

Abstract
To reduce time and cost, virtual ligand screening (VLS) often precedes experimental ligand screening in modern drug discovery. Traditionally, high-resolution structure-based docking approaches rely on experimental structures, while ligand-based approaches need known binders to the target protein and only explore their nearby chemical space. In contrast, our structure-based FINDSITEcomb2.0 approach takes advantage of predicted, low-resolution structures and information from ligands that bind distantly related proteins whose binding sites are similar to the target protein. Using a boosted tree regression machine learning framework, we significantly improved FINDSITEcomb2.0 by integrating ligand fragment scores as encoded by molecular fingerprints with the global ligand similarity scores of FINDSITEcomb2.0. The new approach, FRAGSITE, exploits our observation that ligand fragments, e.g., rings, tend to interact with stereochemically conserved protein subpockets that also occur in evolutionarily unrelated proteins. FRAGSITE was benchmarked on the 102 protein DUD-E set, where any template protein whose sequence identify >30% to the target was excluded. Within the top 100 ranked molecules, FRAGSITE improves VLS precision and recall by 14.3 and 18.5%, respectively, relative to FINDSITEcomb2.0. Moreover, the mean top 1% enrichment factor increases from 25.2 to 30.2. On average, both outperform state-of-the-art deep learning-based methods such as AtomNet. On the more challenging unbiased set LIT-PCBA, FRAGSITE also shows better performance than ligand similarity-based and docking approaches such as two-dimensional ECFP4 and Surflex-Dock v.3066. On a subset of 23 targets from DEKOIS 2.0, FRAGSITE shows much better performance than the boosted tree regression-based, vScreenML scoring function. Experimental testing of FRAGSITE’s predictions shows that it has more hits and covers a more diverse region of chemical space than FINDSITEcomb2.0. For the two proteins that were experimentally tested, DHFR, a well-studied protein that catalyzes the conversion of dihydrofolate to tetrahydrofolate, and the kinase ACVR1, FRAGSITE identified new small-molecule nanomolar binders. Interestingly, one new binder of DHFR is a kinase inhibitor predicted to bind in a new subpocket. For ACVR1, FRAGSITE identified new molecules that have diverse scaffolds and estimated nanomolar to micromolar affinities. Thus, FRAGSITE shows significant improvement over prior state-of-the-art ligand virtual screening approaches. A web server is freely available for academic users at http://sites.gatech.edu/cssb/FRAGSITE.
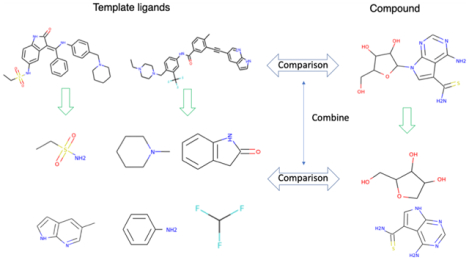
Graphical Abstract
INTRODUCTION
Accurate modeling of protein-ligand interactions is not only of fundamental importance for understanding the biological processes in living cells but also has practical applications to drug discovery.1–6 Earlier methods for modeling protein-ligand interactions can be classified into two broad categories: (1) structure-based and (2) ligand-based. Structure-based methods use high-resolution experimental or high-accuracy homology-modeled structures and physicochemical principles to dock the ligand into the protein’s structure. The binding pose(s) is (are) then used to calculate absolute or relative protein-ligand binding affinities.1,7–15 The advantages of docking methods are the possibility of discovering new binding ligands not similar to known binders and that they provide the binding position/pose for use in subsequent ligand optimization.11,16 However, these methods suffer from the unavailability of high-resolution experimental structures (e.g., membrane bound proteins: a majority of G-protein coupled receptors, ion channels, etc.17) and the inaccuracy of their scoring functions.18 In principle, one can apply homology-modeled structures for docking methods,15 but in addition to inaccurate scoring functions, the inaccuracy of low-resolution target structures significantly diminishes their performance.19 Ligand-based methods require at least one ligand known to bind to the target. A virtual model for the ligand is derived from the set of known binders, and the likelihood of an unknown ligand binding to the target is inferred based on similarity to known binding ligands.20–25 The advantages of ligand-based methods are their inexpensive computational cost and the lack of requirement of the target protein’s structure. In practice, ligand-based methods are often more robust and accurate than docking methods.26 Their disadvantage is the prerequisite of at least one known binder; this is often not the case for many protein targets.27 However, docking-based methods have the distinct advantage over ligand-based methods (except for the 3D shape-based ROCS methods) in that they can potentially discover novel molecules not similar to any in the library of all existing binders of any protein.
To address the limitations of earlier methods that required high-resolution structures to achieve optimal performance or known ligand binders, recently developed methods use homology-modeled, coarse-grained structures. They show comparable performance as that when high-resolution structures are used. Furthermore, homology modeling (LHM) does not require known binders to the target protein. Thus, LHM expands the scope of ligand similarity-based approaches.19,28–30 Ligand homology modeling transfers information about ligands that bind to similar pockets in the template protein to those in the target protein, regardless of their evolutionary relationship.19,31 LHM works because the number of stereochemically known distinct small-molecule ligand binding pockets is remarkably small, about 500.32,33 LHM approaches can be assigned to two classes: (1) low-resolution structure-based docking and (2) structure/threading ligand similarity-based methods. Representatives of low-resolution docking methods include Q-DOCK,34 Q-DOCKLHM,35 and BSP-SLIM.36 So far, Q-DOCK and Q-DOCKLHM have been tested for ligand pose prediction using low-resolution homology-modeled target structures and demonstrate comparable results to high-resolution structure-based methods. BSP-SLIM36 has only been tested on 6 randomly selected targets of the 40 DUD (A Directory of Useful Decoys37) set for virtual screening (ligand ranking). Thus, how well low-resolution structure-based docking methods perform in a large benchmarking set like DUD or DUD-enhanced (DUE-E)5 for virtual screening is not clear. In contrast, the latest version of structure/threading ligand similarity-based methods is FINDSITEcomb2.0.31 FINDSITEcomb2.0 focuses on ligand ranking (instead of pose prediction) and has been shown to yield quite reliable ligand binding predictions. FINDSITEcomb2.031 applies the ideas of homology modeling to binding site prediction and virtual ligand screening.
FINDSITEcomb2.0 is a hybrid of structure-based and ligand-based approaches. It utilizes pockets in the target structure to search for similar pockets and their ligands by structure–pocket and structure–structure comparison in templates. The selected ligands are then used to build a ligand model for the target protein.19,31 It then uses ligand model similarity as done in ligand-based methods to search for similar ligands in the screened compound library. The target structure can be experimentally determined or homology-modeled. Benchmarking shows that the performance of FINDSITEcomb2.0 is quite insensitive to whether an experimental or modeled target structure is used. Thus, FINDSITEcomb2.0 does not require high-resolution structures nor a known set of ligands for a given target. As such, it is different from pure ligand-based methods that require at least one known binder of the given target.
Possibly due to the recent significant advances of high-accuracy protein structure prediction,38 the major limitation of docking methods is now their lack of an accurate scoring function for ligand ranking;18 this makes them usually less accurate than ligand similarity-based methods in terms of successfully identifying binding ligands. In high-resolution docking, the physics-based or empirical/knowledge-based scoring function relies on predefined functional forms and is sensitive to small structural distortions/flexibilities. Meanwhile, in low-resolution docking, scoring functions are knowledge-based and usually are target/family-specific. Thus, they are not universally applicable.34–36 To improve the scoring function accuracy and tolerance to structural distortion/flexibility for high-resolution structure docking, Ballester and Mitchell employed machine learning using random forest and support vector regression approaches and features that depend on the occurrence counts of atomic contacts to predict binding affinities18,39 (RF-score). Their approach shows a very high correlation (R = 0.774) of predicted with experimental binding affinities and demonstrates that using only atomic contacts (which is insensitive to small structure distortions/flexibility) captures the essential features of protein-ligand binding. A new version of the RF-score called RF-score-VS has been developed by including decoys in training to correct a limitation of the RF-score.40 This approach was shown to be much more robust for VS than the original RF-score. Recently, convolutional neural network (CNN) or deep CNN technology was applied to VLS.41,42 CNN methods also require high-resolution protein structures and docked ligands and are basically a method for ranking ligands. There are issues with the small databases used for training and testing CNNs, which lead to memorization artifacts. For example, hidden biases in the DUD-E5 VLS benchmark set caused CNNs to perform similarly to AutoDock Vina,43 a traditional docking algorithm. A more recent study in ref 44 developed a new scoring function called vScreenML that included the RF-score features and trained on their newly developed training set. vScreenML performs similarly to RF-score-VS when tested on the DEKOIS 2.0 set.45 Both RF-score-VS and vScreenML scores show the importance of decoys in training.
While recognizing the importance of decoys in training, in this work, we exploit a new approach for improving ligand virtual screening, which exploits the fact that the number of stereochemically distinct known small-molecule ligand binding pockets is small.32,33 We recently observed that specific ring substructures in a given chemical structure tend to interact with quite unique protein subpockets that occur across evolutionarily unrelated proteins. These substructures explain the frequently used privileged substructures/scaffolds in drug discovery.46
The rest of the paper is organized as follows: In the Methods section, we describe the boosting tree scoring function for ligand protein-binding score prediction. Then, in the Results section, on the DUD-E5 set, we compare the performance of FRAGSITE predictions and scoring functions for virtual ligand screening to FINDSITEcomb2.031 and AutoDock Vina.11 We then present the results for experimental testing of the precision and ligand diversity of FRAGSITE’s and FINDSITEcomb2.0 predictions for two proteins, DHFR and ACVR1. DHFR is a key enzyme in the one carbon carrier folate pathway that occurs across the domains of life and provides the building blocks for DNA synthesis. ACVR1 is a possible therapeutic target relevant to aggressive pediatric brain cancer, diffuse intrinsic pontine glioma (DIPG),47 and the connective tissue disorder fibrodysplasia ossificans progressiva (FOP).48 Finally, in the Discussion section, we discuss some of the advantages and shortcomings of FRAGSITE.
METHODS
The flowchart of FRAGSITE is shown in Figure 1. Given an input target’s protein amino acid sequence and a small-molecule compound library, we employ the three components of FINDSITEcomb2.031 (i.e., FINDSITEfilt using PDB ligand-protein complex structures, FINDSITEX30 using DrugBank49 drug-target information, and FINDSITEX using ChEMBL compound-protein binding data50) to independently select the template ligands associated with the target protein. As in FINDSITEcomb2.0,31 a template protein must have a TM-score51 > 0.6 to the target protein’s structure, and at least 80% of the template sequence must be aligned to the target sequence. A sequence cutoff is applied in benchmarking mode to exclude templates whose sequence identity > cutoff for selecting template ligands. Then, template ligands are selected as they are in FINDSITEcomb2.031 as follows: (1) up to the top 100 ligands from PDB ligand-protein complex structures52 (if there are more than 100 ligands, then the top 100 ligands are selected; if there are fewer ligands, then all are selected), (2) drugs from up to the top 20 targets from DrugBank drug-target information,49 and (3) compounds from up to the top 20 proteins from ChEMBL compound-protein binding data.50 Then, for each set of template ligands, we generate a fingerprint profile. For a given target, these three profiles will be independently combined with each screened molecule’s fingerprint and mTC score as calculated in FINDSITEcomb2.0 to generate three feature vectors. Finally, a machine learning, boosting regression tree method (independently trained on each set of template ligands) predicts three scores for each screened molecule. The FRAGSITE score is the maximal score of the three machine learning scores. We detail each of the steps below.
Figure 1.
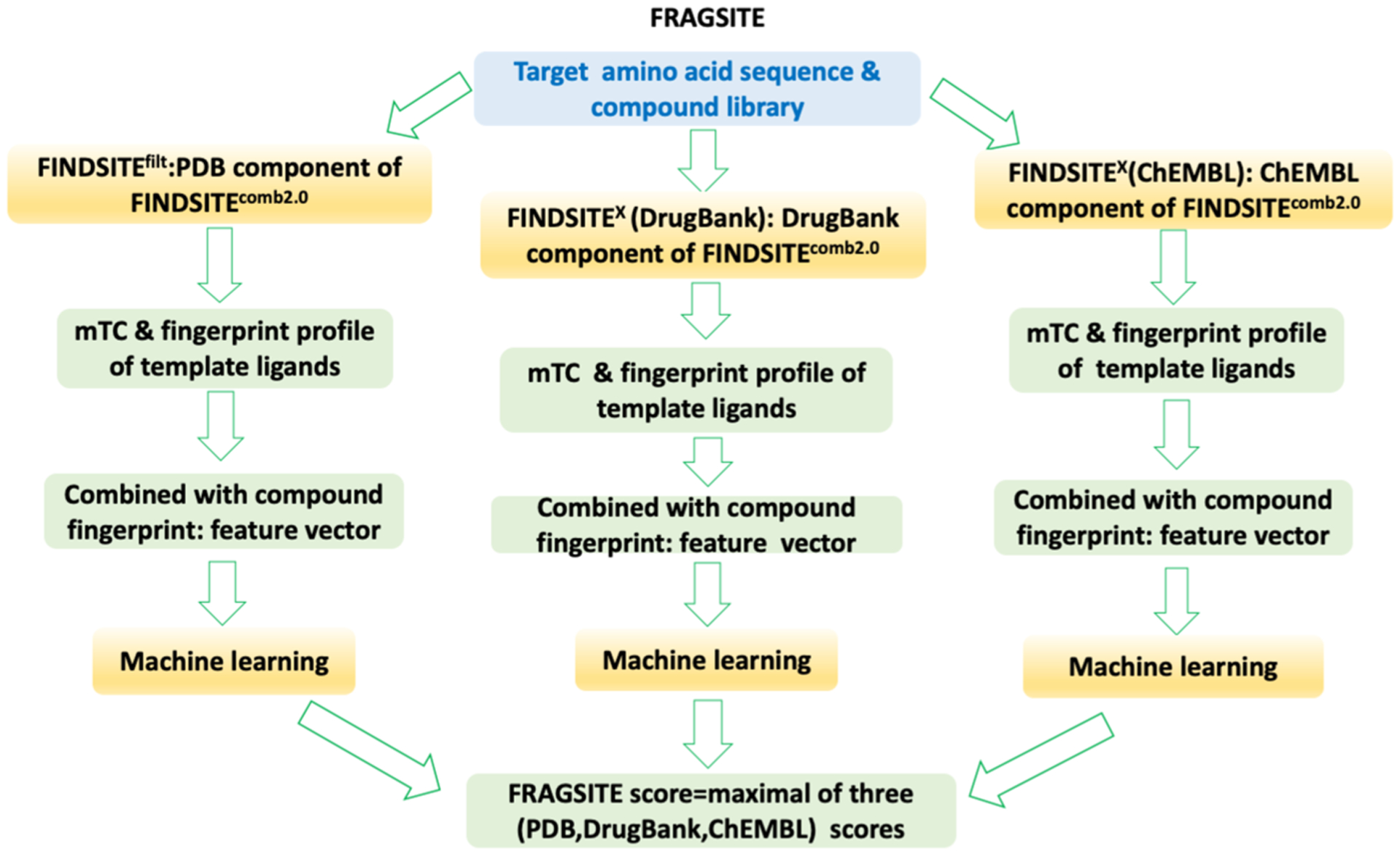
Flowchart of the FRAGSITE approach.
Feature Vector Generation.
Feature vectors are generated based on fragmentation of both the template ligands and screened compounds. For each ligand in the three template ligand sets and for the screened compounds, fragmentation is done using the substructure-based PubChem fingerprint.53 The PubChem fingerprint encodes molecular fragment information using 881 binary digits that can be represented as an 881 dimensional vector. The list of substructures encoded in each bit can be accessed at ftp://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.txt. For PubChem fingerprint computation, we employed the free PaDEL-Descriptor package.54
Generally, there are usually multiple template ligands for a given protein target. We therefore build a profile P of their fingerprints using the mean value of each of the 881 bits (elements)
| (1) |
where Pj is the jth element of profile P, is the jth bit (element) of template ligand i, and N is the number of ligands in the template ligand set. This profile is an 881 dimensional vector whose elements assume values between 0 and 1. The element representing the most conserved fragment will have the largest value. Representing the 881 dimensional fingerprint vector of a to be screened compound as L with element values of either 0 or 1, we construct the 883 dimensional feature vector for each component of FRAGSITE as
| (2) |
The overall similarity score mTC computed from the FP2 fingerprints55 takes into account the topology or sequential order of different fragments of a ligand, P·L takes into account the fragment composition of a ligand regardless of their topology, and the 881 dimensional Hadamard product (constructed by element-wise multiplication) takes into account an individual fragment’s role with emphasis on the most conserved template ligand fragments. Each element of X ranges between 0 and 1. For a given screened molecule, three feature vectors are constructed based on template ligands from the PDB,52 DrugBank,49 and ChEMBL,50 respectively.
Boosting Regression Tree Scoring Function for Ligand-Protein Binding.
The boosting regression tree involves generating a sequence of decision trees, each grown on the residuals of all previous trees.56,57 A decision tree regression is implemented with a maximal depth of eight. The scoring function is represented as boosting decision trees57
| (3) |
where Tm is a decision tree, ε is the shrinkage factor or learning rate, Ntree is the number of trees or iterations, and X is the feature vector defined in eq 2.
In the training of the boosting tree function (3) for ligand-protein binding, the objective function value will be assigned as 1 if the molecule is a true binder of the target (in the DUD-E benchmarking set, the active ligands) and 0 if the molecule is not a binder (decoys in DUD-E).
Training and Benchmarking of the Boosting Regression Tree Scoring Function.
To benchmark FRAGSITE and train the model for future applications, we utilized the DUD-E5 ligand virtual screening benchmark dataset. We conduct a modified leave one out cross-validation (LOOCV). Conventionally, LOOCV will use all other targets than the tested one in the dataset for training. However, this will favor those targets having close homologues in the dataset. Here, in training, we use stricter benchmarking than just LOOCV by excluding all targets having a sequence identity of >30% to the given tested target; we term this LOOCV30%. Since the total number of molecules (actives + decoys) for all 102 targets is around 1.4 million, with mostly decoys, and the dimension of the feature space is 883, this would require quite large memory and cause too much unbalance between positive and negative samples to use all of them for training. We thus only randomly sample ~10% of the decoys and all actives for training. Based on the average training size, we use the following empirical parameters for the boosting regression tree (see eq 3): the number of boosted trees, Ntree = 150, and the learning rate, ε = 0.05.
Assessment.
In modern drug discovery, the compound library could be immense; thus, the top few percent of ligands could contain a large number of molecules, and the cost of experimentally screening all of them could be significant.6 For example, Stein et al. docked 150 million molecules to an MT1 crystal structure;6 1% or even 0.01% of molecules are still too many for experimental testing. Therefore, for cutoff-independent evaluation, we prefer AUPR, the area under the precision-recall curve58 to AUC (area under the ROC curve) to compare FRAGSITE with FINDSITEcomb2.0 and AutoDock Vina. AUPR is a better measure than AUC to distinguish the ability of methods to rank positives in the very top ranks when true positives are rare and only the very top ranked ones are tested as is the case in virtual ligand screening.58 Also, for cutoff-dependent assessment, we examine the precision and recall per target within the top 100 ranked molecules rather than the enrichment factor of the top ranked few percent.
We use the AUC for cutoff-independent assessment and the enrichment factor to compare with some other approaches such as AtomNet41 and CNN scoring.41 The enrichment factor is defined as
| (4) |
at a fraction of x screened molecules.
AtomNet uses a randomly selected set of 30 targets for testing and the rest for training. The CNN scoring method clustered the 102 DUD-E targets with an 80% sequence identity threshold and divided the clusters into 3 sets in a 3-fold cross-validation test. For fair comparison to AtomNet and CNN scoring, we apply an 80% sequence cutoff to templates for template ligand selection: i.e., ligands from any template in the PDB, DrugBank, and ChEMBL having a sequence identity of >80% will be ignored.
In practice, as with FINDSITEcomb2.0, we also report the predicted precision and recall based on the machine learning score Sfrg of FRAGSITE
| (5a) |
| (5b) |
The precision is useful for judging if the prediction is confident or not. To derive the predicted precision and recall using eq 5, we merge all predictions for actives and decoys of all targets from the DUD-E dataset5 and bin the score Sfrg from 0 to 1 using ΔSfrg = 0.05.
Experimental Testing of FRAGSITE.
We experimentally tested two protein targets to assess the ability of FRAGSITE to discover new hits. We then compare their chemical diversity to FINDSITEcomb2.0. FRAGSITE not only utilizes the global ligand structure similarity based on their mTC scores but also fragment similarity through their fragmentation fingerprint score; thus, it might be expected to find more diverse ligands. We applied FRAGSITE and FINDSITEcomb2.0 to predict ligands that bind Escherichia coli (E. coli) dihydrofolate reductase (E. coli DHFR) and human ACVR1 receptor kinase. A sequence identity cutoff of 30% for protein templates was applied. We screened against molecules from the National Cancer Institute (NCI) diversity set (https://dtp.cancer.gov/organization/dscb/obtaining/available_plates.htm). The NCI diversity set consists of 1597 molecules from the Diversity Set III, 97 from the Approved Oncology Drugs Set IV, and 118 from the Natural Product Set II (total 1812 NCI molecules). Predictions from FRAGSITE and FINDSITEcomb2.0 whose expected precision > 0.5 are selected for experimental validation by a Thermofluor assay, a sensitive fluorescence-based thermal shift method indicative of ligand binding to protein targets, using reported protocols.31,59 Then, a few top ranked true binders of E. coli DHFR identified by FRAGSITE were subsequently confirmed to inhibit catalysis of DHFR via steady-state kinetics inhibition assays. Details of the experimental methods are found in the Supporting Information.
RESULTS
Fragment Frequencies.
First, we analyzed the ligand fragments defined by PubChem in the PDB database52 that were used by FINDSITEcomb2.0 for ligand homology modeling. To avoid over-represented protein chains in the PDB protein-ligand structures, we clustered 142,483 sequences of these protein chains using 30% sequence identity cutoff into 11,615 clusters. Using the ligands associated with the cluster centroid chains, we calculate the frequency of each of the 881 fragments. In Table 1, we list the top 40 most frequent fragments with frequencies ranging from 35 to 99% of the PDB ligands. There are nine elements, two types of rings, five simple atom pairs, 12 simple atom nearest neighbors, two detailed atom neighborhoods, and 10 simple SMARTS patterns (https://www.daylight.com/dayhtml/doc/theory/theory.smarts.html). Except for complex SMARTS patterns, all the other six types of fragments defined in PubChem are present in the top 40. Overall, on average, there are 115 atomic element counts (individual chemical atoms) whose frequency is 7.4%, 148 rings whose frequency is 2.6%, 64 simple atom pairs whose frequency is 7.5%, 89 atom nearest neighbors whose frequency is 16.0%, 44 detailed atom neighborhoods whose frequency is 10.3%, 253 simple SMARTS patterns whose frequency is 9.5%, and 168 complex SMARTS patterns whose frequency is 0.2%. We note that the ring fragment “≥ 1 any ring size 6” (i.e., at least one ring with size of six atoms) has a frequency of 43%. If we use DrugBank49 drugs, then the same frequency is 81%. Interestingly, these mainly correspond to the most frequently used “privileged scaffolds” in drug discovery.46 In Table S1 in the Supporting Information, the top 40 most frequent fragments of the 6505 DrugBank49 drugs are listed.
Table 1.
Top 40 Most Frequent Fragments in PDB Ligands
| index | fraction of ligands | PubChem fragment |
|---|---|---|
| 0 | 0.993599 | ≥4 H |
| 9 | 0.975919 | ≥2 C |
| 284 | 0.974334 | C–C |
| 283 | 0.974151 | C–H |
| 344 | 0.970676 | C(~C)(~H) |
| 18 | 0.958361 | ≥1 O |
| 286 | 0.935804 | C–O |
| 352 | 0.933122 | C(~C)(~O) |
| 19 | 0.93172 | ≥2 O |
| 1 | 0.929098 | ≥8 H |
| 308 | 0.923185 | O–H |
| 406 | 0.878803 | O(~C)(~H) |
| 332 | 0.84765 | C(~C)(~C) |
| 366 | 0.828568 | C(~H)(~O) |
| 346 | 0.823081 | C(~C)(~H)(~O) |
| 571 | 0.769737 | [#1]–C–O–[#1] |
| 617 | 0.758215 | C–C–C–O–[#1] |
| 567 | 0.723831 | O–C–C–O |
| 663 | 0.706029 | O–C–C–O–[#1] |
| 10 | 0.701884 | ≥4 C |
| 341 | 0.691642 | C(~C)(~C)(~O) |
| 639 | 0.664756 | O–C–C–C–O |
| 339 | 0.643846 | C(~C)(~C)(~H)(~O) |
| 582 | 0.528684 | C–C–C–C–C |
| 20 | 0.527647 | ≥4 O |
| 637 | 0.499238 | O–C–C–C–C |
| 680 | 0.448881 | O–C–C–C–C–C |
| 405 | 0.443151 | O(~C)(~C) |
| 11 | 0.441931 | ≥8 C |
| 614 | 0.432726 | C–C–O–C–C |
| 178 | 0.43236 | ≥1 any ring size 6 |
| 662 | 0.41846 | O–C–C–O–C |
| 14 | 0.401024 | ≥1 N |
| 285 | 0.398829 | C–N |
| 351 | 0.397488 | C(~C)(~N) |
| 420 | 0.367677 | C=O |
| 374 | 0.361336 | C(~H)(~H)(~H) |
| 443 | 0.361153 | C(–C)(=O) |
| 390 | 0.360056 | N(~C)(~C) |
| 181 | 0.35335 | ≥1 saturated or aromatic heteroatom-containing ring size 6 |
Comparison to Other Methods.
To compare FRAGSITE to other state-of-the art methods, FINDSITEcomb2.0,31 docking-based methods,11,16 and the state-of-the-art deep learning-based methods AtomNet41 and CNN scoring,41 we employed the DUD-E dataset5 using the modified LOOCV30% test with a sequence identify cutoff of 30%. For comparison to AtomNet41 and CNN scoring,41 the sequence identity cutoff for excluding targets in training is 80%. Table 2 summarizes the virtual screening performance of FRAGSITE in comparison to FINDSITEcomb2.0,31 AutoDock Vina,11 AtomNet,41 and CNN scoring41 methods. AutoDock Vina results are obtained locally using its default setting for both experimental and modeled target structures. AtomNet41 and CNN scoring41 results are taken from their respective publications. Since FRAGSITE and FINDSITEcomb2.0 perform similarly with experimental and modeled target structures, we present in Table 2 their results with modeled structures, except for the comparison to AtomNet41 and CNN scoring41 where we provide results of FRAGSITE and FINDSITEcomb2.0 on both modeled and experimental structures. To explore the sensitivity to fingerprint definition, we also implemented two alternative methods using slightly different molecular fingerprints FRAGSITE_MACCS and FRAGSITE_FP2 using the SMARTS pattern-based 256 bit MACCS and a path-based 1024 bit fingerprint FP2, which indexes small-molecule fragments based on linear segments of up to 7 atoms (somewhat similar to the Daylight fingerprints55) generated by Open Babel.60 We concluded that the currently used fingerprint definition is the best. In addition, we examined the contribution from different components of the feature vector from three methods FRAGSITE_no-mTC, FRAGSITE_no-DOT, and FRAGSITE_no-HADA excluding the mTC score, dot product, and Hadamard production in the feature vector, respectively.
Table 2.
Performance of Various Virtual Screening Methods on the DUD-E Set
| Comparison to FINDSITEcomb2.0 and AutoDock Vinaa | ||||
|---|---|---|---|---|
| method | EF0.01 | AUPR | top 100 precisionb | top 100 recall |
| FRAGSITE | 30.20 | 0.397 | 0.475 0.557 | 0.305 |
| FRAGSITE_MACCSc | 28.28 | 0.367 | 0.459 | 0.282 |
| FRAGSITE_FP2d | 29.79 | 0.387 | 0.476 | 0.297 |
| FRAGSITE_no-mTC | 22.12 | 0.283 | 0.355 | 0.227 |
| FRAGSITE_no-DOT | 27.23 | 0.358 | 0.438 | 0.284 |
| FRAGSITE_no-HADA | 23.42 | 0.283 | 0.386 | 0.240 |
| FINDSITEcomb2.0 | 25.22 | 0.321 | 0.416 0.557 | 0.257 |
| AutoDock Vina:11 experimental target structure | 9.13 | 0.093 | 0.151 | 0.093 |
| AutoDock Vina: modeled target structure | 3.57 | 0.045 | 0.063 | 0.045 |
| Comparison to AtomNet and CNN Scoringe | ||
|---|---|---|
| method (no. of targets) | AUC | no. of targets having an AUC > 0.9 (%) |
| FRAGSITE (102) | 0.910 | 73 (71.6%) |
| FRAGSITE (102) (experimental target structure) | 0.924 | 77 (75.5%) |
| FINDSITEcomb2.0 (102) | 0.874 | 61 (59.8%) |
| FINDSITEcomb2.0 (102) (experimental target structure) | 0.892 | 65 (63.7%) |
| CNN scoring (102) | 0.868 | 49 (48.0%) |
| FRAGSITE (randomly selected 30)f | 0.915 | 20 (66.7%) |
| FRAGSITE (randomly selected 30, experimental target structure) | 0.916 | 21 (70.0%) |
| FINDSITEcomb2.0 (randomly selected 30)f | 0.881 | 16 (53.3%) |
| FINDSITEcomb2.0 (randomly selected 30, experimental target structure) | 0.888 | 18 (60.0%) |
| AtomNet (30) | 0.855 | 14 (46.7%) |
Since FRAGSITE and FINDSITEcomb2.0 perform similarly on experimental and modeled target structures, we present only results with modeled target structures. We have generated AutoDock Vina results locally using its default settings.
The second number is the precision of consensus prediction of FRAGSITE and FINDSITEcomb2.0.
FRAGSITE using the 256 bit MACCS fingerprint generated by Open Babel.60
FRAGSITE using the 1024 bit FP2 fingerprint generated by Open Babel.60
A sequence identity cutoff of 80% is used by both FINDSITEcomb2.0 and FRAGSITE for target structure modeling and template ligand selection and training in boosted tree regression.
Since AtomNet was only tested on 30 DUD-E targets and their identities are not known, we randomly selected 30 targets for comparison to AtomNet.
Clearly, FRAGSITE and FINDSITEcomb2.0 perform significantly better than AutoDock Vina using both experimental and modeled structures. When modeled target structures are used, AutoDock Vina’s performance is much worse than when experimental target structures are used. Within the top 100 ranked molecules, FRAGSITE has a precision and recall increase of 14.3 and 18.5% relative to FINDSITEcomb2.0 (precision from 41.6 to 47.5%, recall from 25.7 to 30.5%, respectively). When considering the consensus set of the top 100 predictions from both methods, the precision increases further to 55.7%. Moreover, the mean top 1% enrichment factor increased from 25.22 to 30.20 (an average increase of 19.8%). Notably, the relative increase of AUPR from 0.321 to 0.397 is 23.7%. Both FRAGSITE_MACCS and FRAGSITE_FP2 are only slightly worse than FRAGSITE, which uses PubChem substructures. We note that FRAGSITE_no-mTC performs somewhat worse than FINDSITEcomb2.0. This means that the global similarity characterized by mTC score is a very important contribution to FRAGSITE’s performance. The observation that the elimination of the Hadamard product FRAGSITE_no-HADA is slightly better than FRAGSITE_no-mTC suggests that the Hadamard product is the next most important contributor to FRAGSITE following mTC. The fact that FRAGSITE_no-DOT is better than FRAGSITE_no-HADA indicates that the dot product is the least important contributor to FRAGSITE. By including fragment similarity as assessed by machine learning, FRAGSITE is better than FINDSITEcomb2.0. Thus, FRAGSITE demonstrates a significant and robust improvement over FINDSITEcomb2.0.31
Table 2 also shows that both FRAGSITE and FINDSITEcomb2.0 are both better than the state-of-the-art deep learning-based CNN scoring and AtomNet,41,42 with FRAGSITE being the best. For all 102 targets, using modeled structures, FRAGSITE has a mean AUC of 0.910, with the number of targets having an AUC > 0.9 of 73, respectively, compared to 0.868 and 49, respectively, by CNN scoring using experimental target structures.42 When experimental target structures are used by FRAGSITE, its performance slightly increases from a mean AUC of 0.910 to 0.924, with the number of targets having an AUC > 0.9 increasing from 73 to 77. For a randomly selected set of 30 targets, with modeled target structures, FRAGSITE has a mean AUC and number of targets having an AUC > 0.9 of 0.915 and 20, compared to 0.855 and 14 by AtomNet using experimental target structures.41 Again, FRAGSITE has slightly improved performance for these 30 targets when experimental as opposed to modeled target structures are used.
To examine the improvement on individual targets, Figure 2 shows the scatter plot comparison of EF0.01 for FRAGSITE and FINDSITEcomb2.0. Table S2 presents the performance (EF0.01 and AUPR) of FRAGSITE for individual targets in comparison to FINDSITEcomb2.031 and AutoDock Vina using modeled target structures. Overall, for EF0.01, FRAGSITE performs better for 72 targets, worse for 26 targets, and is tied for 4 targets. FRAGSITE has 91 targets having an EF0.01 > 1 (meaning better than random performance), whereas FINDSITEcomb2.0 and AutoDock Vina have an EF0.01 > 1 for 86 and 59 targets, respectively.
Figure 2.
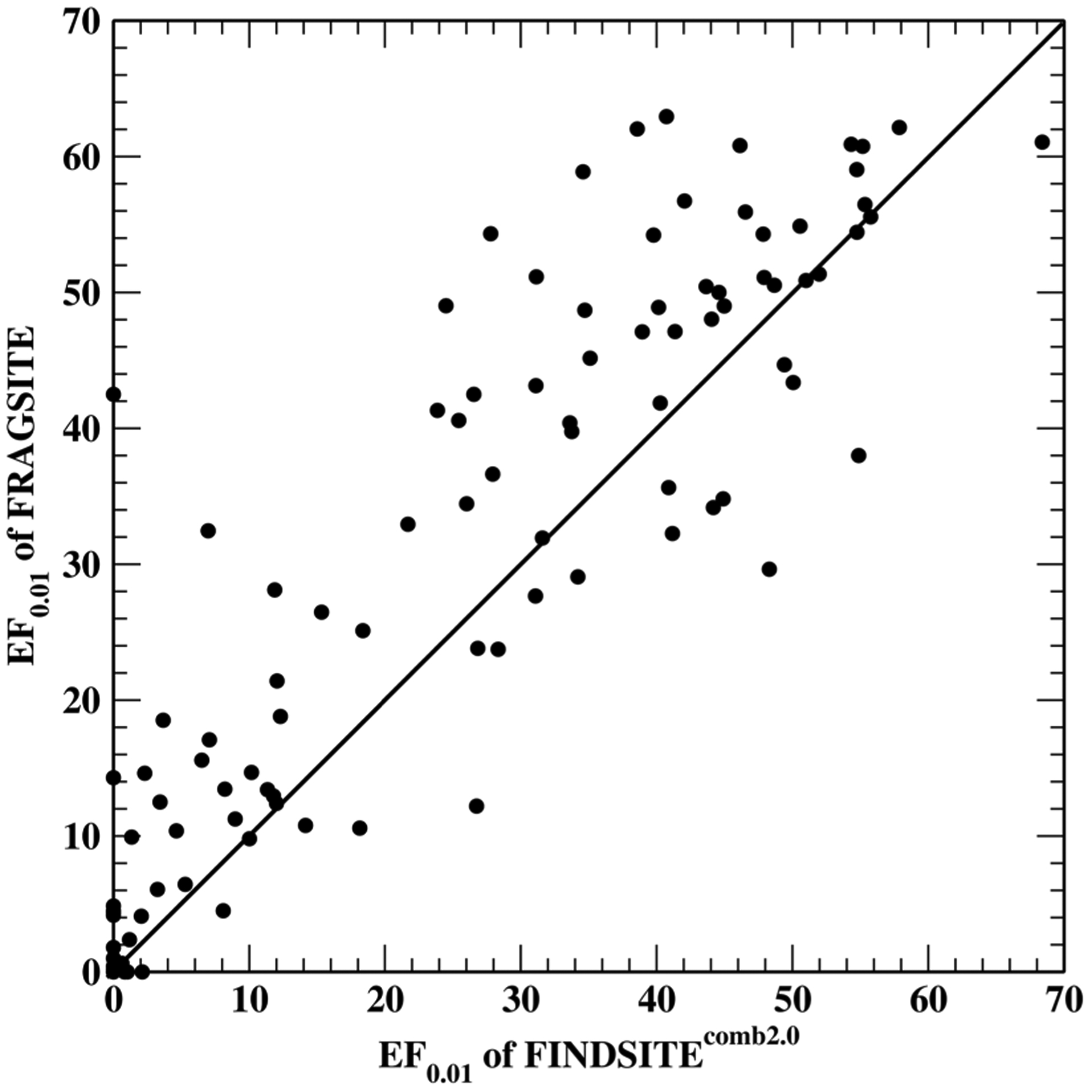
Scatter plot comparison of EF0.01 for FRAGSITE and FINDSITEcomb2.0 for the DUD-E set.
Comparing the performance of the three methods on individual targets shows that FRAGSITE has the best performance for 69 (78) targets as assessed by EF0.01 (AUPR), while FINDSITEcomb2.0 and AutoDock Vina are the best performing on 25 (27) and 7 (8) targets, respectively. Thus, FRAGSITE’s improvement is not just for few targets that show a large improvement; rather, it shows improvement for the majority of targets. Of note is target cxcr4, a GPCR receptor, where FRAGSITE has an EF0.01 of 42.5 compared to 0.0 by FINDSITEcomb2.0 and AutoDock Vina. Another example is pygm. FRAGSITE has an EF0.01 of 14.29, whereas FINDSITEcomb2.0 and AutoDock Vina have an EF0.01 of 0.0 and 10.43, respectively. FINDSITEcomb2.0 failed for this target, but AutoDock Vina seems to have good performance. These results further demonstrate the superior performance of FRAGSITE over FINDSITEcomb2.0 and AutoDock Vina; this might be due to its capture of the physical interaction pattern of the ligand fragments with the receptor protein.
To trace back the sources of improvement and suggest directions for future improvement, we now turn to the analysis of the improvement of the individual FRAGSITE components over FINDSITEcomb2.0 (see Methods and Figure 1), i.e., the improvement using template ligands from the PDB,52 DrugBank,49 and ChEMBL50 (see Figure 1). Table 3 summarizes the results for each of the components. The relative increases of EF0.01 and AUPR by FRAGSITE over FINDSITEcomb2.0 for the PDB component are 42.8 and 51.7%, for the DrugBank component 52.3 and 60.2%, and 18.2 and 21.2% for the ChEMBL component, respectively. This indicates that all components of FRAGSITE show significant improvement. For 62 targets, the FINDSITEcomb2.0 result is contributed mostly by the ChEMBL component; ~20% of the relative increase of the final FRAGSITE improvement is dictated by the ChEMBL component. Thus, future method improvements in FRAGSITE should focus on improving the performance of the ChEMBL component.
Table 3.
Performance of Individual FRAGSITE Components
| FRAGSITE | FINDSITEcomb2.0 | |||
|---|---|---|---|---|
| componenta | EF0.01 | AUPR | EF0.01 | AUPR |
| PDB(102) | 22.64 | 0.300 | 15.85 | 0.198 |
| DrugBank(77) | 23.05 | 0.299 | 15.13 | 0.187 |
| ChEMBL(62) | 36.42 | 0.474 | 30.82 | 0.391 |
Numbers in parentheses are the numbers of targets assessed. For the DrugBank and ChEMBL components, some targets have no template ligands.
Predicted Precision and Recall.
Next, we plot the dependence of predicted precision/recall on the machine learning score of FRAGSITE Sfrg in Figure 3. Sfrg is expected to be between 0 and 1, with 1 being the best value. However, since machine learning has errors, the actual values of Sfrg sometimes can be <0 or >1. For example, among the 1,419,743 predictions for the DUD-E set, the largest value can be 1.3 with 1384 (0.097%) predictions having Sfrg > 1, and the lowest Sfrg value is −0.08. This figure clearly shows that when the score is below 0.5, the precision is below 10%. A 20% precision requires a cutoff score of 0.65, where the recall is ~32%. The maximal precision is 85.8% with score Sfrg ≤ 1. For scores Sfrg > 1, we simply assign a precision of 90.0%. Thus, we have a confidence index that can tell when it is worthwhile to perform an experiment based on the FRAGSITE ranking score.
Figure 3.
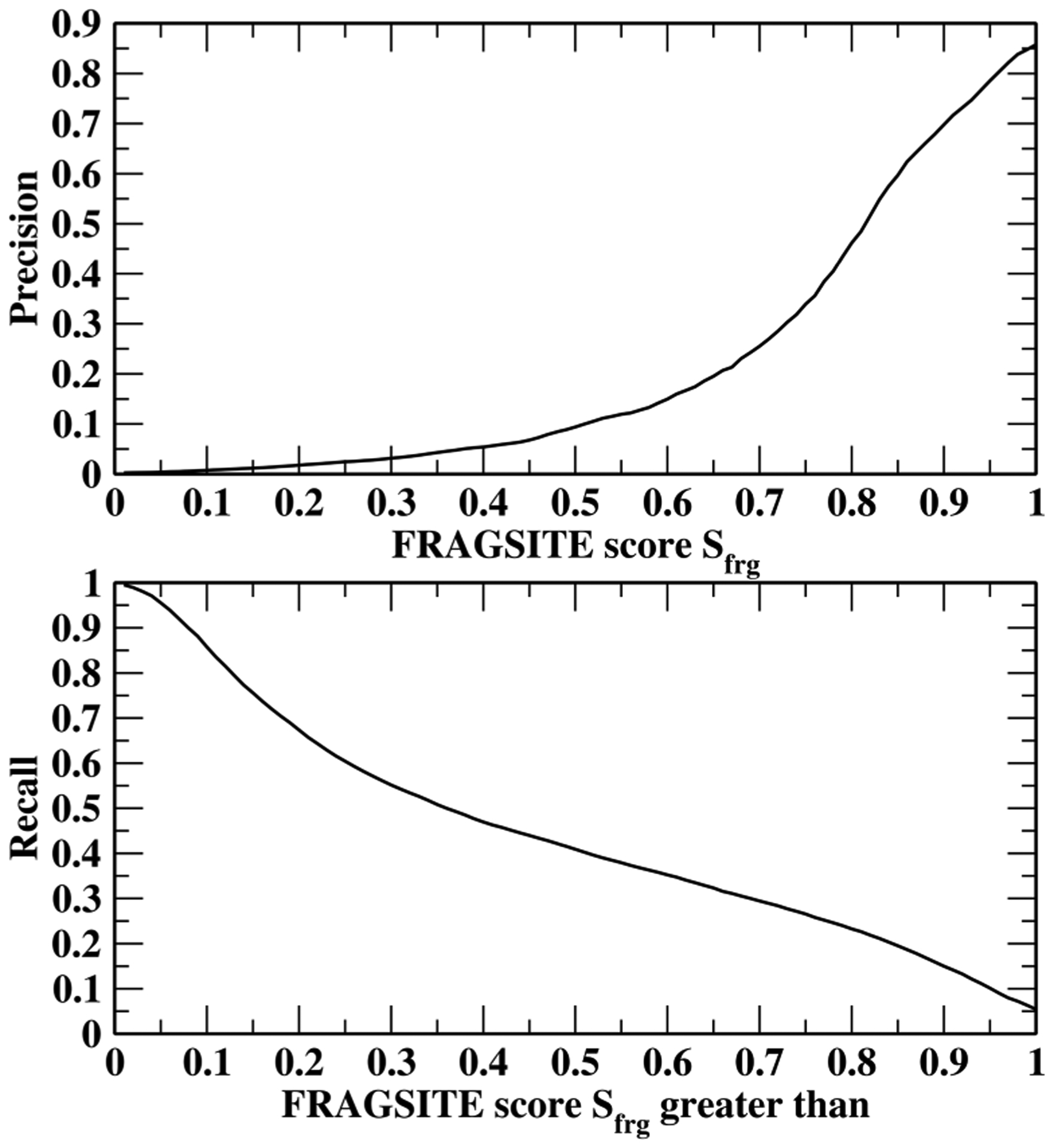
Dependence of the predicted precision (up) and recall (down) on FRAGSITE machine learning score Sfrg.
Comparison to Other Methods.
A number of studies have pointed out that hidden biases in the DUD-E set could favor similarity-based methods over docking-based methods.43,61 In practice, FINDSITEcomb2.0 has a similar step to ligand similarity-based approaches, with the fundamental difference being that FINDSITEcomb2.0 does not use any information from the known ligands that bind to the target. However, FRAGSITE, due to its machine learning component, could learn the hidden biased patterns from its training set that are similar to the testing target. Therefore, it is important to benchmark completely different test sets from the training set.
Here, we conduct tests using a recently developed unbiased benchmarking set LIT-PCBA61 to compare FRAGSITE and FINDSITEcomb2.0 to ligand similarity and docking methods. LIT-PCBA is designed to solve the problems faced by classically used benchmarking sets, e.g., DUD, DUD-E, and MUV that are biased by obvious and hidden chemical biases.44 These biases could favor machine learning and/or similarity-based approaches over docking methods. LIT-PCBA has 15 targets. Each has multiple PDB structures (for a total of 129 structures). FRAGSITE and FINDSITEcomb2.0 use a sequence identity cutoff of 30% for target structure modeling and template ligand selection. Moreover, FRAGSITE employed a 30% identity cutoff for training the machine leaning model on the DUD-E set. The results are compiled in Table 4. Since multiple sequences/structures for each target are used, only the mean values are reported for each target. The average EF0.01 of FRAGSITE for the 15 targets is 4.78, which is 57% better than the EF0.01 = 3.04 for FINDSITEcomb2.0. Both methods are better than two-dimensional (2D) ECFP4 similarity search, 3D shape similarity search, or the molecular docking program Surflex-Dock v.3066 with average EF0.01s of 2.49, 0.96, and 1.70, respectively.61 We note that FRAGSITE has only two targets worse than random (EF0.01 < 1), whereas each of FINDSITEcomb2.0 and 2D ECFP4 similarity search has six, 3D shape similarity search has ten, and Surflex-Dock v.3066 has seven targets with EF0.01 < 1.
Table 4.
Performance (EF0.01) of FRAGSITE for LIT-PCBA Using the Modeled Target Structure
| target | 2D ECFP4 similarity searcha | 3D shape similarity searcha | molecular docking (Surflex-Dock v.3066)a | FINDSITEcomb2.0 | FRAGSITE |
|---|---|---|---|---|---|
| ADRB2 | 0 | 0 | 0 | 11.76 | 9.56 |
| ALDH1 | 1.58 | 1.08 | 1.25 | 0.68 | 0.75 |
| ESR1_ago | 0 | 0 | 0 | 7.69 | 0.51 |
| ESR1_ant | 2.67 | 1.07 | 1.6 | 0 | 2.61 |
| FEN1 | 1.09 | 0 | 3.26 | 1.36 | 17.07 |
| GBA | 1.63 | 0.81 | 4.47 | 1.81 | 2.71 |
| IDH1 | 1.59 | 0.79 | 0.79 | 0 | 3.11 |
| KAT2A | 0.69 | 0.69 | 4.17 | 0.52 | 2.06 |
| MAPK1 | 0.95 | 1.39 | 1.99 | 6.77 | 9.81 |
| MTORC1 | 0 | 0 | 1.52 | 2.62 | 3.00 |
| OPRK1 | 16.67 | 0 | 0 | 8.33 | 4.17 |
| PKM2 | 1.31 | 2.13 | 0.9 | 0.77 | 1.69 |
| PPARG | 5.56 | 5.56 | 5.56 | 1.23 | 5.93 |
| TP53 | 0 | 0.88 | 0 | 0 | 6.96 |
| VDR | 3.64 | 0 | 0 | 1.98 | 1.70 |
| average | 2.49 | 0.96 | 1.70 | 3.04 | 4.78 |
We then conducted another test on a 23 target subset of the DEKOIS 2.0 set45 to compare FRAGSITE and FINDSITEcomb2.0 to the recently developed vScreenML scoring function that has similar performance to RF-score-VS.44 In fact, vScreenML uses the same RF-score as a component of its features and the same boosted tree regression method as FRAGSITE. vScreenML uses the majority of the DEKOIS 2.0 set45 for training and a 23 target subset for testing. Thus, this comparison will test if FRAGSITE has better features than vScreenML. Since vScreenML performs similarly to RF-score-VS, this comparison will also serve as an indirect comparison of RF-score-VS to FRAGSITE and FINDSITEcomb2.0. Once again, we used a sequence identity cutoff of 30% for target structure modeling and template ligand selection. To have a fair comparison to vScreenML whose training set had up to 77% sequence identity to the testing set, FRAGSITE employed an 80% identity cutoff for the training machine learning model on the DUD-E set. The results of EF0.01 are shown in Table 5 where the vScreenML results were taken from ref 44. Table 5 shows that FRAGSITE with an average of EF0.01 of 15.2 and FINDSITEcomb2.0 with an average EF0.01 of 13.9 are both significantly better than vScreenML whose average EF0.01 is 6.7. FRAGSITE shows a 9% improvement over FINDSITEcomb2.0. FRAGSITE has five targets worse than random (EF0.01 < 1), whereas FINDSITEcomb2.0 and vScreenML have six targets with an EF0.01 < 1. Thus, FRAGSITE performs better due to its better features and possibly better training set.
Table 5.
Performance (EF0.01) of FRAGSITE for the 23 Target DEKOIS 2.0 Subset Using the Modeled Target Structure
| target | vScreenMLa | FINDSITEcomb2.0 | FRAGSITE |
|---|---|---|---|
| 3hng | 10.3 | 25.8 | 20 |
| 1hov | 2.5 | 31 | 30 |
| 3ny9 | 0 | 0 | 22.5 |
| 3kk6 | 10.8 | 5.2 | 20 |
| 1nhz | 5.4 | 28.4 | 27.5 |
| 1xp0 | 8.1 | 31 | 22.5 |
| 1z11 | 0 | 10.3 | 7.5 |
| 3tfq | 8.6 | 0 | 0 |
| 2oo8 | 5.1 | 28.4 | 25 |
| 1b8o | 7.5 | 28.4 | 27.5 |
| 2w3l | 5.5 | 15.5 | 10 |
| 1hw8 | 24.6 | 5.2 | 30 |
| 2afx | 0 | 0 | 0 |
| 3ewj | 2.7 | 31 | 30 |
| 3v8s | 18 | 15.5 | 22.5 |
| 3eml | 7.7 | 0 | 10 |
| 2z94 | 0 | 0 | 0 |
| 1uze | 21.4 | 10.3 | 5 |
| 3klm | 5.4 | 5.2 | 5 |
| 2wcg | 2.6 | 15.5 | 12.5 |
| 1w4r | 0 | 20.7 | 0 |
| 1r4l | 8.1 | 12.9 | 22.5 |
| 1uou | 0 | 0 | 0 |
| average | 6.7 | 13.9 | 15.2 |
Experimental Validation.
Finally, we present the results for experimental testing on two proteins: the enzyme Escherichia coli dihydrofolate reductase (E. coli DHFR) and the human ACVR1 receptor kinase screened against the NCI molecules from the National Cancer Institute (NCI) diversity set (https://dtp.cancer.gov/organization/dscb/obtaining/available_plates.htm). A sequence identity cutoff of 30% for protein templates was applied. For prediction, FRAGSITE was trained on the whole DUD-E set excluding targets having a sequence identity of >30% to E. coli DHFR or ACVR1. To be fair to both methods, we tested molecules predicted by both FRAGSITE and FINDSITEcomb2.0 methods with an estimated precision of >0.5 using the thermal shift method (see the Supporting Information).
Table 6 summarizes the results. Overall, with a precision cutoff of 0.5, for E. coli DHFR, FRAGSITE predicted 21 ligands, whereas FINDSITEcomb2.0 predicted 7 ligands. For human ACVR1, FRAGSITE predicted 50 ligands, whereas FINDSITEcomb2.0 predicted 12 ligands. The experimental results show that 7 more hits for DHFR are found by FRAGSITE than FINDSITEcomb2.0; FINDSITEcomb2.0 only has 2 within the top ranked 20 ligands (~1% of 1812 molecules). For ACVR1, FRAGSITE has 14 more hits, including the strongest, new binder NSC105827 with a 9.7 °C thermal shift; only 2 ligands are within the top 20 of FINDSITEcomb2.0. On average, the mean ranks of all binders by FRAGSITE for DHFR and ACVR1 are 10.4 and 29.5, whereas those by FINDSITEcomb2.0 are much lower at 20.0 and 61.2, respectively.
Table 6.
List of Experimentally Validated Top 20 True Binder Hits of E. coli DHFR and Human ACVR1 Ranked by Thermal Shift ΔTm Values Categorized as Either New Binders/Inhibitors or Known Binders/Inhibitors of the Corresponding Enzyme Family with an Expected Precision Cutoff of 0.5 as Predicted by FRAGSITE and Compared with FINDSITEcomb2.0
| Drug NSC#a | Maximal Tc to known inhibitorsb | Thermal shift ΔTm values (°C) (rank)c | FRAGSITE predicted precision (rank)d | FINDSITEcomb2.0 predicted precision (rank)d | Chemical structure of binders |
|---|---|---|---|---|---|
| New true binders of E. coli DHFRe | |||||
| 715055* | 0.59 | 8.0 (1) | 0.86(4) | 0.01(49) | 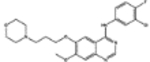 |
| 760766* | 0.59 | 6.7 (2) | 0.82(8) | 0.01(56) | 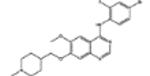 |
| 369100 | 0.77 | 4.0 (3) | 0.52(21) | 0.52(7) |  |
| 106570 | 0.68 | 1.7 (4) | 0.73(10) | 0.16(10) |  |
| 280594 | 0.56 | 1.7 (5) | 0.62(15) | 0.01(51) |  |
| 130801 | 0.60 | 1.7 (6) | 0.60(17) | 0.02(26) |  |
| 274905 | 0.68 | 1.3 (7) | 0.79(9) | 0.11(11) |  |
| 34488 | 0.70 | 1.3 (8) | 0.60(16) | 0.02(33) |  |
| Known inhibitors of the DHFR familye | |||||
| 309401 | 21.0 (1) | 0.90(3) | 0.88(3) | 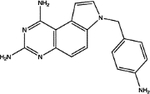 |
|
| 740 | 19.0 (2) | 0.85(5) | 0.81(1) | 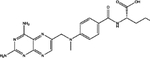 |
|
| 339578 | 18.3 (3) | 0.84(7) | 0.84(2) | 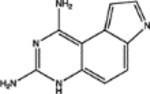 |
|
| 754230 | 18.0 (4) | 0.90(1) | 0.84(6) | 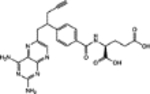 |
|
| 698037 | 14.0 (5) | 0.57(19) | 0.89(5) | 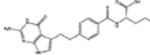 |
|
| New true binders of human ACVR1e | |||||
| 105827 | 0.54 | 9.7 (1) | 0.66(36) | 0.03(117) | 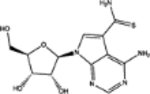 |
| 63701 | 0.56 | 6.0 (2) | 0.70(27) | 0.05(75) | 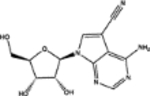 |
| 274905 | 0.65 | 4.7 (3) | 0.70(29) | 0.01(212) | 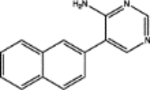 |
| 19125 | 0.64 | 4.0 (4) | 0.68(30) | 0.28(21) | 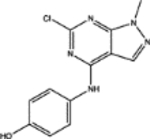 |
| 62609 | 0.58 | 4.0 (5) | 0.57(43) | 0.07(58) | 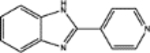 |
| 71795 | 0.63 | 4.0 (6) | 0.52(48) | 0.02(159) | 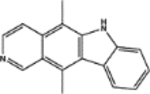 |
| 155703 | 0.59 | 4.0 (7) | 0.52(49) | 0.09(48) | 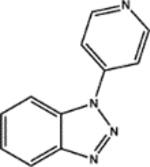 |
| 128737 | 0.57 | 3.0 (8) | 0.66(35) | 0.04(79) | 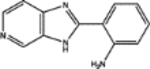 |
| 317605 | 0.61 | 3.0 (9) | 0.52(47) | 0.02(178) | 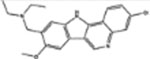 |
| 11668 | 0.62 | 2.0 (10) | 0.60(39) | 0.17(29) | 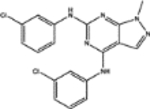 |
| 34488 | 0.58 | 2.0(11) | 0.57(42) | 0.04(85) | 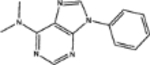 |
| 280594 | 0.56 | 2.0 (12) | 0.55(44) | 0.14(34) | 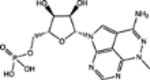 |
| Known inhibitors of ACVR1 and kinasese | |||||
| 760766 | 8.0 (1) | 0.90(1) | 0.62(11) | 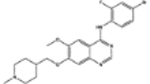 |
|
| 749005 | 8.0 (2) | 0.90(10) | 0.86(7) | ||
| 750690 | 5.0 (3) | 0.64(37) | 0.85(9) | 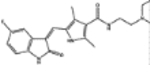 |
|
| 732517 | 4.0 (4) | 0.90(9) | 0.86(3) | 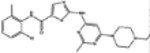 |
|
| 737754 | 3.0 (5) | 0.90(6) | 0.88(5) | ||
| 715055 | 3.0 (6) | 0.90(3) | 0.50(13) |  |
|
Bold NCI# indicates molecules with predicted precision ≥ 0.5 by FRAGSITE but not FINDSITEcomb2.0.
Used FP2 fingerprints generated by Open Babel.60
The ΔTm values of true binders (ΔTm > 1 °C at 500 μM concentration) indicated by bold numbers.
Ranks are among the 1812 NCI molecules by mTC score for FINDSITEcomb2.0 and by machine learning score Sfrg for FRAGSITE. Note that the rank is determined by mTC score in FINDSITEcomb2.0 and precision is fitted into an mTC window of ±0.05. Thus, sometimes, higher mTC might have slightly lower precision. This problem does not happen in FRAGSITE whose Sfrg score has a range of 0–1.
Among all tested drug predictions for DHFR and ACVR1, a few did not show a quantifiable melting curve. They either displayed a continuous decrease in the fluorescence signal of the protein sample or an artifact of significant increase in the fluorescence signal of the drug alone control along the curves. They are excluded from further consideration for plotting or analysis for true hit rates. See Methods for details.
Two of the new true binders of E. coli DHFR, which are top ranked by thermal shift ΔTm of 8.0 and 6.7 °C, respectively, are confirmed by functional assays to inhibit E. coli DHFR steady-state kinetics (Figure 8). They have not been previously reported to bind or inhibit the DHFR family, and their scaffolds do not belong to known inhibitors of the DHFR family or antifolates in general. Their proposed inhibitory poses are described in Figure 6.
We found that the experimentally observed precision (the overall hit rate of true binders defined as compounds displaying a thermal shift over 1 °C at 500 μM final concentration) of the FRAGSITE predictions with an expected precision cutoff of ≥0.5 is generally consistent with the average expected precision based on the DUD-E benchmark set when both protein targets tested here are considered (see the summary in Table 6 and the melting curves in Figures 4 and 5). The true positive binder hit rate of the E. coli DHFR predictions was higher than that of the ACVR1 drug predictions. Consistent with benchmarking results (see Table 2), when comparing FRAGSITE with FINDSITEcomb2.0, the common set predictions for both proteins of the two methods have a much higher observed precision of 0.85 (85% true positive hit rate) than the unique set predictions of each method. These results suggest a strategy for identifying high-confidence predictions by combining the two methods. The observed precision of the unique set predictions is 47 and 0% for FRAGSITE and FINDSITEcomb2.0, respectively, when both E. coli DHFR and human ACVR1 are considered. Overall, FRAGSITE is more powerful than FINDSITEcomb2.0 in identifying true binders with diverse scaffolds. It also expands the chemical space of known binders (Figures 6 and 7). We next examined the drug predictions of the common set vs unique sets of each protein.
Figure 4.
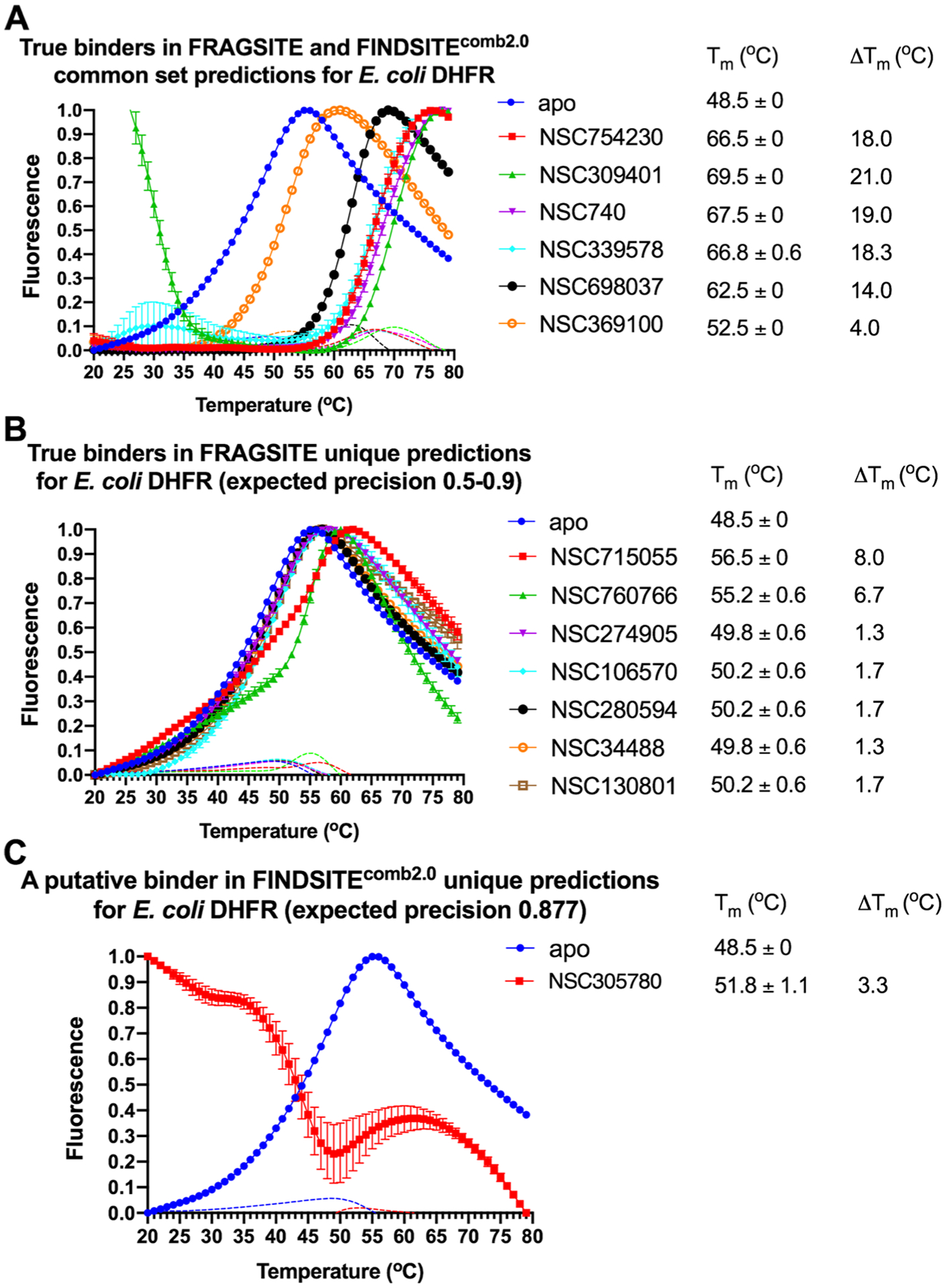
(A–C) Thermal shift assay melting curves of E. coli DHFR with the top ranked drug binders predicted by FRAGSITE and FINDSITEcomb2.0 with an expected precision above 0.5. The slope of each curve is also plotted as dotted lines with the corresponding color coding. The final drug concentration is 500 μM. The reaction buffer contains 50 mM HEPES pH 7.3 and 100 mM NaCl. Each reaction condition has three replicates. ΔTm = Tm (protein with drug) − Tm (apo protein). See details in Methods.
Figure 5.
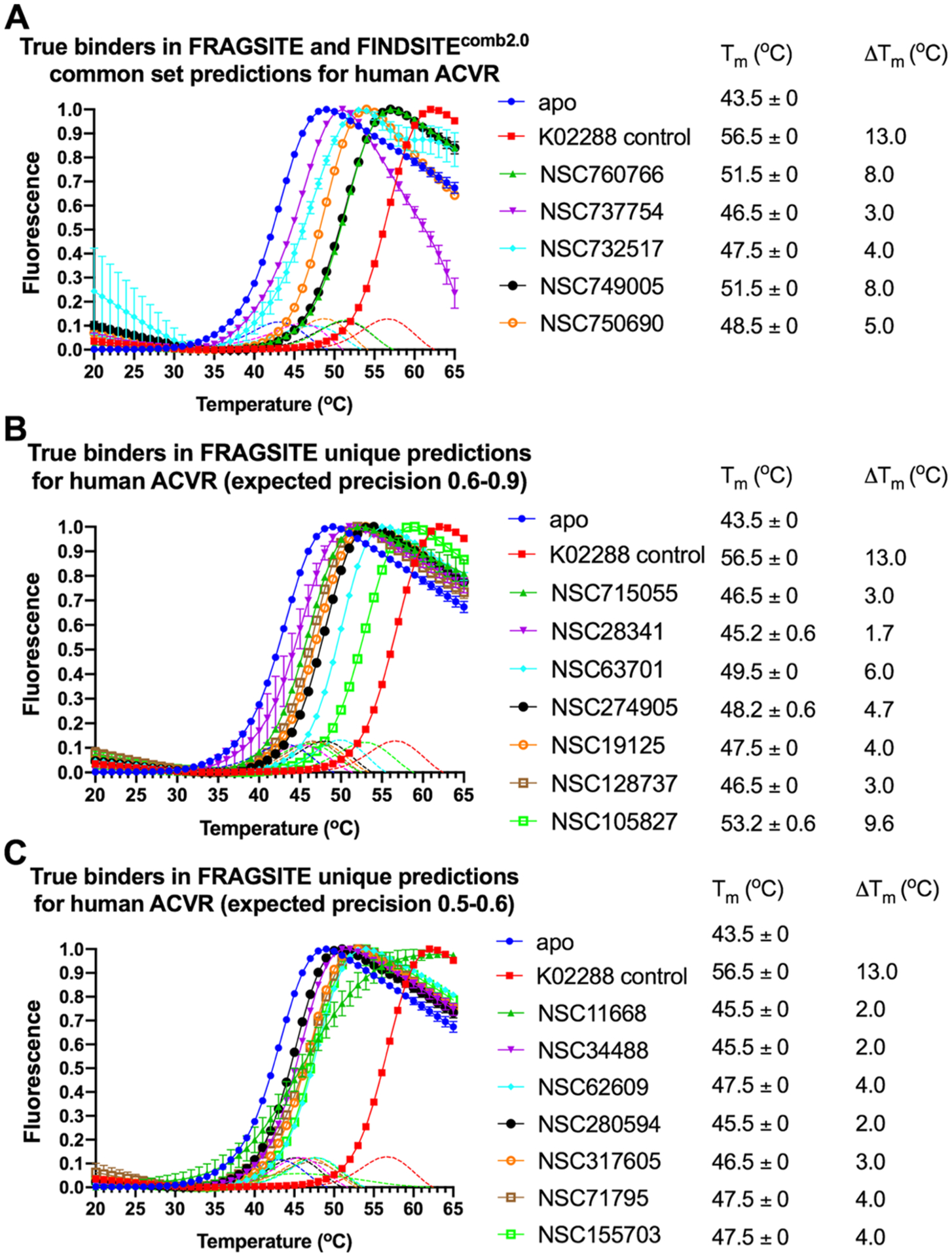
(A–C) Thermal shift assay melting curves of the human ACVR1 receptor kinase cytosolic domain with top ranked drug binders predicted by FRAGSITE and FINDSITEcomb2.0 with expected precision above 0.5. The slope of each curve is also plotted as dotted lines with the corresponding color coding. The reaction buffer contains 50 mM HEPES pH 7.3 and 100 mM NaCl. Each reaction condition has three replicates. ΔTm = Tm (protein with drug) − Tm (apo protein). The final drug concentration is 500 μM except for K02288 (5 μM), which is a known nanomolar inhibitor of ACVR1 and tested in parallel as a positive control.70 Only melting curves displaying a quantifiable increase in the fluorescence signal of protein above that of the drug alone are shown here and considered for further analysis. See details in Methods.
Figure 6.
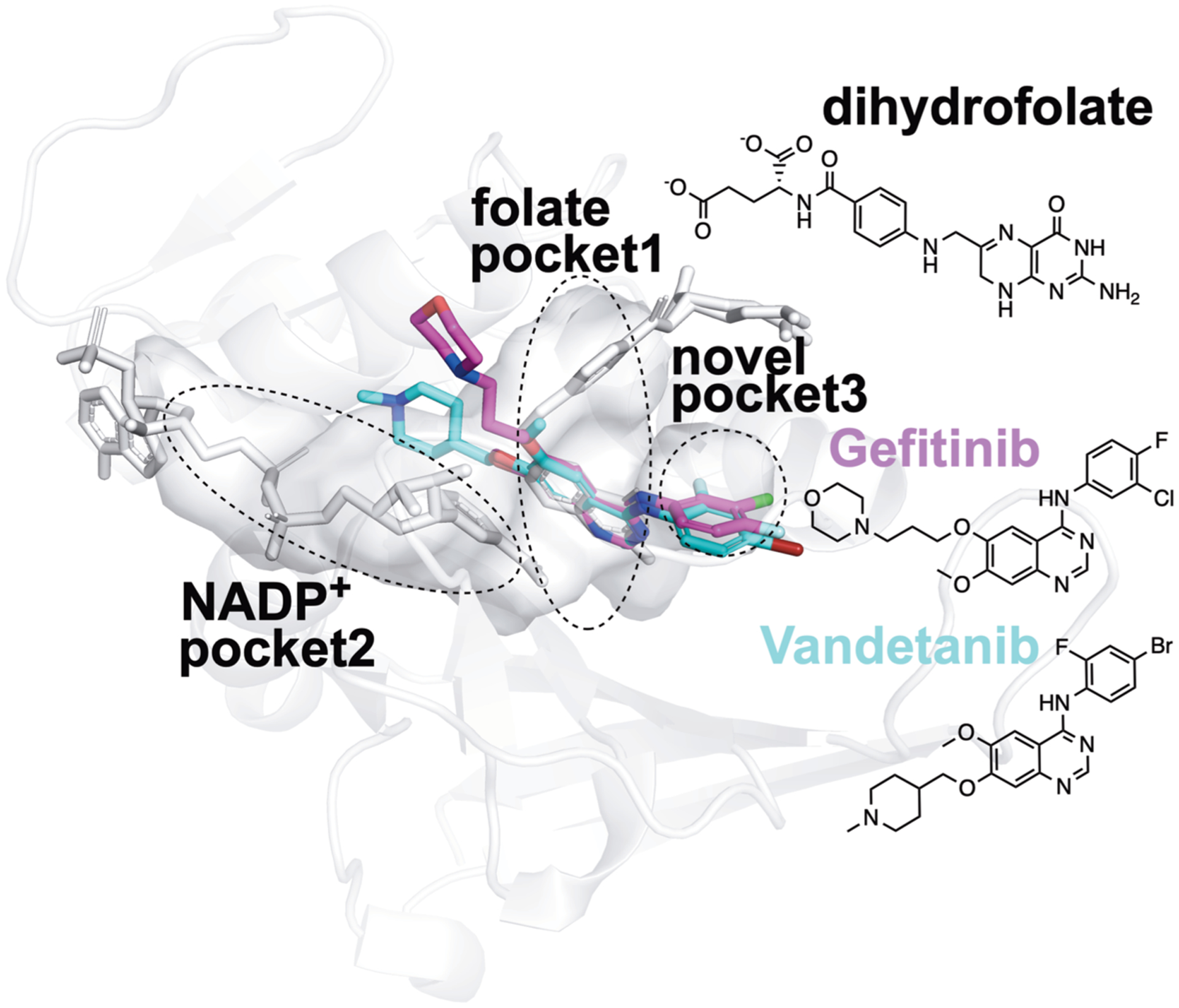
Proposed structural basis of the new true binders of E. coli DHFR identified by FRAGSITE. Using the PyMOL Molecular Graphics System Version 2.3.2 (Schrödinger, LLC), the PDB ligand model structures of NSC715055 (Gefitinib, PDB ligand code IRE, shown as purple sticks) and NSC760766 (Vandetanib, PDB ligand code ZD6, shown as cyan sticks) were manually and rigidly docked into ligand binding pockets in the E. coli DHFR:folate:NADP+ ternary complex crystal structure (PDB entry 4PSY,65 pockets shown as the van der Waals surface in white color) based on spatial alignment of the bicyclic core of the new true binders identified by FRAGSITE to the pterin moiety of the folate ligand in the crystal structure. Based on the structural alignment and the conceivably rotatable O and N-linkages extended from the bicyclic core of these new ligands, their proposed binding modes in E. coli DHFR may result in occupying not only the dihydrofolate pocket but also NADPH and a third new pocket. The protein secondary structures are shown as white ribbons, and the original crystallographically identified ligands (NADP+ and dihydrofolate) in PDB entry 4PSY65 are shown as white sticks. The chemical structures of Gefitinib, Vandetanib, and dihydrofolate are also shown for comparison.
Figure 7.
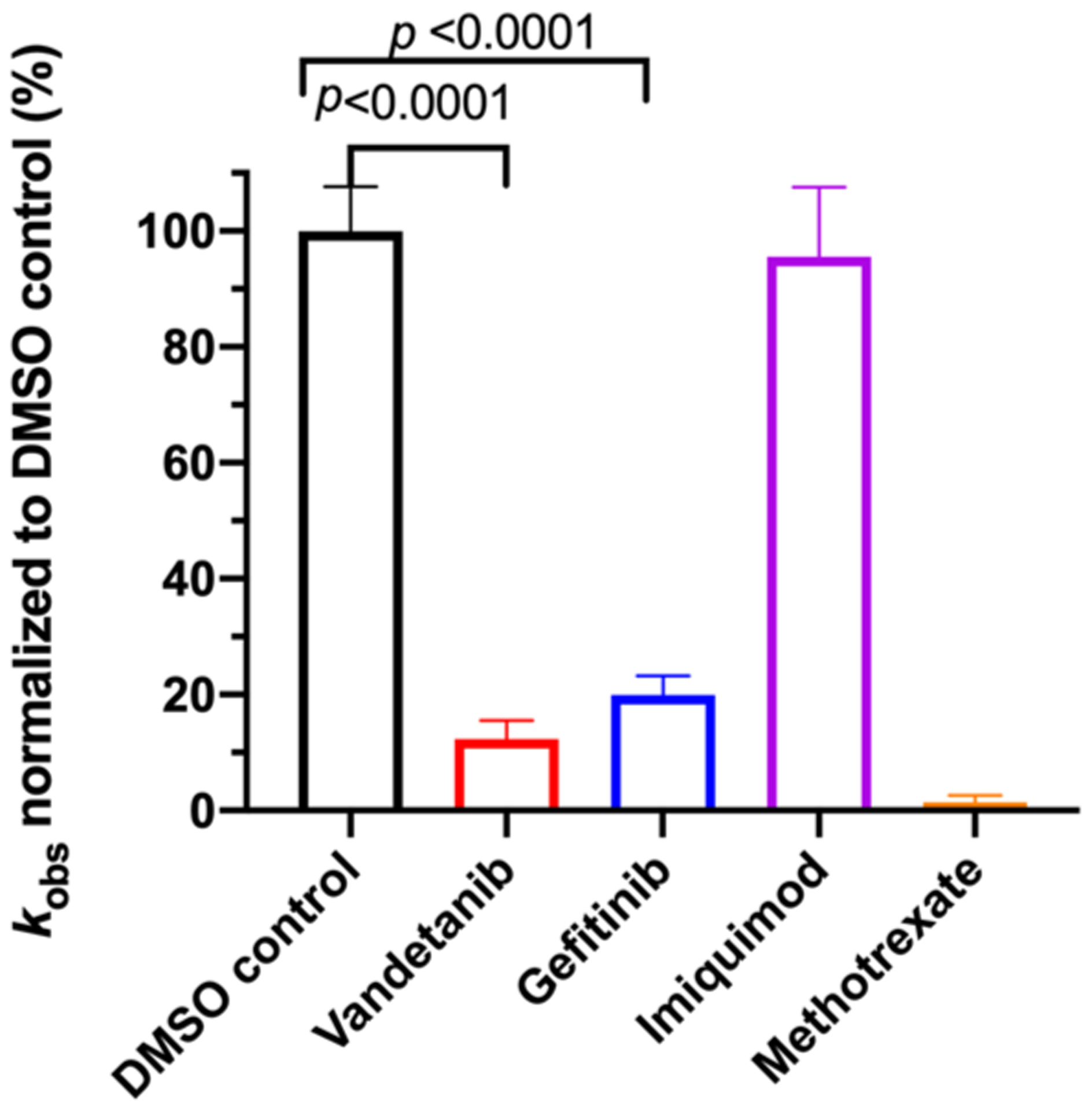
Steady-state kinetics inhibition assays of E. coli DHFR. The reaction at each condition was replicated 4 times and presented as the average value and standard deviation along with unpaired t-test p values. All drugs tested were at a final concentration of 100 μM plus a trace amount of DMSO (1%) residual from stock solution. The reactions were performed under a constant room temperature of 18 °C at steady-state conditions with a catalytic amount of E. coli DHFR (93 nM) and saturated electron acceptor dihydrofolate (50 μM) and electron donor NADPH (100 μM) levels. The oxidation of NADPH was followed at 340 nm for 60 s. The observed initial linear rate was calculated using the published delta molar extinction coefficient Δε of 11.8 mM−1 cm−1 71 and normalized against the rate of the DMSO control sample. DMSO at a final concentration of 1% was included as the negative control for inhibition, and Methotrexate at 100 μM was included as the positive control of inhibition.
For, E. coli DHFR drug binder predictions, in the common set, 5 (of 6) high-confidence predictions with very large thermal shifts of 14–21 °C are known nanomolar DHFR inhibitors (Table 6). For example, Pralatrexate (NCI ID: NSC754230, DrugBank ID: DB06813), Methotrexate (NSC740, DB00563), and Premetrexed (NSC698037, DB00642) are approved anticancer drugs of the antifolate family.49 AMPQD (NSC309401) and PQD (NSC339578) are experimental compounds also reported as DHFR inhibitors62 with the tricyclic core scaffold distinct from and the diaminopyrimidine moiety observed in methotrexate. In contrast, Imiquimod (NSC369100) lacks the diaminopyrimidine moiety and shows a smaller thermal shift of 4 °C; it is likely a weak binder. On the other hand, NSC715055 (Gefitinib, DB00317) and NSC760766 (Vandetanib, DB05294) identified by FRAGSITE but not FINDSITEcomb2.0 belong to the kinase inhibitor families of chemotherapy drugs (according to DrugBank49) that have not been previously reported to bind to DHFR. Both bind to E. coli DHFR with relatively large thermal shifts of 6.7 and 8.0 °C (Figure 4B).
The DHFR enzyme family is a well-studied model system for enzyme catalysis and dynamics that is the target of antimicrobial and chemotherapeutic drugs. DHFR catalyzes the stereospecific reduction of 7,8-dihydrofolate (FH2) to (6s)-5,6,7,8-tetrahydrofolate (FH4) via hydride transfer from NADPH.63,64,71 Structural inspection of Gefitinib, Vandetanib, and an E. coli DHFR Michaelis complex mimic (Protein Data Bank, PDB entry 4PSY65) suggests a new ligand scaffold that may not only occupy the electron acceptor dihydrofolate pocket but also the electron donor NADPH pocket and a third new unexplored pocket adjacent to the dihydrofolate pocket; see Figure 6. The estimated Kd values of Vandetanib and Gefitinib with E. coli DHFR are at a 2–10 μM level based on thermal shift analysis66 and comparison to reference thermal shifts of Methotrexate (NSC740, Kd of ~9.5 nM)67 (Figure 4). We attribute these new findings to FRAGSITE’s scaffold diversification.31 Thus, despite the fact that DHFR has been extensively studied, FRAGSITE discovered a new class of binders that may be clinically relevant and will be pursued in subsequent work.
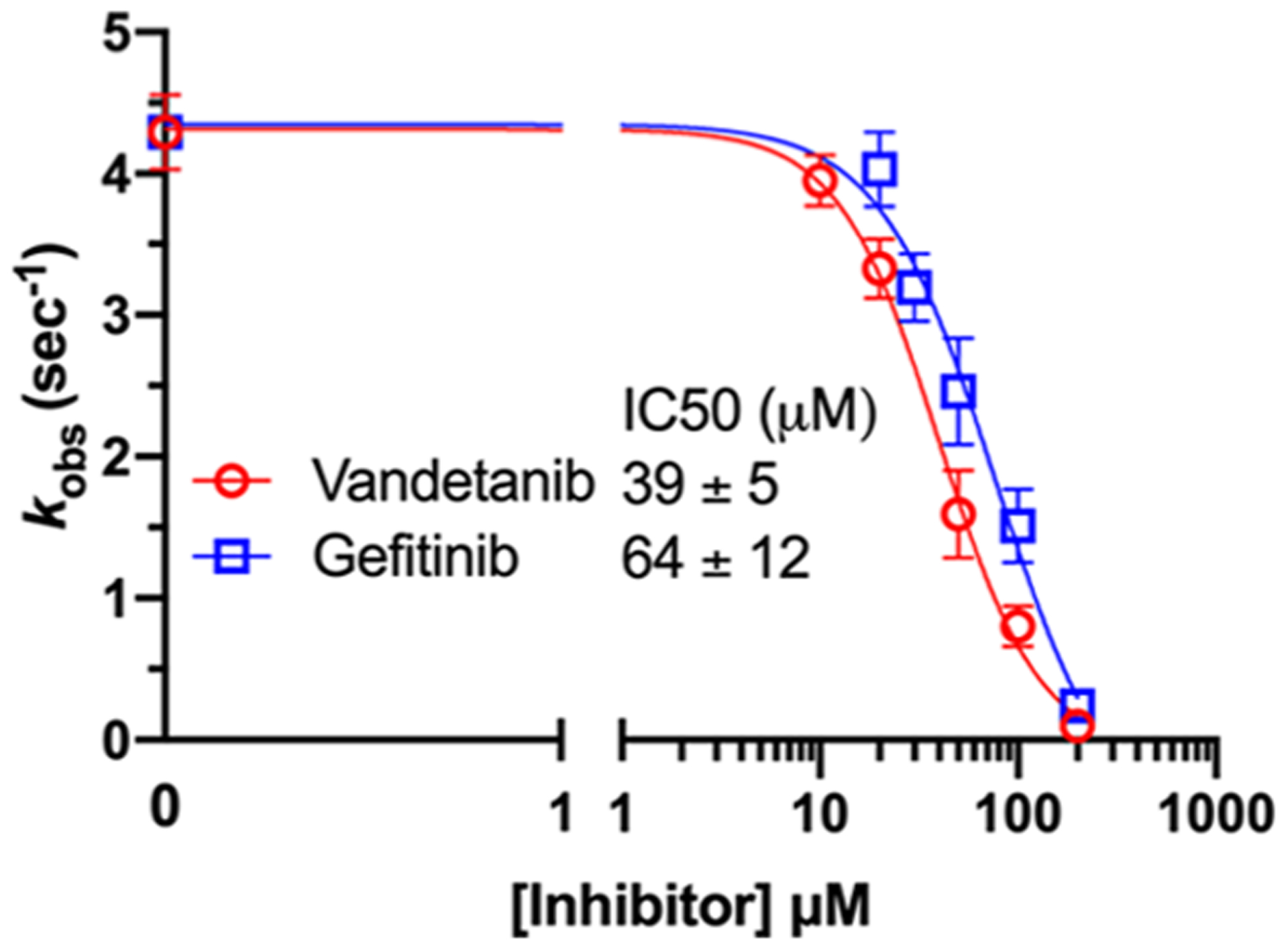
Functional assays confirmed that the above top ranked E. coli DHFR binders are true inhibitors of the enzyme (Figure 7). Compared to the reaction under the DMSO control condition, Vandetanib (NSC760766) and Gefitinib (NSC715055) identified by FRAGSITE but not FINDSITEcomb2.0 showed strong inhibition with an approximately 5-fold decrease in kobs (Figure 7). In contrast, Imiquimod (NSC369100) identified by both VLS methods showed no inhibition at the 100 μM level. This is consistent with the trend of larger ΔTm values of Vandetanib and Gefitinib, 8.0 and 6.7 °C, respectively (estimated Kd of 2–10 μM), compared to the potentially weak binder Imiquimod, 4 °C (see Table 6). Methotrexate (NSC740, Kd of ~9.5 nM)67 completely blocked the enzyme activity as expected with kobs reaching the detection limit of ~0.05 s−1. The inhibitory dose response assays further confirmed that the top ranked true binders Vandetanib and Gefitinib (NSC715055) representing new scaffolds as identified by FRAGSITE indeed inhibit DHFR catalysis with estimated IC50 of 39 and 64 μM, respectively (Figure 8). The slightly stronger inhibition (smaller apparent IC50) of Vandetanib over Gefitinib is consistent with the larger ΔTm value of Vandetanib over Gefitinib (8.0 vs 6.7 °C) as measured in the thermal shift assays. The IC50 calculated from the inhibitory dose response assays and Kd estimated from thermal shift assays are generally consistent at low to mid μM levels, confirming that Vandetanib and Gefitinib are true micromolar affinity inhibitors of E. coli DHFR. Their exact inhibition constants and mechanism of inhibition require further studies.
Figure 8.
Inhibitory dose response of E. coli DHFR steady-state kinetics to Vandetanib and Gefitinib. The reaction was performed under a constant room temperature of 18 °C under steady-state conditions with a catalytic amount of E. coli DHFR (93 nM) and saturated electron acceptor dihydrofolate (50 μM) and electron donor NADPH (100 μM) levels. The oxidation of NADPH was followed at 340 nm for 30–60 s. The observed initial linear rate was calculated using the published delta molar extinction coefficient Δε of 11.8 mM−1 cm−1.71 The reaction at each condition was replicated 3–4 times and is presented as the average value and standard deviation. Inhibitory dose response curve fitting was carried out using the “Absolute IC50” method of Prism software with the default setting and a baseline parameter value set to be 0, consistent with the control data of complete inhibition by Methotrexate and the current detection limit of kobs.
For human ACVR1 drug binder predictions, we observed the same trend that the common set of FRAGSITE and FINDSITEcomb2.0 predictions has higher true positive hit rates (precision of 71.4%) than unique set predictions by individual methods (see Table 6). FRAGSITE’s precision is 42.0% versus 0.0% by FINDSITEcomb2.0. This is an interesting test given that most of the true positive hits in the common set are known kinase inhibitors not originally designed to target ACVR1. This is in principle expected because of the known similarity of kinase ligand binding pockets, which results in the promiscuity of many kinase inhibitors.68 Unexpectedly, without prior knowledge, FRAGSITE was able to enrich the number of high affinity binders in the top ranked drug predictions. Of note, the FDA-approved drugs from our virtual ligand screening results that showed the relatively large positive thermal shifts (Table 6) are among the few high affinity kinase inhibitors of ACVR1 previously identified by the large-scale, brute force high-throughput experimental target screening among 182 different clinical kinase inhibitors across the human kinome.68 These include NSC760766 (Vandetanib, Kd of 0.15 μM),68 NSC732517 (Dasatinib, Kd of 0.62 μM),68 and NSC749005 (Crizotinib, Kd of 0.44 μM),68 consistent with our observed trend of ΔTm values of 8.0, 4.0, and 8.0 °C, respectively, of human ACVR1. However, neither FINDSITEcomb2.0 nor FRAGSITE identified the known potent ACVR1 inhibitor K02288 (IC50 of 1.1 nM),70 which was used as the positive control in ACVR1 thermal shift assays.
In particular, FRAGSITE appears to be more effective in finding true hit binders as reflected in the empirical observation that the no. 1 ranked Vandetanib prediction for ACVR1 identified by FRAGSITE (expected precision of 0.9) is ranked no. 11 (among 1812 molecules) by FINDSITEcomb2.0 (expected precision of 0.62) (Table 6). Of particular interest, in an in vivo mouse model of a rare pediatric brain cancer, diffuse intrinsic pontine glioma (DIPG), Vandetanib (originally designed to inhibit VEGFR/RET/EGFR) when combined with the mTOR inhibitor Everolimus was recently reported to effectively inhibit pharmacodynamic biomarkers of DIPG.69 Considering that there is no current effective treatment and the aggressive nature of DIPG with a median survival rate in child patients of less than a year, our new virtual ligand screening method FRAGSITE is able to identify FDA-approved drugs with repurposing potential for DIPG when inhibition of ACVR1 kinase activity is required. We note that ACVR1 kinase inhibitors may not be effective therapeutics for all forms of DIPG.47 These repurposed drugs might also be applicable to fibrodysplasia ossificans progressiva (FOP),48 an inherent connective tissue disorder due to increased kinase activity that results from a mutation in ACVR1.
Interestingly, FRAGSITE also identified a series of diverse scaffolds as true hit binders for ACVR1 that could serve as a starting point for in silico and experimental fragment-based drug design for high affinity binders and inhibitors, see Figure 9. Moreover, FRAGSITE independently identifies a new, apparent high affinity binder NSC105827 (thiosangivamycin), which FINDSITE could not identify. NSC105827 has a ΔTm of 9.7 °C, which is higher than the above-mentioned FDA-approved kinase inhibitors, which have nanomolar affinity for ACVR1 in the common set. Among the new true hit binders of ACVR1 identified in this study, three compounds NSC63701, NSC274905, and NSC105827 are conservatively estimated to have nanomolar affinity (Figure 9). These ligands will be pursued as new kinase inhibitors in future studies.
Figure 9.
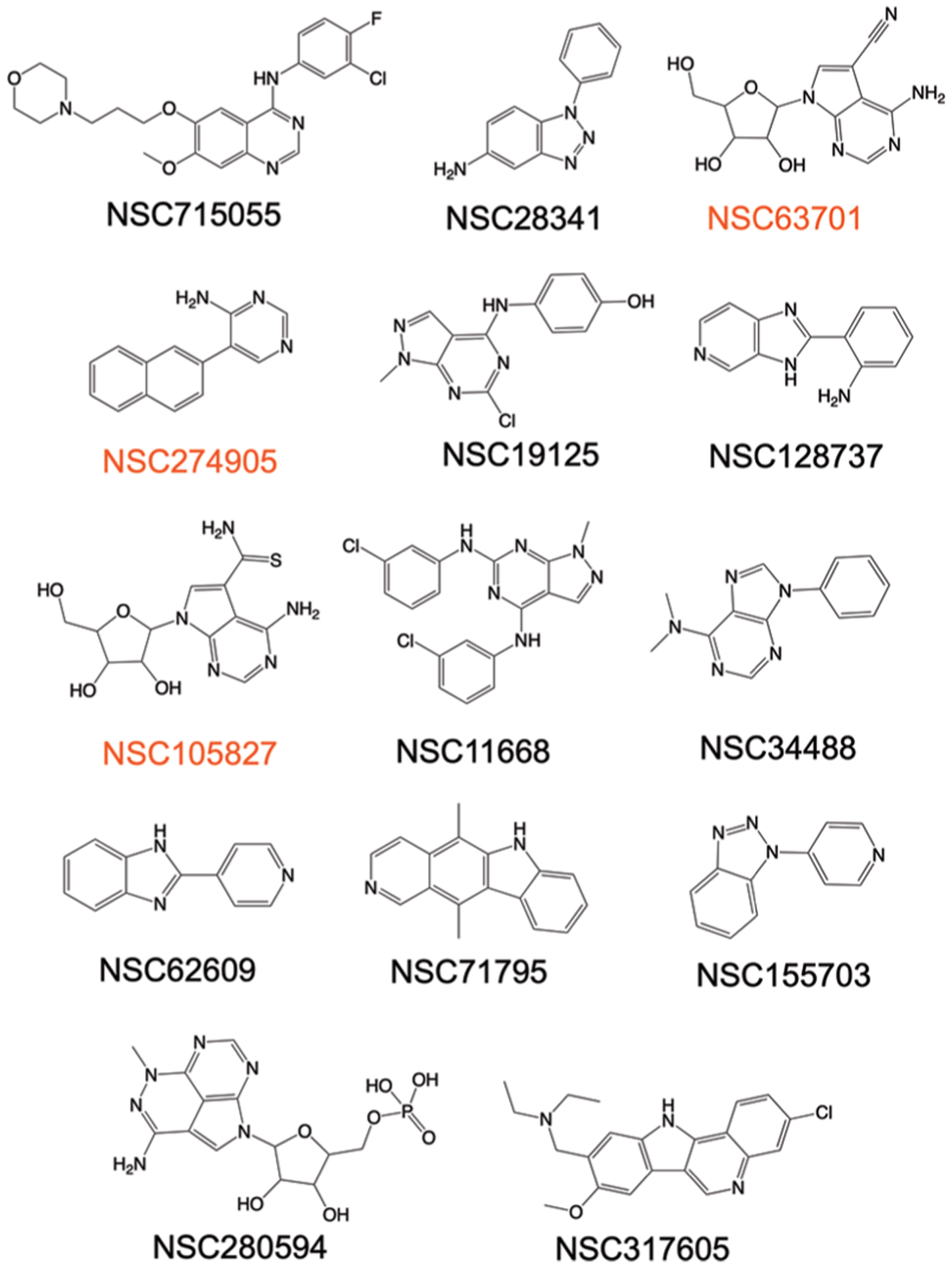
Chemical structures of true binder hits of ACVR1 uniquely identified by FRAGSITE but not FINDSITEcomb2.0 comprising diverse scaffolds for fragment-based drug design. Three compounds labeled red have a conservatively estimated Kd < 1 μM based on the reference ΔTm values observed in this study for the known affinity of kinase inhibitors NSC760766 (Vandetanib, Kd of 0.15 μM),68 NSC732517 (Dasatinib, Kd of 0.62 μM),68 and NSC749005 (Crizotinib, Kd of 0.44 μM)68 and K02288 (IC50 of 1.1 nM),70 8.0, 4.0, 8.0, and 13.0 °C, respectively. The rest of the true hit binders have estimated affinity for ACVR1 at the micromolar level based on the above reference compounds and the reported method of estimating binding affinity based on thermal shift assays.63
Our results corroborate the predictive power of FRAGSITE over FINDSITEcomb2.0. FRAGSITE is a powerful VLS approach that can identify new, diverse scaffolds and potential high affinity true binders for protein targets. FRAGSITE alone or in combination with FINDSITEcomb2.0 should help accelerate experimental drug lead discovery by in silico ligand prescreening and scaffold diversification. These experimental results are also consistent with benchmarking on the DUD-E set, which shows that when a precision cutoff of 0.5 is applied (i.e., predicted precision > 0.5), FRAGSITE has an average recall of 21.9%, whereas FINDSITEcomb2.0 has an average recall of 15.7%.
DISCUSSION
By utilizing fragment information from the template and target ligands, we have developed the boosting tree regression machine learning-based virtual ligand method FRAGSITE. FRAGSITE shows significant improvement over the state-ofthe-art FINDSITEcomb2.0 and is also significantly better than the deep learning technology-based CNN scoring and AtomNet methods. Our results are robust and only depend slightly on the particular choice of fragmentation methods such as MACCS or FP2 fingerprints. Experimental testing on two proteins, DHFR and ACVR1, validated that FRAGSITE discovers more hits with more diverse chemical scaffolds than FINDSITEcomb2.0. Interestingly, despite the fact that DHFR has been extensively studied, FRAGSITE was able to find new hits that are kinase inhibitors that were not previously known as DHFR binders. In addition, for ACVR1, FRAGSITE identified new classes of kinase binders, which will subsequently be assessed for their ability to inhibit kinase activity. With its increase in performance, FRAGSITE is slightly more computationally expensive than FINDSITEcomb2.0. However, once the models are trained and the fingerprints of the ligand library (both template ligands and to be screened compounds) are generated, the additional cost is minimal. For example, for screening 31,980 molecules, the total additional time on a typical single CPU node is 35 s, equivalent to ~1 ms/molecule or just 20 min for one million molecules. Thus, it is still much more efficient than docking-based methods.11,16,31 Overall, FRAGSITE is a powerful new VLS approach that exploits the insight that ligand fragments bind to rather unique protein subpockets to identify new and diverse scaffolds. FRAGSITE will be employed in future work to not only identify new ligands but as part of an approach to predict drug mode of action, efficacy, and drug side effects.
Supplementary Material
ACKNOWLEDGMENTS
This project was funded by R35GM118039 of the Division of General Medical Sciences of the NIH (J.S.) and a seed grant of The Andrew McDonough B + Foundation (J.S.). We thank the National Cancer Institute (NCI), Division of Cancer Treatment and Diagnosis (DCTD), Developmental Therapeutics Program (DTP) (http://dtp.cancer.gov) for providing the NCI small-molecule compounds for thermal shift assays. We also thank Drs. Eugene Shakhnovich and João Rodrigues from the Harvard University for generously providing purified E. coli DHFR, Dr. Yizhi Jane Tao and Xiaotong Lu from the Rice University for kindly providing ACVR1, and Dr. John McDonald at Georgia Tech for sharing the CFX96 real-time PCR machine for the thermal shift assays.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c01160.
Additional information for experimental methods and (Tables S1 and S2) top 40 most frequent fragments in DrugBank drugs and performance of methods for individual DUD-E targets using the modeled target structure (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.0c01160
The authors declare no competing financial interest.
REFERENCES
- (1).Gilson MK; Zhou H-X Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct 2007, 36, 21–42. [DOI] [PubMed] [Google Scholar]
- (2).Hermann JC; Marti-Arbona R; Fedorov AA; Fedorov E; Almo SC; Shoichet BK; Raushel FM Structure-based activity prediction for an enzyme of unknown function. Nature 2007, 448, 775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Reddy AS; Pati SP; Kumar PP; Pradeep HN; Sastry GN Virtual Screening in Drug Discovery – A Computational Perspective. Curr. Protein Pept. Sci 2007, 8, 329–351. [DOI] [PubMed] [Google Scholar]
- (4).Cases M; Mestres J A chemogenomic approach to drug discovery: focus on cardiovascular diseases. Drug Discovery Today 2009, 14, 479–485. [DOI] [PubMed] [Google Scholar]
- (5).Mysinger MM; Carchia M; Irwin JJ; Shoichet BK Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem 2012, 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Stein RM; Kang HJ; McCorvy JD; Glatfelter GC; Jones AJ; Che T; Slocum S; Huang X-P; Savych O; Moroz YS; Stauch B; Johansson LC; Cherezov V; Kenakin T; Irwin JJ; Shoichet BK; Roth BL; Dubocovich ML Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 2020, 579, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Abagyan R; Totrov M; Kuznetsov D ICM–a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem 1994, 15, 488–506. [Google Scholar]
- (8).Joseph-McCarthy D; Baber J; Feyfant E; Thompson D; Humblet C Lead optimization via high-throughput molecular docking. Curr. Opin. Drug Discovery Dev 2007, 10, 264–274. [PubMed] [Google Scholar]
- (9).Brozell SR; Mukherjee S; Balius TE; Roe DR; Case DA; Rizzo RC Evaluation of DOCK 6 as a pose generation and database enrichment tool. J. Comput.-Aided Mol. Des 2012, 26, 749–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Jain AN Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem 2003, 46, 499–511. [DOI] [PubMed] [Google Scholar]
- (11).Trott O; Olson AJ AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (13).Kroemer RT Structure-based drug design: docking and scoring. Curr. Protein Pept. Sci 2007, 8, 312–328. [DOI] [PubMed] [Google Scholar]
- (14).Kramer B; Rarey M; Lengauer T Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins 1999, 37, 228–241. [DOI] [PubMed] [Google Scholar]
- (15).Laurini E; Col VD; Mamolo MG; Zampieri D; Posocco P; Fermeglia M; Vio L; Pricl S Homology Model and Docking-Based Virtual Screening for Ligands of the σ1 Receptor. ACS Med. Chem. Lett 2011, 2, 834–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Allen WJ; Balius TE; Mukherjee S; Brozell SR; Moustakas DT; Lang PT; Case DA; Kuntz ID; Rizzo RC DOCK 6: Impact of new features and current docking performance. J. Comput. Chem 2015, 36, 1132–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Carpenter EP; Beis K; Cameron AD; Iwata S Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. Biol 2008, 18, 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ballester PJ; Mitchell JBO A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhou H; Skolnick J FINDSITEcomb: A Threading/Structure-Based, Proteomic-Scale Virtual Ligand Screening Approach. J. Chem. Inf. Model 2013, 53, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Willett P Similarity-based virtual screening using 2D fingerprints. Drug Discovery Today 2006, 11, 1046–1053. [DOI] [PubMed] [Google Scholar]
- (21).Nikolova N; Jaworska J Approaches to Measure Chemical Similarity – a Review. QSAR Comb. Sci 2003, 22, 1006–1026. [Google Scholar]
- (22).Flower DR On the Properties of Bit String-Based Measures of Chemical Similarity. J. Chem. Inf. Comput. Sci 1998, 38, 379–386. [Google Scholar]
- (23).Glen RC; Adams SE Similarity Metrics and Descriptor Spaces - Which Combinations to Choose? QSAR Comb. Sci 2006, 25, 1133–1142. [Google Scholar]
- (24).Kogej T; Engkvist O; Blomberg N; Muresan S Multi-fingerprint Based Similarity Searches for Targeted Class Compound Selection. J. Chem. Inf. Model 2006, 46, 1201–1213. [DOI] [PubMed] [Google Scholar]
- (25).Keiser MJ; Roth BL; Armbruster BN; Ernsberger P; Irwin JJ; Shoichet BK Relating protein pharmacology by ligand chemistry. Nat. Biotechnol 2007, 25, 197–206. [DOI] [PubMed] [Google Scholar]
- (26).Hawkins PCD; Skillman AG; Nicholls A Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem 2007, 50, 74–82. [DOI] [PubMed] [Google Scholar]
- (27).Xia J; Jin H; Liu Z; Zhang L; Wang XS An Unbiased Method To Build Benchmarking Sets for Ligand-Based Virtual Screening and its Application To GPCRs. J. Chem. Inf. Model 2014, 54, 1433–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Brylinski M; Skolnick J A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Brylinski M; Skolnick J FINDSITELHM: a threading-based approach to ligand homology modeling. PLoS Comput. Biol 2009, 5, No. e1000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhou H; Skolnick J FINDSITEX: A Structure-Based, Small Molecule Virtual Screening Approach With Application to All Identified Human GPCRs. Mol. Pharmaceutics 2012, 9, 1775–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhou H; Cao H; Skolnick J FINDSITEcomb2.0: A New Approach for Virtual Ligand Screening of Proteins and Virtual Target Screening of Biomolecules. J. Chem. Inf. Model 2018, 58, 2343–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Skolnick J; Gao M Interplay of physics and evolution in the likely origin of protein biochemical function. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 9344–9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gao M; Skolnick J A comprehensive survey of small-molecule binding pockets in proteins. PLoS Comput. Biol 2013, 9, No. e1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Brylinski M; Skolnick J Q-Dock: Low-resolution flexible ligand docking with pocket-specific threading restraints. J. Comput. Chem 2008, 29, 1574–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Brylinski M; Skolnick J Q-DockLHM: Low-resolution refinement for ligand comparative modeling. J. Comput. Chem 2010, 31, 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lee HS; Zhang Y BSP-SLIM: A blind low-resolution ligand-protein docking approach using predicted protein structures. Proteins 2012, 80, 93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Huang N; Shoichet BK; Irwin JJ Benchmarking Sets for Molecular Docking. J. Med. Chem 2006, 49, 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).AlQuraishi M AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ballester PJ Machine Learning Scoring Functions Based on Random Forest and Support Vector Regression. In Pattern Recognition in Bioinformatics, Tokyo, 2012; Shibuya T, Ed. Springer-Verlag Berlin Heidelberg: Tokyo, 2012; Vol. 7632; pp. 14–25. [Google Scholar]
- (40).Wójcikowski M; Ballester PJ; Siedlecki P Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep 2017, 7, 46710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wallach I; Dzamba M; Heifets A AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-Based Drug Discovery. arXiv preprint arXiv: 1510.02855, 2015. [Google Scholar]
- (42).Ragoza M; Hochuli J; Idrobo E; Sunseri J; Koes DR Protein–Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model 2017, 57, 942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Chen L; Cruz A; Ramsey S; Dickson CJ; Duca JS; Hornak V; Koes DR; Kurtzman T Hidden bias in the DUD-E dataset leads to misleading performance of deep learning in structure-based virtual screening. PLoS One 2019, 14, No. e0220113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Adeshina YO; Deeds EJ; Karanicolas J Machine learning classification can reduce false positives in structure-based virtual screening. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 18477–18488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bauer MR; Ibrahim TM; Vogel SM; Boeckler FM Evaluation and optimization of virtual screening workflows with DEKOIS 2.0–a public library of challenging docking benchmark sets. J. Chem. Inf. Model 2013, 53, 1447–1462. [DOI] [PubMed] [Google Scholar]
- (46).Barreiro EJ Privileged Scaffolds in Medicinal Chemistry: An Introduction. In Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation, Brase S, Ed.; Royal Society of Chemistry: 2016; Chapter 1, pp. 1–15. [Google Scholar]
- (47).Cao H; Jin M; Gao M; Zhou H; Tao YJ; Skolnick J Differential kinase activity of ACVR1 G328V and R206H mutations with implications to possible TβRI cross-talk in diffuse intrinsic pontine glioma. Sci. Rep 2020, 10, 6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Machiya A; Tsukamoto S; Ohte S; Kuratani M; Fujimoto M; Kumagai K; Osawa K; Suda N; Bullock AN; Katagiri T Effects of FKBP12 and type II BMP receptors on signal transduction by ALK2 activating mutations associated with genetic disorders. Bone 2018, 111, 101–108. [DOI] [PubMed] [Google Scholar]
- (49).Wishart D; Knox C; Guo A; Shrivastava S; Hassanali M; Stothard P; Chang Z; Woolsey J DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Gaulton A; Bellis LJ; Bento AP; Chambers J; Davies M; Hersey A; Light Y; McGlinchey S; Michalovich D; Al-Lazikani B; Overington JP ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids. Res 2012, 40, D1100–D1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Zhang Y; Skolnick J scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [DOI] [PubMed] [Google Scholar]
- (52).Bernstein FC; Koetzle TF; Williams GJB; Meyer EF Jr.; Brice MD; Rodgers JR; Kennard O; Shimanouchi T; Tasumi M The Protein Data Bank: A Computer-based Archival File for Macromolecular Structures. J. Mol. Biol 1977, 112, 535–542. [DOI] [PubMed] [Google Scholar]
- (53).Kim S; Chen J; Cheng T; Gindulyte A; He J; He S; Li Q; Shoemaker BA; Thiessen PA; Yu B; Zaslavsky L; Zhang J; Bolton EE PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Yap CW PaDEL-Descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem 2011, 32, 1466–1474. [DOI] [PubMed] [Google Scholar]
- (55).Toolkit D. Daylight Chemical Information Systems, Inc: Aliso Viejo, CA: 2007. [Google Scholar]
- (56).Friedman JH Greedy function approximation: a gradient boosting machine. Ann. Stat 2001, 29, 1189–1232. [Google Scholar]
- (57).Roe BP; Yang H-J; Zhu J Boosted Decision Trees, A Powerful Event Classifier. In Statistical Problems in Particle Physics, Astrophysics and Cosmology; 2006; Vol. 1, p 139. [Google Scholar]
- (58).Davis J; Goadrich M The Relationship Between PrecisionRecall and ROC Curves. In ICML ‘06 Proceedings of the 23rd International Conference on Machine Learning, Pittsburgh, 2006; ACM; New York, NY, USA Pittsburgh, 2006; pp. 233–240. [Google Scholar]
- (59).Cao H; Walton JD; Brumm P; Phillips GN Jr. Structure and substrate specificity of a eukaryotic fucosidase from Fusarium graminearum. J. Biol. Chem 2014, 289, 25624–25638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).O’Boyle NM; Banck M; James CA; Morley C; Vandermeersch T; Hutchison GR Open Babel: An open chemical toolbox. Aust. J. Chem 2011, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Tran-Nguyen V-K; Jacquemard C; Rognan D LIT-PCBA: An Unbiased Data Set for Machine Learning and Virtual Screening. J. Chem. Inf. Model 2020, 60, 4263–4273. [DOI] [PubMed] [Google Scholar]
- (62).Srinivasan B; Skolnick J Insights into the slow-onset tight-binding inhibition of Escherichia coli dihydrofolate reductase: detailed mechanistic characterization of pyrrolo [3,2-f] quinazoline-1,3-diamine and its derivatives as novel tight-binding inhibitors. FEBS J. 2015, 282, 1922–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Boehr DD; McElheny D; Dyson HJ; Wright PE The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313, 1638–1642. [DOI] [PubMed] [Google Scholar]
- (64).Cao H; Gao M; Zhou H; Skolnick J The crystal structure of a tetrahydrofolate-bound dihydrofolate reductase reveals the origin of slow product release. Commun. Biol 2018, 1, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Wan Q; Bennett BC; Wilson MA; Kovalevsky A; Langan P; Howell EE; Dealwis C Toward resolving the catalytic mechanism of dihydrofolate reductase using neutron and ultrahigh-resolution X-ray crystallography. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 18225–18230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Pantoliano MW; Petrella EC; Kwasnoski JD; Lobanov VS; Myslik J; Graf E; Carver T; Asel E; Springer BA; Lane P; Salemme FR High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screening 2001, 6, 429–440. [DOI] [PubMed] [Google Scholar]
- (67).Rajagopalan PTR; Zhang Z; McCourt L; Dwyer M; Benkovic SJ; Hammes GG Interaction of dihydrofolate reductase with methotrexate: ensemble and single-molecule kinetics. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 13481–13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Fabian MA; Biggs WH III; Treiber DK; Atteridge CE; Azimioara MD; Benedetti MG; Carter TA; Ciceri P; Edeen PT; Floyd M; Ford JM; Galvin M; Gerlach JL; Grotzfeld RM; Herrgard S; Insko DE; Insko MA; Lai AG; Lelias JM; Mehta SA; Milanov ZV; Velasco AM; Wodicka LM; Patel HK; Zarrinkar PP; Lockhart DJ A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- (69).Carvalho D; Olaciregui NG; Ruddle R; Donovan A; Pal A; Raynaud F; Richardson PJ; Carcaboso AM; Jones C DIPG-29. PRECLINICAL EFFICACY OF COMBINED ACVR1 AND PI3K/mTOR INHIBITION IN DIFFUSE INTRINSIC PONTINE GLIOMA (DIPG). Neuro-Oncology 2018, 20, i54–i55. [Google Scholar]
- (70).Sanvitale CE; Kerr G; Chaikuad A; Ramel MC; Mohedas AH; Reichert S; Wang Y; Triffitt JT; Cuny GD; Yu PB; Hill CS; Bullock AN A new class of small molecule inhibitor of BMP signaling. PLoS One 2013, 8, No. e62721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Stone SR; Morrison JF Dihydrofolate Reductase from Escherichia coli: The Kinetic Mechanism with NADPH and Reduced Acetylpyridine Adenine Dinucleotide Phosphate as Substrates. Biochemistry 1988, 27, 5493–5499. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.