

Abstract
Simian immunodeficiency virus (SIV) insert-expressing, 68–1 Rhesus Cytomegalovirus (RhCMV/SIV) vectors elicit major histocompatibility complex (MHC)-E- and -II-restricted, SIV-specific CD8+ T cell responses, but the basis of these unconventional responses and their contribution to demonstrated vaccine efficacy against SIV challenge in the rhesus monkeys (RMs) has not been characterized. We show that these unconventional responses resulted from a chance genetic rearrangement in 68–1 RhCMV that abrogated the function of eight distinct immunomodulatory gene products encoded in two RhCMV genomic regions (Rh157.5/Rh157.4 and Rh158–161), revealing three patterns of unconventional response inhibition. Differential repair of these genes with either RhCMV-derived or orthologous human CMV (HCMV)-derived sequences (UL128/UL130; UL146/UL147) leads to either of two distinct CD8+ T cell response types – MHC-Ia-restricted-only, or a mix of MHC-II- and MHC-Ia-restricted CD8+ T cells. Response magnitude and functional differentiation are similar to RhCMV 68–1, but neither alternative response type mediated protection against SIV challenge. These findings implicate MHC-E-restricted CD8+ T cell responses as mediators of anti-SIV efficacy and indicate that translation of RhCMV/SIV vector efficacy to humans will likely require deletion of all genes that inhibit these responses from the HCMV/HIV vector.
One sentence summary:
Eight RhCMV gene products control induction of unconventionally restricted CD8+ T cells and RhCMV/SIV vaccine efficacy
INTRODUCTION
CMVs differ from other known viruses in their ability to elicit and indefinitely maintain high frequency CD4+ and CD8+ T cell responses with broad tissue distribution and dominant effector-memory differentiation (1–3). CMV-based vaccine vectors were developed to exploit this immunobiology, addressing the hypothesis that such circulating and tissue-based, effector-differentiated memory T cell responses, directed at heterologous pathogens via vaccination, would provide for a rapid immune effector intercept of nascent infections, prior to effective pathogen immune evasion (4, 5). This concept has been tested in the SIV-RM model of AIDS, where, across multiple studies, 50–60% of rhesus macaques (RMs) vaccinated with strain 68–1 RhCMV/SIV vectors show early stringent control with replication arrest of highly pathogenic SIVmac239, with the vast majority of protected RMs going on to eventually clear the infection (6–8). This “control and clear” pattern of anti-SIV efficacy, which has not been documented with any other vaccine modality, is consistent with our hypothesis that an early intercept of SIV infection by effector-differentiated memory CD8+ T cells might mediate superior viral control. However, unexpectedly, we found that, in contrast to natural RhCMV infection, the epitopes targeted by strain 68–1 RhCMV vector-elicited CD8+ T cells are restricted by major histocompatibility complex (MHC)-II or MHC-E molecules, and not classical polymorphic MHC-Ia, and include universal epitopes (“supertopes”) of both restriction types (9, 10). This unusual finding suggests that an early pathogen interception by pre-established effector-differentiated memory CD8+ T cells may not be the only mechanism underlying the efficacy of these vectors, but that unconventional CD8+ T cell epitope targeting might play a key additional role.
The 68–1 RhCMV strain was extensively passaged on fibroblasts prior to cloning of the viral genome as a bacterial artificial chromosome for vector construction, and during this passage developed genetic mutations similar to fibroblast-adapted HCMV strains, resulting in inactivation of the pentameric complex, a CMV entry receptor for non-fibroblast cell types that is selected against during fibroblast passage (11, 12). In the case of 68–1 RhCMV, pentameric complex inactivation occurred via deletion of the genes Rh157.5 and Rh157.4, RhCMV orthologs of HCMV UL128 and UL130, due to a deletion in the flanking region of an inverted genome segment (11). Repair of Rh157.5 and Rh157.4 gene expression in clone 68–1.2 of RhCMV (13) restored conventional MHC-Ia-restricted-only CD8+ T cell recognition (10), suggesting a role of the pentameric complex in this unique reprogramming of vector immunogenicity. The pentameric complex facilitates CMV entry into endothelial, epithelial, and myeloid-derived cells such as macrophages and dendritic cells which are critical for T cell priming. Inactivation of the pentameric complex reduces infection of these cell types (12), thus it is reasonable to hypothesize that such modulation of viral tropism affects CD8+ T cell priming by altering direct antigen presentation by infected cells.
Here we sought to characterize the genetic and mechanistic basis of 68–1 RhCMV’s unconventional CD8+ T cell response programing, in particular the role of the pentameric complex, and determine whether unconventional MHC restriction is necessary for vaccine efficacy, with the ultimate goal of defining the vector genotype necessary for clinical development of an efficacy-optimized HCMV-vectored HIV vaccine. Surprisingly, our analyses revealed that tropism modulation by pentameric complex abrogation played only a partial role in programming the MHC restriction of vector-elicited CD8+ T cells, with the major regulation mediated by non-pentameric complex-related functions of two pentameric complex components and 6 other viral gene products with chemokine homology that were also affected by the tissue culture-acquired 68–1 genomic rearrangement. When starting from a wildtype RhCMV genome, abrogation of all 8 genes was necessary for vector induction of MHC-E-restricted CD8+ T cells, and RhCMV/SIV vaccine efficacy was only observed when such responses were present. Taken together these results strongly link MHC-E-restricted CD8+ T cell response with RhCMV/SIV efficacy and point to the importance of these response programming mechanisms to the translation of RhCMV/SIV efficacy to an HCMV/HIV vector design suitable for clinical development.
RESULTS
Role of the pentameric complex and vector tropism in response programming
The inversion/deletion event within the genetic region encoding the pentameric complex that occurred during in vitro passage of strain 68–1 RhCMV [corresponding to the ULb’ region of HCMV (11)] disrupted 2 sets of genes at either end of the inverted segment: Rh157.5/Rh157.4 and Rh158–161 (Fig. 1A). Repair of Rh157.5/Rh157.4 expression in strain 68–1.2, leaving the Rh158–161 region unchanged, restored pentameric complex function (fig. S1) and reverted CD8+ T cell immunogenicity to conventional MHC-Ia-restricted responses (Fig. 1B), implicating the pentameric complex in this transition. Moreover, administration of both 68–1 and 68–1.2 RhCMV/SIVgag vectors to RMs resulted in a mixed SIVgag-specific, CD8+ T cell restriction phenotype, including both conventionally (MHC-Ia-) restricted and unconventionally (MHC-E- and MHC-II-) restricted responses (Fig. 1B). This observation suggests that response programming of a given RhCMV vector is regulated independently and likely locally, consistent, as hypothesized above, with programming resulting from differences in vector tropism and direct antigen presentation. However, other mechanisms can apply locally, and it is possible that the response programming effect of Rh157.5/Rh157.4 deletion was mediated by non-pentameric complex-related functions of Rh157.5/Rh157.4 gene products or by modulation of non-Rh157.5/Rh157.4 gene expression.
Figure 1. RhCMV strain differences.
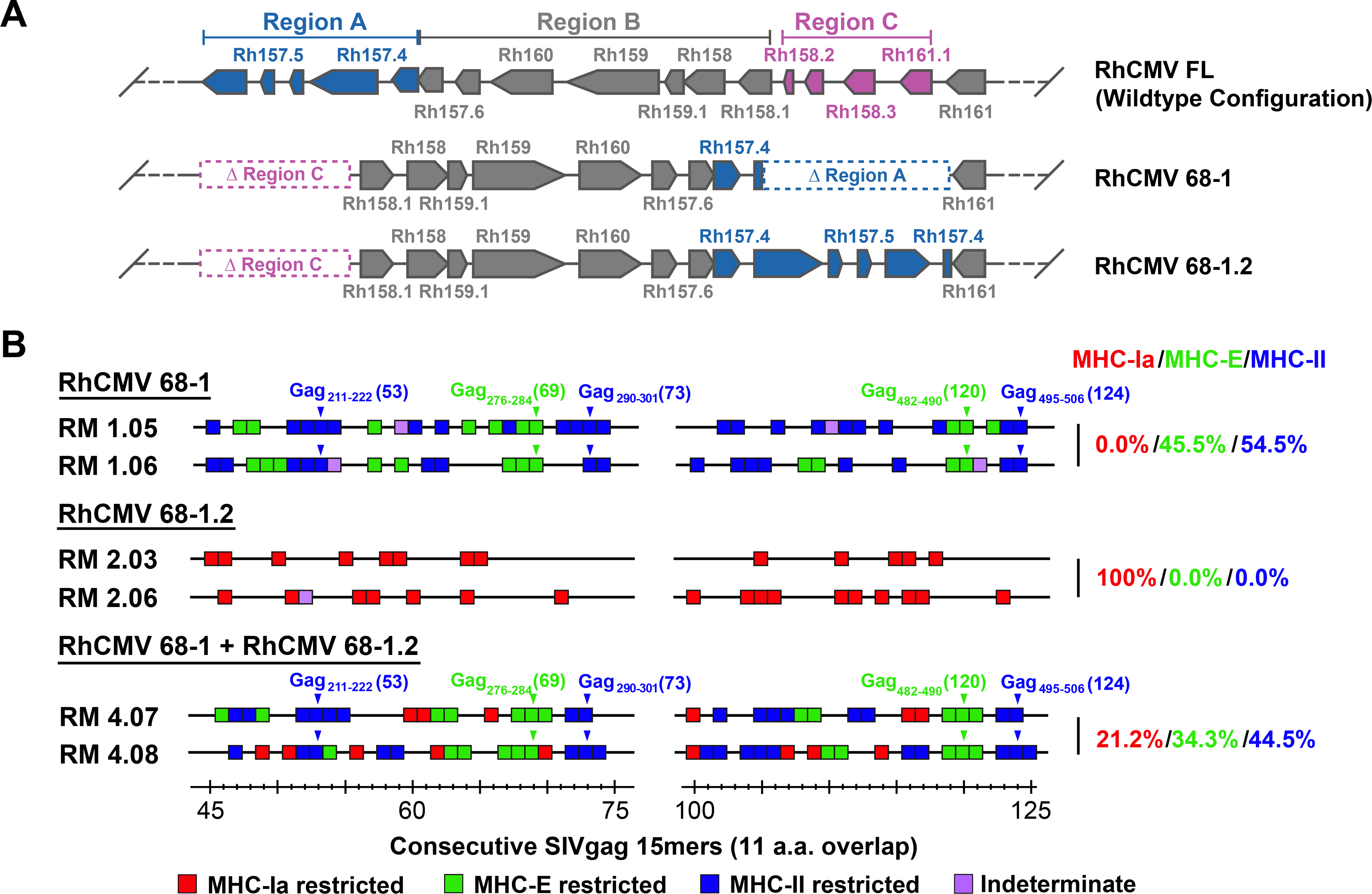
(A) Schematic of the genetic differences between 68–1, 68–1.2, and FL (wildtype) RhCMV in the region of the genome encoding the pentameric complex (analogous to the ULb’ region of HCMV), showing the inversion-deletion event that occurred during in vitro passage of the 68–1 strain and its partial repair in 68–1.2 (with pointed boxes representing distinct exons, the point indicating the 3’ end of the ORF). The genomic segments of inversion and deletion indicated by the letters A, B and C were adapted from Oxford at al. (11). (B) Representative MHC restriction analysis of SIVgag-specific CD8+ T cell responses elicited by strain 68–1 vs. 68–1.2 RhCMV/SIVgag vectors, alone and in combination, assessed by ICS analysis of consecutive 15mer peptides with 11 amino acid overlap comprising the SIVgag protein sequence. Boxes reflect any above-threshold single 15mer response, which were then color-coded based on MHC restriction type analysis (see Methods). In addition, responsiveness to the designated MHC-E and MHC-II optimal supertope peptides are indicated by green and blue arrowheads, respectively. The figure shows only the SIVgag regions from amino acid 45–75 and amino acid 100–125 (the regions including the supertopes) of 2 representative RMs, but complete epitope analysis results are presented in table S1. These overall data were used to calculate the % of the total MHC restriction-assignable SIVgag epitopes that were MHC-Ia-, MHC-E-, and MHC-II-restricted, shown at right in red/green/blue, respectively.
To address this issue, we constructed and tested a series of pentameric complex-modified 68–1.2 RhCMV/gag vectors, asking whether these modifications resulted in changes in the epitope targeting of vaccine-induced SIVgag-specific CD8+ T cell responses (Fig. 2A). First, we demonstrated that incorporation of stop mutations into the Rh157.5 and Rh157.4 genes converted CD8+ T cell responses from targeting MHC-Ia-restricted epitopes (wildtype and 68–1.2 immunotype) to MHC-II- and MHC-E-restricted epitopes (68–1 immunotype), which demonstrates that protein expression, not alteration of genomic configuration by the strain 68–1 inversion-deletion, is responsible for response programming (Fig. 2B). We then tested 68–1.2 vectors with genetic modifications that abrogate pentameric complex function without complete abrogation of both Rh157.5 or Rh157.4 expression, including deletion (Δ) of Rh157.5 alone, Rh157.4 alone, and functional inactivation of the pentameric complex by alanine-substitution mutation of two charged cluster (CC) regions of the UL128 ortholog Rh157.5 (14) (fig. S1). Surprisingly, the CD8+ T cell responses elicited by these 3 vectors did not recapitulate either the 68–1 or 68–1.2 CD8+ T cell response types, but instead resulted in a different response type characterized by mixed CD8+ T cell targeting of both MHC-Ia- and MHC-II-restricted epitopes, the latter notably without supertopes (Fig. 2B).
Figure 2. Role of the pentameric complex in RhCMV vector CD8+ T cell response programming.

(A) Schematic of the genetic configuration of 68–1.2 RhCMV/SIVgag vectors with modified Rh157.5 and Rh157.4 genes. (B) Representative MHC restriction analysis of SIVgag-specific CD8+ T cell responses elicited by the designated Rh157.5 and/or Rh157.4 gene-modified RhCMV vectors, as described in Fig. 1 legend. Overall epitope analysis results are shown in table S1.
To determine whether this new response type was due to alteration of vector tropism, we engineered a pentameric complex-independent tropism restriction into the 68–1.2 vector by incorporating target sites for the myeloid-specific microRNA (miR)-142–3p into the 3’ untranslated region (UTR) of the viral Rh108 and Rh156 genes (orthologs to HCMV UL79 and UL122, respectively) to exploit miR-mediated gene expression suppression to change vector tropism (15, 16). By suppressing expression of these essential viral genes, this miR target site insertion impedes the vector’s ability to productively infect miR-142–3p-expressing myeloid cells, thus simulating one possible effect of pentameric complex inactivation relevant to CD8+ T cell response priming – decreased infection efficiency of myeloid-derived antigen presenting cells (Figs. 3A–C). Notably, this miR-142–3p-restricted 68–1.2 vector elicited a very similar pattern of CD8+ T cell epitope targeting (MHC-Ia- + MHC-II-restriction, without supertopes) as the ΔRh157.5, ΔRh157.4, and CC1+CC2 mutant-Rh157.5 68–1.2 vectors (Figs. 3D), except that the MHC-II-restricted CD8+ T cell priming of the former vector was more efficient (88% of total epitopes) than the latter vectors (57% of total epitopes; p<0.001 by binomial exact test). Taken together, these data indicate that pentameric complex inactivation alone is not responsible for the full development of the exclusively unconventionally targeted CD8+ T cell responses manifested by 68–1 vectors, but does provide for elicitation of non-supertope, MHC-II-restricted CD8+ T cells, an effect that is likely attributable to reduced infection of myeloid-derived cells driven by pentameric complex inactivation. Additionally, these data indicate that both the Rh157.5 and Rh157.4 gene products have a pentameric complex-independent activity that interferes with elicitation of CD8+ T cell responses to all MHC-E-restricted epitopes and to MHC-II-restricted supertopes, potentially related to their N-terminal homology with host chemokines (fig. S2).
Figure 3. Role of myeloid cell tropism in RhCMV vector CD8+ T cell response programming.
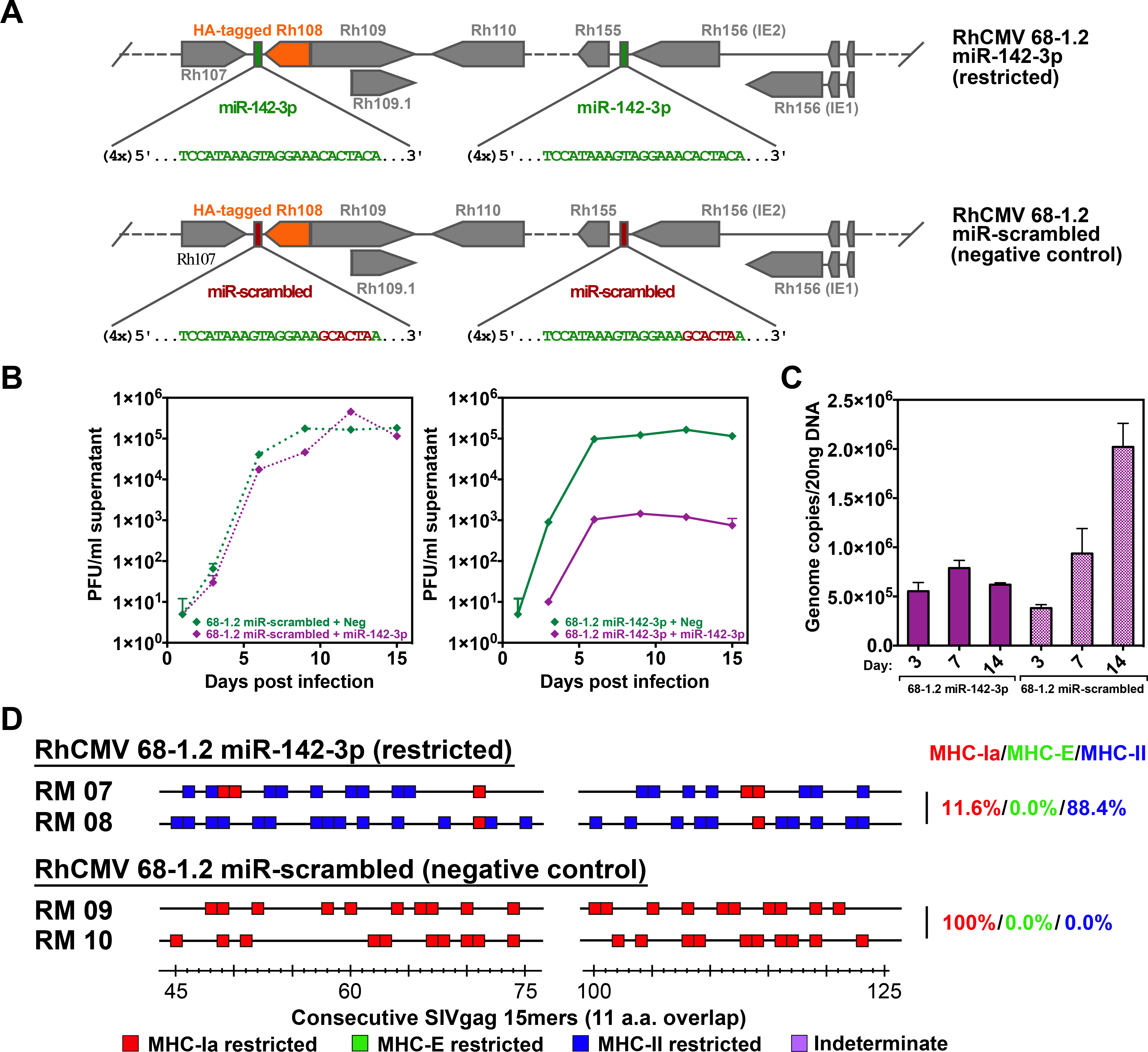
(A) Schematic of the miR-142–3p target sites inserted downstream of both Rh156 and Rh108 (homologs of HCMV IE2 and UL79), which are essential for viral replication. Green sequences represent insertion of four miR-142–3p recognition sites. Red sequences indicate scrambled nucleotides of the miR-142–3p sequence used to create a control vector. Orange boxes represent the Rh108 ORF with a hemagglutinin (HA) epitope-tag. (B) Growth analysis of the 68–1.2 control (scrambled sequence) RhCMV vector (left) vs. 68–1.2 miR-142–3p RhCMV vector (right) in the presence or absence of miR-142–3p expression. Primary rhesus fibroblasts were transfected with negative control or miR-142–3p mimic and infected 24 hours later with the 68–1.2 miR-142–3p or control RhCMV vectors at an MOI of 0.01. Cell supernatants were harvested at the indicated timepoints and titered on primary rhesus fibroblasts. Results are representative of 2 independent experiments. (C) Analysis of 68–1.2 miR-142–3p vs control RhCMV vector replication in primary macrophages. Macrophages were differentiated in vitro from peripheral blood of 3 RMs and infected with 68–1.2 miR-142–3p or scrambled RhCMV at MOI = 5. At the indicated times post-infection viral DNA was isolated and total DNA copies were determined using qPCR (mean + SEM of 3 independent experiments shown). (D) Representative MHC restriction analysis of SIVgag-specific CD8+ T cell responses elicited by 68–1.2 miR-142–3p vs. scrambled RhCMV/SIVgag vectors, as described in Fig. 1. Overall epitope analysis results are shown in table S1.
Identification of non-pentameric complex-related modulators of response programming
We next asked the question of whether abrogating both the pentameric complex- and non-pentameric complex-related immune programming activities of Rh157.5 and Rh157.4 was sufficient to convert a primary isolate-like RhCMV, with conventional MHC-Ia-restricted CD8+ T cell targeting, to a 68–1-like vector with exclusively MHC-II- and MHC-E-restricted CD8+ T cell responses. Using sequence information from primary RhCMV isolates, including the original 68–1 isolate (17), we reconstructed and produced a full length RhCMV clone (RhCMV FL) in which all suspected deletions, inversions, frameshifts and premature termination codons affecting viral open reading frames were repaired and the Rh13.1 open reading frame (ORF) was replaced by an SIVgag insert (Fig. 4A) (18). RhCMV FL has robust in vivo spread (18), and as expected for an essentially wildtype genetic configuration (9), elicits MHC-Ia-restricted, CD8+ T cell responses (Fig. 4B). To determine whether inactivation of Rh157.5 and Rh157.4 was sufficient for unconventional CD8+ T cell response programming, we deleted both genes from RhCMV FL. Unexpectedly, the resulting recombinant vector (RhCMV FL ΔRh157.5/Rh157.4) elicited CD8+ T cell responses showing the same conventional MHC-Ia-restriction as the parent RhCMV FL vector (Fig, 4B), suggesting that genes outside the Rh157 region additionally interfere with the induction of unconventionally restricted CD8+ T cell responses. Since the inversion/deletion event in strain 68–1 RhCMV also affected the viral CXC chemokine-like proteins with similarity to CXCL-1 encoded by the Rh158–161 genes, which are orthologous to HCMV UL146 and UL147 (11) (Fig. 1A; fig. S2), we constructed RhCMV FL vectors with deletions of Rh158-Rh161, either alone or together with deletion of Rh157.5 and Rh157.4 (Fig. 4A). Deletion of the Rh158-Rh161 region alone (RhCMV FL ΔRh158–161) also did not change the MHC-Ia restriction of RhCMV FL elicited CD8+ T cell responses; however, deletion of both Rh157.5/Rh157.4 and Rh158-Rh161 [double-deleted (dd) RhCMV FL] did result in the same MHC-II- + MHC-E-restricted CD8+ T cell targeting (with supertopes) as the original 68–1 RhCMV vector (Fig. 4B), demonstrating that deletion of both these distinct regions from a typical circulating RhCMV strain is required for unconventional response programming.
Figure 4. Regulation of RhCMV vector CD8+ T cell response programming by non-pentameric complex-related genes in the same RhCMV genomic region.
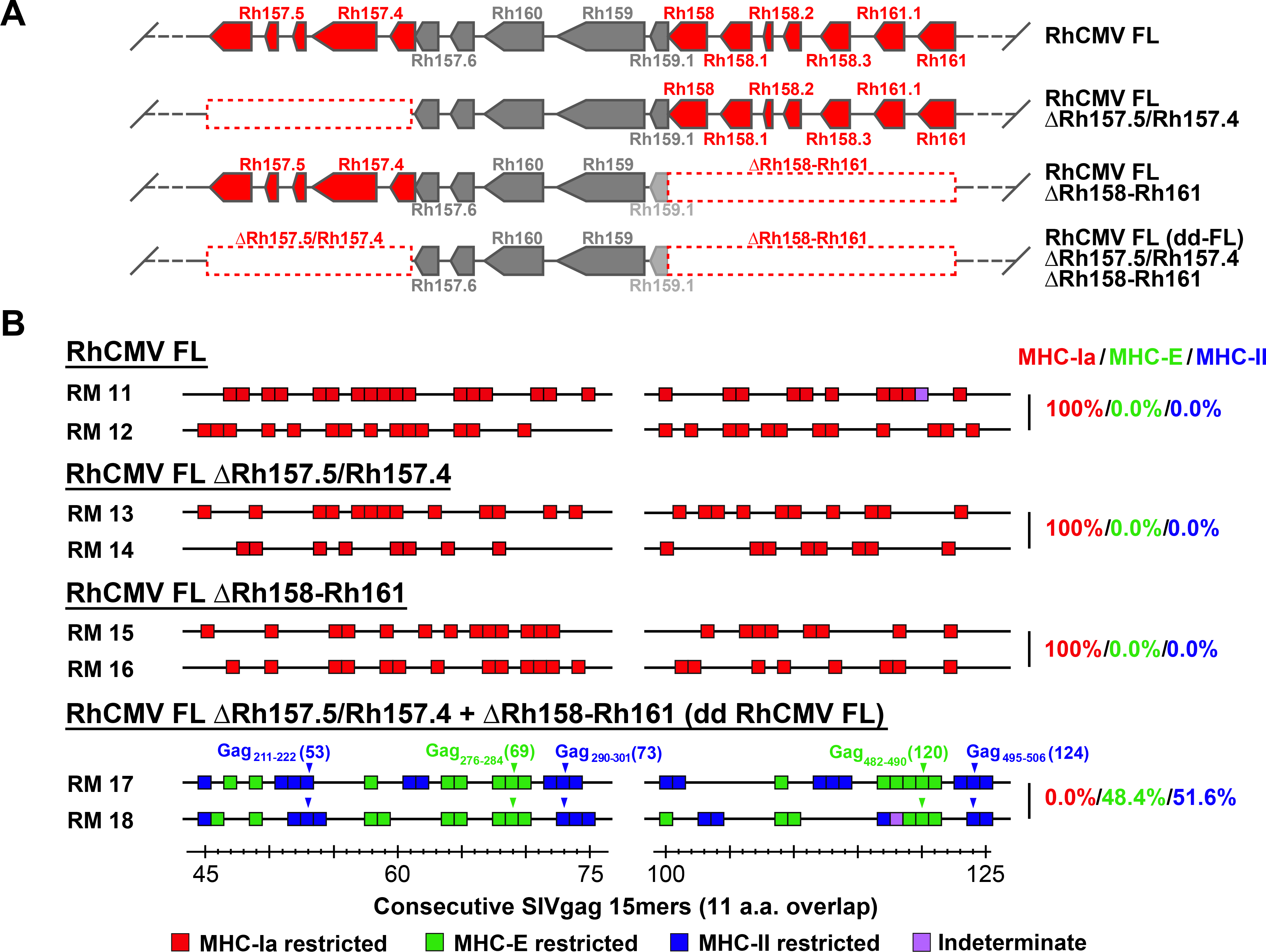
(A) Schematic representation of the configuration of FL-RhCMV vector in the genetic region of the 68–1 inversion/deletion, compared to modified FL-RhCMV vectors in which the gene segments involved in each end of the 68–1 inversion/deletion (Rh157.5/Rh157.4 and Rh158–162) are separately deleted (single-deleted vectors) or both deleted (e.g., double-deleted or dd vector). (B) Representative MHC restriction analysis of SIVgag-specific CD8+ T cell responses elicited by FL, double-deleted and both single-deleted RhCMV/SIVgag vectors, as described in Fig. 1. Overall epitope analysis results are shown in table S1.
To determine the effect of each of the eight genes missing from dd RhCMV FL (Rh157.5, Rh157.4, Rh158, Rh158.1, Rh158.2, Rh158.3, Rh161.1, Rh161) on CD8+ T cell response programming, we engineered their individual expression in the Rh161 locus of the dd RhCMV FL vector (fig. S3). We also evaluated adding back dual expression of the pentameric complex components Rh157.5 and Rh157.4 to this vector. Adding back expression of Rh157.5 alone or Rh157.4 alone to dd RhCMV FL resulted in CD8+ T cell targeting of both MHC-Ia + MHC-II-restricted epitopes, whereas adding expression of both resulted in MHC-Ia-only epitope targeting (Fig. 5A), independently confirming the findings reported above using the 68–1.2, 68–1.2 ΔRh157.5, and 68–1.2 ΔRh157.4 RhCMV vectors. In contrast, adding back expression of each of the six Rh158–161 genes individually to dd RhCMV FL resulted in reversion to the MHC-Ia-only epitope targeting pattern (Fig. 5B). Although only three of these genes are completely deleted in 68–1 RhCMV (Fig. 1A), qPCR analysis indicates that the remaining three transcripts are expressed at substantially lower levels in 68–1 compared to RhCMV FL, presumably as a consequence of the inversion event resulting in altered transcriptional regulation or ORF truncation (fig. S4). Taken together, these data indicate that wildtype RhCMV encodes highly redundant mechanisms to prevent elicitation of unconventionally restricted CD8+ T cell responses, particularly MHC-E-restricted CD8+ T cells. These include eight chemokine-related gene products (fig. S2), six of which (Rh158–161) inhibit both MHC-E and MHC-II-restricted CD8+ T cell priming, and two of which (Rh157.5 and Rh157.4) individually inhibit MHC-E- and MHC-II supertope CD8+ T cell priming and that when expressed together with Rh157.6, Rh104/gH, and Rh147/gL (orthologs of HCMV UL131A, UL75 and UL115, respectively) create the pentameric complex which additionally inhibits MHC-II non-supertope CD8+ T cell priming by tropism modulation.
Figure 5. Identification of CMV-encoded inhibitors of unconventionally restricted CD8+ T cell response priming.
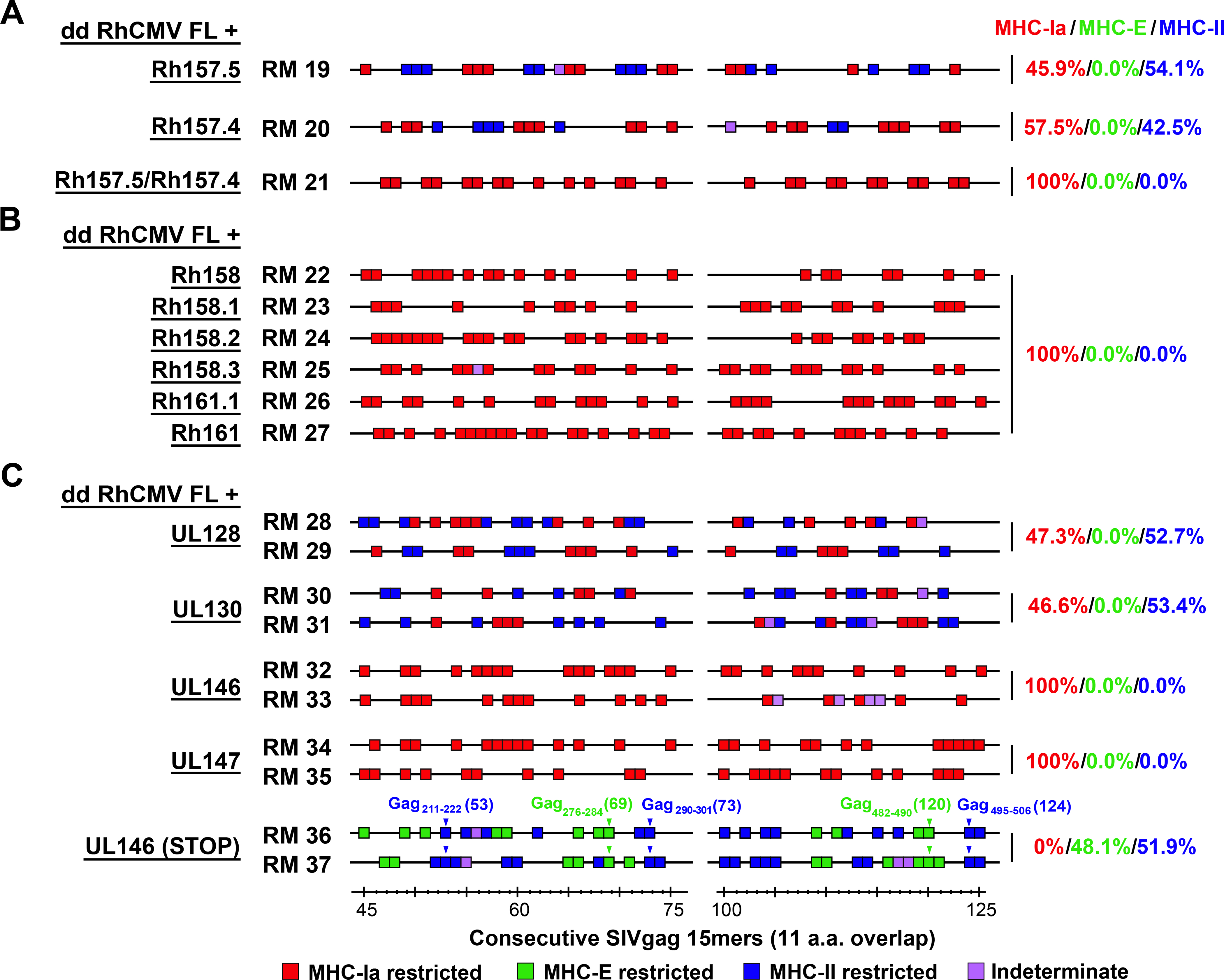
(A,B) Representative MHC restriction analysis of SIV-specific CD8+ T cell responses elicited by double-deleted (dd) FL-RhCMV/SIVgag vectors which were engineered to express Rh157.5 and Rh157.4 alone, Rh157.5 + Rh157.4, or each of the Rh158-Rh161 alone (see fig. S3A–C for genetic configurations), as described in Fig. 1. (C) Representative MHC restriction analysis of SIV-specific CD8+ T cell responses elicited by double-deleted FL-RhCMV/SIVgag vectors which are engineered to individually express UL128, UL130, UL146 and UL147, the HCMV orthologs of the Rh157.5-Rh157.4 and Rh158-Rh161 genes (see fig. S3D for genetic configurations). Note that MHC restriction analysis of the parent dd FL-RhCMV/SIVgag vector-elicited CD8+ T cell response is shown in Fig. 4B (bottom panel). For A-C, overall epitope analysis results are shown in table S1.
Conservation of response programming function by HCMV orthologs
Since our ultimate aim is to develop an HCMV-based vaccine for human use (19), we next asked whether the functions that are inhibitory to unconventional response generation are conserved in the HCMV orthologs of these gene products. As described for the RhCMV genes above, we engineered dd RhCMV FL vectors to individually express HCMV UL128, UL130, UL146, and UL147. Strikingly, these HCMV orthologs, though considerably sequence diverse from their RhCMV counterparts (fig. S2), showed the same pattern of inhibition of unconventional CD8+ T cell priming exhibited by the corresponding RhCMV gene products (Fig. 5C). Of note, stop mutations abrogated the inhibitory activity of HCMV UL146, consistent with a function of the UL146 protein being responsible for the observed inhibitory activity (Fig. 5C). This remarkable conservation of function strongly suggests that unconventional CD8+ T cell response inhibition is also a priority for HCMV infection in humans, and that deletion of at least these 4 genes (UL128, UL130, UL146, UL147) from HCMV would be required to elicit unconventionally restricted CD8+ T cells. These observations likely explain the lack of unconventionally restricted CD8+ T cell responses in humans vaccinated with a pentameric complex negative, but UL130-, UL146- and UL147-expressing Towne/Toledo chimeric HCMV (20).
Immunogenicity of response-programmed RhCMV vectors with different immunotypes
The ability of 68–1 RhCMV to elicit unconventionally restricted CD8+ T cell responses resulted from a complex genetic event that precisely abrogated the activity of 8 genes in two non-contiguous, multi-gene regions. These data, however, do not indicate whether unconventional CD8+ T cell response priming, though present in the 68–1 RhCMV/SIV vectors with demonstrated efficacy (6–8), is required for, or associated with efficacy. To address this question, we initiated a vaccine-challenge study to compare the T cell immunogenicity and efficacy of RhCMV/SIV vaccines (vector sets comprised of 3 vectors of the same genetic configuration, individually expressing SIVgag, retanef, and 5’-pol) that elicit CD8+ T cell responses of the three identified CD8+ T cell targeting response types: 1) MHC-E + MHC-II, with supertopes (68–1), 2) MHC-Ia-only (68–1.2), and 3) MHC-Ia + MHC-II, no supertopes (68–1.2 ΔRh157.5) (Fig. 6A). To ascertain whether a vaccine regimen eliciting CD8+ T cell responses targeting a broad combination of all types of CD8+ T cell epitopes (MHC-Ia- and MHC-E- and MHC-II-restricted, including supertopes) was possibly more efficacious than individual vector sets, we also studied an additional cohort of RMs that were vaccinated with both the 68–1 and 68–1.2 vector sets.
Figure 6. Immunogenicity of differentially CD8+ T cell response-programmed RhCMV vectors.
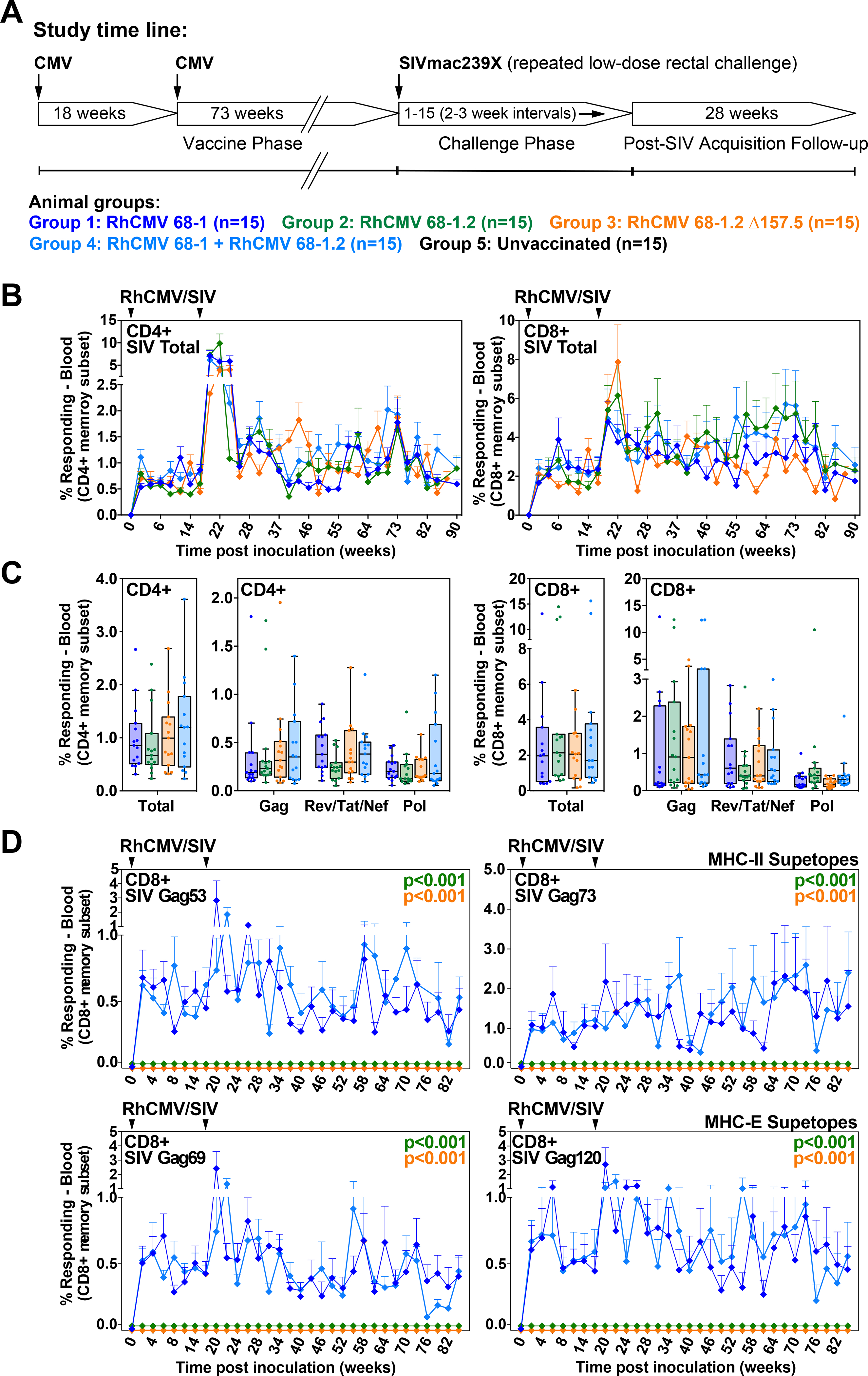
(A) Protocol for the comparison of the immunogenicity and efficacy of 68–1, 68–1.2, and ΔRh157.5 68–1.2 RhCMV/SIV vector sets (each set comprised of 3 vectors individually expressing SIV Gag, Rev/Tat/Nef, and 5’-Pol inserts), and the combination of 68–1 and 68–1.2 vector sets (n = 15 RMs per group). (B,C) Longitudinal and plateau-phase analysis of the vaccine-elicited SIV Gag-, Rev/Tat/Nef-, and 5’-Pol-specific CD4+ and CD8+ T cell responses in peripheral blood of the RMs vaccinated with the designated vector sets. In B, the background-subtracted frequencies of cells producing TNF and/or IFN-γ by flow cytometric ICS assay to peptide mixes comprising each of the SIV inserts within the memory CD4+ or CD8+ T cell subsets were summed for overall responses with the figure showing the mean (+ SEM) of these overall responses at each time point (area-under-the-curve was used to quantitatively compare longitudinal response profiles). In C, boxplots compare the total and individual SIV insert-specific CD4+ and CD8+ T cell response frequencies between the vaccine groups during the vaccine phase plateau (each data point is the mean of response frequencies in all samples from weeks 61–90 post-first vaccination). (D) Longitudinal analysis of the vaccine-elicited CD8+ T cell responses to MHC-E-restricted [Gag276–284 (69) and Gag482–490 (120)] and MHC-II-restricted [Gag211–222 (53) and Gag290–301 (73)] SIVgag supertopes in peripheral blood of each vaccine group by ICS assay. Wilcoxon p-values for comparison of all response parameters shown in panels B-D for the 68–1-only vaccine to all other vaccines (which are individually designated by the color code shown in panel A) are shown where significant (adjusted for multiple comparisons in panels C and D).
Longitudinal analysis of SIV-specific CD4+ and CD8+ T cell responses from these RM cohorts (n = 15 each) showed that the 68–1.2, 68–1.2 ΔRh157.5, and 68–1+68–1.2 RhCMV/SIV vaccines manifested equivalent or greater SIV insert-specific T cell response magnitudes and similar response longevity compared to the 68–1 reference vaccine in both blood and bronchoalveolar lavage fluid (BAL, representing an effector site) (Figs. 6B,C; fig. S5). Epitope restriction analysis revealed the expected MHC restriction patterns for all four RhCMV/SIV vaccines, including supertope recognition only in the RMs vaccinated with 68–1 vectors (Fig. 6D; fig. S6). Importantly, in all vaccine groups, the plateau phase SIVgag-specific CD4+ and CD8+ T cells manifested the characteristic effector-memory bias of RhCMV vector-elicited T cell responses, as well as broadly similar cytokine profiles, indicating that the genetic differences between these vectors did not impact the unique characteristics of RhCMV-induced T cell immunogenicity (Figs. 7A,B). We also compared the magnitude of SIVgag-specific CD4+ and CD8+ T cell responses in tissues of RMs vaccinated with 68–1 vs. 68–1.2 RhCMV/gag vectors studied at necropsy, revealing comparable responses across lymphoid and non-lymphoid tissues (fig. S7). Taken together, these data indicate that these vaccines elicit SIV-specific CD8+ T cells with different epitope targeting, but the magnitude, longevity, differentiation and tissue distribution of these CD8+ T cells, and of the SIV-specific CD4+ T cells, are broadly similar, allowing assessment of the contribution of unconventional response targeting to protective efficacy.
Figure 7. Differentiation of CD8+ T cells elicited by differentially response-programmed RhCMV vectors.
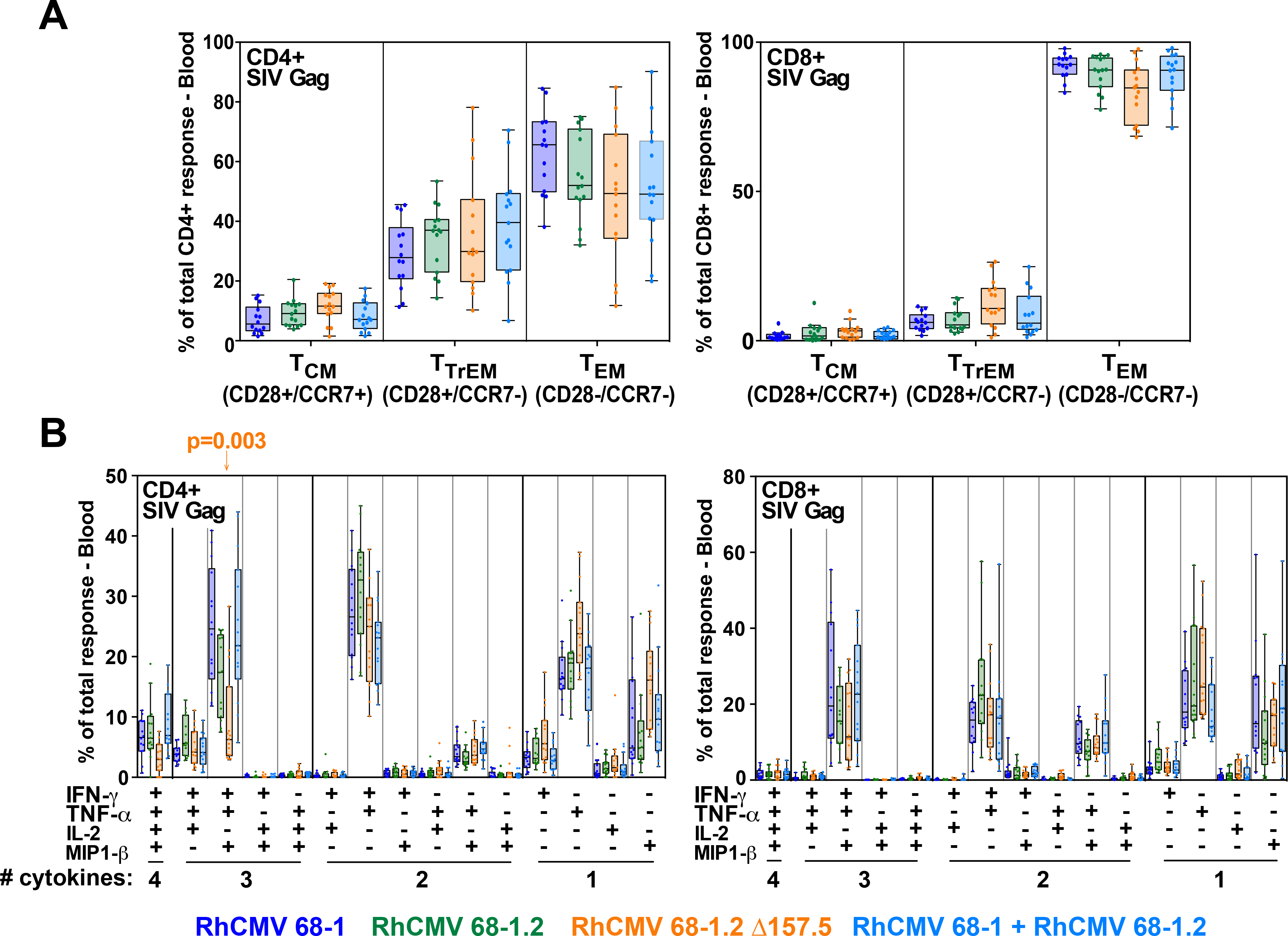
(A) Boxplots compare the memory differentiation phenotype of the vaccine-elicited CD4+ and CD8+ memory T cells in peripheral blood of the same RM cohorts reported in Fig. 6 responding to overall SIV Gag 15mer peptide mix with TNF and/or IFN-γ production during the vaccine phase plateau (24–85 weeks post-first vaccination). Memory differentiation state was based on CD28 and CCR7 expression, delineating central memory (TCM), transitional effector-memory (TTrEM), and effector-memory (TEM), as designated. (B) Boxplots compare the frequency of vaccine-elicited CD4+ and CD8+ memory T cells in peripheral blood responding to the overall SIV Gag 15mer peptide mix with TNF, IFN-γ, IL-2, and MIP-1β production, alone and in all combinations, in the same samples as panel A. Wilcoxon p-values for comparison of all response parameters shown in panels A and B for the 68–1-only vaccine to all other vaccines are shown where significant (adjusted for multiple comparisons).
Efficacy of response-programmed RhCMV vectors with different immunotypes
Starting ninety-one weeks after first vaccination, all vaccinated RM cohorts, and an unvaccinated RM cohort (n = 15), were subjected to repeated, limiting dose, intrarectal SIVmac239 challenge, with take of infection monitored by de novo induction of SIVvif-specific T cell responses (SIVvif is not present in the vaccine inserts and thus such responses are derived from the SIV infection) and plasma viral load (pvl), as previously described (6–8). In this well-characterized challenge model, unvaccinated controls and non-protected RMs manifest typical peak and plateau-phase SIVmac239 pvl in concert with development of SIVvif-specific T cells, whereas protected RMs show development of SIVvif-specific responses in the absence of plasma viremia or with non-sustained pvl blips, and manifest cell-associated SIV DNA and/or RNA in tissues consistent with replication arrest (6–8). As expected, all unvaccinated RMs showed typical progressive infection, whereas 8 of 15 (53%) of the 68–1 vector vaccinated RMs showed protection (Figs. 8A,B). However, none of the RMs vaccinated with the 68–1.2 vectors or 68–1.2 ΔRh157.5 vectors manifested protection, but notably, protection was still observed in 6 of the 15 (40%) RMs vaccinated with both 68–1 and 68–1.2 vectors (p = not significant relative to 68–1) (Figs. 8C–E). Protected RMs in both the 68–1 and 68–1 + 68–1.2 RhCMV/SIV vector vaccinated groups manifested cell-associated SIV DNA and/or RNA in bone marrow (BM) and lymph node (LN), confirming take of infection, and given the absence of measurable viremia, replication arrest (Fig. 8F). Thus, although the RhCMV vectors programmed for the alternative CD8+ T cell targeting response types (MHC-Ia- ± MHC-II-restricted) elicited CD8+ T cell responses with similar magnitude and qualitative characteristics as 68–1 vectors, these responses were unable to provide protection against SIV challenge.
Figure 8. Efficacy of differentially programmed RhCMV vectors.
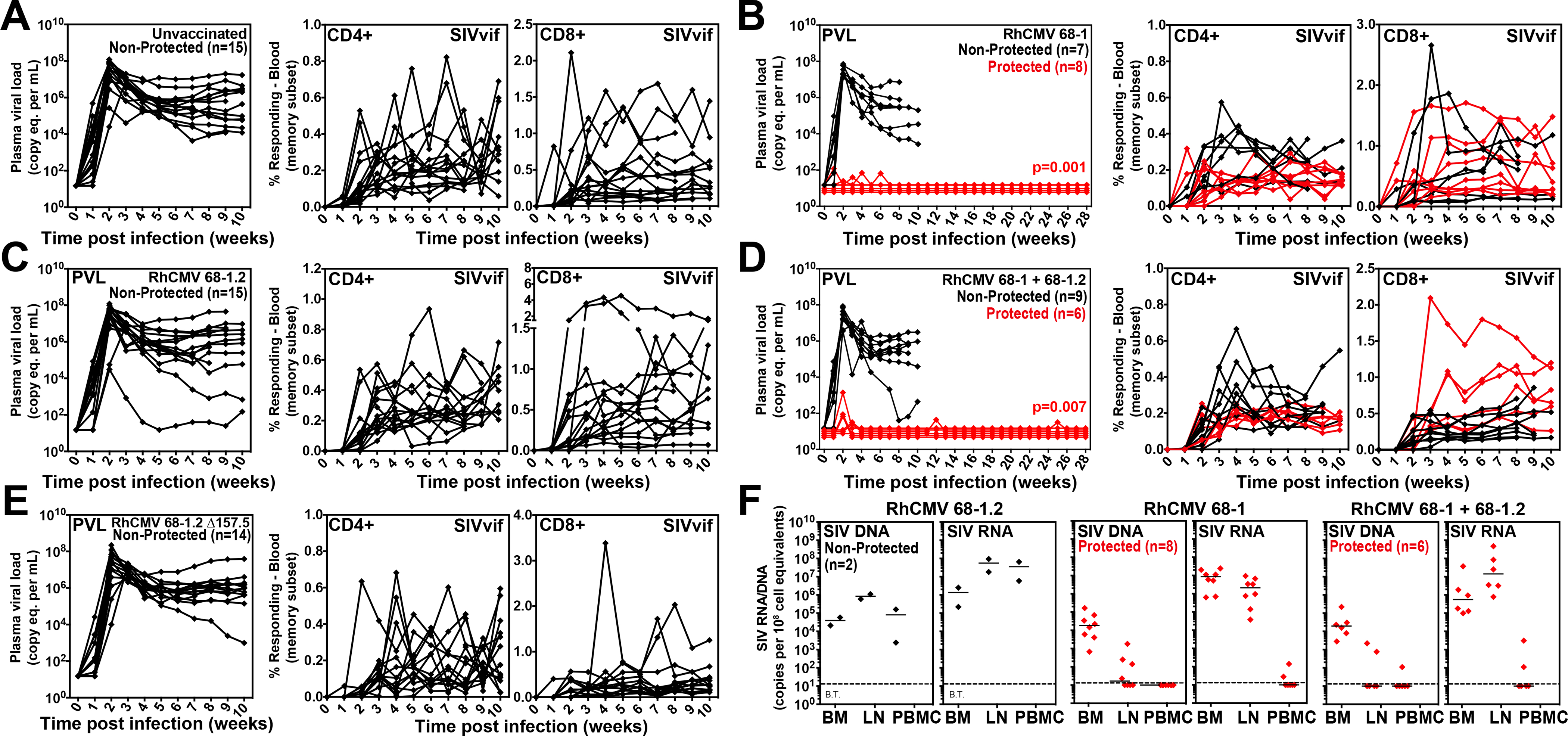
(A-E) Assessment of the outcome of SIV infection after repeated, limiting dose SIVmac239 challenge (see Fig. 6A) of the designated vaccine groups by longitudinal analysis of plasma viral load (left panels) and de novo development of SIVvif-specific CD4+ (middle panels) and CD8+ (right panels) T cell responses. RMs were challenged until the onset of any sustained above-threshold SIVvif-specific T cell response, with the SIV dose administered 2 or 3 weeks prior to the initial response detection considered the infecting challenge (week 0). The n in each panel reflects the total number of RMs with such documented take of SIV infection during the challenge period. RMs with sustained viremia were considered non-protected (black); RMs with no or transient viremia but demonstrating sustained above-threshold SIVvif-specific T cell responses were considered protected (red) (6–8). Binomial exact p-values are shown where the proportion of protected RMs in a vaccine group differs significantly from the unvaccinated group. (F) Bone marrow (BM), peripheral lymph node (LN) and peripheral blood mononuclear cell (PBMC) samples from all vaccine-protected RMs (red) and 2 non-protected RMs (black) for comparison (left panel), collected from between day 28 and 56 post-SIV infection, were analyzed by nested, quantitative PCR/RT-PCR for cell-associated SIV DNA and RNA. The dotted line indicates the threshold of detection (B.T. = below threshold) with data points below this line reflecting no positive reactions across all replicates.
Discussion
These data demonstrate that the deletion of 8 genes – Rh157.5/Rh157.4 and Rh158–161 – from an intact, circulating isolate-like RhCMV genetic configuration is both necessary and sufficient for converting RhCMV vector-elicited CD8+ T cell responses from targeting conventional, MHC-Ia-restricted epitopes to targeting unconventional, MHC-E- or MHC-II-restricted epitopes, with deletion or functional inactivation of these 8 genes by a chance genetic rearrangement accounting for the unconventional CD8+ T cell priming originally observed in RMs vaccinated with 68–1 RhCMV vectors (9, 10). Three activity patterns were identified, including pentameric complex (tropism)-dependent and -independent inhibition of MHC-E and MHC-II supertope priming by Rh157.5/Rh157.4, and pan-unconventional response inhibition by Rh158–161. It is notable that these activities are all inhibitory, in particular having in common inhibition of MHC-E-restricted CD8+ T cell priming. Given that conventional MHC-Ia-restricted CD8+ T cells are the default response in wildtype RhCMV and essentially all other virus-induced CD8+ T cell responses, these data raise the question of why RhCMV encodes multiple redundant inhibitors of what can only be considered an unnatural response.
In this regard, CMV is the epitome of sophisticated host modulation and immune evasion by a virus (21), and it is likely that the answer to this question lies in this host modulation/immune evasion biology. Indeed, in a companion manuscript (22), we demonstrate that 68–1 RhCMV vectors deleted for Rh67, the gene product which is responsible for intracellular transport of MHC-E in RhCMV-infected cells (UL40 in HCMV), specifically fail to prime MHC-E-restricted CD8+ T cells, leaving CD8+ T cell responses which are entirely MHC-II-restricted. Thus, MHC-E-restricted CD8+ T cell responses appear to be one consequence of intracellular transport with enhanced surface expression of MHC-E mediated by the Rh67 gene product. Since MHC-E expression on the surface of infected cells sends a dominant inhibitory signal through NKG2A receptors of NK cells, thereby down-modulating NK cell anti-viral activity, maintaining high MHC-E expression on the infected cell surface is thought to be an important CMV strategy for NK cell evasion, especially since CMV efficiently down-regulates MHC-Ia, increasing susceptibility to NK cell-mediated lysis (21, 23–27). However, this NK cell evasion benefit would appear to come at the price of enhanced (MHC-E-mediated) CD8+ T cell recognition of CMV-infected (MHC-Ehigh) cells, especially since efficient MHC-Ia down-regulation by different CMV immune evasins precludes efficient recognition of infected cells by classical CD8+ T cells (22). Thus, the 8 gene inhibitory system described here likely reflects CMV adaptation to this deleterious side-effect of MHC-E upregulation, allowing the virus to evade NK cells without the hindrance of enhanced CD8+ T cell recognition mediated by MHC-E-restricted responses. The basis for MHC-II-restricted CD8+ T cell priming by RhCMV vectors remains to be determined but given the potential for such responses to mediate CD8+ T cell recognition of infected MHC-II+ cells such as monocyte/macrophages and dendritic cells, it may not be surprising that CMV co-evolved mechanisms to prevent priming of these responses as well.
Taken together, these data demonstrate that RhCMV has evolved the ability to control the nature of its own recognition by CD8+ T cells. To our knowledge, given the capabilities of this virus, this may the only virus-based vaccine vector system at this time that is genetically programmable with respect to CD8+ T cell epitope targeting. In the companion manuscript we document that exclusively MHC-II-restricted SIV-specific CD8+ T cell responses, with MHC-II supertope recognition, elicited by the Rh67-deleted 68–1 RhCMV/SIV vectors are unable to provide protection against SIV challenge (22). Thus, neither MHC-Ia-restricted CD8+ T cell responses, nor MHC-II-restricted CD8+ T cell responses (with or without supertopes) are able to mediate protection against SIV challenge, even though these responses are of similar magnitude and differentiation than the protective responses elicited by 68–1 RhCMV/SIV vectors. CD4+ T cell responses are also comparable across the differentially programmed RhCMV/SIV vaccines and antibody responses are largely absent in 68–1 RhCMV/SIV vaccinated RMs (6–8, 28), thus the presence of MHC-E-restricted CD8+ T cell responses are the only clear difference in adaptive (SIV-specific) immunity between efficacious and non-efficacious RhCMV/SIV vectors. Since SIV inserts (and thus SIV-specific immunity) are demonstrably required for efficacy (7), these data strongly suggest that the MHC-E-restricted CD8+ T cell response is the crucial adaptive immune component of RhCMV/SIV vector immunogenicity that mediates “control and clear” protection. The specific immune functions mediating this efficacy in vivo remain to be elucidated, especially delineation of why MHC-E-restricted CD8+ T cell responses are uniquely efficacious. Nonetheless, these data indicate that not only is epitope-targeting programmability a characteristic of RhCMV vectors, the ability of this programming to produce MHC-E-restricted CD8+ T cell responses appears to be necessary for efficacy against SIV.
One important caveat of these findings is that while RhCMV infection of RMs is biologically analogous to HCMV infection of humans (29), these are different infectious agents adapted to different hosts, and it therefore remains uncertain whether the biology underlying the efficacy of RhCMV/SIV vectors in RMs will translate to humans. However, the finding that the HCMV orthologs of the RhCMV genes that mediate CD8+ T cell response programming, including UL128/UL130, UL146/UL147 and UL40 (22), recapitulate the response programming function of their RhCMV counterparts when used to replace these counterparts in RhCMV suggests the conservation of CD8+ T cell response programming in HCMV. Moreover, a recent report has identified MHC-E-restricted, HIV-specific CD8+ T cells in humans and demonstrated that these T cells can recognize HIV-infected cells in vitro and suppress HIV infection (30). Thus, the available data support the possibility that a UL128/UL130 + UL146/147-deleted HCMV/HIV modeled after the dd RhCMV/SIV vectors described here will elicit functional MHC-E restricted, HIV-specific CD8+ T cell responses in humans, an hypothesis that is currently being tested in a Phase 1 clinical trial (31). Since MHC-E is ubiquitously expressed, non-polymorphic and often upregulated in disease (23, 24, 32), an MHC-E-restricted CD8+ T cell response-inducing HCMV-based vaccine vector may also be applicable to other pathogens or malignancies.
MATERIALS AND METHODS
Study Design
This study had two major objectives. First, we sought to determine the molecular genetic basis of unconventional CD8+ T cell response programming by strain 68–1 RhCMV vectors, knowing that naturally occurring RhCMVs elicit conventional MHC-Ia-restricted CD8+ T cells, whereas the genetically distinct 68–1 RhCMV elicits a mixture of MHC-II- and MHC-E-restricted CD8+ T cells (9, 10). We had previously identified the complex inversion/deletion in the genomic region of RhCMV orthologous to the ULb’ region of HCMV as the likely basis of this immune programming (Fig. 1A), but the specific contribution of the affected genes was unclear, as well as the spectrum of immunologic phenotypes (response types) that might arise from different configurations of this gene region. Once the genetic determinants of unconventional CD8+ T cell response priming were identified and the spectrum of different response types established, our second major objective was to both compare the overall immunogenicity of all distinct RhCMV response types and to determine whether the RhCMV vectors eliciting these alternative response types were able to mediate the unique replication arrest form of protective efficacy previously described for 68–1 RhCMV/SIV vectors (6–8). We sought to determine the basis of 68–1 RhCMV/SIV vector efficacy, in particular the dependence of this efficacy on unconventional CD8+ T cell epitope targeting.
Our approach to Objective #1 was to 1) construct SIVgag-expressing RhCMV vectors with defined deletions, additions, or functional inactivations of specific open reading frames (ORFs) in the ULb’ ortholog region, 2) strategically vaccinate monkeys with these variants, 3) determine the effect of each ORF modification on CD8+ T cell recognition of SIV epitopes (with 34 to >500 epitopes analyzed), and 4) confirm gene contributions and response types with multiple distinct, but functionally overlapping, vectors (table S1). For Objective #2, we selected 3 vector backbones and 1 vector backbone combination for comparison, encompassing the 3 response types identified in our genetic modification studies (MHC-Ia-only, MHC-Ia + MHC-II, MHC-E + MHC-II with supertopes) and a combination phenotype that elicited all these responses. Based on previous experience with 68–1 RhCMV/SIV vectors (6–8), we randomly assigned male RMs to one of the vaccine groups or to serve as unvaccinated controls (n=15 per group; n=75 total). This group size was anticipated to allow us to thoroughly compare overall immunogenicity of each vector backbone or combination and to detect per-vaccine-group protection levels of 14% at 90% power without multiplicity adjustment. All results from these experiments are included in the presented associated data (no data were excluded as outliers). Plasma and cell-associated viral load assays were performed by blinded analysis; however, due to logistical constraints, other staff were not blinded to treatment assignments.
Rhesus macaques
These experiments used a total of 124 purpose-bred male and female RMs (M. mulatta) of Indian genetic background, including 1) 43 RMs for immunogenicity analysis of gene-modified 68–1, 68–1.2 and FL RhCMV/gag vectors, 2) 75 RMs for immunogenicity and efficacy analysis of RhCMV vector backbones representing distinct response types (5 groups of n = 15, 4 groups vaccinated; 1 group unvaccinated), and 3) 6 RMs for RhCMV 68–1.2 vs. 68–1 vector immunogenicity analysis at necropsy. Of note, the 68–1 vaccine group from this study serves as a positive control group for the companion manuscript (22). RhCMV vectors were routinely dosed at 106–107 infectious units for immunogenicity analysis and 5×106 infectious units per vector for efficacy analysis, all via subcutaneous administration. At assignment, all study RMs were free of cercopithicine herpesvirus 1, D-type simian retrovirus, simian T-lymphotrophic virus type 1, and Mycobacterium tuberculosis, but all except one (RM38 in fig. S7) were naturally RhCMV-infected. All study RMs were housed at the Oregon National Primate Research Center (ONPRC) in Animal Biosafety level (ABSL)-2 (vaccine phase) and ABSL-2+ rooms (challenge phase) rooms with autonomously controlled temperature, humidity, and lighting. Study RMs were both single- and pair-cage housed. Animals were only paired with one another during the vaccine phase if they were from the same vaccination group. All RMs were single cage-housed during the challenge phase due to the infectious nature of the study. Regardless of their pairing, all animals had visual, auditory and olfactory contact with other animals. Single cage-housed RMs received an enhanced enrichment plan that was designed and overseen by NHP behavior specialists. RMs were fed commercially prepared primate chow twice daily and received supplemental fresh fruit or vegetables daily. Fresh, potable water was provided via automatic water systems. Physical exams including body weight and complete blood counts were performed at all protocol time points. RMs were sedated with ketamine HCl or Telazol for procedures, including intradermal and subcutaneous vaccine administration, venipuncture, bronchoalveolar lavage, BM and LN biopsy, and SIV challenge. At humane or scheduled endpoints, RMs were euthanized with sodium pentobarbital overdose (>50 mg/kg) and exsanguinated via the distal aorta, and tissue collection at necropsy was performed by a certified veterinary pathologist.
RM care and all experimental protocols and procedures were approved by the ONPRC Institutional Animal Care and Use Committee (IACUC). The ONPRC is a Category I facility. The Laboratory Animal Care and Use Program at the ONPRC is fully accredited by the American Association for Accreditation of Laboratory Animal Care (AAALAC) and has an approved Assurance (#A3304–01) for the care and use of animals on file with the NIH Office for Protection from Research Risks. The IACUC adheres to national guidelines established in the Animal Welfare Act (7 U.S.C. Sections 2131–2159) and the Guide for the Care and Use of Laboratory Animals (8th Edition) as mandated by the U.S. Public Health Service Policy.
Generation of recombinant RhCMV vectors
SIV insert (SIVgag, retanef, 5’pol)-expressing vaccine vectors based on RhCMV 68–1 (GenBank #MT157325) have been described previously (6, 33). All other RhCMV constructs used in this study were based on either the partially-repaired RhCMV 68–1.2 strain (GenBank #MT157326) or our fully reconstructed FL RhCMV clone (GenBank #MT157327) (13, 18). Following the original 68–1 vector design, SIV inserts were introduced into the Rh211 open reading frame (ORF) in RhCMV 68–1.2 and transgene expression was driven by an EFIα promoter. For FL-RhCMV vectors, this design was altered and SIV transgenes were inserted into the Rh13.1 ORF using the endogenous promoter and regulatory elements to drive expression. This configuration resulted in increased genome stability as Rh13.1 is selected against in low passage isolates during tissue culture (18). To introduce deletions into these bacterial artificial chromosomes using recombineering we either chose a classical lambda (λ) Red recombination system in combination with flippase recognition target (FRT) sites (34), en passant recombination (35) or galactokinase- (galK-) mediated bacterial artificial chromosome recombination (36). Red recombination is performed by amplifying an aminoglycoside 3-phosphotransferase gene conferring kanamycin resistance (KanR) flanked by FRT sites with primers carrying a 50bp homology arm to the upstream and downstream region of the targeted genomic locus. This PCR product is inserted into the bacterial artificial chromosome through homologous recombination by heat shock induced expression of the lambda prophage encoded red recombination genes. After XmaI restriction digest and Sanger sequencing across the altered sequence, the selection marker is removed through the arabinose inducible expression of a flip recombinase in the EL250 or SW105 strains of E. coli. Final constructs were analyzed by restriction digest and Sanger sequencing of the altered genomic regions. Unintended off-target alterations in the genome sequence were ruled out by next generation sequencing (NGS) of the complete genome using an Illumina MiSeq or iSeq sequencing platform.
The en passant recombination technique utilizes a strategy which includes the introduction of 50bp DNA repeats during primer design flanking the KanR selection marker. An I-SceI homing enzyme target sequence is introduced between the selection marker and the 5’homology arm, which can be used to selectively introduce DNA double strand breaks after arabinose inducible expression of the homing enzyme in E. coli strain GS1783. These breaks can be repaired via recombination of the repeated DNA sequences flanking the selection marker by inducing the expression of the lambda phage derived Red recombination genes through heat shock, resulting in complete removal of the KanR resistance cassette without retaining any DNA sequences introduced during recombineering (scarless removal). Hence, the latter technique was favored for the introduction of point mutations or the alteration of neighboring amino acids so as to not disturb the overall coding region. We also used this technique to introduce HCMV TR3 (#MN075802) or RhCMV FL derived ORFs into the Rh161 locus in dd RhCMV FL using the endogenous regulatory elements to drive gene expression. As described for the lambda Red recombination system, all constructs were analyzed by XmaI restriction digest, Sanger sequencing of the altered genome region as well as full genome analysis by NGS before virus reconstitution.
The RhCMV 68–1.2-miR-142–3p and 68–1.2-scrambled miR control vectors expressing SIVgag were constructed using galactokinase- (galK-) mediated bacterial artificial chromosome recombination. The SW105 E. coli strain carrying the 68–1.2 SIVgag bacterial artificial chromosome was used to introduce an expression cassette encoding the galactokinase as well as the KanR gene flanked by 80bp homology arms to the targeted regions in the Rh156 or Rh108 3’ untranslated regions. Correctly recombined clones were identified based on the production of bright pink colonies on MacConkey agar containing kanamycin and Sanger sequencing of the inserted region. The galK-KanR cassette was replaced by the insert of interest by homologous recombination and correctly recombined clones were identified through negative selection on 2-deoxy-galactose-containing plates. These constructs were also analyzed by restriction digest, Sanger sequencing of the inserted region as well as full genome analysis by NGS before virus reconstitution.
RhCMV vector recovery and characterization
All vectors derived from bacterial artificial chromosomes were reconstituted in primary embryonal rhesus fibroblasts (RFs) via electroporation (250V, 508 950μF) of purified bacterial artificial chromosome-DNA. The cells were maintained in DMEM complete, Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum and antibiotics (1× Pen/Strep; Gibco) and grown at 37°C in humidified air with 5% CO2. Depending on the reconstituted construct, viral plaques became visible within 3–5 days and full cytopathic effect (CPE) was observed within 1–2 weeks after electroporation. At this point, cells and supernatants were harvested and stored at −80°C until final use. Viral stocks were produced on RFs by infecting eight confluent T-175 tissue culture flasks with an approximate MOI of 0.05–0.1. After full CPE was reached within 4–8 days cells and supernatants were harvested and frozen once overnight at −80°C to release intracellular virus from infected cells. Subsequently the supernatant was clarified by centrifugation in two steps, first at 2,000 x g for 10 minutes at 4°C and secondly at 7,500 x g for 15 minutes, after which the virus was purified through a sorbitol cushion (20% D-sorbitol, 50 mM Tris [pH 7.4], 1 mM MgCl2) by centrifugation at 64,000 x g for 1 hour at 4°C in a Beckman SW28 rotor. The pelleted virus was resuspended in complete DMEM, aliquoted and stored at −80°C until use. Virus titers of all RhCMV stocks were determined by fifty-percent tissue culture infective dose (TCID50) assay on RFs and expression of the utilized SIV immunological marker was confirmed by immunoblot.
To determine which RhCMV constructs contained a functional pentameric complex, we compared entry into rhesus retinal pigment epithelial cells (RPE) to entry into primary rhesus fibroblast (RF). RPEs were a kind gift from Dr. Thomas Shenk (Princeton University, USA) and were propagated in a 1:1 mixture of DMEM and Ham’s F12 nutrient mixture with 5% FBS, 1 mM sodium pyruvate, and nonessential amino acids. Both RFs and RPEs were infected with a MOI determined on RF resulting in infection levels of 20–30 percent after 48 hours of infection (MOI of 0.3 for 1RFs and MOI of 10 for RPEs) using the pentameric complex-intact RhCMV strain 68–1.2 as a positive control. Infection levels were determined by flow cytometry using a RhCMV specific antibody (37) and infection levels were expressed by setting infection in RFs to 100 per cent and expressing infection levels in RPEs in relation to infection levels in RFs. RhCMV strains 68–1 and 68–1.2 were included in all assays as negative and positive controls, respectively, and all experiments were performed with triplicate repeats.
To analyze the growth restriction of the RhCMV-miR-142–3p vector, multi-step growth curve assays were performed on primary rhesus fibroblasts transiently transfected with miR-142–3p or a negative control mimic. 12-well plates seeded with RFs were transfected with 20pmol miRNA mimic/well (Dharmacon) using RNAiMax (Invitrogen) according to the manufacturer’s instructions. 24 hours later the cells were infected with RhCMV-miR-142–3p or the scrambled control virus at a multiplicity of infection of 0.01. Supernatants were collected at the indicated timepoints and titered using a standard plaque assay on RFs. To analyze growth restriction in primary rhesus macrophages, rhesus PBMC were isolated from whole blood using Ficoll and monocytes were purified using non-human primate CD14+ microbeads (Miltenyi). The isolated monocytes were cultured in 50% complete RPMI supplemented with 10% FBS, 18ng/mL human GM-CSF (R&D Systems) and 20ng/mL human M-CSF (R&D Systems) and 50% conditioned media from the cell line KPB-m15 (2) for 7 days on Primaria plates (ThermoFisher Scientific). One half media exchange occurred on days 2, 4 and 6. After this time cells had adhered to the Primaria plates and differentiated to a typical macrophage morphology. Macrophages were infected with RhCMV 68–1.2 miR-142–3p or scrambled control vector at MOI = 5 and cell lysates was harvested at the indicated times post-infection using Trizol (Invitrogen). DNA was isolated and used to perform quantitative PCR (qPCR) for RhCMV genomes using custom primers and probe sets (ThermoFisher Scientific) for RhCMV Rh156: RhCMV Rh156 F primer: GGGCATCCTCAGGATCACAG; RhCMV Rh156 R primer: CGACACCAAGAGGGTATGGG; RhCMV Rh156 probe: 6FAM-ACTCCGAAGACCACAAGGACCCACG-BHQ1. Standard curves were prepared using RhCMV bacterial artificial chromosome DNA.
Expression analysis of RhCMV encoded inhibitors of unconventionally restricted CD8+ T-cell priming
Primary rhesus fibroblasts were seeded out in tissue culture plates and infected the next day with either RhCMV FL, 68–1, or 68–1.2 at a MOI of 5. Mock infected wells were included as negative control samples. The cells were harvested at 0, 4, 8, 12, 24, 36, and 48 hours post infection (hpi) and total RNA was isolated using the Quick RNA Microprep kit (Zymo Research) according to the manufacturer’s instructions. 1 μg of total RNA per sample was transcribed into cDNA using the Maxima Reverse Transcriptase (ThermoFischer Scientific). To examine the transcript levels and kinetics of selected RhCMV ORFs, a q-PCR assay was applied to the cDNA using TaqMan Fast Advanced Master Mix (Applied Biosystem). qPCR reactions were performed using QuantStudio 7 Flex Real-Time PCR Systems (Applied Biosystems) and data were collected using the QuantStudio Real-Time PCR Software v1.3. All determined transcript copy numbers were normalized to the housekeeping gene (GAPDH) and results for each gene and time point are expressed as relative mRNA copy numbers. As controls for our kinetic class analysis, we included the known immediate early (IE) gene Rh156 (UL123, IE1), the characterized early (E) gene Rh189 (US11) as well as the described true late (L) gene Rh137 (UL99, pp28). Each forward and reverse primer set was used initially to generate a PCR fragment specific for each gene which was cloned into the pGEM-T Easy Vector (Promega) according to the manufacturer’s instructions. These plasmids were used for the generation of primer/probe set specific standard curves for each indicated gene which were required to calculate viral genome copy numbers. The primer/probe sets used in this study are listed in table S2.
Immunologic assays
SIV-specific CD4+ and CD8+ T cell responses were measured in peripheral blood mononuclear cells (PBMC) or tissue-derived mononuclear cells by flow cytometric intracellular cytokine analysis, as previously described (6–8). Briefly, individual or whole protein mixes of sequential 15-mer peptides (11 amino acid overlap) spanning the SIVmac239 Gag, 5’-Pol, Nef, Rev, Tat, and Vif proteins or individual SIVmac239 Gag supertope peptides [Gag211–222 (53), Gag276–284 (69), Gag290–301 (73), Gag482–490 (120)] were used as antigens in conjunction with co-stimulatory anti-CD28 (CD28.2, Purified 500 ng/test: eBioscience, Custom Bulk 7014–0289-M050) and anti-CD49d mAb (9F10, Purified 500 ng/test: eBioscience, Custom Bulk 7014–0499-M050). Mononuclear cells were incubated at 37°C with individual peptides or peptide mixes and antibodies for 1 hr, followed by an additional 8 hr incubation in the presence of Brefeldin A (5 μg ml−1; Sigma-Aldrich). Stimulation in the absence of peptides served as background control. After incubation, stimulated cells were stored at 4°C until staining with combinations of fluorochrome-conjugated monoclonal antibodies including: anti-CD3 (SP34–2: Alexa700; BD Biosciences, Custom Bulk 624040 and Pacific Blue; BD Biosciences, Custom Bulk 624034), anti-CD4 (L200: AmCyan; BD Biosciences, Custom Bulk 658025, BV510; BD Biosciences, Custom Bulk 624340 and BUV395; BD Biosciences, Custom Bulk 624165), anti-CD8α (SK1: PerCP-eFluor710; Life Tech, Custom Bulk CUST04424), anti-TNF-α (MAB11: FITC; BD Biosciences, Custom Bulk 624046 and PE; BD Biosciences, Custom Bulk 624049), anti-IFN-γ (B27: APC; BD Biosciences, Custom Bulk 624078) and anti-CD69 (FN50: PE; eBioscience, Custom Bulk CUST01282 and PE-TexasRed; BD Biosciences, Custom Bulk 624005) and for polycytokine analyses, anti-IL-2 (MQ1–17H12; PE Cy-7; Biolegend), and anti-MIP-1β (D21–1351, BV421; BD Biosciences). For analysis of memory differentiation (central- vs transitional- vs effector-memory) of SIV Gag-specific CD4+ and CD8+ T cells, PBMC were stimulated as described above, except that the CD28 co-stimulatory mAb was used as a fluorochrome conjugate to allow CD28 expression levels to be later assessed by flow cytometry, and in these experiments, cells were surface-stained after incubation for lineage markers CD3, CD4, CD8, CD95 and CCR7 (see below for mAb clones) prior to fixation/permeabilization and then intracellular staining for response markers (CD69, IFN-γ, TNF-α; note that Brefeldin A treatment preserves the pre-stimulation cell-surface expression phenotype of phenotypic markers examined in this study).
Stained samples were analyzed on an LSR-II or FACSymphony A5 flow cytometer (BD Biosciences). Data analysis was performed using FlowJo software (Tree Star). In all analyses, gating on the lymphocyte population was followed by the separation of the CD3+ T cell subset and progressive gating on CD4+ and CD8+ T cell subsets. Antigen-responding cells in both CD4+ and CD8+ T cell populations were determined by their intracellular expression of CD69 and either or both of the cytokines IFN-γ and TNF-α (or in polycytokine analyses, expression of CD69 and any combination of the cytokines: IFN-γ, TNF-α, IL-2, MIP-1β). Assay limit of detection was determined as previously described (28), with 0.05% after background subtraction being the minimum threshold used in this study. After background subtraction, the raw response frequencies above the assay limit of detection were “memory-corrected” (e.g., % responding out of the memory population), as previously described (6–8, 28), using combinations of the following fluorochrome-conjugated mAbs to define the memory vs naïve subsets CD3 (SP34–2: Alexa700 and PerCP-Cy5.5; BD Biosciences Custom Bulk 624060), CD4 (L200: AmCyan), CD8α (SK-1: PerCP-eFluor710, RPA-T8: APC; BioLegend), TNF-α (MAB11; FITC), IFN-γ (B27; APC), CD69 (FN50; PE), CD28 (CD28.2; PE/Dazzle 594, BioLegend), CD95 (DX2; PE, BioLegend), CCR7 (15053; Biotin, R&D Systems), streptavidin (Pacific Blue, Life Tech and BV605; BD Biosciences, Custom Bulk 624342) and Ki67 (B56; FITC, BD Biosciences, Custom Bulk 624046). For memory phenotype analysis of SIV Gag-specific T cells, all CD4+ or CD8+ T cells expressing CD69 plus IFN-γ and/or TNF-α were first Boolean OR gated, and then this overall Ag-responding population was subdivided into the memory subsets of interest on the basis of surface phenotype (CCR7 vs. CD28). Similarly, for polycytokine analysis of SIV Gag-specific T cells, all CD4+ or CD8+ T cells expressing CD69 plus cytokines were Boolean OR gated and polyfunctionality was delineated with any combination of the four cytokines tested (IFN-γ, TNF-α, IL-2, MIP-1β) using the Boolean AND function. Gating strategies for these ICS analyses are illustrated in fig. S8.
The MHC restriction type (MHC-Ia, MHC-E, MHC-II) of a 15mer peptide response was determined by pre-incubating isolated mononuclear cell aliquots for 1 hr at room temperature (prior to adding peptides or combining effector and target cells and incubating per the standard ICS assay) in the presence (and absence) of each the following specific inhibitors: 1) the pan anti-MHC-I mAb W6/32 (10μg/ml), 2) the MHC-II-blocking mAb G46.6 (10μg/ml), or 3) the MHC-E blocking VL9 peptide (VMAPRTLLL; 20μM). Stimulated cells were fixed, permeabilized, stained and analyzed as described above. To be considered MHC-E-restricted by blocking, the individual peptide response must have been blocked by both anti-pan MHC-I clone W6/32 and MHC-E-binding peptide VL9, and not blocked by anti-MHC-II. MHC-II-restricted responses were blocked by anti-MHC-II but not anti-MHC-I or VL9, and MHC-Ia-restricted responses were blocked by anti-MHC-Ia only (9, 10). Responses that did not meet these inhibition criteria were considered indeterminate. Minimal independent epitope numbers were estimated from the positive responses identified by testing of consecutive 15mer peptides by the following criteria: single positive peptide of same restriction type = 1 independent epitope; 2 adjacent positive peptides of same restriction type = 1 independent epitope; 3 adjacent positive peptides of same restriction type = 2 independent epitopes; 4 adjacent positive peptides of same restriction type = 2 independent epitopes; and 5 adjacent positive peptides of same restriction type = 3 independent epitopes.
SIV detection assays
Plasma SIV RNA levels were determined using an SIVgag-targeted quantitative real time/digital RT-PCR format assay, essentially as previously described, with 6 replicate reactions analyzed per extracted sample for assay thresholds of 15 SIV RNA copies/ml (7, 8, 38). Quantitative assessment of SIV DNA and RNA in cells and tissues was performed using SIVgag targeted, nested quantitative hybrid real-time/digital RT-PCR and PCR assays, as previously described (7, 8, 38). SIV RNA or DNA copy numbers were normalized based on quantitation of a single copy rhesus genomic DNA sequence from the CCR5 locus from the same specimen, as described, to allow normalization of SIV RNA or DNA copy numbers per 108 diploid genome cell equivalents. Ten replicate reactions were performed with aliquots of extracted DNA or RNA from each sample, with two additional spiked internal control reactions performed with each sample to assess potential reaction inhibition. Samples that did not yield any positive results across the replicate reactions were reported as a value of “less than” the value that would apply for one positive reaction out of 10. Threshold sensitivities for individual specimens varied as a function of the number of cells or amount of tissue available and analyzed; for graphing consistency values are plotted with a common nominal sensitivity threshold.
Statistical Analysis
Boxplots show jittered points and a box from 1st to 3rd quartiles (IQR) and a line at the median, with whiskers extending to the farthest data point within 1.5*IQR above and below the box. For all comparisons of T cell response parameters, we performed Wilcoxon rank-sum tests comparing each non-RhCMV 68–1 vaccine group to the RhCMV 68.1 reference group. For longitudinal responses, we calculated the area under the curve (AUC) or the plateau average value for each RM, as denoted in the figure legends. All Wilcoxon P values are based on two-sided tests and unadjusted except where noted. Adjusted P values were computed using the Holm procedure for family-wise error rate control. All P values for analyses of efficacy or epitope restriction frequency were based on two-sided exact tests of binomial proportions. Analyses were performed in R v3.6.0 with the package Exact v2.0 (39).
Supplementary Material
Fig. S1. Functional pentameric complex analysis of 68-1.2-based RhCMV vectors
Fig. S2. Protein sequence comparison of RhCMV/HCMV orthologs RhCMV157.5/UL128, Rh157.4/ UL130 and Rh158-161/UL146-147
Fig. S3. Genetic configuration of gene-modified double-deleted (dd) FL-RhCMV vectors
Fig. S4. Transcriptional analysis of Rh158-Rh161 region genes in RhCMV FL in comparison to RhCMV 68-1 and 68-1.2
Fig. S5. Analysis of SIV-specific T cell responses in bronchoalveolar alveolar lavage fluid (BAL) in RM cohorts destined for SIV challenge
Fig. S6. Epitope analysis of RhCMV vaccine-elicited, SIV-specific CD8+ T cell responses in RM cohorts destined for SIV challenge
Fig. S7. Analysis of SIVgag-specific CD4+ and CD8+ T cell response magnitude in tissues of 68-1 vs. 68-1.2 RhCMV/SIVgag vector vaccinated RMs at necropsy
Fig. S8. General gating hierarchy for ICS analysis
Table S1. Epitope analysis summary of all study RMs
Table S2. Primer/probe sets used for expression analysis of RhCMV encoded inhibitors of unconventionally restricted CD8+ T cell priming
Table S3. RhCMV vector genetic modifications
Table S4. Raw data file: non-quantitative epitope analysis data
Table S5. Raw data file: quantitative data
Acknowledgments:
We thank T. Whitmer, A. Bhusari, L. Bishop, C. Kreklywich, A. Legasse, M. Fischer, C. Shriver-Munsch, T. Swanson, A. Sylwester, S. Hagen, E. McDonald, E. Ainslie, A. Selseth, and K. Rothstein for technical or administrative assistance; B. Keele for providing SIVmac239 challenge virus, and both J. Womack and A. Townsend for figure preparation.
Funding:
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID) grants P01 AI094417, U19 AI128741, UM1 AI124377, and R37 AI054292 to LJP, R01 AI140888 to JBS, the Oregon National Primate Research Center Core grant from the National Institutes of Health, Office of the Director (P51 OD011092); and contracts from the National Cancer Institute (# HHSN261200800001E) to JDL.
Footnotes
Competing interests: OHSU, LJP, SGH, JAN, and KF have a substantial financial interest in Vir Biotechnology, Inc., a company that may have a commercial interest in the results of this research and technology. LJP, SGH, JAN, and KF are also consultants to Vir Biotechnology, Inc., and JBS has received compensation for consulting for Vir Biotechnology, Inc. LJP, SGH, JAN, and KF are co-inventors of patent WO 2011/143650 A2 “Recombinant RhCMV and HCMV vectors and uses thereof” licensed to Vir Biotechnology, Inc. LJP, SGH, KF, and DM are co-inventors of patent US2016/0010112 A1 “Cytomegalovirus vectors enabling control of T cell targeting” licensed to Vir Biotechnology, Inc. JAN, SGH, MHH, LJP and KF are co-inventors of patent US2017/0143809 A1 “CMV vectors comprising microRNA recognition elements” licensed to Vir Biotechnology, Inc. These potential individual and institutional conflicts of interest have been reviewed and managed by OHSU.
Data and materials availability:
All data associated with this study are present in the paper or Supplementary Materials, including raw data files (tables S3–S5). The computer code used to perform statistical analysis is available upon request. RhCMV/SIV vectors can be obtained through an MTA.
References and Notes
- 1.Jarvis MA, Hansen SG, Nelson JA, Picker LJ, Früh K, in Cytomegaloviruses: From Molecular Pathogenesis to Intervention Reddehase MJ, Ed. (Caister Academic Press, 2013), vol. 2, chap. 21. [Google Scholar]
- 2.Vieira Braga FA, Hertoghs KM, van Lier RA, van Gisbergen KP, Molecular characterization of HCMV-specific immune responses: Parallels between CD8(+) T cells, CD4(+) T cells, and NK cells. Eur J Immunol 45, 2433–2445 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Klenerman P, Oxenius A, T cell responses to cytomegalovirus. Nat Rev Immunol 16, 367–377 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Masopust D, Picker LJ, Hidden memories: frontline memory T cells and early pathogen interception. J Immunol 188, 5811–5817 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Picker LJ, Hansen SG, Lifson JD, New paradigms for HIV/AIDS vaccine development. Annu Rev Med 63, 95–111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M Jr., Lifson JD, Picker LJ, Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473, 523–527 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansen SG, Piatak M Jr., Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, Gilliam AN, Xu G, Whizin N, Burwitz BJ, Planer SL, Turner JM, Legasse AW, Axthelm MK, Nelson JA, Fruh K, Sacha JB, Estes JD, Keele BF, Edlefsen PT, Lifson JD, Picker LJ, Immune clearance of highly pathogenic SIV infection. Nature 502, 100–104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen SG, Marshall EE, Malouli D, Ventura AB, Hughes CM, Ainslie E, Ford JC, Morrow D, Gilbride RM, Bae JY, Legasse AW, Oswald K, Shoemaker R, Berkemeier B, Bosche WJ, Hull M, Womack J, Shao J, Edlefsen PT, Reed JS, Burwitz BJ, Sacha JB, Axthelm MK, Früh K, Lifson JD, Picker LJ, A live-attenuated RhCMV/SIV vaccine shows long-term efficacy against heterologous SIV challenge. Sci Transl Med 11, eaaw2607 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, Gilbride RM, Lewis MS, Gilliam AN, Ventura AB, Malouli D, Xu G, Richards R, Whizin N, Reed JS, Hammond KB, Fischer M, Turner JM, Legasse AW, Axthelm MK, Edlefsen PT, Nelson JA, Lifson JD, Früh K, Picker LJ, Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340, 1237874 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansen SG, Wu HL, Burwitz BJ, Hughes CM, Hammond KB, Ventura AB, Reed JS, Gilbride RM, Ainslie E, Morrow DW, Ford JC, Selseth AN, Pathak R, Malouli D, Legasse AW, Axthelm MK, Nelson JA, Gillespie GM, Walters LC, Brackenridge S, Sharpe HR, Lopez CA, Früh K, Korber BT, McMichael AJ, Gnanakaran S, Sacha JB, Picker LJ, Broadly targeted CD8+ T cell responses restricted by major histocompatibility complex E. Science, 351, 714–720 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oxford KL, Eberhardt MK, Yang KW, Strelow L, Kelly S, Zhou SS, Barry PA, Protein coding content of the UL-b’ region of wild-type rhesus cytomegalovirus. Virology 373, 181–188 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerna G, Kabanova A, Lilleri D , Human Cytomegalovirus Cell Tropism and Host Cell Receptors. Vaccines (Basel) 7, 70 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lilja AE, Shenk T, Efficient replication of rhesus cytomegalovirus variants in multiple rhesus and human cell types. Proc Natl Acad Sci U S A 105, 19950–19955 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuessler A, Sampaio KL, Sinzger C, Charge cluster-to-alanine scanning of UL128 for fine tuning of the endothelial cell tropism of human cytomegalovirus. J Virol 82, 11239–11246 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnes D, Kunitomi M, Vignuzzi M, Saksela K, Andino R, Harnessing endogenous miRNAs to control virus tissue tropism as a strategy for developing attenuated virus vaccines. Cell Host Microbe 4, 239–248 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moller R, Schwarz TM, Noriega VM, Panis M, Sachs D, Tortorella D, tenOever BR, miRNA-mediated targeting of human cytomegalovirus reveals biological host and viral targets of IE2. Proc Natl Acad Sci U S A 115, 1069–1074 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gill RB, Jason Bowman J, Krogmann T, Wollenberg K, Asher DM, Cohen JI, Coding potential of UL/b’ from the initial source of rhesus cytomegalovirus Strain 68–1. Virology 447, 208–212 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taher H, Mahyari E, Kreklywich C, Uebelhoer LS, McArdle MR, Moström MJ, Bhusari A, Nekorchuk M, Whitmer T, Scheef EA, Sprehe LM, Roberts D, Hughes CM, Jackson KA, Selseth AN, Ventura AB, Yue Y, Schmidt KA, Shao J, Edlefsen PT, Smedley J, Stanton RJ, Axthelm MK, Estes JD, Hansen SG, Kaur A, Barry PA, Bimber BN, Picker LJ, Streblow DN, Früh K, Malouli D, In vitro and in vivo characterization of a recombinant rhesus cytomegalovirus containing a complete genome. PLoS Pathog 16, e1008666 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caposio P, van den Worm S, Crawford L, Perez W, Kreklywich C, Gilbride RM, Hughes CM, Ventura AB, Ratts R, Marshall EE, Malouli D, Axthelm MK, Streblow D, Nelson JA, Picker LJ, Hansen SG, Früh K, Characterization of a live-attenuated HCMV-based vaccine platform. Sci Rep 9, 19236 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray SE, Nesterenko PA, Vanarsdall AL, Munks MW, Smart SM, Veziroglu EM, Sagario LC, Lee R, Claas FHJ, Doxiadis IIN, McVoy MA, Adler SP, Hill AB, Fibroblast-adapted human CMV vaccines elicit predominantly conventional CD8 T cell responses in humans. J Exp Med 214, 1889–1899 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powers C, DeFilippis V, Malouli D, Früh K, Cytomegalovirus immune evasion. Curr Top Microbiol Immunol 325, 333–359 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Verweij M, Hansen SG, Iyer R, John N, Malouli D, Scholz I, Abdulhaqq S, Gilbride RM, Hughes CM, Ventura AB, Ford JC, Oswald K, Shoemaker R, Berkemeier B, Bosche WJ, Hill M, Sacha JB, Axthelm MK, Edlefsen PT, Lifson JD, Picker LJ, Früh K, Modulation of MHC-E transport by viral decoy ligands is required for RhCMV/SIV vaccine efficacy. Science, in press (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wieten L, Mahaweni NM, Voorter CE, Bos GM, Tilanus MG, Clinical and immunological significance of HLA-E in stem cell transplantation and cancer. Tissue Antigens 84, 523–535 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Iwaszko M, Bogunia-Kubik K, Clinical significance of the HLA-E and CD94/NKG2 interaction. Arch Immunol Ther Exp (Warsz) 59, 353–367 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Lee N, Llano M, Carretero M, Ishitani A, Navarro F, Lopez-Botet M, Geraghty DE, HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A 95, 5199–5204 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang EC, McSharry B, Retiere C, Tomasec P, Williams S, Borysiewicz LK, Braud VM, Wilkinson GW, UL40-mediated NK evasion during productive infection with human cytomegalovirus. Proc Natl Acad Sci U S A 99, 7570–7575 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lodoen MB, Lanier LL, Viral modulation of NK cell immunity. Nat Rev Microbiol 3, 59–69 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Hansen SG, Zak DE, Xu G, Ford JC, Marshall EE, Malouli D, Gilbride RM, Hughes CM, Ventura AB, Ainslie E, Randall KT, Selseth AN, Rundstrom P, Herlache L, Lewis MS, Park H, Planer SL, Turner JM, Fischer M, Armstrong C, Zweig RC, Valvo J, Braun JM, Shankar S, Lu L, Sylwester AW, Legasse AW, Messerle M, Jarvis MA, Amon LM, Aderem A, Alter G, Laddy DJ, Stone M, Bonavia A, Evans TG, Axthelm MK, Früh K, Edlefsen PT, Picker LJ, Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat Med 24, 130–143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Itell HL, Kaur A, Deere JD, Barry PA, Permar SR, Rhesus monkeys for a nonhuman primate model of cytomegalovirus infections. Curr Opin Virol 25, 126–133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang H, Rei M, Brackenridge S, Brenna E, Sun H, Abdulhaqq S, Lui MKP, Ma W, Kurupati P, Xu X, Cerundolo V, Jenkins E, Davis S, Sacha JB, Früh K, Picker LJ, Borrow P, Gillespie G, McMichael AJ, HLA-E restricted, HIV-1 suppressing, Gag specific CD8+ T cells offer universal vaccine opportunities. Science Immunology, in revision (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.VirBiotechnology press release: https://investors.vir.bio/news-releases/news-release-details/vir-biotechnology-announces-initiation-phase-1-clinical-trial
- 32.Pietra G, Romagnani C, Manzini C, Moretta L, Mingari MC, The emerging role of HLA-E-restricted CD8+ T lymphocytes in the adaptive immune response to pathogens and tumors. J Biomed Biotechnol 2010, 907092 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, Legasse AW, Axthelm MK, Oswald K, Trubey CM, Piatak M Jr., Lifson JD, Nelson JA, Jarvis MA, Picker LJ, Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med 15, 293–299 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG, A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73, 56–65 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Tischer BK, Smith GA, Osterrieder N, En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634, 421–430 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG, Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33, e36 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malouli D, Hansen SG, Nakayasu ES, Marshall EE, Hughes CM, Ventura AB, Gilbride RM, Lewis MS, Xu G, Kreklywich C, Whizin N, Fischer M, Legasse AW, Viswanathan K, Siess D, Camp DG 2nd, Axthelm MK, Kahl C, DeFilippis VR, Smith RD, Streblow DN, Picker LJ, Früh K, Cytomegalovirus pp65 limits dissemination but is dispensable for persistence. J Clin Invest 124, 1928–1944 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hansen SG, Piatak M, Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, Gilliam AN, Xu G, Whizin N, Burwitz BJ, Planer SL, Turner JM, Legasse AW, Axthelm MK, Nelson JA, Früh K, Sacha JB, Estes JD, Keele BF, Edlefsen PT, Lifson JD, Picker LJ, Addendum: Immune clearance of highly pathogenic SIV infection. Nature 547, 123–124 (2017). [DOI] [PubMed] [Google Scholar]
- 39.R Development Core Team. R, A Language and Environment for Statistical Computing. http://www.R-project.org (Vienna, Austria, 2019). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Functional pentameric complex analysis of 68-1.2-based RhCMV vectors
Fig. S2. Protein sequence comparison of RhCMV/HCMV orthologs RhCMV157.5/UL128, Rh157.4/ UL130 and Rh158-161/UL146-147
Fig. S3. Genetic configuration of gene-modified double-deleted (dd) FL-RhCMV vectors
Fig. S4. Transcriptional analysis of Rh158-Rh161 region genes in RhCMV FL in comparison to RhCMV 68-1 and 68-1.2
Fig. S5. Analysis of SIV-specific T cell responses in bronchoalveolar alveolar lavage fluid (BAL) in RM cohorts destined for SIV challenge
Fig. S6. Epitope analysis of RhCMV vaccine-elicited, SIV-specific CD8+ T cell responses in RM cohorts destined for SIV challenge
Fig. S7. Analysis of SIVgag-specific CD4+ and CD8+ T cell response magnitude in tissues of 68-1 vs. 68-1.2 RhCMV/SIVgag vector vaccinated RMs at necropsy
Fig. S8. General gating hierarchy for ICS analysis
Table S1. Epitope analysis summary of all study RMs
Table S2. Primer/probe sets used for expression analysis of RhCMV encoded inhibitors of unconventionally restricted CD8+ T cell priming
Table S3. RhCMV vector genetic modifications
Table S4. Raw data file: non-quantitative epitope analysis data
Table S5. Raw data file: quantitative data
Data Availability Statement
All data associated with this study are present in the paper or Supplementary Materials, including raw data files (tables S3–S5). The computer code used to perform statistical analysis is available upon request. RhCMV/SIV vectors can be obtained through an MTA.