

Abstract
The androgen receptor (AR) is a target of interest for endocrine disruption research, as altered signaling can affect normal reproductive and neurological development for generations. In an effort to prioritize compounds with alternative methodologies, the U.S. Environmental Protection Agency (EPA) used in vitro data from 11 assays to construct models of AR agonist and antagonist signaling pathways. While these EPA ToxCast AR models require in vitro data to assign a bioactivity score, Bayesian machine learning methods can be used for prospective prediction from molecule structure alone. This approach was applied to multiple types of data corresponding to the EPA’s AR signaling pathway with proprietary software, Assay Central®. The training performance of all machine learning models, including six other algorithms, was evaluated by internal five-fold cross-validation statistics. Bayesian machine learning models were also evaluated with external predictions of reference chemicals to compare prediction accuracies to published results from the EPA. The machine learning model group selected for further studies of endocrine disruption consisted of continuous AC50 data from the February 2019 release of ToxCast/Tox21. These efforts demonstrate how machine learning can be used to predict AR-mediated bioactivity and can also be applied to other targets of endocrine disruption.
Keywords: Androgen receptor, Bayesian, endocrine disruption, machine learning
TABLE OF CONTENTS GRAPHIC
INTRODUCTION
Endocrine disruption research efforts are currently driven primarily by government regulatory initiatives like the Endocrine Disruptor Screening Program from the U.S. Environmental Protection Agency (EPA), which aims to evaluate any risks to the population from chemical exposures 1. This initiative started evaluating effects on the endocrine system by estrogen and androgen hormones, and intends to expand evaluation to thyroid receptor, aromatase, and general steroidogenesis alteration. The androgen receptor (AR) is of major interest to the program, as androgen hormone imbalances are implicated in rare diseases, bone diseases, metabolic dysfunction and cancers 2–4; altered AR signaling can also affect normal reproductive and neurological development for multiple generations 2, 5. Unsurprisingly, due to the high risks involved in altered signaling from environmental causes, in vitro and in vivo AR assays comprise half of the Endocrine Disruptor Screening Program Tier 1 battery 6.
Low-throughput screening and animal testing for regulatory purposes can take years, thousands of animals, and millions of dollars to complete – but the backlog of chemicals slated for environmental testing still needs to be managed 7. The EPA has periodically released high-throughput screening assay data through the ToxCast program 7–9 as well as the consortium program Toxicity Testing in the 21st Century (Tox21) 10. The ToxCast/Tox21 screening efforts cover a width breadth of biological targets and processes related to endocrine disruption and offer a rich data source for computational modeling. Computational resources and screening offer a time-saving and cost-effective method of prioritizing the multitude of chemicals, and applying in silico tools is a goal of these EPA programs 1.
In 2016, the EPA published on their use of data from 11 in vitro ToxCast/Tox21 assays to construct models of AR-mediated endocrine disrupting signaling pathways, by agonism or antagonism (Table 1) 11. The primary objective of this study was to validate the ToxCast AR pathway models, and thus apply them with confidence to prioritize chemicals for additional testing with these models; a long-term goal is that these results are ultimately accepted as alternatives to Tier 1 assay data like similar studies of the estrogen receptor 1, 12, 13. Predictive performances of the AR pathway models were validated with in vitro reference chemicals curated from literature sources. Later, the EPA published on the development of a Hershberger database 14 and subsequently used the set of in vivo reference chemicals derived from this work to further validate the AR pathway models 15. Another key publication described the generation and assignment of a burst-flag hit-call to these same ToxCast AR pathway model assays, where the authors aimed to eliminate false-positives resulting from loss-of-function assays: an active classification to a chemical was only assigned if the assay’s AC50 fell below the cytotoxicity measurement 16. Finally, similar to the Collaborative Estrogen Receptor Activity Prediction Project 17, the Collaborative Modeling Project for Androgen Receptor Activity (CoMPARA)18 recently used high-throughput screening data and multiple modeling methods to prioritize chemicals for Tier 1 testing 19, 20. Predictions of agonist and antagonist bioactivity through the CoMPARA consensus models are available online through the EPA’s Chemical Dashboard 21.
Table 1:
List of assays used for machine learning models, available in ToxCast/Tox21; equivalent to those listed in Table 1 of Kleinstreuer et al, 2017 11.
| Assay Abbreviation | Assay ToxCast Name | Brief Description |
|---|---|---|
| A1 | NVS_NR_hAR | Cell-free radioligand binding assay with human AR |
| A2 | NVS_NR_cAR | Cell-free radioligand binding assay with chimpanzee AR |
| A3 | NVS_NR_rAR | Cell-free b radioligand binding assay with rat AR |
| A4 | OT_AR_ARSRC1_0480 | Recruitment assay of coregulator c-Src tyrosine kinase at 8 h |
| A5 | OT_AR_ARSRC1_0960 | Recruitment assay of coregulator c-Src tyrosine kinase at 16 h |
| A6 | ATG_AR_TRANS_up | Reporter gene assay measuring mRNA induction after 24 h |
| A7 | OT_AR_ARELUC_AG_1440 | Reporter gene assay measuring luciferase induction |
| A8 | TOX21_AR_BLA_Agonist_ratio | Reporter gene assay measuring ratio of cleaved to uncleaved substrate |
| A9 | TOX21_AR_LUC_MDAKB2_Agonist | Reporter gene assay measuring luciferase induction |
| A10 | TOX21_AR_BLA_Antagonist_ratio | Reporter gene assay measuring ratio of cleaved to uncleaved substrate |
| A11 | TOX21_AR_LUC_MDAKB2_Antagonist | Reporter gene assay measuring luciferase induction |
Despite the impressive performance of all of these in silico AR models spearheaded by the EPA, a major disadvantage to the ToxCast AR pathway modeling technique is that the in vitro data across the 11 high-throughput assays for each chemical are required to generate the bioactivity score. Hence, these pathway models lack the power of prospective prediction for a chemical with unknown in vitro activity. While the recent release of CoMPARA can generate predictions for external molecules, it is based on the k-nearest neighbors method and utilizes predicted AR activity (albeit with high concordance across consensus predictions). Alternatively, machine learning methods have shown their applicability to drug discovery and toxicology using molecule structure alone 22–24, and it is certainly not a novel idea to apply machine learning to endocrine disruption research. Within the CoMPARA project were participants 20 utilizing multiple individual machine learning methods (random forest, k-nearest neighbors, support vector machines, decision trees, etc.) as well as creating consensus models. Grisoni et al. 25 not only presented the advantages of each algorithm but also evaluate the structural features for AR binding, and Manganelli et al. 19evaluated misclassified binding chemicals. Gupta et al. 26 developed a multilevel ensemble model, which first applied a random forest model to classify compounds and then applied multiple activity scores using four methods (linear, decision trees, random forest, neural network) to a Tox21 AR agonism training dataset. Idakwo et al. 27 utilized both agonist and antagonist datasets from Tox21 for random forest and deep learning, and investigated chemical similarity between prediction classes to further analyze accuracy. Chen et al. 28 extracted an AR binding training set from publications and calculated seven molecular fingerprints to build machine learning models from four methods (k-nearest neighbors, decision tree, naïve Bayes, and support vector machine) to identify substructures of endocrine disrupting chemicals.
The application of Assay Central® Bayesian machine learning methods has previously demonstrated for drug discovery (Ebola, tuberculosis, rare disease) 29–31. The current study therefore describes multiple Bayesian machine learning model groups generated from the same 11 assays used in the EPA’s ToxCast AR agonist and antagonist pathway models. These groups are defined by their source and data type: in vitro ToxCast/Tox21 AR bioactivity and hit-call data 32, the area-under-the-curve (AUC) values output from the agonist and antagonist pathway models 11, or burst-flag hit-call data incorporating cytotoxicity considerations 16. The performance of these groups was evaluated first by internal five-fold cross-validation metrics, then by the prediction accuracy of two external test sets utilized in previous EPA publications 11, 15. A further comparison of multiple machine learning algorithms was also conducted on the finalized AR datasets, building on previous work comparing performance with disease and toxicology datasets 24, 30, 33. The goal of this study was two-fold: 1) derive predictive models to prioritize chemicals for future in vitro and in vivo testing of endocrine disruption mediated by AR, and 2) to describe any differences between machine learning methods when using the same underlying descriptor with AR datasets of varying data type and total size.
EXPERIMENTAL SECTION
Datasets
Four data types for each of the 11 assays used in the AR signaling pathway models from the EPA (Table 1) were retrieved from three sources: 1) hit-call or AC50 data from invitroDBv3.1 summary files 32, referred to herein as “ToxCast2019”, 2) AUC output values from the agonist and antagonist pathway models 11, and 3) burst-flag hit-call data from a recent publication 16 referred to herein as “Nelms2018-BFHC”. Each data source-type pair was considered as one of eight model groups (including AUC models at two thresholds) and abbreviations for these groups are presented in Table 2.
Table 2:
Summary of AR machine learning model groups created in this study. Binary data was assigned by sources.
| Action | No. Assays | Data Source | Data Type | Group Alias | Reference | Threshold |
|---|---|---|---|---|---|---|
| Agonist | 9 | ToxCast/Tox21 2019 release | hit-call | ToxCast2019-HC | - | binary |
| Antagonist | 7 | |||||
| Agonist | 9 | ToxCast/Tox21 2019 release | AC50 | ToxCast2019AC50full | - | automated |
| Antagonist | 7 | |||||
| Agonist | 9 | ToxCast/Tox21 2019 release | (AC50 < 1e6μM) | ToxCast2019AC50active | - | automated |
| Antagonist | 7 | |||||
| Agonist | 9 | ToxCast/Tox21 modified 2014 release | burstflag hit-call | Nelms2018BFHC | 16 | binary |
| Antagonist | 7 | |||||
| Agonist | 1 | EPA AR pathway model | AUC | AUC-0.1 | 11 | 0.1 |
| Agonist | 1 | AUC-0.01 | 11 | 0.01 | ||
| Antagonist | 1 | EPA AR | AUC | AUC-0.1 | 11 | 0.1 |
| Antagonist | 1 | pathway model | AUC-0.01 | 11 | 0.01 |
The Nelms2018-BFHC datasets are similar to the cytotoxicity considerations taken by the EPA’s ToxCast AR model AUC or bioactivity score. When generating the ToxCast AR pathway models, the authors removed chemicals from the two Tox21 antagonist datasets (Table 1) that produced an AC50 below the parallel viability measurement. Nelms et al. 16 applied the same methodology to all assays, but the Nelms2018-BFHC datasets retained these cytotoxic chemicals and considered then inactive.
Bayesian models require a classification of active and inactive chemicals prior to their generation by applying a bioactivity threshold to continuous data. Hit-call and burst-flag hit-call classifications were set by Nelms et al. and the ToxCast pipeline. Models built with AC50 data utilized a calculated threshold that is unique to each dataset, as described in the next section. The EPA’s ToxCast AR model publication describes that an AUC score greater than 0.1 are defined as active, scores between 0.001 and 0.1 are inconclusive, and scores below 0.001 were truncated to zero and classified as inactive 11. The 0.1 threshold was used for models built from AUC values, as well as a lower threshold of 0.01; this is described as an acceptable means to limit false positives by a publication for the Collaborative Estrogen Receptor Activity Project 17.
Data from each source were curated into a single file using a proprietary application called Bleach (Molecular Materials Informatics, Montreal Canada). After downloading invitroDBv3.1 summary files 32, CAS identifiers were used to curate structures and quality control notes with the EPA’s Chemical Dashboard for all 9214 substances provided. Substances that lacked structures or had a valid quality control note (i.e. water samples, mixtures, ill-defined) were removed, as were similarly problematic chemicals (i.e. polymers) that included a structure. This central source of 8645 substances was combined with various data by “code” identifiers from the same summary file into two central sources, one for ToxCast/Tox21 AC50 data 32 and the work done by Nelms et al. 16, and the other for AUC data from Kleinstreuer et al. 11. The final step prior to generating models was to standardize structures for machine learning (i.e. removing salts and balancing charges) within the proprietary software Assay Central®. Further curation included removal of compounds with a molecular weight greater than 750 after removing salts to exclude antibiotics and larger macromolecules that are not as pertinent to consumer product ingredients, with an exception for chemicals with less than 50 non-hydrogen atoms, so as to include chemotypes dominated by halogen atoms like dyes.
External predictions from chemical structure alone required creating new training models that excluded testing chemicals; these new training datasets were generated with a proprietary workflow. Two test sets of in vitro (Table S1A) or in vivo (Table S1B) classifications of AR agonist and antagonist activity were used in previous evaluations of the EPA’s ToxCast AR pathway models by Kleinstreuer et al. 11, 15, and are available for download from the NTP Interagency Center for the Evaluation of Alternative Toxicology Methods website 34. The machine learning model prediction accuracy for each test set was then compared to results published around the ToxCast AR models’ validation 11, 15. Furthermore the CoMPARA 18 consensus model predictions of agonism and antagonism were downloaded from the CompTox Chemistry Dashboard 21 and are also briefly compared to the work herein. The CoMPARA evaluation set of >4,800 chemicals with binding, agonist and antagonist classifications was considered for comparison, however, due to the inclusion of all possible data from ToxCast/Tox21 assays, removing testing chemicals from the training datasets would have resulted in heavily diminished models and would not yield a fruitful and accurate comparison.
It is important to note that the EPA did not evaluate in vitro reference chemicals that did not have data present in ToxCast October 2015 release, as they were unable to assign an AUC score 11. These eight chemicals (Table S1A) were removed from training models as a part of the test set, but were not considered for the performance comparison. In addition to the 54 in vitro reference chemicals, four other chemicals were removed from the machine learning training models due to identical skeletal structures (Table S1A). The number of in vivo reference chemicals in the original test set (n = 39) was also reduced herein, as the mixture abamectin was excluded due to incompatibility with machine learning methods. Test set chemicals were curated similarly to the training set (i.e. removing polymers and salts) to facilitate compatible predictions. The final test sets discussed in this study consist of 46 in vitro reference chemicals and 38 in vivo reference chemicals (Tables S1). These test sets were evaluated in-depth by the EPA with the analysis of an additional confirmatory assay as well as a confidence scoring system; herein this work only compares machine learning model predictions to the raw AUC outputs from the AR agonist and antagonist pathway models.
Assay Central®
The Assay Central® framework 35, 36, including metrics to evaluate model performance and its application to drug discovery and toxicology projects 30, 37, 38, as well as generation and interpretation of prediction scores and applicability domain values 35 have been previously described. Briefly, a series of scripts employ standardized rules for the detection of problematic molecular structures that can be corrected by multiple means, and the output is a high-quality dataset and a Bayesian model that can be used to predict chemical activity. Machine learning models utilize only extended-connectivity fingerprints of maximum diameter 6, generated from the Chemistry Development Kit 39. Each model has a series of internal five-fold cross-validation metrics output by default: receiver operator characteristics, recall or sensitivity, specificity, F1-score, Cohen’s kappa 40, 41, and Matthews Correlation Coefficient; balanced accuracy has also been included in the analysis of models. An automated method to select the activity threshold was applied to optimize individual model performance 35, 36. While inconsistent across datasets, it was important for this study to evaluate if automatically chosen activity thresholds produced reasonable predictions. Prediction scores also include an applicability domain score, where higher values suggest more chemical property space is covered in the model, ensuring a given prediction is within the scope of the training data. The prediction accuracy of machine learning models was compared to the results described previously 11, 15. Predictions were evaluated using the standard probability cutoff, where a prediction score of 0.5 or greater designates a chemical as active 35. Sets of either nine agonist-pathway assays or seven antagonist-pathway assays were utilized for predictions: if more than half of the models in a group (i.e. at least five agonist assay models or four antagonist assay models) predicted the compound as active, it was considered to be an overall active designation. The exception to the majority-rule designation was stand-alone models created with AUC data from the AR agonist or antagonist pathways. Machine learning predictions were then compared to the reported activities of reference chemicals 34 without potency considerations (i.e. “yes” or “no” rather than “strong” or “weak”).
Comparison of machine learning algorithms
The complete (i.e., full training and testing) datasets output by Assay Central® were used for comparison of other machine learning algorithms (random forest, k-Nearest Neighbors, support vector classification, naïve Bayesian, AdaBoosted decision trees, and deep learning architecture with three hidden layers - all previously described 37). Deep learning models were generated with Keras (https://keras.io) and a Tensorflow (www.tensorflow.org, GPU for training) backend, and all other algorithms used Scikit-learn (http://scikit-learn.org/stable/, CPU for training) machine learning python library. All algorithms utilize extended-connectivity fingerprints of maximum diameter 6 generated from RDKit (http://www.rdkit.org), and were also subjected to five-fold cross-validation. A single set of hyperparameters was utilized for deep learning, as determined in previous studies 30, 37.
This study compared the differences in the five-fold cross-validation scores between each of the machine learning algorithms with a rank normalized metric. This group 33, 38 and several others 42 have previously used this rank normalized score as a useful performance criterion. First, all metrics for each model were range-scaled to [0, 1] before designating the mean as the rank normalized score. When rank normalized scores for each machine learning algorithm were not normally distributed then nonparametric comparisons were used. Such rank normalized scores can be evaluated pairwise (machine learning comparison per training set) or independently (to give a general machine learning comparison). An additional measure was recently devised to compare models called “difference from the top” (ΔRNS) metric, which gives a rank normalized score for each algorithm subtracted from the highest rank normalized score from a specific training set 33. This is useful because it maintains the pairwise results from each training set cross-validation score by algorithm, enabling a direct assessment of the performance of two machine learning algorithms whilst also maintaining information from the other machine learning algorithms.
RESULTS
Several groups of Bayesian machine learning models were created using Assay Central® software, and data from 11 in vitro ToxCast/Tox21 assays previously used by the EPA to construct pathways of AR-mediated endocrine disrupting signaling. Specifically, machine learning model groups consisted of hit-call and continuous AC50 data from invitroDB_v3.1, the AUC scores outputted from the EPA’s AR agonist and antagonist pathway models, as well as burst-flag hit-calls which address cytotoxicity. Five-fold cross-validation statistics of final and full Bayesian machine learning models (i.e. including test set chemicals) are summarized in Table S2.
Models built with AC50 data applied inconsistent activity thresholds across individual assays in an automated fashion which optimizes performance metrics, but generally these thresholds produced a reasonable ratio of active to inactive chemicals. The Nelms-BFHC group was most sparse in active chemicals across assays, understandable due to the cytotoxicity considerations of the hit-call. However, this imbalance generated extremely high-performance metrics (Table S2D) that did not translate into external prediction accuracy, as described below. While balanced accuracy, and other metrics varied across individual datasets, averages over groups were fairly consistent with the Nelms-BFHC group being slightly higher overall. We also noted that the ToxCast AUC score models (Table S2E) have performance metrics that were not as good as many of the individual models (Table S2A-D).
Five-fold cross-validation metrics generated by six additional machine learning algorithms (Figure S1) were also evaluated. Generally, Assay Central® Bayesian models had similar performance metrics to other methods, but AdaBoosted decision trees and naïve Bayesian algorithms were consistently outperformed by the other methods (Figure 1). In addition, two of the three comparisons (ΔRNS and rank normalized score with pairwise-comparison) show Assay Central® outperformed both deep learning architecture and k-nearest neighbors in a statistically significant manner (Table S4).
Figure 1.

Machine learning algorithm comparisons across multiple five-fold cross-validation metrics. A) Rank normalized scores and B) ΔRNS. Box and whisker plots show individual points for those values that fall outside of the 5–95 percentile. Abbreviations: AC = Assay Central® (Bayesian), rf = Random Forest, knn = k-Nearest Neighbors, svc = Support Vector Classification, bnb = Naïve Bayesian, ada = AdaBoosted Decision Trees, DL = Deep Learning Architecture.
Two external test sets, which included both agonist and antagonist reference chemicals, were utilized to evaluate the predictive performance of Bayesian machine learning model groups. New training models were generated by removing reference chemicals from each of the 11 assays in each group with a proprietary script. Each test set chemical was assigned a probability-like prediction score from either nine agonist or seven antagonist assays 11, and a majority-rule method was used to assign a binary classification to each chemical. These classifications were then compared to the results of EPA’s ToxCast AR agonist and antagonist pathway model validation studies reported by Kleinstreuer et al. 11, 15 as well as the CoMPARA 18 agonist and antagonist consensus predictions available through the EPA Chemistry Dashboard 21.
The test set of in vitro agonist reference chemicals totaled 29 (Figure 2A), with eight being active and 21 inactive (Table S1A). All Bayesian machine learning models produced zero false negatives, and were able to correctly predict all eight active agonist chemicals with varying frequency of false positives. Particularly, these false positive designations were consistently observed for 17α-estradiol, finasteride, and fulvestrant (Tables 4 and Table S3A); these substances are steroids, a chemical class common to androgenic compounds 43. The CoMPARA 18 consensus agonist model also had false positive designations for these same three chemicals, which may indicate these types of inaccuracies persist across methods. None of the machine learning model groups were able to match the predictive power of EPA’s ToxCast AR agonist pathway model as it was able to predict 8/8 active chemicals and 19/21 inactive chemicals accurately or 27/29 chemicals overall 11. This ToxCast AR model did not produce any false negatives but was associated with one false positive, 17α-estradiol, and assigned an inconclusive score to tamoxifen. The Nelms2018-BFHC group performed best of the machine learning model groups for this test set: it correctly predicted the most chemicals overall (26/29), the most inactive chemicals (18/21), and also produced the fewest false positive predictions.
Figure 2:


Results for the in vitro agonist (A) and antagonist (B) test set across all machine learning model groups, in comparison to Kleinstreuer et al. 11 and CoMPARA consensus classifications 18. Navy bars indicate number of chemicals classified as active by the model group, blue bars indicate the number of correctly classified active chemicals, red bars indicate the number of chemicals classified as inactive by the model group, orange bars indicate the number of correctly classified inactive chemicals, and green bar represents inconclusive scores.
Table 4:
Chemicals that were frequently predicted inaccurately by machine learning model groups.
| Structure | Name (CASRN) | Test Set | Reference Classification (List) | Prediction (Mode of Action) |
|---|---|---|---|---|
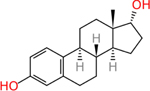 |
17a-estradiol (57-91-0) | in vitro | Inactive (agonist) | Active (agonist) |
 |
Androstenedione (63-05-8) | in vitro | Moderate (agonist) Inactive (antagonist) | Active (agonist) Active (antagonist) |
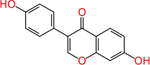 |
Daidzein (486-66-8) | in vitro | Inactive (antagonist) | Active (antagonist) |
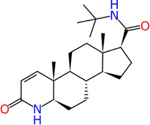 |
Finasteride (98319-26-7) | in vitro | Inactive (agonist) | Active (agonist) |
| Fulvestrant (129453-61-8) | in vitro | Inactive (agonist) | Active (agonist) | |
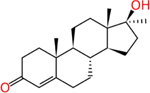 |
Methyl testosterone (58-18-4) | in vitro | Active - Strong (agonist) Inactive (antagonist) | Active (agonist) Active (antagonist) |
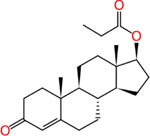 |
Testosterone propionate (57-85-2) | in vitro | Active -Strong (agonist) Inactive (antagonist) | Active (agonist) Active (antagonist) |
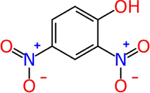 |
2,4-dinitrophenol (51-28-5) | in vivo | Negative | Inactive (agonist) Active (antagonist) |
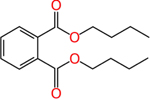 |
Dibutyl phthalate (84-74-2) | in vivo | Anti- androgenic | Inactive (antagonist) |
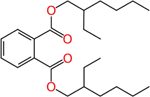 |
Diethylhexyl phthalate (117-81-7) | in vivo | Anti- androgenic | Inactive (antagonist) |
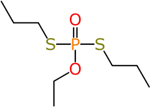 |
Ethoprop (13194-48-4) | in vivo | Anti- androgenic | Inactive (antagonist) |
The in vitro antagonist reference chemicals consisted of 20 active and eight inactive chemicals (Table S1A). At the cost of false positive predictions, 4/6 machine learning model groups were able to correctly predict more active antagonists than the EPA’s ToxCast AR antagonist pathway model (Figure 2B and Table S3B). However, none of the machine learning model groups were able to accurately predict inactive chemicals from the antagonist reference list as well as Kleinstreuer et al. 11. The Nelms2018-BFHC and ToxCast2019-AC50active groups were especially poor predictors of inactive chemicals, as each produced over ten false negative classifications. False positive predictions were prominent in machine learning model groups for testosterone propionate, methyl testosterone, daidzein, and 4-androstenedione (Table 4, Table S3B); all but daidzein are steroid structures, a similar trend to what was seen for in vitro agonist reference chemicals. The EPA’s ToxCast AR antagonist pathway model accurately predicted 17/20 active chemicals as well as all inactive chemicals, with only one false negative (zearalenone), but two active antagonists, methoxyclor and fenarimol, were scored as inconclusive 11. The CoMPARA 18 consensus antagonist model produced two false positive designations for daidzein and 4-androstenedione, but zearalenone was not assigned as consensus score (Table S3B).
The in vivo agonist reference chemicals consisted of three active and 15 inactive chemicals (Table S1B). The ToxCast2019-AC50full, ToxCast2019-HC, and Nelms2018-BFHC groups were able to accurately predict all in vivo reference agonists accurately, similar to what was seen in the previous estrogen receptor predictions 44 (Figure 3A). Total accuracy for this test set was produced by both the ToxCast AR agonist pathway model 15 as well as the CoMPARA 18 consensus agonist model (Table S3C). This test set was the most unbalanced (i.e. lacking a similar number of active and inactive compounds) of the four and was the least informative for evaluating predictive performances of model groups in this study.
Figure 3:


Results for the in vivo agonist (A) and antagonist (B) test set across all machine learning model groups, in comparison to Kleinstreuer et al. 15 and CoMPARA consensus classifications 18. Navy bars indicate number of chemicals classified as active by the model group, blue bars indicate the number of correctly classified active chemicals, red bars indicate the number of chemicals classified as inactive by the model group, orange bars indicate the number of correctly classified inactive chemicals, and green bar represents inconclusive scores.
The in vivo antagonist reference chemicals consisted of 20 active and 15 inactive chemicals (Table S1B). As observed for in vitro antagonist chemicals the Nelms2018-BFHC and ToxCast2019-AC50active groups classified the greatest number of false negatives; however, in this case the EPA’s ToxCast AR antagonist model produced 12 false negatives as well (Figure 3B). The ToxCast-AC50full, AUC-0.1, and ToxCast2019-HC groups yielded the highest overall accuracy with 27/35 antagonist chemicals correctly predicted (Figure 3B). While Nelm2018-BFHC technically predicted the fewest false positives it also had the fewest active designations, similar to for AUC-0.01 for false negatives. When considering both the number of false positives and negatives in addition to the number of correct predictions overall, both ToxCast2019-AC50full and AUC-0.1 models are the optimal performers. All machine learning model groups consistently assigned false negative designations to diethylhexyl phthalate, dibutyl phthalate, and ethoprop. Additionally, 2,4-dinitrophenol was consistently assigned a false positive classification by these groups, but interestingly was assigned the correct inactive classification by the agonist counterparts (Table 4 and Table S3D). The EPA’s ToxCast AR antagonist pathway model assigned false negative AUC values to the following active antagonists 15: finasteride, benfluralin, permethrin, diethylhexyl phthalate, noflurazon, ethoprop, cyfluthrin, iprodione, pronamide, triflualin, dibutyl phthalate, and metolachlor. This model also assigned false positive scores to folpet and chlorothalonil; inconclusive scores were assigned to active antagonist fenarimol and inactive chemicals tetrachlorvinfos and mgk-264 15 (Table S3D). Overall, the ToxCast AR model correctly predicted 18/25 in vivo antagonist reference chemicals. The CoMPARA 18 consensus antagonist model assigned false negative AUC values to the following active antagonists: diethylhexyl phthalate and dibutyl pthalate, cyfluthrin, ethoprop, finasteride, iprodione, noflurazon, permethrin, and propargite, and assigned false positive classifications to folpet and tetrachlorvinfos (Table S3D).
DISCUSSION
Endocrine disruption prediction has become a major area of research with the rising availability of data associated with chemical exposure 45, 46. Decades of quantitative structure-activity relationship research have been undertaken by various research groups globally 25, 47–50, but the most reliable data are generated by governmental organizations pursuing new methods to evaluate endocrine disruption. While the EPA’s ToxCast AR pathway modeling efforts are extensive, they require the generation of substantial in vitro data to assign AUC values for potential bioactivity prediction, while Bayesian machine learning methods do not.
Bayesian machine learning model groups produced higher overall accuracies when predicting external in vitro reference chemicals relative to in vivo reference chemicals. In the former test set, all false positive predictions were steroid structures with the exception of the isoflavone daidzein; in the latter, issues with false negative predictions are prominent regardless of the models utilized. The Bayesian machine learning models generated with Assay Central® were more accurate at predicting AR-mediated agonism than antagonism overall. In contrast to publications by the EPA 11, 15, inaccurate classifications were not resolved as the EPA did with their confidence scores and a confirmation assay (i.e. zearalenone), and only the raw AUC scores were analyzed. Despite this simplification, it is apparent that machine learning models can limit the prevalence of false negatives for in vivo antagonist reference chemicals without the additional confirmatory testing or literature evaluation done by Kleinstreuer et al. 11.
The lack of accuracy, particularly for the in vivo antagonist test set, by multiple in silico methods (i.e. Assay Central®, EPA’s ToxCast AR pathway and CoMPARA models) suggests that improvements can be made in order to extrapolate in vivo results from in vitro data. Evaluation of other test sets shows that machine learning models can be as, or more effective at accurate prediction of potential AR-mediated endocrine disruption. Overall, the ToxCast2019-AC50full group performed well and are the ideal primary prediction set for further validation studies. Interestingly, these model groups were not those with the highest five-fold cross-validation metrics (Table S2), demonstrating the necessity for examining dataset balance and model validation with external test sets.
An important limitation of this study was the consistent prediction of steroid chemicals as active regardless of the test set, but this trend was quite prominent for the in vitro test set and spanned both agonist and antagonist chemicals (Table 4). This is a common limitation of models that use fingerprints as molecular descriptors and can be addressed by analyzing other descriptors in future studies. Similar limitations were observed in a recent study for the estrogen receptor 44. Training set chemical space coverage does not appear to be the source of these limitation, as shown by high average applicability values for incorrectly predicted chemicals (Table S5). Rather, steroids are often active chemicals across these assays and are a well-established class of compound that disrupt the endocrine system 44; Kleinstreuer et al. 11 notes classifying 17α-estradiol as inactive may need to be revised, for example. Both phthalates present in the in vivo test set are classified as active antagonists and predicted as inactive. Kleinstreuer et al. 11 describes that the metabolite of phthalates is the anti-androgenic component rather than the parent compound. Consequently, inaccurate predictions could result from an alternative mechanism. Other explanations of inaccurate predictions include in vitro experimental inconsistencies between the ToxCast/Tox21 training data and the reference data (i.e. concentrations tested and solubility constraints) as well as intrinsic variation of in vivo data 11, 13–15, 18. This is a general limitation of machine learning and other in silico prediction methods that can potentially be addressed with more advanced experimental techniques 29–31,37, 38.
Other limitations to this study are similar to those discussed in the recent estrogen receptor work 44, namely the overall system of activity designation from Bayesian models with probability-like scores and lack of an “inconclusive” designation. However, the majority-rule method utilized herein to classify chemicals for potential AR-mediated endocrine disruption appears be effective for external predictions. Additionally, while the EPA’s ToxCast AR pathway models utilized the same 1855 compounds to cover the same chemical space across assays, all chemicals tested in each assay at the time were included in the model. The Assay Central® software applied inconsistent activity thresholds to AC50 models, which could lead to disagreement between individual assay models of which chemical features translate to bioactivity across a group and hence may skew the resulting predictions. Finally, the EPA’s ToxCast AR pathway models allow for speculation on mechanisms of endocrine disruption or technology-specific assay interference. Despite these limitations, Assay Central® model groups performed similarly to or better than the corresponding ToxCast AR pathway model (Figures 1 and 2).
This study predominantly focused on using Assay Central® to develop Bayesian models, but there is continuing interest in how other machine learning approaches perform. It has compared six additional machine learning algorithms to Assay Central®, adding to the earlier work 30, 37, 38 showing the different algorithms perform similarly: the performance five-fold cross-validation metrics do vary but appear comparable for most algorithms (Figure S2, Table S4). Based on this comparison the focus was on using the Bayesian machine learning models generated with the Assay Central® framework for external test set validation. This study provides further evidence that Bayesian methods may be most suitable for bioactivity prediction 30, 37, 38, especially considering the computational cost of more advanced methods. It is important to note that all these machine learning models only utilized a single molecular descriptor (extended-connectivity fingerprints), and future studies will expand this evaluation with the use of different descriptor classes. Additional goals include evaluating alternative machine learning algorithms with external predictions as well as five-fold cross-validation, and generating consensus predictions across multiple methods for a more accurate comparison to CoMPARA 18 and other consensus models 19, 25.
This study has demonstrated that prospective prediction of external reference chemicals with Bayesian machine learning models has accuracies that rival the EPA’s published ToxCast models for AR-mediated signaling pathways 11, 15 without requiring in vitro data. Without this testing requirement, there are significant savings of time and resources by using machine learning models in place of the ToxCast AR pathway models. We have also demonstrated that the ToxCast AR model AUC score may not be optimal for training machine learning models as we have shown herein. While the CoMPARA consensus model is capable of external predictions, the use of all of the assay data in the Assay Central® models offers an important advantage in the transparency of the training data. This study therefore represents an approach for evaluating machine learning techniques for predicting endocrine disrupting bioactivity potential on the basis of molecular structure alone. Future experiments could include more in-depth comparisons of molecular descriptors and additional machine learning algorithms, and as new AR-related data is published there will be further opportunities for evaluating predictions and testing models.
Supplementary Material
ACKNOWLEDGMENTS
Grant information
We kindly acknowledge SC Johnson and Son, Inc. for funding. We also acknowledge NIH NIGMS funding to develop the software from R44GM122196-02A1. “Research reported in this publication was supported by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number R43ES031038. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.” We are also grateful to the EPA for providing the Tox21/ToxCast datasets, Dr. Alex M. Clark (Molecular Materials Informatics, Inc.) for Assay Central® support and Dr. William C. Kershaw for constructive criticism. We acknowledge Dr. Daniel P. Russo for the development of the alternative machine learning algorithm pipeline and support.
ABBREVIATIONS USED
- AC50
50% maximal response
- AUC
Area under the curve
- AR
androgen receptor
- EPA
U.S. Environmental Protection Agency
- CoMPARA
Collaborative Modeling Project for Androgen Receptor Activity
Footnotes
Competing interests:
S.E. is owner, K.M.Z., D.H.F, and T.R.L. are employees of Collaborations Pharmaceuticals, Inc. All others are SC Johnson and Son, Inc. employees.
SUPPORTING INFORMATION
Table S1 summarizes all test set chemicals used in this study and their reported activities. Table S2: summarizes the five-fold cross-validation results from final machine learning models generated in Assay Central®. Table S3 shows the prediction accuracies of each chemical across machine learning models, as well as CoMPARA and the EPA’s AR agonist and antagonist pathway models, separated by test set. Table S4: Average applicability scores from Assay Central® Bayesian models for each test set chemicals. Table S5 summarizes the applicability scores generated with external predictions by Assay Central®, separated by test set. Figure S1 presents radar plots of the training performance metrics of six machine learning algorithms and Assay Central®. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.EPA, U., Use of High Throughput Assays and Computational Tools: Endocrine Disruptor Screening Program; Notice of Availability and Opportunity for Comment, 80 Fed. Reg. 118. In 2015. [Google Scholar]
- 2.Mooradian AD; Morley JE; Korenman SG, Biological actions of androgens. Endocr Rev 1987, 8, (1), 1–28. [DOI] [PubMed] [Google Scholar]
- 3.NORD National Organization for Rare Diseases. https://rarediseases.org
- 4.Manolagas SC; O’Brien CA; Almeida M, The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol 2013, 9, (12), 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schug TT; Janesick A; Blumberg B; Heindel JJ, Endocrine disrupting chemicals and disease susceptibility. J Steroid Biochem Mol Biol 2011, 127, (3–5), 204–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.EPA, U. EPA Endocrine Disruptor Screening Program Tier 1 Battery of Assays. https://www.epa.gov/endocrine-disruption/endocrine-disruptor-screening-program-tier-1-battery-assays
- 7.Judson RS; Houck KA; Kavlock RJ; Knudsen TB; Martin MT; Mortensen HM; Reif DM; Rotroff DM; Shah I; Richard AM; Dix DJ, In vitro screening of environmental chemicals for targeted testing prioritization: the ToxCast project. Environ Health Perspect 2010, 118, (4), 485–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dix DJ; Houck KA; Martin MT; Richard AM; Setzer RW; Kavlock RJ, The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol Sci 2007, 95, (1), 5–12. [DOI] [PubMed] [Google Scholar]
- 9.Kavlock R; Chandler K; Houck K; Hunter S; Judson R; Kleinstreuer N; Knudsen T; Martin M; Padilla S; Reif D; Richard A; Rotroff D; Sipes N; Dix D, Update on EPA’s ToxCast program: providing high throughput decision support tools for chemical risk management. Chem Res Toxicol 2012, 25, (7), 1287–302. [DOI] [PubMed] [Google Scholar]
- 10.Sun H; Xia M; Austin CP; Huang R, Paradigm shift in toxicity testing and modeling. AAPS J 2012, 14, (3), 473–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleinstreuer NC; Ceger P; Watt ED; Martin M; Houck K; Browne P; Thomas RS; Casey WM; Dix DJ; Allen D; Sakamuru S; Xia M; Huang R; Judson R, Development and Validation of a Computational Model for Androgen Receptor Activity. Chem Res Toxicol 2017, 30, (4), 946–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Judson RS; Magpantay FM; Chickarmane V; Haskell C; Tania N; Taylor J; Xia M; Huang R; Rotroff DM; Filer DL; Houck KA; Martin MT; Sipes N; Richard AM; Mansouri K; Setzer RW; Knudsen TB; Crofton KM; Thomas RS, Integrated Model of Chemical Perturbations of a Biological Pathway Using 18 In Vitro High-Throughput Screening Assays for the Estrogen Receptor. Toxicol Sci 2015, 148, (1), 137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Browne P; Judson RS; Casey WM; Kleinstreuer NC; Thomas RS, Screening Chemicals for Estrogen Receptor Bioactivity Using a Computational Model. Environ Sci Technol 2015, 49, (14), 8804–14. [DOI] [PubMed] [Google Scholar]
- 14.Browne P; Kleinstreuer NC; Ceger P; Deisenroth C; Baker N; Markey K; Thomas RS; Judson RJ; Casey W, Development of a curated Hershberger database. Reprod Toxicol 2018, 81, 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleinstreuer NC; Browne P; Chang X; Judson R; Casey W; Ceger P; Deisenroth C; Baker N; Markey K; Thomas RS, Evaluation of androgen assay results using a curated Hershberger database. Reprod Toxicol 2018, 81, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelms MD; Mellor CL; Enoch SJ; Judson RS; Patlewicz G; Richard AM; Madden JM; Cronin MTD; Edwards SW, A mechanistic framework for integrating chemical structure and high-throughput screening results to improve toxicity predictions. Comp Toxicol 2018, 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mansouri K; Abdelaziz A; Rybacka A; Roncaglioni A; Tropsha A; Varnek A; Zakharov A; Worth A; Richard AM; Grulke CM; Trisciuzzi D; Fourches D; Horvath D; Benfenati E; Muratov E; Wedebye EB; Grisoni F; Mangiatordi GF; Incisivo GM; Hong H; Ng HW; Tetko IV; Balabin I; Kancherla J; Shen J; Burton J; Nicklaus M; Cassotti M; Nikolov NG; Nicolotti O; Andersson PL; Zang Q; Politi R; Beger RD; Todeschini R; Huang R; Farag S; Rosenberg SA; Slavov S; Hu X; Judson RS, CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ Health Perspect 2016, 124, (7), 1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mansouri K; Kleinstreuer N; Abdelaziz AM; Alberga D; Alves VM; Andersson PL; Andrade CH; Bai F; Balabin I; Ballabio D; Benfenati E; Bhhatarai B; Boyer S; Chen J; Consonni V; Farag S; Fourches D; Garcia-Sosa AT; Gramatica P; Grisoni F; Grulke CM; Hong H; Horvath D; Hu X; Huang R; Jeliazkova N; Li J; Li X; Liu H; Manganelli S; Mangiatordi GF; Maran U; Marcou G; Martin T; Muratov E; Nguyen DT; Nicolotti O; Nikolov NG; Norinder U; Papa E; Petitjean M; Piir G; Pogodin P; Poroikov V; Qiao X; Richard AM; Roncaglioni A; Ruiz P; Rupakheti C; Sakkiah S; Sangion A; Schramm KW; Selvaraj C; Shah I; Sild S; Sun L; Taboureau O; Tang Y; Tetko IV; Todeschini R; Tong W; Trisciuzzi D; Tropsha A; Van Den Driessche G; Varnek A; Wang Z; Wedebye EB; Williams AJ; Xie H; Zakharov AV; Zheng Z; Judson RS, CoMPARA: Collaborative Modeling Project for Androgen Receptor Activity. Environ Health Perspect 2020, 128, (2), 27002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manganelli S; Roncaglioni A; Mansouri K; Judson RS; Benfenati E; Manganaro A; Ruiz P, Development, validation and integration of in silico models to identify androgen active chemicals. Chemosphere 2019, 220, 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mansouri K; Kleinstreuer NC; Watt ED; Harris J; Judson R CoMPARA: Collaborative Modeling Project for Androgen Receptor Activiy;. 10.23645/epacomptox.5176876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams AJ; Grulke CM; Edwards J; McEachran AD; Mansouri K; Baker NC; Patlewicz G; Shah I; Wambaugh JF; Judson RS; Richard AM, The CompTox Chemistry Dashboard: a community data resource for environmental chemistry. J Cheminform 2017, 9, (1), 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ekins S, Progress in computational toxicology. J Pharmacol Toxicol Methods 2014, 69, (2), 115–40. [DOI] [PubMed] [Google Scholar]
- 23.Bender A, Bayesian methods in virtual screening and chemical biology. Methods Mol Biol 2011, 672, 175–96. [DOI] [PubMed] [Google Scholar]
- 24.Minerali E; Foil DH; Zorn KM; Lane TR; Ekins S, Comparing Machine Learning Algorithms for Predicting Drug-Induced Liver Injury (DILI). Mol Pharm 2020, 17, (7), 2628–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grisoni F; Consonni V; Ballabio D, Machine Learning Consensus To Predict the Binding to the Androgen Receptor within the CoMPARA Project. J Chem Inf Model 2019, 59, (5), 1839–1848. [DOI] [PubMed] [Google Scholar]
- 26.Gupta VK; Rana PS, Toxicity prediction of small drug molecules of androgen receptor using multilevel ensemble model. J Bioinform Comput Biol 2019, 17, (5), 1950033. [DOI] [PubMed] [Google Scholar]
- 27.Idakwo G; Thangapandian S; Luttrell J. t.; Zhou Z; Zhang C; Gong P, Deep Learning-Based Structure-Activity Relationship Modeling for Multi-Category Toxicity Classification: A Case Study of 10K Tox21 Chemicals With High-Throughput Cell-Based Androgen Receptor Bioassay Data. Front Physiol 2019, 10, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen S; Zhou D; Hsin LY; Kanaya N; Wong C; Yip R; Sakamuru S; Xia M; Yuan YC; Witt K; Teng C, AroER tri-screen is a biologically relevant assay for endocrine disrupting chemicals modulating the activity of aromatase and/or the estrogen receptor. Toxicol Sci 2014, 139, (1), 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anantpadma M; Lane T; Zorn KM; Lingerfelt MA; Clark AM; Freundlich JS; Davey RA; Madrid PB; Ekins S, Ebola Virus Bayesian Machine Learning Models Enable New in Vitro Leads. ACS Omega 2019, 4, (1), 2353–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lane T; Russo DP; Zorn KM; Clark AM; Korotcov A; Tkachenko V; Reynolds RC; Perryman AL; Freundlich JS; Ekins S, Comparing and Validating Machine Learning Models for Mycobacterium tuberculosis Drug Discovery. Mol Pharm 2018, 15, (10), 4346–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ekins S; Puhl AC; Zorn KM; Lane TR; Russo DP; Klein JJ; Hickey AJ; Clark AM, Exploiting machine learning for end-to-end drug discovery and development. Nat Mater 2019, 18, (5), 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.EPA, U. EPA ToxCast & Tox21 Summary Files from invitrodb_v3.1. https://www.epa.gov/chemical-research/toxicity-forecaster-toxcasttm-data (April 16, 2019),
- 33.Korotcov A; Tkachenko V; Russo DP; Ekins S, Comparison of Deep Learning With Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Data Sets. Mol Pharm 2017, 14, (12), 4462–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anon NTP NICEATM Reference Chemical Lists for Test Method Evaluations. https://ntp.niehs.nih.gov/whatwestudy/niceatm/resources-for-test-method-developers/refchem/index.html.
- 35.Clark AM; Dole K; Coulon-Spektor A; McNutt A; Grass G; Freundlich JS; Reynolds RC; Ekins S, Open Source Bayesian Models. 1. Application to ADME/Tox and Drug Discovery Datasets. J Chem Inf Model 2015, 55, (6), 1231–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark AM; Ekins S, Open Source Bayesian Models. 2. Mining a “Big Dataset” To Create and Validate Models with ChEMBL. J Chem Inf Model 2015, 55, (6), 1246–60. [DOI] [PubMed] [Google Scholar]
- 37.Russo DP; Zorn KM; Clark AM; Zhu H; Ekins S, Comparing Multiple Machine Learning Algorithms and Metrics for Estrogen Receptor Binding Prediction. Mol Pharm 2018, 15, (10), 4361–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zorn KM; Lane TR; Russo DP; Clark AM; Makarov V; Ekins S, Multiple Machine Learning Comparisons of HIV Cell-based and Reverse Transcriptase Data Sets. Mol Pharm 2019, 16, (4), 1620–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willighagen EL; Mayfield JW; Alvarsson J; Berg A; Carlsson L; Jeliazkova N; Kuhn S; Pluskal T; Rojas-Cherto M; Spjuth O; Torrance G; Evelo CT; Guha R; Steinbeck C, The Chemistry Development Kit (CDK) v2.0: atom typing, depiction, molecular formulas, and substructure searching. J Cheminform 2017, 9, (1), 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carletta J, Assessing agreement on classification tasks: The kappa statistic. Computational Linguistics 1996, 22, 249–254. [Google Scholar]
- 41.Cohen J, A coefficient of agreement for nominal scales. Education and Psychological Measurement 1960, 20, 37–46. [Google Scholar]
- 42.Caruana R; Niculescu-Mizil A In An empirical comparison of supervised learning algorithms, 23rd International Conference on Machine Learning, Pittsburgh, PA, 2006; Pittsburgh, PA, 2006. [Google Scholar]
- 43.Wang Y; Han R; Zhang H; Liu H; Li J; Liu H; Gramatica P, Combined Ligand/Structure-Based Virtual Screening and Molecular Dynamics Simulations of Steroidal Androgen Receptor Antagonists. Biomed Res Int 2017, 2017, 3572394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zorn KM; Foil DH; Lane TR; Russo DP; Hillwalker W; Feifarek DJ; Jones F; Klaren WD; Brinkman A; Ekins S, Machine Learning Models for Estrogen Receptor Bioactivity and Endocrine Disruption Prediction. In press 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takemura H; Sakakibara H; Yamazaki S; Shimoi K, Breast cancer and flavonoids - a role in prevention. Curr Pharm Des 2013, 19, (34), 6125–32. [DOI] [PubMed] [Google Scholar]
- 46.Rodgers KM; Udesky JO; Rudel RA; Brody JG, Environmental chemicals and breast cancer: An updated review of epidemiological literature informed by biological mechanisms. Environ Res 2018, 160, 152–182. [DOI] [PubMed] [Google Scholar]
- 47.Loughney DA; Schwender CF, A comparison of progestin and androgen receptor binding using the CoMFA technique. J Comput Aided Mol Des 1992, 6, (6), 569–81. [DOI] [PubMed] [Google Scholar]
- 48.Waller CL; Juma BW; Gray LE Jr.; Kelce WR, Three-dimensional quantitative structure--activity relationships for androgen receptor ligands. Toxicol Appl Pharmacol 1996, 137, (2), 219–27. [DOI] [PubMed] [Google Scholar]
- 49.Bohl CE; Chang C; Mohler ML; Chen J; Miller DD; Swaan PW; Dalton JT, A ligand-based approach to identify quantitative structure-activity relationships for the androgen receptor. J Med Chem 2004, 47, (15), 3765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vinggaard AM; Niemela J; Wedebye EB; Jensen GE, Screening of 397 chemicals and development of a quantitative structure--activity relationship model for androgen receptor antagonism. Chem Res Toxicol 2008, 21, (4), 813–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.