

Abstract
Background and Aims:
Both existing clinical criteria and genetic testing have significant limitations for the diagnosis of Wilson’s Disease (WD) often creating ambiguities in patient identification leading to delayed diagnosis and ineffective management. ATP7B protein concentration, indicated by direct measurement of surrogate peptides from patient dried blood spot (DBS) samples, could provide primary evidence of WD. ATP7B concentrations were measured in patient samples from diverse backgrounds, diagnostic potential is determined, and results are compared to biochemical and genetic results from individual patients.
Methods:
264 samples from biorepositories at three international and two domestic academic centers and 150 normal controls were obtained after IRB approval. Genetically or clinically confirmed WD patients with a Leipzig score over 3 and obligate heterozygote (carriers) from affected family members were included. ATP7B peptide measurements were made by immunoaffinity enrichment mass spectrometry.
Results:
Two ATP7B peptides were used to measure ATP7B protein concentration. ROC curve analysis generates an AUC of 0.98. ATP7B peptide analysis of the sequence ATP7B 887 was found to have a sensitivity of 91.2%, specificity of 98.1%, positive predictive value (PPV) of 98.0%, and a negative predictive (NPV) value of 91.5%. In patients with normal ceruloplasmin concentrations (> 20 mg/dL), 14/16 (87.5%) were ATP7B deficient. In patients without clear genetic results, 94% were ATP7B deficient.
Conclusions:
Quantification of ATP7B peptide effectively identified WD patients in 92.1% of presented cases and reduced ambiguities resulting from Cp and genetic analysis. Clarity is brought to patients with ambiguous genetic results, significantly aiding in non-invasive diagnosis. A proposed diagnostic score and algorithm incorporating ATP7B peptide concentrations can be rapidly diagnostic and supplemental to current Leipzig scoring systems.
Keywords: Wilson disease, Leipzig Score, Immuno-SRM, ATP7B
Graphical Abstract
“LAY SUMMARY”
A proposed diagnostic score and algorithm incorporating ATP7B peptide concentrations in DBS can be rapidly diagnostic and supplemental to current Leipzig scoring systems.
INTRODUCTION:
Wilson disease (WD), named after Dr. Samuel Alexander Kinnier Wilson, who first described the disorder in his 1912 doctoral thesis. Since then, treatments have been developed for WD and, importantly, WD has become a preventable disease. Wilson Disease (WD) is an autosomal recessive disorder of copper metabolism due to mutations in the ATP7B gene that encodes copper-transporting P-type ATPase (EC # 7.2.2.8).1–3 WD has an estimated prevalence of 1:30,000 and a carrier frequency of 1:90 with regional variation.4, 5 Although guidelines for the diagnosis of WD have been developed,6, 7 patient identification remains a challenge resulting in delayed diagnosis and development of irreversible severe complications, such as permanent brain or liver damage, which render treatments ineffective.5
The key features of WD are liver disease, neuropsychiatric abnormalities, and Kayser-Fleischer (KF) rings. The presence of KF rings with neurological manifestation and/or low serum ceruloplasmin (Cp) is considered enough to establish WD diagnosis. However, most cases require a combination of clinical symptoms and laboratory evaluations.8 Currently, no single test permits de novo WD diagnosis in every potential patient.7 Serum Cp is decreased in neurological WD but can be in the low normal range in up to 50% of adult patients with active liver disease 9, 10 and the positive predictive value of serum Cp for diagnosis of WD is poor.9, 11,12 In children with WD, 15–36% had Cp in the normal range.13 Serum Cp alone is not sufficient to diagnose or exclude WD. A diagnostic score (Leipzig score 2003) was proposed to guide clinical diagnosis and has been adopted in the clinical practice guideline for the European Association for the Study of the Liver.14 While recent advances in clinical molecular diagnosis have greatly improved the accuracy of WD diagnosis in affected patients and their siblings, traditional Sanger sequencing cannot detect large deletion or duplications. Further, there are many single nucleotide polymorphisms (SNPs) and variants of unknown significance (VUS) in the ATP7B gene. Interpretation of genetic sequencing results, particularly in the presence of only one identified mutation or VUS, create ambiguity in WD patient identification.
Here, we evaluate the direct measurement of ATP7B protein from WD patient dried blood spots (DBS), through surrogate ATP7B peptides, as diagnostic tool.15 As shown in our previous studies for multiple primary immunodeficiency conditions, peptide measurements are made using immunoaffinity enrichment coupled to selected reaction monitoring (immuno-SRM) mass spectrometry.16, 17 This method utilizes anti-peptide antibodies to concentrate and quantify extremely low concentration peptide targets from complex matrices, including DBS.15–21 Analysis of ATP7B concentration in DBS from WD patients with a broad range of genetic backgrounds show that ATP7B peptide levels are greatly reduced. Analysis of ATP7B protein concentration can identify WD with high diagnostic accuracy.
MATERIALS AND METHODS
Dried Blood Spot Samples
This protocol was approved by the institutional review board of Seattle Children’s Hospital (SCH) and each of participating institutes. All subjects gave written informed consent. Patient and carrier samples were provided by the SCH, WA, U.S.A., Medical University of Vienna, Austria, Medical University Innsbruck, Austria, University of Medicine and Pharmacy “Iuliu Hatieganu” Cluj-Napoca, Romania, Wilhelmina Children’s Hospital, University Medical Center, Utrecht, The Netherlands, Yale University, CT, U.S.A., University of Heidelberg, Germany and Asan Medical Center, Seoul, South Korea. Samples were prepared either by fingerstick or by pipetting 70 μL of blood (per 12 mm spot) onto filter paper cards (Protein Saver 903, Piscataway, NJ). The samples were then dried overnight at room temperature, delivered to SCH, and stored at −80 °C until use. 150 normal control DBS samples (BioIVT, Westbury, NY) were analyzed to establish the normal reference range and cut-off.
Immuno-SRM reagents:
Triton X-100 (T9284–100mL) and Ammonium bicarbonate (Ambic) (A6141–25G) was purchased from Sigma Aldrich (St. Louis, MO). TPCK-treated Worthington Trypsin (LS003740) was purchased from Worthington (Lakewood, NJ). 3-[3-cholamidopropyl = dimethylammonio]-1-propanesulfonate (CHAPS) (no. 28300), Acetonitrile (ACN) (LC/MS grade), Acetic Acid (AA) (LC/MS grade), water (Optima LC/MS grade), formic acid (FA) (Optima LC/MS grade), phosphate-buffered saline (1× PBS, no. 10010–023), Dithiothreitol (DTT) (no. 20290) and 1M Tris-(hydroxymethyl)aminomethane pH 8 (TRIS) (no. 15568–025) buffer were obtained from Fisher Scientific (Waltham, MA). Protein G coated magnetic beads (Dynabeads, no. 10004D) were purchased from Invitrogen (Carlsbad, CA)
Isotope-labeled internal standard (IS) peptides were purchased from either Atlantic Peptides (Lewisburg, PA) or Life Technologies Corporation (Carlsbad, CA). IS peptides were >95% pure and incorporated heavy stable isotope-labeled (13C and 15N) C-terminal lysine (+8 Da) or arginine (+10 Da). IS stock solutions are stored frozen as 500× mixtures in 1× PBS + 15% ACN + 0.1% FA + 0.03% CHAPS in H2O and diluted to 1× immediately before use.
Selection of Signature Peptides and Antibody Production:
Selection and production of ATP7B 1056 peptide and antibody has been described in previous report15–17.
Using the same guidelines, ATP7B 887 was selected. Antibody production was performed by Excel Biopharm (San Francisco, CA) and Pacific immunology (Ramona, CA)-ExonBio (San Diego, CA). ELISA assays were performed by above mentioned companies in bleed samples after immunization and in supernatant samples after monoclonal antibody production. Selections were further confirmed at each step by the immuno-SRM method.22
Antibody Bead Reagent Production:
Monoclonal Antibody (mAb) beads were produced by overnight 4 °C incubation of Protein G dynabeads with mAb as previously reported.15–17
DBS Extraction, Trypsin Digestion and Immunoaffinity Enrichment:
Protein extraction and tryptic digestion were performed as previous reported with slight modifications.16 One 6.35-mm diameter DBS punch was placed into 96-well plates (Thermo Scientific, Chicago, IL), covered with an adhesive seal (Genesee Scientific, San Diego, CA), and extracted using 0.1% Triton X-100 in 50 mM Ammonium Bicarbonate (200 μL) with Dithiothreitol (final concentration 0.2M). After 30 min incubation at 37 °C with agitation, trypsin (37.5 μg) was added and incubated for 2 hours at 37 °C. For enrichment, 10 μL of 1 M TRIS (pH 8) and 10 μL of 1x ATP7B IS mix were added (Final concentrations = 0.25 nM). Extracted supernatant (200 μL) was transferred to a new plate and incubated with 2.5 μL of each mAb-bead overnight at 4 °C with agitation.
After incubation, mAb-beads were isolated using a 96-well magnetic plate (Alpaqua Magnum EX, Beverly, MA), washed twice with 1×PBS+ 0.01% CHAPS, and magnetically isolated. Peptides were eluted with 30 μL of H2O with 5% acetic acid and 3% ACN for 5 min and transferred to a new 96-well plate for analysis (Abegene, Chicago, IL).
Liquid Chromatography Mass Spectrometry (LC-MS/MS):
LC-MS/MS was performed using a Waters Xevo TQ-XS with Ionkey source and dual M-Class chromatography pumps (Milford, MA). Chromatographic solvents were A: H2O + 0.1% FA and B: ACN + 0.1% FA. Peptides are loaded onto an M-Class Trap Symmetry C18 column (300 μM x 25 mm, 100A, 5 μM) for three minutes with a constant flow of 98:2 A:B at 20 μL/min. After loading, the flow is reversed and peptides are separated using a 150 μM x 100 mm BEH C18 ionkey (130Å, 1.7 μM). The gradients used are summarized in Supplemental Table 1 and reported previously16. Precursor mass, fragment mass, and collision energy were tuned to optimize the generated signal (Supplemental Table 2). Representative chromatograms for both ATP7B 887 and ATP7B 1056 peptides were shown in Supplemental Figure 1A–D.
Concentration Calculation and Data Analysis:
Selected reaction monitoring data captured in the MS were analyzed using Skyline (MacCoss Lab, Seattle, WA, https://skyline.ms/project/home/begin.view).23 Specificity was assured by monitoring retention times and relative transition intensities of endogenous and IS peptides. Concentrations of endogenous signature peptides were calculated using endogenous/IS signal ratio. DBS spots are assumed to contain 70 μL of evenly distributed whole blood. The volume of blood in the punch area is calculated as 17.5 μl. Concnetrations are calculated from blood volume, ratio and IS concentration. Statistical analyses and Receiver Operating Characteristics (ROC) curves were generated using Graphpad Prism (San Diego, CA).
Method Performance Assessment:
Response curves were generated for each peptide to establish assay linearity and determine the lower limits of detection (LLOD) and quantification (LLOQ). Seven concentrations (0×, 0.05×, 0.1×, 0.5×, 1×, 5×, 50×) of IS were added across the set of pooled digest samples in triplicate. LLOD was calculated where LLOD = Meanblank + 3 x SDLow (Meanblank: mean signal from a triplicate blank injection, SDLow: SD of IS injection below the LLOQ). LLOQ is the lowest concentration with a coefficient of variation (CV) of < 20%. The linearity curves of ATP7B 1056 and ATP7B 887 were shown in Supplemental Figure 1E&F.
The assay precision and accuracy were evaluated by within-day (intra-) and between-day (inter-) assay CV, respectively. The intra- and inter-assay CV were determined using 5 replicates of an identical pooled blood DBS sample each day over a course of 5 days (Table 1).
Table 1:
Analytical and diagnostic performance for ATP7B peptides.
| Peptide | LLOD (pmol/L) | LLOQ (pmol/L) | Intra-assay CV (%) | Inter-assay CV (%) | PPV (%) | NPV (%) | AUC |
|---|---|---|---|---|---|---|---|
| ATP7B 1056 | 3.81 | 71.43 | 12.9 | 15.3 | 96.1 | 91.3 | 0.98 |
| ATP7B 887 | 2.17 | 7.14 | 11.0 | 13.0 | 98.0 | 91.5 | 0.98 |
LLOD = Lower Limit of Detection, LLOQ = Lower Limit of Quantification, CV = Coefficient of Variation, PPV = Positive Predictive Values, NPV = Negative Predictive Value, AUC = Area Under the Curve.
Internal Sample Quality Control:
Measurement of endogenous peptides unrelated to Wilson Disease, and therefore assumed to be present at normal concentrations, was utilized as an internal quality control to monitor the successful extraction, digestion, and enrichment of target peptides. These peptides are ADA 93, representing Adenosine Deaminase, CD42 128, representing glycoprotein Ib, and IDUA 462, representing alpha-L-Iduronidase.16 A sample run was assumed to be of sufficient quality if the measured concentration of two of these three peptides was within 1.75SD of the mean for the cohort (Supplemental Table 3–5). Samples failing these acceptance requirements were repeated. With confirmatory test failure, the DBS was removed from the sample cohort due to inadequate matrix and an additional sample was requested.
RESULTS
Characteristics of Patient Cohort
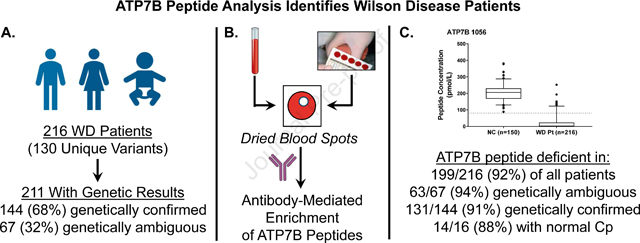
WD DBS samples were obtained from 198 Caucasian, 18 Far Eastern Asian WD patients (114 male and 102 female) and 48 obligate carriers (Supplemental Table 3&4). All carriers are obligate heterozygote from the family members of index case patients with two confirmed variants. Patient age range spanned from 2 months to 73 years old. For the purpose of stability validation, samples from 11 patients and 1 healthy normal were collected from both fresh and blood samples stored up to 11 years prior (Supplemental Table 7). Clinical information including Cp concentrations, Leipzig scores, liver copper content, presence of KF rings, initial presentation, and presence of cirrhosis are presented where available (Supplemental Table 3). Control DBS samples (N = 150) were obtained from healthy subjects ranging from 18–73 years of age.
An analysis of the specific variants in the sample set showed that the cohort contained 130 unique variants (Figure 1B, Supplemental Table 6) including 83 pathogenic or likely pathogenic variants, 43 VUS, 3 benign or likely benign variants and 1 with conflicting interpretations (VCI). In affected patient samples, 145 exhibited only pathogenic or likely pathogenic mutations according to public database (ClinVar, gnomAD [Genome Aggregation Database]), including 31 patients homozygous for p.H1069Q, the most common variant in WD patients (Figure 1A, Supplemental Table 3). In addition, 20 patients were homozygotes for other variants. Seven patients exhibited two VUS (two of them are homozygotes). In addition, 37 patients were compound heterozygous for one VUS and one pathogenic or likely pathogenic mutation. Eighteen patients had only one variant detected by Sanger sequencing. No second variant was detected Three clinically suspected patients were compound heterozygotes, one variant with likely benign variants according to gnomAD and the second variant with likely pathogenic, pathogenic, and unknown, respectively.
Figure 1:

Patient cohort characteristics. Analysis of the genotypes of patients (A) and the characteristics of the variants present (B) show the diversity of variants and variant combinations present.
Forty-eight samples from obligate carriers, all of them being family members of affected patients presented with two variants, had a single pathogenic variant and a single wild-type allele or a benign variant (Supplemental Table 4).
Surrogate Peptide Markers for ATP7B
The signature peptide concentration in normal control was 257.7 ± 57.5 (mean ± SD) pmol/L for ATP7B 887 (Range: 136.4–447.0 pmol/L, 5–95 percentile range: 165.0–359.6 pmol/L) and 203.0 ± 48.9 for ATP7B 1056 (Range: 88.2–381.3 pmol/L, 5–95 percentile range: 129.7–287.3 pmol/L). These cutoffs were set at −2.5 (114.0 pmol/L) and −2.5 SD (80.8 pmol/L) below the mean normal concentration for ATP7B 887 and ATP7B 1056, respectively.
Method Analytical Performance
The analytical figures of merit for immuno-SRM quantification of ATP7B peptides are given in Table 1. The LODs of ATP7B quantification were determined to be 3.81 pmol/L and 2.17 pmol/L for ATP7B 1056 and ATP7B 887, respectively. LLOQs were determined to be 71.43 and 7.14 pmol/L. The intra-assay CVs were 12.9% and 11.0% for ATP7B 1056 and ATP7B 887. The inter-assay CVs were 15.3% and 13.0%, respectively.
ATP7B Concentration Measurements and Primary Diagnostic Performance
Signature peptide levels in patient DBS were below cutoff in 195/216 (90.2%) of samples for both ATP7B 1056 and ATP7B 887 (Figure 2A&B). There were 17 (7.8%) WD patients that had ATP7B level above the cutoff for both ATP7B peptides. There were 2 WD patients had only ATP7B 887 and 2 WD patients had only ATP7B 1056 levels above the cutoff. In all, 199/216 (92.1%) of patients head least one peptide below cutoff. In WD carriers, 8/48 (16.7%) and 4/48 (8.3%) samples were below diagnostic cutoffs for ATP7B 1056 and ATP7B 887, respectively. These patient samples generate potential false positives.
Figure 2:

Diagnostic performance of ATP7B peptide analysis. Comparison of ATP7B peptide measurements for ATP7B 1056 (A) and ATP7B 887 (B) peptides in normal control patients (NC), patients, homozygotes (H), compound heterozygotes (CH) and carriers. Dotted lines represent diagnostic cut-offs for each peptide. ROC curves show the diagnostic performance of ATP7B 1056 (C) and ATP7B 887 (D). WD patients with genetic test results are readily identified even in subgroups where genetic results are ambiguous (E).
As a primary diagnostic, ROC curve analysis (Figure 2C&D) constructed from this DBS sample cohort show that both ATP7B 1056 and ATP7B 887 peptide analysis have an AUC of 0.98 (ATP7B 1056 [SE = 0.006, 95% CI: 0.97–0.99, p<0.0001], ATP7B 887 [SE = 0.007, 95% CI: 0.96–0.99, p<0.0001]). ATP7B 887 analysis was found to have a sensitivity of 91.2%, specificity of 98.1%, positive predictive value (PPV) of 98.0%, and a negative predictive (NPV) value of 91.5%. ATP7B 1056 showed PPV of 96.1% and NPV of 91.3% (Table 1).
Effects of Common Variants
In the 216 WD patient cohort, a total of 130 variants were identified (Supplemental Table 6). Many common pathogenic variants including p.H1069Q (AF = 0.103%), p.R778L (AF = 0.013%), p.M645R (AF = 0.047%), and p.E1064A (AF = 0.015%)were associated with either undetectable or significantly reduced level of ATP7B (Figure 3A–D).
Figure 3:

ATP7B peptide concentrations in patients with common variants are often reduced and variants causing false negatives are rare. Patients homozygous and heterozygous for p.H1069Q (A), R778L (B), M645R (C) and E1064A (D) are largely reduced. False negatives within the patient cohort are possible with the presence of specific variants including G710S (E) and R969Q (F). Patients can have variable peptide concentrations depending on the second variant. H = Homozygote, CH = Compound Heterozygote. Dotted Lines represent peptide diagnostic cutoffs.
Variants Leading to Potential “False” Negatives
In this cohort, 17 out of 216 (7.8%) patients have ATP7B concentrations above the cutoff for both signature peptides. The genetic information is summarized in Table 2. In these 17 WD patients, 13 variants were commonly involved and could contribute to normal levels of ATP7B. According to gnomAD (https://gnomad.broadinstitute.org), these variants are rare with an allele frequency less than 0.0089%; the remaining variant, p.M665I, has a VCI designation. Of note, four variants above (p.R616W, p.G710S p.M769V, and p.R969Q) have been reported to show the ATP7B protein distribution similar to WT in in vitro study (Figure 3E&F).24
Table 2.
Selected Patients from Cohort. Peptide concentrations are given in pmol/L. The diagnostic cutoff for ATP7B 1056 is 80.8 pmol/L. The diagnostic cutoff for ATP7B 887 is 114.0 pmol/L.
| Patient | Gender | Age | Variant1 | Annotation 1 | Variant2 | Annotation 2 | ATP7B 1056 (pmol/L) | ATP7B 887 pmol/L | CPL (mg/dl) | Leipzig Score | Liver Copper (ug/g Tissue) | KF Ring | Presentation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| False Negative (n=17) | |||||||||||||
| 110 | F | 17 | p.G710A | Path | p.G710S | Path | 88.9 | 133.0 | 14 | 7 | - | N | - |
| 10 | M | 34 | p.G710S | Path | p.G710S | Path | 120.4 | 155.3 | <9 | 8 | - | Y | N |
| 11 | M | 17 | p.G710S | Path | p.G710S | Path | 193.2 | 264.3 | 12 | 10 | - | Y | B* |
| 12 | F | 44* | p.G710S | Path | p.G710S | Path | 151.1 | 193.6 | 9.1 | 11 | 164 | Y | N |
| 13 | M | 19 | p.G710S | Path | p.G710S | Path | 252.4 | 381.7 | 20.7 | 8 | 1243 | Y | B |
| 111 | M | 47 | p.M665I | Conflicting | p.G710S | Path | 133.9 | 213.0 | 15.5 | 7 | 324 | N | H |
| 112 | M | NA | p.M665I | Conflicting | p.G710S | Path | 94.9 | 144.4 | NA | 6 | 324 | N | H |
| 140 | F | 29 | p.R616Q | Path | p.L1305P | Path | 105.5 | 151.7 | 2 | 8 | - | Y | B |
| 149 | F | 17 | p.H1069Q | Path | p.M769V | Likely Path | 130.2 | 180.1 | 10 | 8 | 2047 | N | H |
| 150 | F | 31 | p.N41S /p.I1021V | Likely Path | p.M996T | Likely Path | 129.1 | 218.6 | 4 | 6 | >250 | N | H |
| 153 | F | 37 | p.H1069Q | Path | p.P1273L | Path | 105.3 | 188.5 | 8 | 10 | - | Y | N |
| 154 | F | 18 | p.M769H-fs | Path | p.P1273L | Path | 121.4 | 170.4 | 5 | 10 | - | Y | N |
| 170 | M | NA | p.H1069Q | Path | p.R969Q | Path | 90.5 | 131.4 | NA | 4 | - | - | - |
| 124 | M | 17 | p.Q7D-fs*14 | VUS | p.H1069Q | Path | 135.8 | 242.5 | 19 | 5 | - | N | H |
| 171 | F | 9 | p.H1069Q | Path | p.R969Q | Path | 145.6 | 220.9 | 16 | 5 | - | N | H |
| 176 | M | NA | p.R616W | Path | p.R969Q | Path | 134.2 | 134.2 | 25 | 7 | - | N | H |
| 183 | F | 31 | p.T977M | Path | p.T991A | VUS | 137.9 | 191.7 | 12 | 3 | - | Y | H |
| Patients with One Pathogenic/Likely Pathogenic variant in Combination with one VUS (n=37) | |||||||||||||
| 124 | M | 17 | p.H1069Q | Path | p.Q7D-fs*14 | VUS | 135.8 | 242.5 | 19 | 5 | - | N | H |
| 125 | M | 9 | p.H1069Q | Path | p.Q7D-fs*14 | VUS | 79.4 | 176.3 | 16 | 5 | - | N | H |
| 131 | F | 13 | p.H1069Q | Path | p.I1007T-fs | VUS | ND | 5.4 | 14 | 5 | - | Y | H |
| 55 | M | 59 | p.H1069Q | Path | arr[GRCh37] 13q14.3(52541594_52548863)x1 | VUS | ND | 16.1 | 13 | 5 | - | Y | H |
| 87 | F | 19 | p.H1069Q | Path | p.D1447G-fs | VUS | 67.5 | 130.7 | 13 | 6 | - | N | H |
| 159 | M | 40 | p.R827W | Likely Path | p.R1320T | VUS | 58.7 | 93.4 | 13 | 7 | - | N | N |
| 133 | M | 18 | p.H1069Q | Path | p.K1028S-fs | VUS | ND | 3.9 | 12 | 7 | 842 | N | H* |
| 183 | F | 31 | p.T977M | Path | p.T991A | VUS | 137.9 | 191.7 | 12 | 3 | - | Y | H |
| 164 | F | NA | p.H1069Q | Path | p.R778P | VUS | ND | ND | 11 | 9 | 1332 | N | H |
| 118 | M | 6 | p.H1069Q | Path | p.G1011X | VUS | ND | 10.0 | 10 | 6 | - | N | H |
| 157 | F | 8 | p.R1041W | Likely Path | p.D765Y | VUS | 6.8 | 20.7 | 10 | 8 | 1211 | N | H |
| 163 | M | 2 months | p.R778L | Path | p.V1106I | VUS | 4.0 | 9.7 | 10 | - | - | N | A |
| 182 | M | 40 | p.T850I | Likely Path | p.G515S | VUS | 11.6 | 13.4 | 10 | 6 | - | Y | B |
| 67 | F | 17 | c.2865+1G>A | Path | p.Ser135* | VUS | ND | ND | <13 | 9 | - | N | H |
| 167 | F | 19 | p.H1069Q | Path | p.R919L | VUS | 3.6 | 22.1 | 9 | 10 | - | Y | N |
| 95 | M | 53 | p.H1069Q | Path | p.F1343dup | VUS | ND | ND | 9 | 3 | - | Y | H |
| 108 | M | 15 | p.H1069Q | Path | p.G1341E | VUS | ND | 10.4 | 6.7 | 8 | - | Y | H |
| 181 | F | 41 | p.G1266R | Path | p.T807I | VUS | 8.3 | 5.9 | 6 | 9 | 291 | N | H |
| 193 | F | 15 | p.S1365C-fs*12 | Path | p.Y743I-fs*19 | VUS | ND | ND | <6 | 7 | - | N | H |
| 80 | M | 40 | p.A874V | Path | c.2299InsC | VUS | 6.0 | 10.5 | 5.5 | 10 | 1575 | Y | H* |
| 120 | F | 13 | p.H1069Q | Path | p.Lys269* | VUS | ND | ND | 5 | 5 | 923 | N | H |
| 142 | M | 39 | p.T977M | Path | p.L1350P | VUS | 22.6 | 32.6 | 5 | 10 | - | Y | B |
| 76 | M | 5 | p.A1018V | Path | p.E458X | VUS | 26.4 | 47.5 | 4.1 | 8 | 793 | N | H |
| 141 | F | 46 | p.H1069Q | Path | p.L1333P | VUS | ND | 6.3 | 4 | 7 | - | Y | H |
| 184 | M | 10 | p.A874V | Path | p.V1106I | VUS | 20.1 | 15.5 | 4 | - | - | - | - |
| 119 | M | 38 | p.H1069Q | Path | p.K844E-fs*10 | VUS | ND | 8.7 | <4 | 4 | - | N | N |
| 79 | M | 59 | p.L1088X | Likely Path | p.A1135Q-fs*13 | VUS | ND | ND | <4 | 6 | - | Y | H |
| 165 | M | 15 | p.R778W | Path | p.K35N-fs*6 | VUS | 21.8 | 25.1 | <3 | 4 | - | N | H |
| 143 | F | 26 | p.R778L | Path | p.L770L | VUS | ND | ND | <3 | 9 | - | Y | H |
| 69 | F | 16 | p.T1029I | Path | c.3060+5G>C | VUS | 2.8 | ND | <3 | 8 | - | Y | H |
| 74 | F | 12 | p.H1069Q | Path | IVS19–1C>G | VUS | ND | 13.5 | 3 | 9 | - | Y | N |
| 188 | F | 8 | p.M645R | Path | p.V997-fs | VUS | 38.7 | 72.4 | 2.4 | 8 | >250 | N | H* |
| 102 | M | 9 | p.G1341D | Path | p.F1026F-fs | VUS | ND | ND | 2 | 10 | - | Y | N |
| 134 | F | 10 | p.M769H-fs | Path | p.K1028S-fs | VUS | ND | ND | 2 | 10 | - | Y | N |
| 72 | F | 24 | p.G710S | Path | c.3400delC | VUS | 57.7 | 96.6 | 0.6 | 12 | 448 | Y | B |
| 68 | M | 11 | c.1708–1g>c | Likely Path | c.2866–3c>g | VUS | 22.4 | 37.7 | NA | 6 | - | Y | H |
| 88 | F | 8 | p.M769H-fs | Path | p.D1460Y | VUS | 20.3 | 39.3 | low | 6 | - | N | H |
| Patients with Two VUS’s (n=7) | |||||||||||||
| 86 | F | 68 | p.E332K | VUS | p.D1047V | VUS | 51.7 | 79.3 | 27 | 6 | 26 | N | B |
| 3 | M | 18 | p.G1335E | VUS | p.G1335E | VUS | ND | ND | <2 | - | - | Y | N |
| 9 | F | 15 | p.G1341E | VUS | p.G1341E | VUS | 2.2 | ND | 1.4 | 6 | - | N | H |
| 84 | M | NA | p.I1336V | VUS | p.C709T | VUS | 10.5 | 15.5 | 10 | 8 | 552 | N | H |
| 85 | F | NA | p.I1336V | VUS | p.C709T | VUS | ND | 6.3 | 13 | 7 | 900 | N | H |
| 186 | M | NA | p.S932L | VUS | p.V1364V-fs | VUS | ND | ND | 10 | 9 | - | N | B |
| 130 | M | 20 | p.T59H-fs*19 | VUS | p.H1247Q | VUS | 23.6 | 27.2 | <4 | 9 | 54 | Y | N |
| Patients with Only One Variant Found (No 2nd variant detected) (n=18) | |||||||||||||
| 208 | M | 17 | c.2299delC | VUS | unknown | - | ND | ND | <2 | - | - | N | H |
| 209 | M | 19 | c.2299delC | VUS | unknown | - | ND | ND | <2 | - | 502 | N | N |
| 195 | F | 43 | c.51+4a>t | Path | unknown | - | 25.6 | 37.6 | 4 | 8 | - | Y | N |
| 196 | F | 28 | p.A1049A-fs | VUS | unknown | - | 2.2 | 9.1 | 3 | 7 | - | Y | N |
| 194 | M | 50 | p.D765N | Path | unknown | - | 20.4 | 21.4 | <4 | 8 | - | Y | N |
| 197 | M | 22 | p.G1176R | Path | unknown | - | 3.6 | 7.3 | 3 | 5 | - | N | H |
| 199 | M | 17 | p.H1069Q | Path | unknown | - | 8.6 | 32.9 | <10 | 5 | - | N | N |
| 201 | M | 14 | p.H1069Q | Path | unknown | - | 31.4 | 29.0 | 3.4 | - | - | N | N |
| 198 | F | NA | p.H1069Q | Path | unknown | - | ND | 5.0 | 12 | 6 | - | N | H |
| 200 | M | 14 | p.H1069Q | Path | unknown | - | ND | 6.7 | 13 | 4 | - | Y | H |
| 211 | F | 22 | p.H1069Q | Path | unknown | - | ND | 7.9 | 2 | 5 | - | N | N |
| 210 | M | NA | p.L1305P | Path | unknown | - | ND | 6.0 | NA | 3 | - | - | - |
| 202 | F | 16 | p.M769H-fs | Path | unknown | - | ND | ND | 13.8 | 6 | 525 | Y | H |
| 203 | M | 37 | p.R1319X | Path | unknown | - | 3.0 | 5.2 | <10 | 9 | 1042 | Y | B |
| 204 | M | 12 | p.R778L | Path | unknown | - | ND | 9.5 | <3.0 | 5 | - | N | H |
| 205 | M | 25 | p.T1220M | Likely Path | unknown | - | 67.7 | 77.0 | <10 | 4 | 191 | N | B |
| 207 | F | 18 | p.W779X | Path | unknown | - | 38.0 | 65.6 | 15.3 | 6 | 258 | N | H |
| 206 | M | 45 | p.W779X | Path | unknown | - | ND | ND | <^4 | 6 | - | Y | H |
KF ring: Y=Present, N=Not present
Presentation: H=Hepatic, N=Neurologic, A=Asymptomatic, B=Both Hepatic and Neurologic,
=Cirrhosis
ATP7B Analysis and Variant Pathogenicity
Of the 216 WD patients, 211 had available genetic test results (Figure 2E). 144 were genetically confirmed to be WD patients by being compound heterozygous or homozygous for known pathogenic or likely pathogenic mutations (Figure 1A). 131 of these patients (91.0%) had concentrations of at least one signature peptide below established cutoffs (Figure 4D). Alternatively, 67 patients had ambiguous genetic test results preventing straightforward genetic identification. Sixty-three (94%) of these patients were deficient in ATP7B by peptide analysis (Figure 2E). Seven patients were compound heterozygous or homozygous for two VUS. All samples (100%) contained significant reduction in ATP7B peptides (Table 2). Thirty-seven patients were compound heterozygous for one VUS and one known pathogenic or likely pathogenic variant (Table 2, Figure 4E). ATP7B concentrations were below cutoff in 35/37 (94.6%) of these cases. One VCI, p.M665I, was found in five patients. In this case, three patients had peptide concentrations below established cutoffs and the remaining two were compound heterozygous p.G710S, known to cause false negatives. Three patients had known benign or likely benign in combination with known pathogenic or likely pathogenic mutations. Two are likely carriers with normal ATP7B, but one patient with a likely benign mutation in combination with a known pathogenic mutation had non-detectable ATP7B indicating possible misannotation (#64). Finally, in 18 WD cases that have only one variant with no second mutation detected, their ATP7B peptide levels were all (100% of samples) reduced below the cutoff (Table 2, Figure 4F).
Figure 4:

ATP7B peptide concentration analysis can provide clear results where ceruloplasmin results and genetic analysis are ambiguous. Patients with significantly (A), moderately (B), and normal (C) Cp were readily identified. ATP7B concentrations are reduced regardless of variant status including in patients with two pathogenic or likely pathogenic variants (D), at least one VUS (E), or where no second variant was found (F). Cp = Ceruloplasmin. Dotted Lines represent peptide diagnostic cutoffs. Patients with liver copper above (G) or below 256 ug/g (H) are shown. In three samples from non-WD patients with elevated liver copper, ATP7B concentrations are normal (I).
ATP7B Analysis and Cp Concentration
The 200 patients in whom had Cp values were provided were stratified into three subgroups (Figure 4A–C): (1) 107 patients with Cp < 10 mg/dL, (2) 77 patients with Cp between 10–20 mg/dL, and (3) 16 patients with Cp > 20 mg/dL. Within these groups, 101/107 (94.3%) with Cp < 10 mg/dL, 70/77 (90.9%) with Cp 10–20 mg/dL, and 14/16 (87.5%) with Cp > 20 mg/dL had DBS ATP7B peptide concentrations below diagnostic cutoffs.
ATP7B Analysis and Other Clinical Indications
Where possible, clinical information including age, Leipzig scores, liver copper content, presence of KF rings, initial presentation, and presence of cirrhosis are presented (Supplemental Table 3). The exact number of treated patients and their regimens is unknown. No differences in ATP7B concentration were found amongst the patients based on any of these factors. Fifty-nine patients had hepatic copper measurements available (Figure 4G–I). Fifty-one patients (86.4%) had elevated liver copper (>250 μg/g), 46 of them (90.2%) had deficient ATP7B levels. Eight patient had liver copper ranged from 25 to 248 ug/g, 7 of them with deficient ATP7B levels. Of note, three samples were received with suspicion of WD by elevated liver copper but with no ATP7B variants identified indicating they are not WD patients. As expected, these samples had normal ATP7B concentrations and were not contained within the final 216 WD patient cohort.
DISCUSSION
This is the first large cohort study directly measuring ATP7B from DBS of WD patients with diverse genetic backgrounds. It showed ATP7B peptide concentrations have a high diagnostic potential. This test is a novel approach to WD testing and screening with a high sensitivity and specificity. It can be successfully applied in patients who do not present with clear clinical and laboratory criteria for WD as a first “second” line test and expand the clinician’s ability to non-invasively diagnose WD by reducing the need for liver biopsy. As the assay measures the ATP7B peptides produced in the peripheral bloods, liver synthetic dysfunction would have little or no effect on the concentration of ATP7B peptides in DBS, which is another advantage of the assay. In suspected cases, no single test is diagnostic, and combination of laboratory tests and clinical investigation are required to establish the diagnosis. These include ophthalmologic testing for KF rings, Cp measurement, 24-hour urine copper measurement, liver biopsy to determine copper content, and ATP7B gene sequencing. In patients with hepatic WD, KF-rings may be absent and Cp in the low normal range contributing to diagnostic ambiguity.
Genetic sequencing can give a definitive diagnosis when two known pathogenic variants are found. However, many mutations found in the ATP7B gene are VUS, VCI, or are extremely rare. Now, more than 1300 variants in ATP7B gene are listed in Varsome (varsome.com);25 649 of them pathogenic/likely pathogenic, while 692 of them were classified as VUS. Variant interpretation remains a challenge for clinical laboratories.26, 27 The recommended Leipzig score for diagnosis of WD assigns numerical values to the number of disease-causing mutations to give probable WD diagnoses. The definition of “disease” causing variant is based on a variety of databases, which may give conflicting answers. 28
We hypothesized that direct measurement of ATP7B could identify WD patients as majority of pathogenic mutations often result in protein misfolding, absence of decay of mRNA and enhanced degradation. To explore this goal, two wild type (WT) ATP7B peptides were chosen for anti-peptide antibody generation. Several factors influence peptide selection as they must be unique to ATP7B, detectable by mass spectrometry, and elicit specific antibodies for isolation. ATP7B 1056 contains the most common WD-causing mutation, p.H1069Q. If the patient is homozygous for this mutation, WT ATP7B 1056 will not be found because the WT sequence is not present. Having a second peptide, ATP7B 887, builds a redundancy into the assay to ensure accurate performance.
As a primary diagnostic test, quantification of ATP7B from DBS effectively identified WD patients (Figure 2A&B, Table 1). Reduction of ATP7B concentrations below diagnostic cutoffs for at least one ATP7B peptide was evident in 92.1% of WD patients. Because the cutoffs set are based on the number of NCs analyzed, ROC curves were constructed showing ATP7B analysis to be a highly sensitive and specific for diagnosis. The calculated AUC for the dataset is 0.98 regardless of peptide quantified (Figure 2C&D). Here, 211 patients with available genetic results (Figure 2E). WD is genetically confirmed in 144 patients with two evident pathogenic or likely pathogenic variants. This leaves 67 (32%) patients without straightforward genetic diagnosis. Within the genetically confirmed subgroup, 91% were ATP7B deficient agreeing with sequencing results. More importantly, 94% of patients without clear genetic results (containing VUS, VCI, or missing variants upon sequencing) were ATP7B deficient. ATP7B peptide concentration analysis can be highly useful in these patients.
ATP7B peptide concentrations were measured in patient samples collected up to 11 years prior to study whether ATP7B degradation impacts stored samples (Supplemental Table 7). Individuals with reduced ATP7B in freshly or recently collected samples had reduced concentrations across time. This suggests that older samples are not being identified as patients due to ATP7B degradation. No diagnosis in this group changed due to date of sampling. This includes one normal control with three separate samples taken over six months. All measured concentrations clearly identified this individual as normal and the CV for the measurements in these samples was ~11.4% and 12.4% for ATP7B 1056 and 887, respectively.
Certain variants are highly prevalent in the population and more commonly seen in clinic. These represent important test cases for the discriminatory ability of ATP7B quantification by immuno-SRM. These variants had predominantly low or undetectable peptide concentrations supporting the hypothesis that protein levels are reduced in vivo. Three of these high frequency variants (H1069Q, R778L, E1064A, and M645R) have significantly reduced ATP7B concentrations in both homozygotes and compound heterozygotes (Figure 3A–D). Patients with these common mutations, and therefore a significant percentage of patients overall, should be readily discriminated by the use of immuno-SRM as an index test.
Direct measurement of ATP7B peptides means that disease causing mutations that affect protein activity but not protein concentration will generate false negative results. In each of the 17 false negative patients both ATP7B 887 and ATP7B 1056 are above diagnostic cutoffs indicating significant production of non-functional protein. Identifying these variants and their frequency will be important in interpreting immuno-SRM in the context of other clinical results. If ATP7B concentrations in patient DBS are in the established normal ranges but clinical suspicion for WD is high, continued patient workup is obligatory to confirm the diagnosis. Several such variants were found here and ATP7B levels often depended on the nature of the second variant (Table 2). The variants found in these patients have allele frequencies (AF) ranging from unknown to 0.0089%, and therefore would represent a small percentage of the overall patient population.
The most common variants ATP7B levels above the cutoff are p.G710S and R969Q (Table 2; Figure 3E&F). There were 13 WD patients carrying the p.G710S variant and 8 patients with p.R969Q. Seven p.G710S patients had normal concentrations of ATP7B peptides when they were either homozygous for p.G710S or compound heterozygous with p.G710A and p.M665I. Three p.R969Q patients were false negatives. In two of these cases p.R969Q is in combination with p.H1069Q, which is shown to significantly reduce ATP7B concentrations. p.G710S is a variant associated with severe liver disease, 2 patients required an emergency liver transplantation due to fulminant hepatic failure.29 These variants are known pathogenic by causing significantly impaired copper transport activity while maintaining normal ATP7B trafficking and phosphorylation in vitro.24, 30, 31 Patients with these variants could be missed by immuno-SRM when in combination with variants producing significant ATP7B. When in combination with a variant severely affecting ATP7B concentrations, as in the p.H1069Q cases, ATP7B levels may give false negatives (Table 2). The mechanisms of variant interaction in vivo are currently under study. Diagnostic potential will therefore depend on the specific genotype of these patients. However, knowing these variants have the potential to generate normal levels of ATP7B will be useful when evaluating genetic analysis following ATP7B measurement.
Like the method-dependent cut-offs used for Cp, 24-hour urinary copper32 and hepatic copper for diagnosing WD33, 34, measurement of ATP7B peptides require validation in the population in which it will be used. This will aid in generating clearly defined cutoffs. A true description of the final diagnostic performance of ATP7B concentration measurement will come with conducting a large cohort validation or pilot study such as newborn screening.
Cp is not useful as a screening test for WD,9 but levels of Cp <10 mg/dL are regarded as useful for diagnosis (score 2 in Leipzig score) of WD. A recent Chinese study showed that Cp levels <12 mg/dL are strongly indicative of a diagnosis of WD.32 Here, 200 patients had Cp values available, among them 77 (38.5%) had moderate Cp values of 10–20 mg/dL and an additional 16 (8%) had normal Cp levels >20 mg/dL. 92.9% of patients with Cp <10 mg/dL had ATP7B concentrations below diagnostic cutoffs (Figure 4A–C). When Cp levels were moderately reduced, 91.6% of patients would be identified by ATP7B analysis. Even in those with normal Cp values ATP7B identified 87.5% of cases. Measurement of DBS ATP7B can provide clarity when Cp levels are ambiguous. This is likely because ATP7B is not a measurement of secondary disease effects. Cp and copper measurements are often confounded by external processes or disease influences including liver cirrhosis and malnutrition. As Cp is an acute-phase reactant possessing ferroxidase activity, the concentration can be elevated by acute inflammation. A prospective study on serum Cp as a screening test for WD in patients referred with liver disease showed positive predictive value of only 6%.9 Here, immuno-SRM analysis of ATP7B clearly outperforms Cp measurement.
Clinical information including age, Leipzig scores, liver copper content, presence of KF rings, initial presentation, and presence of cirrhosis were obtained where possible (Supplemental Table 3). No significant differences in ATP7B concentration were found based on age (pediatric vs. adult), presence of KF ring, presentation (hepatic, neurologic, both hepatic and neurologic, or asymptomatic), or presence of cirrhosis. The ATP7B 887 peptide concentration in hepatic and neurologic presentation was 33.8 ± 51.7 pmol/L (mean ± SD) and 38.2 ± 57.1 pmol/L, respectively. This appears aligned with previous observations in a large cohort showing the absence of any phenotype-genotype correlation regarding initial symptomatic manifestation.35 Fifty-nine patients had hepatic copper measurements and 86.4% of these had elevated liver copper above 250 μg/g. This includes five with normal ATP7B levels, three of them with p.G710S variant, and supports their diagnosis as WD patients despite negative immuno-SRM results (Figure 4G–I). Of interest, three samples were received with suspicion of WD by elevated liver copper but with no ATP7B variants identified. These samples had normal ATP7B concentrations, supporting their status as non-WD patients and providing an example of immuno-SRM analysis in ruling out WD (Figure 4I). Finally, it is unknown how many patients were being treated and what their treatment regimens were. Current WD therapies are focused on copper depletion and not restoration of ATP7B protein. As such, treatments would not affect the measured levels of ATP7B.
Examples from data set where diagnosis is simplified
The ability of ATP7B measurement to clarify case results extends to ambiguities in genetic analysis and contribute significantly to advancing non-invasive clinical diagnosis of WD (Figure 2E, Figure 4D–F). There are seven cases presented where patients were compound heterozygous or homozygous for two VUS indicating a lack of strong understanding of how these variants will contribute to phenotype (Table 2). All these patients had significantly reduced ATP7B peptides levels which suggesting affected patient status. Three patients in this group had only moderately reduced Cp between 10 and 20 mg/dL. In one case (#186), sequencing returned one VUS computationally predicted to be pathogenic and one (p.V1364V-fs) predicted to be benign. This patient had entirely non-detectable DBS ATP7B indicating the consequence of this genetic combination is disease-causing. One patient (#86) presented an interesting clinical situation in that a VUS was found on sequencing and Cp concentrations were clearly within the normal range (27 mg/dL) (Table 2). Here, ATP7B measurement provided clear indication that these patients severely lacked ATP7B protein and were very likely affected by WD in a way that genetic analysis, prediction, and Cp measurement could not.
Similarly, there were 37 patients where a known pathogenic or likely pathogenic variant was found in combination with a VUS (Figure 4E, Table 2). Thirty-five (94.6%) of these had reduced ATP7B concentrations of at least one peptide indicating positive identification as a WD patient despite a lack of knowledge of the consequences of their specific mutations. In 14 of these cases, Cp was found to be ≥10 mg/dL. Here, a significant reduction in patient ATP7B is particularly valuable in assigning a clinical designation and treatment course.
Of concern are situations where Sanger sequencing and/or NGS identifies only one variant. Sequencing analysis can be robust for targeting known variants but can return negative results if disease causing mutations are small or large deletions, duplications, in the deep intronic or promoter regions, or in poly-A tails. These situations can have significant impact on patient identification and place greater emphasis on Cp measurements and other WD diagnostics while the genetic basis for disease is not known. In 18 patients from our cohort with high suspicion for WD, only one ATP7B variant was identified (Figure 4F, Table 2). All of them had ATP7B measurement with peptide concentrations below established cutoffs. This includes four patients with Cp concentrations between 10 and 20 mg/dL, where a single variant and a moderate Cp reduction would be insufficient to establish the diagnosis. In three patients the single variant detected from sequencing was a VUS causing a further complication in genetic interpretation. ATP7B measurement can provide direct evidence of the consequences of existing variants even if the second variant was not detected from sequencing workflows.
It is notable that some carriers with ATP7B peptide concentrations below cut-off create potential false positives. ATP7B 887 analysis showed 4/48 carriers had the peptide levels below diagnostic cut-offs. The mechanisms by which a single affected allele reduce ATP7B concentrations are unknown and require study. Polymorphisms affecting protein concentration or factors affecting possible co-translated protein interactions may exist but are currently unknown.
Finally, we propose a new algorithm for WD diagnosis incorporating ATP7B measurement findings for each peptide (Figure 5). Here we define four possible patient groups based on ATP7B peptide level: [1] directly established WD (< 32 pmol/L for ATP7B 1056 and < 56 pmol/L for ATP7B 887); [2] probable WD (ATP7B between group 1 and the diagnostic cutoff); [3] possible WD (ATP7B 1056: Cutoff - 154 pmol/L and ATP7B 887: Cutoff - 200 pmol/L); and [4] unlikely WD (> 154 pmol/L for ATP7B 1056 and > 200 pmol/L for ATP7B 887). In validating the algorithm on the current retrospectively collected patients, we found that 172 out of 216 (79.6%) showed ATP7B within the range of patient group 1 making their diagnosis very highly likely. Applying this method to the remaining cases along with mutation analysis show that all are able to reach WD diagnosis except for two cases in which diagnosis or carrier status cannot be determined. Full validation of this proposed algorithm will be necessary through a large cohort prospective study.
Figure 5:

Proposed Wilson Disease Diagnostic Algorithm
Limitations
The study is limited in that data are predominantly Caucasian patients. Patients from the Indian subcontinent, Africa and South America may have different genotype distributions and possibly differing ATP7B peptide levels. There is also an unavoidable selection bias due to the analysis of only well-described WD cases. A significant limitation comes from the fact that these samples have been collected in a retrospective manner. It will be important to prospectively analyze a large cohort samples before genetic analysis in future applications. Full validation in a large prospective study will allow for more accurate definition of ATP7B variability in healthy normal samples and diagnostic parameters including sensitivity, specificity, PPV and NPV. The current statistical measures of predictive significance may change upon conducting a validating study. Furthermore, very few cases of fulminant WD and hemolysis were investigated. Patients presenting with hemolysis may have differing ATP7B peptides concentrations due to RBC lysis. In addition, ATP7B analysis is unable to delineate patient groups based on phenotype, i.e., hepatic or neurological types or disease severity. Finally, only two signature ATP7B peptides have been analyzed. Alternative peptide sequences may have different discriminating capability such that study of additional candidate peptides may be helpful.
LC-MS/MS is considered highly specialized technology though it has been used in clinical laboratories in a wide array of fields such as toxicology, drug monitoring and newborn screening. Following the standard validation process in the clinical laboratory, we anticipate the assay can be successfully implemented into clinical practice.
CONCLUSION
ATP7B peptide analysis identified WD patients in a large majority cases and reduced ambiguities resulting from genetic analysis and Cp levels. This non-invasive assay can serve as an adjunctive test for the diagnosis of WD and is expected to fundamentally advance the use of proteomic technology for a rapid screening tool, an area that holds great promise but is largely untapped.
Supplementary Material
“WHAT YOU NEED TO KNOW”.
BACKGROUND AND CONTEXT:
WD patient identification remains a challenge resulting in delayed diagnosis and development of irreversible severe complications, such as permanent brain or liver damage, which render treatments ineffective
NEW FINDINGS:
This first large cohort study directly measuring ATP7B from DBS of WD patients with diverse genetic backgrounds showed ATP7B peptide concentrations have a high diagnostic potential.
LIMITATIONS:
The study is limited in that data are mostly obtained from Caucasian patients. Sensitivity and specificity may be variable geographically.
IMPACT:
ATP7B peptide analysis identifies most WD patients reducing ambiguities resulting from genetic analysis and is expected to advance the use of proteomics, a promising technology but largely untapped.
Acknowledgement:
This study was supported by the grants from NIH-R21HD097558, and R01HD098180 and Wilson Disease Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Bull PC, Thomas GR, Rommens JM, et al. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet 1993;5:327–37. [DOI] [PubMed] [Google Scholar]
- 2.Chang IJ, Hahn SH. The genetics of Wilson disease. Handbook of clinical neurology 2017;142:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terada K, Schilsky ML, Miura N, et al. ATP7B (WND) protein. Int J Biochem Cell Biol 1998;30:1063–7. [DOI] [PubMed] [Google Scholar]
- 4.Reilly M, Daly L, Hutchinson M. An epidemiological study of Wilson’s disease in the Republic of Ireland. J Neurol Neurosurg Psychiatry 1993;56:298–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hahn SH. Population screening for Wilson’s disease. Ann N Y Acad Sci 2014;1315:64–9. [DOI] [PubMed] [Google Scholar]
- 6.Saroli Palumbo C, Schilsky ML. Clinical practice guidelines in Wilson disease. Annals of translational medicine 2019;7:S65–S65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol 2012;56:671–85. [DOI] [PubMed] [Google Scholar]
- 8.Chapter Ferenci P. 14 - Diagnosis of Wilson disease. In: Członkowska A, Schilsky ML, eds. Handbook of Clinical Neurology. Volume 142: Elsevier, 2017:171–180. [DOI] [PubMed] [Google Scholar]
- 9.Cauza E, Maier-Dobersberger T, Polli C, et al. Screening for Wilson’s disease in patients with liver diseases by serum ceruloplasmin. J Hepatol 1997;27:358–62. [DOI] [PubMed] [Google Scholar]
- 10.Steindl P, Ferenci P, Dienes HP, et al. Wilson’s disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology 1997;113:212–8. [DOI] [PubMed] [Google Scholar]
- 11.Korman JD, Volenberg I, Balko J, et al. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008;48:1167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong A, Leitold S, Uhanova J, et al. Non-Wilson’s Disease-Associated Hypoceruloplasminemia. J Clin Exp Hepatol 2020;10:284–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merle U, Eisenbach C, Weiss KH, et al. Serum ceruloplasmin oxidase activity is a sensitive and highly specific diagnostic marker for Wilson’s disease. J Hepatol 2009;51:925–30. [DOI] [PubMed] [Google Scholar]
- 14.Ferenci P, Caca K, Loudianos G, et al. Diagnosis and Phenotypic Classification of Wilson Disease. Liver International 2003;23:139–142. [DOI] [PubMed] [Google Scholar]
- 15.Jung S, Whiteaker JR, Zhao L, et al. Quantification of ATP7B Protein in Dried Blood Spots by Peptide Immuno-SRM as a Potential Screen for Wilson’s Disease. J Proteome Res 2017;16:862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins CJ, Yi F, Dayuha R, et al. Multiplexed Proteomic Analysis for Diagnosis and Screening of Five Primary Immunodeficiency Disorders From Dried Blood Spots. Front Immunol 2020;11:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins CJ, Chang IJ, Jung S, et al. Rapid Multiplexed Proteomic Screening for Primary Immunodeficiency Disorders From Dried Blood Spots. Frontiers in Immunology 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson NL, Anderson NG, Haines LR, et al. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J Proteome Res 2004;3:235–44. [DOI] [PubMed] [Google Scholar]
- 19.Kuhn E, Whiteaker JR, Mani DR, et al. Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol Cell Proteomics 2012;11:M111.013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoenherr RM, Zhao L, Whiteaker JR, et al. Automated screening of monoclonal antibodies for SISCAPA assays using a magnetic bead processor and liquid chromatography-selected reaction monitoring-mass spectrometry. J Immunol Methods 2010;353:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whiteaker JR, Paulovich AG. Peptide immunoaffinity enrichment coupled with mass spectrometry for peptide and protein quantification. Clin Lab Med 2011;31:385–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoofnagle AN, Whiteaker JR, Carr SA, et al. Recommendations for the Generation, Quantification, Storage, and Handling of Peptides Used for Mass Spectrometry-Based Assays. Clin Chem 2016;62:48–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacLean B, Tomazela DM, Shulman N, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010;26:966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huster D, Hoppert M, Lutsenko S, et al. Defective cellular localization of mutant ATP7B in Wilson’s disease patients and hepatoma cell lines. Gastroenterology 2003;124:335–45. [DOI] [PubMed] [Google Scholar]
- 25.Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics 2019;35:1978–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reeve JLV, Frayling IM, Twomey PJ. Challenges in molecular diagnosis of Wilson disease. Journal of Clinical Pathology 2020;73:181. [DOI] [PubMed] [Google Scholar]
- 27.Poon K-S, Teo ZH, Yap JH, et al. Challenges in molecular diagnosis of Wilson disease: viewpoint from the clinical laboratory. Journal of Clinical Pathology 2020;73:231. [DOI] [PubMed] [Google Scholar]
- 28.Tang N, Sandahl TD, Ott P, et al. Computing the Pathogenicity of Wilson’s Disease ATP7B Mutations: Implications for Disease Prevalence. Journal of Chemical Information and Modeling 2019;59:5230–5243. [DOI] [PubMed] [Google Scholar]
- 29.Stättermayer AF, Entenmann A, Gschwantler M, et al. The dilemma to diagnose Wilson disease by genetic testing alone. Eur J Clin Invest 2019;49:e13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huster D, Kühne A, Bhattacharjee A, et al. Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology 2012;142:947–956.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997;61:317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong Y, Wang RM, Yang GM, et al. Role for Biochemical Assays and Kayser-Fleischer Rings in Diagnosis of Wilson’s Disease. Clin Gastroenterol Hepatol 2020. [DOI] [PubMed] [Google Scholar]
- 33.Yang X, Tang XP, Zhang YH, et al. Prospective evaluation of the diagnostic accuracy of hepatic copper content, as determined using the entire core of a liver biopsy sample. Hepatology 2015;62:1731–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferenci P, Steindl-Munda P, Vogel W, et al. Diagnostic Value of Quantitative Hepatic Copper Determination in Patients With Wilsons Disease. Clinical Gastroenterology and Hepatology 2005;3:811–818. [DOI] [PubMed] [Google Scholar]
- 35.Ferenci P, Stremmel W, Członkowska A, et al. Age and Sex but Not ATP7B Genotype Effectively Influence the Clinical Phenotype of Wilson Disease. Hepatology 2019;69:1464–1476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.