

Abstract
Objective:
Abnormal deposition of the antimicrobial peptide amyloid beta (Aβ) is a characteristic of Alzheimer’s Disease (AD). The objective of this study was to elucidate risk factors for brain Aβ in a cohort enriched for human immunodeficiency virus (HIV) and other neurotropic pathogens.
Design:
Cross-sectional cohort study.
Methods:
We examined autopsy brains of 257 donors with a mean age of 52.8 years; 62% were male; and 194 were HIV+ and 63 HIV-. Hyperphosphorylated tau (p-tau) and Aβ were identified in frontal and temporal regions by immunohistochemistry. APOE genotyping was performed. Clinical and neuropathological predictors for Aβ were identified in univariate analyses, and then tested in multivariate regressions.
Results:
Cortical Aβ was identified in 32% of the sample, and active brain infection in 27%. Increased odds of Aβ were seen with increasing age and having an APOE ε4 allele; for the overall sample, HIV+ status was protective and brain infection was not a predictor. Within the HIV+ population, predictors for Aβ were duration of HIV disease and APOE alleles, but not age. When HIV disease duration and other HIV parameters were introduced into models for the entire sample, HIV disease duration was equivalent to age as a predictor of Aβ.
Conclusion:
We hypothesize that dual aspects of immune suppression and stimulation in HIV, and beneficial survivor effects in older HIV+ individuals, account for HIV+ status decreasing, and HIV duration increasing, odds of Aβ. Importantly, with HIV, disease duration replaces age as an independent risk for Aβ, suggesting HIV-associated accelerated brain senescence.
Keywords: HIV, beta amyloid, brain infection
Introduction
The role of neuroinflammation in Alzheimer’s Disease (AD) has renewed interest in the potential for neurotropic infection to participate in AD pathogenesis (1; 2; 3; 4; 5). There are concerns that human immunodeficiency virus (HIV) may pre-dispose to accumulation of abnormal proteins characteristic of AD neurodegeneration (6; 7). People with HIV constitute an informative population to explore impacts of neurotropic infection on neurodegeneration: they are routinely monitored for immunologic and virologic status, and the timing and mechanisms by which HIV enters brain and impacts central nervous system (CNS) immunity have been elaborated (8; 9). Thus, unlike other viral infections, the duration, clinical severity, and CNS impacts of HIV disease can be uniformly ascertained and examined in the context of neurodegenerative pathology.
Cortical deposition of amyloid beta (Aβ) is a hallmark of AD, and in some research nosologies, is considered the earliest manifestation of disease, preceding clinical deficits by up to 20 years (10). Aβ peptides have anti-microbial properties, and many infectious agents, including HIV, bias cellular processing of amyloid precursor protein to enhance formation of Aβ in the innate immune response (11; 12). Thus, middle-aged HIV+ populations, harboring a potentially amyloidogenic microbe that stimulates innate immunity, and that cannot be eradicated but can be measured, provide opportunity to examine associations between infection and Aβ deposition. Accordingly, small groups of predominantly middle aged, HIV+ individuals have been examined with positron emission tomography (PET), revealing no evidence of brain amyloid in excess of age-matched HIV- controls (13; 14; 15). However, conventional PET imaging may be insensitive to early stages of amyloid deposition, and neuroimaging lacks the resolution afforded by direct examination of brain tissue by light microscopy (16).
The Manhattan HIV Brain Bank (MHBB) has operated a longitudinal, observational cohort study, brain donation program, and biorepository since 1999. MHBB also conducts brain donation programs with HIV- individuals whose medical and psychiatric morbidities are prevalent in HIV disease. Utilizing densely annotated brain tissues from HIV+ and demographically-matched HIV- donors, we investigated risks for Aβ deposition in mid-frontal and inferior temporal cortex and hippocampus, neuroanatomical regions characteristic of early AD involvement.
Materials and methods
Patient population:
Brains were obtained between 1999 and 2019 from MHBB donors. The MHBB operates under oversight of the Icahn School of Medicine at Mount Sinai Institutional Review Board. All brain donations were obtained after approved consent, either from decedents during pre-mortem study via anatomical gift documentation, or from primary next of kin via authorization for autopsy and medical research. At the time of analysis, the demographic composition of the MHBB was: 61% male, 39% female; 45% black, 30% Hispanic, 24% white; and mean age of 53.0 +/− 11.0 years at study termination. There were no demographic differences (age, sex, race/ethnicity) in the HIV+ and HIV- donor populations for this study.
Clinical annotation:
For participants who completed assessments in prospective study, medical illnesses and medications were elucidated by interview and medical record review, and laboratories including CD4 T-cell counts and plasma HIV RNA loads were obtained. Substance use (SUD) and mood disorders (depression, dysthymia, bipolar) were elaborated by psychiatric interview (the Composite International Diagnostic Interview v 2.1 or the Psychiatric Research Interview for Substance and Mental Disorders v 1.9B). For all donors, a review of the electronic medical record was conducted at the time of autopsy, inclusive of medical, psychiatric and social worker assessments. 56% of the sample came from the prospective study (75% of HIV+ donors, and none of the HIV-). For HIV+ donors, the duration of HIV disease, last CD4 T-cell count and plasma HIV load prior to death, and last combination antiretroviral therapy (cART) regimen taken within 2 weeks of demise, were recorded. For all donors, presence or absence of SUD, mood disorder, hepatitis B virus (HBV), hepatitis C virus (HCV), herpes virus (simplex, zoster, or cytomegalovirus), and anti-herpes therapy, were recorded.
Brain processing and analysis:
The entire brain was removed at autopsy, and half fixed in 10% phosphate-buffered formalin for routine processing and generation of a minimum of 50 tissue blocks for histologic assessment by a board-certified neuropathologist with expertise in neuro-infectious disorders (SM). Brain pathologies were categorized using standard data dictionaries (17). Neuropathologies recorded for this study included: HIV-associated pathology (HIV leptomeningitis, microglial nodule encephalitis, and leukoencephalopathy), other brain infections (bacterial, mycobacterial, viral, fungal, and protozoan), infarcts, and other focal lesions (tumor, intracranial hemorrhage, contusion, demyelination, optic degeneration, aneurysm, shunt tract). Terminal neuropathologies (anoxic and metabolic changes) were also noted.
Blocks of mid frontal gyrus (primarily Brodman’s area 9) and medial temporal lobe with representation of hippocampus, entorhinal- and neo-cortices (over 90% mid-hippocampus) were sectioned at 5 microns and stained with hematoxylin and eosin, Luxol fast blue, and Bielschowsky techniques. Immunohistochemistry was performed on a Ventana Benchmark XT autostainer (Roche Tissue Diagnostics, Tucson, Arizona), utilizing primary antibodies to detect Aβ (clone 4G8, recognizing the Aβ17–24 fragment, Biolegend, San Diego, California) and p-tau (clone AT8, recognizing the ser202/thr205 phosphorylated epitope of tau, Thermo Fisher Scientific, Waltham, MA). Slides were independently reviewed by three neuropathologists (SM, EC, JFC) and scored for the presence and location of Aβ and p-tau. If scores did not agree, differences were adjudicated by consensus.
Double label IHC was manually performed on selected cases using the ImmPRESS Duet Double Staining Polymer Kit (Vector Laboratories, Burlingame, CA). A mixture of primary antibodies to detect Aβ and either GFAP (Dako #Z0334, Santa Clara, CA), Iba1 (Invitrogen #PA5–27436, Carlsbad, CA), or CD68 (SP 251, Sigma-Aldrich, St. Louis, MO) was used. ImmPACT SG (blue/gray) was applied to detect Aβ, followed by ImmPACT Vector Red to detect GFAP, Iba1, or CD68.
Genotyping:
APOE genotyping was performed using DNA extracted from peripheral blood mononuclear cells or brain tissue with the Qiagen DNeasy blood and tissue kit according to manufacturer’s instructions (Qiagen USA, Germantown, MD). Taqman Assays-on-demand (Applied Biosystems, Foster City, CA) were used to target rs429358 and rs7412 to determine APOE alleles. Assays were performed in triplicate according to manufacturer’s protocols using the Applied Biosystems 7900HT system.
Statistical procedures:
Presence or absence of cortical Aβ deposition was modeled as a categorical variable in multivariate (nominal) logistic regression. Initially, univariate analyses to predict amyloid were conducted to determine candidates for multivariate models; factors with association at p<0.1000 were entered into logistic regression, and odds ratios and confidence intervals calculated. Additional modeling used χ2, Fisher’s exact test, or analysis of variance to investigate associations between HIV status and duration of HIV infection on clinical and neuropathologic factors. Analyses were generated using JMP version 9.0 for Macintosh, and SAS/STAT Version 9.4 of the SAS System for Windows (SAS Institute Inc., Cary, NC, USA).
Results
Population characteristics and clinical predictors of Aβ:
Clinical characteristics of the overall population, and analyses for associations with extracellular Aβ deposition in frontal and/or temporal gray matter are presented in table 1. The average age of the sample was 52.8 years, with a range of 21 to 86 years. The population was predominantly black and Hispanic, with slightly more men than women. The distribution of APOE genotypes was: 0.4% ε2/ε2; 13.6% ε2/ε3; 4.7% ε2/ε4; 56.0% ε3/ε3; 23.0% ε3/ε4; and 2.3% ε4/ε4; thus, allelic frequencies (9.5% ε2, 74.3% ε3, 16.1% ε4) were consistent with other racially heterogeneous New York City populations (18). 30% had one or more ε4 alleles, and 19% had one or more ε2 alleles.
Table 1.
Clinical characteristics of the overall study population, and analyses for associations with Aβ deposition
| Total sample (n=257) | Aβ absent (n=174) | Aβ present (n=83) | Association with Aβ | ||
|---|---|---|---|---|---|
| Univariate, P value | Multivariate1, P value, (OR; 95% CI) | ||||
| Mean age (SD) | 52.8 (12.1) | 50.3 (12.4) | 58.0 (9.8) | <0.0001 | <0.0001 (1.07; 1.04,1.11) |
| Male gender, n (%) | 160 (62%) | 116 (67%) | 44 (53%) | 0.0356 | |
| Race/ethnicity, n (%) | 0.8512 | ||||
| Hispanic | 86 (33%) | 60 (34%) | 26 (31%) | ||
| Other | 7 (3%) | 5 (3%) | 2 (2%) | ||
| HIV positive, n (%) | 194 (75%) | 139 (80%) | 55 (66%) | 0.0187 | 0.0274 (0.47; 0.23,0.92) |
| APOE ε4, n (%) | 77 (30%) | 40 (23%) | 37 (45%) | 0.0005 | <0.0001 (3.97; 2.10,7.69) |
| APOE ε2, n (%) | 48 (19%) | 38 (22%) | 10 (12%) | 0.0633 | |
| HCV, n (%) | 120 (47%) | 78 (45%) | 42 (51%) | 0.3859 | |
| HBV, n(%) | 111 (43%) | 79 (45%) | 32 (39%) | 0.3006 | |
| Any viral hepatitis, n (%) | 166 (65%) | 111 (64%) | 55 (66%) | 0.6985 | |
| Any herpes infection, n (%) | 102 (40%) | 70 (40%) | 32 (39%) | 0.7974 | |
| On herpes therapy, n (%) | 65 (25%) | 45 (26%) | 20 (24%) | 0.7608 | |
| Hypertension | 119 (46%) | 73 (42%) | 46 (55%) | 0.0429 | |
| Diabetes mellitus | 68 (26%) | 37 (21%) | 31 (37%) | 0.0063 | |
Statistically significant (p<0.05) terms shown.
Age, sex, HIV status, hypertension, diabetes, and both APOE alleles had significant or trend-level association with Aβ deposition in univariate analyses. Age, HIV status, and APOE ε4 remained significant predictors of Aβ when entered into nominal logistic regression, with r2 = 0.1724, χ2 = 55.76, p<0.0001 for the overall model. Odds ratios (OR) and 95% confidence intervals (CI) for significant predictors were: for age, 1.07 [1.04, 1.11] for every year change, χ2 = 19.57; for APOE ε4, 3.97 [2.10, 7.69], χ2 = 17.40; and for HIV+ status, 0.47 [0.23, 0.92], χ2 = 4.87.
Within HIV analysis for clinical predictors of Aβ:
As HIV was a protective factor for amyloid deposition, we next examined clinical predictors of Aβ within this group. Variables assessed included demographic factors, HIV-specific indices of immunologic/virologic status, presence and type of cART at death, and factors with significantly different prevalence in HIV+ and HIV- individuals. These factors included SUD (present in 77% of HIV+ and 43% of HIV-, χ2 = 26.35, p<0.0001), mood disorders (68% of HIV+, 11% of HIV-, χ2 = 62.33, p<0.0001), HBV (50% of HIV+, 22% of HIV-, χ2 = 14.96, p=0.0001), and herpesvirus infection (46% of HIV+, 19% of HIV-, χ2 = 14.86, p=0.0001).
Within the HIV+ population, significant or trend-level univariate associations with Aβ were seen for age, sex, APOE alleles, HIV disease duration, diabetes, hypertension, plasma HIV load, cART, and CD4 T-cell count (table 2). While many variables were related, variable inflation factors were all under 5. When entered into nominal logistic regression to predict Aβ, the overall model was significant with r2 = 0.2381, χ2 = 50.60, p<0.0001. Predictors that remained significant were APOE ε4 and ε2 alleles, and duration of HIV disease; age and other immunovirologic variables were not significant, although sex and undetectable viral load remained at trend level (p values 0.0626 and 0.0777, respectively). Odds ratios and 95% CI for significant predictors were: HIV duration, 1.07 [1.01, 1.31] for every year change, χ2 = 4.60; APOE ε4, 4.76 [2.07, 11.63], χ2 = 12.77; and APOE ε2, 0.23 [0.05, 0.84], χ2 = 4.08.
Table 2.
Clinical characteristics of the HIV+ study population, and analyses for associations with Aβ deposition
| HIV pos (n=194) | Aβ absent (n=139) | Aβ present (n=55) | P value for association with Aβ | ||
|---|---|---|---|---|---|
| Univariate, P value | Multivariate1, P value, (OR; 95% CI) | ||||
| Mean age (SD) | 52.6 (0.9) | 50.6 (11.1) | 57.7 (9.4) | <0.0001 | |
| Male gender, n (%) | 121 (62%) | 93 (67%) | 28 (51%) | 0.0396 | |
| SUD (any), n (%) | 150 (77%) | 109 (78%) | 41 (75%) | 0.5620 | |
| Depression spectrum disorder1, n (%) | 131 (68%) | 95 (68%) | 36 (65%) | 0.6985 | |
| APOE ε4, n (%) | 59 (30%) | 32 (23%) | 27 (49%) | 0.0005 | 0.0004 (4.76; 2.07,11.63) |
| APOE ε2, n (%) | 37 (19%) | 31 (22%) | 6 (11%) | 0.0748 | 0.0434 (0.23; 0.05,0.84) |
| HBV, n (%) | 97 (50%) | 73 (53%) | 24 (44%) | 0.2658 | |
| Any herpes infection, n (%) | 90 (46%) | 62 (45%) | 28 (51%) | 0.4279 | |
| Hypertension | 84 (43%) | 55 (40%) | 29 (53%) | 0.0955 | |
| Diabetes mellitus | 42 (22%) | 24 (17%) | 18 (33%) | 0.0184 | |
| cART at death2, n (%) | 148 (77%) | 101 (73%) | 47 (85%) | 0.0634 | |
| Protease Inhibitor in cART regimen, n (%) | 84 (57% of treated) | 59 (58% of treated) | 25 (53% of treated) | 0.5506 | |
| INI* in cART regimen, n (%) | 38 (26% of treated) | 22 (22% of treated) | 16 (34% of treated) | 0.1120 | |
| NNRTI** in cART regimen, n(%) | 50 (34% of treated) | 36 (36% of treated) | 14 (30% of treated) | 0.4832 | |
| Mean duration HIV3 (SD) | 15.5 (7.5) | 14.0 (7.0) | 19.2 (7.5) | <0.0001 | 0.0321 (1.07; 1.01,1.31) |
| Log Plasma VL4, Median (Q1, Q3) | 1.93 [1.69, 4.81] | 2.34 [1.69,5.06] | 1.69 [1.69,2.59] | 0.0064 | |
| Undetectable VL4, n (%) | 92 (48%) | 57 (41%) | 35 (64%) | 0.0050 | |
| CD4 count1, Median (Q1, Q3) | 156 (56,394) | 133 (35,298) | 188 (103,537) | 0.0014 | |
| CD4>200 cells/mm31, n (%) | 82 (43%) | 55 (40%) | 27 (49%) | 0.2274 | |
INI: Integrase inhibitor;
NNRTI: non-nucleoside reverse transcriptase inhibitor
Statistically significant (p<0.05) terms shown
Data not available for 2 cases
Data not available for 1 case
Data not available for 14 cases (10 Aβ absent, 4 Aβ present)
Data not available for 3 cases
With the complex relationship of HIV status (protective in the overall population for Aβ) and HIV disease duration (longer duration a risk for Aβ in the HIV population), we wished to further explore these variables in the entire population. Accordingly, HIV- individuals were coded as “0” years duration HIV, as having undetectable viral loads, and as not being on cART; then, HIV status, age, sex, APOE alleles, HIV duration, HIV load, and cART were entered into nominal logistic regression to predict Aβ for the entire population. The model was significant (p<0.0001) with r2 = 0.1932, and χ2 = 58.91. Significant predictors for amyloid were: HIV status (OR 0.19, CI [0.034, 0.92], χ2 = 4.02, p=0.0449), age (OR 1.04, CI [1.01, 1.08], χ2 = 7.06, p=0.0079), sex (OR for males 0.51, CI [0.27, 0.97], χ2 =4.34, p=0.0373), APOE ε4 (OR 3.95, CI [2.02, 7.98], χ2 =15.44, p<0.0001), APOE ε2 (OR 0.39, CI [0.14, 0.94], χ2 =3.98, p=0.0462), and HIV disease duration (OR 1.07, CI [1.01, 1.14], χ2 =4.66, p=0.0309); again, while having HIV+ status was protective, longer duration HIV disease constituted an independent risk for Aβ.
Histological patterns of Aβ:
We examined patterns of Aβ deposition, to determine if distribution differed between HIV+ and HIV- individuals. The HIV+ group was split into short and long duration HIV for this analysis, to aid in understanding the complex HIV risk associations for Aβ. The grouping of HIV+ individuals was based on the median (15 years) for HIV duration, which was an inflection point for Aβ deposition (figure 1).
Figure 1.
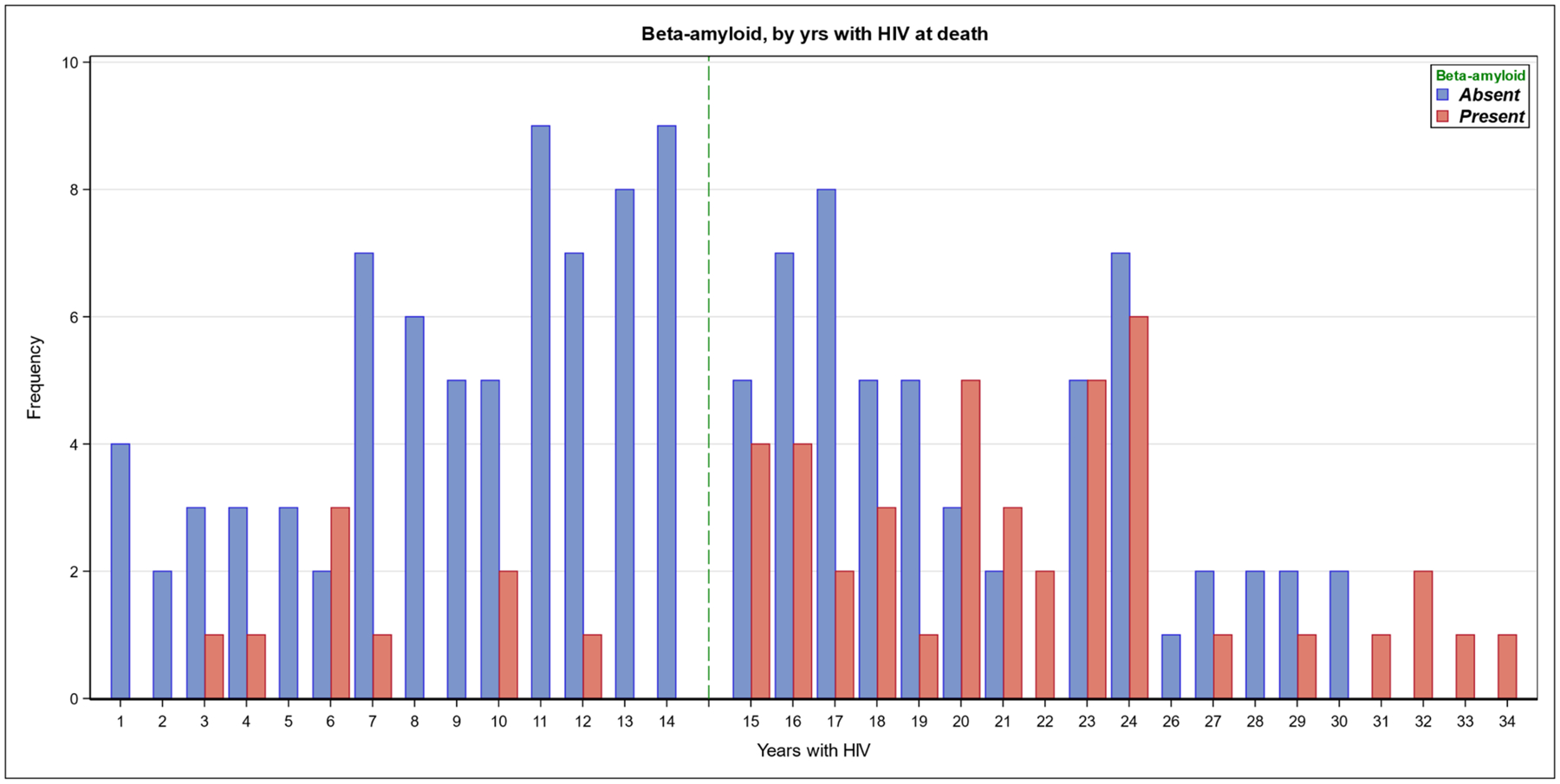
Aβ accumulation by years duration of HIV infection Numbers of HIV+ individuals with (red bars) or without (blue bars) cortical Aβ deposition, displayed by the number of years HIV disease duration. The median of the distribution for HIV duration is 15 years, denoted by a dotted line. The frequency of Aβ deposition demonstrates a significant increase at and above the median.
The following patterns of Aβ were recorded (figure 2): extracellular deposits in gray matter (used for analyses described above; the majority were diffuse “lake-like” or “fleecy” accumulations with variable size, and in 22 patients, also dense plaque cores) (19); extracellular deposition in the sub-pial molecular layer (a subset of gray matter Aβ, recorded because of the superficial gliosis commonly encountered with infectious processes that circulate through cerebrospinal fluid); deposits in white matter (recorded because of historical observations of WM amyloid precursor protein accumulations in untreated disease); cerebral congophilic angiopathy (CAA); and sharply-demarcated staining within glial processes. In a subset of cases, double IHC staining for Aβ and either GFAP, CD68, or Iba1 revealed that glial deposits were in variable proportions of astrocytes and microglial cells (figure 2). Intraneuronal Aβ staining, although previously reported in HIV (20), was not analyzed, as it showed predictable neuroanatomic specificity (for example, it was present routinely in lateral geniculate nucleus regardless of HIV status), as well as batch staining effects.
Figure 2.
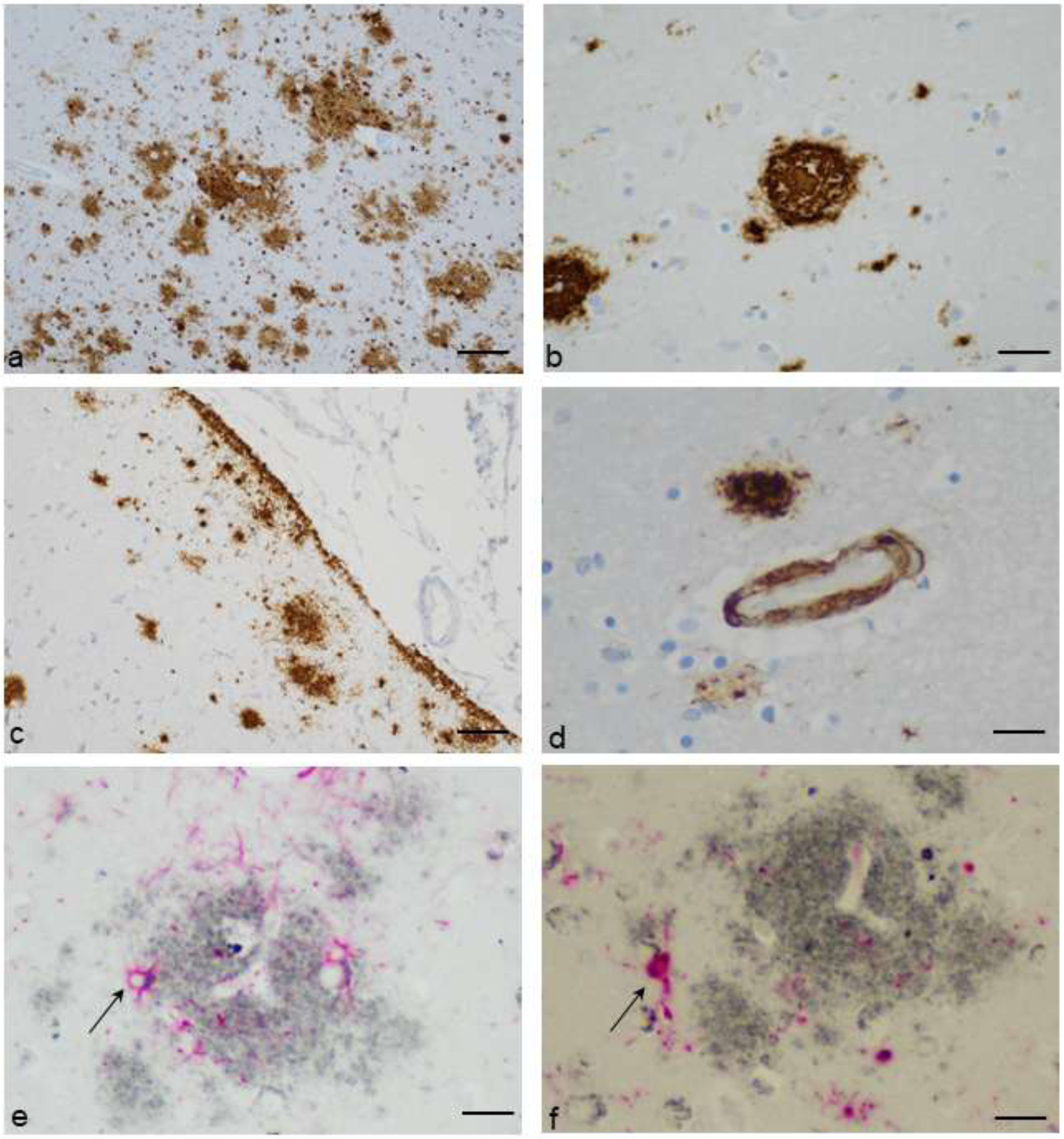
Patterns of Aβ deposition. a. Diffuse amyloid plaques of varying sizes in cerebral cortex, with and without paravascular locations (Aβ immunostain with diaminobenzidine chromogen and hematoxylin counterstain, original magnification 100X, scale bar = 100um). b. A dense core plaque (Aβ immunostain with diaminobenzidine chromogen and hematoxylin counterstain, original magnification 400X, scale bar = 25um). c. Subpial, diffuse deposits of amyloid (Aβ immunostain with diaminobenzidine chromogen and hematoxylin counterstain, original magnification 200X, scale bar = 50um). d. Cerebral congophilic angiopathy, with adjacent diffuse plaques and glial Aβ (Aβ immunostain with diaminobenzidine chromogen and hematoxylin counterstain, original magnification 600X, scale bar = 16um). e. Aβ plaque (gray) and reactive astrocytes identified with immunohistochemical stain for glial fibrillary acidic protein (GFAP) (red). Aβ is seen within astrocyte cytoplasm (representative arrow). (Immunohistochemical stain for Aβ and GFAP with ImmPACT SG and ImmPACT Vector Red chromogens, original magnification 600X, scale bar = 16um). f. Aβ plaque (gray) and reactive microglia identified with immunohistochemical stain for Iba1 (red). Aβ is seen within microglial cell cytoplasm (representative arrow). (Immunohistochemical stain for Aβ and Iba1 with ImmPACT SG and ImmPACT Vector Red chromogens, original magnification 600X, scale bar = 16um).
Participants who were HIV- and those with long duration HIV were older than those with short duration HIV; accordingly, in all patterns or distributions except white matter deposition, both HIV- and HIV long duration groups showed greater frequencies of Aβ in pairwise comparisons (table 3). Individuals with long duration HIV and those who were HIV- showed trend level differences in age (Student’s t test p=0.0676); there was no difference in frequency of cortical Aβ deposition or CAA between these two groups. Individuals with long duration HIV had more white matter and less glial Aβ than the HIV- group at trend level (white matter amyloid, χ2=3.348 p =0.0673; glial amyloid, χ2=2.751 p=0.0972). Thus, there was a suggestion that HIV might alter the relationship between glia and amyloid in both gray and white matter, consistent with concepts of altered microglial and astroglial biology in HIV infection; however, this is only speculative, as significance was only at trend level.
Table 3.
Histologic patterns of Aβ accumulation in HIV-, HIV+ short duration, and HIV+ long duration participants
| HIV negative (n=63) | HIV duration<15 (n=82) | HIV duration>=15 (n=98) | P-value | |
|---|---|---|---|---|
| Mean Age1,2,3 (SD) | 53.3 (14.9) | 46.6 (9.8) | 56.8 (9.9) | <0.0001 |
| Gray matter Aβ1,2, n (%) | 28 (44%) | 9 (11%) | 42 (43%) | <0.0001 |
| White matter Aβ3 , n (%) | 2 (3.2%) | 4 (4.9%) | 11 (11.2%) | 0.1516 |
| Aβ at pia limitans1,2 (molecular layer), n (%) | 8 (12.7%) | 1 (1.2%) | 16 (16.3%) | 0.0009 |
| Glial Aβ1,2,3, n (%) | 38 (60%) | 20 (24%) | 46 (47%) | <0.0001 |
| Congophilic angiopathy1,2, n (%) | 6 (9.5%) | 1 (1.2%) | 9 (9.2%) | 0.0344 |
| Frontal cortex Aβ1,2, n (%) | 25 (40%) | 8 (10%) | 40 (41%) | <0.0001 |
| Temporal cortex Aβ1,2 n (%) | 15 (24%) | 5 (6.1%) | 27 (28%) | 0.0003 |
Duration of HIV disease data not available for 14 individuals. Analyses by Fisher’s exact test with exception of age, which used simple ANOVA.
Pairwise comparison between HIV- and short duration HIV+ significant (p<0.05)
Pairwise comparison between long duration HIV+ and short duration HIV+ significant (P<0.05)
Pairwise comparison between HIV- and long duration HIV+ at trend level (P<0.10)
Other neuropathologies and Aβ:
There was a high rate of significant neuropathologies in this autopsy sample, with 72% of brains displaying abnormalities, inclusive of terminal anoxic and metabolic changes. The frequency of infectious brain pathologies was higher in HIV+ (34%) than HIV– (6.4%); 24% of the HIV+ sample had active HIV and 14% other infections. Brain infection had trend level significance in association with Aβ, driven by HIV pathology (p values for Aβ association with: any brain infection 0.0951; active brain HIV 0.0777; other brain infection 0.6789). The direction of association was negative; that is, with infection there was less Aβ deposition. Similarly, other focal brain pathologies were more common in HIV+ (45%) than HIV- (21%), with negative trend-level associations for Aβ (p = 0.0862). Neuronal accumulations of p-tau were seen with similar frequency in HIV+ (60%) and HIV- (68%); this was the only histology demonstrating a significant positive association with Aβ (p<0.0001). When all pathologies were introduced into multivariate analysis, only neuronal p-tau remained a significant predictor of Aβ. Neuronal p-tau was roughly twice as frequent as Aβ in the entire sample. Only 17 (6.6%) of 257 brains had Aβ deposition in the absence of neuronal p-tau; 14 (82%) of these were from HIV+ donors.
Finally, for the entire sample, we added neuronal p-tau to clinical predictors of Aβ (age, HIV status, sex, APOE alleles, with duration of HIV, viral load, and cART set at the null level for HIV-). The model was significant (p<0.0001) with r2 = 0.2212 and χ2 = 67.448. HIV status (OR 0.18, CI [0.03, 0.90], p=0.0417), male sex (OR 0.46, CI [0.24, 0.87], p=0.0181), both APOE ε2 (OR 0.37, CI [0.13, 0.89], p=0.0353) and ε4 (OR 4.90, CI [2.40, 10.49], p<0.0001) alleles, the duration of HIV disease (OR 1.08 for every year, CI [1.02, 1.15], p=0.0131), and neuronal p-tau (OR 3.31, CI [1.47, 7.75], p=0.0045) were significant predictors; age was not a significant predictor of Aβ.
Discussion
Abnormal accumulations of Aβ and p-tau are central to current nosologies of AD, and essential to its histologic diagnosis (21). Clinical manifestations of AD begin an exponential rise in the 7th decade, and many models of pathogenesis presume accumulation of abnormal proteins predates symptom onset by one to two decades (10; 22). Some models postulate amyloid deposition is the earliest biochemical lesion, preceding neuronal degeneration (10; 23). Thus, disease initiation and amyloid deposition might occur in the 5th and 6th decades of life, rendering middle-aged populations ideal for studying factors that increase risks for abnormal proteostasis. At age 50, 10% or fewer individuals demonstrate brain amyloid by PET imaging, dependent on APOE genotype (24; 25). However, neuroimaging is less sensitive than light microscopy, and the earliest stages of brain Aβ accumulation are missed in PET studies (16). Our autopsy population, with a mean age of 52.8 years, is well suited to investigating risks for the earliest stages of Aβ deposition. We examined neuroanatomical regions typical of early Aβ and p-tau accumulation in AD. The finding that 32% of our population had parenchymal Aβ is consistent with the premise that this early biochemical lesion occurs in middle age. 72% of our sample died at 60 years of age or younger; of this subgroup, Aβ was present in 26%.
Neuronal p-tau was present in 62% of our sample, suggesting Aβ deposition is not the earliest event in AD pathogenesis, although it is unclear what aspects of either amyloid or p-tau contribute to future development of AD. These proteins cannot be reliably placed on a timeline for developing AD, and may represent “benign” accumulations of aging, or indicators of other disorders such as traumatic brain injury. Morphologically, most parenchymal amyloid in our cohort was diffuse, and there is no human data to suggest diffuse amyloid transforms into neuritic plaque. Many cognitively normal individuals demonstrate amyloid on PET, and neuronal p-tau in individuals coming to autopsy in the 2nd and 3rd decade is well documented (25; 26). However, when Aβ is detected by PET, future risk for incident dementia increases (27). It is unclear if this predictive power applies in the context of a chronic, neurotropic infection with immunologic consequence, such as HIV.
HIV has a bivalent relationship to systemic immunity: while untreated disease results in immunosuppression, under cART there may be immune stimulation (28). Magnetic resonance spectroscopy (MRS) demonstrates HIV-associated neuroinflammation in all disease stages, with and without therapy, albeit waning over time with cART (29; 30). Thus, the duration of HIV’s CNS impact can be estimated through ascertaining a date of initial diagnosis, and disease severity can be measured by CD4 counts and viral loads. The ability to assess a longitudinal burden of HIV stands in contrast to other neurotropic viruses implicated in AD, where point-in-time estimates of viral activity are apparent only with symptomatic activation or in autopsy brain tissue. For example, herpesviruses are implicated in AD due to brain virus detection in proximity to amyloid, and identification of brain anti-microbial molecular signatures with elevated viral transcripts (5; 4). Many non-herpes microbes have been similarly implicated, raising questions about causal inferences (5; 4; 31; 3).
While our autopsy study is cross-sectional, it is advantaged by having measures of HIV duration and activity, allowing examination of these factors in concert with traditional risks for Aβ. In our cohort, traditional risk factors behaved in the expected manner: increasing age, positive APOE ε4 status, and female sex increased risk for Aβ, and APOE ε2 was protective. Furthermore, another characteristic of neurodegeneration – neuronal p-tau – also constituted an independent risk. It has been observed that the most important genetic risk for Aβ, APOE ε4, also conveys risk for p-tau (32). The association of both neurodegenerative proteins may reflect a common biological senescence with shared genetic susceptibility.
An important aspect of our study was the complex relationship between HIV status, infection duration, age, and Aβ. Within the HIV+ sample, longer HIV duration predicted Aβ, and undetectable viral load was a trend-level positive predictor. This is partially consistent with a previous autopsy study, where higher plasma HIV loads predicted lower likelihood of Aβ (7). This prior within-HIV study also demonstrated association of Aβ with increasing age, but did not examine HIV duration. The fact that HIV duration replaced age as a predictor in our study is striking, as it is widely accepted that age is the strongest risk for neurodegeneration (33). Age is a surrogate for unclear biological processes, as multiple genetic and environmental factors contribute to senescence. One study examining the brain methylome observed that longer duration HIV correlated with epigenetic evidence of accelerated aging (34). Thus, HIV duration may be a critical variable in advancing brain biologic age, and our study suggests disease duration replaces chronologic age as the pertinent time-dependent variable for predicting deposition of proteins associated with senescence and neurodegeneration.
Another aging change is the development of dysregulated immunity with a pro-inflammatory bias, termed immunosenescence (35). In treated HIV disease, elevated inflammatory markers may occur after years of cART, including mediators of innate immunity, such as IFNα and TNFα (36). Relevant to neurodegeneration, some literature suggests autoimmune diseases characterized by innate immunity confer increased risk for AD, and that exposure to anti-TNFα agents mitigates risk (37; 38). Increased risk for Aβ in long duration HIV may be predicated on persistent immune stimulation, with duration of inflammation more critical to amyloid production than chronologic age. This hypothesis could explain enhanced risk for Aβ with undetectable viral load, as control of viremia is a predicate for restored immune function.
However, accelerated senescence is seemingly counter to the protective effect of HIV status on amyloid in the entire sample. Explanations for this may be two-fold. The majority (72%) of younger, HIV+ individuals with shorter duration disease had CD4 counts <200 cells/mm3 and detectable viral loads; thus, immunocompromise may have decreased risk. In the older, longer duration HIV group, survivor bias may have pertained, with factors like advantageous health behaviors mitigating risk. While a prior American autopsy study described increased risk for amyloid with HIV, its HIV- controls were a historical sample from Germany, with sampling of brain regions not typical for early stages of Aβ deposition (39; 26). Differences in Aβ frequency (29.3% of HIV+, 25.8% of historical HIV-) may have reflected demography, geographic location, and neuroanatomical sites examined. In contrast, our study used a contemporaneous HIV- group enriched for HIV-associated comorbidities, with uniform neuroanatomic sampling.
Finally, Aβ was not associated with active brain infection in our study. In animal models demonstrating infectious associations, therapy lessens Aβ deposition (11). 77% of our HIV+ population was on cART at death, and 25% of the overall population received anti-herpetic medication; it is possible that therapies were mitigating factors.
There are limitations to our study. Amyloid was assessed with an antibody to the Aβ17–24 fragment, which recognizes longer peptides in the beta secretase pathway; while abnormal, these may not be fibrillar. As an autopsy study, donors had serious medical morbidity, and findings might not pertain to healthier individuals. Regardless, the intriguing relationships between HIV and Aβ in our population support the need for further investigation into the immunologic and epigenetic bases of this association, as well as neuroimaging confirmation of the impacts of disease duration in healthier HIV+ populations.
Acknowledgements:
The authors thank the participants and staff of the Manhattan HIV Brain Bank for their contributions to this work, and Dr. Lisa Spielman for helpful discussions regarding the analytic plan.
Supported by grants from the National Institutes of Health: U24MH100931 (The Manhattan HIV Brain Bank), RF01AG060961, R01AG054008, R01NS095252, P30AG066514.
Footnotes
Conflict of interest: The authors declare that they have no conflicts of interest.
References
- 1.Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol 2015; 14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komaroff AL. Can infections cause Alzheimer’s Disease? JAMA 2020; 324 (3):239–240. [DOI] [PubMed] [Google Scholar]
- 3.Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, et al. Microbes and Alzheimer’s disease. J Alzheimers Dis 2016; 51:979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Readhead B, Haure-Mirande J-V, Funk CC, Richards MA, Shannon P, Haroutunian V, et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 2018; 99 (1):64–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris SA, Harris EA. Molecular mechanisms for herpes simplex virus type 1 pathogenesis in Alzheimer’s Disease. Front Aging Neurosci March 2018; 10 (48): doi: 10.3389/fnagi.2018.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ortega M, Ances BM. Role of HIV in amyloid metabolism. J Neuroimmune Pharmacol 2014; 9:483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soontornniyomkij V, Moore DJ, Gouaux B, Soontornniyomkij B, Sinsheimer JS, Levine AJ. Associations of regional amyloid-b plaque and phospho-tau pathology with biological factors and neuropsychological functioning among HIV-infected adults. J Neurovirol 2019; 25:741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez-Scarano F, Martin-Garcia J The neuropathogenesis of AIDS. Nat Rev Immunol 2005; 5;69–81. [DOI] [PubMed] [Google Scholar]
- 9.Ginsberg SD, Alldred MJ, Gunnam SM, Schiroli C, Lee SH, Morgello S, Fischer T Expression profiling suggests microglial impairment in human immunodeficiency virus neuropathogenesis. Ann Neurol 2018; 83:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013; 12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gosztyla ML, Brothers HM, Robinson SR. Alzheimer’s amyloid-b is an antimicrobial peptide: a review of the evidence. J Alzheimers Dis 2018; 62:1495–1506. [DOI] [PubMed] [Google Scholar]
- 12.Bourgade K, Dupuis G, Frost EH, Fulop T Jr. Anti-viral properties of amyloid-b peptides. J Alzheimers Dis 2016; 54:859–878. [DOI] [PubMed] [Google Scholar]
- 13.Ances BM, Benzinger TL, Christensen JJ, Thomas J, Venkat R, Teshome M, et al. 11C-PiB imaging of human immunodeficiency virus-associated neurocognitive disorder. Arch Neurol 2012; 69 (1):72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howdle GC, Quide Y, Kassem MS, Johnson K, Rae CD, Brew BJ, Cysique LA. Brain amyloid in virally suppressed HIV-associated neurocognitive disorder. Neurol Neuroimmunol Neuroinflamm 2020; 7:e739 doi: 10.1212/NXI.0000000000000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohamed M, Skolasky RL, Zhou Y, Ye W, Brasic JR, Brown A, et al. Beta-amyloid (ab) uptake by PET imaging in older HIV+ and HIV- individuals. J Neurovirol 2020; 26:382–390. [DOI] [PubMed] [Google Scholar]
- 16.Grothe MJ, Barthel H, Sepulcre J, Dyrba M, Sabri O, Teipel SJ, for ADNI. In vivo staging of regional amyloid deposition. Neurology 2017; 89:2031–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgello S, Gelman BB, Kozlowski PB, Vinters HV, Masliah E, Cornford M, et al. The National NeuroAIDS Tissue Consortium: a new paradigm in brain banking with an emphasis on infectious disease. Neuropathol Appl Neurobiol 2001. 27:326–335. [DOI] [PubMed] [Google Scholar]
- 18.Tang MX, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, et al. The APOE-e4 allele and the risk of Alzheimer disease among african americans, whites, and hispanics. JAMA 1998; 279:751–755. [DOI] [PubMed] [Google Scholar]
- 19.Thal DR, Capetillo-Zarate E, DelTredici K, Braak H The development of amyloid beta protein deposits in the aged brain. Sci Aging Knowledge Environ March 8, 2006; 6 (re1) doi: 10.1126/sageke.2006.6.re1. [DOI] [PubMed] [Google Scholar]
- 20.Achim CL, Adams A, Dumaop W, Everall IP, Masliah E, HNRC. Increased accumulation of intraneuronal amyloid b in HIV-infected patients. J Neuroimmune Pharmacol 2009; 4:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012; 123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeStrooper B, Karran E The cellular phase of Alzheimer’s disease. Cell 2016; 164:603–615. [DOI] [PubMed] [Google Scholar]
- 23.Thomas KR, Bangen KJ, Weigland AJ, Edmonds EC, Wong CG, Cooper S, et al. Objective subtle cognitive difficulties predict future amyloid accumulation and neurodegeneration. Neurology 2020; 94:e1–e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jack CR Jr, Wiste HJ, Weigand SD, Rocco WA, Knopman DS, Mielke MM, et al. Age-specific population frequencies of cerebral b-amyloidosis and neurodegeneration among people with normal cognitive function aged 50–89 years: a cross-sectional study. Lancet Neurol 2014; 13:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FRJ, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 2015; 313 (19):1924–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braak H, Thal DR, Ghebremedhin E, DelTredici K Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011; 70 (11):960–969. [DOI] [PubMed] [Google Scholar]
- 27.Donohue MC, Sperling RA, Petersen R, Sun CK, Weiner MW, Aisen PS, for ADNI. Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA 2017; 317 (22):2305–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cha L, Berry CM, Nolan D, Castley A, Fernandez S, French MA Interferon-alpha, immune activation and immune dysfunction in treated HIV infection. Clin Transl Immunology 2014; 3:e10 doi: 10.1038/cti.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harezlak J, Buchthal S, Taylor M, Schifitto G, Zhong J, Daar E, et al. Persistence of HIV-associated cognitive impairment, inflammation, and neuronal injury in era of highly active antiretroviral treatment. AIDS 2011; 25:625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang L, Ernst T, Leonido-Yee M, Witt M, Speck O, Walot I, Miller EN. Highly active antiretroviral therapy reverses brain metabolite abnormalities in mild HIV dementia. Neurology 1999; 53:782–789. [DOI] [PubMed] [Google Scholar]
- 31.Itzhaki RF. Herpes and Alzheimer’s disease: Subversion in the central nervous system and how it might be halted. J Alzheimers Dis 2016; 54:1273–1281. [DOI] [PubMed] [Google Scholar]
- 32.Therriault J, Benedet AL, Pascoal TA, Mathataarachchi S, Chamoun M, Savard M, et al. Association of apolipoprotein E e4 with medial temporal tau independent of amyloid-b. JAMA Neurol 2020; 77 (4):470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blennow K, deLeon MJ, Zetterberg H Alzheimer’s disease. Lancet 2006; 368:387–403. [DOI] [PubMed] [Google Scholar]
- 34.Levine AJ, Quach A, Moore DJ, Achim CL, Soontornniyomkij V, Masliah E, et al. Accelerated epigenetic aging in brain is associated with pre-mortem HIV-associated neurocognitive disorders. J Neurovirol 2016: 22 (3): 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eikelenboom P, vanExel E, Hoozemans JJM, Veerhuis R, Rozemuller AJM, vanGool WA. Neuroinflammation - an early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegenerative Dis 2010; 7:38–41. [DOI] [PubMed] [Google Scholar]
- 36.French MA, King MS, Tschampa JM, daSilva BA, Landay AL. Serum immune activation markers are persistently increased in patients with HIV infection after 6 years of antiretroviral therapy despite suppression of viral replication and reconstitution of CD4+ t cells. J Infect Dis 2009; 200:1212–1215. [DOI] [PubMed] [Google Scholar]
- 37.Chou RC, Kane M, Ghimire S, Gautam S, Gui J Treatment for rheumatoid arthritis and risk of Alzheimer’s disease: A nested case-control analysis. CNS Drugs 2016; 30:1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou M, Xu R, Kaelber DC, Gurney ME. Tumor necrosis factor (TNF) blocking agents are associated with lower risk for Alzheimer’s disease is patients with rheumatoid arthritis and psoriasis. PLoS One 2019; 15 (3):e0229819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Umlauf A, Soontornniyomkij B, Sundermann EE, Gouaux B, Ellis RJ, Levine AJ, et al. Risk of developing cerebral b-amyloid plaques with post-translational modification among HIV-infected adults. AIDS 2019; 33:2157–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gama L, Abreu CM, Shirk EN, Price SL, Li M, Laird GM, et al. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS 2017; 31:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson KA, Cherry CL, Bell JE, McLean CA. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol 2011; 179 (4):1623–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ko A, Kang G, Hattler JB, Galadima HI, Zhang J, Li Q, Kim WK. Macrophages but not astrocytes harbor HIV DNA in the brains of HIV-1-infected aviremic individuals on suppressive antiretroviral therapy. J Neuroimmune Pharmacol 2019; 14:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.An SF, Giometto B, Groves M, Miller RF, Beckett AAJ, Gray F, et al. Axonal damage revealed by accumulation of b-APP in HIV-positive individuals without AIDS. 11, 1997, J Neuropathol Exp Neurol 1997; 56 (11):1262–1268. [DOI] [PubMed] [Google Scholar]