

Summary
Expansion microscopy is a sample preparation technique in which fixed and immunostained cells or tissues are embedded in a cross-linked network of swellable polyelectrolyte hydrogel that expands isotropically upon addition of deionized water. We utilize the X10 method for tenfold expansion of U2OS cells with concurrent DNA staining. A custom 3D-printed gel cutter and chambered slides minimize gel drift, facilitating analysis of the components of nuclear structures at nanoscale resolution by conventional microscopy or Airyscan confocal imaging.
For complete information on the generation and use of this protocol, please refer to Do et al. (2020).
Subject areas: Cell Biology, Microscopy
Graphical abstract
Highlights
-
•
Tenfold expansion of fixed and stained cultured cells with concurrent DNA staining
-
•
Custom 3D-printed gel cutter to minimize drift in chambered slide during imaging
-
•
Airyscan and Imaris-based imaging and volume rendering of nuclear structures
-
•
Approach to calculate expansion factor using Fiji open-source software
Expansion microscopy is a sample preparation technique in which fixed and immunostained cells or tissues are embedded in a cross-linked network of swellable polyelectrolyte hydrogel that expands isotropically upon addition of deionized water. We utilize the X10 method for tenfold expansion of U2OS cells with concurrent DNA staining. A custom 3D-printed gel cutter and chambered slides minimize gel drift, facilitating analysis of the components of nuclear structures at nanoscale resolution by conventional microscopy or Airyscan confocal imaging.
Before you begin
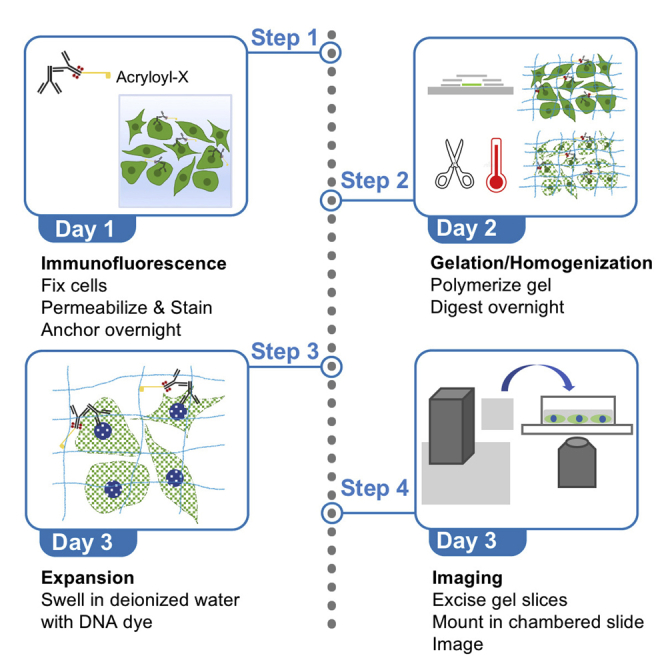
This protocol provides a detailed workflow for performing expansion microscopy (ExM) on cultured human cells. The original technique, which turned the concept of super-resolution imaging on its head by simply “making the specimen bigger”, was developed by the Boyden group (Chen et al., 2015). Further optimization by their group and others over the past several years has increased its applicability and also the original 4.5× expansion factor (Wassie et al., 2019). The protocol described here utilizes the X10 workflow, developed by Truckenbrodt and Rizzoli, that enables a single step 10× expansion (Truckenbrodt et al., 2019, 2018). We have added a gel excision/mounting strategy based on a custom 3D-printed cutter to minimize drift during imaging, and pair ExM with both conventional and higher resolution (Zeiss Airyscan) imaging. We also describe a simplified method for calculating the expansion factor using the open source software Fiji (ImageJ).
There are several experimental factors that should be considered before beginning this protocol. Below, we describe these experimental considerations in greater detail.
Choice of fluorophores
Preservation of signal intensity in ExM is a challenge, due to the obligate signal dilution that occurs in all 3 spatial dimensions during expansion (i.e., same signal spread across a greater area in the expanded gel) and to the potential damage/loss of fluorophores and dyes during the polymerization and digestion steps. Although the technique is compatible with a wide range of commercial fluorescently-conjugated secondary antibodies and genetically encoded fluorophores, certain caveats apply. Most fluorescent dye-conjugated antibodies and β-barrel scaffolded fluorophores (i.e., GFP- and RFP-type) exhibit up to 50% signal retention. Key exceptions include cyanine family dyes such as Cy5 and Alexa Fluor 647 that are degraded during the polymerization step (CF633 and Atto647N are alternate far red dye choices) and the bacteriophytochrome IR protein iRFP that is degraded during the proteinase K digestion step.(Tillberg et al., 2016)
Optimization of fluorescent signal strength
-
1.
Choose your cell line and optimize fixation and immunostaining of the desired antigen(s) to obtain the best possible signal-to-noise ratio with conventional imaging.
-
2.
For stably or transiently expressed genetically-encoded fluorophore tags such as GFP or RFP, immunostaining (using a dye with similar spectral properties) may be required to boost a weak signal. We use primary antibodies paired with either Alexa Fluor 488- or AlexaFluor 555-conjugated secondary antibodies to stain GFP and RFP fusion proteins, respectively.
Validation
The principle of ExM is that anchored biomolecules/labels are pulled apart evenly while preserving their spatial organization relative to each other. The rigorous empirical validation and registration-based analysis of distortion to which current ExM protocols have been subjected have consistently confirmed that the level of distortion is low (i.e., a few percent over length scales of tens to hundreds of micrometers; (Wassie et al., 2019)). When using a well-tested protocol with a previously validated cell type or tissue, further validation is not absolutely necessary unless obvious cracks or deformations are observed or precise distance measurements are required. Ultimately, the most accurate way to calculate the expansion factor and determine distortions is to image the same structure before and after expansion and compare their alignment (see (Truckenbrodt et al., 2019) for a detailed discussion and Python script).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken anti-GFP (green fluorescent protein) | Aves | Cat#GFP-1020 RRID:AB_10000240 |
| Alexa Fluor® 488-AffiniPure F(ab'')2 Fragment Donkey Anti-Chicken IgY (IgG) | Jackson ImmunoResearch | Cat#703-546-155 RRID:AB_2340376 |
| Rat anti-RFP (red fluorescent protein), clone 5F8 | ChromoTek (Proteintech) | Cat#5F8-100 RRID:AB_2336064 |
| Rabbit anti-Fibrillarin | Abcam | Cat#ab5821 RRID:AB_2105785 |
| Rabbit anti-Histone H3, trimethyl (Lys 9) | Abcam | Cat#ab8898 RRID: AB_306848 |
| Alexa Fluor™ 555 goat anti-rat IgG (H+L) | Thermo Fisher Scientific | Cat#A21434 RRID:AB_2535855 |
| Goat anti-mouse IgG (H+L) Alexa Fluor™ Plus 488 | Thermo Fisher Scientific | Cat#A32723 RRID:AB_2633275 |
| Goat anti-mouse IgG (H+L) Alexa Fluor™ Plus 555 | Thermo Fisher Scientific | Cat#A32727 RRID:AB_2633276 |
| Goat anti-rabbit IgG (H+L) Alexa Fluor™ Plus 488 | Thermo Fisher Scientific | Cat#A32731 RRID:AB_2633280 |
| Goat anti-rabbit IgG (H+L) Alexa Fluor™ Plus 555 | Thermo Fisher Scientific | Cat#A32732 RRID:AB_2633281 |
| Goat anti-rabbit CF633 | Biotium | Cat#20122 RRID:AB_10853773 |
| Goat anti-mouse CF633 | Biotium | Cat#20120 RRID:AB_10853296 |
| Chemicals, peptides, and recombinant proteins | ||
| bisBenzimide H 33342 trihydrochloride (Hoechst 33342) | Millipore Sigma | Cat#B2261 |
| PEI (Polyethylenimine) | Polysciences Inc. | Cat#23966 |
| Paraformaldehyde powder, 95% | Millipore Sigma | Cat#158127 |
| Triton™ X-100 | Millipore Sigma | Cat#T8787 |
| Tween-20 | Millipore Sigma | Cat#P2287 |
| Normal goat serum | Abcam | Cat#Ab7481 RRID:AB_2716553 |
| PBS, 1× pH 7.4, without calcium and magnesium | Wisent | Cat#311-010-CL |
| Acryloyl-X (6-((acryloyl)amino)hexanoic acid) | Thermo Fisher Scientific | Cat#A20770 |
| DMAA (N,N-dimethylacrylamide) | Millipore Sigma | Cat#274135 |
| Sodium acrylate | Millipore Sigma | Cat#408220 |
| Proteinase K | Millipore Sigma | Cat#P4850 |
| Potassium persulfate | Millipore Sigma | Cat#216224 |
| TEMED | Millipore Sigma | Cat#T7024 |
| Experimental models:Cell lines | ||
| U-2 OS cells (human osteosarcoma epithelial; female derived) | ATCC | Cat#ATCC® HTB-96™ RRID:CVCL_0042 |
| Recombinant DNA | ||
| pEGFP(C1)-RepoMan | Addgene | Cat#44212 RRID_Addgene44212 |
| Software and algorithms | ||
| ZEN 3.0 (blue edition), Airyscan module | Zeiss | ZEN 3.0 |
| Imaris Surfaces (3D Volume Rendering) | Oxford Instruments | IMARIS |
| Fiji/Image J image processing package (open source) | https://imagej.net/Fiji | FIJI |
| Other | ||
| μ-Slide 8-well chambered coverslip | ibidi | Cat#80826 |
| μ-Slide 8-well chambered coverslip, glass bottom | ibidi | Cat#80827 |
| Gold Seal Cover Glass, 18 × 18 mm, No. 1.5 | Thermo Fisher Scientific | Cat#3405 |
| Microscope cover glasses, 22 × 22 mm, No. 2 | VWR | Cat#16004-304 |
| N-EVAP Nitrogen Evaporator | Organomation | Cat#11106-O |
| Paragon sterile scalpels | Ultident | Cat#02-90000-24 |
| Bemis™ Parafilm™ M Laboratory Wrapping Film | Fisher Scientific | Cat#ACAPM999 |
| Sterile water, molecular grade | Wisent | Cat#809-115-CL |
| Petri dishes, 110 mm × 15 mm | Fisher Scientific | Cat#FB0875712 |
| Cell culture dishes, non-treated, 35 mm | Millipore Sigma | Cat#EP0030700115 |
CRITICAL: Acryloyl-X (6-((acryloyl)amino)hexanoic acid), DMAA (N,N-Dimethylacrylamide), Sodium Acrylate, Proteinase K, Potassium persulfate, TEMED.
Alternative: 4′,6-diamidino-2-phenylindole (DAPI) can be substituted for Hoechst 33342 to stain DNA during expansion. Cell lines, plasmids, transfection reagent, fixative and antibodies are experiment-specific.
ExM gel solution (weigh and add to 50 mL tube in the order indicated below)
| Reagent | Mol % | Weight (g) |
|---|---|---|
| DMAA | 80 | 1.335 |
| Sodium Acrylate | 20 | 0.32 |
| ddH2O | n/a | 2.850 |
Materials and equipment
Custom ExM Gel Cutter (3D printing files)
| Equipment | 3D Printable file (Data S1) |
|---|---|
| ExM gel slice cutter (FreeCAD) | LTM-cutter.FCStd NOTE: cutter and pusher in this editable file |
| ExM gel slice cutter (standard triangle language) | Cutter.stl |
| ExM gel slice pusher (standard triangle language) | Pusher.stl |
Step-by-step method details
Fixation, permeabilization, and immunostaining
Timing: [4–5 h]
This is a standard immunocytochemistry workflow for labeling proteins in fixed cells on glass coverslips with primary antibodies and secondary antibodies conjugated to fluorophores.
-
1.Seed cells on coverslips and transfect if desired
-
a.On day one, passage cells according to standard protocol and seed at 30–50% confluency on 18 × 18 mm glass coverslips, No. 1.5. We maintain U2OS cells in DMEM supplemented with 10% fetal bovine serum and 1× penicillin/streptomycin and passage every 2–3 days.
-
b.On day two, cells should be 50–70% confluent. At this point they can be fixed for immunostaining of endogenous proteins, or transfected for transient overexpression of a fluorophore-tagged protein prior to fixation on day three.
-
i.For transfection of U2OS cells in a 6 cm dish, combine 5 μg of plasmid DNA with 12.5 μL of PEI solution (1 mg/mL) and 300 μL of serum-free DMEM. Vortex and leave at room temperature for 15 min.
-
ii.Drip the transfection solution onto the 5 mL of media in the dish and swirl to mix. Leave in incubator at 37°C for 18–20 h.
-
i.
-
a.
-
2.Fix cells
-
a.Remove media and rinse cells 2× with 5 mL of warm PBS.
-
b.Add 5 mL of warm, freshly-prepared PFA Fixation Solution (4% PFA in PBS or buffer of choice) to the dish and let sit at 20°C–22°C for 10 min.
-
c.Remove fixation buffer and rinse cells 2× with 5 mL of PBS.
-
a.
-
3.Permeabilize cells and block non-specific binding sites
-
a.Remove PBS and add 5 mL of Permeabilization Solution (1% Triton X-100 in PBS). Let sit at 20°C–22°C for 10 min. This solution can be stored at 20°C–22°C for up to 3 months.
-
b.Rinse cells 2× with 5 mL of PBS.
-
c.Using fine forceps, transfer coverslips to a Humidified Chamber (box lined with parafilm and containing damp paper towels to maintain humidity when lid is closed). Touch each coverslip carefully against a Kimwipe to remove excess liquid before placing cell side-up on the parafilm.
-
d.Gently pipette 50–100 μL freshly-prepared Blocking Buffer (PBS containing 1% goat serum and 0.1% Tween-20) on top of the cells, ensuring that the entire surface of the coverslip is covered.
-
e.Place lid on box and let sit at 20°C–22°C for 10 min.
-
f.During the blocking step, prepare the primary and secondary antibody dilutions in Blocking Buffer. Clear by centrifuging at full speed (~21,000 × g) in a refrigerated microcentrifuge for 10 min to pellet any aggregates and store on ice.
-
a.
-
4.Immunostain cells (all steps are done sequentially)
-
a.Pick up each coverslip and gently touch the edge to a Kimwipe to drain off excess blocking buffer, then pipette 50–100 μL of primary antibody solution (prepared in Blocking Buffer) on top, ensuring that the entire surface is covered. Place lid on box and leave for 1 h at 20°C–22°C. Note that if the volume of antibody solution is limited, the coverslips can instead be inverted (placed cell side-down) on top of a 20–50 μL drop of antibody solution.
-
b.Use forceps to transfer each coverslip to a well in a 6-well plate. Rinse 3 × 5 min with 2 mL PBS.
-
c.Remove coverslip from well using forceps, touch edge to Kimwipe to drain off excess PBS and set cell side-up on parafilm inside humidified chamber. Gently pipette 50–100 μL of secondary antibody solution (prepared in Blocking Buffer) on top, ensuring that the entire coverslip is covered. Place lid on box and leave for 1 h at 20°C–22°C.
-
d.Transfer coverslips back to 6-well plate and repeat PBS wash steps.
-
e.Repeat these primary and secondary antibody incubation steps (with washes) for additional targets, if desired, ensuring that there is no potential species cross-reactivity.
-
i.To stain endogenous proteins, we use the relevant primary antibodies at the same concentration that we use for standard immunofluorescence analysis. We detect them using species-specific Alexa Fluor® Plus 488 or 555-conjugated secondary antibodies at a higher concentration than normal (1:250 dilution, which is 8 μg/mL). If a Far red fluorophore is required, we use CF633-conjugated secondary antibodies, as this dye is compatible with ExM.
-
ii.To amplify the signal of GFP-tagged proteins, we use a chicken anti-GFP polyclonal primary antibody (1:500 dilution) and an Alexa Fluor® 488-conjugated donkey anti-chicken IgY secondary antibody (1:500 dilution).
-
iii.To amplify the signal of RFP-tagged proteins, we use a rat anti-RFP monoclonal primary antibody (1:500 dilution) and an Alexa Fluor® Plus 555-conjugated goat anti-rat IgG secondary antibody (1:500 dilution).
-
i.
-
a.
Note: Genetically encoded fluorophores can be imaged directly, provided a sufficiently strong signal is retained following anchoring/gelation/digestion. Another option is the ProExM (protein-retention expansion) technique, which employs a less robust homogenization step that better preserves direct fluorescent signals and achieves ~4.5-fold expansion (Tillberg et al., 2016).
Note: Although formaldehyde is used here, ExM is also compatible with workflows that utilize alternate fixatives such as methanol and glutaraldehyde (Truckenbrodt et al., 2019).
Pause point: Cells can be stored for 1–3 days after fixation (in PBS at 4°C) prior to permeabilization and immunostaining.
Anchoring
In this step, the succinimidyl ester of Acryloyl-X reacts with proteins in the fixed/stained cells, adding an acrylate group that integrates into the polylacrylamide matrix to anchor the proteins to the gel.
-
5.
Prepare (or thaw) Anchoring Reagent Stock (10 mg/mL Acryloyl-X in anhydrous DMSO) and dilute to 0.1 mg/mL in PBS pH7.4. This is the Anchoring Solution.
-
6.
Drain excess PBS from coverslips and place cell side-up on the parafilm in the humidified chamber. Gently pipette an 80 μL drop of Anchoring Solution on top, ensuring that the entire surface is covered.
-
7.
Place lid on humidified chamber and incubate 18–20 h at 20°C–22°C.
-
8.
Transfer coverslips back to 6-well plate and wash 3 × 5 min with PBS.
Gelation
In this step, a gelation solution of monomers is poured across the fixed/stained/anchored cells and allowed to polymerize into a swellable hydrogel.
-
9.Prepare the Gelation Solution in a fume hood, weighing all components using a micro-balance and adding them in this order:
-
a.Weigh out 1.335 g of DMAA and place in a 50 mL conical bottom tube.
-
b.Weigh out 0.32 g of Sodium Acrylate, add to the tube and vortex.
-
c.Weigh out 2.85 g of ddH2O, add to the tube and vortex. The solution may be slightly turbid, but there should be no visible granules.
-
a.
-
10.
Purge the O2 in the Gelation Solution by bubbling with N2 for 40 min at 20°C–22°C. This is most easily done using a nitrogen evaporator, as it offers a controlled bubbling rate (see Figure 1).
-
11.
Prepare Potassium Persulfate (KPS) Solution by weighing out 0.036 g KPS and placing it in a 1.5 mL Eppendorf tube. Immediately before use, add 1 mL of ddH2O and vortex for 1–2 min.
-
12.
Transfer 2.7 mL of purged Gelation Solution to a 15 mL conical bottom tube. Add 0.3 mL of KPS Solution and quickly vortex for 1–3 s.
-
13.
Purge the O2 in the Gelation Solution by bubbling with N2 for 15 min ON ICE. Keep on ice from this step onwards.
-
14.During this 2nd purging step, prepare the Gelation Chambers (Figure 2).
-
a.Place a glass slide on a sheet of paper towel. This will be the base of the chamber.
-
b.Place 2 blank coverslips (18 × 18 mm, No 1.5) on the slide, a coverslip-width apart (with a drop of water under each to secure it to the slide).
-
c.Pick up one of the stained/anchored coverslips, touching it carefully against a Kimwipe to remove excess liquid before placing it cell-side up between the 2 blank coverslips on the slide (moving them inwards to ensure that the cell-containing coverslip is secured tightly between them).
-
d.Carefully place another blank coverslip (18 × 18 mm, No 1.5) on the left side of the cell-containing coverslip, securing it with a drop of water and allowing it to overlap the cells by ~3 mm. Repeat on the right side. These coverslips act as spacers.
-
a.
-
15.
Transfer 500 μL of the Gelation Solution to a 1.5 mL Eppendorf tube. Add 2 μL of TEMED, vortex 1–3 s, pipette 75 μL on top of the cell-containing coverslip and carefully lower a 22 × 22 mm, No 2 coverslip on top, pushing gently to force out any air bubbles.
-
16.
Allow gels to polymerize for 2–3 h by incubating the Gelation Chambers at 20°C–22°C in a Humidified Chamber. For issues with gelation, see Troubleshooting section.
-
17.
To remove the gel from the Gelation Chamber, use fine forceps to gently pry up the top coverslip. We use a larger (22 × 22) and thicker (No 2) coverslip on top, as it is easier to handle and less prone to cracking. Sliding a scalpel blade lengthwise under the edge and gently levering the coverslip upwards can also help to avoid breakage. The gel usually remains attached to the top coverslip (see Figure 3A). Transfer the coverslip to a 35 mm dish, with the gel facing up. In the event that the gel remains attached to the original coverslip, carefully prise that coverslip off the slide and put it in the 35 mm dish with the gel facing up. Note that in either case, the gel will separate from the coverslip during the subsequent digestion step.
Figure 1.

Purging O2 from the gelation solution
We use a nitrogen evaporator for this step, as it offers a controlled rate of bubbling (A). First, the gel solution in a 50 mL conical tube is purged at 20°C–22°C for 40 min. For purging of the smaller aliquot with added KPS in a 15 mL conical tube for 15 min, the tube is kept on ice throughout (B). In both cases, the top of the tube is sealed with parafilm to minimize re-oxygenation. When setting up the evaporator, adjust the flow rate carefully using a tube of ddH2O before transferring to the gel solution (C). The needles should be carefully cleaned before and after use.
Figure 2.

Setting up the gelation chamber
Steps 1–8 walk through the set-up of a gelation chamber for a single 18 × 18 mm coverslip containing fixed/stained/anchored cells.
Figure 3.

Digestion and expansion of the gel following removal from the gelation chamber
(A). When the top coverslip is removed from the Gelation Chamber following polymerization, the gel usually remains attached to it (A). Place the coverslip gel side-up in a 35 mm dish. If the gel stays on the original coverslip, transfer that coverslip gel side-up to a 35 mm dish. Once the digestion buffer is added, the lid should be secured to the dish using parafilm to prevent evaporation and the dish transferred to a humidified chamber containing a damp tissue. Seal the chamber and place it in a water bath set to 50°C (B). Close the lid and leave 18–20 h. Transfer the digested gel to a 10 cm Petri dish (C). At this point we normally observe some initial expansion (D). Add ddH2O with Hoechst dye to start expansion/DNA staining. After ~1 h (and at least 3 changes of ddH2O), the gel will be significantly larger than the original coverslip, which is shown on top of it for comparison (E).
Digestion
In this step, the proteolytic enzyme proteinase K is used to digest structural proteins in the hydrogel-embedded cells to remove any resistance to expansion that could lead to distortion or rupture of the sample.
-
18.
Prepare (or thaw) Digestion Buffer (50 mM Tris, 800 mM guanidine HCl, 1 mM EDTA and 0.5% Triton X-100 in ddH2O, pH 8.0) and add Proteinase K at 8 U/mL. This is the Digestion Solution.
-
19.
Add 2 mL of Digestion Solution to each 35 mm dish containing a gel. Use a strip of parafilm to seal the lid to the dish (Figure 3A).
-
20.
Place the dishes in a Humidified Chamber for 18–20 h incubation at 50°C (Figure 3A). We use a glass container with an air-lock lid and carefully place it in a 50°C water bath so that the lower third of the container is submerged (Figure 3B).
Note: The digestion step may need to be optimized if ruptures or distortions are observed in the gels or if the retained fluorescent signal is weak (see Troubleshooting section).
Note: Digestion Buffer can be stored at −20°C for up to 6 months. Proteinase K can be stored at 2°C–8°C for up to 2 years.
Expansion and DNA staining
In this step, immersion in ddH2O washes out the ions in the gel matrix that have kept it constricted through ionic interactions, allowing up to 10-fold isotropic expansion. Inclusion of the intercalating dye Hoechst 33342 stains nuclear DNA during expansion.
-
21.
Remove gel from Digestion Solution by inverting the 35 mm dish onto a 100 mm dish (Figures 3C and 3D).
-
22.
Prepare sufficient Expansion/Staining Buffer (2 μL of 25 mg/mL Hoechst 33342 stock in 30 mL ddH2O) for the number of gels to be processed (10 mL per dish).
-
23.
Add the Expansion/Staining Buffer to the 100 mm dish with the gel and incubate for 20 min at 20°C–22°C (can monitor nuclear staining and expansion using an inverted fluorescence microscope).
-
24.
Carefully remove the Expansion/Staining Buffer from the dish and replace with ddH2O.
-
25.
Continue to incubate at 20°C–22°C for 30–40 min, with at least 3 changes of water, until no further expansion is observed (Figure 3E).
Note: For solution changes, a pipette can be used to carefully remove liquid from the dish. Tilt the dish to keep the gel away from the tip while suctioning. The gel is fairly resilient, so if it is partially suctioned into the tip, just expel it slowly.
Gel excision
In this step, a custom 3D-printed gel cutter is used to excise gel segments that fit snugly into a glass-bottom chambered slide to prevent movement during imaging.
-
26.
To excise gel segments for imaging, first remove all of the ddH2O from the 100 mm dish.
-
27.We use a 3D-printed custom Gel Cutter (Figure 4A) to excise segments that are the internal dimensions of an ibidi 8-well μ-Slide (Figure 4). The cutter was designed using Freecad (.FCStd and .stl files provided as Data S1) and took 4 h to print using a fine tip nozzle (see Note below). A less robust cutter can be printed in 40 min using a normal nozzle.
-
a.Lower the Gel Cutter onto the expanded gel and push down firmly until it touches the bottom of the dish. Use a scalpel blade to cut from the Gel Cutter to the edge of the gel so that the segment can be removed (Figure 4B). If the segment remains in the cutter, it can be gently pushed out using the 3D-printed Gel Pusher (Figure 4A). After use, we clean and store the Gel Cutter with the Gel Pusher inserted in it, to protect the cutting edge from damage.
-
b.Slide a bent metal spatula blade under the gel segment and lift it out of the dish (Figure 4C).
- c.
-
d.Repeat for a second well, but this time invert the gel segment. This ensures that one of the 2 wells that you set up will have the gel segment in the correct orientation (cell-side down) for imaging with a high resolution/short working distance objective.
-
e.Using the microscope system of your choice, identify which well has the gel segment in the correct orientation. If you cannot bring the Hoechst-stained nuclei into focus using a high resolution/short working distance objective, the gel is upside-down (Figure 5). Note that this is not an issue when using a lower resolution/longer working distance objective.
-
a.
Note: If you do not have access to a 3D printer, the top opening of a P1000 pipette tip can also be used to excise gel segments that fit in an ibidi 8-well μ-Slide (Figures 4E–4G. The fit is not as snug (Figures 4H–4I), so be sure to remove excess liquid to minimize gel movement during imaging.
Note: If imaging for a long time, add a small amount of ddH2O to the excised gel slice in the well to ensure that it does not dry out. Excised gel slices can be stored in PBS at 4°C for a week (re-swell before imaging by washing 3× with ddH2O for at least 30 min).
Note: A Fused Deposition Modeling (FDM)-based 3D printer was used to print the cutter and the pusher using 1.75 mm Polylactic acid (PLA) filament. The cutter STL model was sliced in the vertical orientation with the cutting end pointing upwards. For printing, the best results were obtained using a 0.2 mm printer nozzle, 0.05 mm layer height, 0.6 mm walls and 0.6 mm top and bottom thickness, and 15% infill. On our printer, print time was approximately 4 hours with these settings. The pusher STL model was also sliced vertically but printed using a 0.4 mm nozzle and 0.2 mm layer height (all other settings were the same). Print time for the pusher was approximately 30 minutes. As not all printers have the options to change nozzle and printing material, other common settings were also tested. Printing the cutter using a 0.4 mm nozzle and 0.1 mm layer height will produce a workable cutter; however the cutting edge is not as sharp. With these printer settings, the cutter and the pusher can be printed together in approximately 3 hours. We also printed the cutter using Polyethylene terephthalate glycol (PETG) and Acrylonitrile Butadiene Styrene (ABS) filament. Cutters printed with PETG were durable using 0.2 mm and 0.4 mm nozzles whereas the cutting edge of cutters printed with ABS was fragile and snapped easily with the 0.2 mm nozzle, and rigid enough for use but still brittle with the 0.4 mm nozzle.
Figure 4.

Excision of gel segments for imaging
(A). Diagram of the custom 3D printed gel cutter and pusher. Press the gel cutter firmly down on the gel until it touches the bottom of the plate, and then use a scalpel to slice from the cutter to the edge of the gel so that it can be moved carefully to a clear section of the dish for removal (B). If the segment remains in the cutter when it is lifted from the dish, the pusher can be used to eject it. Using a bent metal spatula to handle the gel segment (C), transfer it carefully to an ibidi μ-Slide 8 Well Chambered Coverslip (D). If a gel cutter is not available, another option is to use the top of a P1000 pipette tip as a gel punch (E), transferring the round segment to the ibidi chambered coverslip (F and G). Although not as snug a fit as the segments excised using the custom cutter (which match the internal dimensions of the well), movement of the gel segment during imaging can be minimized by removing any excess liquid from the well (H and I).
Figure 5.

Imaging expansion gels
When using a lower magnification objective with a longer working distance, the orientation of the cells embedded in the gel with respect to the bottom of the well is not critical (A). The increase in size and observable detail is demonstrated here by imaging U2OS cells pre- and post-expansion using a Zeiss AxioObserver D1 widefield system with a 20×/0.8 NA air objective. In addition to the Hoechst-stained DNA (blue), the cells express GFP-G3BP2, boosted by staining with anti-GFP and AlexaFluor488 secondary antibodies (green), which has accumulated in cytoplasmic stress granules due to sodium arsenite treatment. Nucleoli have been stained using anti-fibrillarin (1:50 dilution) and AlexaFluor555 secondary antibodies (red). When using a higher magnification objective with a much shorter working distance, the cells embedded in the gel can only be brought into focus if they are lying directly on the bottom of the well (B). More detail can be observed for smaller structures (in this case, nuclear stress foci at which GFP-tagged RepoMan accumulates in response to sodium arsenite treatment) following expansion when a 60×/1.4 NA oil objective is used on a DeltaVision Core restoration deconvolution system. The white boxes mark the regions that have been enlarged to demonstrate the increased detail. Although this is already super-resolution imaging, we routinely combine ExM with Airyscan imaging on a Zeiss LSM880 laser confocal scanning system, which provides a further increase in resolution (C). Volume rendering of the 3D z-stacks using Imaris helps to visualize the nuclear substructure (D), confirming that nuclear stress foci (containing GFP-RepoMan, green) overlap and are surrounded by localized accumulations of the heterochromatic epigenetic histone marker H3K9me3 (1:50 dilution; red; E).
Imaging
Here we describe imaging of expansion gels using a standard widefield system, a restoration deconvolution system and a confocal system with a higher resolution imaging mode
-
28.
The expansion gels can be imaged using your fluorescence imaging system of choice, bearing in mind that the orientation of the gel (i.e. cell-side down) is critical when using high resolution/short working distance objectives. As shown in Figure 5A, gels could be imaged in either orientation using a Zeiss AxioObserver D1 widefield system with a 20×/0.8 NA air objective. The combination of ExM with this relatively simple system already provides a significant increase in the amount of detail that can be observed.
-
29.
When using a shorter working distance oil objective (60×/1.4 NA) on a DeltaVision Core restoration deconvolution system, the embedded cells can only be brought into focus when oriented toward the bottom of the chambered slide (Figure 5B). For the small nuclear foci that we wanted to resolve, expansion allowed us to observe a much higher level of detail.
-
30.
We routinely acquire very high resolution images of our ExM samples using a Zeiss LSM880 AxioObserver Z1 confocal laser scanning microscope equipped with a 63×/1.4 NA Plan-Apochromat oil immersion objective in Airyscan mode. For the image shown in Figure 5C, the multichannel Z-stack had a pixel size of 0.04 μm x 0.04 μm x 0.14 μm and the dwell time per pixel was 1.21 μs, with no averaging. The 405 nm, 488 nm and 561 nm lasers were used at 2% power.
-
31.
For 3D visualization of the Z-stacks (Figure 5D), we use Imaris 9.6.0 (Bitplane). The images are first processed by background subtraction to ease the rendering steps. A 3D surface rendering for each channel is then created, using a smoothing of 2× the pixel size (0.08 μm), a threshold for background subtraction of the diameter of the smallest nucSF (~0.35 μm in this case) and a surface filter to keep only the more specific regions of the image. This allowed us to confirm that the histone modification signal (anti-H3K9me3, red) spreads out and around the centers of the nuclear stress foci marked by accumulations of GFP-RepoMan (green), as shown in the enlarged inset (Figure 5E; (Do et al., 2020)).
Optional: To assess distortion, the same region/structural component in a cell is compared directly in images taken pre- and post-expansion. To do this, it is necessary to excise a smaller segment of the gel, e.g. using a scalpel blade, so that it can be expanded directly in the same imaging chamber (we use a 35 mm glass-bottom dish). This should be done when testing the protocol for the first time, and whenever any deformations or cracks are observed.
Expected outcomes
Using the X10 protocol with PFA-fixed U2OS cells, we routinely obtain an expansion factor of 7–10×. This allows us to study the spatial relationships between fluorophore-labeled proteins in subnuclear structures at super-resolution using either a conventional widefield imaging system or laser scanning confocal system (Do et al., 2020).
Quantification and statistical analysis
To calculate the expansion factor, we compare the mean area of Hoechst-stained nuclei in paired expansion gel (Figure 6B) and non-expanded control coverslips from the same dish (Figure 6A) imaged using a 10× objective. Although it will vary depending on confluency, a typical field-of-view for our non-expanded samples contains 70–120 cells, which drops to ~6–12 for our expanded gels. We typically image 4–6 fields of view for each pre-expansion sample and 8–10 fields of view for each post-expansion sample. In the example shown here, the calculated expansion factor was 10.4 (Figure 6C).
Figure 6.

Calculating the expansion factor
We take images of multiple fields of view in pre-expansion (A) and post-expansion (B) samples, and use Fiji/ImageJ to segment nuclei based on their Hoechst signal and output tables with nuclear areas that are exported to Microsoft Excel for analysis. In the sample shown here (C), we imaged 6 pre-expansion fields of view containing 681 nuclei and 15 post-expansion fields of view containing 147 nuclei. The graph plots the areas measured for all nuclei (gray X), along with the mean ± SD (black bars). For the experiment shown here the calculated expansion factor (post-ExM nuclear area/pre-ExM nuclear area) was 10.4.
The freely available Fij/ImageJi open source software (Schindelin et al., 2012) can be used to segment images and calculate nuclear area. Our workflow is as follows:
-
•
Open image file
-
•
Image > Type > 16-bit
-
•
Image > Adjust > Threshold (in dropdown menus choose “Default” and “Red” and then tick “Dark background”; adjust sliders so that everything outside the main intensity histogram peak is black and the nuclei are red)
-
•
Edit Selection > Create Mask (nuclei should now appear white)
-
•
Process > Binary > Watershed (touching nuclei will be separated)
-
•Analyze > Analyze Particles
-
ochoose a range that accurately segments individual nuclei
-
oin the dropdown Show menu select “Outlines”
-
oselect “Display results” and “Exclude on edges” (will remove any nuclei not fully within the field of view)
-
o
Click OK to generate a drawing of mask window that shows the identified particles (numbered) and a table that lists their areas. Save the results as a .csv file that can be analyzed using other software (e.g., Microsoft Excel). This workflow can be automated by creating a Macro (Plugins > Macro > Record).
Limitations
Although ExM is a powerful approach that has been applied to the analysis of a range of different cell and tissue types, its main limitation is that it can only be carried out on fixed samples. Sample fragmentation during expansion is a more common artifact when working with tissue sections, as they can have varying amounts of connective tissue. The digestion step must then be further optimized to ensure complete homogenization. Additional caveats of ExM include a significant loss of fluorophore signal compared to pre-expansion, which is due to a combination of dilution and quenching/loss during the anchoring, gelation and digestion steps. This is an important consideration when using the X10 technique, as it employs a harsher homogenization step. For weaker signals (or direct fluorophore imaging), the ProExM approach is a better choice, as it employs milder homogenization conditions (LysC and heat) that optimize signal preservation while achieving 4.5× expansion (Tillberg et al., 2016). Post-expansion staining is also an option, provided the epitopes survive the gelation/digestion steps (Asano et al., 2018; Zwettler et al., 2020). We have also observed better preservation of fluorescence signal in X10 using the brighter GFP and RFP variants mGreenLantern (Campbell et al., 2020) and mScarlet (Bindels et al., 2017).
Another issue that has become more relevant as imaging moves to the nanoscale level is label displacement. When proteins are detected using antibodies, their large sizes can place the fluorophore reporter molecule up to 30 nm away from the antigen whose localization it’s reporting. The much smaller single chain nanobodies reduce this displacement significantly, however they have not yet proven to be sufficiently retained in ExM protocols (Thevathasan et al., 2019)(Gao et al., 2018). A promising alternative is the development of tri-functional probes that incorporate a targeting molecule, fluorophore and acryloyl anchoring moiety for direct incorporation into the gel (Wen et al., 2020).
Although ExM has been successfully applied to the analysis of proteins, RNA transcripts (ExFISH; (Chen et al., 2016)) and gene loci (post-expansion DNA FISH; (Zhao et al., 2017)), the visualization of other biomolecules such as lipids has remained elusive due to the technical challenge of retaining them during sample preparation. This was addressed in the recently developed Click-ExM strategy by metabolically (or genetically) labeling a biomolecule of interest with a clickable alkyne or azide functional group. This group is then reacted with alkyne- or azide-biotin and stained with fluorescently labeled streptavidin prior to expansion (Sun et al., 2021). Different clickable labels can be combined to target multiple biomolecules (alongside immunostained proteins, if desired) for simultaneous imaging within a given sample.
Importantly, the optimization and extension of ExM will continue as more labs adopt this convenient and accessible strategy for super-resolution imaging.
Troubleshooting
Problem 1
The gel solution starts to polymerize too early, e.g., in the tube, or while transferring it to the coverslip (step 15).
Potential solution
Work with small volumes at a time and ensure that everything is kept chilled (including the tubes and pipette tips). We routinely store tubes and pipette tip boxes at 4°C for use in ExM.
Problem 2
The gel is overly viscous/gooey (step 17). This is an indication that it has not polymerized fully. Although there may be a problem with the reagents, another possibility is that too much liquid was left on the surface of the cells on the coverslip when the gel was poured, which can dilute the gelation solution.
Potential solution
Before pouring the gel, gently press a Kimwipe to the side of the coverslip to drain off any remaining liquid. If this does not solve the issue, prepare new reagents for the gelation solution.
Problem 3
The gel is too dry/fragile (step 17). This may indicate that there was insufficient humidity in the Humidified Chamber during polymerization.
Potential solution
This can be addressed by adding a few more damp Kimwipes along the sides of the humidified chamber (and ensure that the lid fits snugly).
Problem 4
The cells show visible rupture/distortion following expansion (step 23). This is an indication that the digestion time/conditions are not optimal.
Potential solution
Higher concentrations of Proteinase K and longer digestion times can be tested, although this may exacerbate signal loss. In that case, a 4.5× expansion protocol would be a better option (Asano et al., 2018).
Problem. 5
Significant loss of signal post-expansion (step 23). While there is an expected loss of signal in ExM, there may be additional loss due to incomplete anchoring or over-digestion.
Potential solution
The anchoring and digestion steps can be further optimized, as detailed in the original X10 protocol (Truckenbrodt et al., 2019). Alternatively, the ProExM approach may be a better choice (Tillberg et al., 2016). For genetically-encoded fluorophores, the use of brighter green and red variants such as mGreenLantern (Campbell et al., 2020) and mScarlet (Bindels et al., 2017) may help, with or without additional staining.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Laura Trinkle-Mulcahy (ltrinkle@uottawa.ca).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The 3D printing files for the Custom ExM Gel cutter and Pusher are provided as Data S1.
Acknowledgments
This work was funded by a Cancer Research Society/uOttawa Alliance (23484). We thank Chloë van Oostende-Triplet and Skye Greene at the University of Ottawa Faculty of Medicine Cell Biology and Image Acquisition Core Facility for technical support.
Author contributions
A.G.-L. carried out visualization, methodology, investigation, and writing; K.M. and J.C.-B. provided methodology and resources; L.T.-M. was responsible for visualization, methodology, supervision, formal analysis, and writing.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100630.
Contributor Information
Antoine Gaudreau-Lapierre, Email: agaud100@uottawa.ca.
Laura Trinkle-Mulcahy, Email: ltrinkle@uottawa.ca.
Supplemental information
References
- Asano S.M., Gao R., Wassie A.T., Tillberg P., Chen F., Boyden E.S. Expansion microscopy: protocols for imaging proteins and RNA in cells and tissues. Curr. Protoc. Cell Biol. 2018;80:e56. doi: 10.1002/cpcb.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindels D.S., Haarbosch L., van Weeren L., Postma M., Wiese K.E., Mastop M., Aumonier S., Gotthard G., Royant A., Hink M.A. mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods. 2017;14:53–56. doi: 10.1038/nmeth.4074. [DOI] [PubMed] [Google Scholar]
- Campbell B.C., Nabel E.M., Murdock M.H., Lao-Peregrin C., Tsoulfas P., Blackmore M.G., Lee F.S., Liston C., Morishita H., Petsko G.A. mGreenLantern: a bright monomeric fluorescent protein with rapid expression and cell filling properties for neuronal imaging. PNAS. 2020;117:30710–30721. doi: 10.1073/pnas.2000942117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Tillberg P.W., Boyden E.S. Expansion microscopy. Science. 2015;347:543–548. doi: 10.1126/science.1260088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Wassie A.T., Cote A.J., Sinha A., Alon S., Asano S., Daugharthy E.R., Chang J.-B., Marblestone A., Church G.M. Nanoscale imaging of RNA with expansion microscopy. Nat. Methods. 2016;13:679–684. doi: 10.1038/nmeth.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do T.Q.-T., Gaudreau-Lapierre A., Palii C.G., Resende V.M.F., Campuzano D., Aeschimann C.S., Brand M., Trinkle-Mulcahy L. A nuclear stress pathway that parallels cytoplasmic stress granule formation. iScience. 2020;23:101664. doi: 10.1016/j.isci.2020.101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Maraspini R., Beutel O., Zehtabian A., Eickholt B., Honigmann A., Ewers H. Expansion stimulated emission depletion microscopy (ExSTED) ACS Nano. 2018;12:4178–4185. doi: 10.1021/acsnano.8b00776. [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D., Fan X., Shi Y., Zhang H., Huang Z., Cheng B., Tang Q., Li W., Zhu Y., Bai J. Click-ExM enables expansion microscopy for all biomolecules. Nat. Methods. 2021;18:107–113. doi: 10.1038/s41592-020-01005-2. [DOI] [PubMed] [Google Scholar]
- Thevathasan J.V., Kahnwald M., Cieśliński K., Hoess P., Peneti S.K., Reitberger M., Heid D., Kasuba K.C., Hoerner S.J., Li Y. Nuclear pores as versatile reference standards for quantitative superresolution microscopy. Nat. Methods. 2019;16:1045–1053. doi: 10.1038/s41592-019-0574-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillberg P.W., Chen F., Piatkevich K.D., Zhao Y., Yu C.-C., English B.P., Gao L., Martorell A., Suk H.-J., Yoshida F. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat. Biotechnol. 2016;34:987–992. doi: 10.1038/nbt.3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truckenbrodt S., Maidorn M., Crzan D., Wildhagen H., Kabatas S., Rizzoli S.O. X10 expansion microscopy enables 25-nm resolution on conventional microscopes. EMBO Rep. 2018;19:e45836. doi: 10.15252/embr.201845836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truckenbrodt S., Sommer C., Rizzoli S.O., Danzl J.G. A practical guide to optimization in X10 expansion microscopy. Nature Protocols. 2019;14:832–863. doi: 10.1038/s41596-018-0117-3. [DOI] [PubMed] [Google Scholar]
- Wassie A.T., Zhao Y., Boyden E.S. Expansion microscopy: principles and uses in biological research. Nat. Methods. 2019;16:33–41. doi: 10.1038/s41592-018-0219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen G., Vanheusden M., Acke A., Valli D., Neely R.K., Leen V., Hofkens J. Evaluation of Direct Grafting Strategies via Trivalent Anchoring for Enabling Lipid Membrane and Cytoskeleton Staining in Expansion Microscopy. ACS Nano. 2020;14:7860–7867. doi: 10.1021/acsnano.9b09259. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Bucur O., Irshad H., Chen F., Weins A., Stancu A.L., Oh E.-Y., DiStasio M., Torous V., Glass B., Stillman I.E., Schnitt S.J., Beck A.H., Boyden E.S. Nanoscale imaging of clinical specimens using pathology-optimized expansion microscopy. Nat. Biotechnol. 2017;35:757–764. doi: 10.1038/nbt.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwettler F.U., Reinhard S., Gambarotto D., Bell T.D.M., Hamel V., Guichard P., Sauer M. Molecular resolution imaging by post-labeling expansion single-molecule localization microscopy (Ex-SMLM) Nat. Commun. 2020;11:3388. doi: 10.1038/s41467-020-17086-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 3D printing files for the Custom ExM Gel cutter and Pusher are provided as Data S1.