

Abstract
Energy-dependent Lon proteases play a key role in cellular regulation by degrading short-lived regulatory proteins and misfolded proteins in the cell. The structure of the catalytically inactive S679A mutant of Escherichia coli LonA protease (EcLon) has been determined by cryo-EM at the resolution of 3.5 Å. EcLonA without a bound substrate adopts a hexameric open-spiral quaternary structure that might represent the resting state of the enzyme. Upon interaction with substrate the open-spiral hexamer undergoes a major conformational change resulting in a compact, closed-circle hexamer as in the recent structure of a complex of Yersinia pestis LonA with a protein substrate. This major change is accomplished by the rigid-body rearrangement of the individual domains within the protomers of the complex around the hinge points in the interdomain linkers. Comparison of substrate-free and substrate-bound Lon structures allows to mark the location of putative pivotal points involved in such conformational changes.
Keywords: AAA+ proteins, ATPase module, Lon protease, Cryo-EM
Graphical abstract
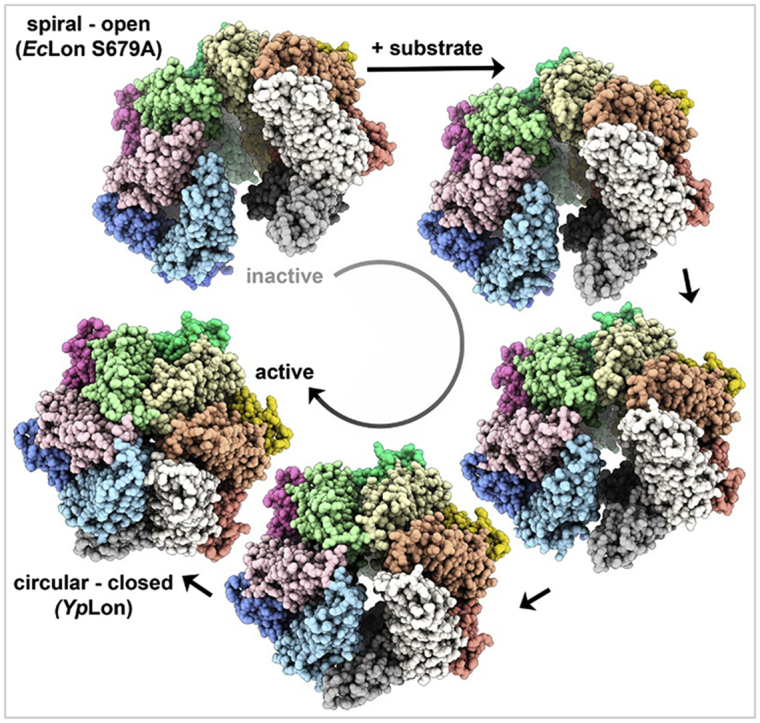
In Brief The structure of the E. coli LonA protease was determined by cryo-EM. The unliganded hexameric complex adopts an open spiral quaternary structure. Upon interaction with substrate it likely undergoes a rigid-body conformational change resulting in a compact, closed-circle hexamer as reported in the Yersinia pestis Lon structure.
Highlights
-
•
E. coli LonA(S679A) protease adopts an open spiral quaternary structure.
-
•
This conformation might represent the resting state of the complex.
-
•
Upon substrate binding the open spiral will close into a circular hexamer.
1. Introduction
Lon proteases (Lons) are ubiquitous enzymes involved in regulation of cellular activity. They require ATP for their function and are responsible for degradation of improperly folded or short-lived proteins. Lons contain an ATPase AAA+ module and a proteolytic domain that is a unique serine-lysine hydrolase. At least two subfamilies of Lons have been classified as LonA and LonB (Adams et al., 2010, Bieniossek et al., 2009). The most prevalent, subfamily A, includes mainly bacterial and eukaryotic enzymes, with archaeal Lons assigned to subfamily B. In addition to the ATPase module and protease domains, LonA proteases also contain a two-domain extra N-terminal region, with all domains being parts of a single polypeptide chain (consisting of 784 amino acids in the Escherichia coli LonA protease (EcLonA) studied here). The N-terminal region has been implicated in oligomerization of LonA proteases and is involved in the binding of protein substrates, as well as in the support of the processive mechanism of their hydrolysis (Botos et al., 2004, Botos et al., 2004, Botos et al., 2005, Brown et al., 2018, Cha et al., 2010, Cole et al., 2008, de la Pena et al., 2018).
Whereas crystal structures of various fragments of Lon proteases have been determined (Rotanova et al., 2006), no structures of the full-length enzymes have been reported. Structures of one- or two-domain constructs of EcLonA include its catalytic domain (residues 596–784) (Botos et al., 2004), partial and complete structures of the ATPase module (residues 326–583) consisting of a large α/β nucleotide-binding domain (α/β) and a small α-helical domain (α) (Garcia-Nafria et al., 2010, Gasteiger et al., 2003), as well as the N-terminal domain (residues 1–116) (Li et al., 2005) and the extended fragment of the N-terminal region (residues 1–245) (Li et al., 2010). Medium resolution crystal structures of the closely related Bacillus subtilis LonA (BsLon) covered the N-terminal domain and separately a construct consisting of the ATPase module and the proteolytic domain (Duman & Lowe, 2010). Although crystal structures of the fragments of Lons from several other bacterial sources and of the proteolytic domain of human mitochondrial LonA (HumLon) (Garcia-Nafria et al., 2010) have also been published, crystallization efforts of full-length Lon have not been successful so far.
Electron microscopy can provide structural data without any need of crystallization of the target proteins and it has been also successfully applied in the studies of Lon proteases. Negative staining of the samples yields low-resolution structures that can show only global features, whereas cryo-electron microscopy (cryo-EM) is capable of generating high-resolution models. The first cryo-EM study of Lon utilized mitochondrial LonA from Saccharomyces cerevisiae, reporting the presence of a flexible ring-shaped heptamer (Stahlberg et al., 1999). A low-resolution model of EcLon, obtained by negative staining of the wild-type enzyme, was presented by Vieux et al. (Vieux et al., 2013). The authors reported simultaneous existence of two oligomeric states of the enzyme, hexameric and dodecameric. The latter assemblies were interpreted as involving head-to-head interactions between the N-terminal domains, with the proteolytic domains sandwiching the AAA+ and N-terminal parts of the complexes. Whereas it was possible to fit to the maps the crystallographic coordinates of the peptidase and ATPase domains of BsLon, as well as the N-terminal domain of EcLon, the low resolution of the maps prevented more detailed fitting. The observed dodecamer was further stabilized in a V217A/Q220A EcLon mutant (Brown et al., 2018).
Low-resolution cryo-EM structures of full-length HumLon (Kereiche et al., 2016) show the presence of a hexamer with six-fold symmetry for a complex of the enzyme with AMPPNP, but imperfect (lock-washer) symmetry for the complex with ADP. The difference between these two structures was interpreted as being due to ATP hydrolysis, but the low resolution of these structures (15–21 Å) precluded detailed description of the N-terminal domains. Removal of the N-terminal domain resulted in significant structural heterogeneity of the enzyme, including very large flexibility of its sixth protomer.
A 3.4 Å-resolution cryo-EM structure of the hexameric form of the Yersinia pestis LonA (YpLon) complexed with Y2853, an 18 kDa protein that is a Lon substrate, was recently published (Shin et al., 2019). The primary structure of this enzyme is 91.5% identical to that of EcLon, although no crystal structure of YpLon has been reported. It was possible to model only part of the tertiary structure, since no density for most of the N-terminal region (residues 1–252 in each protomer) could be seen in the map. All six proteolytic domains are related by almost perfect six-fold symmetry, whereas the arrangement of the ATPase domains is helical with a very small pitch (not exceeding 3 Å). Only seven residues of the substrate, all modeled as alanines, have been fitted to the density, and a single Mg2+ ion and an ATP (or ADP) molecule were seen bound to the ATPase module of each protomer.
With the aim of determining the structure of full-length EcLonA in the absence of a substrate we first characterized by negative-staining the proteolytically inactive S679A mutant of the enzyme (in what follows, the term EcLon refers to EcLonA(S679A), unless explicitly mentioned otherwise). We also determined the cryo-EM structure of this Lon construct complexed with only Mg2+ ions, with no explicitly added nucleotides. The latter structure was compared with the structure of the substrate complex of YpLon in which both ATP and ADP nucleotides were present.
2. Materials and methods
2.1. Cloning, expression, and purification
The catalytically inactive S679A mutant of E. coli (strain K12) Lon protease (EcLon(S679A)) was prepared from the wild-type gene cloned into pET-30, using the QuikChange Lightning Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA). The mutant was expressed in the Lon-deficient E. coli strain BL21(DE3) (EMD Millipore Corporation, Billerica, MA). Cells containing expression plasmid were grown to mid-log phase (OD600 ~ 0.5) at 37 °C in Luria broth containing 30 μg/mL kanamycin and 0.2% glucose. Overproduction of protein was induced with isopropyl-β-d-thiogalactopyranoside at a final concentration of 1 mM for 4 h at 30 °C. The cells were pelleted by centrifugation and stored at −80 °C.
All procedures were performed at 4–8 °C. E. coli cell paste from 4 L of culture was suspended in 50 mM potassium phosphate, pH 6.7, 2 mM EDTA buffer (buffer A) and disrupted with an APV-1000 homogenizer (SPX Corporation, Charlotte, NC) at 10,000 psi. The homogenate was centrifuged at 27,000×g for 30 min, filtered through a 0.45 μm polyethersulfone membrane, and the supernatant was applied to a 100 mL P11 cellulose phosphate column (Whatman Inc., Clifton, NJ) equilibrated in buffer A. The column was washed to baseline with buffer A and bound protein was eluted with two steps to 50% buffer B and to 100% buffer B (300 mM potassium phosphate, pH 6.7 buffer). Fractions containing EcLon were pooled, adjusted with Tris pH 8 buffer to a final concentration of 25 mM, and applied to a MonoQ HR 16/5 column (GE Healthcare Life Sciences, Pittsburgh, PA) equilibrated in 25 mM Tris, pH 7.5 buffer (buffer C). The column was washed to baseline with equilibration buffer and then eluted with a linear gradient to 100% buffer D (25 mM Tris, pH 7.5, 1 M NaCl buffer). Fractions containing target protein were pooled, diluted with buffer C to reduce the NaCl concentration to 125 mM, and applied to a HiPrep 16/10 heparin column (GE Healthcare Life Sciences, Pittsburgh, PA) equilibrated in buffer C. The column was washed to baseline with buffer C, then eluted with a linear gradient to 100% buffer D. Fractions containing recombinant protein were pooled, concentrated using an Ultracel® 30 kDa ultrafiltration disc (EMD Millipore Corporation, Billerica, MA), and applied to a HiPrep 26/60 Sephacryl S-400 HR column (GE Healthcare Life Sciences, Pittsburgh, PA) equilibrated in 25 mM Tris pH 7.2, 150 mM NaCl, 2 mM tris(2-carboxyethyl) phosphine (TCEP) buffer (buffer E). Fractions containing pure recombinant EcLon were pooled and either used directly for grid preparation or concentrated as above to 25–30 mg/mL (estimated at 280 nm using molar extinction coefficients derived from the Expasy ProtParam tool) (Gasteiger et al., 2003). Aliquots were flash-frozen in liquid nitrogen and stored at −80 °C. The final product was judged to be >95% pure, based on silver staining after sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The molecular weight of the recombinant Lon protease subunits were confirmed using the Agilent 6520 Accurate-Mass Q-TOF liquid chromatography/mass spectrometer (Agilent Technologies, Santa Clara, CA). Dynamic light scattering using the DynaPro Titan instrument (Wyatt Technologies, Santa Barbara, CA) showed that the protein was monodisperse, and the mass was consistent with a homohexamer (Fig. S1).
2.2. Sedimentation velocity analytical ultracentrifugation
Sedimentation velocity experiments were carried out at 20 °C on a Beckman Optima XL-A or a Beckman Coulter ProteomeLab XI-I analytical ultracentrifuge following standard protocols (Zhao et al., 2013). Samples of full-length EcLon prepared by dilution of stock solutions at ~28 mg/mL, were studied at different loading concentrations in buffer E. Samples were loaded in 2-channel sector shaped cells with 3 or 12 mm pathlength, depending on the concentration, and analyzed at 30,000 rpm using the absorbance (280 nm) and interference (655 nm, when available) optical detection systems. Sedimentation data were time-corrected (Ghirlando et al., 2013) and analyzed in SEDFIT 16.1c (Schuck, 2000) in terms of a continuous c(s) distribution of Lamm equation solutions, with a maximum entropy regularization confidence level of 0.68. The solution density and viscosity, and protein partial specific volume were calculated based on the composition in SEDNTERP (Cole et al., 2008). Experimental sedimentation coefficients s were corrected to s20,w values at standard conditions (Fig. S2).
2.3. Negative stain electron microscopy
For negative stain experiments the samples were prepared as follows. EcLon was diluted to 1 mg/mL with buffer E and incubated with 1 mM MgCl2 on ice for one hour. Prior to applying onto the grid, the sample was further diluted to 0.01 mg/mL with buffer E (also containing 1 mM MgCl2). 3 μL of protein solution was applied onto a 200 mesh carbon-coated copper grid (Electron Microscopy Sciences, Protochips, Inc.) that had been glow discharged for 30 s at 30 mA (Pelco easiGlow, Ted Pella, Inc.), blotted with filter paper, and stained with 0.7% (w/v) uranyl formate. Images were collected using a FEI Tecnai T20 electron microscope operating at 200 kV with a magnification of 80,000× that resulted in a pixel size of 2.74 Å at the specimen plane. Images were acquired with an Eagle 2 K × 2 K camera using a nominal defocus of 1 μm. 600 micrographs were processed using Relion (Zivanov et al., 2018) and 22,993 particles were selected in total. 8,146 2D-classified hexamer particles and 14,847 dodecamer particles were used for 3D reconstruction (Fig. 1).
Fig. 1.

Negative stain electron microscopy results. (A) Representative negative stain raw micrograph. (B) Negative stain EcLon hexamer 2D classes and (C) dodecamer 2D classes. (D) Low resolution reconstruction of EcLon hexamer and (E) dodecamer.
2.4. Cryo-electron microscopy
For cryo-EM data collection, protein was diluted to 1 mg/mL in buffer E and incubated with 1 mM MgCl2 for 1 h prior to grid preparation. 3 μL of EcLon solution was applied onto a glow-discharged Quantifoil R1.2/1.3200 mesh grid, after which the grid was plunge frozen using a Vitrobot (Thermo Fisher, Inc). The freezing conditions were as follows: 100% humidity, 4 °C, 15 s wait time, 4 s blot time, and 0 blot force. Micrographs were collected on a FEI Titan Krios operating at 300 kV, coupled with a Gatan K2 Summit direct electron detector via the Latitude software (Gatan, Inc). Each exposure image was collected at 29,000 × nominal magnification resulting in a pixel size of 0.85 Å/pixel in the counting mode, using a dose rate of ~1 e−/Å2/s, and 200 ms exposure per frame. A total of 4,168 micrographs were collected. The total dose in the EM data collection was 50 e−/Å2. The nominal defocus range used was −1.2 to −2.5 μm.
2.5. Cryo-electron microscopy data analysis
Data were processed using MotionCorr2 (Zheng et al., 2017) for motion correction, Ctffind4 (Rohou & Grigorieff, 2015) for CTF estimate, EMAN2 (Tang et al., 2007) for binning the micrographs and/or picking particles to form a test data for later Relion auto particle picking, and Relion for 2D classification, 3D initial model, 3D classification, 3D refinement, Bayesian Polishing, and evaluation of local resolution. Molecular graphics and analyses were performed with UCSF Chimera (Pettersen et al., 2004) developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. All computational jobs were performed on the NIH Biowulf cluster. The details are illustrated in Fig. S3.
2.6. Model building and refinement
The protomer of EcLon was built from the crystal structures of the EcLon proteolytic (P) domain and nucleotide-binding domain (PDB:1RR9 (Botos et al., 2004) and 6N2I (Rotanova et al., 2019), respectively). Six protomers of this AP domain model were docked into the EcLon 3.5 Å cryo-EM map with UCSF Chimera (Pettersen et al., 2004) and real-space refined in Phenix (Adams et al., 2010). The resulting structure was rebuilt in Coot (Emsley et al., 2010) using a Phenix auto-sharpened 3.7 Å map and further refined in Phenix.
3. Results and discussion
3.1. Characterization of LonA construct
Purified full-length EcLon runs as a single band on SDS-PAGE and elutes as a homogenous peak on a size exclusion column, having a Mass Q-TOF mass of 87,523 Da (Fig. S1). Sedimentation velocity experiments were conducted on a sample of EcLon to determine its oligomeric state. Experiments at loading concentrations of 1.84, 0.92 and 0.46 mg/mL show the presence of a species at 22.92 S that accounts for ~82% of the loading signal. Based on the value of the sedimentation coefficient and the estimated molar mass of 1.1 MDa, obtained using the interference sedimentation data, this species represents the Lon protease dodecamer (Fig. S2). Evidence for a slower sedimenting Lon protease hexamer at 15.5 S (~11% of the loading signal) and faster oligomer at ~30 S (~3% of the loading signal) is also presented. DLS analysis reveals a monodisperse species at 9.9 nm consistent with sedimentation velocity data (Fig. S1).
3.2. Negative staining
Negative-stain screening of EcLon under various conditions was conducted to find the best quality sample for cryo-EM experiments. Purified EcLon was first analyzed without additional additives and then was also incubated with 1 mM MgCl2, 1 mM MgCl2 + 1 mM ADP, or 1 mM MgCl2 + 1 mM AMPPNP (Table S1). Test data sets were collected and processed to evaluate the quality of the particles. In all samples, the collected images showed rather heterogeneous dodecamer populations and smaller populations of hexamers (Fig. 1). EcLon imaged in the presence of Mg2+ showed 2D-classes with the most homogenous population of dodecamers (14,847 particles) and with few hexamers (8,146 particles) (Fig. 1). 3D-reconstruction and refinement at ~20 Å resolution shows two LonA hexamers facing each other with their N-termini interacting, forming an asymmetric V-shaped complex (Fig. 1). The shape of individual protomers can be distinguished in the low resolution maps, but the flexible N-terminal domains are less clear. In our structures the two hexamers do not assemble into a head-to-head symmetrical dodecamer, as previously suggested by the Baker lab (Brown et al., 2018, Lee et al., 2019).
3.3. Cryo-EM structures
Purified EcLon in the presence of 1 mM MgCl2 or 1 mM MgCl2 + 1 mM AMPPNP was used for grid preparation. Test images showed that EcLon in the presence of 1 mM MgCl2 yielded the best particles and micrographs and was chosen for cryo-EM data collection (Table 1). This procedure yielded cryo-EM micrographs with mainly hexameric particles (Fig. 2, Fig. S3). 2D-classification was carried out and 3D reconstruction and refinement to 3.53 Å resolution showed a spiral open-ring hexamer, with five well-defined protomers and a partially-defined, more flexible sixth one at the periphery (Fig. 3). Previous crystallographic and cryo-EM studies of Lon from different organisms (Li et al., 2010, Schuck, 2000) also noted the high flexibility of the N-terminal region of the molecule up to the last third of the CC-region (residue 246), which in our case might also be destabilized by the ice-air interface on the grid.
Table 1.
Cryo-EM data collection and analysis.
| Construct | S679A |
|---|---|
| Deposition code | EMDB-20659, PDB: 6U5Z |
| Data collection and processing | |
| Magnification | 29,000 (nominal) |
| Voltage (kV) | 300 |
| Electron exposure (e−/Å2) | 50 |
| Defocus range (μm) | −0.7 to −2.5 |
| Pixel size (Å) | 0.858 |
| Symmetry imposed | C1 |
| Initial particle images (no.) | 889,189 |
| Final particle images (no.) | 274,765 |
| Map resolution (Å) | 3.5 |
| FSC threshold | 0.143 |
| Map resolution range (Å) | 3.5–19.0 |
| Refinement | |
| Initial model used (PDB code) | 6N2I, 1RR9 |
| Map sharpening B factor (Å2) | −100.4 |
| Model composition | |
| Non-hydrogen atoms | 24,606 |
| Protein residues | 3174 |
| B factors (Å2) | |
| Protein | 268.16 |
| R.m.s. deviations | |
| Bond length (Å) (# > 4σ) | 0.002 (0) |
| Bond angles (°) (# > 4σ) | 0.527 (0) |
| Validation | |
| Refined model CC | 0.71 |
| MolProbity score | 1.69 |
| Clashscore | 6.6 |
| Poor rotamers (%) | 0.0 |
| Ramachandran plot | |
| Favored (%) | 95.26 |
| Allowed (%) | 4.74 |
| Disallowed (%) | 0.0 |
Fig. 2.

Cryo-EM analysis of EcLon. (A) Representative raw micrograph. (B) Hexamer 2D classes. (C) local resolution map. (D) FSC graph.
Fig. 3.

Structure of the EcLon(S679A) open-ring hexamer. (A) Structure of the fragment (235–784) of EcLon(S679A). The AAA+ module, consisting of the nucleotide-binding and α-helical domains, the proteolytic domain, the connecting inter-domain linker, as well as the 3-helix bundle preceding the AAA+ module are marked. Localization of consensus fragments (Walker motifs A and B, pore-loops 1 and 2, as well as residues Arg finger, Sensor-1 and Sensor-2) is given according to Ref. (Rotanova et al., 2019). (B) Top view cartoon representation with the individual protomers color-coded. (C–D) ¾-tilt and top view CPK representations of the spiral hexamer.
In contrast, negative staining preserved the dodecameric form that is abundant in solution, as shown by analytical ultracentrifugation experiments (see Section 3.1). An EcLon protomer in the cryo-EM map can be fitted with the crystal structures of the EcLon P domain (PDB:1RR9 (Botos et al., 2004)) and AAA+ module (PDB:6N2I (Rotanova et al., 2019)) and accounts for residues 245–775, with the N terminus not visible in the maps (Fig. 4). The three-helix bundle from the coiled-coil region immediately before the AAA+ module is well defined, despite the absence of the very flexible N-terminal fragment comprising the N domain and a larger part of the α-helical HI(CC) domain (Rotanova et al., 2019). Pairwise comparison of the corresponding domains from the protomers in the real-space refined cryo-EM hexamer indicates that the conformation of their main chains is virtually identical (r.m.s.d. less than 0.59 Å). Individual protomers also retain the same conformation as in the recently determined crystal structure of EcLon in the presence of ADP (PDB:6N2I (Rotanova et al., 2019)). Pairwise comparison of the corresponding domains between the EcLon structure and the crystal structure of Meiothermus taiwanensis LonA (MtLon; PDB:4YPL (Lin et al., 2016)) reveals larger differences (Table S2). The small α domain of the AAA+ module is the most similar (average r.m.s.d. 3.07 Å), whereas the three-helix bundle is the most different (average r.m.s.d. 8.49 Å, Table S2). This difference originates from the helices adopting slightly different angles relative to each other in the three-helix bundle.
Fig. 4.

Cryo-EM map with real-space refined EcLon hexamer (residues 247–775). (A) The model accounts for all the map density, the N terminus of the molecule is not visible. (B) Representative fit of a fragment comprising residues 539–557 into the map.
3.4. Comparison to the substrate-bound structure of YpLon
In the absence of substrate, full-length EcLon adopts an open hexameric spiral structure. This is in contrast with the closed-ring, hexameric complex of YpLon (Shin et al., 2019) (Fig. 5). In our structure the individual EcLon protomers are arranged in a spiral around a central axis, showing step-like displacement along the axis while spiraling away from the axis when viewed on the axis. This displacement can be visualized by modeling nucleotides into each protomer and viewing the hexamer on the axis (Fig. S4). The height of the EcLon hexamer viewed perpendicular to the axis is 185 Å, compared to 145 Å for YpLon, while the width of both hexamers is ~130 Å. The transition from the EcLon open conformation into the YpLon closed one is analogous to the closing of a palm of a hand (Supplemental movie). This major conformational change involves displacements as large as 54.2 Å (from YpLon molecule D, A253 Cα to EcLon molecule D, A247 Cα).
Fig. 5.

Comparison of EcLon to the substrate-bound YpLon structure. (A) Top views of spiral open-ring hexamer EcLon and (B) closed-ring hexamer YpLon (PDB: 6ON2). (C) Side views of EcLon and (D) YpLon, with the dimensions of the complexes marked.
The proteolytic domains in YpLon form a C6-symmetric closed ring, which was also observed in previous X-ray structures of the individual proteolytic domains of EcLon and Lons from other bacteria (Emsley et al., 2010, Schuck, 2000, Shin et al., 2019, Stahlberg et al., 1999, Tang et al., 2007). In EcLon cryo-EM structure the hexamers of these domains are not planar, but form an open hexameric ring thus breaking the C6-symmetry. The AAA+ domains in YpLon form an asymmetric spiral staircase with a slight vertical displacement, whereas in EcLon the steps of the spiral staircase have much larger displacement along the vertical axis, and the protomers are placed along a logarithmic spiral, centered on the vertical axis. In the MtLon crystal structure these domains form a symmetric trimer-of-dimers quaternary structure, more similar to the hexamer of YpLon. The spiral quaternary structure is a common feature of AAA+ translocases, as reported in recent cryo-EM structures (Vasilyeva et al., 2004, Vieux et al., 2013, Wohlever et al., 2014, Zhao et al., 2013). Our structure shows that this spiral protomer organization also defines the unliganded, “resting state” of EcLon, poised to bind a substrate.
Differences in the orientation of the individual domains within the protomers of EcLon and YpLon can be visualized by pairwise alignment of their corresponding domains, one at a time (Fig. S5). This comparison finds small α-helical domain (residues 493–583) to be the most similar between the two structures (average r.m.s.d. 0.82 Å). and the proteolytic domain as the most different (average r.m.s.d. 2.23 Å) (Table S3). Thus each EcLon domain is much more similar to YpLon than MtLon.
Rigid body movements of individual domains within their protomers, facilitating rearrangements of the protomers in the structures of substrate-bound vs. substrate-free hexamers of Lon, suggests the presence of “hinge” points in the linker regions. Comparison of the inter-domain linker region (residues 583–593) that connects the α-domain to the proteolytic domain reveals a different conformation of the linker in the structures of EcLon and YpLon (Fig. 6A). This region contains a conserved glycine residue (Gly580, Fig. 6A) shown to be involved in substrate translocation (Vasilyeva et al., 2004, Zheng et al., 2017). Mutating this residue in YpLon reduces ATP hydrolysis and subtrate degradation (Shin et al., 2019), while an equivalent change in Thermotoga maritima FtsH eliminates ATPase activity, reduces proteolytic activity, and prevents the formation of hexamers (Bieniossek et al., 2009). Comparing the positions of the centers of gravity of each domain in the structures of EcLon and YpLon we observe an approximate 30° relative rotation between the proteolytic domain and the rest of the molecule. This local conformational change can translate into a fairly large (~20 Å) shift of the P domain and might have possible functional implications.
Fig. 6.

The inter-domain linker region after the conserved G580 residue adopts a different conformation in EcLon than in YpLon and MtLon. (A) Alignment based on the α domain of EcLon with YpLon (PDB: 6ON2) (both residues marked in black are identical in type and sequence number). (B) An analogous alignment of EcLon with MtLon (PDB: 4YPL) (residues of interest color coded). All figures made with Chimera (Pettersen et al., 2004) and Chimera X (Goddard et al., 2018).
An important role of this linker region was previously suggested for the function of MtLon (Lin et al., 2016). A comparison of the α domain/proteolytic domain junction in the cryo-EM structure of YpLon and crystal structure of MtLon with the cryo-EM structure of EcLon shows a similar shift in the position of the proteolytic domains of YpLon and MtLon with respect to its position in EcLon (Fig. 6A and B). While the conformation of the main chain around Gly580 is surprisingly similar in all compared structures, this comparison allows to pinpoint the residue from which the conformation of the linking region starts to change. Gly587 marks the starting point (Fig. 6A and B) where the random coil linkers in YpLon and MtLon adopt a helical turn in EcLon, initiating changes in the conformation of the linker, that in turn will alter the relative position of the two domains. Gly587 is not highly conserved in the Lon family (Fig. S6), but the substitutions in the residue type (charged) are very similar to the ones for Gly580 in the group of bacterial outliers (Fig. S6). We propose to consider this residue to be a putative hinge point in the structures of Lons. Support of this idea comes from the structure of MtLon, where Gly587 of EcLon is substituted by Asp586 (Fig. S6). Such a substitution does not prevent the large change in the conformation of the linker at this point (Fig. 6B) in a manner very similar to YpLon (Fig. 6A), that leads to the formation of the close ring hexamers. Another argument comes from the difference distance matrix plot of EcLon vs. YpLon prepared with the program DDMP (Fig. S7) that also places the hinge point around Gly587 in an unbiased way.
The surface area for each protomer in the cryo-EM structures is comparable: 24,087 Å2 and 25,894 Å2 for EcLon and YpLon, respectively. The EcLon protomers interact with each other only in specific areas, burying an average 6.9% of their surface areas per protomer, whereas the YpLon protomers bury as much as 20.4% per protomer (Table S4, Fig. S8). This is a threefold increase in buried surface area upon substrate binding and closing of the hexamer. The number of hydrogen bonds and salt bridges also increases as much as five-fold for some protomers upon substrate binding. The EcLon protomers do not interact at all with each other in their areas between residues 245–493 that include the nucleotide-binding domains of their AAA+ modules.
4. Conclusions
Cryo-EM has shown that the proteolytically inactive EcLonA protease adopts an open-spiral hexameric quaternary structure in the absence of a substrate, significantly different from the closed hexamer of YpLon observed when the substrate is present. The protomers at the core of the hexamer are well-defined at 3.5 Å resolution, whereas the ones at the periphery are more flexible and are resolved only at lower contour levels. In the absence of a substrate the N-terminal part of EcLon is not visible in the EM maps. Such disorder was also observed in the recently determined YpLon cryo-EM structure in the presence of a substrate that completely lacks the N-terminal part of the protomers. A comparison of the two structures reveals significant conformational changes in their oligomeric state achieved by rigid-body movement of the domains in a protomer upon binding of the substrate, yielding a closed-ring hexamer with pseudo-C6 symmetry that is very compact. The structure of substrate-free E. coli LonA(S679A) represents the resting state of the hexameric complex, poised to bind a substrate, and may be used as a reference to identify the crucial points for the functioning of such highly dynamic biological machineries as Lon proteases.
Declaration of Competing Interest
Nothing declared.
Acknowledgments
We thank the Biophysics Resource of the Structural Biophysics Laboratory, NCI at Frederick, for use of the LC/ESMS instrument and Marzena Dyba for collecting the DLS data.
This project has been funded in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E, by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by the Russian Foundation for Basic Research (project no. 19-04-00646). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products or organizations imply endorsement by the US Government.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.crstbi.2019.10.001.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C., Richardson J.S., Terwilliger T.C., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniossek C., Niederhauser B., Baumann U.M. The crystal structure of apo-FtsH reveals domain movements necessary for substrate unfolding and translocation. Proc. Natl. Acad. Sci. U. S. A. 2009;106:21579–21584. doi: 10.1073/pnas.0910708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botos I., Melnikov E.E., Cherry S., Tropea J.E., Khalatova A.G., Rasulova F., Dauter Z., Maurizi M.R., Rotanova T.V., Wlodawer A., Gustchina A. The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser-Lys dyad in the active site. J. Biol. Chem. 2004;279:8140–8148. doi: 10.1074/jbc.M312243200. [DOI] [PubMed] [Google Scholar]
- Botos I., Melnikov E.E., Cherry S., Khalatova A.G., Rasulova F.S., Tropea J.E., Maurizi M.R., Rotanova T.V., Gustchina A., Wlodawer A. Crystal structure of the AAA+ α domain of E. coli Lon protease at 1.9 Å resolution. J. Struct. Biol. 2004;146:113–122. doi: 10.1016/j.jsb.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Botos I., Melnikov E.E., Cherry S., Kozlov S., Makhovskaya O.V., Tropea J.E., Gustchina A., Rotanova T.V., Wlodawer A. Atomic-resolution crystal structure of the proteolytic domain of Archaeoglobus fulgidus Lon reveals the conformational variability in the active sites of Lon proteases. J. Mol. Biol. 2005;351:144–157. doi: 10.1016/j.jmb.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Brown B.L., Vieux E.F., Kalastavadi T., Kim S., Chen J.Z., Baker T.A. N domain of the Lon AAA+ protease controls assembly and substrate choice. Protein Sci. 2018 doi: 10.1002/pro.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha S.S., An Y.J., Lee C.R., Lee H.S., Kim Y.G., Kim S.J., Kwon K.K., De Donatis G.M., Lee J.H., Maurizi M.R., Kang S.G. Crystal structure of Lon protease: molecular architecture of gated entry to a sequestered degradation chamber. EMBO J. 2010;29:3520–3530. doi: 10.1038/emboj.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole J.L., Lary J.W., Moody P., Laue T.M. Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol. 2008;84:143–179. doi: 10.1016/S0091-679X(07)84006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Pena A.H., Goodall E.A., Gates S.N., Lander G.C., Martin A. Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science. 2018;362 doi: 10.1126/science.aav0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman R.E., Lowe J. Crystal structures of Bacillus subtilis Lon protease. J. Mol. Biol. 2010;401:653–670. doi: 10.1016/j.jmb.2010.06.030. [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Nafria J., Ondrovicova G., Blagova E., Levdikov V.M., Bauer J.A., Suzuki C.K., Kutejova E., Wilkinson A.J., Wilson K.S. Structure of the catalytic domain of the human mitochondrial Lon protease: proposed relation of oligomer formation and activity. Protein Sci. 2010;19:987–999. doi: 10.1002/pro.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E., Gattiker A., Hoogland C., Ivanyi I., Appel R.D., Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirlando R., Balbo A., Piszczek G., Brown P.H., Lewis M.S., Brautigam C.A., Schuck P., Zhao H. Improving the thermal, radial, and temporal accuracy of the analytical ultracentrifuge through external references. Anal Biochem. 2013;440:81–95. doi: 10.1016/j.ab.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kereiche S., Kovacik L., Bednar J., Pevala V., Kunova N., Ondrovicova G., Bauer J., Ambro L., Bellova J., Kutejova E., Raska I. The N-terminal domain plays a crucial role in the structure of a full-length human mitochondrial Lon protease. Sci. Rep. 2016;6:33631. doi: 10.1038/srep33631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudzhaev A.M., Andrianova A.G., Dubovtseva E.S., Serova O.V., Rotanova T.V. Role of the inserted α-helical domain in E. coli ATP-dependent Lon protease function. Acta Naturae. 2017;9:75–81. [PMC free article] [PubMed] [Google Scholar]
- Kudzhaev A.M., Dubovtseva E.S., Serova O.V., Andrianova A.G., Rotanova T.V. Effect of the deletion of the (173–280) fragment of the inserted α-helical domain on the functional properties of АТР-dependent lon protease from E. coli. Russ. J. Bioorganic Chem. 2018;44:518–527. [Google Scholar]
- Lee S., Roh S.H., Lee J., Sung N., Liu J., Tsai F.T.F. Cryo-em structures of the Hsp104 protein disaggregase captured in the ATP conformation. Cell Rep. 2019;26:29–36. doi: 10.1016/j.celrep.2018.12.037. e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Rasulova F., Melnikov E.E., Rotanova T.V., Gustchina A., Maurizi M.R., Wlodawer A. Crystal structure of the N-terminal domain of E. coli Lon protease. Protein. Sci. 2005;14:2895–2900. doi: 10.1110/ps.051736805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Gustchina A., Rasulova F.S., Melnikov E.E., Maurizi M.R., Rotanova T.V., Dauter Z., Wlodawer A. Structure of the N-terminal fragment of E. coli Lon protease. Acta Crystallogr. 2010;D66:865–873. doi: 10.1107/S0907444910019554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.C., Su S.C., Su M.Y., Liang P.H., Feng C.C., Wu S.H., Chang C.I. Structural insights into the allosteric operation of the Lon AAA+ protease. Structure. 2016;24:667–675. doi: 10.1016/j.str.2016.03.001. [DOI] [PubMed] [Google Scholar]
- Lupas A.N., Martin J. AAA proteins. Curr. Opin. Struct. Biol. 2002;12:746–753. doi: 10.1016/s0959-440x(02)00388-3. [DOI] [PubMed] [Google Scholar]
- Melnikov E.E., Andrianova A.G., Morozkin A.D., Stepnov A.A., Makhovskaya O.V., Botos I., Gustchina A., Wlodawer A., Rotanova T.V. Limited proteolysis of E. coli ATP-dependent protease Lon - a unified view of the subunit architecture and characterization of isolated enzyme fragments. Acta Biochim Pol. 2008;55:281–296. [PMC free article] [PubMed] [Google Scholar]
- Monroe N., Han H., Shen P.S., Sundquist W.I., Hill C.P. Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. Elife. 2017;6 doi: 10.7554/eLife.24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishii W., Kukimoto-Niino M., Terada T., Shirouzu M., Muramatsu T., Kojima M., Kihara H., Yokoyama S. A redox switch shapes the Lon protease exit pore to facultatively regulate proteolysis. Nat. Chem. Biol. 2015;11:46–51. doi: 10.1038/nchembio.1688. [DOI] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Puchades C., Rampello A.J., Shin M., Giuliano C.J., Wiseman R.L., Glynn S.E., Lander G.C. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science. 2017;358 doi: 10.1126/science.aao0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A., Grigorieff N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotanova T.V., Melnikov E.E., Khalatova A.G., Makhovskaya O.V., Botos I., Wlodawer A., Gustchina A. Classification of ATP-dependent proteases Lon and comparison of the active sites of their proteolytic domains. Eur. J. Biochem. 2004;271:4865–4871. doi: 10.1111/j.1432-1033.2004.04452.x. [DOI] [PubMed] [Google Scholar]
- Rotanova T.V., Botos I., Melnikov E.E., Rasulova F., Gustchina A., Maurizi M.R., Wlodawer A. Slicing a protease: structural features of the ATP-dependent Lon proteases gleaned from investigations of isolated domains. Protein. Sci. 2006;15:1815–1828. doi: 10.1110/ps.052069306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotanova T.V., Andrianova A.G., Kudzhaev A.M., Li M., Botos I., Wlodawer A., Gustchina A. New insights into structural and functional relationships between LonA proteases and ClpB chaperones. FEBS Open Bio. 2019 doi: 10.1002/2211-5463.12691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudiak S.G., Shrader T.E. Functional role of the N-terminal region of the Lon protease from Mycobacterium smegmatis. Biochemistry. 1998;37:11255–11263. doi: 10.1021/bi980945h. [DOI] [PubMed] [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M., Asmita A., Puchades C., Adjei E., Wiseman R.L., Karzai A.W., Lander G.C. Distinct structural features of the lon protease drive conserved hand-over-hand substrate translocation. BioRxiv. 2019:1–21. doi: 10.1101/61715. [DOI] [Google Scholar]
- Stahlberg H., Kutejova E., Suda K., Wolpensinger B., Lustig A., Schatz G., Engel A., Suzuki C.K. Mitochondrial Lon of Saccharomyces cerevisiae is a ring-shaped protease with seven flexible subunits. Proc. Natl. Acad. Sci. U. S. A. 1999;96:6787–6790. doi: 10.1073/pnas.96.12.6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G., Peng L., Baldwin P.R., Mann D.S., Jiang W., Rees I., Ludtke S.J. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Vasilyeva O.V., Martynova N.I., Potapenko N.A., Ovchinnikova T.V. Isolation and characterization of fragments of ATP-dependent protease Lon from Escherichia coli obtained by limited proteolysis. Russ. J. Bioorg. Chem. 2004;30:306–314. [PubMed] [Google Scholar]
- Vieux E.F., Wohlever M.L., Chen J.Z., Sauer R.T., Baker T.A. Distinct quaternary structures of the AAA+ Lon protease control substrate degradation. Proc. Natl. Acad. Sci. U. S. A. 2013;110:E2002–E2008. doi: 10.1073/pnas.1307066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlever M.L., Baker T.A., Sauer R.T. Roles of the N domain of the AAA+ Lon protease in substrate recognition, allosteric regulation and chaperone activity. Mol. Microbiol. 2014;91:66–78. doi: 10.1111/mmi.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Brautigam C.A., Ghirlando R., Schuck P. Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr. Protoc. Protein Sci. 2013 doi: 10.1002/0471140864.ps2012s71. [Chapter 20], Unit 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S.Q., Palovcak E., Armache J.-P., Verba K.A., Cheng Y., Agard D.A. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J., Nakane T., Forsberg B., Kimanius D., Hagen W.J.H., Lindahl E., Scheres S.H.W. RELION-3: new tools for automated high-resolution cryo-EM structure determination. BioRxiv. 2018:421123. doi: 10.1101/421123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.