

Summary
Owing to spatial segregation of tumor subclones, solid tumor sampling using formalin-fixed, paraffin-embedded blocks is often inadequate to represent the genomic heterogeneity of solid tumors. We present an approach, representative sampling, to dissect and homogenize leftover residual surgical tissue prior to sequencing. We also detail optional tumor cell enrichment and DNA preparation. This method, applicable only to surgically removed tumors with leftover tissue, facilitates robust sampling to avoid missing or over-representing actionable variants.
For complete details on the use and execution of this protocol, please refer to Litchfield et al. (2020).
Subject areas: Cancer, Clinical Protocol, Flow Cytometry/Mass Cytometry, Genomics, Sequencing
Graphical abstract
Highlights
-
•
Leftover surgical tumor tissue from patients with cancer is collected for sampling
-
•
This tissue can be dissected and homogenized to create a representative tumor sample
-
•
Multiple tumor types are amenable to dissection and homogenization
-
•
NGS is more accurate and can be further improved by tumor enrichment
Owing to spatial segregation of tumor subclones, solid tumor sampling using formalin-fixed, paraffin-embedded blocks is often inadequate to represent the genomic heterogeneity of solid tumors. We present an approach, representative sampling, to dissect and homogenize leftover residual surgical tissue prior to sequencing. We also detail optional tumor cell enrichment and DNA preparation. This method, applicable only to surgically removed tumors with leftover tissue, facilitates robust sampling to avoid missing or over-representing actionable variants.
Before you begin
The protocol below describes steps to create Representative Samples of tumor tissue for sequencing and molecular studies. In preparation for dissection of fixed surgical samples, please take note of our detailed guidance for dissecting specific tumor types.
Preparation for dissection of fixed surgical samples
Timing: 1 day to 1 week
-
1.
In the weeks/days before carrying out the dissection step, identify potential cases that are amenable to homogenization (greater than an estimated 1 g of tumor tissue remaining after samples are taken for TNM staging).
-
2.
Follow all relevant guidelines for completion of the diagnosis (Amin et al., 2017), and check that the case has been formally signed off by the reporting Consultant Pathologist.
-
3.
As needed according to study protocols, allocate a patient tracking number and prepare any required documentation (see Figure 1A for an example).
-
4.On the day of dissection, prepare the work area for specimen dissection. See Figure 1 and Methods video S1 for an example work station.
-
a.Collect and label appropriately sized containers for tumor, tumor-adjacent, and distant normal tissues.
-
b.Pair empty containers with specimen buckets and paperwork in an area that is accessible to the workbench.
-
c.Ensure bench is equipped with dissection board, forceps, scalpel, ruler, tissue paper and a set of weighing scales (reading to an accuracy of 0.1 g).
-
d.Ensure there is a sufficient supply of 1× PBS in which to submerge the samples for 24 h following dissection.
-
a.
Figure 1.

Example of a work station for preparing representative samples
(A) Example of paperwork for recording dissection notes.
(B) Dissection area containing a scale, scalpels, ruler, cutting board, strainer, and paper towels.
(C) Trolley or cart for holding collection containers, other extra supplies and PBS.
(D) Work area for filling out paperwork and writing dissection notes.
-
5.
Consult the histopathology report, as needed, to ascertain the size of the tumor and areas that were initially sampled for embedding into paraffin blocks. This can be useful when trying to reconstruct tumors or when establishing the number of focal nodules when sampling pathological remnants of tissue.
CRITICAL: The Representative Sampling workflow is not currently standard clinical practice. As such, first check that appropriate consents and approvals are in place prior to initiating any study.
Note: It is helpful to have a cart or trolley on which to place prepared specimens, labeled containers, and accompanying paperwork ahead of dissection (Figure 1C).
Note: General dissection notes can be on hand, detailing how to define and separate tumor from tumor adjacent tissue and preferences for designated normal tissue.
Detailed guidance for specific tumor types
Tissue with multifocal tumor deposits (e.g., metastatic tumors, primary tumors with multiple separate nodules): Studies may include specimens of multiple related tumors from different areas in the body collected into different containers (e.g., from debulking surgery in gynecologic cancers). They may also include specimens with multiple metastatic nodules contained within a single specimen, such as multifocal breast carcinoma treated with wide local excision, or multifocal breast carcinoma treated with unilateral mastectomy. These cases are treated in the same way as tissues without multifocal deposits. It may be necessary to assign a number to each separate tumor nodule, and to collect both tumor and tumor-adjacent tissue for each (i.e., Tumor 1; Tumor-Adjacent 1; Tumor 2; Tumor-Adjacent 2 etc.). However, a single piece of distant normal, with distance measured from the closest tumor nodule only, is taken. The distance between the distant normal and the closest tumor nodule is recorded. Map each tumor designation and area of distant normal on the sketch diagram, if applicable.
Large tumor specimens (weighing > 1 kg): These samples are dissected as described in the protocol. However, due to manual handling concerns we limit each tumor sample to a maximum weight of 1 kg per bucket of PBS. Each segment (up to 1 kg) of the tumor is assigned as Tumor Section 1; Tumor Section 2; Tumor Section 3 etc. Although these are each weighed and separated in multiple containers for incubation in PBS for 24 h, the portions of the tumor will be mixed later during homogenization to form the representative sample. The tumor-adjacent and distant normal tissues are collected into a single bucket each, as usual.
Invasive ductal carcinoma (IDC) of the breast: These lesions are generally stellate in appearance with an irregular border. When selecting the tumor sample, we have focused on the macroscopic border of the lesion at the point that meets the surrounding tissue (adipose tissue / fibrous stroma). We are aware that some of the tumor striations may extent into the tumor -adjacent normal tissue sample; however, we are focusing on what is macroscopically attainable and achieving a tumor sample with minimal normal tissue content.
Invasive lobular carcinoma (ILC) of the breast: These lesions can present as macroscopically similar-to Invasive Ductal Carcinoma; in this case, follow the guidance for IDC of the breast. However, these lesions are more commonly irregular in shape and can grow in irregular patterns between stroma and normal breast adipose tissue. With these cases, we often use palpation to identify the contours of the tumor in trying to separate it from the tumor-adjacent tissue. This can be difficult in highly fibrotic samples or samples with a high ratio of stroma to adipose tissue. Macroscopic assessment is also required to aid in sampling these specimens. This can be difficult if inks have leached from the primary tumor assessment. If it is difficult to obtain a tumor sample with minimal normal tissue content, it is possible to enrich tumor cells ahead of sequencing applications using the optional tumor enrichment step described in this protocol.
Mucinous adenocarcinoma of the breast: These lesions are usually well defined with a clear border. Some of the gelatinous contents may separate from the specimen slices after sampling due to its fragility. It is therefore important to check the remaining formalin in the specimen bucket to ensure that any tumor tissue that has detached from the slices is retrieved for sampling. If the formalin is contaminated with residual ink it may be necessary to pour the formalin through a sieve to check for left over tumor tissue.
Sarcoma excisions: These samples are often relatively straightforward, with the majority of sarcomas being well circumscribed and easily distinguished from the background tissues. However, liposarcoma cases excised with surrounding adipose tissue are a notable exception. A primary difficulty or consideration is that the thin excision margins (sometimes just a thin fascial layer <1 mm in thickness) can influence the ability to attain adjacent normal and distant normal tissue (Figure 2). If this is not possible, the entire tumor is submitted without a normal adjacent or distant normal sample. Liposarcoma cases present the same issues as above, with the added difficulty of trying to identify whether the surgeon has removed any normal surrounding adipose tissue. This border between tumor and normal can be very difficult to identify macroscopically. The pathological report can be consulted to ascertain whether the tumor is present at the margins to aid in this decision. If in doubt, use judgment and experience to assess the peripheral border.
Figure 2.

Tumors with minimal resection margins
(A) Re-assembled retroperitoneal sarcoma tissue after routine pathological sampling.
(B) Tissue slices of the same tumor, arranged from lateral to medial. Note the minimal resection margins, which make it difficult to attain tumor-adjacent tissue samples for all aspects of the tumor.
Sarcoma retroperitoneal resections: These tumors often abut the surgical resection margins and it may be difficult to differentiate between tumor and peri-nephric adipose tissue in some samples where a nephrectomy has been included. The additional issue with these specimens is that the tumors can be very large, often weighing more than 1 kg, and sometimes up to 15–20 kg. With these larger specimens, it is often better to work in sections governed by the size of the dissection area. By working in smaller segments of 1–2 kg at a time it is possible to separate out the components and weigh them as individual segments, collating the segment weights of each component at the end. Tumor-adjacent and distant normal are usually taken from the surrounding organs (nephrectomy, hemi-colectomy) for these specimens.
Endometrial tumors: Some of these tumors can present difficulty in trying to determine the macroscopic peripheral border. In cases where the tumor was not macroscopically defined, we identified pieces of the uterine cavity and tried to remove the endometrial tissue from the underlying myometrium. The endometrial tissue is then submitted in its entirety as the tumor sample with the closest myometrium submitted as the tumor-adjacent tissue and the distant myometrium towards the serosa (usually at the fundus) or cervix taken as the distant normal. This has been our approach based on the difficulty of macroscopically separating the tumor from the normal tissue. Microscopic myometrial invasion may therefore be present in the tumor-adjacent sample. Some endometrial tumors will be macroscopically defined and therefore, easier to separate (Figure 3).
Figure 3.

Sketches showing variation in macroscopic appearance amongst cases of endometrial cancer
(A) Example of a macroscopically straightforward polypoid lesion lying within the uterine cavity.
(B) Macroscopic appearance of inflamed/irregular endometrial lining.
(C) Macroscopic appearance of infiltrative tumor passing into the myometrium.
(D) Macroscopic appearance of infiltrative tumor with border further complicated by presence of multiple fibroids. Note the position of the tumor, tumor-adjacent, and distant normal tissue samples taken for each case.
Tubo-ovarian tumors: These samples often present themselves as a large multi-cystic mass. The majority of these samples are composed of cyst wall epithelium with papillary projections entering the cystic cavities. In these cases, we commonly find striations of tumor along the lining of the cyst wall, so we have adopted the approach of submitting all of the cystic tissue as our tumor sample. We submit any directly adjacent ovary surrounding the cystic structure as the tumor-adjacent tissue component. Distant normal tissue consists of any remains of macroscopically normal fallopian tube present within the specimen.
Renal tumors: These samples are often comprised of well-defined tumors that are easily distinguishable from the normal background kidney. One possible difficulty with these lesions can be the ability to separate them from the surrounding tissue, as no separating layer (e.g., capsule or facial layer) exists between the tumor and tumor-adjacent tissue. It is therefore accepted that there will be small quantities of adjacent normal renal tissue contained within the tumor sample. In our experience, separate tumor nodules can also further complicate these specimens.
Melanoma (lymph nodes): These samples are usually sampled in their entirety with the remnant of the lymph node taken as the tumor sample and any peripheral adipose tissue taken as tumor-adjacent and distant normal tissue samples.
Lung carcinoma (adenocarcinoma, squamous cell carcinoma, and small cell carcinoma): These lesions are usually well defined with a clear border. These tumors can have significant necrosis leading to fragility of the tissue, so it is important to check the specimen container for any residual pieces of tumor. The macroscopic border of the lesion is the point that meets the surrounding tissue (lung parenchyma/adipose tissue/fibrous stroma, etc.) and its entirety is submitted for tumor sample (with minimal normal tissue content).
Adenocarcinoma of the colon: These tumors may either present as macroscopically well-defined lesions or may be infiltrative with an irregular, invasive pattern. If the tumor is well defined it is sampled similar to a lung carcinoma. However, if the tumor is infiltrative, palpation is used to identify the contours of the tumor as much as possible while trying to minimize the amount of normal tissue.
Metastatic liver nodules: These samples present the same issues as Renal Cell Carcinomas (See above).
Preparation for homogenization of dissected samples
-
6.Prepare the area to be used for homogenization.
-
a.Line the workbench with absorbent material.
-
b.Ensure blender base and scale are accessible.
-
a.
-
7.Weigh the tissue and determine which blender container is required.
-
a.Remove tissue sample from PBS and weigh on an electronic scale.
-
b.Select the appropriate sized blender container and place the tissues inside:
- i.150 g or less – use a 16 oz container.
- ii.150 g to 1 kg – use a 72 oz container.
- iii.1 kg or more – use two 72 oz containers or one 72 oz container and one 16 oz container; see optional “Process for blending and sub-sampling tumors larger than 1 kg”.
-
a.
-
8.Determine the volume of 1× PBS required during homogenization:
-
a.50 g or less: prepare to add up to 100 mL volume.
- i.e.g., to 1 g tissue, add 99 mL 1× PBS
- ii.e.g., to 50 g tissue, add 50 mL 1× PBS
-
b.50 g and more: prepare to add an equal volume of 1× PBS and tissue.
- i.e.g., to 700 g tissue, add 700 mL 1× PBS
-
a.
-
9.
Label appropriate sized storage tubes with unique patient sample identifier, typically 50 mL conical tubes.
Preparation for optional enrichment of tumor nuclei
-
10.
Adjust heat blocks or water baths to 80°C, 50°C, and 37°C.
-
11.
Prepare pepsin and proteinase K solutions, DAPI, Antibody staining wash buffer, Nuclei stabilization buffer.
Preparation for DNA extraction
-
12.
Adjust heat block to 56°C for proteinase K digestion
-
13.
Add ethanol to buffers from QIAamp FFPE kit.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-cytokeratin 8/18 antibody | Ventana Medical Systems | cat # 760-4344; RRID: AB_379877 |
| Goat anti-mouse antibody, Alexa Fluor 647 conjugate | Invitrogen | cat # A-21236; RRID:AB_2535805 |
| Chemicals, peptides, and recombinant protein | ||
| CC1 buffer | Ventana Medical Systems | cat # 950-124 |
| Antibody diluent buffer | Ventana Medical Systems | cat # 251-018 |
| Phosphate-buffered saline (for nuclei isolation) | Fisher Scientific | cat # 14190 |
| Phosphate-buffered saline tablets (for tissue processing) | Sigma | cat # P4417 |
| Neutral buffered formalin | Sigma | cat # HT501320 |
| Tween-20, 100% | Fisher Scientific | cat # AC233362500 |
| DAPI | Sigma | cat #D9542 |
| Proteinase K | VWR | cat # 0706 |
| Pepsin | Sigma | cat # P7012 |
| 5 M NaOH | Sigma | cat #S8263 |
| 1 M HCl | Sigma | cat # H9892 |
| NaCl | Sigma | cat # S9888 |
| Sodium azide 5% (w/v) in aqueous solution | VWR | cat # BDH7465-2 |
| Ethanol, molecular biology grade | Sigma | cat # E7023 |
| Other | ||
| 20-micron Cell strainer | pluriSelect | cat # 43-50020-03 |
| Connector Ring | pluriSelect | cat # 41-50000-03 |
| 30 mL Luer lock syringe | VWR | cat # 75801-380 |
| Round bottom tubes with 35-micron cell strainer cap | VWR | cat # 21008-948 |
| BD FACS Aria | Becton Dickinson | cat # 656700 |
| Ninja Professional Countertop Blender with 1100-Watt Base, 72 oz and 16 oz blender containers | Amazon | part # BL660 |
| 2100 Bioanalyzer Instrument | Agilent | cat # G2939BA |
| Qubit 4 fluorimeter | Thermo Fisher Scientific | cat # Q33238 |
| Pipette controller, Pipet-Aid XP | VWR | cat # DRUM4-000-201-E |
| Magnetic stirrer | VWR | cat# 444-0609 |
| Cut-up board | CellPath | cat# CGB-0302-51A |
| Disposable Scalpel | Swann-Morton | cat # 0509 |
| 10 inch (250 mm) Lung Knife | CellPath | cat # CAA-1001-01A |
| Ruler | VWR | cat # 500024-324 |
| Tree Electronic Precision Balance HRB-E 20001 | VWR | cat # 611-2373 |
| Specimen containers | CellPath | Tissuestor Tubs -various sizes |
| Stainless steel strainer | Amazon | https://www.amazon.com/Stainless-Micro-Perforated-Strainer-2-5-Quart-4-5-Quart/dp/B01G1CDRIC/ref=sr_1_6?dchild=1&keywords=stainless+steel+strainer&qid=1610475428&sr=8-6 |
| Conical tubes, 50 mL | VWR | cat # 89039-660 |
| Biological samples | ||
| Leftover surgical tissue | N/A | N/A |
| Critical commercial assays | ||
| QIAamp DNA FFPE Tissue Kit | QIAGEN | cat# 56404 |
| Agilent DNA 12000 Kit | Agilent | cat # 5067-1508 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | cat# Q32851 |
Materials and equipment
Antibody staining wash buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| BSA | 0.1% | 0.5 g |
| Tween 20 | 0.1% | 0.5 mL |
| Phosphate buffered saline | 1× | 500 mL |
Nuclei stabilization buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| BSA | 0.1% | 0.1 g |
| Tween 20 | 0.1% | 0.1 mL |
| Spermine Tetrahydrochloride | 1.5 mM | 1 mL of a 150 mM frozen stock solution |
| Phosphate buffered saline | 1× | 100 mL |
| Reagents and solutions | Preparation |
|---|---|
| 1 mg/mL proteinase K | 100 mg proteinase K, add 10 mL 1× phosphate buffered saline (PBS) for 10× stock solution. Freeze and store at −20°C in 1 mL aliquots. Thaw 1 mL aliquot on ice and add 9 mL 1× PBS for working concentration of 1 mg/mL proteinase K. Use immediately and discard any un-used working solution. |
| 150 mM NaCl, pH 1.5 | Dilute 1.5 mL of 5 M NaCl into 48.5 mL double distilled H2O (ddH2O), and adjust the pH to 1.5 using HCl. Store at 20°C–22°C. |
| 5 mg/mL pepsin | 250 mg pepsin, add 10 mL of ddH2O for 5× stock solution. Freeze and store at −20°C in 1 mL aliquots. Thaw 1 mL aliquot on ice and add 4 mL 150 mM NaCl, pH 1.5, for the working concentration of 5 mg/mL pepsin. Use immediately and discard any un-used working solution. |
| Antibody staining wash buffer | Add 0.5 g BSA and 0.5 mL Tween 20–500 mL 1× PBS. Filter through a 0.2 μm filter and store at 4°C. |
| Nuclei stabilization buffer | 1 g Spermine Tetrahydrochloride in 19.2 mL ddH2O for a 150 mM stock solution. Freeze and store at −20°C in 1 mL aliquots. Dilute 1 mL 150 mM Spermine Tetrahydrochloride into 100 mL Antibody staining wash buffer to create Nuclei stabilization buffer. Store at 4°C. |
| DAPI | 10 mg DAPI in 9.5 mL DMSO as a 3 mM stock solution. Freeze and store at −20°C in 1 mL aliquots. Dilute 1 μL stock solution into 1 mL antibody diluent for a 3 μM working solution. Use immediately and discard any un-used working solution. |
Alternatives: Reagents should be molecular biology-grade. Alternative DNA prep kits suitable for FFPE tissue may also be used, with similar considerations as outlined in this protocol. Alternative blenders can be used for homogenization; recommended output is at least 1000 watts.
Step-by-step method details
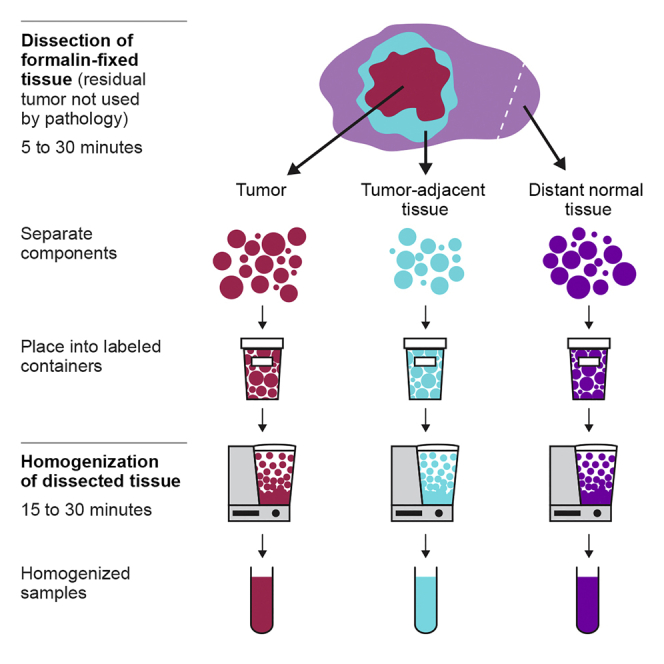
Dissection of formalin-fixed tissue
Leftover formalin fixed tissue from the surgical resection of a solid tumor is dissected into three portions – tumor, tumor-adjacent, and distant normal tissue.
This describes the major steps of tissue dissection (Figure 4 and Methods video S1, Residual tissue dissection). Please see above for tissue-specific information.
-
1.
For a single patient sample, place the specimen bucket and empty containers for collection of tumor, tumor-adjacent, and distant normal tissue on the dissection board.
-
2.
Collect all material from the specimen bucket and place on the dissection board, blotting any excess formaldehyde. Use a sieve to collect any dispersed remnant tissue.
-
3.
Work systematically to identify tumor, tumor-adjacent, and distant normal tissues. Using the scalpel, separate these areas based on the macroscopic assessment of the peripheral tumor bordering the surrounding tissue.
Note: See troubleshooting 1.
-
4.Arrange the tissue into 3 piles on the dissection board. Each pile should be comprised as follows:
-
a.Tumor: Sample the tumor in its entirety to include all necrotic, mucinous, hemorrhagic, and cystic areas. Amount of tissue will vary from 1 g up to 20 kg, depending on the tumor type.
-
b.Tumor-adjacent: The tumor-adjacent tissue should comprise of predominantly normal tissue immediately adjacent to the macroscopic tumor perimeter, usually 5–10 mm in width for most specimens (depending upon tumor size and thickness of surgical resection margins). Depending on the surgical procedure performed, the tumor may lie asymmetrically within the specimen. If the tumor lies very close to one or more of the surgical margins, it is acceptable to take what tissue is available (i.e., some tumor- adjacent tissue may be very thin, capsular or absent at the surgical resection margins. Amount of tumor-adjacent tissue will vary, but may often be in the range of 5–10 g.
-
c.Distant normal: Distant normal should ideally be a piece of normal tissue from the same organ from which the tumor has originated (e.g., Normal kidney from furthest pole in cases of renal carcinoma). If this is not possible, then a piece of macroscopically normal tissue from an adjacent organ contained within the specimen is taken (e.g., Adrenal Gland in cases of renal carcinoma), and the tissue of origin noted on the dissection paperwork. If possible, avoid skin, colonic mucosa and lipomatous tissues as distant normal (See detailed guidance notes in Preparation for Dissection of Fixed Tissue). Collect 5–10 g of tissue minimum, if possible; however, it is of higher importance to ensure that the distant normal does not contain contaminating tumor tissue.
-
a.
-
5.
Once separated, record the weight of each sample and place in the empty buckets labeled appropriately as tumor, tumor-adjacent, or distant normal tissue.
-
6.Add 1× PBS to each of the sample buckets.
-
a.Completely submerge the tissue in PBS. Currently, there is no recommended size of tissue pieces for submersion in PBS; it is sufficient to simply collect the tissues that have previously been segmented prior to formalin fixation and further dissected as described above.
-
b.Ensure the lid is fitted correctly and remove from the dissection area.
-
c.Soak tissue in PBS for at least 24 h to dissipate the formalin.
-
a.
-
7.
Finalize any documentation prior to moving to the next case.
Pause point: The dissected, formalin-fixed samples can be stored for 24–48 h at 20°C–22°C in PBS prior to homogenization.
Note: Take utmost care to ensure correct labeling of sample buckets.
Note: It may be helpful to draw a rough sketch demonstrating the macroscopic relationship between the tumor, tumor-adjacent, and distant normal tissues, and keep this with the documentation for each specific case (Figure 5).
Note: Some tumors will have a well-circumscribed border or encapsulation making it easier to separate tumor from tumor-adjacent tissue (Figure 6). Others with more irregular borders, such as stellate lesions, will require personal judgment. The fundamental principle is to use expert judgement to separate the leftover surgical tissue macroscopically into the three key components described above (tumor, tumor-adjacent, and distant normal tissues). Although the goal is for each component to be as pure as possible, it is acceptable for each of these to contain small amounts of the other components. For example, small quantities of macroscopically normal tissue may be included in the tumor tissue sample in these cases. Likewise, some striations of tumor on the periphery may be included in the tumor-adjacent tissue sample if they are difficult to macroscopically identify. The most critical aspect is for distant normal tissue to avoid contamination with tumor tissue.
Note: When targeting distant normal tissue, try to target stromal areas that will have a high degree of cellularity. If this is not possible, other sources of germline DNA, such as PBMCs, are acceptable.
Note: When selecting an area of the specimen from which to sample distant normal tissue, try to focus on areas of high or dense cellularity with the exception of colonic mucosa and skin. Lipomatous areas should be avoided (if possible) as these often yield low quantities of genetic material. Colonic mucosa and skin possess high quantities of strong elastic fibers. These fibers tend to wrap themselves around the blender blades and create a physical barrier that prevents the blade from producing a homogenate. If skin or colonic mucosa is the only tissue available for distant normal sampling, first dissect the tissue into small segments approximately 1 × 1 cm to reduce the length of the fibers and their ability to wrap around the blades.
Note: Take care to examine the dissected tissue for any material that may not be suitable for blending. These materials include, but are not limited to: suture materials, staples, heavily calcified areas of the tumor (more often encountered in tumors from gynecologic cancers, breast cancers, and sarcomas).
Note: It is accepted that institutional practices in both surgery and the pathological examination of surgical tissue will vary according to local protocols and requirements. This study has primarily been focused on specimens received and examined at the Royal Marsden NHS Foundation Trust (London, UK). Local protocols relating to surgical orientation of specimens and subsequent differential marking of resection margins makes it easier to re-construct specimens following histological sampling. Other institutions may not implement these practices, and thus reconstructing left over surgical tissue may prove more challenging. Please see the previous notes describing our experience with the dissection of specific tumor tissue types.
Figure 4.

Dissection workflow for left-over surgical tissue
(A) Lay out collection containers and retrieve surgical specimen from formalin.
(B) Examine the tissue to plan separation into required components.
(C) Using a scalpel, separate tumor, tumor-adjacent, and distant normal components.
(D) Place each component in a separate pile.
(E) Place each tissue pile into a separate labeled container and record the tissue weight.
(F) Submerge tissues in PBS to further remove formalin prior to homogenization.
Figure 5.

Colorectal tumor treated by Abdominal Perineal Resection
(A) Single slice through the circumference of the tumor, showing tumor tissue situated between normal prostate and bowel.
(B) Dissection data captured prior to tissue homogenization, including the location of the tumor, tumor-adjacent, and distant normal tissue samples, and the weight of each collected tissue sample. Note: the image was taken following typical pathological sampling.
Figure 6.

Examples of tumors with different borders
(A) A recurrent invasive breast carcinoma showing irregular infiltrative edges extending deep into adipose tissue.
(b) A well-circumscribed liposarcoma with a smooth capsule surrounding the tumor.
Homogenization of dissected tissue
Dissected tumor, tumor-adjacent, and distant normal tissues are homogenized into three individual well-mixed representative samples (Methods video S2, Residual tissue homogenization).
-
8.
Add 1× PBS to tissue in appropriate sized blender and close container.
-
9.
Place blender container on blender base and secure properly on the base.
-
10.
Blend for 30 s on the highest setting of a 1000 watt output blender (on the suggested BL660 Ninja blender, use setting 3).
Note: See troubleshooting 2 and 3.
-
11.Transfer sample to labeled 50 mL conical storage tube.
-
a.Pipette using a 10–50 mL open tip pipette, or pour if too dilute to pipette.
-
b.If sample in excess of desired amount is generated, dispose of excess sample in appropriate waste receptacle.
-
c.Add 1 mL of 5% sodium azide to each 50 mL storage tube to reach a final concentration of 0.1% sodium azide.
-
a.
-
12.
Each blender container is single use. Dispose of blender containers in a sealed clinical waste bin, or other appropriate waste bin, in accordance with hospital policies for formalin fixed waste tissue.
Optional: Process for blending, and proportionally sub-sampling, tumors larger than 1 kg in order to preserve the relative abundance of subclonal genomic alterations. Separate the sample into sections weighing 1 kg or smaller. Process the first section at a 1:1 ratio of tissue (g) to 1× PBS using the 72 oz blender container. Take an aliquot of blended tissue equal to 1/10th of the starting weight of the tissue section. Add the aliquot of blended tissue to a second 16 oz or 72 oz mixing blender container, as appropriate. The 16 oz blender container will hold a maximum of 400 mL. Pour off the remaining blended tissue from the first blender into a waste container. To the first blender container, add the second tissue section and an equal volume of PBS. Blend and sub-sample the second tissue section as before, adding the aliquot of blended tissue to the same mixing blender container. Blend and sub-sample any additional tissue sections, each time adding a 1/10 aliquot to the same mixing blender container. Once the last tissue section has been sub-sampled, place the second mixing blender containing all of the aliquots on the blender base. Blend for 30 s to homogenize the aliquots and create a final homogenized representative sample. Transfer the final homogenized representative sample to storage tubes for downstream applications or tissue archival (Figure 7).
Figure 7.

Examples of representative samples from different tumors
Representative Samples consist of dissected leftover formalin fixed tissues that have been homogenized to create a well-mixed liquid sample that is capable of being pipetted or poured. The sample color varies due to surgical dyes, varying cell type composition, or other tissue features.
Note: Homogenization times are generally no longer than 30 s, but in some instances the blending time will be determined empirically. The solid tissue fragments must be sufficiently blended to create a liquid sample capable of being aspirated by a 10 mL pipette or poured into a storage tube (Figure 7). The suggested blender model will only function if the lid of the blender is properly closed and the blender base is properly situated.
Note: In our experience, blender bases will need to be replaced periodically in clinical practice (about every 3–6 months).
Optional enrichment of tumor nuclei
Enrichment of tumor nuclei improves detection of low frequency variants. This step can be skipped, if desired (e.g., if tumor cellularity is assumed to be high, or if the workflow must be kept simple).
-
13.Collect tumor tissue.
-
a.Transfer 1 mL of homogenized tumor tissue from the 50 mL conical tube into a 15 mL conical tube using a 1 mL pipette tip. It will be necessary to widen the opening in the pipette tip by cutting off about 0.25 cm from the narrow end using scissors or a razor blade.
-
b.Centrifuge at 1000 × g for 2 min.
-
c.Discard liquid, and keep tissue. Weigh tissue and record weight for yield calculation. Add more tissue as needed.
-
a.
-
14.De-crosslink tissue.
-
a.Resuspend tissue in CC1 buffer, 5:1 volume to mass.
-
b.Heat at 80°C for 30 min.
-
a.
-
15.Digest tissue with Proteinase K.
-
a.Remove from heat block, centrifuge at 500 × g for 3 min and discard supernatant.
-
b.Resuspend digested tissue in 10 mL 1× PBS to wash, centrifuge at 500 × g for 3 min and remove PBS.
-
c.Resuspend in 1 mg/mL proteinase K solution (1:1 volume to mass).
-
d.Incubate at 50°C for 10 min.
-
e.While tissue is incubating, prepare 5 mg/mL pepsin in 150 mM NaCl, pH 1.5.
-
f.Centrifuge at 500 × g for 3 min to collect partially digested tissues; remove supernatant.
-
a.
-
16.Digest tissue and quench digestion with Pepsin.
-
a.Resuspend tissue in 5 mg/mL pepsin, pH 1.5. Adjust pH to 1.5–2, if necessary, using 1–5 μL 1M HCl per 1 mL digestion; pH strips can be used to estimate the pH.
-
b.Incubate 30 min at 37°C. Gently agitate the tube every 10 min if tissue settles during the incubation.
-
c.Adjust pH to above 8 with 5 M NaOH. For a 1 mL digestion, this typically requires 6 μL 5 M NaOH.
-
d.Centrifuge at 500 × g for 3 min to collect partially digested tissues; remove supernatant.
-
a.
Note: See troubleshooting 4
-
17.Collect nuclei and determine yield.
-
a.Exchange into 1 mL wash buffer.
-
b.Filter through a 20 μm pluriselect filter, washing through the filter with 5 mL wash buffer, to collect nuclei. Use a 30 mL syringe to draw the liquid through the filter using a pluriselect filter ring.Note: See troubleshooting 5
-
c.Collect nuclei by centrifugation at 300 × g for 3 min
-
d.Count nuclei on coulter counter, hemacytometer, or other quantification system to determine yield. Typical yields are 50–300 × 106 nuclei per gram of tissue.
-
a.
-
18.FACS sort to enrich tumor nuclei.
-
a.Measure out desired number of nuclei (suggested 10–20 × 106 nuclei per tube) into a fresh microcentrifuge tube, including tubes for both negative staining control and stained nuclei.
-
b.Centrifuge at 300 × g for 3 min to collect nuclei. Exchange into 300 μL of antibody diluent as a blocking buffer for 10 min at 20°C.
-
c.Centrifuge at 300 × g for 3 min to collect nuclei. Remove blocking buffer and exchange into 100 μL of mouse anti-cytokeratin 8/18 antibody for 1 h at 4°C (or 100 μL of antibody diluent for negative control).
-
d.Centrifuge at 300 × g for 3 min to collect nuclei. Remove antibody solution and resuspend cells in 500 μL of antibody staining wash buffer. Repeat this step twice to remove any residual antibody.
-
e.Centrifuge at 300 × g for 3 min to collect nuclei. Remove antibody staining wash buffer and exchange both positive and negative control cells into 100 μL of goat anti-mouse antibody conjugated to Alexa Fluor 647 (2 μg/mL) and DAPI (3 μM) in antibody diluent for 30 min at 4°C.
-
f.Centrifuge at 300 × g for 3 min to collect nuclei. Remove antibody solution and resuspend nuclei in 500 μL of antibody wash buffer. Repeat this step twice to remove any residual antibody.
-
g.Pipette through a 40 μm filter cap tube prior to analysis on a BD FACS Aria or similar flow cytometer.
-
h.Enrich tumor nuclei by gating on the cytokeratin positive population with high side scatter (Figure 8). Use the DAPI fluorescence for doublet discrimination, and use the negative control sample to help determine the position of the gating for the CK-positive stain.
-
i.Collect 0.3–1 × 106 nuclei in order to prepare around 0.3–1 μg of DNA for sequencing library prep.
-
j.Centrifuge at 1000 × g for 10 min to collect nuclei. Remove all but ∼50–100 μL buffer. Transfer to a 1 mL tube and freeze, if not used immediately for preparation of DNA.
-
a.
Note: This enrichment strategy is only useful for carcinomas, which retain cytokeratin from their epithelial cell of origin. Other nuclear markers may be used for tumors of mesenchymal origin.
Figure 8.

Flow cytometry gating strategy for enriched tumor nuclei
(A) Control un-stained nuclei.
(B) CK staining intensity (AF647) vs. side scatter (SSC) for nuclei stained with anti-CK 8/18 antibody, visualized with Alexa Fluor 647. Gates were established for CK-positive (purple) and CK-negative, low-SSC (blue) nuclei. (c) DAPI staining intensity for the CK-negative (blue) and –positive (purple) populations defined by the gates in (b).
Preparation of DNA from representative samples or enriched tumor nuclei
Extraction of DNA ahead of standard DNA sequencing library prep.
-
19.Collect tissue or nuclei by centrifugationNote: See troubleshooting 6
-
a.Tissue collection from homogenate samples: using a pipette tip with the end cut to widen the tip, pipette 100 μL of tissue homogenate into a 1.5 mL microcentrifuge tube. Centrifuge at 1000 × g to collect the tissue, and discard the supernatant layer from the top of the sample. If the collected tissue is less than 50 μL, measure out and collect an additional aliquot of homogenate until the collected tissue measures in the range of 50–100 μL.
-
b.Nuclei collection from tumor-enriched samples: see above optional enrichment of tumor nuclei.
-
a.
-
20.
Digest tissue or nuclei with proteinase K for 24 h at 56°C. Buffers from a Qiagen QIAamp FFPE kit can be used for this step. Add 180 μL of buffer ATL and 20 μL of proteinase K for DNA purification from nuclei. Add 360 μL of buffer ATL and 40 μL of proteinase K for DNA purification from homogenate; following digestion, split the digested material into two columns and process in parallel.
-
21.
Incubate digested tissue at 90°C for 1 h to inactivate proteinase K.
-
22.
Allow sample to cool to 20°C–22°C. Add 2 μL of RNase A (100 mg/mL) for 2 min at 20°C–22°C.
-
23.
Add 200 μL buffer AL to each sample; mix by vortexing. Add 200 μL ethanol and mix again by vortexing.
-
24.
Carefully transfer the lysate to the center of a QIAamp minelute column in a 2 mL collection tube. From this step, follow the QIAamp manufacturer’s protocol to bind and wash DNA on the column.
-
25.
Check the DNA concentration and fragment size distribution using standard Qubit and Bioanalyzer methods.
-
26.
Proceed to sequencing library preparation for the selected method of analysis and platform, or store DNA at −20 to −80°C for use at a later date.
Note: It is critical to digest both tissue and nuclei with proteinase K for 24 h. Using shorter digest times may appear to completely digest the tissue or nuclei, but it will significantly decrease the genomic DNA yield upon completion of the protocol.
Note: In order to extract the maximal amount of DNA from tumor-enriched nuclei, collect a second elution from the column, as optionally described in the QIAamp protocol.
Expected outcomes
A successful preparation of representative samples should yield three homogenized tissue samples: tumor, tumor-adjacent, and distant normal tissues. These samples should be sufficiently homogenized to be transferrable with a blunt-ended pipette, and the homogenates may be either dilute or thick, depending on the ratio of tissue to PBS (Figure 7).
Preparation of DNA from a representative sample should yield DNA comparable to de-paraffinized FFPE tissue (Litchfield et al., 2020).
Clinical sequencing from tissue prepared by Representative Sampling will be expected to represent an accurate relative variant allele frequency for all variants in the sample. When compared to sequencing results from a traditional FFPE sample from the same tumor, it is possible that the variant allele frequency for the homogenized sample is lower than that of the traditional FFPE sample. This is due to an over-representation of the subclonal variants in the blocks relative to their true frequency in the tumor, i.e., “clonal illusion”. Conversely, it is also possible that we do not find variants in the FFPE sample that we can clearly identify in the Representative Sample because, due to spatial segregation of tumor subclones, they were not present in the region sampled for the FFPE block (Litchfield et al., 2020).
It is possible that the sensitivity of the representative sample is lower than that of an FFPE sample due to a higher percentage of contaminating normal cells. We have included an optional step between homogenization of the tissue and preparation of the DNA sequencing libraries to enrich tumor nuclei using flow cytometry. We have demonstrated that this step improves the number of variant reads detected by the representative sample because the contaminating normal cells are removed (Litchfield et al., 2020). In addition, it is possible that low frequency variants that were over-represented and detected in an FFPE block fall under the threshold of detection when diluted to a more accurate frequency in a Representative Sample; these low frequency variants may require higher depth of sequencing for detection (Litchfield et al., 2020).
As described herein, the Representative Sampling methods can be applied to both primary and metastatic tumor deposits; indeed, these methods can likely be applied to any tissue sample that is removed by surgical resection. Metastases are generally anticipated to have lower clonal diversity than primary tumors, particularly if sampled following drug treatment; however, metastatic deposits may also contain multiple subclonal drivers (Hu et al., 2020). The clinical utility of improved sampling across tumor regions, and longitudinally throughout the patient journey, remains an active area of study (Swanton, 2020).
Limitations
This protocol applies only to tumors having greater than 1 gram of tissue remaining after the process of taking traditional samples for FFPE blocks. In some cases, an equal or greater amount of tissue will be embedded compared to the tissue left over, leaving a relatively small amount of tissue behind for further sampling; for these tumors, the tissue having the highest tumor content is often selected for FFPE blocks. In these cases, homogenized samples taken from the remainder of the tumor would likely be less representative of the entire tumor. As such, the sampling of some small tumors will not be improved by this methodology. Additionally, tumors for which standard practice is to embed the entire tumor (e.g., primary cutaneous melanoma, prostate carcinoma) will not be amenable to Representative Sampling.
This protocol describes tissues we have encountered through a clinical feasibility study of Representative Sampling at the Royal Marsden NHS Foundation Trust (NCT03832062), or through tissue samples we have purchased from external vendors. Figure 9 shows the numbers of different tumor types we have processed to Representative Samples for full transparency.
Figure 9.

Tumor types, by tissue of origin, sampled by representative sampling
Samples were created using the workflow described herein. Protocol limitations and troubleshooting are based on experiences with these specific samples.
Despite the detailed nature of this protocol, it is likely that additional specialist training in anatomical pathology is required to confidently dissect the leftover tissue into the three components described in this protocol. In our study, the dissection was undertaken by advanced practitioners in anatomical dissection.
Due to the remnant nature of some of the pathological tissue post sampling, it may not always be possible to match small tumor fragments to larger fragments in cases where there are multiple foci. For these cases, the smaller tumor fragments may be discarded, and the decisions noted on the study paperwork. Alternative strategies may be devised in the future to attribute these tissue fragments to the appropriate tumor foci.
Some surgical procedures remove tumor with only a thin fascial margin. These tumor samples are rarely sampled in their entirety for TNM staging and diagnosis. However, the limitation here is the lack of a distant normal tissue sample. Other sources of normal genomic DNA may substitute in these cases, e.g., DNA from PBMCs.
It is possible that the optional step of enriching tumor nuclei by FACS may bias the representation of the sample by omitting tumor populations that do not retain expression of cytokeratin. Virtually all of the variants from the bulk representative sample should be present in the FACS-enriched tumor sample, but at a higher frequency. The FACS enriched sample should contain additional low-frequency variants that were not discovered in the bulk representative sample. If key variants are lost upon sorting a specific sample, then the enrichment strategy should not be used for this sample. Over time, we anticipate a better understanding of which disease states benefit most from the optional tumor enrichment step.
Lastly, some genetically homogeneous tumors may have the same sequencing results from an FFPE block and a representative tumor sample. However, it is not currently possible to detect the level of heterogeneity from a macroscopic or microscopic evaluation of the tumor prior to sequencing.
Troubleshooting
Problem 1
Before surgical excision specimens are dissected for pathological analysis, key surgical resection margins are often preserved using tissue marking dyes. These dyes are applied to the surgical resection margin and either air dried or fixed using a mildly acidic solution. This prevents leaching of the dyes during slicing, and provides a clean cut surface for inspection. However, when the sampled specimen is placed back into the container, the dyes often leach out into the formalin, particularly in lipomatous specimens. The amount of leaching often corresponds to the amount of ink applied and the time spent in formalin. When the ink leaches onto the cut surface, it obscures clear macroscopic identification of the residual tumor, particularly in areas of fibrosis or dense background stroma. This is commonly a problem for breast specimens, which are often inked on all planes of excision (dissection of formalin fixed tissue, step 3).
Potential solution
All residual tissue can be placed in a stainless steel strainer and rinsed briefly under running water. The tissue should then be blotted to try to remove as much of the excess dye as possible. This may remove some on the leached dye. If the macroscopic residual tumor is still not easily identified following this process, then a combination of slice palpation and further slicing with a clean knife blade, halving the thickness of the slices, can be employed to provide a clean cut surface from which to identify the tumor borders.
Problem 2
Some tissues may have fatty areas, and it can be difficult to get a fully blended liquid homogenate or create soapy mixtures. (homogenization of dissected tissue, step 10).
Potential solution
Blending 2–3 times in 30 s intervals will help break down tissues having a high amount of fat.
Problem 3
Certain tissue types with high elasticity (skin, colonic mucosa, etc.) can wrap around the blender blade and prevent efficient blending (homogenization of dissected tissue, step 10).
Potential solution
Cut tissue into cubes to break up long filaments before blending. The shorter filaments will be less efficient at coating the blender blade, allowing the blender blades to break up the tissue. However, these tissues will always suffer from inefficient blending and should be avoided when possible.
Problem 4
Since the preparation of tumor nuclei involves the use of commercially available proteases, it is possible that lot-to-lot variability in the activity of the proteases results in more robust digestion than expected. This could lead to the partial loss of cytokeratin positivity during preparation of nuclei for tumor cell enrichment, because the proteases could destroy the antigen recognized by the anti-cytokeratin antibody (optional enrichment of tumor nuclei, steps 15 and 16).
Potential solution
Consider keeping a reference representative sample that is used to calibrate the process of preparing tumor nuclei. This can be a tissue sample that is not of tumor origin, such as tonsil or another normal tissue, and it can also be a mixture of tissues. New lots of proteinase K or pepsin can be used to prepare nuclei from the reference sample, and the percentages of cytokeratin positive nuclei should be consistent with nuclei from the reference sample that were prepared using proteases of previous lots.
Problem 5
While filtering the tumor nuclei away from the remaining partially digested tissue, it is possible that the pluriselect filter can become clogged (optional enrichment of tumor nuclei, step 17).
Potential solution
If the pluriselect filter becomes clogged with undigested tissue while filtering tumor nuclei, gently use a pipette tip to dislodge the tissue that is clogging the filter, and continue to draw the liquid containing tumor nuclei through the filter with the 30 mL syringe and filter ring. For some samples, it may be necessary to dislodge undigested tissue from the filter several times while drawing the tumor nuclei through the filter.
Problem 6
Breast and other tissue samples containing high amounts of fat may be difficult to collect by centrifugation prior to the Proteinase K digestion for nuclei enrichment or DNA preparation. This is because the fat may separate and rest on top of the aqueous layer, making it difficult to ascertain when supernatant removal is complete (preparation of DNA from representative samples or enriched tumor nuclei, step 19).
Potential solution
Prior to removal of the supernatant containing the buffer used to homogenize the sample, it may be helpful to first remove the fatty layer that rests on top of the aqueous supernatant using 1 mL pipette tip that has been cut about 0.25 cm from the narrow end using scissors or a razor blade to widen the opening.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nelson Alexander, nelson.alexander@roche.com.
Materials availability
This protocol does not require unique reagents.
Data and code availability
This protocol does not require unique data or code.
Acknowledgments
We acknowledge Nirali Patel, Lee Gumble, Lyra Del Rosario, Nahid Shaikh, Dilruba Kabir, Kim Edmonds, Tim Slattery, Yasir Khan, Benjamin Shum, Isla Leslie, Lewis Au, James Larkin, Scott Shepherd, and all others who have contributed to the Homogenisation of leftover surgical tissue across multiple cancer types: A feasibility study (HoLST-F) (NCT03832062), as well as funding from the Royal Marsden Renal and Skin Unit Research Fund, which enabled the authors of this study to develop the methods detailed in this protocol. We acknowledge Alan Shimato for preparation of the graphical abstract and Eric Walk, Eslie Dennis, Patrick Brunhoeber, Fiona Byrne, Husayn Pallikonda, and Charlotte Spencer for review of the manuscript. S.T. is funded by Cancer Research UK (grant reference number A29911); the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC10988), the UK Medical Research Council (FC10988), and the Wellcome Trust (FC10988); the National Institute for Health Research (NIHR) Biomedical Research Centre at the Royal Marsden Hospital and Institute of Cancer Research (grant reference number A109); the Royal Marsden Cancer Charity; the Rosetrees Trust (grant reference number A2204); Ventana Medical Systems Inc (grant reference numbers 10467 and 10530); the National Institutes of Health (U01 CA247439);, and Melanoma Research Alliance (Award Ref no 686061).
Author contributions
L.L.G. developed methods for homogenization and optional tumor enrichment, was responsible for project administration to obtain tissue samples that were used to develop the methods, and wrote and revised the manuscript. A.G. developed methods for dissection of all tissue types, captured video footage, and wrote and revised the manuscript. L.S. was responsible for project administration to obtain the tissue samples that were used to develop the methods. S.S., S.M.H., and and A.B. developed methods for homogenization and DNA purification. V.P. and C.J. contributed to the development of dissection methods. S.A. validated methods for homogenization and contributed to project administration. Z.T. was responsible for analysis of case data for tumor categorization. G.N.-S. was responsible for project supervision. N.R.A. was responsible for conceptualization of methods, securing funding, project supervision, and editing the manuscript. S.T. was responsible for project supervision, securing funding, and editing the manuscript. All authors reviewed the manuscript.
Declaration of interests
L.L.G., S.S., S.M.H., C.J., A.B., and N.R.A. are employees of Roche Diagnostics. L.L.G., S.S., and N.R.A. are shareholders of Roche. Funding was provided by Roche. Roche has submitted patent applications for methods, workflows, and applications downstream of representative sampling. S.T. has received speaking fees from Roche, AstraZeneca, Novartis, and Ipsen. S.T. has the following patents filed: Indel mutations as a therapeutic target and predictive biomarker PCTGB2018/051892 and PCTGB2018/051893 and Clear Cell Renal Cell Carcinoma Biomarkers P113326GB.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100624.
Contributor Information
Lisa L. Gallegos, Email: lisa.gallegos@roche.com.
Nelson R. Alexander, Email: nelson.alexander@roche.com.
Samra Turajlic, Email: samra.turajlic@crick.ac.uk.
References
- Amin M.B., Edge S.B., American Joint Committee on Cancer . Springer; 2017. AJCC cancer staging manual. [Google Scholar]
- Hu Z., Li Z., Ma Z., Curtis C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat Genet. 2020;52:701–708. doi: 10.1038/s41588-020-0628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litchfield K., Stanislaw S., Spain L., Gallegos L.L., Rowan A., Schnidrig D., Rosenbaum H., Harle A., Au L., Hill S.M. Representative sequencing: unbiased sampling of solid tumor tissue. Cell Rep. 2020;31:107550. doi: 10.1016/j.celrep.2020.107550. [DOI] [PubMed] [Google Scholar]
- Swanton C. Take lessons from cancer evolution to the clinic. Nature. 2020;581:382–383. doi: 10.1038/d41586-020-01347-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This protocol does not require unique data or code.