

Abstract
Most U‐rich small nuclear ribonucleoproteins (snRNPs) are complexes that mediate the splicing of pre‐mRNAs. U7 snRNP is an exception in that it is not involved in splicing but is a key factor in the unique 3′ end processing of replication‐dependent histone mRNAs. However, by introducing controlled changes in the U7 snRNA histone binding sequence and in the Sm motif, it can be used as an effective tool for gene therapy. The modified U7 snRNP (U7 Sm OPT) is thus not involved in the processing of replication‐dependent histone pre‐mRNA but targets splicing by inducing efficient skipping or inclusion of selected exons. U7 Sm OPT is of therapeutic importance in diseases that are an outcome of splicing defects, such as myotonic dystrophy, Duchenne muscular dystrophy, amyotrophic lateral sclerosis, β‐thalassemia, HIV‐1 infection and spinal muscular atrophy. The benefits of using U7 Sm OPT for gene therapy are its compact size, ability to accumulate in the nucleus without causing any toxic effects in the cells, and no immunoreactivity. The risk of transgene misregulation by using U7 Sm OPT is also low because it is involved in correcting the expression of an endogenous gene controlled by its own regulatory elements. Altogether, using U7 Sm OPT as a tool in gene therapy can ensure lifelong treatment, whereas an oligonucleotide or other drug/compound would require repeated administration. It would thus be strategic to harness these unique properties of U7 snRNP and deploy it as a tool in gene therapy.
Keywords: adenoassociated virus, gene‐editing, gene‐therapy, HIV, muscular dystrophy, neurodegenerative disease, RNA‐technologies, stem/progenitor cell research
Involvement of modified U7 small nuclear ribonucleoprotein in splicing activity and gene therapy.
1. INTRODUCTION
Small nuclear ribonucleoproteins (snRNPs) are complexes composed of small nuclear RNA (snRNA) and proteins in specific structures. Initially, the term snRNA was introduced by Weinberg and Penman in 1968. 1 It was further observed that some of these snRNAs were uridine rich compared to ribosomal or messenger RNAs and thus were described as U snRNAs. These U snRNAs were further numbered (e.g. U1 snRNA) by their order of discovery, and not by size, location or abundance. Valuable research was published by the group led by Joan Steitz in 1980, where they initially reported the role of snRNPs in splicing. 2 The spliceosomes, comprising large complexes that catalyse splicing, are divided into major and minor spliceosomes: U1, U2, U4, U5, U6 and U11, U12, U4atac, U5atac and U6atac snRNP, respectively. Subsequently, our knowledge about U snRNPs that interact with pre‐messenger RNA (pre‐mRNA) and other proteins forming the spliceosome has expanded.
In simple terms, splicing is the removal of introns and joining of exons from pre‐mRNA. Splicing is divided into constitutive and alternative splicing (Figure 1) with alternative splicing considered responsible for protein diversity. 3 , 4 , 5 , 6 The interaction between cis‐acting elements and trans‐acting factors during alternative splicing define the exons that would be in the mature mRNA. 3 Exonic and intronic splicing enhancers are cis‐acting elements bound by positive trans‐active factors such as the nuclear phosphoprotein family (serine/arginine‐rich). By contrast, exonic and intronic splicing silencers are bound by negatively acting factors such as heterogeneous ribonucleoproteins (hnRNP). 3 In constitutive splicing, the enhancing elements are dominant, whereas the silencers play a prominent role in controlling alternative splicing. Collaboration between these elements results in the promotion or inhibition of spliceosome assembly at the weak splice sites. 3 , 7 , 8
FIGURE 1.
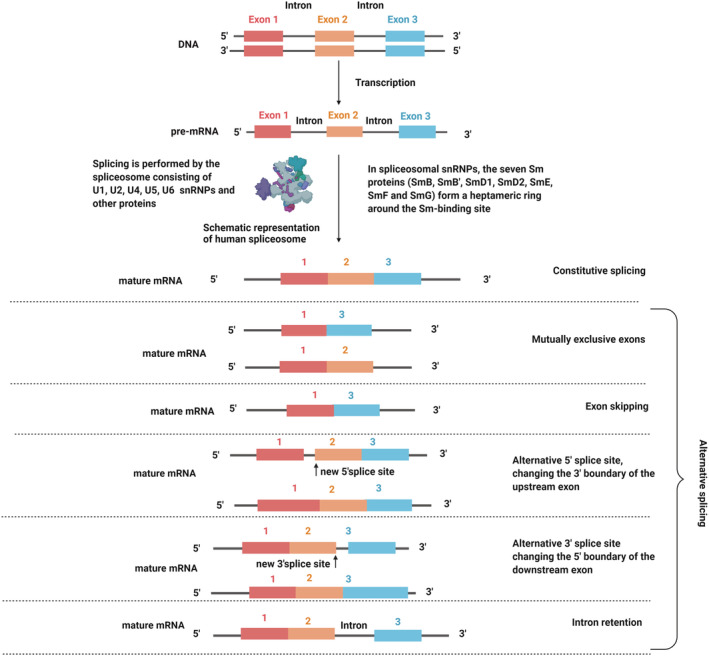
Constitutive and alternative splicing
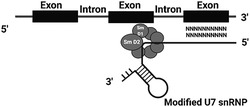
Exceptionally, one of the U snRNPs, U7 snRNP, is not involved in splicing but is a key factor in the unique 3′ end processing of replication‐dependent histone (RDH) pre‐mRNAs. Moreover, recent studies showed U7 snRNA as an important tool in therapeutic studies, with reports based on modified U7 snRNP (U7 Sm OPT) targeting splicing to induce efficient skipping or inclusion of selected exons. U7 Sm OPT is designed by changing the histone binding sequence at the 5′ region of U7 snRNA to the complementary sequence of the gene to be modified. Further modifications include changing of U7 snRNP specific proteins, Lsm10 and Lsm11, to the consensus protein ring of spliceosomal snRNPs.
In U7 Sm OPT‐based therapy, an antisense oligonucleotide is incorporated into the U7 snRNA. Antisense oligonucleotide and its importance was published for the first time by Stephenson Zamecnik in 1978. 9 Antisense oligonucleotides are short single‐stranded nucleotide sequences binding the mRNA to alter gene expression by degradation of the transcript or by inhibition of translation. 10 They do so by various mechanisms, which include ribonuclease H mediated decay of pre‐mRNA or steric hindrance. An alternative way of using antisense oligonucleotides is splicing modulation in which these short nucleotides can bind to pre‐mRNA splicing elements and disrupt the recognition of splicing regulators. 11 For this purpose, as a tool of manipulation of pre‐mRNA splicing, antisense oligonucleotides are used in the U7 Sm OPT therapeutic strategy. Although directly using antisense oligonucleotide has certain benefits, it has some limitations because it is sensitive to degradation, may cause immunoreactivity and usually needs repeated dosage. Thus, incorporating antisense oligonucleotides into U7 snRNP and delivering it using viral vectors overcomes many limitations. U7 Sm OPT snRNP is compact in size, accumulates in the nucleus, is non‐toxic to the cells and overcomes the issue of repeated administration of oligonucleotides. Therefore, using U7 snRNA as a tool in gene therapy enables the desired splicing correction to be achieved along with stable antisense oligonucleotide levels. All of these factors are the basis of gene therapy and U7 snRNP clearly appears to have the upper hand compared to other candidates.
The use of U7 snRNP as a tool for gene therapy has shown promise in pre‐clinical studies and in human clinical trials. The representative diseases include myotonic dystrophy, Duchenne muscular dystrophy (DMD), amyotrophic lateral sclerosis (ALS), β‐thalassemia, HIV‐1 infection, and spinal muscular atrophy (SMA). The aim of this review is to discuss the role of U7 snRNA as a tool for gene therapy.
2. HISTONE GENES AND THEIR PROCESSING
Histone proteins comprise two major classes: canonical RDHs and proteins called histone variants. The genes of RDH are in clusters and are intronless. Interestingly, RDH transcripts are the only mRNAs in metazoan cells that are not polyadenylated. 12 , 13 By contrast, the genes encoding histone variants contain introns, are not arranged in clusters and produce polyadenylated mRNAs. 13 , 14 The expression of RDH genes is activated at the S phase of the cell cycle. 15 During the 3′ end processing, RDH pre‐mRNAs undergo a single endonucleolytic cleavage after a specific stem‐loop structure, located at the 3′ end and recognized by the stem‐loop binding protein (SLBP). 16 , 17 Downstream of the stem‐loop structure is a purine‐rich conserved sequence, known as the histone downstream element (HDE). HDE is recognized by the 5′ end of U7 snRNA. 18 , 19 , 20 The stem‐loop structure, HDE, SLBP and U7 snRNP are respective cis and trans elements in the post‐transcription regulation of RDH mRNAs. These elements determine the cell‐cycle regulated expression of canonical histone genes. 15 , 21 , 22 The binding of U7 snRNP to HDE aids in the recruitment of other factors involved in processing, known as the histone cleavage complex. Cleavage occurs between the 3′ stem‐loop and the HDE, and is catalysed by endonuclease CPSF73 22 , 23 , 24 , 25 , 26 , 27 (Figure 2). It remains unclear which of the mechanisms, polyadenylation or RDH 3′ end processing, developed first in evolution, although it is agreed that U7 snRNP based RDH 3′ end processing was lost in protozoa, plants and fungi, thus making it unique to metazoan cells. 13
FIGURE 2.
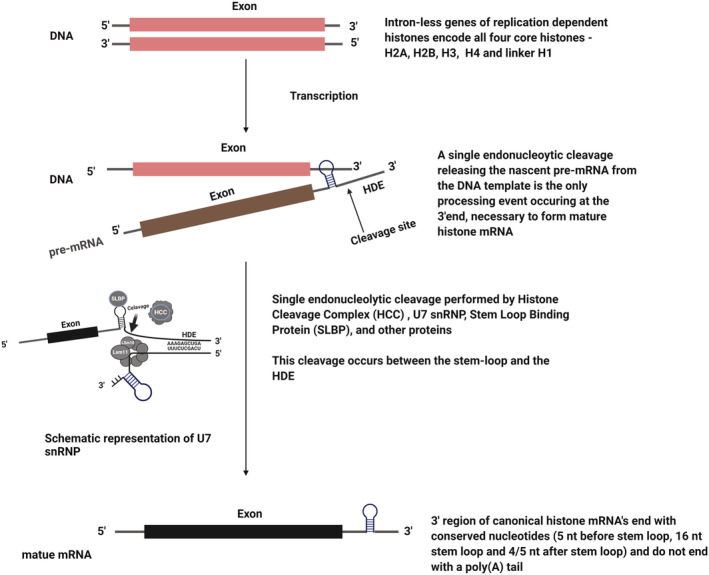
Formation of mature RDH transcripts
3. U7 SNRNP
In humans, U7 snRNP comprises 63 nucleotides of U7 snRNA and a protein core consisting of five common Sm proteins, SmB/B′, SmD3, SmE, SmF and SmG, as well as two U7 snRNP‐specific Sm‐like proteins, Lsm10 and Lsm11. 28 Lsm10 and Lsm11 replace SmD1 and SmD2, respectively, which are found in other U snRNPs. 28 , 29 , 30
The 5′ part of the U7 snRNA is complementary to the HDE motif of the 3′ UTR of RDH pre‐mRNA, and the 3′ region is occupied by a non‐canonical Sm binding site (recognized by the Sm/Lsm protein core), followed by a conserved stem‐loop secondary structure required for its stability 31 (Figure 2). It has been reported in a Drosophila model that blocking the 5′ end of the U7 snRNA with a complementary oligonucleotide specifically blocks the processing of RDH pre‐mRNA. 32 This is one such primary finding that is further implemented with respect to using U7 snRNA as a tool for gene therapy.
U7 snRNP follows the same maturation pathway as spliceosomal U snRNPs. During biogenesis, newly transcribed U7 snRNA is exported to the cytoplasm, where it is assembled with core proteins already present in the cytoplasm (SmB/B′, SmD3, SmE, SmF, SmG and Lsm10 and Lsm11) in a process mediated by the survival motor neuron (SMN) complex and protein arginine methyltransferase‐5 (PRMT5) complex. 33 , 34 After assembly, the U7 snRNP is imported into the nucleus and localizes in histone locus bodies. 28 , 33 , 35
In comparison with major spliceosomal snRNPs, the level of U7 snRNP in the cell is very low, reaching approximately 500 molecules. 36 The non‐canonical Sm binding site and Lsm10 and Lsm11 are assumed to be critical factors of U7 snRNP, and replacement of the U7 Sm binding site with the consensus sequence leads to a higher production of U7 Sm OPT, reaching a level comparable to that of other U snRNPs. 28 , 36
4. U7 SNRNA IN GENE THERAPY
Factors that make U7 snRNA such an interesting choice as a tool for treating genetic disorders are its small size, good stability and ability to accumulate in the nucleus. However, to further improve its activity, the wild‐type U7 snRNA needs to be modified.
Schuemperli and colleagues have shown that the non‐canonical Sm binding site of U7 snRNA (AAUUUGUCUAG; U7 Sm WT) can be converted into the consensus sequence derived from major spliceosomal U snRNPs (AAUUUUUGGAG; U7 Sm OPT), leading to the formation of a spliceosomal‐type heptameric protein core wrapped around U7 Sm OPT. This change results in the augmented expression of U7 Sm OPT particles, which efficiently accumulate in the nucleus and can be redirected to the sites of pre‐mRNA splicing. Moreover, because U7 Sm OPT is unable to bind U7‐specific proteins, Lsm10 and Lsm11, U7 Sm OPT particles are non‐functional in RDH mRNA processing and thus do not affect this process. 28 , 36 , 37 Next, modification of the sequence motif of U7 snRNA, which is complementary to HDE within RDH pre‐mRNA, can make the snRNP particle hybridize to almost any RNA sequence within the nucleoplasm. U7 Sm OPT, even when expressed permanently, does not elicit immunological reactions or toxic effects in the cells. This modification enables U7 snRNA, with its original 3′ elements, to be used in gene therapy (Figure 3).
FIGURE 3.
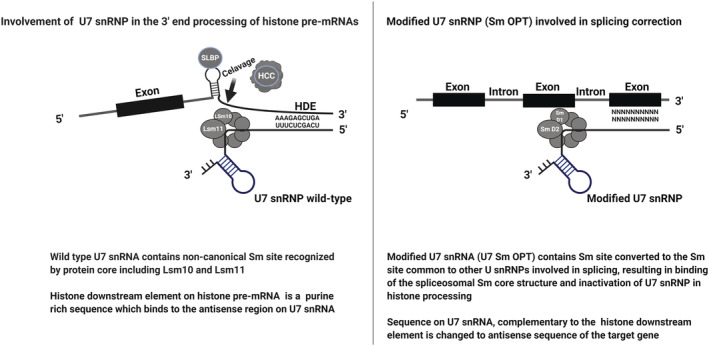
Modifications made to U7 snRNP to be used as a tool in gene therapy
The diseases targeted for U7 snRNA gene therapy are predominantly an outcome of splicing defects. Mutations disrupt splice sites, leading to the creation of cryptic acceptor or donor sites, and aberrant splicing leads to the synthesis of premature terminated or truncated proteins. One disease can be caused by different mutations, thus requiring different therapeutic approaches. Therefore, using antisense oligonucleotides incorporated into U7 Sm OPT proves to be advantageous because it has varied therapeutic applications, such as exon skipping to restore expression, exon skipping to introduce a stop codon, and exon inclusion or displacement of mRNA binding factors (Figure 4).
FIGURE 4.
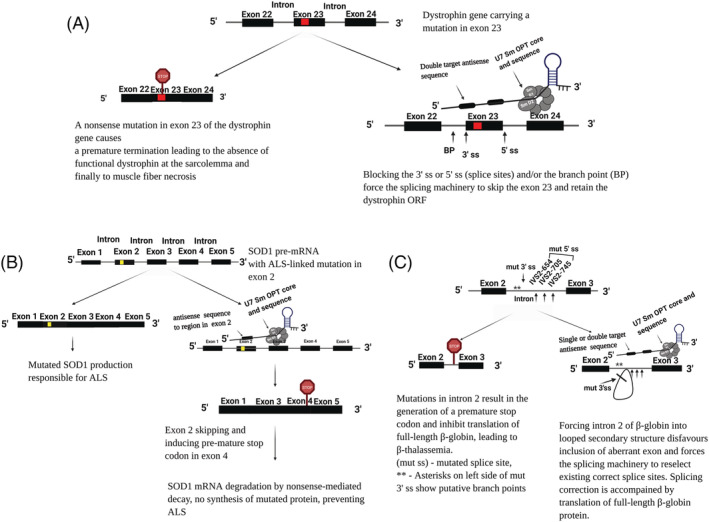
Examples of U7 snRNA‐based splicing modulation for therapy of (a) DMD, (B) ALS and (C) β‐thalassemia
In modifications made to U7 snRNP for blocking splicing, the binding sites to U1 and U2 snRNP are known to comprise the most effective targets. 38 A stable stem‐loop structure as short as 7 bp in an RNA transcript has been shown to abolish enhancer activity, and an exonic splicing enhancer was found to act as intronic splicing enhancer when located in the intron instead of the exon. 39 , 40 , 41 It is further shown that a looping out pre‐mRNA leads to exonic sequestration from the rest of pre‐mRNA transcript. 3 , 42
An initial report based on data obtained from experiments conducted on cell lines mentioned the use of double‐target antisense U7 Sm OPT in the treatment of β‐thalassemia. This treatment was achieved by forcing the aberrant exon into a looped secondary structure and strongly promoting its exclusion from the mRNA. 43 This approach is the fundamental observation reported by the Schuemperli group. Research was conducted on myoblasts obtained from DMD patients, 44 as well as fibroblasts from an SMA patient, 45 and extended to a SMA mouse model. 46 The efficiency of U7 Sm OPT was increased by adding sequences acting as splicing silencers or enhancers. 44 , 45 , 46
The delivery of engineered constructs is an important aspect in gene therapy and is rapidly evolving subsequent to the study of viral vectors. Adeno‐associated viruses (AAV) are non‐enveloped, single‐stranded DNA, replication defective viruses from the Parvoviridae family. 47 , 48 , 49 AAV are currently not known to cause any disease and thus adeno‐associated viral vectors can be used to deliver U7 Sm OPT to the nervous system, as well as other targets organs, as a result of the availability of numerous serotypes with different tissue tropism. 50 , 51 The delivery of U7 Sm OPT by AAV is now broadly used because it ensures high efficiency gene transfer and relatively stable expression.
5. DISEASES TARGETED BY U7 SNRNA GENE THERAPY
U7 Sm OPT is of therapeutic importance in diseases that are an outcome of splicing defects, such as myotonic dystrophy, DMD, ALS, β‐thalassemia, HIV‐1 infection and SMA, as described in detail below. Apart from this, using U7 Sm OPT for correction of aberrant splicing of three genes, PTCH1, BRCA1 and CYP11A, where mutations cause nevoid basal cell carcinoma syndrome, breast cancer and congenital adrenal insufficiency, respectively, is also reported. 52 A brief summary of the diseases targeted by U7 snRNA gene therapy is provided in Table 1.
TABLE 1.
Summary of different research studying the use of U7 snRNP in gene therapy
| Condition | Description |
|---|---|
| Nevoid basal cell carcinoma syndrome 52 |
Cause – mutation c.584G>A in the PCTH1 gene at the 3′ end of exon 3 leading to insertion of a 37 bp intronic sequence between exon 3 and exon 4 and premature termination of PCTH1 protein Study 52 – used HeLa cell line. U7 Sm OPT contains a sequence complementary to the cryptic donor splice site of PCTH1 intron 3. U7 Sm OPT was transfected along with a minigene containing the sequence for exon 3, intron 3 and exon 4. Authentic splicing restored |
| Breast cancer 52 |
Cause – mutation IVS16+6 T>C, located in intron 16 of BRCA1 gene leading to a 65 bp insertion of the 5′ end of intron 16 and premature termination of BRCA1 protein Study 52 – used HeLa cell line. U7 Sm OPT contains a complementary sequence encompassing activated cryptic splice site in intron 16. Transfected along with a minigene containing the sequence of exon 16, intron 16 and exon 17. Observed partial correction of splicing and premature termination. |
| Congenital adrenal insufficiency 52 |
Cause – mutation c.566C>T in the exon 3 of CYP11A gene leading to a 61‐bp deletion of exon 3 Study 52 – used HeLa cell line. U7 Sm OPT contains anti‐CYP11A sequence targeting the cryptic donor splice site in exon 3. Transfected along with minigenes containing exon 3. Targeted exon deletion to various degrees rather than restoring splicing was observed |
| Myotonic dystrophy 1 (DM1) 57 |
Cause – expanded CTG repeats in the 3′ UTR of the DMPK gene Study 57 – used skeletal muscle cells isolated from individuals with DM1, containing various CTG expansions, and myoblasts. U7 Sm OPT contains antisense oligonucleotide with 15 CAG repeats. Long‐lasting selective destruction of deleterious CUGexp RNAs in DM1 cells and in transduced wild‐type myoblasts in a dose‐dependent manner was observed |
| DMD 44 , 59 , 60 , 64 , 65 , 70 , 71 |
Cause(s) – mutations in the dystrophin gene resulting in a premature stop codon leading to the absence of functional dystrophin at the sarcolemma and finally to muscle fibre necrosis. Mutations include, for example: (i) deletion or duplication of exon 2 53 ; (ii) nonsense mutation in exon 23 in mdx mice 53 (iii) C7360A mutation in exon 51 53 ; and (iv) single base change (A to G) in the 3′ splice site of intron 6 of the dystrophin gene, which cause skipping of exon 7 and termination of the ORF within N‐terminus of exon 8 62 Study 64 – used HEK 293 cells, HSMM cells (wild‐type or with exon 2 duplication), fibroblast cell lines, myoblasts and DMD mouse model (Dup2). U7 Sm OPT targeted exon 2 of the dystrophin gene contains antisense oligonucleotides targeting splice donor and splice acceptor sites. Exon 2 skipping resulted in generation of a truncated reading frame upstream of the IRES, which led to the synthesis of a functional N‐truncated isoform in both human subject–derived cell lines (HEK293, HSMM) and in Dup2 model. Expression of the truncated isoform protected the muscle from contraction‐induced injury and corrected muscle force to the same level as that observed in control mice Study 59 – used dystrophin deficient mouse models, mdx mice. Double target U7 Sm OPT (AAV‐U7‐SD23/BP22) 59 contains: (i) a 24‐nucleotide sequence located across the splicing branching point in intron 22 (BP22) and (ii) a 20‐nucleotide sequence in intron 23 that corresponds to the U1 binding region at the donor site (SD23). Reported significant stability of exon 23 skipping in comparison to other U7 snRNA‐based constructs in myoblasts or oligonucleotides injected in vivo Study 65 – used myoblasts from H‐2Kb‐tsA58 mdx mice and C2C12 cells. Double target U7 Sm OPT targeted to block the 5′ splice site and the branch point upstream of exon 23. Using a double target molecule yielded a maximum effect but not an additive effect as in the case of β‐thalassemia 65 Study 60 – used mdx mice and dystrophin/utrophin double‐knockout mice. Used the construct as described by Le et al. 60 for treatment and showed that arrest of dystrophin process is crucial for maintaining viral genomes. Further reported that nontherapeutic U7 Sm OPT resulted in the loss of AAV genome within three weeks which correlates with dystrophin loss but except in the heart Study 44 – bifunctional U7 Sm OPT containing antisense sequences to exon 51, targeting either the acceptor or donor splice site, was transduced into immortalized myoblasts from healthy individuals and from DMD patient carrying a deletion of exons 49 and 50 to restore ORF and dystrophin expression. The second construct carried a 20‐nucleotide sequence complementary to exon 51 and a free tail harboring high‐affinity binding site for hnRNPA1. 59 The study suggests that either the exonic splicing enhancer, proper internal secondary RNA structure, or combination of two play an important role in correct splicing of exon 51. Remarkably, the hnRNPA1‐tailed U7 Sm OPT induced complete exon 51 skipping in patient cells with restoration of dystrophin expression to almost wild‐type level Study 70 – used double knockout utrophin/dystrophin mice, single dose injected intravenously. The construct described in reference 59 was used. Restored near‐normal levels of dystrophin in all muscles, including heart; the treated muscles showed dystrophin detection even 1 year after injection Study 71 – Golden retriever muscular dystrophy dog (GRMD), injected intramuscularly with proximal muscles as a test and with contralateral muscles as control. Three‐week olf GRMD puppies were injected intramuscularly in their whole left cranial leg muscle compartment. Two GRMD dogs were given high pressure intravenous injection in one of the forelimbs by locoregional delivery. Used constructs AAV1‐U7E6 and AAV1‐U7E8ref. 71 The constructs contained 2′ O‐methylated 22 bp and 24 bp antisense oligonucleotides against exon‐splicing enhancers of exon 6 and exon 8, respectively. Partially functional truncated dystrophin was restored |
| ALS 80 |
Cause – single base substitutions such as H48Q, 77 insertion mutation such as 132insTT, 77 CA dinucleotide repeat (D21S223) in exon 2, 78 C to A substitution at codon 41 in exon 2 79 in human SOD1 gene Study 80 – used ALS mouse model B6SJLTg (SOD1*G93A)1Gur/J. U7 Sm OPT contains two steric blocking RNA‐based antisense oligonucleotides masking the splicing acceptor site in intron 1 or the exon splicing enhancer in exon 2 to promote efficient skipping of exon 2. U7 Sm OPT was administered using AAV vector by intravenous and intracerebroventricular routes combined. Therapy at birth or at 50 days of age delayed disease onset, prevented weight loss, prevented the decline of neuromuscular junction, and increased life expectancy by 92% and 58%, respectively |
| β‐Thalassemia/HbE disorder HbE; α2β2 26Glu to Lys 83 |
Cause – G to A mutation (glutamic acid is changed to lysine) at codon 26 in exon 1 of the human β‐globin gene, leading to the activation of the cryptic 5′ splice site at codon 25 and generation of aberrantly spliced β E ‐globin mRNA with a premature termination codon at position 55, leading to the reduction of βE‐globin chains Study 83 – used HeLa βE‐cell model and erythroid progenitor cells from β‐thalassemia/HbE patients. The U7 Sm OPT (U7 βE4+1) (UCCACUUGCACCUACUUCAACCACC) targeted 102–127 nucleotides of exon 1. Observed near complete splicing correction for 5 months in HeLa cells. Furthermore, observed improved erythroid cell pathology |
| β‐Thalassemia 43 , 85 , 86 , 88 , 91 |
Cause – mutation at position 654 (C to T), or 705 (T to G), or 745 (C to G) in intron 2 of β‐globin gene creates an aberrant 5′ splice site at different positions but a common cryptic 3′ splice site at the nucleotide 579 in the β‐globin intron 2 Study 43 – used HeLa cell line stably expressing plasmids carrying β‐globin gene with each of the three mutations. Efficient and permanent correction of aberrant splicing and production of β‐globin levels similar to cells expressing wild‐type gene could be obtained by stable expression of a double target construct U7‐BP+5′654. U7–3′c and U7–3′/24c are other potent candidates Study 88 – used HeLa cell line carrying the thalassemic IVS2–705 human β‐globin gene and cell lines stably expressing U7 Sm OPT, containing a sequence antisense to either the 5′ splice site created by the 705 mutation (U7.5) or to the cryptic 3′ splice site activated in the aberrant splicing pathway (U7.3 and its derivatives). The approach was effective at restoring correct splicing. 65% and 55% correction in cell lines Study 86 – used iPSCs derived from mesenchymal stromal cells from a patient with IVS2–654 β‐thalassemia mutation. U7 Sm OPT (U7.623) (reference 91 ) carrying antisense oligonucleotide (UGUUAUUCUUUAGAAUGGUGCAAAG) targeted the 623 position of intron 2 of the IVS2–654 β‐globin pre‐mRNA. Erythroblasts generated from these iPSCs expressed ~80% restoration in splicing compared to healthy cells. Initial report of combined use of U7 Sm OPT with patient‐specific iPSCs together to treat patients Study 91 – used HeLa cells expressing IVS2–654, IVS2–705 and IVS2–745 human thalassemic β ‐globin genes. U7 Sm OPTU7.623 targeted the 623 position of intron 2 of β‐globin mRNA and U7.324 targeted cryptic 3′ splice site activated by IVS2–654 mutation in the β ‐globin gene. It increased the levels of correctly spliced β ‐globin mRNA and protein by for at least 6 months. It showed therapeutic potential in haematopoietic stem cells and erythroid progenitor cells from a patient with IVS2–745/IVS2–1 thalassemia. 25‐fold correction in patient cells after 12 days of transduction was observed Study 85 – used HeLa IVS2–654 cells and erythroid progenitor cells from patients carrying βIVS2–654 thalassemia mutation. U7. BP+623 targeted the cryptic branch point site and the exonic splicing enhancer in intron 2 of β‐globin pre‐mRNA. Therapeutic potential was shown in both cell models |
| HIV‐1 infection/AIDS 93 , 94 , 95 |
Cause – HIV type 1 Study 93 – used HEK 293 T, HeLa and human T‐cell lines CEM‐SS or CEM. U7 Sm OPT constructs contain antisense sequences targeting flanking internal HIV‐1 exons to reduce tat and rev expression. A merged construct with an additional exonic splicing enhancer and upstream splice donor, named as ESE/SD4, proved 40–50% effectiveness in the context of ‘real’ HIV‐1 replication in human T cells of CEM‐SS line Study 94 – used HEK 293 T, HeLa, CEM‐SS, CEM, P4.2 and CD4+ T cells. Triple combination therapy was used: (i) shRNA targeting nucleotides 330–348 of human cyclophilin A mRNA; (ii) shRNA targeting nucleotides 423–443 and 479–498 of the vif ORF of HIV; and (iii) U7 Sm OPT ESE/SD4 as in reference. 93 Complete inhibition of viral multiplication in semi‐permissive CEM T cells was observed Study 95 – used cell lines HEK 293 T, HeLa and human T‐cell lines Jurkat and CEM‐SS. U7 Sm OPT double target constructs targeted 5′ and 3′ splice site of exon 3 or exon 4 of cyclophilin A gene to eliminate either exon or both and inhibit the interaction of cyclophilin A with the capsid protein of the virus. In addition, siRNAs targeting the region between nucleotides 265 to 283 and 330 to 348 of cyclophilin A mRNA were used. Treated CEM‐SS cell line showed delayed and reduced HIV‐1 multiplication |
| SMA 45 , 46 , 104 , 107 , 108 |
Cause – homozygous exon 7 deletion or inactivation of SMN1 gene. Furthermore, C to T transition in SMN2 gene located at position 6 of exon 7 causes exon 7 deletion in mRNA, leading to the synthesis of truncated protein Study 104 – used HeLa cells. U7 Sm OPT contains antisense nucleotide complementary to 36 nucleotides upstream and 34 nucleotides downstream of the intron7/exon 8 junction to target the 3′ splice site of exon 8. Anti‐SMN U7 Sm OPT G as the most potent along with anti‐SMN U7 Sm OPTs targeting intronic silencer or exon 7 was found. These potent molecules contained ~20 nucleotides hybridization regions with high G/C content. The length of the most potent anti‐SMN RNAs was between 18 to 22 nucleotides. In addition to length and G/C content, competing secondary structures of anti‐SMN U7 snRNAs are important. Inclusion of exon 7 is present as long as the expression cassette is retained in the cell Study 45 – used HEK 293 T cells, HeLa cells and immortalized human fibroblasts from SMA type 1 patient and a healthy individual. bifunctional U7 Sm OPT (U7‐ESE‐B) (sequence B – GUGCUCACAUUCCUUAAAU) 54 , 55 carries an antisense sequence to exon 7 of SMN2 gene together with an exon splicing enhancer or serine arginine repeat. The approach prolonged SMN protein restoration ensuring its localization in gems Study 46 – used SMA mice strain FVB.Cg‐Tg (SMN2)89Ahmb smn1tm1Msd/J, stock number: 005024 from Jackson laboratories. Used U7‐ESE‐B construct from reference. 45 Introducing therapeutic U7 snRNA by germline transgenesis resulted in efficient rescue of exon 7 in the most severe SMA mouse model Study 105 – used severe SMA mouse strain (Burghes severe model) with stillborn or death by postnatal 4–6 days, stock number: 005204 from Jackson laboratories. Neuromuscular junctions of the diaphragm and soleus muscles having the discrete function in breathing and locomotion were selected for study. Used U7‐ESE‐B construct from reference. 45 Neuromuscular junction in treated mice showed correct SMN2 splicing, with delayed or no SMA symptoms Study 107 – used HeLa cells and SMA1‐patient‐derived fibroblast. Five U7 Sm OPT constructs were designed to target exon 8 of SMN gene. Upregulation of SMN levels similar to control cells was observed Study 108 – used HeLa S2 and HeLa cells stably transformed with human SMN2 minigene and SMN Δ7 mice, stock number: 00525 from Jackson laboratories. Used U7‐ESE‐B construct from reference, 45 delivered by intracerebroventricular injection. Introduction into motoneurons significantly increased life span and improved muscle function. Therapeutic U7 snRNA was also observed to be expressed in the heart and liver |
5.1. U7 snRNA therapy for treating myotonic dystrophy
Myotonic dystrophy type 1 caused by expanded CTG repeats in the 3′ UTR of DMPK (myotonic dystrophy protein kinase) gene is the most prevalent form of adult muscular dystrophy. 56 Research was conducted on skeletal muscle cells isolated from individuals with myotonic dystrophy 1 using U7 Sm OPT containing 15 CAG repeats. The construct targeted the expanded CUG repeats of mutant DMPK transcripts in myotonic dystrophy. 60 This resulted in the subsequent release of mRNA binding factors and MBNL1 from the foci, improving splicing abnormalities and differentiation defects. Thus, permanent targeted degradation of pathogenic DMPK mRNA was ensured without affecting the products of wild‐type DMPK allele over subsequent cell divisions.
5.2. U7 snRNA therapy for treating DMD
DMD is an X‐linked recessive muscle‐wasting severe muscular disorder caused by mutations in the dystrophin gene, leading to aberrant or reduced levels of the dystrophin protein. The majority of mutations causing DMD are deletion mutations, which disrupt the open reading frame (ORF) and lead to the synthesis of prematurely terminated proteins. 58 Approximately 70% of the mutations lead to the absence of dystrophin protein and thus to severe DMD phenotype. 59 The presence of qualitatively and quantitively altered dystrophin protein results in Becker muscular dystrophy, a mild form of DMD. In Becker muscular dystrophy, by preserving the reading frame, a truncated, partially functional dystrophin protein is produced that contains deletions in the rod domain with an intact N‐ and C‐terminus. For treatment, research is being carried out that aims to use antisense oligonucleotides to induce specific exon skipping at the dystrophin pre‐mRNA level. The target for therapy is the central rod‐domain because it is known to tolerate large internal deletions. The aim is to convert the “out‐of‐frame” mutation into an “in frame” mutation, giving rise to internally deleted but still functional dystrophin. 60 , 61 Moreover, deletion or duplication of exon 2, nonsense mutation in exon 23, C7360A mutation in exon 51, and A to G mutation in the 3′ splice site of intron 6 of dystrophin gene leading to exon 7 skipping are some of the examples of the cause of DMD. 62
Patients with exon 2 deletions are either asymptomatic or show mild symptoms as a result of the expression of N‐truncated isoform of dystrophin. 63 For treatment, two copies each of antisense oligonucleotides targeting splice donor and splice acceptor were introduced into U7 Sm OPT and delivered using AAV. Research conducted on patient fibroblasts, myoblasts and mice using this construct showed efficient skipping of exon 2 resulting in alternative translation initiation in exon 6 (via an internal ribosome entry sequence), leading to the expression of a functional N‐truncated protein. 64 Based on these studies, a recent clinical trial “AAV9 U7 snRNA gene therapy to treat boys with DMD exon 2 duplications” is now underway, starting from January 2020, registered at NIH with the clinical trials.gov identifier NCT04240314. This clinical trial focuses on skipping of exon 2 leading to either mRNA containing only one copy of exon 2 mRNA (error‐free) or no copy of exon 2, thus giving rise to a highly functional isoform.
One of the earliest research studies on the rescue of DMD via U7 snRNA‐mediated exon skipping reported using a ‘double‐target’ U7 Sm OPT to skip exon 23 of mdx dystrophin mRNA 59 (Figure 4A). In this study, Goyenvalle and colleagues used U7 Sm OPT equipped with two sequences: (i) a 24‐nucleotide sequence complementary to the splicing branch point within intron 22 and (ii) a 20‐nucleotide sequence complementary to U1 snRNA binding region within intron 23. 65 , 66 The engineered U7 snRNA was introduced into skeletal muscles of mdx mouse using AAV vector. 67 The observed level and stability of exon skipping were significantly higher than those obtained using other U7 snRNA‐based constructs in myoblast cultures or using oligonucleotides injected in vivo. Histology performed after treatment showed healthy morphology in corrected muscles. 59 In another study performed on mdx mice, AAV vector was encoded with U7 Sm OPT as reported previously by Goyenvalle et al. 59 This construct allowed efficient exon 23 skipping, therefore rescuing dystrophin. However, it was observed that, over the course of time, loss of AAV vector genome from muscle fibres led to a decrease in exon‐skipping therapeutic effect. 60
Next, Goyenvalle and colleagues reported bifunctional U7 Sm OPT as a promising tool for DMD therapy. 44 , 68 The bifunctional U7 Sm OPT was constructed as follows: first, the U7 Sm OPT was equipped with a splicing silencer sequence complementary to the exonic splicing enhancer located within exon 51 of the human dystrophin gene, aiming to promote efficient skipping of exon 51. Furthermore, the U7 Sm OPT carrying splicing silencer was extended by a tail harbouring canonical binding sites for the heterogeneous ribonucleoprotein A1 (hnRNPA1), which naturally contributes to exon skipping. Enhanced exon 51 skipping, induced by this bifunctional U7 Sm OPT, was observed to restore a near wild‐type level of dystrophin expression in myoblasts derived from DMD patients. The efficacy of the U7 Sm OPT construct was further confirmed by the observation of positive results after subsequent injection into the tibialis anterior muscle of a mouse model transgenic for the entire human dystrophin locus. 44 , 69 Based on this strategy, gene therapy employing bifunctional U7 Sm OPT showed positive results with respect to treating muscular dystrophy in both mdx mice and golden retriever muscular dystrophy dogs. 61 , 70
A study performed on a golden retriever muscular dystrophy dog reported sustained correction of the dystrophic phenotype with U7 Sm OPT carrying antisense oligonucleotides designed for exon skipping therapy. In golden retriever muscular dystrophy dogs, a single base mutation changes the 3′ end of intron 6, leading to skipping of exon 7 and termination within the N‐terminal domain of exon 8. 71 , 72 Using antisense sequence to skip exons 6 and 8, it is possible to restore partially functional truncated dystrophin. The researchers constructed two U7 Sm OPT cassettes for targeting exon 6 or exon 8 skipping. These two different constructs were delivered using AAV vector to treat the forelimb of a golden retriever muscular dystrophy dog by locoregional delivery. No immune rejection was observed after a 5‐year follow‐up. 71
The research performed on DMD is very successful and medication such as eteplirsen (brand name Exondys 51) has been approved in the USA to treat DMD. 73 Ataluren is another drug that has been approved for use in certain cases. 74 Another study sheds light on using the utrophin gene as therapy for the treatment of DMD in animal models. 75 In this case, miniaturized utrophin, a highly functional and non‐immunogenic substitute for dystrophin, is used, thus preventing the most deleterious histological and physiological aspects of DMD in small and large animal models. 75
5.3. U7 snRNA therapy for treating ALS
ALS is a neurodegenerative disease involving mostly mutations in SOD1 (superoxide dismutase 1) gene. The human SOD1 gene is located on chromosome 21 and encodes the superoxide dismutase 1 enzyme responsible for destroying potentially toxic free superoxide radicals in the body. Over 180 mutations in the SOD1 gene are described as being involved in ALS. 76 Some of the mutations are single base substitutions such as H48Q, 77 insertion mutation such as 132insTT, 77 CA dinucleotide repeat (D21S223) in exon 2 78 and C to A substitution at codon 41 in exon 2. 79
One study mentions using U7 Sm OPT to restore the function of SOD1 and prolong the survival of mice in an ALS mouse model. 80 For this, high copy SOD1G93A mice (Jackson SN 2726) were used. 80 U7 Sm OPT containing an antisense sequence targeting exon 2 of the human SOD1 pre‐mRNA was embedded in AAV vectors and administered by intravenous and intracerebroventricular routes combined. 80 It was designed to mask the splicing acceptor site in intron 1 or the exonic splicing enhancer in exon 2 to promote exon 2 skipping. This led to the production of mRNA with a premature stop codon, subjecting it to a RNA degradation pathway and thus reducing the level of toxic protein 80 , 81 (Figure 4B). This treatment, when initiated at birth or at 50 days of age, lead to a delay of disease onset and an increase in life expectancy of 92% and 58%, respectively, thus indicating the effectiveness of an exon‐skipping approach in SOD1‐ALS mice. Therefore, in ALS studies, exon 2 is targeted with the aim of producing mature mRNA containing a premature stop codon to inhibit protein synthesis and thus ultimately reduce the level of toxic proteins. This is in contrast to the discussed therapy for DMD (related to exon 2), in which exon 2 carrying the mutation was skipped to produce a wild‐type protein or its isoform.
5.4. U7 snRNA therapy for treating β‐thalassemia
β‐thalassemia is an autosomal recessive hereditary disease caused by defects in the β‐ chain, which affect hemoglobin (Hb) production. The hemoglobin E (HbE) α2β226 variant, where glutamic acid is changed to lysine as a result of G to A mutation at codon 26 in exon 1 of the human β‐globin (HBB) gene, is a widespread HbE variant found among the Southeast Asian population. 82 , 83 This substitution activates the cryptic 5′ splice site at codon 25, which generates an aberrantly‐spliced β E ‐globin mRNA containing a premature termination codon at position 55, leading to 16‐nucleotide deletion of the 3′ end in exon 1 of β‐globin pre‐mRNA. In a study that focused on the treatment of β‐thalassemia patients using engineered U7 snRNA, antisense oligonucleotides, spanning nucleotides from 102 to 130 of β‐globin mRNA exon 1, were designed to target the 5′ cryptic splice site created by HbE mutation. 83 These oligonucleotides were incorporated into U7 Sm OPT and transiently introduced into cells to test the optimal target site for U7 Sm OPT to attain maximum splicing correction of β‐globin mRNA. From the series of vectors tested, one of the most active engineered U7 Sm OPT vectors, U7 βE4+1 snRNA, was selected. It was then transduced into the HeLa‐βE model cell line and effectively restored the correctly spliced β E ‐globin mRNA for at least 5 months. The U7 βE4+1 snRNA was further used to correctly splice β E ‐globin mRNA in erythroid progenitor cells from β‐thalassemia patients, resulting in phenotypic improvements of β‐thalassemic erythroid cells. 83
Aberrant splicing is also associated with mutations in intron 2 of the β‐globin gene at nucleotide positions 654, 705 or 745. These mutations are not the same (in mutation IVS2–654, C is substituted to T; in mutation IVS2–705, T is substituted to G; in mutation IVS2–745, C is substituted to G) and generate aberrant 5′ splice sites at different positions while activating the same cryptic 3′ splice site at the 579 position, leading to the inclusion of intronic sequences in β‐globin pre‐mRNA. 84 , 85 , 86 Mutations in intron 2 result in the generation of a premature stop codon and inhibit translation into full‐length β‐globin, leading to β‐thalassemia. 84 Antisense oligonucleotides targeting either 5′ or 3′ splice site were shown to repair β‐globin mRNA in mammalian cells. 87 Schuemperli and colleagues first reported the use of the U7 Sm OPT‐mediated approach in splicing correction of β‐globin genes carrying mutations IVS2–654, IVS2–705 88 and IVS2–745, expressed from exogenous plasmids in HeLa cells. 43 Some key points from this foundational research were (i) an optimum sequence of approximately 24 nucleotides influenced the efficiency of antisense sequence and (ii) combining two antisense sequences directed against different target sites in intron 2 significantly enhanced the efficiency of splicing correction 43 (Figure 4C).
Mutation IVS2–654 creates an intron inclusion [nucleotides 580–652] 89 , 90 and patients with this IVS2–654 mutation produce a minor amount of correctly spliced β‐globin mRNAs with severe symptoms of β‐thalassemia. An initial report mentioned combining U7 Sm OPT and induced pluripotent stem cells (iPSCs), leading to successful aberrant splicing reduction of the β‐globin gene in IVS2–654 β‐thalassemia disease. 86 In this study, the U7 Sm OPT, called U7.623 snRNA, was constructed as reported by Vacek et al. 91 It contained a 25‐bp antisense oligonucleotide targeting the 623 position of intron 2 of β‐globin mRNA 91 to restore correct splicing and protein levels. This U7 Sm OPT was transduced into iPSCs derived from mesenchymal stromal cells from a patient with the IVS2–654 mutation using a lentivirus system. The U7 Sm OPT stably integrated into the genome and maintained splicing correction over many passages. Erythroblasts were further generated from these transduced iPSCs, which expressed high levels of correctly spliced β‐globin mRNA. These erythroblasts were further differentiated into haematopoietic stem cells and transplanted back into the same patient with a reduced chance of rejection. 86
Another study reported the restoration of correct β IVS2–654 ‐globin mRNA splicing and hemoglobin production using U7 Sm OPT (U7.BP+623). 85 U7 BP+623 targeted the cryptic branch point and an exonic splicing enhancer. Therapy was performed using thalassemia patient erythroid progenitor cells, as well as HeLa cells carrying the βIVS2–654 thalassemic mutation. Altogether, this approach showed a positive response by restoring correctly spliced β‐globin mRNA and correcting thalassemic erythroid cell pathology. 85
5.5. U7 snRNA therapy for treating HIV infection
HIV is a single‐stranded, positive sense, enveloped RNA retrovirus. 49 HIV cellular tropism is immune cells of the body, with CD4+ T cells being the preferred target. 49 , 92 HIV‐1 exploits alternative splicing, which makes it stand out from other retroviruses. In addition to the standard retroviral genes gag, pol and env, HIV‐1 also codes for tat, rev and nef, expressed at an early stage from the integrated provirus genome along with some other proteins. Nef attunes the physiological status of the host cell, whereas Tat and Rev are RNA‐binding proteins important for the synthesis of full‐length genomic RNA, the regulation of expression of other viral genes and the production of progeny virions. 49 HIV‐1 uses alternative splicing in which Tat strongly activates transcription and Rev channels unspliced and partially spliced RNAs into a nucleo‐cytoplasmic export. 49
In a study on HIV‐1 inhibition, Schuemperli and colleagues employed U7 Sm OPT equipped with two antisense sequences directed against the tat and rev pre‐mRNA internal exons. 93 Targeting the transcripts leads to exon skipping within tat and rev ORFs, inhibition of expression of both proteins and thus inhibition of transition into the late phase of the viral replication cycle. 43 , 93 This U7 snRNA‐based approach resulted in the suppression of HIV‐1 multiplication by up to 50% in human T cells. It was further suggested that the use of other antivirals or siRNA‐based methods in combination with the U7 snRNA‐based approach will lead to increased effects, ensuring a robust response and preventing the evolution of viral escape mutants. 93
The inhibitory effect of the U7 Sm OPT was observed to be stronger for a viral infectivity factor (Vif)‐deficient virus. Therefore, a new approach combining RNAi and U7 Sm OPT strategies was developed. 94 Three different cassettes containing antiviral RNAs were expressed from one triple lentiviral vector: shRNA against the host factor cyclophilin A (CyPA), U7 Sm OPT to modulate exon skipping of tat and rev viral pre‐mRNA, and shRNA targeting the Vif ORF. This approach dramatically affects HIV‐1 infection and allows for a strong inhibition of HIV‐1 multiplication in human T cell lines. Moreover, all three therapeutic RNAs exhibit antiviral effects at early stages of the viral replication cycle. Using these strategies, no changes in cell proliferation or morphology have been observed, suggesting a low toxicity to cells. 94
Inhibition of HIV‐1 replication by targeting the cellular protein cyclophilin A is another strategy based on U7 Sm OPT. 95 Cyclophilin A is known to engage Gag polyprotein in HIV‐1 replication and is essential for HIV‐1 infectivity. 96 Importantly, this protein is neither crucial for early development, nor essential for cell survival. 97 In this research, U7 Sm OPT was exploited as an alternative splicer by inserting appropriate antisense sequences directed against the 3′ and 5′ splice sites of exons 3 and 4 of cyclophilin A pre‐mRNA, respectively. Using this approach, efficient skipping of these exons was achieved, which in turn disturbed the protein ORF. As a consequence, a significantly reduced level of cyclophilin A protein and delayed multiplication of HIV‐1 in T cells were observed. 95 The ability of the cells to sustain HIV‐1 replication was impaired. In the same study, RNAi was used in addition to U7 Sm OPT to reduce cyclophilin A. Adding RNAi along with U7 Sm OPT further improved the survival of T cells. It was further hypothesized that using the aforesaid strategy to modify haematopoietic stem cells instead of T cells would be beneficial to achieve a long‐lasting effect. 95
5.6. U7 snRNA therapy for treating SMA
SMA is an autosomal recessive neuromuscular disorder characterized by the degeneration of anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. 98 SMA is linked to a genetic mutation in the SMN1 gene, 99 which is unable to correctly code for the SMN protein, either as a result of a deletion at exon 7 or other point mutations. 100 Simultaneously, a SMN1 paralogous gene, the SMN2 gene, has identical coding potential but, because of single point mutation in exon 7, an exonic splicing enhancer is disrupted or an exon silencer element is created. This favours alternative splicing at the junction of intron 6 to exon 8, resulting in truncated protein (SMNΔ7). 101 , 102 It is thus hypothesized that correcting the splicing of exon 7 would help in the treatment of SMA. Because exclusion of exon 7 is a major reason for the pathogenesis of SMA and individuals with SMA usually lack the correct SMN1 gene and have a functional SMN2 gene, modulating the inclusion of exon 7 in the human SMN2 gene represents an attractive therapeutic outlook. 103
Exchange of the anti‐histone region of U7 snRNA with a region complementary to the intron7/exon 8 junction of SMN2 pre‐mRNA increases the inclusion of exon 7. 104 Indeed, the administration of such anti‐SMN U7 Sm OPT to a HeLa cell line expressing the SMN2 minigene favoured exon 7 inclusion in SMN2 mRNA and resulted in a higher concentration of full‐length SMN protein. 104
Next, a more permanent strategy using bifunctional U7 snRNA therapy to correct SMN2 pre‐mRNA splicing was developed. 45 In this approach, the U7 Sm OPT was modified both with an antisense sequence binding to exon 7 of SMN2 pre‐mRNA and with the splicing enhancer sequence, improving the recognition of the targeted exon. Addition of the splicing enhancer sequence to the U7 Sm OPT strongly stimulated SMN2 exon 7 recognition and boosted exon inclusion, performing an almost complete re‐inclusion of SMN2 exon 7 in all systems tested. In fibroblasts from a type I SMA patient, this bifunctional approach induced prolonged expression of SMN protein, which was correctly localized within the cell. 45
Schuemperli and colleagues confirmed the hypothesis that promoting SMN2 exon 7 inclusion might help to prevent or benefit treatment of SMA. 46 They used a U7 snRNA‐mediated splicing modulation approach to rescue a severe mouse model of SMA. The U7 Sm OPT construct was developed to target the 3′ end of exon 7 and carried an exon splicing enhancer to attract stimulatory splicing factors. When introduced into the mouse by germline transgenesis, this U7 Sm OPT construct proved efficient in rescuing a severe SMA mouse model. This process reversed the pathology to milder forms and, in some cases, restored neuromuscular functionality and life expectancy. 46
U7 Sm OPT construct capable of stimulating the inclusion of SMN2 exon 7 in severe SMA mouse models was also designed by Voigt et al. 105 They checked the effectiveness of the therapy using a severe mouse model where the mice contained two copies of the human SMN2 transgene and one copy of the mouse Smn gene. 107 For this study, the diaphragm and soleus muscle, which have discrete functions in breathing and locomotion, respectively, were selected. It was observed that the diaphragm displayed prominent adverse morphological alterations in a severe mouse model. No significant changes were observed in either muscle of SMA mice undergoing treatment. Research was further extended by investigating neuromuscular junctions (NMJs). Unfortunately, the development of NMJs was incomplete in a severe SMA mouse model, as well as in treated individuals. 105
To restore full‐length SMN, Geib and Hertel 107 modified the histone binding region of the U7 snRNA to a sequence complementary to the 3′ splice site of SMN exon 8 to inhibit its recognition and induce exon 7 inclusion. The experiments were carried out in both HeLa cells and fibroblasts derived from SMA‐1 patients. AAV vector delivered U7 Sm OPT therapy resulted in a higher rate of exon 7 inclusion, as well as an increase in the level of full‐length SMN protein from 33% to 60%. Moreover, the percentage of gems significantly increased after AAV/U7 Sm OPT therapy, ensuring proper SMN nuclear localization. 107
Recently, Schuemperli and colleagues performed somatic therapy on an SMNΔ7 mouse model (SMN1−/−, SMN2+/+, and SMNΔ7+/+). 108 The mice that would normally survive for only 14–17 days survived for a median of 150 days when treated with the U7 Sm OPT construct, prepared as described previously. 45 , 46 The SMNΔ7 mice, when injected with U7 Sm OPT, showed therapeutic U7 Sm OPT expressed in both the heart and liver. The mice grew smaller in size compared to wild‐type mice and showed mild SMA symptoms, although they were able to feed and drink without any assistance. Additionally, the neuromuscular junctions were less affected compared to SMA models. In conclusion, somatic gene therapy using U7 Sm OPT is feasible for correcting the skipped exon and significantly improving the SMA phenotype. 108
The research examples described above demonstrate that splicing correction of SMN mRNA by the U7 Sm OPT construct works for both transgenic and somatic gene therapy. Nusinersen (SPINRAZA™), an antisense oligonucleotide that acts as a modulator for alternative splicing of the SMN2 gene and converts its functionality to the SMN gene, has been approved against SMA. 109 , 110 , 111 , 112 In May 2019, the US Food and Drug Administration approved Zolgensma, which represents the first gene therapy that has been approved to treat children aged less than 2 years who are diagnosed with SMA. 113 , 114
5.7. Limitations and benefits
U7 snRNP is not a spliceosomal U snRNP and thus requires additional modifications to function as a splicing modifier. For example, in U7 Sm OPT, the binding of unique Lsm proteins is changed to canonical Sm proteins. However, it can generate errors in the 3′ end processing of U7 snRNA and lead to the production of a truncated (20 nucleotides shorter) version of U7 snRNA that is unlikely to mediate correct pre‐mRNA splicing. Because the higher level of truncated U7 snRNA was observed in mice compared to humans, it has been suggested that the processing of U7 Sm OPT differs in these two species and thus should be taken into consideration. 115 , 116 It must also be considered that the snRNA processing capacity of the cell is limited, and that going past this limit may lead to additional U7 snRNA by‐products, which comprise both 3′ processed and unprocessed species. Understanding the formation of these potentially inactive by‐products is necessary to improve the potency of U7 snRNP‐based gene therapy. 116 A limitation for using U7 snRNP in gene therapy is also the amount of vector that can be administered. Administering modified snRNA using the AAV vector requires very high doses that are potentially toxic. 116 Moreover, a study performed on mdx mice emphasizes how the immune response against AAV leads to the production of neutralizing antibodies and thus forbids reinjection, indicating it as a major drawback of the AAV vector approach. 60 Successful delivery is not only a hurdle for administering U7 Sm OPT but for all antisense therapies in general. 117 , 118 Even though all these difficulties exist, viral vectors including AAV vectors and bioreducible lipid nanoparticles comprise useful candidates for delivery, creating a fertile ground for antisense oligonucleotide incorporated U7 snRNA‐based therapeutics. 50 , 119 , 120 , 121
Some questions that remain to be answered are, for example, how to decide a time point to start the therapy? In the case of diseases that develop in a later stage of life, would individual screening for genetic diseases be practised? Nowadays, screening for breast cancer is performed for individuals with genetic background, and maybe this approach can be expanded to other diseases as well. The economic constraint is a hurdle that needs to be overcome. A further consideration is the number of U7 snRNA copies required to achieve the desired correction. High dosage would ensure improved correction but can pose a threat of potential side effects if the dosage increases beyond the cellular processing capacity. Low dosage treatment can have no or minimum side effects but, at the same time, it may not provide maximum correction. It can thus lead to repeated visits to the therapy centre. These decisions are very complicated because they involve physical, economical, psychological and, importantly, emotional factors concerning the affected individual and its family.
Neurodegenerative diseases still remain to be fully understood, with a number of different hypotheses describing the process and cause of neurodegeneration. SMN deficiency, found in SMA and ALS, leads to lower levels and defects in U snRNP assembly, including U7 snRNP. Therefore, the biogenesis and function of U7 snRNP can paradoxically be disrupted in diseases, and thus used as a potential tool in gene therapy.
RDH pre‐mRNA 3′ end processing is undoubtedly the fundamental role of U7 snRNP but, by introducing controlled changes at the histone binding region and the binding site of Sm/Lsm proteins, it can be used as an effective tool for gene therapy. A modification such as converting U7 snRNA into U7 Sm OPT facilitates the accumulation of U7 snRNP in the nucleus and, at the same time, the particle is no longer functional for the processing of RDH pre‐mRNAs, making it an advantage. 28 , 36 The benefits of using U7 snRNP in gene therapy include its compact size and the ability to accumulate in the nucleus without causing any toxic effects to the cells. U7 cassettes will fit into smaller vectors and do not carry the risk of transgene misregulation because the therapeutic RNA only corrects the expression of an endogenous gene that is controlled by its own regulatory elements. The benefit of U7 snRNA‐based gene therapy is also that a single treatment can ensure a lifelong therapeutic effect, while an oligonucleotide or other drug/compound would require repeated administration. U7 snRNP is a unique resource available exclusively to metazoan cells and it would be strategic to use our resources to the fullest.
AUTHOR CONTRIBUTIONS
Ankur Gadgil prepared the main text of the manuscript, as well as Figures 1, 2, 3, 4 and Table 1. Katarzyna Dorota Raczynska revised the manuscript.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
ACKNOWLEDGEMENTS
This work was supported by: Polish National Science Centre, Narodowe Centrum Nauki under Grant UMO‐2016/21/B/NZ1/00232 and UMO‐2018/30/E/NZ2/00295 (to KDR).
Gadgil A, Raczyńska KD. U7 snRNA: A tool for gene therapy. J Gene Med. 2021;23:e3321. 10.1002/jgm.3321
Contributor Information
Ankur Gadgil, Email: ankgad@amu.edu.pl.
Katarzyna Dorota Raczyńska, Email: doracz@amu.edu.pl.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article because no datasets were generated or analysed during the current study.
REFERENCES
- 1. Weinberg RA, Penman S. Small molecular weight monodisperse nuclear RNA. J Mol Biol. 1968;38:289‐304. [DOI] [PubMed] [Google Scholar]
- 2. Lerner MR, Boyle JA, Mount SM, Wolin SL, Steitz JA. Are snRNPs involved in splicing? Nature. 1980;283:220‐224. [DOI] [PubMed] [Google Scholar]
- 3. Wang Y, Liu J, Huang BO, et al. Mechanism of alternative splicing and its regulation. Biomed Reports. Mar 2015;3:152‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gilbert W. Why genes in pieces? Nature. 1978;271:501‐501. [DOI] [PubMed] [Google Scholar]
- 5. Consortium CeS . Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282:2012‐2018. [DOI] [PubMed] [Google Scholar]
- 6. International Human Genome Sequencing C . Finishing the euchromatic sequence of the human genome. Nature. Oct 21 2004;431:931‐945. [DOI] [PubMed] [Google Scholar]
- 7. Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Z, Xiao X, Van Nostrand E, Burge CB. General and specific functions of exonic splicing silencers in splicing control. Mol Cell. Jul 7 2006;23:61‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S a. 1978;75:285‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen X, Corey DR. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018;46:1584‐1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dominski Z, Kole R. Restoration of correct splicing in thalassemic pre‐mRNA by antisense oligonucleotides. Proc Natl Acad Sci U S A. 1993;90:8673‐8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marzluff WF. Metazoan replication‐dependent histone mRNAs: a distinct set of RNA polymerase II transcripts. Curr Opin Cell Biol. Jun 2005;17(3):274‐280. [DOI] [PubMed] [Google Scholar]
- 13. Davila Lopez M, Samuelsson T. Early evolution of histone mRNA 3′ end processing. RNA. 2008;14:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ausio J. Histone variants‐‐the structure behind the function. Brief Funct Genomic Proteomic. 2006;5:228‐243. [DOI] [PubMed] [Google Scholar]
- 15. Harris ME, Bohni R, Schneiderman MH, Ramamurthy L, Schumperli D, Marzluff WF. Regulation of histone mRNA in the unperturbed cell cycle: evidence suggesting control at two posttranscriptional steps. Mol Cell Biol. 1991;11:2416‐2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muller B, Schumperli D. The U7 snRNP and the hairpin binding protein: key players in histone mRNA metabolism. Semin Cell Dev Biol. 1997;8:567‐576. [DOI] [PubMed] [Google Scholar]
- 17. Wang ZF, Whitfield ML, Ingledue TC 3rd, Dominski Z, Marzluff WF. The protein that binds the 3′ end of histone mRNA: a novel RNA‐binding protein required for histone pre‐mRNA processing. Genes Dev. 1996;10:3028‐3040. [DOI] [PubMed] [Google Scholar]
- 18. Mowry KL, Steitz JA. Identification of the human U7 snRNP as one of several factors involved in the 3′ end maturation of histone premessenger RNA's. Science. 1987;238:1682‐1687. [DOI] [PubMed] [Google Scholar]
- 19. Bond UM, Yario TA, Steitz JA. Multiple processing‐defective mutations in a mammalian histone pre‐mRNA are suppressed by compensatory changes in U7 RNA both in vivo and in vitro. Genes Dev. 1991;5:1709‐1722. [DOI] [PubMed] [Google Scholar]
- 20. Schaufele F, Gilmartin GM, Bannwarth W, Birnstiel ML. Compensatory mutations suggest that base‐pairing with a small nuclear RNA is required to form the 3′ end of H3 messenger RNA. Nature. 1986;323:777‐781. [DOI] [PubMed] [Google Scholar]
- 21. Luscher B, Stauber C, Schindler R, Schumperli D. Faithful cell‐cycle regulation of a recombinant mouse histone H4 gene is controlled by sequences in the 3′‐terminal part of the gene. Proc Natl Acad Sci U S a. 1985;82:4389‐4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marzluff WF, Wagner EJ, Duronio RJ. Metabolism and regulation of canonical histone mRNAs: life without a poly(a) tail. Nat Rev Genet. 2008;9:843‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dominski Z, Yang XC, Marzluff WF. The polyadenylation factor CPSF‐73 is involved in histone‐pre‐mRNA processing. Cell. 2005;123:37‐48. [DOI] [PubMed] [Google Scholar]
- 24. Yang XC, Sabath I, Debski J, et al. A complex containing the CPSF73 endonuclease and other polyadenylation factors associates with U7 snRNP and is recruited to histone pre‐mRNA for 3′‐end processing. Mol Cell Biol. 2013;33:28‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marzluff WF, Koreski KP. Birth and death of histone mRNAs. Trends Gen. 2017;33:745‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Romeo V, Schumperli D. Cycling in the nucleus: regulation of RNA 3′ processing and nuclear organization of replication‐dependent histone genes. Curr Opin Cell Biol. 2016;40:23‐31. [DOI] [PubMed] [Google Scholar]
- 27. Ruepp MD, Schweingruber C, Kleinschmidt N, Schumperli D. Interactions of CstF‐64, CstF‐77, and symplekin: implications on localisation and function. Mol Biol Cell. 2011;22:91‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schumperli D, Pillai RS. The special Sm core structure of the U7 snRNP: far‐reaching significance of a small nuclear ribonucleoprotein. Cell Mol Life Sci. 2004;61:2560‐2570. [DOI] [PubMed] [Google Scholar]
- 29. Pillai RS, Will CL, Luhrmann R, Schumperli D, Muller B. Purified U7 snRNPs lack the Sm proteins D1 and D2 but contain Lsm10, a new 14 kDa Sm D1‐like protein. EMBO j. 2001;20:5470‐5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pillai RS, Grimmler M, Meister G, et al. Unique Sm core structure of U7 snRNPs: assembly by a specialized SMN complex and the role of a new component, Lsm11, in histone RNA processing. Genes Dev. 2003;17:2321‐2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gilmartin GM, Schaufele F, Schaffner G, Birnstiel ML. Functional analysis of the sea urchin U7 small nuclear RNA. Mol Cell Biol. 1988;8:1076‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dominski Z, Yang XC, Purdy M, Marzluff WF. Cloning and characterization of the drosophila U7 small nuclear RNA. Proc Natl Acad Sci U S a. 2003;100:9422‐9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Azzouz TN, Pillai RS, Dapp C, et al. Toward an assembly line for U7 snRNPs: interactions of U7‐specific Lsm proteins with PRMT5 and SMN complexes. J Biol Chem. 2005;280:34435‐34440. [DOI] [PubMed] [Google Scholar]
- 34. Tisdale S, Lotti F, Saieva L, et al. SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3′‐end formation of histone mRNAs. Cell Rep. 2013;5:1187‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dominski Z, Marzluff WF. Formation of the 3′ end of histone mRNA: getting closer to the end. Gene. 2007;396:373‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grimm C, Stefanovic B, Schumperli D. The low abundance of U7 snRNA is partly determined by its Sm binding site. EMBO j. 1993;12:1229‐1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stefanovic B, Hackl W, Luhrmann R, Schumperli D. Assembly, nuclear import and function of U7 snRNPs studied by microinjection of synthetic U7 RNA into Xenopus oocytes. Nucleic Acids Res. 1995;23:3141‐3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morcos PA. Achieving targeted and quantifiable alteration of mRNA splicing with Morpholino oligos. Biochem Biophys Res Commun. 2007;358:521‐527. [DOI] [PubMed] [Google Scholar]
- 39. Jin Y, Yang Y, Zhang P. New insights into RNA secondary structure in the alternative splicing of pre‐mRNAs. RNA Biol. 2011;8:450‐457. [DOI] [PubMed] [Google Scholar]
- 40. Liu W, Zhou Y, Hu Z, et al. Regulation of splicing enhancer activities by RNA secondary structures. FEBS Lett. Nov 5 2010;584:4401‐4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McManus CJ, Graveley BR. RNA structure and the mechanisms of alternative splicing. Curr Opin Genet Dev. 2011;21:373‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nasim FU, Hutchison S, Cordeau M, Chabot B. High‐affinity hnRNP A1 binding sites and duplex‐forming inverted repeats have similar effects on 5′ splice site selection in support of a common looping out and repression mechanism. RNA. 2002;8:1078‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suter D, Tomasini R, Reber U, Gorman L, Kole R, Schumperli D. Double‐target antisense U7 snRNAs promote efficient skipping of an aberrant exon in three human beta‐thalassemic mutations. Hum Mol Genet. 1999;8:2415‐2423. [DOI] [PubMed] [Google Scholar]
- 44. Goyenvalle A, Babbs A, van Ommen GJ, Garcia L, Davies KE. Enhanced exon‐skipping induced by U7 snRNA carrying a splicing silencer sequence: promising tool for DMD therapy. Mol Ther. 2009;17:1234‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marquis J, Meyer K, Angehrn L, Kampfer SS, Rothen‐Rutishauser B, Schumperli D. Spinal muscular atrophy: SMN2 pre‐mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol Ther. 2007;15:1479‐1486. [DOI] [PubMed] [Google Scholar]
- 46. Meyer K, Marquis J, Trub J, et al. Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA‐mediated splicing modulation. Hum Mol Genet. 2009;18:546‐555. [DOI] [PubMed] [Google Scholar]
- 47. Lai CM, Lai YK, Rakoczy PE. Adenovirus and adeno‐associated virus vectors. DNA Cell Biol. 2002;21:895‐913. [DOI] [PubMed] [Google Scholar]
- 48. Akli S, Caillaud C, Vigne E, et al. Transfer of a foreign gene into the brain using adenovirus vectors. Nat Genet. 1993;3:224‐228. [DOI] [PubMed] [Google Scholar]
- 49. Fields BN, Knipe DM, Howley PM. Chapter 49 ‐ Human Immunodeficiency Viruses: Replication. In: Fields virology. Vol. 2. 6th ed. 2013:1502‐1560. [Google Scholar]
- 50. Balakrishnan B, Jayandharan GR. Basic biology of adeno‐associated virus (AAV) vectors used in gene therapy. Curr Gene Ther. 2014;14:86‐100. [DOI] [PubMed] [Google Scholar]
- 51. Nonnenmacher M, Weber T. Intracellular transport of recombinant adeno‐associated virus vectors. Gene Ther. 2012;19:649‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Uchikawa H, Fujii K, Kohno Y, et al. U7 snRNA‐mediated correction of aberrant splicing caused by activation of cryptic splice sites. J Hum Genet. 2007;52:891‐897. [DOI] [PubMed] [Google Scholar]
- 53. Vieitez I, Gallano P, Gonzalez‐Quereda L, et al. Mutational spectrum of Duchenne muscular dystrophy in Spain: study of 284 cases. Neurologia. 2017;32:377‐385. [DOI] [PubMed] [Google Scholar]
- 54. Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans‐acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci U S a. 2003;100:4114‐4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cartegni L, Krainer AR. Correction of disease‐associated exon skipping by synthetic exon‐specific activators. Nat Struct Biol. 2003;10:120‐125. [DOI] [PubMed] [Google Scholar]
- 56. Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799‐808. [DOI] [PubMed] [Google Scholar]
- 57. Francois V, Klein AF, Beley C, et al. Selective silencing of mutated mRNAs in DM1 by using modified hU7‐snRNAs. Nat Struct Mol Biol. 2011;18:85‐87. [DOI] [PubMed] [Google Scholar]
- 58. Goyenvalle A, Davies KE. Engineering exon‐skipping vectors expressing U7 snRNA constructs for Duchenne muscular dystrophy gene therapy. Methods Mol Biol. 2011;709:179‐196. [DOI] [PubMed] [Google Scholar]
- 59. Goyenvalle A, Vulin A, Fougerousse F, et al. Rescue of dystrophic muscle through U7 snRNA‐mediated exon skipping. Science. 2004;306:1796‐1799. [DOI] [PubMed] [Google Scholar]
- 60. Le Hir M, Goyenvalle A, Peccate C, et al. AAV genome loss from dystrophic mouse muscles during AAV‐U7 snRNA‐mediated exon‐skipping therapy. Mol Ther. 2013;21:1551‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Harper SQ, Hauser MA, DelloRusso C, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253‐261. [DOI] [PubMed] [Google Scholar]
- 62. White SJ, Aartsma‐Rus A, Flanigan KM, et al. Duplications in the DMD gene. Hum Mutat. 2006;27:938‐945. [DOI] [PubMed] [Google Scholar]
- 63. Lattanzi A, Duguez S, Moiani A, et al. Correction of the exon 2 duplication in DMD myoblasts by a single CRISPR/Cas9 system. Mol Ther Nucleic Acids. 16 2017;7:11‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wein N, Vulin A, Falzarano MS, et al. Translation from a DMD exon 5 IRES results in a functional dystrophin isoform that attenuates dystrophinopathy in humans and mice. Nat Med. 2014;20:992‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Brun C, Suter D, Pauli C, et al. U7 snRNAs induce correction of mutated dystrophin pre‐mRNA by exon skipping. Cell Mol Life Sci. 2003;60:557‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mann CJ, Honeyman K, McClorey G, Fletcher S, Wilton SD. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J Gene Med. 2002;4:644‐654. [DOI] [PubMed] [Google Scholar]
- 67. Chao H, Liu Y, Rabinowitz J, Li C, Samulski RJ, Walsh CE. Several log increase in therapeutic transgene delivery by distinct adeno‐associated viral serotype vectors. Mol Ther. 2000;2:619‐623. [DOI] [PubMed] [Google Scholar]
- 68. Goyenvalle A. Engineering U7snRNA gene to reframe transcripts. Methods Mol Biol. 2012;867:259‐271. [DOI] [PubMed] [Google Scholar]
- 69. Bremmer‐Bout M, Aartsma‐Rus A, de Meijer EJ, et al. Targeted exon skipping in transgenic hDMD mice: a model for direct preclinical screening of human‐specific antisense oligonucleotides. Mol Ther. 2004;10:232‐240. [DOI] [PubMed] [Google Scholar]
- 70. Goyenvalle A, Babbs A, Wright J, et al. Rescue of severely affected dystrophin/utrophin‐deficient mice through scAAV‐U7snRNA‐mediated exon skipping. Hum Mol Genet. 2012;21:2559‐2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vulin A, Barthelemy I, Goyenvalle A, et al. Muscle function recovery in golden retriever muscular dystrophy after AAV1‐U7 exon skipping. Mol Ther. 2012;20:2120‐2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sharp NJ, Kornegay JN, Van Camp SD, et al. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics. 1992;13:115‐121. [DOI] [PubMed] [Google Scholar]
- 73. Syed YY. Eteplirsen: first global approval. Drugs. Nov 2016;76(17):1699‐1704. [DOI] [PubMed] [Google Scholar]
- 74. D'Ambrosio P, Orsini C, Nigro V, Politano L. Therapeutic approach with Ataluren in Duchenne symptomatic carriers with nonsense mutations in dystrophin gene. Results of a 9‐month follow‐up in a case report. Acta Myol. 2018;37:272‐274. [PMC free article] [PubMed] [Google Scholar]
- 75. Song Y, Morales L, Malik AS, et al. Non‐immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat Med. 2019;25:1505‐1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pansarasa O, Bordoni M, Diamanti L, Sproviero D, Gagliardi S, Cereda C. SOD1 in amyotrophic lateral sclerosis: ‘ambivalent’ behavior connected to the disease. Int J Mol Sci. 2018;19:1345‐1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Orrell RW, Habgood JJ, Malaspina A, et al. Clinical characteristics of SOD1 gene mutations in UK families with ALS. J Neurol Sci. 1999;169:56‐60. [DOI] [PubMed] [Google Scholar]
- 78. Rosen DR, Siddique T, Patterson D, et al. Mutations in cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59‐62. [DOI] [PubMed] [Google Scholar]
- 79. Niu Q, Yi Y, Sun X, et al. The G41D mutation in the superoxide dismutase 1 gene is associated with slow motor neuron progression and mild cognitive impairment in a Chinese family with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2016;87:788‐789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Biferi MG, Cohen‐Tannoudji M, Cappelletto A, et al. A new AAV10‐U7‐mediated gene therapy prolongs survival and restores function in an ALS mouse model. Mol Ther. 2017;25:2038‐2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ward AJ, Norrbom M, Chun S, Bennett CF, Rigo F. Nonsense‐mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic Acids Res. 2014;42:5871‐5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fucharoen S, Weatherall DJ. The hemoglobin E thalassemias. Cold Spring Harb Perspect Med. 2012;2:a011734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Preedagasamzin S, Nualkaew T, Pongrujikorn T, et al. Engineered U7 snRNA mediates sustained splicing correction in erythroid cells from beta‐thalassemia/HbE patients. Biochem Biophys Res Commun. 2018;499:86‐92. [DOI] [PubMed] [Google Scholar]
- 84. Cheng TC, Orkin SH, Antonarakis SE, et al. Beta‐thalassemia in Chinese: use of in vivo RNA analysis and oligonucleotide hybridization in systematic characterization of molecular defects. Proc Natl Acad Sci U S a. 1984;81:2821‐2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nualkaew T, Jearawiriyapaisarn N, Hongeng S, Fucharoen S, Kole R, Svasti S. Restoration of correct beta (IVS2–654)‐globin mRNA splicing and HbA production by engineered U7 snRNA in beta‐thalassaemia/HbE erythroid cells. Sci Rep. 2019;9:7672‐7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Phanthong P, Borwornpinyo S, Kitiyanant N, et al. Enhancement of beta‐globin gene expression in Thalassemic IVS2‐654 induced pluripotent stem cell‐derived erythroid cells by modified U7 snRNA. Stem Cells Transl Med. 2017;6:1059‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sierakowska H, Sambade MJ, Agrawal S, Kole R. Repair of thalassemic human beta‐globin mRNA in mammalian cells by antisense oligonucleotides. Proc Natl Acad Sci U S a. Nov 12 1996;93:12840‐12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gorman L, Suter D, Emerick V, Schumperli D, Kole R. Stable alteration of pre‐mRNA splicing patterns by modified U7 small nuclear RNAs. Proc Natl Acad Sci U S a. 1998;95:4929‐4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang JZ, Cai SP, He X, et al. Molecular basis of beta thalassemia in South China. Strategy for DNA analysis. Hum Genet. 1988;78:37‐40. [DOI] [PubMed] [Google Scholar]
- 90. Huang SZ, Zeng FY, Ren ZR, et al. RNA transcripts of the beta‐thalassaemia allele IVS‐2‐654 C‐‐>T: a small amount of normally processed beta‐globin mRNA is still produced from the mutant gene. Br J Haematol. 1994;88:541‐546. [DOI] [PubMed] [Google Scholar]
- 91. Vacek MM, Ma H, Gemignani F, Lacerra G, Kafri T, Kole R. High‐level expression of hemoglobin a in human thalassemic erythroid progenitor cells following lentiviral vector delivery of an antisense snRNA. Blood. 2003;101:104‐111. [DOI] [PubMed] [Google Scholar]
- 92. Cunningham AL, Donaghy H, Harman AN, Kim M, Turville SG. Manipulation of dendritic cell function by viruses. Curr Opin Microbiol. 2010;13:524‐529. [DOI] [PubMed] [Google Scholar]
- 93. Asparuhova MB, Marti G, Liu S, Serhan F, Trono D, Schumperli D. Inhibition of HIV‐1 multiplication by a modified U7 snRNA inducing tat and rev exon skipping. J Gene Med. 2007;9:323‐334. [DOI] [PubMed] [Google Scholar]
- 94. Asparuhova MB, Barde I, Trono D, Schranz K, Schumperli D. Development and characterization of a triple combination gene therapy vector inhibiting HIV‐1 multiplication. J Gene Med. 2008;10:1059‐1070. [DOI] [PubMed] [Google Scholar]
- 95. Liu S, Asparuhova M, Brondani V, Ziekau I, Klimkait T, Schumperli D. Inhibition of HIV‐1 multiplication by antisense U7 snRNAs and siRNAs targeting cyclophilin a. Nucleic Acids Res. 2004;32:3752‐3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Thali M, Bukovsky A, Kondo E, et al. Functional association of cyclophilin a with HIV‐1 virions. Nature. 1994;372:363‐365. [DOI] [PubMed] [Google Scholar]
- 97. Colgan J, Asmal M, Luban J. Isolation, characterization and targeted disruption of mouse ppia: cyclophilin A is not essential for mammalian cell viability. Genomics. 2000;68:167‐178. [DOI] [PubMed] [Google Scholar]
- 98. Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat. 2000;15:228‐237. [DOI] [PubMed] [Google Scholar]
- 99. Brzustowicz LM, Lehner T, Castilla LH, et al. Genetic mapping of chronic childhood‐onset spinal muscular atrophy to chromosome 5q11.2–13.3. Nature. 1990;344:540‐541. [DOI] [PubMed] [Google Scholar]
- 100. Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell. 1995;80:155‐165. [DOI] [PubMed] [Google Scholar]
- 101. Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S a. 1999;96:6307‐6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lewin B. Genes for SMA: multum in parvo. Cell. 1995;80:1‐5. [DOI] [PubMed] [Google Scholar]
- 103. Nlend Nlend R, Meyer K, Schumperli D. Repair of pre‐mRNA splicing: prospects for a therapy for spinal muscular atrophy. RNA Biol. 2010;7:430‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Madocsai C, Lim SR, Geib T, Lam BJ, Hertel KJ. Correction of SMN2 pre‐mRNA splicing by antisense U7 small nuclear RNAs. Mol Ther. 2005;12:1013‐1022. [DOI] [PubMed] [Google Scholar]
- 105. Voigt T, Meyer K, Baum O, Schumperli D. Ultrastructural changes in diaphragm neuromuscular junctions in a severe mouse model for spinal muscular atrophy and their prevention by bifunctional U7 snRNA correcting SMN2 splicing. Neuromuscul Disord. 2010;20:744‐752. [DOI] [PubMed] [Google Scholar]
- 106. Monani UR, Sendtner M, Coovert DD, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in SMN(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333‐339. [DOI] [PubMed] [Google Scholar]
- 107. Geib T, Hertel KJ. Restoration of full‐length SMN promoted by adenoviral vectors expressing RNA antisense oligonucleotides embedded in U7 snRNAs. PLoS One. 2009;4:e8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Odermatt P, Trub J, Furrer L, Fricker R, Marti A, Schumperli D. Somatic therapy of a mouse SMA model with a U7 snRNA gene correcting SMN2 splicing. Mol Ther. 2016;24:1797‐1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hoy SM. Nusinersen: first global approval. Drugs. 2017;77:473‐479. [DOI] [PubMed] [Google Scholar]
- 110. Zanetta C, Nizzardo M, Simone C, et al. Molecular therapeutic strategies for spinal muscular atrophies: current and future clinical trials. Clin Ther. 2014;36:128‐140. [DOI] [PubMed] [Google Scholar]
- 111. Bennett CF, Krainer AR, Cleveland DW. Antisense oligonucleotide therapies for neurodegenerative diseases. Annu Rev Neurosci. 2019;42:385‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in later‐onset spinal muscular atrophy. N Engl J Med. 2018;378:625‐635. [DOI] [PubMed] [Google Scholar]
- 113. Zolgensma ‐ one‐time gene therapy for spinal muscular atrophy. Med Lett Drugs Ther. 2019;61:113‐114. [PubMed] [Google Scholar]
- 114. Mendell JR, Al‐Zaidy S, Shell R, et al. Single‐dose gene‐replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377:1713‐1722. [DOI] [PubMed] [Google Scholar]
- 115. d'Arqom A, Nualkaew T, Jearawiriyapaisarn N, Kole R, Svasti S. Engineered U7 small nuclear RNA restores correct beta‐globin pre‐mRNA splicing in mouse beta (IVS2–654)‐Thalassemic erythroid progenitor cells. Hum Gene Ther. 2020. 10.1089/hum.2020.145. Online ahead of print [DOI] [PubMed] [Google Scholar]
- 116. Eckenfelder A, Tordo J, Babbs A, Davies KE, Goyenvalle A, Danos O. The cellular processing capacity limits the amounts of chimeric U7 snRNA available for antisense delivery. Mol Ther Nucleic Acids. 2012;1:e31‐e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Weyermann J, Lochmann D, Zimmer A. Comparison of antisense oligonucleotide drug delivery systems. J Control Release. 2004;100:411‐423. [DOI] [PubMed] [Google Scholar]
- 118. Juliano R, Alam MR, Dixit V, Kang H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008;36:4158‐4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Yang L, Ma F, Liu F, Chen J, Zhao X, Xu Q. Efficient delivery of antisense oligonucleotides using bioreducible lipid nanoparticles in vitro and in vivo. Mol Ther Nucleic Acids. 2020;19:1357‐1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Imbert M, Dias‐Florencio G, Goyenvalle A. Viral vector‐mediated antisense therapy for genetic diseases. Gene. 2017;8:51‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19:673‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article because no datasets were generated or analysed during the current study.