

Abstract
An advanced aerosol treatment, with a focus on semivolatile nitrate formation, is introduced into the Community Atmosphere Model version 5 with interactive chemistry (CAM5‐chem) by coupling the Model for Simulating Aerosol Interactions and Chemistry (MOSAIC) with the 7‐mode Modal Aerosol Module (MAM7). An important feature of MOSAIC is dynamic partitioning of all condensable gases to the different fine and coarse mode aerosols, as governed by mode‐resolved thermodynamics and heterogeneous chemical reactions. Applied in the free‐running mode from 1995 to 2005 with prescribed historical climatological conditions, the model simulates global distributions of sulfate, nitrate, and ammonium in good agreement with observations and previous studies. Inclusion of nitrate resulted in ∼10% higher global average accumulation mode number concentrations, indicating enhanced growth of Aitken mode aerosols from nitrate formation. While the simulated accumulation mode nitrate burdens are high over the anthropogenic source regions, the sea‐salt and dust modes respectively constitute about 74% and 17% of the annual global average nitrate burden. Regional clear‐sky shortwave radiative cooling of up to −5 W m−2 due to nitrate is seen, with a much smaller global average cooling of −0.05 W m−2. Significant enhancements in regional cloud condensation nuclei (at 0.1% supersaturation) and cloud droplet number concentrations are also attributed to nitrate, causing an additional global average shortwave cooling of −0.8 W m−2. Taking into consideration of changes in both longwave and shortwave radiation under all‐sky conditions, the net change in the top of the atmosphere radiative fluxes induced by including nitrate aerosol is −0.7 W m−2.
Keywords: Aerosol model, anthropogenic emissions, climate change, climate model, nitrate aerosol, radiative effects
Key Points
A modal version of the advanced aerosol chemistry module MOSAIC is developed and introduced in a climate model to simulate nitrate aerosol
MOSAIC provides an accurate and efficient treatment for dynamically partitioning semivolatile gases over to entire aerosol size distribution
The modeled global distribution of nitrate is in good agreement with observations and its impact on the radiative effects is quantified
1. Introduction
Atmospheric aerosols influence climate by scattering and absorbing solar radiation and by serving as nuclei for cloud formation (Boucher et al., 2013). Coarse aerosols (diameter larger than 1 µm) such as wind‐driven sea salt and mineral dust dominate the globally averaged particulate mass concentrations. However, it is the much more numerous fine aerosols (smaller than 1 µm) that are crucial in modulating the Earth's radiative forcing. Fine aerosols include primary particles, such as soot directly emitted during fossil fuel combustion and biomass burning; and secondary particulate species such as sulfate (SO4), nitrate (NO3), ammonium (NH4), and organics, respectively formed in the atmosphere via gas‐to‐particle conversion of sulfur dioxide (SO2), nitrogen oxides (NOx), ammonia (NH3), and volatile organic compounds (VOCs). While the precursor gases for secondary aerosols are released from a variety of natural sources, human activity during the industrial era has added significantly to their emissions inventory in many regions around the world (Bouwman et al., 1997; Hoesly et al., 2018). Field observations and global modeling studies indicate that fine aerosol mass is presently dominated by SO4 and organics, with NO3 estimated at about a quarter of SO4 in terms of the overall global burden (Myhre, Samset, et al., 2013). However, NO3 competes with SO4 for the available NH3 to exist as stable ammonium nitrate in the fine mode since ammonium sulfate is thermodynamically favored due to its extremely low volatility. Consequently, the relative contribution of NO3 is predicted to increase as anthropogenic SO2 emissions and the resulting SO4 aerosols are projected to decline faster than NOx while anthropogenic NH3 emissions are expected to rise through the 21st century (Bauer et al., 2007; Bellouin et al., 2011; Hauglustaine et al., 2014; Li et al., 2015; Myhre, Shindell, et al., 2013; Paulot et al., 2016).
The size distribution, mixing state, physicochemical properties, and atmospheric lifetimes of aerosols strongly depend on the formation and loss mechanisms of secondary species, which must be faithfully represented in global models for skillful predictions of aerosol climate impacts. Gas‐phase oxidation of SO2 by the hydroxyl radical (OH) forms extremely low volatile sulfuric acid (H2SO4) vapor that can nucleate to form nanometer‐sized new particles as well as irreversibly condense on preexisting aerosols. In contrast, gas‐phase oxidation of NOx by OH during the day and heterogeneous oxidation of dinitrogen pentoxide (N2O5) at night forms nitric acid (HNO3) vapor, which is semivolatile under typical atmospheric conditions, and the mechanisms leading to nitrate aerosol formation are rather complex. For instance, HNO3 can combine with excess NH3 (left after fully neutralizing sulfate) and reversibly condense on preexisting aerosols to form ammonium nitrate (NH4NO3). In deliquesced aerosols, dissociation of NH4NO3 into ammonium and nitrate ions greatly increases partitioning of HNO3 and NH3 to the particle phase as aerosol liquid water content increases with ambient relative humidity (RH). Increased aerosol water associated with NH4NO3 is also available for dissolving soluble semivolatile organic gases (SOAGs), some of which can react further to form low‐volatility secondary organic aerosol (SOA; Ervens et al., 2011). The extent of HNO3 and NH3 partitioning also increases with decrease in temperature due to lowering of their volatilities. Recent laboratory experiments show that supersaturated HNO3 and NH3 vapors, sustained by source inhomogeneity, can rapidly grow freshly nucleated particles via nonequilibrium condensation at temperatures below 5°C (M. Wang et al., 2020). And at temperatures below −15°C, HNO3 and NH3 vapors can become supersaturated enough to even nucleate to form new NH4NO3 particles through an acid‐base stabilization mechanism (M. Wang et al., 2020). The importance of fine mode NH4NO3 therefore generally increases with proximity to the precursor sources and with colder temperatures typically encountered at higher altitudes and during wintertime. NH4NO3 is more hygroscopic than sulfate salts of ammonium and deliquesce at lower relative humidity levels than pure ammonium sulfate, thus increasing the particle size, its scattering efficiency, and its ability to serve as cloud condensation nucleus.
However, particulate NH4NO3 is semivolatile and can evaporate back to HNO3 and NH3 as the polluted air mass undergoes dilution or experiences warmer temperatures. Complex chemical interactions occur when polluted air rich in NH4NO3 aerosol mixes with marine air containing sea‐salt aerosol. In the case of deliquesced sea‐salt aerosol, HNO3 partially displaces particulate Cl as HCl gas until both gases reach an equilibrium with their particle‐phase counterparts (Hildemann et al., 1984). In the case of solid sea‐salt particles, HNO3 can continue to condense until it completely displaces Cl from solid NaCl to form solid NaNO3. The resulting depletion of HNO3 in the gas phase due to uptake by sea‐salt aerosol causes particulate NO3 aerosol to evaporate until it reaches a new equilibrium. However, because sea‐salt aerosols typically constitute a much larger reservoir of particulate Cl compared to the total particulate NO3 in Aitken and accumulation modes, it is thermodynamically favorable for most of the particulate NH4NO3 to evaporate and partition the resulting HNO3 to sea‐salt aerosol.
Similar chemical interactions occur when polluted air mixes with continental air containing soil dust aerosol containing insoluble calcium carbonate (CaCO3). In this case, HNO3 undergoes heterogeneous reaction on dust to completely displace carbonate as CO2 gas to form particulate Ca(NO3)2 (Hanisch & Crowley, 2001; Hwang & Ro, 2006; Laskin et al., 2005; Tobo et al., 2010). Again, due to the relatively much larger reservoir of carbonates in fine and coarse mode dust aerosols, the NH4NO3 in the Aitken and accumulation modes continues to evaporate as the gas‐phase HNO3 gradually partitions to the dust aerosol (Fairlie et al., 2010). Moreover, in regions where polluted air mixes with both dust and sea‐salt aerosols, such as polluted Asian outflow over the Pacific Ocean, the HCl displaced by HNO3 from sea‐salt aerosol can also react with dust to form particulate CaCl2. Both Ca(NO3)2 and CaCl2 are highly soluble and can deliquesce at low RH, thus converting the relatively less hygroscopic dust aerosol into aqueous droplets with aging (Tobo et al., 2010). HCl can also combine with excess NH3 to form ammonium chloride (NH4Cl) in fine mode aerosols (Du et al., 2010).
The characteristic time for gas‐phase HNO3 to equilibrate with fine mode aerosols is on the order of a few seconds to 30 min (Dassios & Pandis, 1999; Fountoukis et al., 2009) and ranges from a few hours to days in the case of coarse sea‐salt and dust aerosols (Fridlind & Jacobson, 2000; Meng & Seinfeld, 1996). Even when the gas‐phase HNO3 may appear to have equilibrated with the bulk of the fine mode aerosols, particles of different sizes within the fine mode may still be out of equilibrium when they grow from condensation of supersaturated HNO3 and NH3 vapors (M. Wang et al., 2020). A size‐resolved dynamic mass transfer treatment is therefore necessary to reliably simulate the competitive partitioning of HNO3 along with H2SO4, HCl, and NH3 to particles across the entire size distribution. However, simulating dynamic mass transfer of these gases to size‐distributed aerosol is numerically challenging and computationally expensive due to the coupled stiff differential equations that govern the process. Consequently, many previous global modeling studies of inorganic aerosols have adopted various ad hoc bulk equilibrium partitioning treatments for nitrate for either the entire aerosol size distribution or for only the fine aerosol mode while ignoring the coarse mode dust and/or sea‐salt aerosols (Adams et al., 1999, 2001; An et al., 2019; Bauer et al., 2007; Bellouin et al., 2011; Bian et al., 2017; Gong et al., 2003; Li et al., 2015; Liao & Seinfeld, 2005; Liao et al., 2009; Metzger & Lelieveld, 2007; Mezuman et al., 2016; Myhre et al., 2006; Pye et al., 2009; Zhou et al., 2012). Several hybrid approaches have also been used in global models to reduce the computational cost by assuming equilibrium partitioning for the fine mode NH4NO3 aerosol while using kinetic mass transfer for the coarse mode dust and/or sea‐salt aerosols (Benduhn et al., 2016; Bian et al., 2017; Fairlie et al., 2010; Feng & Penner, 2007; Hauglustaine et al., 2014; Karydis et al., 2016; Pozzer et al., 2012; Pringle et al., 2010; Rodriguez & Dabdub, 2004; Xu & Penner, 2012). Although an improvement over the bulk equilibrium assumption, the hybrid approaches are still prone to significant errors and biases (Hu et al., 2008) and underestimate the contribution of HNO3 to nanoparticle growth in inhomogeneous source environments (M. Wang et al., 2020). Due to the high computational cost, only a few previous global modeling studies have used a fully dynamic treatment for partitioning semivolatile gases over the entire size distribution (Jacobson, 2001; Matsui, 2017).
In this paper, we introduce a computationally efficient dynamic gas‐particle partitioning treatment into the Community Atmosphere Model version 5 with interactive chemistry (CAM5‐chem) global chemistry‐aerosol‐climate model (Tilmes et al., 2015) by coupling the comprehensive aerosol chemistry module, Model for Simulating Aerosol Interactions and Chemistry (MOSAIC; Zaveri et al., 2008), with the 7‐mode Modal Aerosol Module (MAM7) (Liu et al., 2012). We apply the model in the free‐running climate mode with prescribed historical climatological forcing conditions to simulate global aerosol distributions with a focus on nitrate. We first evaluate the model against observations of key gas and aerosol species. We then evaluate the impact of nitrate aerosols on the direct and indirect radiative effects.
2. Model and Methods
2.1. Model Description
The CAM5, which is the atmosphere component of the Community Earth System Model (CESM) version 1.2, is used in this study. CAM5 includes updated treatments of several major physical processes in the atmosphere, particularly clouds and aerosols (Neale et al., 2012). Microphysical conversions among cloud liquid droplets, ice crystals, rain and snow are treated by the two‐moment stratiform cloud microphysics with a prognostic precipitation scheme (Gettelman et al., 2015), interacting with aerosols predicted by the MAM (Liu et al., 2012, 2016) for the indirect effects through droplet activation (Abdul‐Razzak & Ghan, 2000) and ice nucleation (Liu & Penner, 2005; Liu et al., 2007). The radiative effects of aerosol and clouds are taken into account by the Rapid Radiative Transfer Model for General Circulation Models radiation scheme (Iacono et al., 2008). A unified parameterization of boundary‐layer turbulence, shallow convection, and cloud macrophysics is provided by the Cloud Layers Unified by Binormals scheme (Bogenschutz et al., 2013; Larson et al., 2002). Deep convection in CAM5 is represented following a modified version of the Zhang‐McFarlane (ZM) parameterization (G. J. Zhang & McFarlane, 1995). The other model components include the Community Land Model (CLM) version 4.5 (Lawrence et al., 2011) for terrestrial ecosystems, biogenic, and dust emissions, and the Community Ice Code version 4 for sea ice and snow (Hunke & Lipscomb, 2008).
2.1.1. Gas Chemistry Scheme
The CAM5‐chem used in this study includes extensive trace‐gas chemistry for both the troposphere and stratosphere, based on the MOZART (Model for Ozone and Related chemical Tracers) chemical mechanism (Emmons et al., 2010; Tilmes et al., 2016). The version of the chemical mechanism used here includes additional updates that have been released in CESM2.0, including expansion of aromatic oxidation, isoprene and terpene oxidation, and more detailed representation of organic nitrates, as described in Emmons et al. (2010). The MOZART‐T1 tropospheric chemistry scheme includes 130 gas‐phase species, with updated reaction rates following recommendations by Burkholder et al. (2015). Hydrolysis of N2O5 is treated in the mechanism as heterogeneous uptake on aerosols, with an uptake coefficient (gamma) of 0.1 and the surface area density calculated from the simulated sulfate, nitrate, hydrophilic organic, and black carbon aerosols (Tilmes et al., 2016). This study uses the simple SOA scheme that is part of MAM (described in Section 2.1.2) and is not connected to the MOZART gas‐phase chemistry.
2.1.2. Aerosol Scheme
The 7‐mode version of MAM is used to predict the mass mixing ratios of major aerosol species (i.e., sulfate, black carbon, organic carbon [OC], dust, and sea salt) and number concentration of aerosols in each aerosol mode (i.e., Aitken, accumulation, primary carbon, fine sea salt, fine dust, coarse sea salt, and coarse dust). MAM represents the aerosol size distribution and internal/external mixing of aerosol species within aerosol modes and between modes (Liu et al., 2012, 2016). Aerosol processes include emission, transport, dry and wet removal, gas‐ and aqueous‐phase chemistry, water uptake, and aerosol microphysics (nucleation, coagulation, and condensation). The transfer of aerosol mass and number between aerosol modes due to the growth of aerosol particles is also considered. Ammonium aerosol is explicitly treated, while nitrate is not included in the default MAM7. There are 33 transported aerosol component‐mass and number species in the default MAM7 used in this study (Table 1).
Table 1.
List of Chemical Component Species in the MAM7 Aerosol Modes
| Species | Accum. (a1) | Aitken (a2) | Primary carbon (a3) | Fine sea salt (a4) | Coarse sea salt (a6) | Fine dust (a5) | Coarse dust (a7) |
|---|---|---|---|---|---|---|---|
| BC | X | X | |||||
| POM | X | X | |||||
| SOA | X | X | |||||
| SO4 | X | X | X | X | X | X | |
| NH4 | X | X | X | X | X | X | |
| NO3 | O | O | O | O | O | O | |
| Cl | O | O | O | O | O | O | |
| Na | X | X | X | X | |||
| Dst | X | X | X | X | |||
| Ca | O | O | O | O | |||
| CO3 | O | O | O | O | |||
| Total | 7/11 | 5/9 | 2/2 | 3/5 | 3/5 | 3/7 | 3/7 |
Note. “X” indicates species that are present in both the default and MOSAIC model versions; “O” indicates additional species that are present only in the MOSAIC version. The Na species is bulk primary sea salt in the default model version but is sodium in the MOSAIC version. The Dst species is bulk primary soil dust in the default model version but is other inorganics in the MOSAIC version. (Note that the Liu et al. [2012] MAM7 did not have Dst in the accumulation and Aitken modes.) The last row is number of chemical species in each mode for the default and MOSAIC model versions.
The MOSAIC (Zaveri et al., 2008) is coupled with MAM7 to treat dynamic gas‐particle partitioning of all condensable aerosol species, including sulfate, nitrate, chloride, ammonium, and secondary organics and the related aerosol thermodynamics. The standalone box model version of MOSAIC is a comprehensive aerosol chemistry and microphysics module that treats new particle formation, aerosol thermodynamics, dynamic gas‐particle mass transfer, particle‐phase chemistry, coagulation, and sectional growth dynamics. It includes all major inorganic salts and electrolytes composed of H+, NH4 +, Na+, Ca2+, SO4 2−, HSO4 −, CH3SO3 −, NO3 −, Cl−, and CO3 2− ions. Relatively minor ions such as K+ and Mg2+ are represented by equivalent amounts of Na+ while other unspecified inorganic species such as silica, other inert minerals, and trace metals found in soil dust aerosols are lumped together as “other inorganic mass” (OIN). MOSAIC also includes carbonaceous species such as black carbon, primary organics, and secondary organics. While organic–inorganic interactions are not presently treated explicitly in MOSAIC, organics and OIN species can absorb water.
At a given time step within MOSAIC, the thermodynamics submodule MESA (Multicomponent Equilibrium Solver for Aerosols) (Zaveri, Easter, & Peters, 2005) first determines the equilibrium particle‐phase state and water content in each size section as a function of particle composition, particle size (accounting for the Kelvin effect), relative humidity, and temperature. The dynamic gas‐particle partitioning submodule ASTEM (Adaptive Step Time‐split Euler Method) then calculates the driving forces for mass transfer of the gas‐phase species over each size section and simultaneously integrates the set of coupled differential equations (Zaveri et al., 2008). The mean stoichiometric activity coefficients of electrolytes necessary for the equilibrium phase state and mass transfer driving force calculations are estimated using the Multicomponent Taylor Expansion Method (Zaveri, Easter, & Wexler, 2005). A sectional version of MOSAIC has been implemented and extensively evaluated in the chemistry version of the regional Weather Research and Forecasting model, WRF‐Chem (Fast et al., 2006). Although MOSAIC has been used in the WRF‐Chem model using distributed‐memory parallelization software, significant refactorization of the MOSAIC code was required to use it within CAM and CESM, which allow a hybrid model supporting both distributed‐ and shared‐memory parallelization. At the same time, CESM simulations exposed MOSAIC to a wider range of meteorological and chemical conditions that were not previously encountered in WRF‐Chem simulations. Consequently, some numerical treatments in MOSAIC were revised slightly for robust performance and stability.
Many global models based on bulk equilibrium or hybrid partitioning approaches rely on aerosol thermodynamics modules such as ISORROPIA‐I (Nenes et al., 1998), ISORROPIA II (Fountoukis & Nenes, 2007), and EQSAM3 (Metzger & Lelieveld, 2007). While MOSAIC and these thermodynamics modules are comparable in terms of chemical complexity, the gas‐particle partitioning approach in MOSAIC is fundamentally different. The thermodynamics modules are designed with the assumption of instantaneous equilibrium between the gas and particle phases and are therefore ideally applicable for bulk equilibrium calculations. In contrast, the MESA module in MOSAIC performs equilibrium phase state calculations for the particle phase only, while the ASTEM module is designed for dynamic gas‐particle mass transfer to reliably simulate the competitive partitioning of all semivolatile gases (HNO3, HCl, NH3, and SOA) to particles of different sizes and compositions while considering size‐dependent processes such as the Kelvin effect.
In the version of MOSAIC in CAM5‐chem, the ASTEM submodule for dynamic gas‐particle mass transfer and the MESA submodule for phase state and aerosol water uptake replace the corresponding default‐MAM treatments. The existing MAM code for renaming (intermode transfer due to particle growth), ageing, nucleation, coagulation, and other processes is unchanged except as noted. For computational efficiency, the MESA submodule in MOSAIC was constrained to the deliquesced metastable state at relative humidities greater than 40%. The MOSAIC SOA submodule was modified to use the simple one‐species SOA scheme of MAM (Liu et al., 2012). The scheme utilizes a lumped condensable SOAG that is directly emitted at the surface, with emissions based on five precursor VOC categories and the following estimated mass yields applied without considering the intermediate gas‐phase oxidation steps: 5% for BIGALK (lumped butanes and larger alkanes), 5% for BIGENE (lumped butenes and larger alkenes), 15% for aromatics, 4% for isoprene, and 25% for monoterpenes (Liu et al., 2012; Tilmes et al., 2019). SOAG is dynamically partitioned to the Aitken and accumulation modes of MAM according to the temperature‐dependent volatility of SOAG and primary organic mass concentration.
Nitrate aerosol can exist in the form of ammonium nitrate in the accumulation and Aitken modes while it exists as sodium nitrate in sea salt and as calcium nitrate in dust particles. Partitioning of nitrate to sea salt and dust displaces chloride and carbonate, respectively. Thus, the MOSAIC implementation added five new aerosol chemical components (nitrate, chloride, sodium, calcium, and carbonate) and 20 transported aerosol tracers to MAM7 (see Table 1). The default‐MAM bulk primary sea‐salt chemical component is replaced by a mixture of sodium, chloride, and sulfate with emitted/primary mass fractions of 0.385441717, 0.537558283, and 0.077, respectively (Pilson, 1998). The sulfate value is based on experimental sea water composition, and the sodium and chloride value are obtained from electroneutrality. This three‐species primary sea‐salt treatment gives a somewhat more accurate estimate of chloride content with no added computational cost. The default‐MAM bulk soil dust chemical component is replaced with a mixture of calcium, carbonate, and “other inorganics” with emitted/primary mass fractions of 0.020021541, 0.029978459, and 0.95, respectively, following (Zaveri et al., 2008). The HNO3 accommodation coefficient onto dust‐containing particles is based on the parameterization used in Fairlie et al. (2010). Refractive indices and hygroscopicities (used only for aerosol activation calculations) of the new chemical components are set as follows: nitrate uses default‐MAM sulfate values; sodium and chloride use default‐MAM bulk sea‐salt values; and calcium, carbonate, and other inorganics use default‐MAM bulk soil dust values. The default‐MAM aqueous chemistry routine was modified to treat reversible uptake of HNO3 and HCl to cloud droplets. Also, the MAM code for gas‐aerosol exchange, renaming, nucleation, coagulation, and aging was replaced with the newer version in the Energy Exascale Earth System Model Version 1 (H. Wang et al., 2020). This was used in all simulations, and its main impact is to increase nucleation and Aitken mode particle number.
2.1.3. Emissions
The emissions used in these simulations are based on the anthropogenic and biomass burning inventories specified for CMIP6 (Coupled Model Intercomparison Project Phase 6). Anthropogenic emissions for 1750–2014 have been compiled with the Community Emissions Data System (CEDS) (Hoesly et al., 2018). Biomass burning emissions are described by van Marle et al. (2017). Additional emissions (e.g., oceanic, soil NOx) are taken from the POET (Precursors of Ozone and their Effects in the Troposphere) inventory (Granier et al., 2005). Biogenic emissions are calculated online using the MEGANv2.1 algorithms incorporated in the CLM (Guenther et al., 2012). Continuous volcanic outgassing emissions of SO2, with 2.5% emitted as sulfate aerosols, are from the GEIA inventory (Andres & Kasgnoc, 1998). Dust (Zender et al., 2003) and sea‐salt (Mårtensson et al., 2003; Monahan, 1986) emissions are calculated online in the model, dependent on surface wind speed, and are tuned to match satellite observations of aerosol optical depth (AOD; Mahowald, Lamarque, et al., 2006; Mahowald, Muhs, et al., 2006). Dust emission factor (the dust_emis_fact parameter) is set to 1.2 and the sea‐salt emission scaling parameter is set to 1.6.
2.1.4. Simulations
As listed below, three simulations (i.e., Control, MOSAIC, and MOSAIC_no_nitrate) were conducted to study the sensitivity of aerosol properties and its radiative effects to the MOSAIC treatment and the role of nitrate, in particular. The simulations were run at 1.9° × 2.5° horizontal resolution for 11 years (1995–2005) in the free‐running mode with prescribed present‐day climatological forcing conditions (i.e., sea surface temperature, sea ice concentration, and greenhouse gases). Monthly mean fields from the last 10 years are used for analyses.
-
(a)
Control: CAM5‐chem‐MAM7—base case with the MOZART default gas chemistry, MAM7 aerosol scheme, emissions, and other model parameters/configurations as described in the sections above.
-
(b)
MOSAIC: CAM5‐chem‐MAM7‐MOSAIC—same as (a) but with MOSAIC aerosol chemistry (including HNO3 partitioning to particles) and aerosol water content.
-
(c)
MOSAIC_no_nitrate: same as (b) but excluding HNO3 partitioning to particles.
2.2. Observational Data Sets
2.2.1. Trace‐Gas Observations
Measurements of reactive nitrogen species that are precursors of nitrate aerosols have been made in the troposphere from aircraft in many regions of the world, allowing for detailed model evaluation of gas‐phase species. A climatology of these observations between 1995 and 2010 (Tilmes et al., 2015) is used to evaluate the simulations of ozone (O3), NOx, and the reactive nitrogen reservoirs, including peroxyacetyl nitrate (PAN) and nitric acid (HNO3).
2.2.2. Aerosol Observations
Modeled aerosol species, including sulfate, nitrate, and ammonium (SNA), are validated against several ground‐based observations of aerosol mass concentrations. For validating the SNA over United States, we adopt observations from IMPROVE (Interagency Monitoring of Protected Visual Environments), network data (http://vista.cira.colostate.edu/Improve) (Malm et al., 2004). The IMPROVE samplers collect 24‐h aerosols every 3 days. The aerosol samples are further analyzed for PM2.5 mass, elemental composition, ions, and OC and elemental carbon (EC). In this study, SNA fine particle masses obtained from 162 sites are averaged and further compared against annual mean SNA fine particle mass. EMEP (The European Monitoring and Evaluation Programme, http://www.emep.int) network monitors the surface aerosol mass concentrations of SNA over Europe. The data from 40 sites of EMEP network are available for the simulation period for comparison. In order to validate the model simulation in East Asia and China specifically, we adopt observations from 37 sites of EANET (Acid Deposition Monitoring Network in East Asia) and 14 sites of CAWNET (China Atmosphere Watch Network) (X. Y. Zhang et al., 2008), respectively. It should be noted that, EMEP and EANET data sets are not quality assured across different countries in Europe and East Asia.
3. Results and Discussion
3.1. Evaluation Against Observations
3.1.1. Key Trace Gases
The three model experiments are evaluated against an aircraft climatology for the free troposphere for 2–7 km. The climatology has been compiled using various aircraft campaigns performed between 1995 and 2010, sorted by different regions (Southern Hemisphere [SH], Tropics, Northern Hemisphere [NH] midlatitudes, NH Polar) and the four seasons. Averages of profiles of high occurrence were produced for various chemical species and binned together over 1 km altitude bins, as further described in Tilmes et al. (2015). Figure 1 compares model results that have been interpolated to the location of each profile for the aircraft campaigns used and averaged over regions and season as for the climatology.
Figure 1.
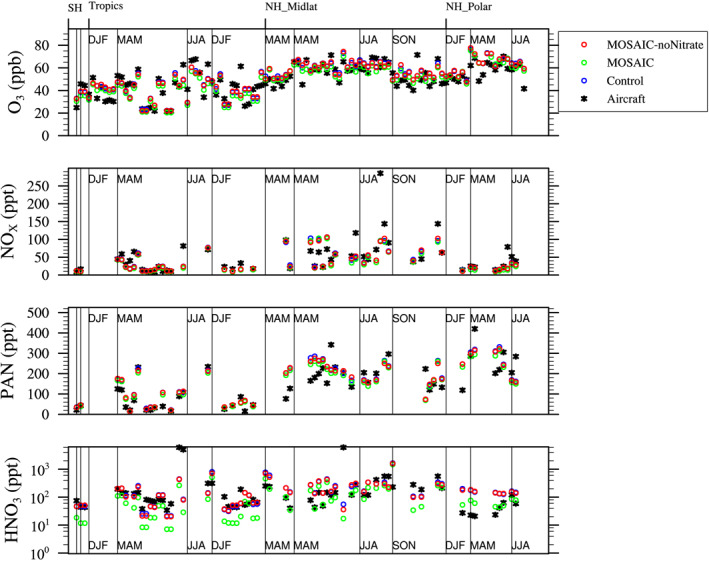
Comparison of O3, NOx, PAN, and HNO3 simulated by the three model experiments against an aircraft climatology for the free troposphere for 2–7 km sorted by different regions (Southern Hemisphere [SH], Tropics, Northern Hemisphere [NH] midlatitudes, NH Polar) and the four seasons between 1995 and 2010.
Comparisons with ozone (Figure 1, top row) show a very good representation of ozone in the model compared to aircraft observations. The MOSAIC simulation shows a slight reduction of ozone, most pronounced in the NH polar region in spring and summer. The reduction in ozone goes along with a consistent reduction in ammonium nitrate in the MOSAIC simulation compared to the MOSAIC_no_nitrate simulation (Figure 1, panel 4). The MOSAIC simulation shows smaller HNO3 mixing ratios, resulting in some improvement compared to observations in the NH polar region and a slight underestimation in the tropics. However, in general, HNO3, NOx, and PAN in the model agree very well within the range of the observed values, and differences between the simulations are of similar range to the observational variability. Very small changes between the simulations occur for NOx and only small reductions in PAN. There are almost no differences between the Control and the MOSAIC_no_nitrate case.
3.1.2. Key Aerosol Species
The simulated annual mean surface aerosol nitrate (NO3), sulfate (SO4), and ammonium (NH4) are evaluated against the surface observations from the IMPROVE network in the U.S., the EMEP network in Europe, the EANET network in East Asia, and the CAWNET network in China. As shown in Figure 2, the model captures the spatial distribution patterns of NO3 reasonably well, with relatively high concentrations over polluted regions across western Europe, northeastern China, and southern California in the U.S. However, the significant underprediction of NO3 in the agricultural areas of the Midwestern U.S., despite the large amounts of NH3 emitted there, is likely due to underestimated soil NOx emissions in the POET inventory. The spatial distribution patterns of SO4 and NH4 are shown in Figures S1 and S2, respectively, in the supporting information.
Figure 2.
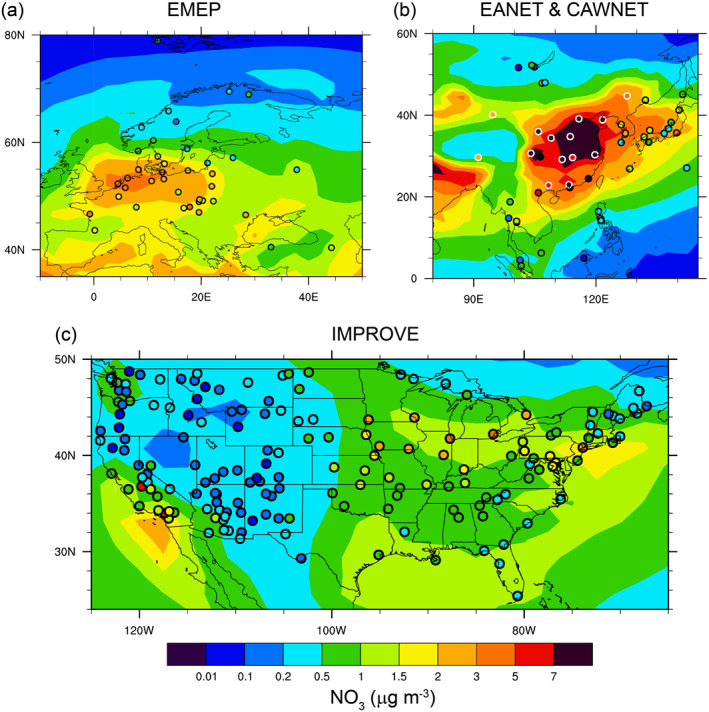
Simulated and observed (filled circles) spatial distributions of the annual mean aerosol NO3 mass concentrations for (a) EMEP network in Europe; (b) EANET and CAWNET networks in East Asia and China, respectively; and (c) IMPROVE network in the United States.
Figure 3 shows the scatter plots of the simulated versus observed annual mean surface mass concentrations of NO3, SO4, and NH4. The simulated NO3 agrees reasonably well with the observations over the U.S. (IMPROVE), with the model slightly underestimating the annual average fine aerosol NO3 mass concentrations. The ratios between the modeled and observed NO3 mass concentration fall within the 1:10 and 10:1 lines with the exception of the two observation sites in California, where the model's coarse horizontal resolution of 2° is unable to adequately resolve the emissions from polluted urban areas. Similar results are seen for Europe (EMEP) and China (CAWNET) where the model predicts lower average NO3 mass concentration compared to observations, especially for China where the model underestimates by about 70%. Again, this discrepancy is due to the coarse model resolution, which is too large to resolve the differences between urban and rural areas in China. The model performance over Europe (EMEP) is relatively better as it underestimates NO3 by about 25%. In contrast, the model overpredicts NO3 by about 100% over East Asia (EANET). As shown in Figure 2, this is caused by stations over South Korean and Japan, which are influenced by the outflow of NO3 aerosols from China. It is worth mentioning that if CMIP5 emissions are used instead of CEDS emission, the model will tend to overestimate NO3 mass concentrations over the U.S. and Europe and will perform even worse in China (CAWNET). This is because compared to CMIP5 emissions, CEDS has significantly lower NH3 emissions in the U.S. and Europe but higher NH3 emissions in China and produces higher NOx emissions in all three regions (Hoesly et al., 2018).
Figure 3.
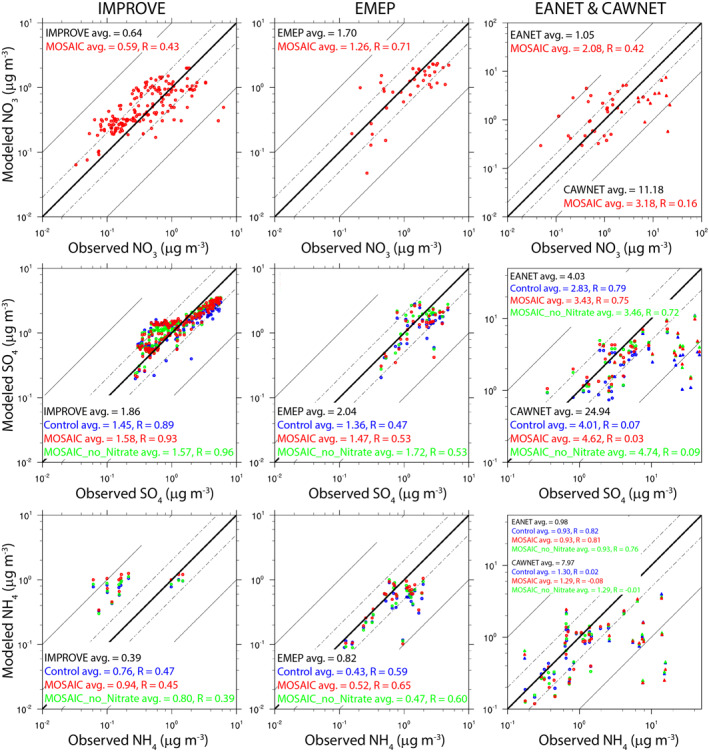
Scatter plots of modeled versus observed annual mean aerosol NO3 (top row), SO4 (middle row), and NH4 (bottom row) surface mass concentrations at IMPROVE (left column), EMEP (middle column), and EANET and CAWNET (left column) network sites.
Similar results were obtained with regards to aerosol SO4, wherein the MOSAIC simulation slightly underestimates the surface SO4 mass concentrations by about 15%–28% over the U.S., Europe, and East Asia (Figure 3). In general, the SO4 mass concentrations predicted by the MOSAIC and MOSAIC_no_nitrate simulations are much closer to observations than the Control case. However, all three simulations underestimate the SO4 mass concentrations by about 80% over China (CAWNET). This discrepancy is likely due to biases in emission inventory in China or missing representation of chemical mechanisms in the model (Fan et al., 2018; G. Wang et al., 2016). As also shown in Figure 3, the simulated NH4 mass concentrations are in a relatively reasonable agreement with the observations in Europe (EMEP) and East Asia (EANET), especially for the MOSAIC case. However, the model overestimates NH4 mass concentrations over the U.S. (IMPROVE) and underestimates NH4 over China (CAWNET). Note that only a few sites of the IMPROVE network monitor NH4 mass concentrations in southeast United States.
Figure 4 shows comparison of mean vertical profiles of total (fine + coarse) aerosol NO3, SO4, and NH4 between MOSAIC simulation and aircraft observations made between 0 and 12 km altitudes during three field campaigns: PEM‐TROPICS B 1999 over the tropical Pacific Ocean (Raper et al., 2001); TRACE‐P 2001 over the North Western Pacific Ocean (Jacob et al., 2003); and INTEX‐A 2004 over the Eastern US and the Atlantic Ocean (Singh et al., 2006). Overall, MOSAIC captures the vertical distributions of all three aerosol species well, although it appears to systematically overpredict SO4 and NH4 above 6 km during INTEX‐A 2004.
Figure 4.
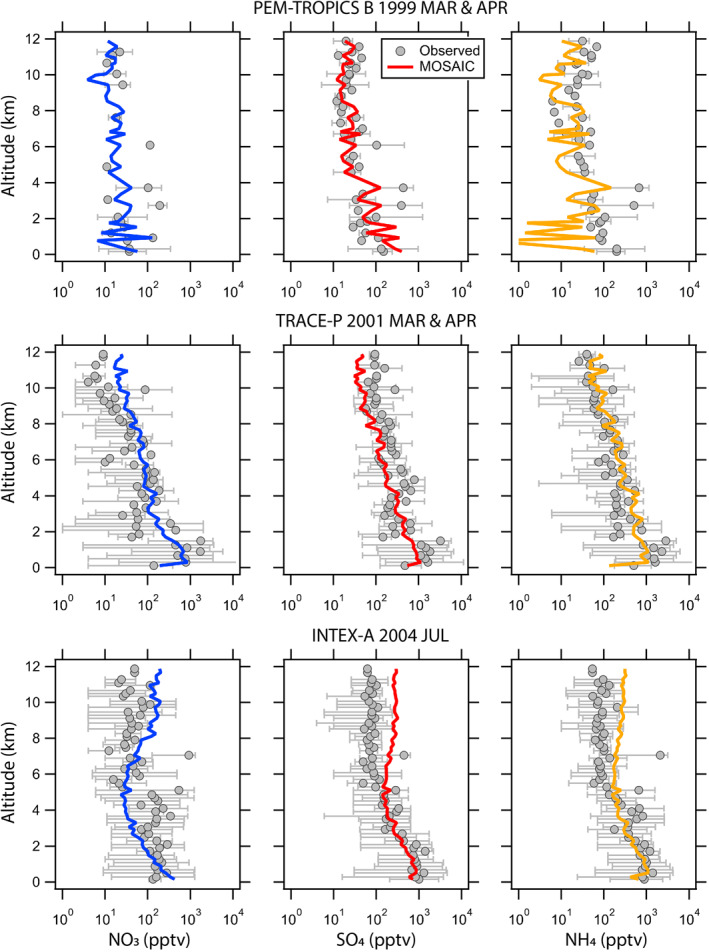
Comparison of modeled (colored lines) and observed (gray circles with error bars representing the range) mean vertical profiles of aerosol NO3 (first column), SO4 (middle column), and NH4 (last column) during PEM‐TROPICS B 1999 over the tropical Pacific Ocean, TRACE‐P 2001 over the North Western Pacific Ocean, and INTEX‐A 2004 over the Eastern U.S. The model results were sampled along the aircraft flight tracks and then the mean vertical profiles were calculated.
3.2. Global Distributions and Burdens
Figures 5, 6, 7 show the seasonal maps of NO3, SO4, and NH4 column burdens in different modes, respectively. Most of the NO3 mass exists in the coarse and fine sea‐salt modes (∼73% of the total annual burden), peaking in the JJA season, followed by NO3 mass in the two soil dust modes. As shown in the top row of Figure 5, large burdens of accumulation mode NO3 occur over heavily polluted regions, such as eastern United States, Europe, China, and India, peaking in the DJF season. These masses are formed from NOx emissions from industrial, power, and transportation sectors. It is noticeable that during DJF, the accumulation mode NO3 over China exceeds the burdens of NO3 in other modes.
Figure 5.
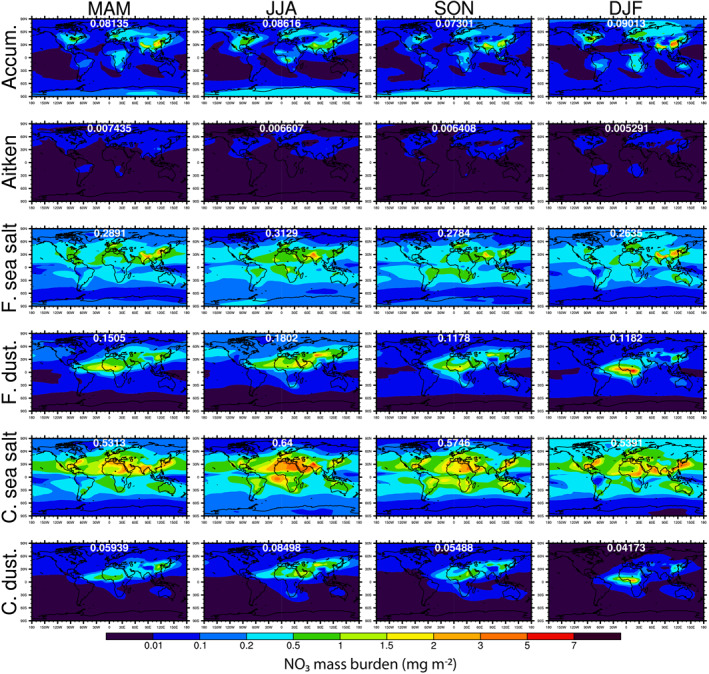
Seasonally averaged spatial distributions of NO3 mass burdens in different aerosol modes. The global mean NO3 burden in each mode and season is shown in white font.
Figure 6.
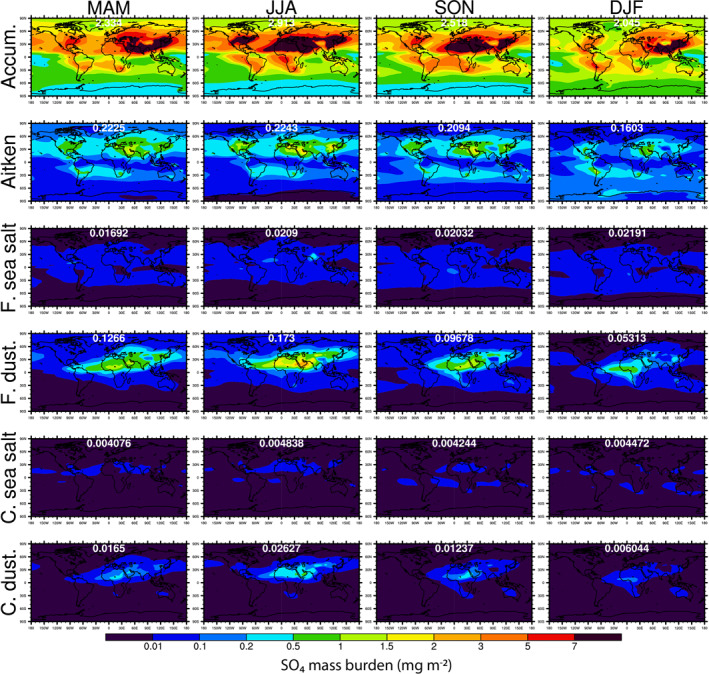
Seasonally averaged spatial distributions of SO4 mass burdens in different aerosol modes. The global mean SO4 burden in each mode and season is shown in white font.
Figure 7.
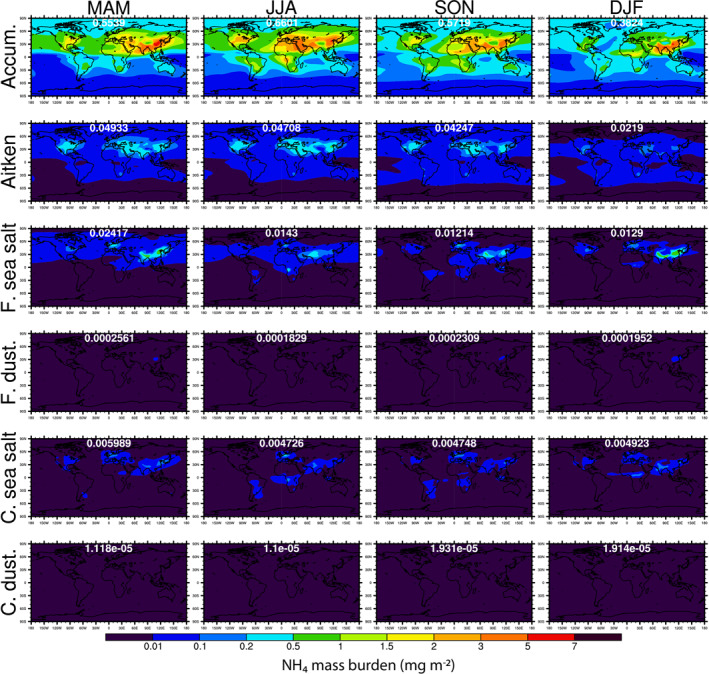
Seasonally averaged spatial distributions of NH4 mass burdens in different aerosol modes. The global mean NH4 burden in each mode and season is shown in white font.
Figure 6 shows the seasonal map of non‐sea‐salt SO4 mass burden. Unlike NO3, the majority of non‐sea‐salt SO4 masses are in accumulation mode (88%) and Aitken mode (7.1%). The accumulation and Aitken mode SO4 peak in JJA season, when the mass burdens predominate over the Sahara Desert, U.S., and China. As shown in Figure 7, the seasonality and spatial distributions of NH4 mass burdens in different modes are very similar to those of SO4—almost all mass is in accumulation mode and Aitken mode, peaking in JJA.
In Figure 8, we show the surface mass mixing ratios, column burdens, and vertical zonal means of aerosol NO3, aerosol NH4, gaseous NH3, and gaseous HNO3. The contour levels and colors are similar to those in Figure 3 of Bian et al. (2017) to facilitate comparisons with AeroCom III results. The NO3 aerosol fields (Figures 8a–8c) are very close to those from the EMEP chemical transport model (Figure 3a in Bian et al. [2017]). The differences are that our simulation produces higher nitrate surface mass mixing ratios and column burdens over oceans (especially over the Southern Ocean), lower nitrate surface mass mixing ratios and column burdens over dust source and outflow regions, lower nitrate concentrations over northern China and India, and lower nitrate aerosol concentrations around 30°N, compared to the EMEP model simulation.
Figure 8.
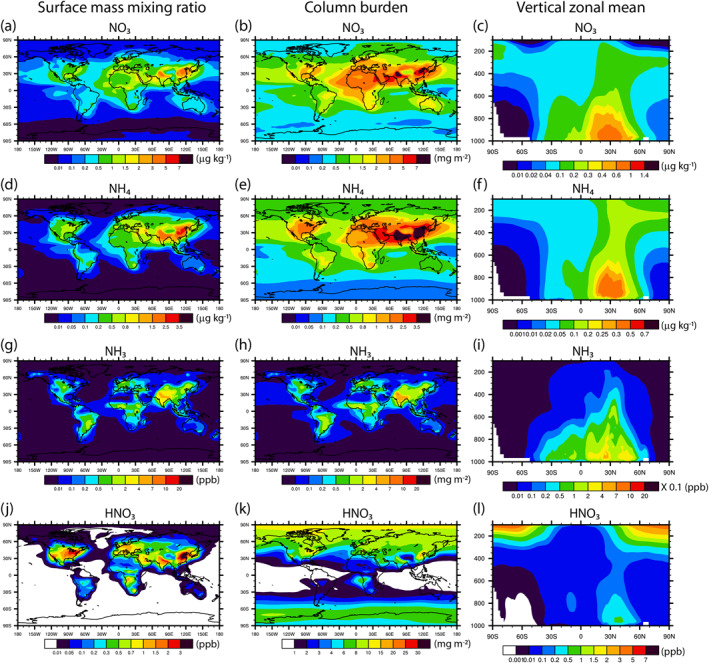
Annually averaged surface mass mixing ratios (left column), column burdens (middle column), and vertical zonal means (right column) of aerosol NO3 (first row), aerosol NH4 (second row), NH3 (third row), and HNO3 (bottom row).
As shown in Figure 8d, the surface mass mixing ratios of NH4 over ocean are below 0.01 µg kg−1, about 1 order of magnitude lower compared to all AeroCom III models. In addition, the value is low over heavy polluted region, such as China and India. By examining the column burden and vertical zonal mean fields as shown in Figures 8e and 8f, we found that the total amount of ammonium produced by MOSAIC case lies in the median of AeroCom III simulations as shown in Table 2, since ammonium aerosols in our simulation are located higher in altitude compared to most AeroCom III models.
Table 2.
Comparison of Global Annual Atmospheric Burdens (TgN or TgS) From This and Previous Studies
| Modeling study | HNO3 | NO3 | NH3 | NH4 | SO2 | Nss‐SO4 |
|---|---|---|---|---|---|---|
| This study (MOSAIC) | (0.42) | 0.14 | 0.070 | 0.24 | 0.35 | 0.49 |
| This study (MOSAIC_no_nitrate) | (0.60) | – | 0.082 | 0.24 | 0.33 | 0.50 |
| This study (Control) | (0.60) | – | 0.069 | 0.26 | 0.37 | 0.49 |
| Bian et al. (2017) | ||||||
| Median | (0.40) | 0.13 | 0.10 | 0.23 | – | 0.52 |
| Range | (0.15–1.3) | 0.03–0.43 | 0.04–0.70 | 0.13–0.58 | – | 0.28–1.1 |
| Median of the other studies below | (0.37) | 0.14 | 0.16 | 0.22 | 0.48 | 0.50 |
| Matsui (2017) | – | 0.03 | – | 0.2 | – | 0.4 |
| Karydis et al. (2016) | (0.37) | (0.10) | (0.68) | (0.13) | – | (0.59) |
| Pozzer et al. (2012) | 1.1 | 0.1 | 0.1 | 0.1 | – | 0.3 |
| Xu and Penner (2012) | (0.30) | 0.17 | 0.07 | 0.26 | – | – |
| Bellouin et al. (2011) | – | 0.1 | – | – | – | 0.5 |
| Pringle et al. (2010) (EQSAM) | (0.58) | 0.11 | 0.19 | 0.20 | 0.55 | 0.51 |
| Pringle et al. (2010) (ISORROPIA) | (0.55) | 0.13 | 0.16 | 0.21 | 0.55 | 0.53 |
| Pye et al. (2009) | – | 0.35 | – | 0.24 | – | 0.28 |
| Myhre et al. (2009) | – | 0.013 | – | – | – | 0.33 |
| Feng and Penner (2007) | (0.37) | 0.16 | 0.084 | 0.29 | – | – |
| Bauer et al. (2007) | 3.88 | 0.52 | 0.17 | 0.27 | 0.48 | 0.56 |
| Rodriguez and Dabdub (2004) | 0.96 | 0.42 | 0.19 | 0.045 | 0.20 | 0.70 |
| Liao et al. (2003) | 0.32 | 0.15 | 0.17 | 0.33 | 0.26 | 0.62 |
Note. SO4 is non‐sea salt. Burdens are mostly for the entire model atmosphere. Because HNO3 mixing ratios above the tropopause can be large, some studies have reported (quasi) tropospheric burdens for HNO3. These values are shown in parentheses and were calculated as pressure > 200 hPa for Feng and Penner (2007) and Xu and Penner (2012); pressure > 150 hPa for Pringle et al. (2010); pressure > 100 hPa for Bian et al. (2017) and this study; and “troposphere” for Karydis et al. (2016) for all species.
Figures 8g–8i show the surface mass mixing ratio, column burden, and vertical zonal mean fields of NH3 modeled by the MOSAIC case. The total burden produced by the MOSAIC case is lower compared to the median value of AeroCom III simulations as shown in Table 2 (0.07 TgN vs. 0.10 TgN). Surprisingly, NH3 burden is even lower than the burdens predicted by two GISS models (GISS‐Matrix and GISS‐OMA), which used CMIP5 emissions, since CEDS emission data set used in the MOSAIC case tuned up NH3 emissions globally compared to CMIP5 emission data set.
HNO3 modeled in the MOSAIC case are very close to GMI and INCA models in AeroCom III simulations in terms of total burden and horizontal and vertical spatial patterns when comparing Figures 8j–8l with Figure 3d in Bian et al. (2017). However, compared to these two models, our simulation produces less HNO3 over tropical ocean but more HNO3 over mid latitude and high latitude in the Northern Hemisphere.
Table 2 shows annual average global burdens for aerosol NO3, NH4, and SO4 and gas HNO3, NH3, and SO2 from this study's simulation, the AeroCom phase III global model intercomparison study (with nine models) (Bian et al., 2017), and 13 other modeling studies from the literature. For all these species, the burdens from the MOSAIC simulation are quite close to the medians for the AeroCom III study and for the 13 other studies. (The medians for the AeroCom III study and for the 13 other studies are also quite close.) The ranges of reported values from the AeroCom III and other studies is quite large. The spread of values for all the previous studies combined can be quantified with a geometric standard deviation, and these range from 2.6 and 1.8 for NO3 and HNO3, 1.7 and 1.8 for NH4 and NH3, and 1.5 for SO4 and SO2. The geometric standard deviation for NO3 is noticeably larger than those for the other species, which is perhaps not surprising since aerosol NO3 depends on NOx sources and gas‐phase chemistry, HNO3 removal, aerosol chemistry/thermodynamics (which is sensitive to overall aerosol composition), and aerosol removal.
Species burdens for the three simulations in this study are mostly very similar, except for HNO3 being lower in the MOSAIC simulation because of its condensation onto aerosol particles. NH3 burdens are small in all simulations, its higher value in MOSAIC_no_nitrate is due to absence of NO3 affecting the NH3 gas‐aerosol partitioning, and its default‐simulation value matches the MOSAIC simulation by chance.
Table 3 shows the tropospheric annual average mixing ratios for NO3, Cl, NH4, and non‐sea‐salt SO4 in the seven aerosol modes. Note that NO3 in the primary‐carbon mode is not carried as a transported species in MAM; any NO3 produced in that mode is immediately transferred to the accumulation mode through the MAM aging process. The sea‐salt modes contain ∼74% of the nitrate. The accumulation mode NO3 accounts for only 9% globally, although it is higher over the pollution source regions, and it dominates in a few regions (e.g., over the Antarctic, and in the PBL over E. Asia and E. N. America). Dust mode NO3 is 17% globally and dominates over Asian dust regions and the southern Sahara (and its outflow).
Table 3.
Tropospheric Annual Average Mixing Ratios, Percentage of All‐Mode Total Mixing Ratio, and Production/Loss for NO 3 , Cl, NH 4 , and Non‐Sea‐Salt SO 4 in the MAM7 Aerosol Modes
| Mode | Mixing ratio (ng kg−1) | Percentage of total mixing ratio | Condensation production (ng kg−1 day−1) | Evaporation loss (ng kg−1 day−1) | Net aqueous production (ng kg−1 day−1) | Overall net production (ng kg−1 day−1) |
|---|---|---|---|---|---|---|
| NO3 | ||||||
| Accumulation | 11.9 | 8.6 | 11.2 | −53.5 | 49.7 | 7.4 |
| Aitken | 0.8 | 0.6 | 1.9 | −2.9 | 3.2 | 2.2 |
| F. sea salt | 33.0 | 23.8 | 16.3 | −7.6 | 0.2 | 8.9 |
| C. sea salt | 68.9 | 49.8 | 38.4 | −2.1 | 0.0 | 36.3 |
| Fine dust | 16.7 | 12.0 | 2.9 | −0.6 | 0.4 | 2.7 |
| Coarse dust | 7.2 | 5.2 | 2.3 | −0.0 | 0.0 | 2.3 |
| All | 138.5 | 100.0 | 73.0 | −66.8 | 53.7 | 59.9 |
| Cl | ||||||
| Accumulation | 4.5 | 0.6 | 2.2 | −23.8 | 19.0 | −2.6 |
| Aitken | 0.3 | 0.0 | 0.9 | −1.8 | 1.7 | 0.7 |
| F. sea salt | 54.8 | 7.4 | 4.0 | −9.0 | 0.8 | −4.2 |
| C. sea salt | 678.4 | 91.0 | 5.7 | −25.1 | 0.2 | −19.2 |
| Fine dust | 5.1 | 0.7 | 1.1 | −0.4 | 0.2 | 0.9 |
| Coarse dust | 2.6 | 0.4 | 0.9 | −0.0 | 0.0 | 0.9 |
| All | 745.8 | 100.0 | 14.9 | −60.1 | 21.8 | −23.4 |
| NH4 | ||||||
| Accumulation | 58.9 | 89.2 | 19.5 | −11.8 | 2.1 | 9.9 |
| Aitken | 4.6 | 7.0 | 4.3 | −2.4 | 0.1 | 2.1 |
| F. sea salt | 1.9 | 2.8 | 2.2 | −1.5 | 0.0 | 0.7 |
| C. sea salt | 0.6 | 1.0 | 2.1 | −1.4 | −0.0 | 0.7 |
| Fine dust | 0.0 | 0.0 | 0.1 | −0.1 | 0.1 | 0.0 |
| Coarse dust | 0.0 | 0.0 | 0.0 | −0.0 | 0.0 | 0.0 |
| All | 66.0 | 100.0 | 28.2 | −17.1 | 2.3 | 13.3 |
| Non‐sea‐salt SO4 | ||||||
| Accumulation | 273.6 | 86.9 | 14.2 | 0.0 | 42.4 | 56.6 |
| Aitken | 23.6 | 7.5 | 7.6 | 0.0 | 4.0 | 11.6 |
| F. sea salt | 2.5 | 0.8 | 0.4 | 0.0 | 0.8 | 1.1 |
| C. sea salt | 0.5 | 0.2 | 0.3 | 0.0 | 0.2 | 0.5 |
| Fine dust | 12.9 | 4.1 | 1.6 | 0.0 | 0.7 | 2.2 |
| Coarse dust | 1.8 | 0.6 | 0.5 | 0.0 | 0.0 | 0.5 |
| All | 314.9 | 100.0 | 24.6 | 0.0 | 48.1 | 72.7 |
Note. The production/loss columns are gas condensation (HNO3, HCl, NH3, and H2SO4) to interstitial aerosol particles, loss by evaporation from interstitial particles, and net aqueous production (net uptake of these gases to cloud‐droplet‐borne aerosol particles, plus uptake and aqueous oxidation of SO2), and overall net production. Losses from cloud‐droplet‐borne particles are relatively small so are not shown. Single time step production/loss values from the aerosol and aqueous chemistry modules are saved separately to produce these results. Average mixing ratios of HNO3, HCl, NH3, SO2, and H2SO4 gases are 219, 71.1, 19.6, 161, and 0.26 ng kg−1.
Size‐resolved aerosol measurements over the Atlantic Ocean (Huebert et al., 1996; Keene et al., 2009) found nearly all of the aerosol nitrate to be in particles with diameters ≥1 µm. Model results are in agreement with this finding: 85% or more of the nitrate is in supermicrometer particles over most of the areas sampled in those studies. (Supermicrometer nitrate is calculated as that in the coarse sea‐salt and dust modes, and ∼70% of that in the fine dust mode, which gets dust emissions up to 2 µm diameter.) Sixty‐four percent of the global annual nitrate burden is in supermicrometer particles. Submicrometer nitrate dominates in the upper atmosphere (above ∼350 hPa). The externally mixed treatment of submicrometer sea salt and dust in MAM7 appears to have a strong impact on the amount of submicrometer nitrate. Lu et al. (2021) implemented MOSAIC in CAM6‐chem and performed simulations with both 4 and 7 mode versions of MAM. Their supermicrometer nitrate burdens were nearly identical for MAM4‐MOSAIC and MAM7‐MOSAIC, but the submicrometer nitrate was ∼40% lower for MAM4‐MOSAIC, where submicrometer sea salt and dust are internally mixed with other aerosol species.
The simulated non‐sea‐salt SO4 partitioning is completely different from the nitrate partitioning, with 88% of it in the accumulation mode, 7.1% in the Aitken mode, 0.3% in the two sea‐salt modes, and 4.4% in the two dust modes. The preferential partitioning of nitrate to the sea‐salt and dust modes results in a shorter lifetime compared to sulfate and ammonium. The NO3 lifetime, calculated as the ratio of global burden to global wet + dry removal rate, is 2.4 days, which is intermediate between those of sea salt (0.4 days) and SO4 and NH4 (4.4 and 5.3 days). The AeroCom III study reported a median NO3 lifetime of 5.5 days, suggesting that NO3 was predominantly in submicron aerosol particles in most of the participating models, although two of the seven models that reported lifetimes had NO3 lifetimes of about 2 days.
Table 3 also shows the tropospheric annual average chemical production and loss of NO3, Cl, NH4, and non‐sea‐salt SO4 for all modes except primary carbon: condensation to and evaporation from interstitial aerosol particles, and net production by uptake to cloud‐borne aerosol particles (i.e., aqueous chemistry in cloud droplets that contain the cloud‐borne particles). The total production of nitrate in cloud droplets is similar in magnitude to the total production by condensation. Nearly, all of this aqueous production is in the accumulation mode, because it accounts for most of the cloud condensation nuclei and cloud droplets. Most of the cloud‐borne nitrate evaporates when cloud droplets evaporate and the cloud‐borne aerosol material is resuspended, because the moderately acidic cloud droplets become highly acidic interstitial particles due to water loss. For the accumulation mode, the sum of evaporation and net aqueous production (−3.8 ng kg−1 day−1) is 34% of the condensation production, indicating significant evaporation of the interstitial‐particle NO3 produced by condensation. This cycling of nitrate into and out of particles is also strong for the fine sea‐salt mode, where aqueous production is negligible, and evaporation is about 47% of condensation. There is a net loss of Cl from the two sea‐salt modes and the accumulation mode, cause by displacement of primary sea‐salt Cl by secondary SO4 and NO3, and Cl uptake to the accumulation and Aitken modes is primarily aqueous. NH4 also has moderate evaporation loss (∼50% of the condensation production) from many of the modes. SO4 is treated as nonvolatile in this version of the model so has no evaporation loss, and its production is dominated by aqueous uptake and oxidation of SO2. The chemical production and loss of NO3, Cl, NH4, and SO4 have interesting horizontal and vertical spatial distributions, which are further discussed in supporting information S1.
3.3. Nitrate Aerosol Radiative Effects
As shown above, the CAM5‐chem‐MAM7‐MOSAIC produces a significant amount of nitrate aerosol and changes in other aerosols, which in turn have an appreciable impact on aerosol‐radiation interaction. Figure 9 shows the global distribution of changes in annual mean top of the atmosphere (TOA) clear‐sky shortwave flux caused by the additional nitrate aerosol and/or the MOSAIC scheme itself. The inclusion of nitrate (MOSAIC vs. MOSAIC_no_nitrate) produces a small global mean shortwave radiative cooling effect (−0.05 W m−2), which is smaller than the present‐day nitrate direct radiative effect, −0.11 and −0.12 W m−2, estimated by Bauer et al. (2007) and Xu and Penner (2012), respectively, but within the AeroCom II multimodel range from −0.12 to −0.02 W m−2 (Myhre, Samset, et al., 2013). With the experiment setup in the present study, the difference in TOA clear‐sky shortwave flux between MOSAIC and MOSAIC_no_nitrate is due to a 0.4% increase in global mean AOD and a 2.0% decrease in global mean absorption AOD. The nitrate speeds up the aging of primary‐carbon mode aerosol resulting in a lower BC burden, and dust emissions are slightly greater in the simulation with nitrate. The inclusion of nitrate exerts a discernible and statistically significant impact on the regional cooling (up to −5 W m−2) in Asia and the Arctic and produces some areas of regional warming, which is more likely due to interactions with other species than the additional nitrate aerosol. The interactions with other aerosol species, shown in the bottom panel, also cause a regional cooling over continents, especially near major anthropogenic sources and outflow areas but a net warming effect over oceanic areas in the tropics and Southern Hemisphere. The latter is due to a reduction in the sea‐salt burden induced by the MOSAIC scheme (more aerosol water in sea‐salt modes, which increases particle sizes and dry deposition rates, and reduces lifetimes and burdens).
Figure 9.
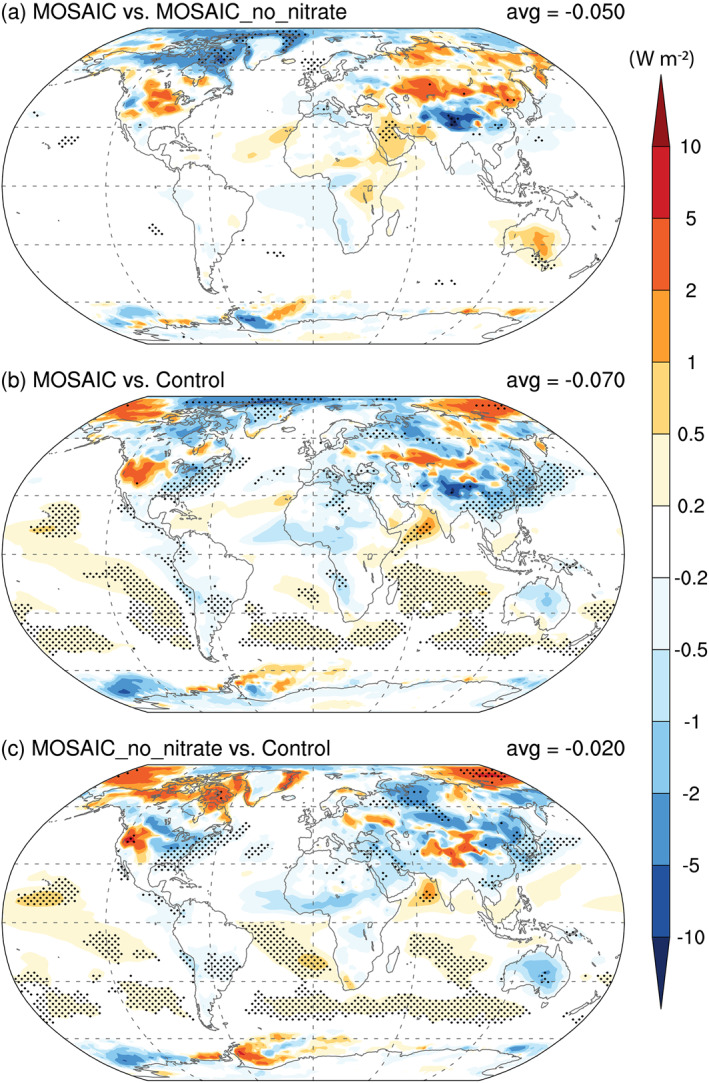
Global distribution of changes in the annual mean clear‐sky shortwave fluxes at the top of the model atmosphere due to (a) including nitrate aerosol (MOSAIC vs. MOSAIC_no_nitrate), (b) all MOSAIC effects (MOSAIC vs. Control), and (c) MOSAIC effects other than nitrate aerosol (MOSAIC_no_nitrate vs. Control). Stippled areas indicate statistical significance (using t test based on the 10 individual annual means).
Including nitrate aerosol in the model produces significant impacts on regional cloud condensation nuclei (CCN) (Figure 10), cloud droplet number (not shown), and liquid water path (LWP; not shown). Concentrations of tropospheric CCN (at 0.1% supersaturation), accumulation mode number, cloud droplet number, and liquid water path are 2.5%, 10.5%, 7.9%, and 2.5% greater on average in the MOSAIC simulation compared to MOSAIC_no_nitrate. The higher accumulation mode number concentration in the MOSAIC simulation with nitrate formation is due to a combination of somewhat more rapid growth of Aitken mode particles and aging of primary‐carbon mode particles into the accumulation mode, and somewhat slower removal of the accumulation mode particles. These differences are compensated to some extent by other effects of the MOSAIC scheme (i.e., MOSAIC_no_nitrate vs. Control). The overall global mean net impact of MOSAIC (i.e., MOSAIC vs. Control) becomes small, but the impact over major source regions such as East Asia, Europe, and North America is enhanced. The increase in the boundary‐layer CCN number concentrations is as high as 50–100 cm−3. Simulated CCN number concentrations were compared to the aircraft campaign measurements in Figure 23 of Liu et al. (2012). For some of the campaigns, the MOSAIC results showed somewhat better agreement with measurements relative to MOSAIC_no_nitrate (not shown). However, the aircraft measurements were not in the CCN enhancement locations, so the comparison does not support the regional CCN increases seen in Figure 10. A comprehensive evaluation of regional CCN changes and the connection to nitrate aerosol require further investigation.
Figure 10.
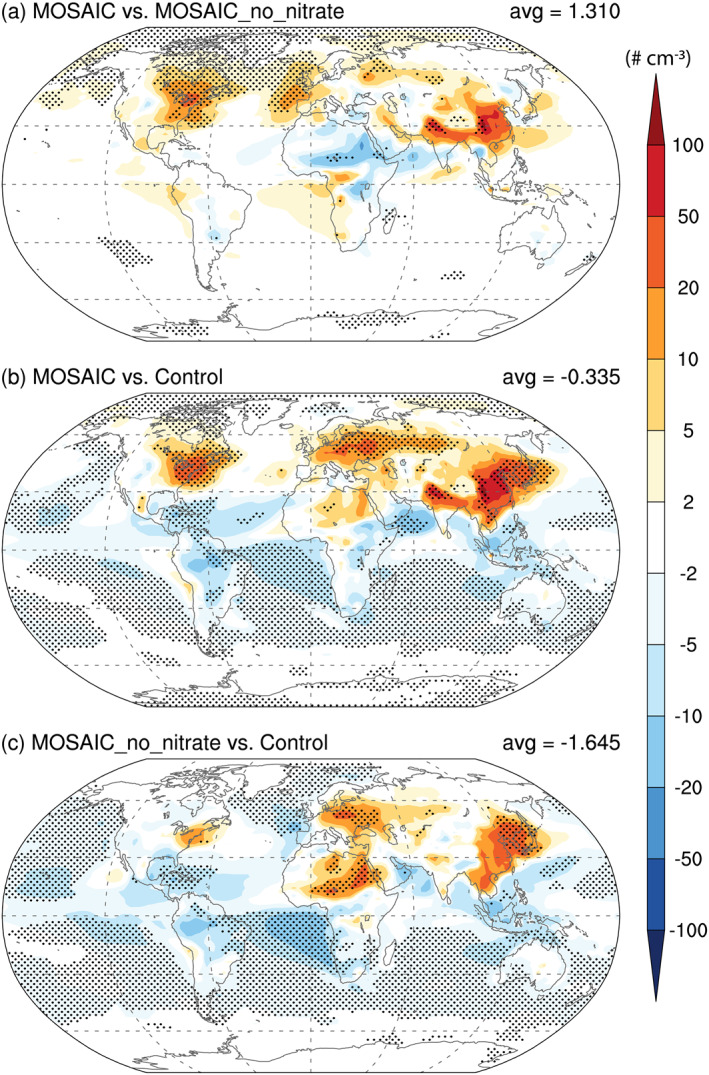
Same as Figure 9 but for changes in CCN (at 0.1% supersaturation) concentrations averaged below 850 hPa.
Large increases are also seen in regional cloud droplet number concentrations, but the net effect on LWP is not as significant. The impact on CCN and clouds by including nitrate aerosol causes a decrease (i.e., additional cooling) in global mean shortwave cloud forcing by −0.8 W m−2 under the year‐2000 conditions, which is however outweighed by the compensating effect through changes in other aerosol species, leading to an overall global mean net warming of +0.2 W m−2, mostly in the Southern Hemisphere. Taking into consideration of changes in both longwave and shortwave radiative fluxes under all‐sky conditions, the radiative effect (induced by including nitrate aerosol) is −0.7 W m−2, while the overall net change in TOA radiative fluxes caused by MOSAIC aerosols is +0.15 W m−2, with discernible amount of cooling (warming) in the Northern (Southern) Hemisphere, as shown in Figure 11. Note that the net change in TOA all‐sky radiative fluxes (−0.7 W m−2), calculated as the difference between the two experiments with (MOSAIC) and without (MOSAIC_no_nitrate) year‐2000 nitrate aerosol, is not comparable to the traditional total anthropogenic nitrate aerosol forcing (i.e., difference between present‐day and preindustrial aerosol conditions), so it is not surprising that the magnitude here is larger than those reported in previous studies (e.g., −0.21 W m−2, Xu & Penner, 2012).
Figure 11.
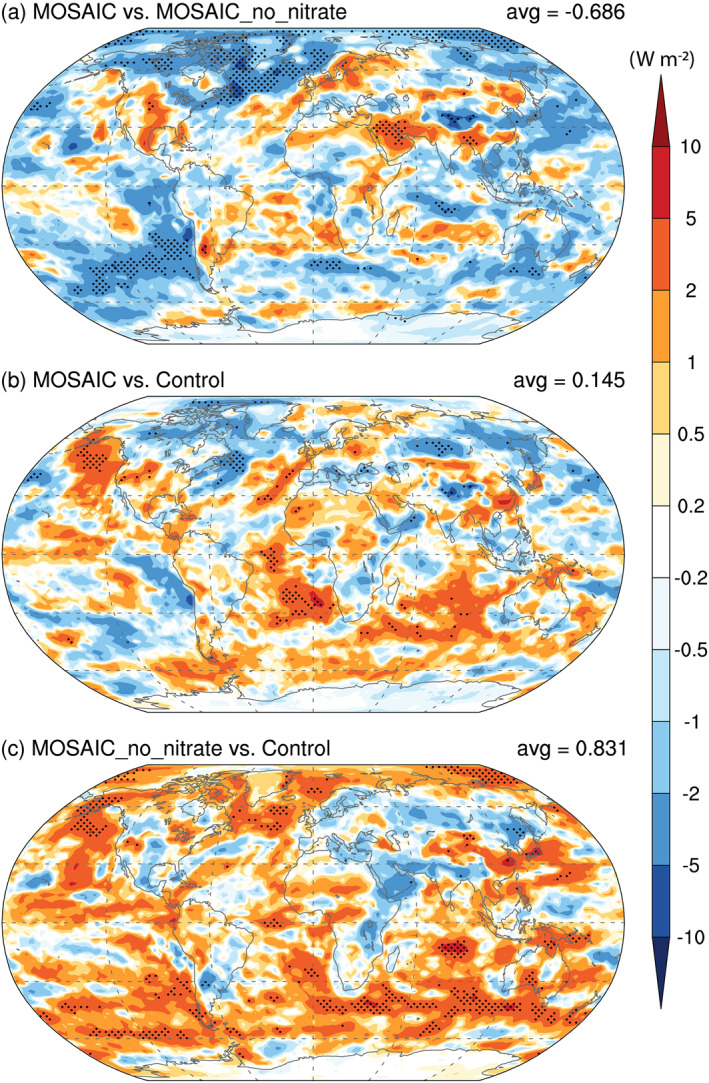
Same as Figure 9 but for changes in TOA net (longwave and shortwave) radiative fluxes. TOA, top of the atmosphere.
3.4. Computational Performance
Table 4 shows Simulated Years Per Day (SYPD) from the three simulations discussed above. Percentage slowdown for the test simulations (MOSAIC_no_nitrate and MOSAIC) is shown in parentheses. These simulations were performed on Environmental Molecular Sciences Laboratory's “Cascade” supercomputer. Cascade is a 3.4 Petaflop machine with 1440 Intel Xeon E52670 nodes. Each Cascade node has 16 CPU cores with 128 GB memory (8 GB per core). All simulations were performed using 528 CPU cores (33 nodes).
Table 4.
Comparison of Computational Performance in Simulated Years per Day (SYPD)
| Simulations | Number of tracers | SYPD (percentage slowdown) |
|---|---|---|
| Control | 223 | 4.98 |
| MOSAIC_no_nitrate | 243 | 4.08 (18.09%) |
| MOSAIC | 243 | 3.94 (20.97%) |
Note. Percentage slowdown with respect to the Control simulation is shown in parentheses.
The major differences between the Control and the test simulations are the number of additional tracers and the additional chemistry computations in the MOSAIC module coupled with MAM7. These two factors directly impact the computational performance of the model. MOSAIC and MOSAIC_no_nitrate simulations have 20 additional advected tracers as compared with the Control simulation. MOSAIC simulation performs additional nitrate computations as compared with the MOSAIC_no_nitrate simulation. MOSAIC is about ∼21% more expensive than the Control simulation. MOSAIC_no_nitrate is about ∼18% more expensive than the Control simulation. Additional nitrate computations in MOSAIC simulation are about ∼3% more expensive.
Tracer advection is accomplished by model dynamics while chemistry calculations take place in MOZART + MAM7 + MOSAIC modules. Table 5 presents a comparison between the time taken by the tracer transport and chemistry (includes advancing dust/sea‐salt emissions and dry deposition of aerosols) calculations by the three simulations—Control, MOSAIC_no_nitrate, and MOSAIC. Percentage slowdown is shown for the test cases in parenthesis. As expected, the MOSAIC simulation is the most expensive among the three, taking ∼33% more time for tracer transport and ∼73% more time for the chemistry calculations as compared to the Control simulation. In comparison, the MOSAIC_no_nitrate simulation takes ∼28% more time for the tracer transport and ∼66% more time for the chemistry calculations.
Table 5.
Comparison of Time Taken by Tracer Advection and Aerosol Chemistry Computations for One Simulated Month
| Simulations | Tracer transport time (s) (percentage slowdown) | Aerosol chemistry time (s) (percentage slowdown) |
|---|---|---|
| Control | 441.11 | 384.1 |
| MOSAIC_no_nitrate | 562.76 (27.58) | 636.61 (65.74%) |
| MOSAIC | 585.07 (32.64) | 664.37 (72.97%) |
Note. Aerosol chemistry includes calculations performed within MOZART, MAM7, MOSAIC (if included) along with dust/sea‐salt emissions and aerosol dry deposition. Percentage slowdown with respect to the Control simulation is shown in parentheses.
It should be noted that the default CAM5 configuration uses the 4‐mode version of MAM aerosol package (MAM4) (Liu et al., 2016). The Control simulation (CAM5 + MOZART + MAM7) is about 3 times more expensive than the default (CAM5 + MAM4) configuration, primarily because of the prescribed oxidants and very limited gas‐phase chemistry in the default configuration.
4. Conclusions
We have introduced an advanced aerosol treatment into the CAM5‐chem global chemistry‐aerosol‐climate model by coupling the MOSAIC aerosol chemistry module with the MAM7 aerosol dynamics module (with seven modes) and the MOZART atmospheric chemical mechanism. Unlike the traditional equilibrium and hybrid partitioning approaches, MOSAIC dynamically partitions condensable inorganic gases (H2SO4, HNO3, HCl, and NH3) to all the fine and coarse mode aerosols, as governed by mode‐resolved thermodynamics and heterogeneous chemical reactions. We applied the model in the free‐running mode from 1995 to 2005 with prescribed transient historical climatological forcing conditions. The simulated annual average global distributions of NO3, NH4, and SO4 aerosol mass concentrations agreed well with surface observations from the IMPROVE network in the U.S., the EMEP network in Europe, and the EANET network in East Asia but were generally underpredicted compared to the CAWNET network in China. The model captured the spatial distribution patterns of NO3 reasonably well, with relatively high concentrations over polluted regions across western Europe, northeastern China, and southern California in the U.S.
The simulated global burdens of NO3, NH4, and SO4 aerosols, and also HNO3, NH3, and SO2 gases, were close to the median values from the AeroCom III intercomparison (Bian et al., 2017) and a group of other previous global nitrate aerosol studies. The simulated accumulation mode NO3 column burden was high over the heavily polluted regions in the U.S., Europe, China, and India, peaking during the winter season in the Northern Hemisphere. The accumulation mode NO3 over China even exceeded the burdens of NO3 in other aerosol modes during winter. These high NO3 burdens resulted from its precursors emission of industrial and/or transportation origins. Globally however, about 74% and 17% of the tropospheric annual average NO3 burden was contained in the sea‐salt and dust modes, respectively, with only about 9% in the accumulation mode. The accumulation mode particles were relatively more acidic as they contain 87% of the tropospheric non‐sea‐salt sulfate, while the sea‐salt and dust mode particles are much less acidic and contain considerable amounts of primary Cl and CO3 that can be displaced by NO3. Also, accumulation mode NO3 formed over polluted regions tended to evaporate due to dilution and/or increase in temperature. Globally, 64% of the NO3 aerosol was in supermicrometer particles (Dp,dry > 1 μm), in general agreement with measurements. The nitrate partitioning was very different from the partitioning of non‐sea‐salt SO4 and NH4, which were primarily in the accumulation mode.
About 69% of the submicrometer NO3 mass was in the fine sea‐salt mode, and the simulated submicrometer nitrate burden was quite sensitive to the model's treatment of submicrometer aerosol mixing state (Lu et al., 2021). NO3 in the accumulation, Aitken, and fine sea‐salt modes showed a strong dynamical behavior, with loss by evaporation equaling about half of the production by condensation and aqueous uptake. For the accumulation and Aitken modes, this was largely due to cycling into and out of clouds, with aqueous uptake when interstitial particles move into clouds and are activated, then evaporation loss when cloud‐borne particles move out of clouds and are resuspended.
The inclusion of NO3 aerosol significantly increased the accumulation mode number concentrations over the polluted regions due to enhanced growth of Aitken mode aerosols and resulted in about 10% higher global average accumulation mode number concentrations. Consequently, aerosol NO3 exerted a discernible and statistically significant impact on the regional shortwave radiative cooling of up to −5 W m−2 in Asia and the Arctic but produced a much smaller global average shortwave radiative cooling of −0.05 W m−2. The NO3 induced growth of accumulation mode aerosols increased the regional boundary‐layer CCN (at 0.1% supersaturation) number concentrations by as high as 50–100 cm−3, with large increases in regional cloud droplet number concentrations as well, which resulted in an additional global average shortwave cooling of −0.8 W m−2. However, the cloud‐related cooling was outweighed by compensating effects via changes in other aerosol species, leading to an overall global average net warming of +0.2 W m−2, mostly in the Southern Hemisphere. Taking into consideration of changes in both longwave and shortwave radiative fluxes under all‐sky conditions, the net change in TOA radiative fluxes induced by including nitrate aerosol was −0.7 W m−2.
Conflict of Interest
The authors declare no conflicts of interest relevant to this study.
Supporting information
Supporting Information S1
Acknowledgments
R. A. Zaveri, R. C. Easter, B. Singh, H. Wang, R. Zhang, S. J. Ghan, and P. J. Rasch acknowledge support by the U.S. Department of Energy (DOE), Office of Science, Office of Biological and Environmental Research (BER), Earth and Environmental System Modeling (EESM) program as part of its Earth System Model Development (ESMD) activity. The Pacific Northwest National Laboratory (PNNL) is operated for DOE by Battelle Memorial Institute under contract DE‐AC05‐76RLO1830. We acknowledge the IMPROVE, EMEP, EANET, CAWNET, and the NASA GTE campaign data sets used in this study. We gratefully acknowledge Dr Susanne Bauer (NASA GISS) for assistance in using the NASA GTE campaign data. X. Liu and Z. Lu acknowledge the partial funding support for this work by the NASA Modeling, Analysis, and Prediction Program (award no. NNX14AC85G). This material is based on work supported by the National Center for Atmospheric Research, which is a major facility sponsored by the NSF under Cooperative Agreement No. 1852977. We acknowledge the use of computational resources at the Environmental Molecular Sciences Laboratory (EMSL), a DOE Office of Science user facility sponsored by BER and located at PNNL, and from Cheyenne supercomputer (http://doi.org/10.5065/D6RX99HX) provided by NCAR's Computational and Information Systems Laboratory (CISL), sponsored by the National Science Foundation.
Zaveri, R. A. , Easter, R. C. , Singh, B. , Wang, H. , Lu, Z. , Tilmes, S. , et al. (2021). Development and evaluation of chemistry‐aerosol‐climate model CAM5‐chem‐MAM7‐MOSAIC: Global atmospheric distribution and radiative effects of nitrate aerosol. Journal of Advances in Modeling Earth Systems, 13, e2020MS002346. 10.1029/2020MS002346
Data Availability Statement
The IMPROVE data set is available at http://vista.cira.colostate.edu/Improve/. The EANET data set is available at https://monitoring.eanet.asia/document/public/index. The EMEP data set is available at http://www.emep.int. INTEX‐A, PEM‐TROPICS B, and TRACE‐P data sets are available at the NASA GTE campaign data archive at https://www-air.larc.nasa.gov/pub/. The gas‐phase species aircraft climatology data set is available as part of the CESM Atmospheric Diagnostics package at http://www.cesm.ucar.edu/working_groups/Atmosphere/amwg-diagnostics-package/. The model CAM5‐chem‐MAM7‐MOSAIC is available at https://github.com/razaveri/CAM5-MAM7-MOSAIC and the simulations are available at https://doi.org/10.25584/DH-172021/1756093.
References
- Abdul‐Razzak, H. , & Ghan, S. J. (2000). A parameterization of aerosol activation: 2. Multiple aerosol types. Journal of Geophysical Research, 105(D5), 6837–6844. 10.1029/1999JD901161 [DOI] [Google Scholar]
- Adams, P. J. , Seinfeld, J. H. , Koch, D. , Mickley, L. , & Jacob, D. (2001). General circulation model assessment of direct radiative forcing by the sulfate–nitrate–ammonium–water inorganic aerosol system. Journal of Geophysical Research, 106(D1), 1097–1111. 10.1029/2000JD900512 [DOI] [Google Scholar]
- Adams, P. J. , Seinfeld, J. H. , & Koch, D. M. (1999). Global concentrations of tropospheric sulfate, nitrate, and ammonium aerosol simulated in a general circulation model. Journal of Geophysical Research, 104(D11), 13791–13823. 10.1029/1999JD900083 [DOI] [Google Scholar]
- An, Q. , Zhang, H. , Wang, Z. , Liu, Y. , Xie, B. , Liu, Q. , et al. (2019). The development of an atmospheric aerosol/chemistry–climate model, BCC_AGCM_CUACE2.0, and simulated effective radiative forcing of nitrate aerosols. Journal of Advances in Modeling Earth Systems, 11, 3816–3835. 10.1029/2019MS001622 [DOI] [Google Scholar]
- Andres, R. J. , & Kasgnoc, A. D. (1998). A time‐averaged inventory of subaerial volcanic sulfur emissions. Journal of Geophysical Research, 103(D19), 25251–25261. 10.1029/98JD02091 [DOI] [Google Scholar]
- Bauer, S. E. , Koch, D. , Unger, N. , Metzger, S. M. , Shindell, D. T. , & Streets, D. G. (2007). Nitrate aerosols today and in 2030: A global simulation including aerosols and tropospheric ozone. Atmospheric Chemistry and Physics, 7(19), 5043–5059. 10.5194/acp-7-5043-2007 [DOI] [Google Scholar]
- Bellouin, N. , Rae, J. , Jones, A. , Johnson, C. , Haywood, J. , & Boucher, O. (2011). Aerosol forcing in the Climate Model Intercomparison Project (CMIP5) simulations by HadGEM2‐ES and the role of ammonium nitrate. Journal of Geophysical Research, 116, D20206. 10.1029/2011JD016074 [DOI] [Google Scholar]
- Benduhn, F. , Mann, G. W. , Pringle, K. J. , Topping, D. O. , McFiggans, G. , & Carslaw, K. S. (2016). Size‐resolved simulations of the aerosol inorganic composition with the new hybrid dissolution solver HyDiS‐1.0: Description, evaluation and first global modelling results. Geoscientific Model Development, 9(11), 3875–3906. 10.5194/gmd-9-3875-2016 [DOI] [Google Scholar]
- Bian, H. , Chin, M. , Hauglustaine, D. A. , Schulz, M. , Myhre, G. , Bauer, S. E. , et al. (2017). Investigation of global particulate nitrate from the AeroCom phase III experiment. Atmospheric Chemistry and Physics, 17(21), 12911–12940. 10.5194/acp-17-12911-2017 [DOI] [Google Scholar]
- Bogenschutz, P. A. , Gettelman, A. , Morrison, H. , Larson, V. E. , Craig, C. , & Schanen, D. P. (2013). Higher‐order turbulence closure and its impact on climate simulations in the community atmosphere model. Journal of Climate, 26(23), 9655–9676. 10.1175/JCLI-D-13-00075.1 [DOI] [Google Scholar]
- Boucher, O. , Randall, D. , Artaxo, P. , Bretherton, C. , Feingold, G. , Forster, P. , et al. (2013). Clouds and aerosols. In Stocker T. F., et al. (Eds.), Climate change 2013: The physical science basis. Contribution of working group I to the fifth assessment report of the Intergovernmental Panel on Climate Change (pp. 571–658). Cambridge, UK/New York, NY: Cambridge University Press. 10.1017/CBO9781107415324.016 [DOI] [Google Scholar]
- Bouwman, A. F. , Lee, D. S. , Asman, W. A. H. , Dentener, F. J. , Van Der Hoek, K. W. , & Olivier, J. G. J. (1997). A global high‐resolution emission inventory for ammonia. Global Biogeochemical Cycles, 11(4), 561–587. 10.1029/97GB02266 [DOI] [Google Scholar]
- Burkholder, J. B. , Sander, S. P. , Abbatt, J. P. D. , Barker, J. R. , Huie, R. E. , Kolb, C. E. , et al. (2015). Chemical kinetics and photochemical data for use in atmospheric studies: Evaluation number 18 (JPL publication 15‐10). Pasadena, CA: Jet Propulsion Laboratory. Retrieved from http://jpldataeval.jpl.nasa.gov/ [Google Scholar]
- Dassios, K. G. , & Pandis, S. N. (1999). The mass accommodation coefficient of ammonium nitrate aerosol. Atmospheric Environment, 33(18), 2993–3003. 10.1016/S1352-2310(99)00079-5 [DOI] [Google Scholar]
- Du, H. , Kong, L. , Cheng, T. , Chen, J. , Yang, X. , Zhang, R. , et al. (2010). Insights into ammonium particle‐to‐gas conversion: Non‐sulfate ammonium coupling with nitrate and chloride. Aerosol and Air Quality Research, 10(6), 589–595. 10.4209/aaqr.2010.04.0034 [DOI] [Google Scholar]
- Emmons, L. K. , Walters, S. , Hess, P. G. , Lamarque, J.‐F. , Pfister, G. G. , Fillmore, D. , et al. (2010). Description and evaluation of the Model for Ozone and Related chemical Tracers, version 4 (MOZART‐4). Geoscientific Model Development, 3(1), 43–67. 10.5194/gmd-3-43-2010 [DOI] [Google Scholar]
- Ervens, B. , Turpin, B. J. , & Weber, R. J. (2011). Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): A review of laboratory, field and model studies. Atmospheric Chemistry and Physics, 11(21), 11069–11102. 10.5194/acp-11-11069-2011 [DOI] [Google Scholar]
- Fairlie, T. D. , Jacob, D. J. , Dibb, J. E. , Alexander, B. , Avery, M. A. , van Donkelaar, A. , & Zhang, L. (2010). Impact of mineral dust on nitrate, sulfate, and ozone in transpacific Asian pollution plumes. Atmospheric Chemistry and Physics, 10(8), 3999–4012. 10.5194/acp-10-3999-2010 [DOI] [Google Scholar]
- Fan, T. , Liu, X. , Ma, P.‐L. , Zhang, Q. , Li, Z. , Jiang, Y. , et al. (2018). Emission or atmospheric processes? An attempt to attribute the source of large bias of aerosols in eastern China simulated by global climate models. Atmospheric Chemistry and Physics, 18(2), 1395–1417. 10.5194/acp-18-1395-2018 [DOI] [Google Scholar]
- Fast, J. D. , Gustafson, W. I. , Easter, R. C. , Zaveri, R. A. , Barnard, J. C. , Chapman, E. G. , et al. (2006). Evolution of ozone, particulates, and aerosol direct radiative forcing in the vicinity of Houston using a fully coupled meteorology–chemistry–aerosol model. Journal of Geophysical Research, 111, D21305. 10.1029/2005JD006721 [DOI] [Google Scholar]
- Feng, Y. , & Penner, J. E. (2007). Global modeling of nitrate and ammonium: Interaction of aerosols and tropospheric chemistry. Journal of Geophysical Research, 112, D01304. 10.1029/2005JD006404 [DOI] [Google Scholar]
- Fountoukis, C. , & Nenes, A. (2007). ISORROPIA II: A computationally efficient thermodynamic equilibrium model for K+–Ca2+–Mg2+–NH4 +–Na+–SO4 2−–NO3 −–Cl−–H2O aerosols. Atmospheric Chemistry and Physics, 7(17), 4639–4659. 10.5194/acp-7-4639-2007 [DOI] [Google Scholar]
- Fountoukis, C. , Nenes, A. , Sullivan, A. , Weber, R. , Van Reken, T. , Fischer, M. , et al. (2009). Thermodynamic characterization of Mexico City aerosol during MILAGRO 2006. Atmospheric Chemistry and Physics, 9(6), 2141–2156. 10.5194/acp-9-2141-2009 [DOI] [Google Scholar]
- Fridlind, A. M. , & Jacobson, M. Z. (2000). A study of gas–aerosol equilibrium and aerosol pH in the remote marine boundary layer during the First Aerosol Characterization Experiment (ACE 1). Journal of Geophysical Research, 105(D13), 17325–17340. 10.1029/2000JD900209 [DOI] [Google Scholar]
- Gettelman, A. , Morrison, H. , Santos, S. , Bogenschutz, P. , & Caldwell, P. M. (2015). Advanced two‐moment bulk microphysics for global models. Part II: Global model solutions and aerosol–cloud interactions. Journal of Climate, 28(3), 1288–1307. 10.1175/JCLI-D-14-00103.1 [DOI] [Google Scholar]
- Gong, S. L. , Barrie, L. A. , Blanchet, J.‐P. , von Salzen, K. , Lohmann, U. , Lesins, G. , et al. (2003). Canadian Aerosol Module: A size‐segregated simulation of atmospheric aerosol processes for climate and air quality models 1. Module development. Journal of Geophysical Research, 108(D1), 4007. 10.1029/2001JD002002 [DOI] [Google Scholar]
- Granier, C. , Lamarque, J. F. , Mieville, A. , Muller, J. F. , Olivier, J. , Orlando, J. , et al. (2005). POET, a database of surface emissions of ozone precursors. Retrieved from http://www.aero.jussieu.fr/projet/ACCENT/POET.php [Google Scholar]
- Guenther, A. B. , Jiang, X. , Heald, C. L. , Sakulyanontvittaya, T. , Duhl, T. , Emmons, L. K. , & Wang, X. (2012). The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): An extended and updated framework for modeling biogenic emissions. Geoscientific Model Development, 5(6), 1471–1492. 10.5194/gmd-5-1471-2012 [DOI] [Google Scholar]
- Hanisch, F. , & Crowley, J. N. (2001). Heterogeneous reactivity of gaseous nitric acid on Al2O3, CaCO3, and atmospheric dust samples: A Knudsen cell study. The Journal of Physical Chemistry A, 105(13), 3096–3106. 10.1021/jp001254+ [DOI] [Google Scholar]
- Hauglustaine, D. A. , Balkanski, Y. , & Schulz, M. (2014). A global model simulation of present and future nitrate aerosols and their direct radiative forcing of climate. Atmospheric Chemistry and Physics, 14(20), 11031–11063. 10.5194/acp-14-11031-2014 [DOI] [Google Scholar]
- Hildemann, L. M. , Russell, A. G. , & Cass, G. R. (1984). Ammonia and nitric acid concentrations in equilibrium with atmospheric aerosols: Experiment vs theory. Atmospheric Environment, 18(9), 1737–1750. 10.1016/0004-6981(84)90349-4 [DOI] [Google Scholar]
- Hoesly, R. M. , Smith, S. J. , Feng, L. , Klimont, Z. , Janssens‐Maenhout, G. , Pitkanen, T. , et al. (2018). Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS). Geoscientific Model Development, 11(1), 369–408. 10.5194/gmd-11-369-2018 [DOI] [Google Scholar]
- Hu, X.‐M. , Zhang, Y. , Jacobson, M. Z. , & Chan, C. K. (2008). Coupling and evaluating gas/particle mass transfer treatments for aerosol simulation and forecast. Journal of Geophysical Research, 113, D11208. 10.1029/2007JD009588 [DOI] [Google Scholar]
- Huebert, B. J. , Zhuang, L. , Howell, S. , Noone, K. , & Noone, B. (1996). Sulfate, nitrate, methanesulfonate, chloride, ammonium, and sodium measurements from ship, island, and aircraft during the Atlantic Stratocumulus Transition Experiment/Marine Aerosol Gas Exchange. Journal of Geophysical Research, 101(D2), 4413–4423. 10.1029/95JD02044 [DOI] [Google Scholar]
- Hunke, E. C. , & Lipscomb, W. H. (2008). CICE: The Los Alamos Sea Ice Model, documentation and software user's manual, version 4.0 (Tech. Rep., 76 pp.). Los Alamos, NM: Los Alamos National Laboratory. [Google Scholar]
- Hwang, H. , & Ro, C.‐U. (2006). Direct observation of nitrate and sulfate formations from mineral dust and sea‐salts using low‐Z particle electron probe X‐ray microanalysis. Atmospheric Environment, 40(21), 3869–3880. 10.1016/j.atmosenv.2006.02.022 [DOI] [Google Scholar]
- Iacono, M. J. , Delamere, J. S. , Mlawer, E. J. , Shephard, M. W. , Clough, S. A. , & Collins, W. D. (2008). Radiative forcing by long‐lived greenhouse gases: Calculations with the AER radiative transfer models. Journal of Geophysical Research, 113, D13103. 10.1029/2008JD009944 [DOI] [Google Scholar]
- Jacob, D. J. , Crawford, J. H. , Kleb, M. M. , Connors, V. S. , Bendura, R. J. , Raper, J. L. , et al. (2003). Transport and Chemical Evolution over the Pacific (TRACE‐P) aircraft mission: Design, execution, and first results. Journal of Geophysical Research, 108(D20), 9000. 10.1029/2002jd003276 [DOI] [Google Scholar]
- Jacobson, M. Z. (2001). Global direct radiative forcing due to multicomponent anthropogenic and natural aerosols. Journal of Geophysical Research, 106(D2), 1551–1568. 10.1029/2000JD900514 [DOI] [Google Scholar]
- Karydis, V. A. , Tsimpidi, A. P. , Pozzer, A. , Astitha, M. , & Lelieveld, J. (2016). Effects of mineral dust on global atmospheric nitrate concentrations. Atmospheric Chemistry and Physics, 16(3), 1491–1509. 10.5194/acp-16-1491-2016 [DOI] [Google Scholar]
- Keene, W. C. , Long, M. S. , Pszenny, A. A. P. , Sander, R. , Maben, J. R. , Wall, A. J. , et al. (2009). Latitudinal variation in the multiphase chemical processing of inorganic halogens and related species over the eastern North and South Atlantic Oceans. Atmospheric Chemistry and Physics, 9(19), 7361–7385. 10.5194/acp-9-7361-2009 [DOI] [Google Scholar]
- Larson, V. E. , Golaz, J.‐C. , & Cotton, W. R. (2002). Small‐scale and mesoscale variability in cloudy boundary layers: Joint probability density functions. Journal of the Atmospheric Sciences, 59(24), 3519–3539. [DOI] [Google Scholar]
- Laskin, A. , Wietsma, T. W. , Krueger, B. J. , & Grassian, V. H. (2005). Heterogeneous chemistry of individual mineral dust particles with nitric acid: A combined CCSEM/EDX, ESEM, and ICP‐MS study. Journal of Geophysical Research, 110, D10208. 10.1029/2004JD005206 [DOI] [Google Scholar]
- Lawrence, D. M. , Oleson, K. W. , Flanner, M. G. , Thornton, P. E. , Swenson, S. C. , Lawrence, P. J. , et al. (2011). Parameterization improvements and functional and structural advances in Version 4 of the Community Land Model. Journal of Advances in Modeling Earth Systems, 3, M03001. 10.1029/2011MS00045 [DOI] [Google Scholar]
- Li, J. , Wang, W.‐C. , Liao, H. , & Chang, W. (2015). Past and future direct radiative forcing of nitrate aerosol in East Asia. Theoretical and Applied Climatology, 121(3), 445–458. 10.1007/s00704-014-1249-1 [DOI] [Google Scholar]
- Liao, H. , Adams, P. J. , Chung, S. H. , Seinfeld, J. H. , Mickley, L. J. , & Jacob, D. J. (2003). Interactions between tropospheric chemistry and aerosols in a unified general circulation model. Journal of Geophysical Research, 108(D1), 4001. 10.1029/2001JD001260 [DOI] [Google Scholar]
- Liao, H. , & Seinfeld, J. H. (2005). Global impacts of gas‐phase chemistry–aerosol interactions on direct radiative forcing by anthropogenic aerosols and ozone. Journal of Geophysical Research, 110, D18208. 10.1029/2005JD005907 [DOI] [Google Scholar]
- Liao, H. , Zhang, Y. , Chen, W.‐T. , Raes, F. , & Seinfeld, J. H. (2009). Effect of chemistry–aerosol–climate coupling on predictions of future climate and future levels of tropospheric ozone and aerosols. Journal of Geophysical Research, 114, D10306. 10.1029/2008JD010984 [DOI] [Google Scholar]
- Liu, X. , Easter, R. C. , Ghan, S. J. , Zaveri, R. , Rasch, P. , Shi, X. , et al. (2012). Toward a minimal representation of aerosols in climate models: Description and evaluation in the Community Atmosphere Model CAM5. Geoscientific Model Development, 5(3), 709–739. 10.5194/gmd-5-709-2012 [DOI] [Google Scholar]
- Liu, X. , Ma, P.‐L. , Wang, H. , Tilmes, S. , Singh, B. , Easter, R. C. , et al. (2016). Description and evaluation of a new four‐mode version of the Modal Aerosol Module (MAM4) within version 5.3 of the Community Atmosphere Model. Geoscientific Model Development, 9(2), 505–522. 10.5194/gmd-9-505-2016 [DOI] [Google Scholar]
- Liu, X. , & Penner, J. E. (2005). Ice nucleation parameterization for global models. Meteorologische Zeitschrift, 14(4), 499–514. 10.1127/0941-2948/2005/0059 [DOI] [Google Scholar]
- Liu, X. , Penner, J. E. , Ghan, S. J. , & Wang, M. (2007). Inclusion of ice microphysics in the NCAR Community Atmospheric Model Version 3 (CAM3). Journal of Climate, 20(18), 4526–4547. 10.1175/JCLI4264.1 [DOI] [Google Scholar]
- Lu, Z. , Liu, X. , Zaveri, R. A. , Easter, R. C. , Tilmes, S. , Emmons, L. K. , et al. (2021). Radiative forcing of nitrate aerosols from 1975 to 2010 as simulated by MOSAIC module in CESM2‐MAM4. Journal of Geophysical Research, Submitted. [Google Scholar]
- Mahowald, N. M. , Lamarque, J.‐F. , Tie, X. X. , & Wolff, E. (2006). Sea‐salt aerosol response to climate change: Last Glacial Maximum, preindustrial, and doubled carbon dioxide climates. Journal of Geophysical Research, 111, D05303. 10.1029/2005JD006459 [DOI] [Google Scholar]
- Mahowald, N. M. , Muhs, D. R. , Levis, S. , Rasch, P. J. , Yoshioka, M. , Zender, C. S. , & Luo, C. (2006). Change in atmospheric mineral aerosols in response to climate: Last glacial period, preindustrial, modern, and doubled carbon dioxide climates. Journal of Geophysical Research, 111, D10202. 10.1029/2005JD006653 [DOI] [Google Scholar]
- Malm, W. C. , Schichtel, B. A. , Pitchford, M. L. , Ashbaugh, L. L. , & Eldred, R. A. (2004). Spatial and monthly trends in speciated fine particle concentration in the United States. Journal of Geophysical Research, 109, D03306. 10.1029/2003JD003739 [DOI] [Google Scholar]
- Mårtensson, E. M. , Nilsson, E. D. , de Leeuw, G. , Cohen, L. H. , & Hansson, H.‐C. (2003). Laboratory simulations and parameterization of the primary marine aerosol production. Journal of Geophysical Research, 108(D9), 4297. 10.1029/2002JD002263 [DOI] [Google Scholar]
- Matsui, H. (2017). Development of a global aerosol model using a two‐dimensional sectional method: 1. Model design. Journal of Advances in Modeling Earth Systems, 9, 1921–1947. 10.1002/2017MS000936 [DOI] [Google Scholar]
- Meng, Z. , & Seinfeld, J. H. (1996). Time scales to achieve atmospheric gas–aerosol equilibrium for volatile species. Atmospheric Environment, 30(16), 2889–2900. 10.1016/1352-2310(95)00493-9 [DOI] [Google Scholar]
- Metzger, S. , & Lelieveld, J. (2007). Reformulating atmospheric aerosol thermodynamics and hygroscopic growth into fog, haze and clouds. Atmospheric Chemistry and Physics, 7(12), 3163–3193. 10.5194/acp-7-3163-2007 [DOI] [Google Scholar]
- Mezuman, K. , Bauer, S. E. , & Tsigaridis, K. (2016). Evaluating secondary inorganic aerosols in three dimensions. Atmospheric Chemistry and Physics, 16(16), 10651–10669. 10.5194/acp-16-10651-2016 [DOI] [Google Scholar]
- Monahan, E. C. (1986). The ocean as a source for atmospheric particles. In P. Buat‐Ménard (Ed.), The role of air–sea exchange in geochemical cycling. Dordrecht, The Netherlands: Springer. 10.1007/978-94-009-4738-2_6 [DOI] [Google Scholar]
- Myhre, G. , Berglen, T. F. , Johnsrud, M. , Hoyle, C. R. , Berntsen, T. K. , Christopher, S. A. , et al. (2009). Modelled radiative forcing of the direct aerosol effect with multi‐observation evaluation. Atmospheric Chemistry and Physics, 9(4), 1365–1392. 10.5194/acp-9-1365-2009 [DOI] [Google Scholar]
- Myhre, G. , Grini, A. , & Metzger, S. (2006). Modelling of nitrate and ammonium‐containing aerosols in presence of sea salt. Atmospheric Chemistry and Physics, 6(12), 4809–4821. 10.5194/acp-6-4809-2006 [DOI] [Google Scholar]
- Myhre, G. , Samset, B. H. , Schulz, M. , Balkanski, Y. , Bauer, S. , Berntsen, T. K. , et al. (2013). Radiative forcing of the direct aerosol effect from AeroCom Phase II simulations. Atmospheric Chemistry and Physics, 13(4), 1853–1877. 10.5194/acp-13-1853-2013 [DOI] [Google Scholar]
- Myhre, G. , Shindell, D. , Shindell, F. M. , Collins, W. , Fuglestvedt, J. , Fuglestvedt, J. , et al. (2013). Anthropogenic and natural radiative forcing. In Stocker T. F., et al. (Eds.), Climate change 2013: The physical science basis. Contribution of working group I to the fifth assessment report of the Intergovernmental Panel on Climate Change (pp. 659–740). Cambridge, UK/New York, NY: Cambridge University Press. 10.1017/CBO9781107415324.018 [DOI] [Google Scholar]
- Neale, R. B. , Chen, C.‐C. , Gettelman, A. , Lauritzen, P. H. , Park, S. , Williamson, D. L. , et al. (2012). Description of the NCAR Community Atmosphere Model (CAM 5.0), NCAR/TN‐486+STR. Retrieved from http://www.cesm.ucar.edu/models/cesm1.0/cam/docs/description/cam5_desc.pdf [Google Scholar]
- Nenes, A. , Pandis, S. N. , & Pilinis, C. (1998). ISORROPIA: A new thermodynamic equilibrium model for multiphase multicomponent inorganic aerosols. Aquatic Geochemistry, 4(1), 123–152. 10.1023/A:1009604003981 [DOI] [Google Scholar]
- Paulot, F. , Ginoux, P. , Cooke, W. F. , Donner, L. J. , Fan, S. , Lin, M.‐Y. , et al. (2016). Sensitivity of nitrate aerosols to ammonia emissions and to nitrate chemistry: Implications for present and future nitrate optical depth. Atmospheric Chemistry and Physics, 16(3), 1459–1477. 10.5194/acp-16-1459-2016 [DOI] [Google Scholar]
- Pilson, M. E. Q. (1998). An introduction to the chemistry of the sea. Upper Saddle River, NJ: Prentice Hall. [Google Scholar]
- Pozzer, A. , de Meij, A. , Pringle, K. J. , Tost, H. , Doering, U. M. , van Aardenne, J. , & Lelieveld, J. (2012). Distributions and regional budgets of aerosols and their precursors simulated with the EMAC chemistry–climate model. Atmospheric Chemistry and Physics, 12(2), 961–987. 10.5194/acp-12-961-2012 [DOI] [Google Scholar]
- Pringle, K. J. , Tost, H. , Message, S. , Steil, B. , Giannadaki, D. , Nenes, A. , et al. (2010). Description and evaluation of GMXe: A new aerosol submodel for global simulations (v1). Geoscientific Model Development, 3(2), 391–412. 10.5194/gmd-3-391-2010 [DOI] [Google Scholar]
- Pye, H. O. T. , Liao, H. , Wu, S. , Mickley, L. J. , Jacob, D. J. , Henze, D. K. , & Seinfeld, J. H. (2009). Effect of changes in climate and emissions on future sulfate–nitrate–ammonium aerosol levels in the United States. Journal of Geophysical Research, 114, D01205. 10.1029/2008JD010701 [DOI] [Google Scholar]
- Raper, J. L. , Kleb, M. M. , Jacob, D. J. , Davis, D. D. , Newell, R. E. , Fuelberg, H. E. , et al. (2001). Pacific exploratory mission in the tropical Pacific: PEM‐TROPICS B, March–April 1999. Journal of Geophysical Research, 106(D23), 32401–32425. 10.1029/2000JD900833 [DOI] [Google Scholar]
- Rodriguez, M. A. , & Dabdub, D. (2004). IMAGES‐SCAPE2: A modeling study of size‐ and chemically resolved aerosol thermodynamics in a global chemical transport model. Journal of Geophysical Research, 109, D02203. 10.1029/2003JD003639 [DOI] [Google Scholar]
- Singh, H. B. , Brune, W. H. , Crawford, J. H. , Jacob, D. J. , & Russell, P. B. (2006). Overview of the summer 2004 Intercontinental Chemical Transport Experiment–North America (INTEX‐A). Journal of Geophysical Research, 111, D24S01. 10.1029/2006JD007905 [DOI] [Google Scholar]
- Tilmes, S. , Hodzic, A. , Emmons, L. K. , Mills, M. J. , Gettelman, A. , Kinnison, D. E. , et al. (2019). Climate forcing and trends of organic aerosols in the Community Earth System Model (CESM2). Journal of Advances in Modeling Earth Systems, 11, 4323–4351. 10.1029/2019MS001827 [DOI] [Google Scholar]
- Tilmes, S. , Lamarque, J.‐F. , Emmons, L. K. , Kinnison, D. E. , Ma, P.‐L. , Liu, X. , et al. (2015). Description and evaluation of tropospheric chemistry and aerosols in the Community Earth System Model (CESM1.2). Geoscientific Model Development, 8(5), 1395–1426. 10.5194/gmd-8-1395-2015 [DOI] [Google Scholar]
- Tilmes, S. , Lamarque, J.‐F. , Emmons, L. K. , Kinnison, D. E. , Marsh, D. , Garcia, R. R. , et al. (2016). Representation of the Community Earth System Model (CESM1) CAM4‐chem within the Chemistry–Climate Model Initiative (CCMI). Geoscientific Model Development, 9(5), 1853–1890. 10.5194/gmd-9-1853-2016 [DOI] [Google Scholar]
- Tobo, Y. , Zhang, D. , Matsuki, A. , & Iwasaka, Y. (2010). Asian dust particles converted into aqueous droplets under remote marine atmospheric conditions. Proceedings of the National Academy of Sciences, 107(42), 17905–17910. 10.1073/pnas.1008235107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marle, M. J. E. , Kloster, S. , Magi, B. I. , Marlon, J. R. , Daniau, A.‐L. , Field, R. D. , et al. (2017). Historic global biomass burning emissions for CMIP6 (BB4CMIP) based on merging satellite observations with proxies and fire models (1750–2015). Geoscientific Model Development, 10(9), 3329–3357. 10.5194/gmd-10-3329-2017 [DOI] [Google Scholar]
- Wang, G. , Zhang, R. , Gomez, M. E. , Yang, L. , Zamora, M. L. , Hu, M. , et al. (2016). Persistent sulfate formation from London Fog to Chinese haze. Proceedings of the National Academy of Sciences, 113(48), 13630. 10.1073/pnas.1616540113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Easter, R. C. , Zhang, R. , Ma, P. L. , Singh, B. , Zhang, K. , et al. (2020). Aerosols in the E3SM Version 1: New developments and their impacts on radiative forcing. Journal of Advances in Modeling Earth Systems, 12, e2019MS001851. 10.1029/2019MS001851 [DOI] [Google Scholar]
- Wang, M. , Kong, W. , Marten, R. , He, X.‐C. , Chen, D. , Pfeifer, J. , et al. (2020). Rapid growth of new atmospheric particles by nitric acid and ammonia condensation. Nature, 581(7807), 184–189. 10.1038/s41586-020-2270-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. , & Penner, J. E. (2012). Global simulations of nitrate and ammonium aerosols and their radiative effects. Atmospheric Chemistry and Physics, 12(20), 9479–9504. 10.5194/acp-12-9479-2012 [DOI] [Google Scholar]
- Zaveri, R. A. , Easter, R. C. , Fast, J. D. , & Peters, L. K. (2008). Model for Simulating Aerosol Interactions and Chemistry (MOSAIC). Journal of Geophysical Research, 113, D13204. 10.1029/2007JD008782 [DOI] [Google Scholar]
- Zaveri, R. A. , Easter, R. C. , & Peters, L. K. (2005). A computationally efficient Multicomponent Equilibrium Solver for Aerosols (MESA). Journal of Geophysical Research, 110, D24203. 10.1029/2004JD005618 [DOI] [Google Scholar]
- Zaveri, R. A. , Easter, R. C. , & Wexler, A. S. (2005). A new method for multicomponent activity coefficients of electrolytes in aqueous atmospheric aerosols. Journal of Geophysical Research, 110, D02201. 10.1029/2004JD004681 [DOI] [Google Scholar]
- Zender, C. S. , Bian, H. , & Newman, D. (2003). Mineral Dust Entrainment and Deposition (DEAD) model: Description and 1990s dust climatology. Journal of Geophysical Research, 108(D14), 4416. 10.1029/2002JD002775 [DOI] [Google Scholar]
- Zhang, G. J. , & McFarlane, N. A. (1995). Sensitivity of climate simulations to the parameterization of cumulus convection in the Canadian climate centre general circulation model. Atmosphere‐Ocean, 33(3), 407–446. 10.1080/07055900.1995.9649539 [DOI] [Google Scholar]
- Zhang, X. Y. , Wang, Y. Q. , Zhang, X. C. , Guo, W. , & Gong, S. L. (2008). Carbonaceous aerosol composition over various regions of China during 2006. Journal of Geophysical Research, 113, D14111. 10.1029/2007JD009525 [DOI] [Google Scholar]
- Zhou, C.‐H. , Gong, S. , Zhang, X.‐Y. , Liu, H.‐L. , Xue, M. , Cao, G.‐L. , et al. (2012). Towards the improvements of simulating the chemical and optical properties of Chinese aerosols using an online coupled model—CUACE/Aero. Tellus B: Chemical and Physical Meteorology, 64(1), 18965. 10.3402/tellusb.v64i0.18965 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1
Data Availability Statement
The IMPROVE data set is available at http://vista.cira.colostate.edu/Improve/. The EANET data set is available at https://monitoring.eanet.asia/document/public/index. The EMEP data set is available at http://www.emep.int. INTEX‐A, PEM‐TROPICS B, and TRACE‐P data sets are available at the NASA GTE campaign data archive at https://www-air.larc.nasa.gov/pub/. The gas‐phase species aircraft climatology data set is available as part of the CESM Atmospheric Diagnostics package at http://www.cesm.ucar.edu/working_groups/Atmosphere/amwg-diagnostics-package/. The model CAM5‐chem‐MAM7‐MOSAIC is available at https://github.com/razaveri/CAM5-MAM7-MOSAIC and the simulations are available at https://doi.org/10.25584/DH-172021/1756093.