAF, based on Student’s non-paired t-test (non-nuclear SERCA2a in panel C, nuclear p-CaMKIIδ in panel D) or Mann-Whitney test (all other comparisons). Histone H3 and GAPDH were loading controls.
IP3R1 Dysregulation and L-type Ca2+-current Remodeling.
L-type Ca2+-current downregulation is a functional hallmark in AF CMs and critical to AF-related electrical remodeling.2,4 To test whether IP3R1 signaling might contribute to ICa,L downregulation in AF, we prevented the pacing-induced IP3R1 upregulation in paced canine atrial CMs by ITPR1 siRNA-induced knockdown. Figure 8A shows ICaL recordings from exemplar CMs from each group studied. Mean data in Figure 8B show that CMs subjected to 24-hour tachypacing at 3 Hz (P3) showed reduced ICaL density compared to CMs paced at 1 Hz (P1). P1 and P3 CMs incubated with siRNA selectively targeting ITPR1 show that ITPR1 knockdown prevented ICaL decreases in P3 CMs Figure 8B). We conclude that IP3R1-dysregulation is a key upstream factor in the ICaL-decreases induced by atrial-CM tachycardia.
Figure 8.

A. ICaL recordings at 0.1 Hz in cardiomyocytes (CMs) paced for 24 hours at 1-Hz (P1) and 3-Hz (P3) with or without ITPR1 knockdown (KD). B. Mean±SEM ICaL density-voltage relations. C. Mean±SEM ITPR1 mRNA relative level, IP3R1protein (along with original immunoblots) and miR-26a expression in isolated CMs from control (CTL) and atrial-fibrillation (AF) dogs. D. Mean±SEM Luciferase relative activities from H9c2 cells transfected with miR-26a (20 nM), miR-NC (20 nM), or AMO-26a (10 nM). E. Mean±SEM ITPR1 mRNA relative level, IP3R1 protein expression from atrial CMs transfected with miR-NC, miR-26a and AMO-26a. F. Original recordings of [Ca2+]Nuc and [Ca2+]Cyto transients in 1-Hz stimulated atrial CMs transfected with miR-NC, miR-26a and AMO-26a. G. Median ± interquartile range violin plots for diastolic [Ca2+], CaT amplitude, time to peak and DT50 of nucleoplasmic and cytosolic CaTs from atrial CMs transfected with miR-NC, miR-26a and AMO-26a. In panel B, P-values in black reflect P1 vs. P3, P-values in blue italics reflect P3 vs. P3 ITPR1 KD based on multilevel mixed effects models. In panel C, P-values reflect CTL vs. AF based on regular mixed effects models. In panels D, E and G, P-values reflect miR-NC vs. miR-26a or AMO-26a based on regular mixed effects models (D, E) or multilevel mixed effects model (G); (N=number of independent experiments, n/N=CMs/dogs).
Mechanism Underlying Dysregulation of Canine Atrial CM IP3R1 in AF.
Analysis of ITPR1 mRNA and IP3R1 protein-expression in canine AF-CMs showed no significant change in mRNA but a substantial increase in IP3R1 protein-expression (Figure 8C). This result points to selective increases in IP3R1-translation, possibly via microRNA mediated regulation. MiR-target prediction indicated a number of miRs potentially targeting ITPR1. Of these, miR-26a was selected for further analysis, based on its strong expression in CMs and known dysregulation in human and canine AF.12,24 MiR-26a was significantly downregulated, by >60%, in AF-dog CMs (Figure 8C). Luciferase assay was used as a readout of ITPR1 translation. Overexpression of miR-26a overexpression significantly decreased luciferase readout, whereas silencing of endogenous miR26a by its antisense inhibitor AMO-26a increased luciferase fluorescence (Figure 8D). Moreover, transfection of miR-26a into dog-atrial CMs downregulated IP3R1 protein, whereas miR-26a silencing with its antisense inhibitor AMO-26a upregulated IP3R1-protein (Figure 8E). No apparent change in ITPR1-mRNA was seen with these manipulations (Figure 8E), consistent with the AF CM data (Figure 8C). Diastolic [Ca2+]Nuc was increased in atrial CMs after miR-26a overexpression, whereas the opposite effect was seen after AMO-26a treatment (Figure 8F, G), confirming the potential functional significance of miR-26a in IP3R1-mediated dysregulation of nuclear Ca2+-handling in AF.
DISCUSSION
In this study, we tested the effects of AF on atrial CM nuclear Ca2+ and related downstream signaling. We found that AF-related remodeling alters atrial CM Ca2+-handling to increase nuclear Ca2+-load by IP3R1-dependent Ca2+-mobilization from the nuclear envelope, with consequent activation of the nuclear CaMKIIδ-HDAC4 pathway.
Nuclear Ca2+-handling and Changes in Heart Disease.
The nucleoplasm is enclosed by a complex double-bilayer structure, the nuclear envelope.8 The nuclear envelope is traversed by NPCs that allow for relatively free diffusion of Ca2+ and small molecules from the cytosol. In addition, the nuclear envelope stores Ca2+ and releases it into the nucleoplasm via specialized Ca2+-release channels (IP3Rs). IP3R-mediated nuclear Ca2+-mobilization can be triggered by the stimulation of Gq-coupled receptors at both the sarcolemma and nuclear envelope that activate phospholipase-C to increase membrane IP3-content via the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2).20,25
Nuclear Ca2+-transients are implicated in Ca2+-dependent signaling that controls gene-transcription and are an integral part of the complex “excitation-transcription” regulatory machinery that governs cardiac remodeling in response to a wide range of stressors.10,26 In spontaneously-hypertensive rats, enhanced IP3R-mediated Ca2+-release is reported to contribute to larger cytosolic Ca2+-transients in hypertrophied CMs; nuclear Ca2+-transients were not altered.27 This observation contrasts with findings that intact nuclear IP3-signaling is essential for the development of neonatal CM hypertrophy in response to endothelin or insulin-like growth factor-1, as well as for pathological growth in response to activation of the calcineurin/nuclear factor of activated T-cells and HDAC5 pathways.28 More recent work in mouse and rabbit models of cardiac hypertrophy/failure and in tissue samples from failing human hearts indicates important remodeling of nuclear Ca2+-signaling, with changes similar to those we observed in AF atrial CMs, including elevated diastolic Ca2+-levels, slowed upstroke and decay and reduced systolic Ca2+-transient amplitudes.29 Increased nuclear CaMKII-activation and evidence for HDAC export into the cytosol were also noted.
In atrial CMs from AF-dogs, we noted here marked changes in nucleoplasmic Ca2+-handling, with the alterations appearing to be due partially related to kinetic factors manifested as a slowing of [Ca2+]Nuc decay (Figure 1F). This finding is compatible with continuing release from upregulated nuclear IP3Rs as a significant contributor to the observed nucleoplasmic Ca2+-changes, a notion supported by the substantial return of nucleoplasmic Ca2+-transients towards control in AF-CMs upon IP3R block with 2APB (Figure 3). Furthermore, 24-hour in vitro tachypacing of atrial-CMs mimicked the changes resulting from AF and allowed us to test the effect of siRNA-mediated IP3R-knockdown, which once again largely eliminated the diastolic Ca2+-increase caused by tachypacing (Online Figure XIII, Figure 4). Nucleoplasmic Ca2+-accumulation was associated with CaMKII autophosphorylation (especially in the nucleus) and consequent HDAC4 phosphorylation and export, all of which were suppressed by IP3R knockdown.
IP3Rs, Nuclear Ca2+-Signaling and AF.
IP3Rs play an important regulatory role in the heart, and are more strongly expressed in atrial than ventricular CMs.30 Biochemical indices suggest particularly brisk IP3 accumulation in atrium,31 and expression-studies show that IP3Rs are several-fold more strongly concentrated in atrium than ventricle.32 Endothelin induces atrial Ca2+-dependent arrhythmias in wild-type mice that are absent with IP3R2 knockout.33 IP3R-upregulation has been noted in both human34 and animal35 models of AF.
All of the components of the phosphoinositide signaling system are present and functional in cardiac nuclei and IP3R signaling is believed to be particularly important in excitation-transcription coupling.30 Here, we noted important changes in nuclear Ca2+-homeostasis both in atrial CMs isolated from AF-dogs and in an in vitro paced-CM model of AF-associated remodeling. These changes occurred concomitantly with upregulation of atrial CM nuclear IP3-expression. Moreover, suppression of the IP3R response by pharmacological or gene-expression manipulation largely eliminated the nucleoplasmic Ca2+-alterations, implicating IP3R remodeling as a primary cause.
IP3R1-CaMKII-mediated HDAC Export.
CaMKII is a multifunctional serine-threonine kinase that plays a prominent role in coupling Ca2+-signals to functional and transcriptional changes in CMs.36 CaMKIIα and CaMKIIβ are predominant in neuronal tissue, whereas CaMKIIδ predominates in CMs (with CaMKIIγ providing the rest).36 CaMKIIδ regulates transcription factors and other DNA-binding proteins including the cAMP response element-binding protein CREB,37 myocyte enhancer factor-2 (MEF2)38 and type-II HDACs.39 HDAC4 contains unique CaMKII docking-sites.23 Phosphorylation of HDAC4 by CaMKII at Ser246, Ser467, and Ser632 enhances nuclear export and prevents nuclear import of HDAC4.40 Overexpression of HDAC4 has profound effects on cardiac development, inhibiting cardiomyogenesis, while inhibition of class-II HDAC activity via CaMKII-induced phosphorylation enhances cardiac muscle development along with upregulation of transcription factors like Nkx2-5, GATA4 and MEF2c.41 Further work is needed to identify the molecular pathways dysregulated by altered CaMKII-HDAC signaling and define their pathophysiological role in AF.
Novelty and Potential Significance.
This is the first study to evaluate nuclear Ca2+-handling changes in AF. We found that AF causes important nuclear Ca2+-dysregulation that results in substantial elevation of nuclear diastolic [Ca2+]. Upregulation of IP3Rs, particularly IP3R1, is prominent and appears to be central to AF-induced nuclear Ca2+-handling changes. Increased nuclear Ca2+-load is associated with increased nuclear CaMKII-phosphorylation and HDAC4-export, which can be prevented by downregulating IP3R-expression with siRNA or by preventing CaMKII-activation through a cell-permeable autocamtide derivative.
Our findings provide new mechanistic insights and a working hypothesis whereby the AF-induced elevated nuclear Ca2+ leads to reduction of miR-26 (involving calcineurin),24 which upregulates IP3R1-expression that further elevates nuclear Ca2+ in a vicious positive feedback cycle. Downregulation of ICaL, a downstream electrophysiological response to this cycle, limits Ca2+-loading to put a brake on the system, at the expense of AF-promotion. Breaking the positive-feedback cycle by inhibiting IP3R1-upregulation might limit nuclear Ca2+-loading, CaMKII activation, HDAC4 nuclear export and depression of ICaL. The initial elevation of nuclear Ca2+ remains unexplained, but might be related to changes in nuclear shape and loss of invagination (Online Fig 3), as seen in ventricular myocytes in hypertrophy and progression to heart failure.29 These changes may increase nuclear transients via existing IP3R1 and reduced Ca2+-diffusion out of the nucleus, which would slow [Ca]Nuc-decline because re-uptake by the SR and nuclear envelope proceeds predominantly via extra-nuclear SR Ca2+-ATPase. In addition, the rapid atrial activation in AF exacerbates nuclear Ca2+-loading and associated remodeling.42
AF is one of the most common cardiac disorders, is increasing in prevalence and constitutes an important factor in cardiovascular morbidity and mortality.1 Presently-available therapies have major drawbacks and the identification of novel therapeutic targets is highly desirable.43 The identification of discrete nuclear remodeling changes associated with defined molecular signaling provides new insights into the pathophysiology of AF, while opening up pathways for the discovery and validation of novel therapeutic targets.
Potential Limitations,
No animal model of AF truly reproduces the complex clinical context. The model we selected here, involving electrically-maintained AF in the dog, produces a clinically-relevant substrate for AF-maintenance. However, these dogs do not manifest spontaneous AF and programmed stimulation is needed to reveal the AF-substrate. Furthermore, the AF-conditioning period is one week, in the range of paroxysmal AF but much shorter than in many cases of persistent AF. It is reassuring that several key findings, like IP3R1-upregulation and reduced nuclear/cytosolic HDAC4-ratio, were also seen in samples from patients with relatively long-standing AF (>6 months, Figures 6C and 7).
CaMKIIδ has multiple isoforms, which can be distinguished based on small differences in molecular mass.44 In the mouse, the “B” isoform is the predominant nuclear form.42,44 In our dog-atrial CM samples, CaMKIIδ ran as a single band and distinct isoforms could not be resolved. In most human atrial samples, two distinct CaMKIIδ-bands could be identified (Online Figure XIX). Quantification indicated that total CaMKIIδB in the nucleus did not change significantly, whereas the phosphorylated moiety was upregulated and both components were increased in non-nuclear fractions, paralleling the canine data (Figure 5). CaMKIIδC-expression did not change significantly.
The present studies differ from past work in which atrioventricular (AV)-block/ventricular pacing was used to control the ventricular rate. We have found that avoiding AV-block, similar to the practice in many other animal models of AF,45,46 produces more reliable AF-promotion. This paradigm is similar to initial AF-presentation in many patients, for whom ventricular rate-control therapy is lacking. The phenotype differs from that onlinn with a tightly-controlled ventricular response, most notably in terms of profibrotic gene-activation and the degree/reliability of AF-promotion.12 Cellular Ca2+-handling changes may differ in dogs with AF and AV-block from those without AV-block. Atrial tachycardia with AV-block typically produces a reduced cellular Ca2+-transient;47 in the present study, cytosolic Ca2+-transients and SR Ca2+-load were increased, consistent with atrial changes associated with cardiac dysfunction.48
We noted an increased nuclear [Ca2+] in AF CMs even at steady-state (Figure 1C), indicating that the [Ca2+] increase was not simply due to a reduced elimination rate of the Ca2+ entering with the preceding activation. Potential factors include a contribution of the increased SR Ca2+-load in AF (Online Figure V) and a contribution of increased IP3R-mediated nuclear Ca2+-entry to the steady-state equilibrium nuclear Ca2+-level, or even elevated [Na+]i that can raise steady-state nuclear and cytosolic Ca2+-levels.
We did not adjust statistical significance for the number of different analyses performed in the study. Each experimental series was based on specific scientific hypotheses and given the detailed and complex nature of our study, many series of studies were required, often with complex design and analysis requirements. We recognize that the more tests done, the higher the chance of observing a false association and that this is a weakness of the study.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Atrial fibrillation (AF) is the most common cardiac rhythm disorder; is associated with major morbidity and mortality; and has a progressive nature that makes it hard to treat.
The mechanisms underlying the progressive nature of AF are poorly understood.
New insights into underlying mechanisms are important for the identification of novel therapeutic targets and innovative treatments.
What Does This Article Contribute?
We show that sustained AF causes nuclear calcium overload in atrial cardiomyocytes.
This nuclear calcium overload is due to the upregulation of nuclear calcium channels called inositol-trisphosphate receptors, at least in part due to escape from translational inhibition by microRNA-26.
Prevention of AF-related nuclear calcium overload suppresses downregulation of cell-membrane calcium channels, known to be an important electrophysiological motif in AF-progression.
Atrial fibrillation (AF) is the most common cardiac rhythm disorder, is associated with major morbidity and mortality (for example, AF is the single most important cause of stroke in the elderly) and has a progressive nature that makes it hard to treat. The mechanisms underlying the progressive nature of AF are poorly understood. New insights into underlying mechanisms are important for the identification of novel therapeutic targets and the development of more effective treatments. The calcium content of cell-nuclei is an important controller of gene transcription. Here, we show that sustained AF causes calcium overload in the nuclei of atrial cardiac myocytes. Furthermore, we demonstrate that this nuclear calcium overload is due to the upregulation of nuclear calcium channels called inositol-trisphosphate receptors, which occurs at least in part due to their escape from translational inhibition by the AF-induced reduction in the expression of an important regulator, microRNA-26. Prevention of AF-related nuclear calcium overload by knocking down inositol-trisphosphate receptors suppressed downregulation of cell-membrane calcium channels, known to be an important electrophysiological motif in AF-progression. These new insights into the molecular mechanisms underlying AF progression may help to design newer and more effective treatments to tame this important clinical problem.
ACKNOWLEDGMENTS
the authors thank Anna Nozza for biostatistical consultation and data analysis, Nathalie L’Heureux, Chantal St. Cyr and Monika Hagedorn for technical help, and Lucie Lefebvre and Jennifer Bacchi for secretarial assistance with the manuscript.
SOURCES OF FNDING
This work was funded by operating funds from the Canadian Institutes of Health Research (SN: 148401), the Heart and Stroke Foundation of Canada (SN), National Institutes of Health (R01-HL142282 and P01-HL141084 to DMB; R01-HL131517, R01-HL136389, and R01-HL089598 to DD). the German Research Foundation (Do 769/4-1 to DD), Austrian Science Fund FWF (V 530 to SLH), and the Netherlands Organization for Scientific Research (ZonMW Veni 91616057 to JH).
Nonstandard Abbreviations and Acronyms:
- AF
atrial fibrillation
- AIP
autocamtide CaMKII inhibitory peptide
- CaMKII
Ca2+/Calmodulin dependent protein kinase-II
- CM
cardiomyocytes
- ERPs
effective refractory periods
- HDAC
histone deacetylase
- HDAC4
histone deacetylase type-4
- ICaL
L-type calcium current
- IP3
inositol-trisphosphate
- IP3R
inositol-trisphosphate-receptor
- miR-26a
microRNA-26a
- NE
nuclear envelope
- RA
right atrial
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Andrade J, Khairy P, Dobrev D, Nattel S. The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ Res 2014;114:1453–1468. [DOI] [PubMed] [Google Scholar]
- 2.Nattel S, Dobrev D. The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur Heart J 2012;33:1870–1877. [DOI] [PubMed] [Google Scholar]
- 3.Greiser M, Lederer WJ, Schotten U. Alterations of atrial Ca2+ handling as cause and consequence of atrial fibrillation. Cardiovasc Res 2011;89:722–733. [DOI] [PubMed] [Google Scholar]
- 4.Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol 2008;1:62–73. [DOI] [PubMed] [Google Scholar]
- 5.Qi XY, Yeh Y-H, Xiao L, Burstein B, Maguy A, Chartier D, Villeneuve LR, Brundel BJJM, Dobrev D, Nattel S. Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ Res 2008;103:845–854. [DOI] [PubMed] [Google Scholar]
- 6.Tavi P, Pikkarainen S, Ronkainen J, Niemelä P, Ilves M, Weckström M, Vuolteenaho O, Bruton J, Westerblad H, Ruskoaho H. Pacing-induced calcineurin activation controls cardiac Ca2+ signalling and gene expression. J Physiol 2004;554:309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cardin S, Libby E, Pelletier P, Le Bouter S, Shiroshita-Takeshita A, Le Meur N, Léger J, Demolombe S, Ponton A, Glass L, et al. Contrasting gene expression profiles in two canine models of atrial fibrillation. Circ Res 2007;100:425–433. [DOI] [PubMed] [Google Scholar]
- 8.Gaborit N, Steenman M, Lamirault G, Le Meur N, Le Bouter S, Lande G, Léger J, Charpentier F, Christ T, Dobrev D, et al. Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation 2005;112:471–481. [DOI] [PubMed] [Google Scholar]
- 9.da Costa Martins PA, Leptidis S, De Windt LJ. Nuclear calcium transients: Hermes Propylaios in the heart. Circulation 2014;130:221–223. [DOI] [PubMed] [Google Scholar]
- 10.Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest 2006;116:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ljubojevic S, Bers DM. Nuclear calcium in cardiac myocytes. J Cardiovasc Pharmacol 2015;65:211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 2012;126:2051–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang D, Wu CT, Qi X, Meijering RA, Hoogstra-Berends F, Tadevosyan A, Cubukcuoglu Deniz G, Durdu S, Akar AR, Sibon OC, et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of α-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 2014;129:346–358. [DOI] [PubMed] [Google Scholar]
- 14.Qi XY, Diness JG, Brundel BJ, Zhou XB, Naud P, Wu CT, Huang H, Harada M, Aflaki M, Dobrev D, et al. Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation 2014;129:430–440. [DOI] [PubMed] [Google Scholar]
- 15.Thomas D, Tovey SC, Collins TJ, Bootman MD, Berridge MJ, Lipp P. A comparison of fluorescent Ca2+ indicator properties and their use in measuring elementary and global Ca2+ signals. Cell Calcium 2000;28:213–223. [DOI] [PubMed] [Google Scholar]
- 16.Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol 1994;40:3–29. [DOI] [PubMed] [Google Scholar]
- 17.Ljubojević S, Walther S, Asgarzoei M, Sedej S, Pieske B, Kockskämper J. In situ calibration of nucleoplasmic versus cytoplasmic Ca2+ concentration in adult cardiomyocytes. Biophys J 2011;100:2356–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tadevosyan A, Maguy A, Villeneuve LR, Babin J, Bonnefoy A, Allen BG, Nattel S. Nuclear-delimited angiotensin receptor-mediated signaling regulates cardiomyocyte gene expression. J Biol Chem 2010;285:22338–22349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XHT, Nattel S, Dobrev D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 2014;129:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merlen C, Farhat N, Luo X, Chatenet D, Tadevosyan A, Villeneuve LR, Gillis M-A, Nattel S, Thorin E, Fournier A, et al. Intracrine endothelin signaling evokes IP3-dependent increases in nucleoplasmic Ca2+ in adult cardiac myocytes. J Mol Cell Cardiol 2013;62:189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeHaven WI, Smyth JT, Boyles RR, Bird GS, Putney JW. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J Biol Chem 2008;283:19265–19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Bers DM, Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res 2003;92:912–919. [DOI] [PubMed] [Google Scholar]
- 23.Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest 2006;116:1853–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo X, Pan Z, Shan H, Xiao J, Sun X, Wang N, Lin H, Xiao L, Maguy A, Qi X-Y, et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest 2013;123:1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tadevosyan A, Xiao J, Surinkaew S, Naud P, Merlen C, Harada M, Qi X, Chatenet D, Fournier A, Allen BG, et al. Intracellular Angiotensin-II Interacts With Nuclear Angiotensin Receptors in Cardiac Fibroblasts and Regulates RNA Synthesis, Cell Proliferation, and Collagen Secretion. J Am Heart Assoc 2017;6. pii: e004965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dewenter M, von der Lieth A, Katus HA, Backs J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circ Res 2017;121:1000–1020. [DOI] [PubMed] [Google Scholar]
- 27.Harzheim D, Movassagh M, Foo RS, Ritter O, Tashfeen A, Conway SJ, Bootman MD, Roderick HL. Increased InsP3Rs in the junctional sarcoplasmic reticulum augment Ca2+ transients and arrhythmias associated with cardiac hypertrophy. Proc Natl Acad Sci U S A 2009;106:11406–11411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arantes LAM, Aguiar CJ, Amaya MJ, Figueiró NCG, Andrade LM, Rocha-Resende C, Resende RR, Franchini KG, Guatimosim S, Leite MF. Nuclear inositol 1,4,5-trisphosphate is a necessary and conserved signal for the induction of both pathological and physiological cardiomyocyte hypertrophy. J Mol Cell Cardiol 2012;53:475–486. [DOI] [PubMed] [Google Scholar]
- 29.Ljubojevic S, Radulovic S, Leitinger G, Sedej S, Sacherer M, Holzer M, Winkler C, Pritz E, Mittler T, Schmidt A, et al. Early remodeling of perinuclear Ca2+ stores and nucleoplasmic Ca2+ signaling during the development of hypertrophy and heart failure. Circulation 2014;130:244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kockskämper J, Zima AV, Roderick HL, Pieske B, Blatter LA, Bootman MD. Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J Mol Cell Cardiol 2008;45:128–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leung E, Johnston CI, Woodcock EA. Stimulation of phosphatidylinositol metabolism in atrial and ventricular myocytes. Life Sci 1986;39:2215–2220. [DOI] [PubMed] [Google Scholar]
- 32.Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA, Blatter LA. IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol 2008;294:H596–604. [DOI] [PubMed] [Google Scholar]
- 33.Li X, Zima AV, Sheikh F, Blatter LA, Chen J. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate (IP3)-receptor type 2-deficient mice. Circ Res 2005;96:1274–1281. [DOI] [PubMed] [Google Scholar]
- 34.Yamda J, Ohkusa T, Nao T, Ueyama T, Yano M, Kobayashi S, Hamano K, Esato K, Matsuzaki M. Up-regulation of inositol 1,4,5 trisphosphate receptor expression in atrial tissue in patients with chronic atrial fibrillation. J Am Coll Cardiol 2001;37:1111–1119. [DOI] [PubMed] [Google Scholar]
- 35.Zhao ZH, Zhang HC, Xu Y, Zhang P, Li XB, Liu YS, Guo JH. Inositol-1,4,5-trisphosphate and ryanodine-dependent Ca2+ signaling in a chronic dog model of atrial fibrillation. Cardiology 2007;107:269–276. [DOI] [PubMed] [Google Scholar]
- 36.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 2011;51:468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci 2001;4:261–267. [DOI] [PubMed] [Google Scholar]
- 38.Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, Overbeek P, Richardson JA, Grant SR, Olson EN. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest 2000;105:1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000;408:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Qin G, Zhao TC. HDAC4: mechanism of regulation and biological functions. Epigenomics 2014;6:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karamboulas C, Swedani A, Ward C, Al-Madhoun AS, Wilton S, Boisvenue S, Ridgeway AG, Skerjanc IS. HDAC activity regulates entry of mesoderm cells into the cardiac muscle lineage. J Cell Sci 2006;119:4305–4314. [DOI] [PubMed] [Google Scholar]
- 42.Ljubojevic-Holzer S, Herren AW, Djalinac N, Voglhuber J, Morotti S, Holzer M, Wood BM, Abdellatif M, Matzer I, Sacherer M, et al. CaMKIIδC Drives Early Adaptive Ca2+ Change and Late Eccentric Cardiac Hypertrophy. Circ Res 2020; August 21. doi: 10.1161/CIRCRESAHA.120.316947. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heijman J, Guichard JB, Dobrev D, Nattel S. Translational Challenges in Atrial Fibrillation. Circ Res 2018;122:752–773. [DOI] [PubMed] [Google Scholar]
- 44.Mishra S, Gray CB, Miyamoto S, Bers DM, Brown JH. Location matters: clarifying the concept of nuclear and cytosolic CaMKII subtypes. Circ Res 2011;109:1354–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995;92:1954–1968. [DOI] [PubMed] [Google Scholar]
- 46.Martins RP, Kaur K, Hwang E, Ramirez RJ, Willis BC, Filgueiras-Rama D, Ennis SR, Takemoto Y, Ponce-Balbuena D, Zarzoso M, et al. Dominant frequency increase rate predicts transition from paroxysmal to long-term persistent atrial fibrillation. Circulation 2014;129:1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wakili R, Yeh YH, Yan Qi X, Greiser M, Chartier D, Nishida K, Maguy A, Villeneuve LR, Boknik P, Voigt N, et al. Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ Arrhythm Electrophysiol 2010;3:530–541. [DOI] [PubMed] [Google Scholar]
- 48.Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kääb S, Ravens U, Coutu P, Dobrev D, Nattel S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol 2008;1:93–102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.