

Abstract
The solution-mediated behavior of lithium-sulfur (Li-S) batteries presents a wide range of opportunity for evaluating and improving the performance at practical lean-electrolyte conditions. Here, we introduce methyl trifluoroacetate (CH3TFA) as an additive to the Li-S electrolyte to evaluate the joint effects of two distinct strategies: high donor number solvents/salts and organosulfur-mediated discharge. CH3TFA is shown to react with lithium polysulfides in-situ to form lithium trifluoroacetate (LiTFA) and dimethyl polysulfides. We find that both the methyl group and trifluoroacetate anion considerably enhance Li-S discharge behavior over the course of cycling, though they have distinctly beneficial effects. The TFA anion impacts solution coordination behavior, improving polarization and discharge kinetics during cycling. Meanwhile, the derivatization to dimethyl polysulfides improves the solubility of intermediate species, enhancing overall utilization under lean-electrolyte conditions. CH3TFA thus represents a new class of additives for Li-S batteries, enabling an in-situ systematic molecular engineering of intermediate species for improved performance.
Graphical Abstract
We have recently started to see tremendous advancements in electrified mobility for a large range of transportation use-cases.1 The incumbent lithium-ion battery chemistry has been the driving enabler behind this dynamic transition, presenting a roadmap towards eliminating carbon dioxide emissions from automobiles and short-range semi-trucks.2–4 While this initial transition is promising, the state-of-the art lithium-ion chemistry lacks the requisite specific energy needed to electrify other transportation sectors, including those within the aerospace sector.5 The electrification of these highly weight-dependent applications will necessitate the implementation of next-generation battery chemistries with higher theoretical specific energy, such as lithium-sulfur (Li-S) batteries.6
The Li-S battery chemistry has been long heralded as a suitable alternative given the high theoretical discharge capacity of the sulfur electrode (1,672 mA h g−1) and overall high specific energy of the Li-S redox couple (~ 2,600 W h kg−1). The potential implementation of such a novel chemistry is uniquely predicated on the battery’s ability to store greater amounts of energy per unit mass. In practice, however, it is quite difficult to achieve the high active material utilizations required under practical cell construction conditions, most notably while using lean electrolyte amounts.6,7 This is due to the inherent solution-mediated reaction paradigm intrinsic to the sulfur cathode material; given the highly electronically and ionically insulating nature of sulfur and the Li2S discharge product, the sulfur cathode undergoes a solid-liquid-solid phase transition, forming intermediate soluble lithium polysulfides (Li2Sx, 2 < x ≤ 8) during the course of discharge.8–10 Suppressing the dissolution of these intermediates has been a major focus in the Li-S literature, often through the use of electrocatalytic polar hosts in the sulfur cathode.11,12 However, these intermediate byproducts can enable facile solution-mediated charge transfer, presenting an avenue to overcoming the unfavorable kinetics presented through a feasibly more direct solid-solid conversion route. This intrinsic property necessitates a certain degree of active material dissolution, and thus presents an additional stipulation on a necessary amount of electrolyte required within the cell in order for the redox couple to operate successfully.7,9 This metric is quantified in the Li-S research community via the electrolyte-to-sulfur ratio (E/S, in μLelectrolyte mg−1sulfur) represented within the cell. Achieving favorable cell performance under lean electrolyte conditions is thus quite difficult in the Li-S battery chemistry and highlights the importance of furthering our mechanistic and kinetic understanding of the solution-mediated reaction paradigm.
Recently, there has been an increased focus within the Li-S research literature on high donor number solvents and salt anions to enhance the solubility of intermediate polysulfide species and improve the kinetic performance and capacity attainment.9,13–19 While this strategy tends to lead to shorter cycling lifetimes due to heightened polysulfide shuttling and subsequent parasitic reaction with the lithium-metal anode, it also leads to higher initial capacities and directly enhances the core enabler of favorable kinetic performance during discharge.16 Additionally, there has been recent work on designing high donor number solvents with increased lithium-metal stability,20 while other approaches have focused exclusively on high donor number salt anions to avoid lithium-metal instability.17,18 Our group built upon this research area by showing how more than just the solubility of polysulfides, the actual coordination environment in solution of polysulfides is highly critical to the kinetic behavior.21 Particularly at low temperature conditions, lithium polysulfides tend to cluster and coordinate with each other, forming dimers and trimers that impede lithiation behavior and curtail active material utilization.22,23 We have shown that introducing a highly binding (high donor number) electrolyte salt like lithium trifluoroacetate (LiCO2CF3, or LiTFA) helps inhibit this clustering behavior, enhancing the kinetics of discharge.21 Beyond improving the solubility and coordination of polysulfides through high donor solvents and salts, there has also been considerable work performed on organosulfur compounds.24 In contrast to lithium polysulfides, organopolysulfides contain sulfur chains capped with organic functional groups, which can dramatically alter and enhance the innate reaction pathways and kinetics during discharge.25,26 Organosulfur species can exhibit heightened stability with the lithium-metal anode, decreasing parasitic side reactions stemming from active material shuttling.27 Furthermore, organosulfur species can enable improved overall active material utilization, stemming from the heightened solubility of covalent organopolysulfides species as well as the decreased tendency to form insulating and passivating Li2S upon discharge.28–30 In fact, organosulfur and organopolysulfide compounds have been demonstrated as both cathode active materials and electrolyte additives in a variety of electrochemical studies to enhance cycling performance and discharge behavior.24,25,27,31,32
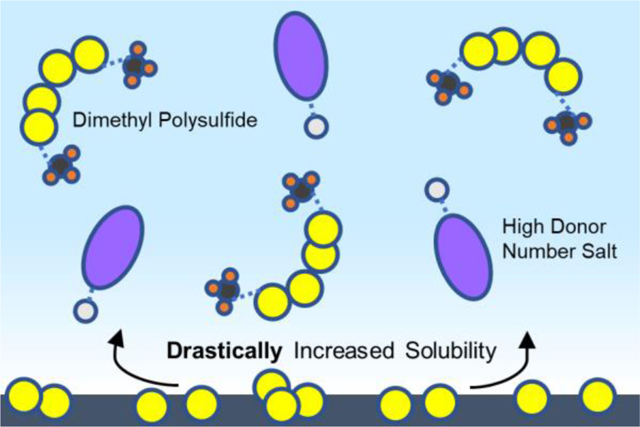
In this work, our driving motivation is to investigate whether these two distinct approaches (high donor number compounds and organosulfur compounds) can be fused and reconciled into a single cohesive strategy for understanding and improving the electrochemical behavior of Li-S batteries. Specifically, we evaluate the introduction of methyl trifluoroacetate (CF3COOCH3, denoted as CH3TFA), containing the organic methyl group and well as the anionic trifluoroacetate (TFA) group, as an additive to the predominantly used electrolyte in Li-S batteries. This additive is employed to not only optimize the solution coordination behavior through the TFA group, but also introduce organic groups to lithium polysulfides via an in-situ derivatization.33 This would theoretically boost the intermediate species solubility to new heights as shown in Figure 1a, inhibiting detrimental clustering behavior, mitigating cathode passivation, and boosting solution-mediated discharge performance. We expect the in-situ methylation of lithium polysulfides to occur according to the reaction in Figure 1b, generating dimethyl polysulfides and the high donor number LiTFA salt in-situ. This additive thus takes advantage of the active material dissolution intrinsic to the Li-S system, augmenting the solution-mediated behavior of Li-S batteries to a new extreme and granting the ability to assess the joint effects of favorable functionalization, coordination, and solubility of polysulfides.
Figure 1.

(a) Illustration of the theorized improvements to the Li-S electrolyte enabled through the presence of high donor number compounds in solution, as well as high donor compounds in conjunction with organosulfur active material. (b) Computational evaluation of the energetics of the reaction between CH3TFA and lithium polysulfides theorized to take place in-situ.
In order to investigate the thermodynamic feasibility of the forward reaction taking place, computational chemistry was used to gain an understanding from first principles. Gas phase hybrid-density functional theory calculations were employed to optimize the structures of all reactants and products in Figure 1b, after which the total change in energy was calculated according to Equation 1 and normalized to a kJ mol−1 basis.34 Energies were calculated using Li2S6 as the starting reactant, which was assumed to nominally represent a spectrum of lithium polysulfides that could exist in solution.
| (1) |
As can be seen in Figure 1b, the use of CH3TFA results in a strongly energetically favorable methylation reaction, resulting in the formation of dimethyl polysulfides and LiTFA salt. Similar calculations were performed on other common methylating agents including methyl triflate (CH3SO2OCH3, or CH3TF), as detailed in the Supporting Information Table S1. As can be seen, the forward reaction with CH3TFA exhibits a minimization of energy on the same order of magnitude as methylating reagents like CH3TF, while producing a favorable and strongly binding LiTFA salt.
The suspected methylation reaction was further corroborated through an assortment of experimental characterization studies. As seen in Figure 2a and Figure S1, it is visually confirmed that 0.1 M of nominal Li2S6 in 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME) solvents spontaneously reacts with 0.2 M CH3TFA in solution. Looking at the ultraviolet-visible (UV-VIS) spectra of 0.1 M Li2S6 with and without 0.2 M CH3TFA in Figure 2b, the reacted solution displays much lower absorbance at higher wavelengths and predominant signals spanning 250 – 340 nm, consistent with past observations on dimethyl polysulfides.32,35 In order to precisely measure the relative change in bond environment, both 1H and 13C nuclear magnetic resonance (NMR) spectroscopy were conducted on samples containing either 0.2 M CH3TFA or the additive in solution with 0.1 M nominal Li2S6. As seen in Figure 2c, the signal associated with CH3TFA at 4 ppm entirely disappears in contact with 0.1 M nominal Li2S6, resulting in two new signals at 2.40 and 2.65 ppm. These two signals correspond to two dominant dimethyl polysulfides whose average polysulfide chain length equates to 6, stemming from the stoichiometry dictated by the initial input of nominal Li2S6.26 Given that this NMR study was conducted in deuterated dimethyl sulfoxide (D6-DMSO) solvent, it is likely that these signals correspond to S62- and S3•− containing intermediates.9 This is further supported by the 13C NMR spectra in Figure 2d. After exposure to 0.1 M Li2S6, the CH3TFA signals corresponding to the trifluoroacetate group at 115 and 157 ppm shift downfield to 118 and 159 ppm, respectively, corresponding to the formation of LiTFA. The direct observation of the LiTFA signal confirms that the reaction is proceeding as expected. Meanwhile, the CH3TFA signal corresponding to the methyl group (55 ppm) is no longer present after exposure to Li2S6, and in its place, two new signals corresponding to dimethyl polysulfides are observed at 22 and 25 ppm. Thus, Figures 2c and d both confirm that CH3TFA completely and spontaneously reacts with lithium polysulfides in solution, presenting a feasibly simple strategy towards simultaneously evoking high donor number-assisted and organosulfur-mediated discharge in Li-S batteries.
Figure 2.

(a) Optical image of vials containing 0.1 M Li2S6 in DOL/DME solvents, with and without the addition of 0.2 M CH3TFA. (b) UV-VIS spectra of the polysulfide-containing samples with and without the CH3TFA additive. (c) 1H NMR and (d) 13C NMR spectra of samples containing either CH3TFA or CH3TFA in combination with 0.1 M Li2S6 in D6-DMSO.
With a firm understanding of the role CH3TFA can play in solution, Li-S cells were constructed and tested with an electrolyte consisting of 0.2 M CH3TFA, 0.2 M LiNO3, and 0.8 M lithium bistriflimide (LiTFSI) in DOL/DME (1:1, by vol.). This formulation is expected to allow for a moderate degree of in-situ polysulfide methylation and LiTFA formation, and was designed with the intent of keeping the total solute concentration consistent with a control electrolyte made up of 1.0 M LiTFSI and 0.2 M LiNO3. Additionally, a halfway-optimized electrolyte consisting of 0.2 M LiTFA, 0.2 M LiNO3, and 0.8 M LiTFSI in DOL/DME was also tested to deconvolute the distinct effects of the strongly binding TFA anion and the dimethyl polysulfide formation on discharge. Cells were assembled with blade cast sulfur cathodes possessing a sulfur loading of 3.2 mg cm−2 and an E/S ratio of 12 μL mg−1. These cells were charged and discharged at a C/10 rate over 50 cycles to deduce the major contributions to electrochemical performance brought about through the CH3TFA additive.
As shown in Figure 3a, the electrolyte containing CH3TFA enabled vastly superior discharge characteristics over 50 cycles. Beyond demonstrating less capacity fade per cycle, it allowed for a capacity in excess of 800 mA h g−1 for a majority of the cycling duration, an almost 60% improvement over the 500 mA h g−1 achieved with the control electrolyte. Furthermore, CH3TFA enabled a sizeable 25% improvement in capacity over the ~ 640 mA h g−1 achieved during cycling in electrolyte containing 0.2 M LiTFA. In order to further understand the source of these capacity improvements, the evolution of the polarization between the average charge and discharge voltages was plotted over the course of cycling. As can be seen in Figure 3b, the average polarization of cells with the CH3TFA and LiTFA additives is significantly less by on the order of 40 mV than that with the control electrolyte, particularly during the early stages of cycling. This performance improvement is understandable given that both solutions contain the kinetically beneficial TFA anion, enabling more ideal solution-coordination behavior and lithium-ion exchange.21 However, one interesting note is that both additives enable the same exact degree of improvement to polarization over the course of cycling, suggesting that presence of the TFA plays a dominant role in improving the electrochemical discharge/charge kinetics rather than the in-situ methylation of polysulfides. The formation of dimethyl polysulfides, rather, seems to bring about non-kinetic improvements to capacity attainment and retention. As visually depicted in Figure 3c, the presence of CH3TFA enables the same performance improvements to discharge kinetics seen by LiTFA, but also demonstrates additional improvements intrinsic to the methylation of polysulfide active material.
Figure 3.

(a) Discharge behavior of Li-S cells with electrolyte containing 1 M LiTFSI and 0.2 M LiNO3 in DOL/DME, or 0.8 M LiTFSI and 0.2 M LiNO3 with the addition of either 0.2 M LiTFA or 0.2 M CH3TFA. (b) The evolution each cell’s voltage polarization, or the difference between the average charge and discharge voltages, plotted over the course of 50 cycles. (c) A magnified view of the discharge behavior of each cell, with the bifunctional improvements presented through the CH3TFA additive deconvoluted. (d) A comparison of the improvements to rate capability brought about by the addition of CH3TFA to the control electrolyte.
This is further supported by Figure 3d, which compares the rate capabilities within the CH3TFA electrolyte and the control electrolytes at rates from C/20 to C/3, normalized to their initial capacity attainment. This experiment was conducted with high loading sulfur cathodes (4.6 mg cm−2) at an E/S ratio of 10 μl mg−1 to understand the kinetic behavior under more practically relevant cell constraints. As can be seen, while the CH3TFA additive does demonstrate superior rate performance, much of this improvement stems from possessing lower intrinsic capacity fade rates over the course of cycling. While this is in part due to the superior polarization behavior, this is likely primarily driven from properties intrinsic to the use of organopolysulfides, including superior lithium-metal stability and decreased tendency to passivate the cathode with insulating Li2S and Li2S2 byproducts, both of which are well explored in the organosulfur electrochemical literature.27,32,36,37 The benefits to kinetic behavior and lithium-metal stability are also explored through cyclic voltammetry, electrochemical impedance spectroscopy, and galvanostatic cycling of lithium symmetric cells shown in Figure S2.
Thus, the implementation of CH3TFA as an additive to the Li-S electrolyte enables a bifunctional improvement to performance, both from a kinetic standpoint as well as a stability standpoint. A final perspective from which to assess performance improvement is by the degree of intermediate polysulfide solubility within the Li-S electrolyte. The active material utilization within Li-S batteries is uniquely predicated on the ability of the electrolyte to solvate intermediate polysulfide species, given the highly solution-dependent reaction pathways involved during discharge of the sulfur cathode.9,18 Thus, heightening the degree to which the electrolyte can solvate intermediate polysulfide species is expected to increase the active material utilization at a set E/S ratio. In other words, as the solubility to intermediate polysulfides increases within the electrolyte, the E/S ratio at which the cell can successfully operate and have ample solution-mediated discharge is expected to decrease. This relation is plotted in Figure 4a, which relates the electrolyte solubility of Li2S6 to the E/S ratio in a cell operating completely under solution-mediated reaction pathways (where all of the active material is present in solution). The solubility limit of Li2S6 in conventional DOL/DME electrolyte (approximately ~ 1 M Li2S6) is represented via the dashed line, demonstrating exactly how the solution mediated pathways are impeded at E/S ratios lower than 5 with the predominantly used Li-S electrolyte.9
Figure 4.

(a) The relationship between the degree of solubility towards lithium polysulfides (Li2S6) and the achievable E/S ratio in a Li-S cell operating completely under solution-mediated pathways. (b) Optical image showing the inability to solvate 1 M Li2S6 in DOL/DME, but the heightened solubility offered through the addition of 0.2 M CH3TFA. (c) The discharge behavior of two Li-S pouch cells operating under a lean E/S ratio of 4.5 μL mg−1, with both formulations also containing 0.2 M LiNO3. An aberrant charge step occurred on Cycle 20 in the CH3TFA-containing cell, after which the upper voltage cutoff was decreased for the remainder of cycling. (d) Discharge and charge behaviors of each pouch cell on Cycle 30.
By incorporating the strategy of high donor number assisted discharge, which can bring about improvements through heightened polysulfide solubility, with the organosulfur approach of using dimethyl polysulfides, which can also drastically boost the solubility of intermediate and final discharge products, the intermediate solubility of the Li-S electrolyte can be boosted to new heights. This is demonstrated in Figure 4b, where 1 M of nominal Li2S6 can be readily accommodated in the solution phase when 0.2 M of CH3TFA additive is present in DOL/DME solvent. As expected in the absence of the additive, however, 1 M Li2S6 cannot be fully solvated in DOL/DME, with excess undissolved material settling at the bottom of the vial. This speaks to the improvements to performance and capacity seen with the CH3TFA in Figure 3 and highlights how this additive may be an excellent candidate towards enabling optimal Li-S performance under challenging lean electrolyte conditions.
In order to assess this, large format Li-S pouch cells were constructed with both a high sulfur loading cathode (4.8 mg cm−2) and a lean amount of either the control electrolyte or the CH3TFA-containing electrolyte (E/S = 4.5 μL mg−1). This lean electrolyte amount lies beyond the cusp of what would be expected to operate successfully using the model assumptions from Figure 4a, and thus serves as a suitable benchmark to evaluate the efficacy of the additive under practical cell constraints.6 As shown in Figure 4c and Figure S3 over the course of 40 cycles, the additive-containing cell achieves a maximum capacity of 750 mA h g−1, or an effective electrolyte-to-capacity (E/C) ratio of 6 μL (mA h)−1, compared to only 475 mA h g−1, or an E/C ratio of 9.5 μL (mA h)−1, in the control cell. Furthermore, the CH3TFA-containing cell displays a capacity that is more than 200% of the capacity of the control pouch cell during a representative discharge like that on Cycle 30, as seen in Figure 4d. The voltage profiles shown here also help illustrate the different beneficial contributions to charge and discharge brought about through the CH3TFA additive. While the discharge curve of CH3TFA-containing cell exhibits slightly less overpotential, there are major differences in the charge profiles. Specifically, the additive-containing cell lacks the large voltage spike at the onset of charge and exhibits much lower overpotential during the charge process. This can be explained by the presence of LiSCH3 as an additional final discharge product to Li2S. LiSCH3 exhibits less tendency than Li2S to passivate the cathode with an insulating layer that obstructs ionic charge transfer.28 This sizeable improvement signifies the degree to which this additive enhances performance under practical constraints.
However, the ability of CH3TFA to methylate polysulfides in-situ is highly concentration dependent. Under lean E/S ratios, the use of CH3TFA heightens solubility to a relatively significant amount compared to electrolytes without the additive. This results in more effective active material utilization during cycling, along with the benefits to lithium metal stability and superior kinetics seen at higher E/S ratios. The performance improvements seen here are brought through a relatively unoptimized electrolyte formulation, and the additive amounts of CH3TFA in future formulations will need to be tuned for the target E/S conditions. While the long-term cycling stability of the CH3TFA-modified pouch cell falls short of what will inevitably be needed for application-oriented implementation, this is an excellent step towards making this battery chemistry a reality. The performance exhibited in this cell illuminates a path towards creating practical Li-S cells under challenging conditions like high areal sulfur loadings and lean electrolyte amounts.
The CH3TFA additive thus serves as a starting point for a previously unconsidered approach in Li-S batteries, combining the advantages of high donor number salts with the benefits of organosulfur-mediated discharge. Here, the in-situ derivatization utilized the simplest and most fundamental organic functional group and enabled encouraging discharge behavior under challenging lean electrolyte conditions. Moreover, the formation of LiTFA in-situ allowed for a further boost to kinetic behavior, reducing polarization over the course of cycling and enabling favorable solution coordination behavior of polysulfides. However, this joint strategy is demonstrated in its earliest iteration. There is a vast, unexplored space for optimization of the functional group by which the polysulfides are derivatized in-situ, as well as the anionic group by which the in-situ salt formation proceeds. Through such a strategy, we choose a path that brings the high specific energy promise of the Li-S battery closer in sight, enabling further discovery into the rich, complex, and dynamic system.
Supplementary Material
Acknowledgements
This work was supported by a NASA Space Technology Research Fellowship (NSTRF) under award number 80NSSC17K0089. We also acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC resources that have contributed to the research results reported within this paper (URL: http://www.tacc.utexas.edu).
Footnotes
Supporting Information
Additional supporting information is available free of charge at the ACS Publications Website. This includes experimental methods, evidence for the reaction occurring with excess lithium salt, cyclic voltammetry and electrochemical impedance spectroscopy of Li-S cells with and without the additive, lithium symmetric cell cycling data with and without dimethyl polysulfides, coulombic efficiencies of cycled pouch cells, computational energy change values for comparable methylating agents, and coordinates and energy values for all molecular structures used in this work.
The authors declare no competing financial interests.
References
- (1).Manthiram A. A Reflection on Lithium-Ion Battery Cathode Chemistry. Nat. Commun. 2020, 11 (1), 1–9. 10.1038/s41467-020-15355-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Li W; Erickson EM; Manthiram A. High-Nickel Layered Oxide Cathodes for Lithium-Based Automotive Batteries. Nat. Energy 2020, 5 (1), 26–34. 10.1038/s41560-019-0513-0. [DOI] [Google Scholar]
- (3).Winter M; Barnett B; Xu K. Before Li Ion Batteries. Chem. Rev. 2018, 118 (23), 11433–11456. 10.1021/acs.chemrev.8b00422. [DOI] [PubMed] [Google Scholar]
- (4).Myung ST; Maglia F; Park KJ; Yoon CS; Lamp P; Kim SJ; Sun YK Nickel-Rich Layered Cathode Materials for Automotive Lithium-Ion Batteries: Achievements and Perspectives. ACS Energy Lett. 2017, 2 (1), 196–223. 10.1021/acsenergylett.6b00594. [DOI] [Google Scholar]
- (5).Krishnamurthy V; Viswanathan V. Beyond Transition Metal Oxide Cathodes for Electric Aviation: The Case of Rechargeable CF X. ACS Energy Lett. 2020, 3330–3335. 10.1021/acsenergylett.0c01815. [DOI] [Google Scholar]
- (6).Bhargav A; He J; Gupta A; Manthiram A. Lithium-Sulfur Batteries: Attaining the Critical Metrics. Joule 2020, 4 (2), 285–291. 10.1016/j.joule.2020.01.001. [DOI] [Google Scholar]
- (7).Betz J; Bieker G; Meister P; Placke T; Winter M; Schmuch R. Theoretical versus Practical Energy: A Plea for More Transparency in the Energy Calculation of Different Rechargeable Battery Systems. Adv. Energy Mater. 2019, 9 (6), 1803170. 10.1002/aenm.201803170. [DOI] [Google Scholar]
- (8).Zhang SS Liquid Electrolyte Lithium/Sulfur Battery: Fundamental Chemistry, Problems, and Solutions. J. Power Sources 2013, 231, 153–162. 10.1016/j.jpowsour.2012.12.102. [DOI] [Google Scholar]
- (9).Gupta A; Bhargav A; Manthiram A. Highly Solvating Electrolytes for Lithium–Sulfur Batteries. Adv. Energy Mater. 2018, 1803096. 10.1002/aenm.201803096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Shen C; Xie J; Zhang M; Andrei P; Hendrickson M; Plichta EJ; Zheng JP Understanding the Role of Lithium Polysulfide Solubility in Limiting Lithium-Sulfur Cell Capacity. Electrochim. Acta 2017, 248, 90–97. 10.1016/J.ELECTACTA.2017.07.123. [DOI] [Google Scholar]
- (11).Wang H; Zhang W; Xu J; Guo Z. Advances in Polar Materials for Lithium–Sulfur Batteries. Adv. Funct. Mater 2018, 28 (38). 10.1002/adfm.201707520. [DOI] [Google Scholar]
- (12).Chen Z; Yang X; Qiao X; Zhang N; Zhang C; Ma Z; Wang H. Lithium-Ion-Engineered Interlayers of V2C MXene as Advanced Host for Flexible Sulfur Cathode with Enhanced Rate Performance. J. Phys. Chem. Lett 2020, 11 (3), 885–890. 10.1021/acs.jpclett.9b03827. [DOI] [PubMed] [Google Scholar]
- (13).Zou Q; Lu Y-C Solvent-Dictated Lithium Sulfur Redox Reactions: An Operando UV–Vis Spectroscopic Study. J. Phys. Chem. Lett 2016, 7 (8), 1518–1525. 10.1021/acs.jpclett.6b00228. [DOI] [PubMed] [Google Scholar]
- (14).Cuisinier M; Hart C; Balasubramanian M; Garsuch A; Nazar LF Radical or Not Radical: Revisiting Lithium-Sulfur Electrochemistry in Nonaqueous Electrolytes. Adv. Energy Mater. 2015, 5 (16), 1401801. 10.1002/aenm.201401801. [DOI] [Google Scholar]
- (15).Li Z; Zhou Y; Wang Y; Lu YC Solvent-Mediated Li 2 S Electrodeposition: A Critical Manipulator in Lithium–Sulfur Batteries. Adv. Energy Mater. 2019, 9 (1), 1802207. 10.1002/aenm.201802207. [DOI] [Google Scholar]
- (16).Shin H; Baek M; Gupta A; Char K; Manthiram A; Choi JW Recent Progress in High Donor Electrolytes for Lithium–Sulfur Batteries. Adv. Energy Mater. 2020, 10 (27), 2001456. 10.1002/aenm.202001456. [DOI] [Google Scholar]
- (17).Chu H; Noh H; Kim Y-J; Yuk S; Lee J-H; Lee J; Kwack H; Kim Y; Yang D-K; Kim H-T Achieving Three-Dimensional Lithium Sulfide Growth in Lithium-Sulfur Batteries Using High-Donor-Number Anions. Nat. Commun. 2019, 10 (1), 188. 10.1038/s41467-018-07975-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chu H; Jung J; Noh H; Yuk S; Lee J; Lee J; Baek J; Roh Y; Kwon H; Choi D; et al. Unraveling the Dual Functionality of High-Donor-Number Anion in Lean-Electrolyte Lithium-Sulfur Batteries. Adv. Energy Mater. 2020, 2000493. 10.1002/aenm.202000493. [DOI] [Google Scholar]
- (19).Yang B; Jiang H; Zhou Y; Liang Z; Zhao T; Lu YC Critical Role of Anion Donicity in Li2S Deposition and Sulfur Utilization in Li-S Batteries. ACS Appl. Mater. Interfaces 2019, 11 (29), 25940–25948. 10.1021/acsami.9b07048. [DOI] [PubMed] [Google Scholar]
- (20).Baek M; Shin H; Char K; Choi JW New High Donor Electrolyte for Lithium–Sulfur Batteries. Adv. Mater. 2020, 2020, 2005022. 10.1002/adma.202005022. [DOI] [PubMed] [Google Scholar]
- (21).Gupta A; Bhargav A; Jones JP; Bugga RV; Manthiram A. Influence of Lithium Polysulfide Clustering on the Kinetics of Electrochemical Conversion in Lithium-Sulfur Batteries. Chem. Mater. 2020, 32 (5), 2070–2077. 10.1021/acs.chemmater.9b05164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Rajput NN; Murugesan V; Shin Y; Han KS; Lau KC; Chen J; Liu J; Curtiss LA; Mueller KT; Persson KA Elucidating the Solvation Structure and Dynamics of Lithium Polysulfides Resulting from Competitive Salt and Solvent Interactions. Chem. Mater. 2017, 29 (8), 3375–3379. 10.1021/acs.chemmater.7b00068. [DOI] [Google Scholar]
- (23).Andersen A; Rajput NN; Han KS; Pan H; Govind N; Persson KA; Mueller KT; Murugesan V. Structure and Dynamics of Polysulfide Clusters in a Nonaqueous Solvent Mixture of 1,3-Dioxolane and 1,2-Dimethoxyethane. Chem. Mater. 2019, 31 (7), 2308–2319. 10.1021/acs.chemmater.8b03944. [DOI] [Google Scholar]
- (24).Zhang X; Chen K; Sun Z; Hu G; Xiao R; Cheng HM; Li F. Structure-Related Electrochemical Performance of Organosulfur Compounds for Lithium-Sulfur Batteries. Energy Environ. Sci. 2020, 13 (4), 1076–1095. 10.1039/c9ee03848e. [DOI] [Google Scholar]
- (25).Chen S; Wang D; Zhao Y; Wang D. Superior Performance of a Lithium-Sulfur Battery Enabled by a Dimethyl Trisulfide Containing Electrolyte. Small Methods 2018, 2 (6), 1800038. 10.1002/smtd.201800038. [DOI] [Google Scholar]
- (26).Chen S; Dai F; Gordin ML; Yu Z; Gao Y; Song J; Wang D. Functional Organosulfide Electrolyte Promotes an Alternate Reaction Pathway to Achieve High Performance in Lithium-Sulfur Batteries. Angew. Chemie - Int. Ed 2016, 55 (13), 4231–4235. 10.1002/anie.201511830. [DOI] [PubMed] [Google Scholar]
- (27).Boateng B; Han Y; Zhen C; Zeng G; Chen N; Chen D; Feng C; Han J; Xiong J; Duan X; et al. Organosulfur Compounds Enable Uniform Lithium Plating and Long-Term Battery Cycling Stability. Nano Lett. 2020, 20 (4), 2594–2601. 10.1021/acs.nanolett.0c00074. [DOI] [PubMed] [Google Scholar]
- (28).Wu M; Cui Y; Bhargav A; Losovyj Y; Siegel A; Agarwal M; Ma Y; Fu Y. Organotrisulfide: A High Capacity Cathode Material for Rechargeable Lithium Batteries. Angew. Chemie Int. Ed 2016, 55 (34), 10027–10031. 10.1002/anie.201603897. [DOI] [PubMed] [Google Scholar]
- (29).Wu M; Bhargav A; Cui Y; Siegel A; Agarwal M; Ma Y; Fu Y. Highly Reversible Diphenyl Trisulfide Catholyte for Rechargeable Lithium Batteries. ACS Energy Lett. 2016, 1 (6), 1221–1226. 10.1021/acsenergylett.6b00533. [DOI] [Google Scholar]
- (30).Bhargav A; Bell ME; Karty J; Cui Y; Fu Y. A Class of Organopolysulfides As Liquid Cathode Materials for High-Energy-Density Lithium Batteries. ACS Appl. Mater. Interfaces 2018, 10 (25), 21084–21090. 10.1021/acsami.8b06803. [DOI] [PubMed] [Google Scholar]
- (31).Bhargav A; Manthiram A. Xanthogen Polysulfides as a New Class of Electrode Material for Rechargeable Batteries. Adv. Energy Mater. 2020, 10 (37), 2001658. 10.1002/aenm.202001658. [DOI] [Google Scholar]
- (32).He J; Bhargav A; Manthiram A. Three-Dimensional Fe3O4/N-Graphene Sponge as an Efficient Organosulfide Host for High-Performance Lithium-Organosulfur Batteries. Energy Storage Mater. 2019, 23, 88–94. 10.1016/j.ensm.2019.05.027. [DOI] [Google Scholar]
- (33).Zheng D; Zhang X; Wang J; Qu D; Yang X; Qu D. Reduction Mechanism of Sulfur in Lithium-Sulfur Battery: From Elemental Sulfur to Polysulfide. J. Power Sources 2016, 301, 312–316. 10.1016/j.jpowsour.2015.10.002. [DOI] [Google Scholar]
- (34).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; et al. Gaussian 16, Revision A.03. Gaussian, Inc.: Wallingford CT: 2016. [Google Scholar]
- (35).Carmack M; Fehnel EA The Ultraviolet Absorption Spectra of Organic Sulfur Compounds. I. Compounds Containing the Sulfide Function. J. Am. Chem. Soc. 1949, 71 (1), 84–93. 10.1021/ja01169a025. [DOI] [PubMed] [Google Scholar]
- (36).Kamphaus EP; Balbuena PB Effects of Dimethyl Disulfide Cosolvent on Li-S Battery Chemistry and Performance. Chem. Mater. 2019, 31 (7), 2377–2389. 10.1021/acs.chemmater.8b04821. [DOI] [Google Scholar]
- (37).Wang DY; Guo W; Fu Y. Organosulfides: An Emerging Class of Cathode Materials for Rechargeable Lithium Batteries. Acc. Chem. Res. 2019, 52 (8), 2290–2300. 10.1021/acs.accounts.9b00231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.