

SUMMARY
Metabolic reprogramming powers and polarizes macrophage functions, but the nature and regulation of this response during infection with pathogens remain controversial. In this study, we characterize the metabolic and transcriptional responses of murine macrophages to Mycobacterium tuberculosis (Mtb) in order to disentangle the underlying mechanisms. We find that type I interferon (IFN) signaling correlates with the decreased glycolysis and mitochondrial damage that is induced by live, but not killed, Mtb. Macrophages lacking the type I IFN receptor (IFNAR) maintain glycolytic flux and mitochondrial function during Mtb infection in vitro and in vivo. IFNβ itself restrains the glycolytic shift of inflammatory macrophages and initiates mitochondrial stress. We confirm that type I IFN acts upstream of mitochondrial damage using macrophages lacking the protein STING. We suggest that a type I IFN-mitochondrial feedback loop controls macrophage responses to mycobacteria and that this could contribute to pathogenesis across a range of diseases.
Graphical Abstract
In brief
The mechanisms controlling macrophage metabolism during bacterial infections remain poorly characterized. Olson et al. show that type I interferon, rather than direct bacterial virulence factors, restrains macrophage metabolism during Mycobacterium tuberculosis infection. IFNβ itself prevents the shift to aerobic glycolysis in inflammatory macrophages and drives mitochondrial dysfunction and stress.
INTRODUCTION
Macrophages patrol every tissue and orchestrate immune responses to diverse threats. A growing body of evidence has underscored the functional importance of metabolic changes downstream of pattern recognition receptors or host cytokine receptors in macrophages. Mammalian cells rely on two interconnected metabolic programs to generate energy for cellular functions: cytosolic glycolysis and mitochondrial oxidative phosphorylation (OXPHOS). Preferential reliance on glycolysis generates ATP rapidly, enables carbons in metabolic intermediates to be shunted to macromolecular biosynthesis, and leads to increased export of lactate. Measuring the resulting extracellular acidification rate (ECAR) can quantify glycolytic flux (O’Neill et al., 2016). Mitochondrial metabolism fully oxidizes carbons and stores the energy in a mitochondrial membrane potential (ΔΨm) that can be used to do work, for example in OXPHOS to efficiently generate ATP. This process requires electrons to be passed to oxygen. Measuring the resulting oxygen consumption rate (OCR) can quantify flux through the electron transport chain (ETC) (O’Neill et al., 2016). Low levels of mitochondrial reactive oxygen species (mROS) are an unavoidable byproduct of healthy mitochondrial metabolism as a small fraction of electrons escape the ETC. However, the accumulation of mROS, concomitant with a decreased OCR and a dissipated ΔΨm, is a hallmark of mitochondrial damage.
Macrophages differentially engage these two energy programs during activation to power and polarize subsequent immune responses. Studies using canonical immune stimuli support a model in which inflammatory macrophages rely on glycolytic metabolism while reparative or tolerogenic macrophages rely on mitochondrial metabolism. For example, Toll-like receptor (TLR) recognition of lipopolysaccharide (LPS) skews macrophages toward aerobic glycolysis, that is, an increased reliance on glycolytic ATP, a repurposing of the tricarboxylic acid cycle for anabolism, and ΔΨm hyperpolarization as OXPHOS decreases (Buck et al., 2017; Loftus and Finlay, 2016; Weinberg et al., 2015). Conversely, interleukin (IL)-4 (a cytokine involved in tissue repair) increases mitochondrial respiration and OXPHOS at the expense of glycolysis (O’Neill et al., 2016; Pearce and Pearce, 2013).
Not only does immune signaling alter metabolism, but metabolic distress also engages immune signaling. Mitochondria—as endosymbionts carrying vestiges of their bacterial past—release potent immunostimulatory molecules when damaged. For example, mitochondrial DNA (mtDNA) released from damaged mitochondria activates the nucleic acid sensor cGAS, which signals through the adaptor protein STING to induce robust expression of the type I interferon (IFN) family of cytokines (Mills et al., 2017; Weinberg et al., 2015).
Although studies with purified bacterial components support the simplified model that macrophages balance glycolysis and OXPHOS (i.e., when one increases, the other decreases), macrophage metabolism during infections with live pathogens displays much more complex patterns (Ayres, 2020; Eisenreich et al., 2019; Gleeson and Sheedy, 2016; Russell et al., 2019). The mechanisms driving these patterns remain largely uncharacterized, in part because the literature does not always agree on the metabolic responses to pathogens.
Many excellent reviews (Cumming et al., 2020; Howard and Khader, 2020; Sheedy and Divangahi, 2021) have highlighted one of the most pressing examples of these inconsistencies, that is, macrophage infection with Mycobacterium tuberculosis (Mtb), a global pathogen that killed more than 1.4 million people in 2019 (World Health Organization, 2020). Although the consensus remains that Mtb infection shifts macrophages toward aerobic glycolysis (Gleeson et al., 2016; Howard et al., 2018; Lachmandas et al., 2016a, 2016b; Marín Franco et al., 2020) (similar to purified inflammatory stimuli), a few recent reports suggest that Mtb infection decreases both glycolytic and mitochondrial metabolism in macrophages, especially at higher multiplicities of infection (MOIs) (Cumming et al., 2018; Rahman et al., 2020). Two variables provide the most likely explanations for the discrepancies: the viability and virulence of the Mtb and the macrophage subset. The initial studies showing a glycolytic shift mainly used Mtb lysates or killed Mtb (Gleeson et al., 2016; Lachmandas et al., 2016a). Whether live, virulent Mtb decreases both metabolic programs of macrophages compared to uninfected controls (Cumming et al., 2018) or only when compared directly to macrophages stimulated with inactivated/killed Mtb (Chen et al., 2006; Cumming et al., 2018; Hackett et al., 2020; Wiens and Ernst, 2016) remains controversial. Macrophages are a heterogeneous group, and two different macrophage subsets play pivotal roles during early Mtb infection. Lung-resident alveolar macrophages (AMs) first engulf Mtb but fail to control bacterial replication. As the Mtb burden grows, AMs initiate the recruitment of inflammatory monocyte-derived macrophages (MDMs) that more effectively control Mtb (Cohen et al., 2018; Huang et al., 2018; Rothchild et al., 2019). It has been proposed that the divergent metabolic responses of AMs and MDMs contribute to their divergent control of Mtb (possibly by altering nutritional availability to Mtb) (Huang et al., 2018; Pisu et al., 2020), but the signals that control the metabolic responses remain elusive.
The type I IFN cytokine family, including >10 IFNα members and the single IFNβ, signals through the receptor IFNAR to change the expression of hundreds of genes. This response is critical for eliciting an antiviral state, but how type I IFN regulates immune responses to intracellular bacterial pathogens (Decker et al., 2005), including Mtb (Moreira-Teixeira et al., 2018), remains contentious. For example, increasing type I IFN can increase the efficacy of vaccination against Mtb (Giacomini et al., 2009; Grode et al., 2005; Gröschel et al., 2017) while also exacerbating disease progression in animal models (Antonelli et al., 2010), in part by suppressing the host-protective cytokine IL-1 (Ji et al., 2019; Mayer-Barber et al., 2011) and establishing an imbalance in eicosanoids, a family of potent signaling lipids (Mayer-Barber et al., 2014). Although recent reports suggest that type I IFN modulates dendritic cell (DC) energy metabolism in a subset-dependent manner—increasing glycolysis in conventional DC (Everts et al., 2014) or increasing OXPHOS in plasmacytoid DC (Wu et al., 2016)—surprisingly little is known about how type I IFNs modulate macrophage energy metabolism.
In this study, we began with the hypothesis that we could exploit the divergent responses of murine bone marrow-derived macrophages (BMDMs) to live and heat-killed (HK) H37Rv (a virulent strain of Mtb) to identify mechanisms controlling macrophage metabolism during Mtb infection. Our initial findings support the recent claim that live Mtb infection decreases both glycolysis and mitochondrial respiration in BMDMs compared to either mock infection or HK Mtb stimulation (Cumming et al., 2018). Interestingly, we identified that activation of type I IFN signaling correlates with these differences. Although it is well known that Mtb virulence is required for type I IFN induction in macrophages (Collins et al., 2015; Manzanillo et al., 2012; Wassermann et al., 2015; Watson et al., 2015), how type I IFN shapes the macrophage metabolic or transcriptional response to Mtb is poorly characterized. To establish a causal relationship between type I IFN and macrophage metabolism in the context of Mtb infection, we show that type I IFN by itself decreases BMDM glycolysis and mitochondrial respiration. We then demonstrate that IFNAR knockout (KO) of BMDMs maintains both glycolytic capacity and mitochondrial health during Mtb infection. Importantly, we confirm that type I IFN decreases macrophage metabolism in vivo during an aerosol infection with Mtb. We then substantiate that type I IFN acts upstream of mitochondrial health in macrophages by studying the metabolic response of STING KO BMDMs to Mtb infection. Consistent with our predictions, STING KO BMDMs maintain glycolytic capacity and mitochondrial health upon Mtb infection, while exogenous type I IFN added during Mtb infection in STING KO BMDMs recapitulates the decreased metabolism seen in wild-type (WT) macrophages.
RESULTS
Live Mtb decreases BMDM metabolism more than does HK Mtb
Live H37Rv infection dramatically decreases OCRs in BMDMs 24 h after infection, while HK H37Rv has little effect at the same MOI (Figures 1A and 1B; see Figure S1 for derivation of parameters). The decrease in mitochondrial respiration in BMDMs is accompanied by hallmarks of mitochondrial stress. After 24 h of infection, live H37Rv dissipates ΔΨm to a greater extent than does HK H37Rv (Figures 1C and 1D), which cannot be explained by a loss of mitochondrial mass (Figure S1D). In addition, mROS accumulate after infection with live H37Rv more than after HK H37Rv (Figure 1E). Accumulation of mROS begins as early as 4 h after infection and continues to increase during the first 48 h in BMDMs infected with live H37Rv (Figure 1F).
Figure 1. Live H37Rv decreases BMDM metabolism more than does heat-killed (HK) H37Rv.

(A) Oxygen consumption rate (OCR) in WT BMDMs 24 h after mock infection or infection with live H37Rv or HK H37Rv at an MOI of 10. A single representative plate is shown.
(B) Quantification of mitochondrial parameters derived from (A). The OCR dedicated to ATP production (ATP) or at maximal respiration (Max) was normalized to mock infection controls on each plate. Each dot represents a single well and bars represent the mean. Data from eight plates across three independent experiments. See also Figure S1.
(C) Representative images (original magnification, x20) of BMDMs either mock infected (left) or infected with live (middle) or HK (right) H37Rv (MOI of 10) and stained 24 h later with the mitochondrial membrane potential (Δψm)-sensitive dye TMRM (tetramethylrhodamine, methyl ester). TMRM intensity depicted with the “Fire” LUT from Fiji (Schindelin et al., 2012). Scale bars, 15 μm.
(D) Quantification of Δψm derived from (C). The TMRM mean fluorescence intensity (MFI) for each field of view was measured in CellProfiler (McQuin et al., 2018) and normalized to mock-infected wells. Each dot represents a single field of view collected across two independent experiments.
(E) Representative histograms of MitoSOX Red (MSR) fluorescence quantified with flow cytometry to measure mitochondrial reactive oxygen species (mROS) in BMDMs either mock infected or infected with an MOI of 10 of live H37Rv or HK H37Rv for 24 h.
(F) Quantification of mROS derived from (E). MSR MFI in each condition was normalized to mock-infected controls at each time point. Means for four technical replicates ± SEM are shown for a representative experiment of three independent experiments.
(G) Fold change in basal glycolysis or glycolytic capacity was calculated relative to mock-infected controls on each plate. Each dot represents a well and bars represent the mean from eight plates across three independent experiments.
Interestingly, BMDMs infected with live H37Rv do not increase glycolysis to compensate for the decreased mitochondrial respiration, as seen with classical inflammatory stimuli (e.g., LPS) (O’Neill et al., 2016); live H37Rv infection decreases BMDM glycolysis, especially by limiting glycolytic capacity, after 24 h of infection (Figures 1G and S1E). Although the lack of glycolytic compensation in the setting of impaired mitochondrial respiration decreases total ATP production in infected BMDMs to ~60% of mock-infected controls at 48 h post-infection (Figure S1F), these levels are sufficient to maintain cellular viability: live H37Rv causes only minimal (~10%) cell death during the first 48 h, as measured by lactate dehydrogenase (LDH) release and violet viability dye exclusion (Figures S1G and S1H). Furthermore, the single-cell measurements of ΔΨm and mROS confirm that live H37Rv specifically alters energy metabolism in viable cells.
Type I IFN signaling dominates the BMDM transcriptional response to live Mtb and is correlated with metabolic changes
To determine how live Mtb rewires macrophage metabolism, we first tested the involvement of TLRs since they recognize mycobacterial components (Mortaz et al., 2015), drastically alter macrophage metabolism (O’Neill et al., 2016), and induce mROS (Figure S2A) (West et al., 2011). We measured mROS in BMDMs lacking key TLR signaling adaptors (MyD88-TRIF double KO) and found that TLR signaling does not explain the induction of mitochondrial stress; mROS accumulation after live H37Rv infection is TLR-independent, and—as in WT BMDMs—live H37Rv induces more mROS than does HK H37Rv in MyD88-TRIF double KO BMDMs across many MOIs (Figure S2A).
To interrogate the myriad signaling pathways that could explain the metabolic changes during infection, we performed global RNA sequencing (RNA-seq) on WT BMDMs at an early (4 h) and late (24 h) time point after mock infection or infection with live or HK Mtb. The transcriptional response to live and HK Mtb diverges early: BMDMs infected with live H37Rv for 4 h differentially express (DE) 1,717 genes compared to BMDMs infected with HK H37Rv (∣log2fold change [FC]∣ > 1, false discovery rate [FDR] < 0.001) (Figures 2A and S2B). Although canonical inflammatory genes such as Nos2 and Il12b are robustly induced by both live and HK H37Rv, a subset of genes are only upregulated by live H37Rv (Figure 2A). Five members of the type I IFN family—including Ifnb1, the gene encoding IFNβ—comprise the most differentially expressed genes in this subset (Figure 2A). We validated that live H37Rv induces more Ifnb1 and downstream IFN signaling (Isg15 expression) than does HK H37Rv using qRT-PCR (Figure S2C) and confirmed that only live H37Rv-infected BMDMs secrete IFNβ using an ELISA (Figure S2D).
Figure 2. Type I IFN dominates BMDM transcriptional response to live Mtb and correlates with metabolic changes.

(A) Scatterplot of log2FC values of the 2,210 genes differentially expressed (DE) (FDR < 0.001, ∣log2FO∣ > 1; gray dotted lines) across both comparisons in WT BMDMs. Magenta points represent genes significantly DE directly comparing the infection conditions (live H37Rv infected – HK H37Rv infected).
(B) As in (A), except showing the 1,712 genes DE across the same comparisons in IFNAR KO BMDMs. See also Figure S2.
(C) Principal-component analysis of WT or IFNAR KO BMDMs at 24 h post-infection with live H37Rv or HK H37Rv at an 10 of MOI. WT BMDMs were either left untreated or treated with 500 U/mL IFNβ. The percent of variance explained by the top two principal components is indicated.
(D) Heatmap showing the cluster number (left) and log2FC of the 6,337 genes differentially expressed in any of the six comparisons in WT BMDMs.
(E) The 1,411 genes from cluster 4 were tested for enrichment in the Hallmark gene sets from MSigDB (Liberzon et al., 2015; Subramanian et al., 2005) using a hypergeometric test for overlap. The 10 gene sets with the smallest FDR are shown.
Although it is known that the induction of type I IFN by Mtb requires active virulence processes (Collins et al., 2015; Lienard et al., 2020; Manzanillo et al., 2012; Wassermann et al., 2015; Watson et al., 2015; Wiens and Ernst, 2016), to the best of our knowledge, the transcriptional response in IFNAR KO cells after infection with virulent Mtb has only been characterized in lung homogenates (Dorhoi et al., 2014), obfuscating the response of any single cell type. To assess how the induction of type I IFN contributes to the differential transcriptional response to live and HK Mtb, we repeated the RNA-seq analysis in BMDMs generated from mice lacking IFNAR. IFNAR KO BMDMs respond remarkably similarly to live and HK H37Rv: at 24 h, IFNAR KO BMDMs differentially express only 250 genes when comparing live H37Rv to HK H37Rv conditions, while WT BMDMs differentially express 2554 genes at the same time point (Figures 2B and S2B). Ifnb1 itself is still induced in IFNAR KO BMDMs, and the remaining most differentially expressed genes are primarily members of the IFIT gene family, which can be upregulated independently of IFN (Diamond and Farzan, 2013) (Figure 2B).
We next performed dimensionality reduction using principal-component analysis (PCA) to confirm that IFNAR KO dampens the response to live H37Rv rather than augmenting the response to HK H37Rv. The transcriptional responses of both genotypes to HK H37Rv cluster together (Figure 2C), verifying that IFNAR signaling plays little role in the BMDM response to HK H37Rv. Strikingly, the transcriptional response of IFNAR KO BMDMs to live H37Rv clusters together with the HK H37Rv response (Figure 2C), suggesting that type I IFN signaling dominates the transcriptional response of BMDMs to live Mtb. If this is indeed the case, then the addition of exogenous type I IFN should make the transcriptional response to HK H37Rv approximate live H37Rv. We chose to test this with exogenous IFNβ, because it is the type I IFN family member that is (1) the most highly induced by H37Rv infection (Figure 2A), (2) the best characterized during Mtb infection (Moreira-Teixeira et al., 2018), and (3) the only member of the β subfamily (as opposed to the >10 α members). Consistent with a critical role for type I IFN signaling, the transcriptional response of WT BMDMs to HK H37Rv in the presence of exogenous IFNβ more closely clusters with the response to live H37Rv than does the response to HK H37Rv at 24 h after stimulation (Figure 2C).
Remarkably, the transcriptional response to IFNβ alone resembles the response to live H37Rv (Figure 2C).Therefore, we performed K-means clustering on the union of the sets of genes differentially expressed after stimulation with HK, live H37Rv, or IFNβ alone compared to mock stimulation. Overall, both the qualitative trends and the quantitative fold changes induced by IFNβ and live H37Rv are remarkably similar across the >6,000 genes used in the clustering, corroborating the PCA analyses (Figures 2C and 2D).
The clustering allowed us to query whether transcriptional regulation correlates with the metabolic changes observed during infection with live H37Rv. The transcriptional pattern of genes in cluster 4 mirrors the metabolic response of BMDMs: a robust downregulation at late time points after live H37Rv infection but not after HK H37Rv (Figure 2D). To identify the pathways represented by this cluster, we performed hypergeometric enrichment on all Hallmark gene sets from the Molecular Signatures Database (MSigDB) (Liberzon et al., 2015; Subramanian et al., 2005). Five of the top 10 gene sets represent nutrient transporters, central carbon metabolism, or ROS metabolism (Figure 2E).
IFNβ restrains BMDM glycolytic machinery
The observation that IFNβ treatment alone also downregulates the metabolic pathways in cluster 4 (Figures 2D and 2E) suggests that induction of type I IFN could explain the metabolic response of BMDMs to live Mtb. We thus explored whether IFNβ alone induces metabolic changes in BMDMs that could explain those seen upon live H37Rv infection. As a control, we compared the metabolic response following IFNβ treatment to the well-characterized shift to aerobic glycolysis described for stimulation with LPS.
As expected, LPS stimulation for 24 h shifts BMDMs toward aerobic glycolysis, which causes an increased ECAR in the presence of glucose (Figures 3A and 3B). In contrast, IFNβ treatment decreases basal glycolysis and dramatically reduces the capacity to increase glycolysis upon mitochondrial inhibition (Figures 3A and 3B). The reduction in glycolytic capacity occurs at two different doses (Figure S3A) and begins as early as 4 h after treatment (Figure S3B).
Figure 3. IFNβ restrains BMDM glycolytic machinery.

(A) Extracellular acidification rate (ECAR) of WT BMDMs either untreated or treated with 500 U/mL IFNβ, 10 ng/mL LPS, or both for 24 h. A single representative plate is shown. See also Figure S3.
(B) Quantification of glycolytic parameters derived from (A). Each dot represents a single well and the bar is the mean from three (LPS alone), five (IFNβ), or four (both) independent experiments.
(C) Log2FC (RNA-seq) in expression of gene families (rectangles) involved in the KEGG glycolysis pathway comparing WT BMDMs treated with 500 U/mL IFNβ for 28 h to untreated BMDMs. Key metabolites (circles) are labeled.
(D) Western blots of cell lysates from two independent WT mice either untreated or treated with 500 U/mL IFNβ for 24 h.
(E) Quantification of (D) showing four biological replicates across two independent experiments. The bars show the mean.
Although we observed that IFNβ treatment does not increase cell death during the first 48 h (assessed by either LDH release [Figure S3C] or violet viability dye exclusion [Figure S3D]), we noticed that there were fewer cells (Figure S3E) and a reduction in total protein (Figure S3F). These data together with the transcriptional repression of gene sets involved in cell-cycle progression (Figure 2E) suggest that IFNβ reduces BMDM proliferation. We confirmed that IFNβ treatment (and H37Rv infection) reduces proliferation after 24 h using 5-ethynyl-2′-deoxyuridine (EdU) incorporation to monitor DNA synthesis and propidium iodide (PI) to monitor DNA content (Figure S3G). While ~23% of untreated BMDMs are in the S-phase, less than 1% of IFNβ-treated or H37Rv-infected BMDMs are (Figure S3H). However, the changes in glycolytic parameters remained statistically significant after we normalized bulk measurements of ECAR to total protein in each well (Figure S3I), supporting a cell-intrinsic effect of IFNβ on glycolysis in addition to the profound anti-proliferative effect. Interestingly, IFNβ-treated or H37Rv-infected BMDMs maintain their total ATP concentration (Figure S3J) despite reduced glycolysis (Figure S3I) and reduced ATP production (Figure S1F), possibly by reducing energy expenditure toward proliferation.
Transcriptional upregulation of glycolytic machinery contributes to the shift toward aerobic glycolysis in LPS-stimulated BMDMs (Cheng et al., 2014; O’Neill et al., 2016). Although the Hallmark glycolysis gene set is enriched in a cluster of genes downregulated by IFNβ (Figure 2E), the hypergeometric test used is agnostic to the direction of gene regulation. We therefore performed a self-contained rotational gene set test (Wu et al., 2010) that confirmed IFNβ leads to a significant transcriptional downregulation of the Hallmark glycolysis pathway after 28 h (FDR of 4.94E–4). Mapping these transcriptional changes onto the Kyoto Encyclopedia of Genes and Genomes (KEGG) glycolysis pathway shows downregulation of genes at multiple steps (Figure 3C) (and see also Figure S4G for qRT-PCR validation). We confirmed that IFNβ also decreases the protein concentrations of two glycolytic enzymes—glucose-6-phosphate isomerase (GPI) and aldolase C (ALDOC)—after 24 h (Figures 3D and 3E). Interestingly, the first step of the pathway (phosphorylation of glucose) is upregulated, suggesting that glucose-6-phosphate might be shunted to non-glycolytic uses through the pentose phosphate pathway in IFNβ-treated BMDMs (Figure 3C). We reasoned that the downregulation of glycolytic enzymes by IFNβ might restrain the LPS-driven glycolytic shift. Indeed, IFNβ treatment prevents BMDMs from increasing glycolysis during LPS stimulation (Figures 3A and 3B). We therefore conclude that prolonged treatment of BMDMs with IFNβ not only reduces glycolysis but also restrains the metabolic reprogramming that powers inflammatory macrophages.
IFNβ impairs BMDM mitochondrial metabolism
We next investigated the effects of IFNβ treatment itself on mitochondria. BMDMs treated with IFNβ dramatically decrease their OCR, indicative of decreased mitochondrial respiration (Figure 4A). In fact, IFNβ decreases the basal respiratory rate, the rate of ATP generation, and the maximum capacity for electron flux through the mitochondrial ETC (Figure 4B). As expected, BMDMs stimulated with LPS for 24 h shift away from mitochondrial metabolism and display a similar reduction in many aspects of mitochondrial function (Figures 4A and 4B). Although the changes in OCR measurements decrease in magnitude after normalizing for total protein, they remain statistically significant (Figure S4A). Since both LPS and type I IFN induce the cytokine IL-10, which plays a critical role in mitochondrial homeostasis (Ip et al., 2017), we investigated whether IL-10 mediates the IFNβ-driven mitochondrial changes by repeating the OCR measurements in IL-10 KO BMDMs; IFNβ decreases mitochondrial respiration in IL-10 KO cells to the same extent as in the WT, suggesting that the observed changes are IL-10-independent (Figure S4B).
Figure 4. IFNβ impairs mitochondrial function and induces mitochondrial stress in BMDMs.

(A) OCR of WT BMDMs untreated or treated with 500 U/mL IFNβ, 10 ng/mL LPS, or both for 24 h. A single representative plate is shown.
(B) Quantification of mitochondrial parameters derived from (A). Each dot represents a single well and the bar is the mean from four (LPS conditions), six (IFNβ), or three (both) independent experiments. See also Figure S4.
(C) Δψm was calculated by normalizing TMRM to MitoTracker Green (MTG) fluorescence measured by flow cytometry. Representative histograms of BMDMs untreated or treated with 500 U/mL IFNβ or 10 ng/mL LPS for 48 h.
(D) Quantification of Δψm derived from (C). The TMRM/MTG ratio was normalized to the mean of uninfected wells. Each point is a single well and the bar is the mean from three experiments.
(E) Log2FC in expression (RNA-seq) of the 13 protein coding genes encoded on mtDNA in WT BMDMs treated with 500 U/mL IFNβ compared to untreated cells for the indicated time.
(F) Quantification of mROS in WT BMDMs untreated or treated with 500 U/mL IFNβ for indicated times. mROS were measured with MSR MFI normalized to untreated controls at each time point. Each point represents a single well and the bar is the mean from five independent experiments.
Interestingly, treatment of WT BMDMs with both IFNβ and LPS together further suppresses mitochondrial respiration (Figures 4A and 4B), suggesting that they might be impairing mitochondrial function through distinct mechanisms. Indeed, IFNβ and LPS change ΔΨm in opposite directions, even though both stimuli increase mitochondrial mass (Figure S4C): LPS stimulation causes mitochondrial hyperpolarization while IFNβ dissipates ΔΨm after 24 (Figure S4D) and 48 h (Figures 4C and 4D).
As mentioned above, the transcriptional response of BMDMs to prolonged IFNβ treatment includes downregulation of many metabolic pathways (Figure 2). Surprisingly, the most downregulated genes in pathways related to OXPHOS include all 13 protein coding genes encoded on mtDNA (Figure 4E). To test whether these mitochondrial changes decreased the tolerance of IFNβ-treated BMDMs to additional mitochondrial insults, we treated WT or IFNAR KO BMDMs with oligomycin A (an inhibitor of the mitochondrial ATP synthase) with and without IFNβ. Exogenous IFNβ increased cell death in oligomycin-treated BMDMs, and IFNAR KO BMDMs better tolerated the insult (Figure S4F).
Since the changes described above suggested profound mitochondrial damage, we directly quantified one common outcome of such damage, the accumulation of mROS. IFNβ treatment of BMDMs increases mROS at 24 and 48 h (Figure 4F) by signaling through IFNAR (Figure S4E). To explore whether mROS signaling mediates the metabolic changes after IFNβ treatment, we pretreated BMDMs with MitoTEMPO, a mitochondrial-targeted ROS scavenger (Trnka et al., 2008). Although we confirmed the RNA-seq data that IFNβ treatment changes expression of key glycolytic and mitochondrial genes, MitoTEMPO did not restore their transcription to baseline (Figure S4G). MitoTEMPO also failed to restore the mitochondrial respiratory capacity of BMDMs either treated with IFNβ or infected with H37Rv (Figure S4H), suggesting that mROS is a consequence rather than cause of the mitochondrial dysfunction observed.
Taken together, the OCR changes in the setting of single-cell measurements of mitochondrial dysfunction support the conclusion that IFNβ signaling through IFNAR leads to many hallmarks of mitochondrial damage in BMDMs.
Type I IFN controls the metabolic response of macrophages to Mtb infection in vitro and in vivo
Next, we tested the causal role of type I IFN signaling during Mtb infection. We reasoned that if type I IFN directly causes the metabolic changes during infection, IFNAR KO BMDMs infected with live H37Rv should maintain the metabolic function of mock-infected cells. Indeed, IFNAR KO BMDMs, in contrast to WT BMDMs, infected with 2 MOIs of live H37Rv consume oxygen at rates indistinguishable from mock-infected cells (Figures 5A and 5B). The reduction in glycolysis during infection with live H37Rv requires IFNAR signaling as well; infected IFNAR KO BMDMs actually have slightly increased glycolysis compared to mock-infected conditions (Figures 5C and 5D). The genotypic differences in metabolic function are much more pronounced after infection; IFNAR KO BMDMs have similar rates of basal mitochondrial respiration and ATP generation before infection (Figure S5A). Although IFNAR KO BMDMs have a slight increase in maximal respiration before infection (1.17-fold higher than mock WT), this is dwarfed by the difference between genotypes after infection (2.45-fold higher in infected IFNAR KO). We confirmed that the differences in metabolism are not caused by differences in bacterial burden (Figure S5B): IFNAR KO BMDMs and WT BMDMs treated with either exogenous IFNβ or a neutralizing antibody against IFNAR harbor similar numbers of Mtb during a 5-day in vitro infection (Figure S5B).
Figure 5. Type I IFN restrains macrophage metabolism during live Mtb infection.

(A) OCR of WT BMDMs (left) or IFNAR KO BMDMs (right) either mock infected or infected with live H37v at an MOI of 1 or 10 for 24 h. A single representative plate is shown.
(B) Quantification of mitochondrial parameters in BMDMs infected with live H37Rv (MOI of 10) derived from (A). Each point represents a single well and the bar is the mean from seven plates across two independent experiments.
(C) ECAR from the same conditions as in (A). A single representative plate is shown.
(D) Quantification of glycolytic parameters in BMDMs infected with live H37Rv (MOI of 10) derived from (C). Each point represents a single well and the bar is the mean from seven plates across two independent experiments.
(E) Log2FC in expression (RNA-seq) of the 13 protein coding genes encoded on mtDNA in WT or IFNAR KO BMDMs infected with live H37Rv (MOI of 10) for 24 h compared to mock-infected cells of each genotype.
(F) mROS in WT or IFNAR KO BMDMs infected with an MOI of 10 live H37Rv or HK H37Rv for either 24 or 48 h. BMDMs were either untreated or treated with 500 U/mL IFNβ. mROS measured with MSR MFI (flow cytometry) normalized to untreated, mock-infected controls at each time point. A representative experiment of two independent experiments is shown. Statistical tests shown comparing genotypes within the same infection and treatment conditions unless otherwise specified.
(G) OCR during a mitochondrial stress test of alveolar macrophages (AMs) isolated by BAL from WT or IFNAR KO mice either uninfected or infected for 15 days with a high-dose aerosol challenge of H37Rv. One of six plates from two independent experiments is shown.
(H) OCR during a mitochondrial stress test of AMs sorted by fluorescence-activated cell sorting (FACS) from WT or IFNAR KO mice 15 days after aerosol infection. Each line is a technical replicate (across three plates) from one of three independent experiments.
(I and J) Basal glycolysis and glycolytic capacity for (I) monocyte-derived macrophages (MDMs) or (J) AMs sorted by FACS 15 days after aerosol infection. Points are technical replicates (across nine plates) and the bar is the mean for three independent experiments. See also Figure S5.
(K) Bacterial burden in the lung and spleen of WT and IFNAR KO mice 14–15 days after high-dose aerosol infection. CFU were enumerated by serial dilution on 7H10 plates. Each dot is an individual mouse from three independent experiments and the bars represent the mean.
Other measures of mitochondrial physiology support the central role of type I IFN signaling. Although WT and IFNAR KO BMDMs express similar levels of all protein coding genes encoded on mtDNA before infection (Figure S5C), only WT BMDMs, not IFNAR KO BMDMs, downregulate expression of these genes after live H37Rv infection (FDR < 0.01) (Figure 5E). Much less mROS accumulates during infection with live H37Rv in IFNAR KO BMDMs compared to the WT (Figure 5F). Furthermore, BMDMs treated with exogenous IFNβ in addition to HK H37Rv accumulate mROS similar to BMDMs infected with live H37Rv, and this requires intact IFNAR signaling, whereas exogenous IFNβ does not change mROS accumulation in WT BMDMs infected with live H37Rv (Figure 5F). Because an intact ESX-1 type VII secretion system in mycobacteria is required for robust IFNβ induction (Lienard et al., 2020; Wassermann et al., 2015), we tested mROS induction in BMDMs infected with a ΔRD1 H37Rv strain that lacks the ESX-1 system. WT BMDMs infected with low MOIs of the ΔRD1 strain had less mROS accumulation than did those infected with parental H37Rv. Although higher MOIs of DRD1 induced mROS in our hands, this induction still correlated with evidence of type I IFN signaling (Isg15 expression) (Figure S5D) and was also dependent on IFNAR signaling (Figure S5E). Collectively, these data demonstrate that the differential induction of type I IFN by Mtb strains accounts for the observed metabolic differences.
We next investigated whether type I IFN decreases macrophage metabolism in vivo during an Mtb infection. We isolated AMs by bronchoalveolar lavage (BAL) from uninfected WT and IFNAR KO mice and from mice infected with a high dose (~2,000 colony-forming units [CFU]) of H37Rv via aerosol for 15 days. Although WT and IFNAR KO AMs from uninfected mice display comparable mitochondrial respiration by extracellular flux analysis, IFNAR KO AMs from infected mice have significantly increased mitochondrial capacity compared to either IFNAR KO AMs from uninfected mice or WT AMs from infected mice (Figures 5G and S5F). In contrast to infected WT BMDMs, WT AMs from infected mice maintain rates of mitochondrial metabolism similar to WT AMs from uninfected mice, suggesting that additional signals received during infection in vivo counter the type I IFN-mediated mitochondrial restraint.
To confirm and extend these findings to other pulmonary macrophage subsets present during infection, we sorted AMs and MDMs from the lungs of WT and IFNAR KO mice 15 days following aerosol infection (see Figure S5G for the gating scheme). IFNAR KO AMs from infected mice have modestly higher mitochondrial respiration than do WT AMs from infected mice (Figures 5H and S5H). Although mitochondrial parameters are not consistently different between genotypes in MDMs (Figure S5H), this could be explained by recent reports (Huang et al., 2018; Pisu et al., 2020) and our own observations (Figure S5I) that MDMs rely very little on OXPHOS, especially compared to AMs. Both MDMs (Figure 5I and S5J) and AMs (Figure 5J) from infected IFNAR KO mice have elevated basal glycolysis and glycolytic capacity compared to WT from infected mice. Differences in bacterial burden in vivo do not explain the observed metabolic differences; the bacterial burden in both the lung and spleen is similar between genotypes under these infection conditions (Figure 5K). Taken together, these results support a role for type I IFN in restraining macrophage metabolism during Mtb infections both in vitro and in vivo.
STING signaling is upstream of mitochondrial damage during Mtb infection
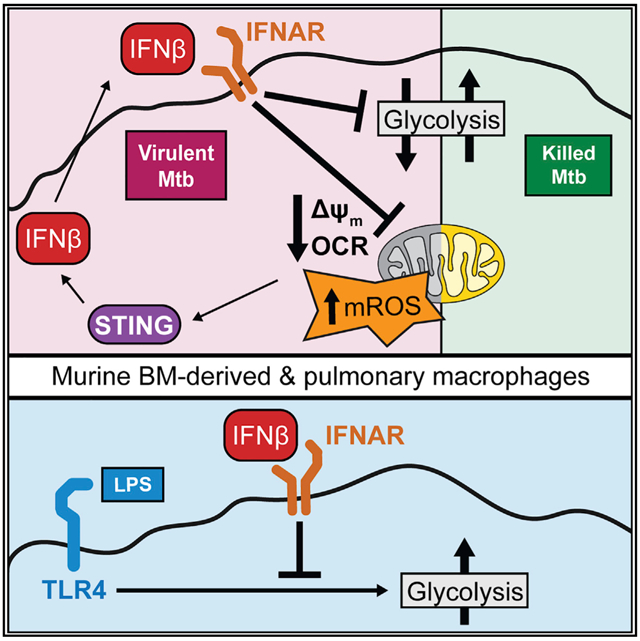
Following Mtb infection, macrophages require the cytosolic DNA sensor cGAS and subsequent signaling through the protein STING to drive robust type I IFN production. Original models described mycobacterial DNA as the main cGAS ligand during infection (Collins et al., 2015; Wassermann et al., 2015; Watson et al., 2015), but more recent reports suggest that mtDNA released by Mtb-induced mitochondrial damage is the main activator of this pathway (Lienard et al., 2020; Wiens and Ernst, 2016). The data presented above suggest that type I IFN itself can induce mitochondrial dysfunction in macrophages. This implies the intriguing possibility that during Mtb infection, type I IFN amplifies mitochondrial damage through a positive feedback loop (Figure 6A). We reasoned that BMDMs lacking STING would allow us to test this model since they have impaired induction of type I IFN during live Mtb infection but retain the ability to signal through IFNAR. If type I IFN is only downstream of mitochondrial damage (as current models claim [Hopfner and Hornung, 2020; Weinberg et al., 2015; West et al., 2015]), then STING KO BMDMs should have mitochondrial damage indistinguishable from WT BMDMs during infection with live Mtb. In contrast, our model—with type I IFN being both upstream and downstream of mitochondrial damage (Figure 6A)—predicts that STING KO BMDMs will have reduced mitochondrial damage during infection. Importantly, exogenous type I IFN added during infection with Mtb in STING KO BMDMs should recapitulate the damage seen in WT macrophages.
Figure 6. STING signaling is upstream of mitochondrial damage during Mtb infection.

(A) Proposed model of positive feedback loop: IFNβ signaling through IFNAR causes mitochondrial damage, which could release mtDNA to be sensed by the cGAS-STING signaling pathway, leading to induction of Ifnb1 expression and IFNβ secretion.
(B) Quantification of glycolytic parameters in BMDMs infected with live H37Rv (MOI of 10). The ECAR representing glycolytic capacity or the glycolytic reserve of infected WT, infected, untreated STING KO, or infected, IFNβ-treated STING KO BMDMs was normalized to mock-infected controls. Each point represents a single well and the bars are the mean from six plates from two independent experiments. See also Figure S6.
(C) Quantification of mitochondrial parameters in BMDMs infected with live H37Rv (MOI of 10). The OCR dedicated to ATP production (ATP) or at maximal respiration (Max) was normalized to mock-infected, untreated controls. Each point represents a single well and the bars are the mean from six plates from two independent experiments.
(D) Quantification of mROS in WT or STING KO BMDMs infected with an MOI of 10 live H37Rv or HK H37Rv for the indicated time. BMDMs were either untreated or treated with 500 U/mL IFNβ. mROS measured with MSR MFI (flow cytometry) normalized to untreated, mock-infected controls at each time point. A representative experiment of two independent experiments is shown.
(E) Principal-component analysis of WT or STING KO BMDMs at 24 h post-infection with an MOI of 10 live H37Rv or HK H37Rv. BMDMs infected with live H37Rv were left untreated or treated with 500 U/mL IFNβ. The percent of variance explained by the top two principal components is indicated.
(F) Log2FC in expression (RNA-seq) of the 13 protein coding genes encoded on mtDNA in BMDMs (either WT or STING KO) infected with live H37Rv at an MOI of 10 for 24 h compared to mock infection. STING KO BMDMs were either left untreated or treated with 500 U/mL IFNβ in addition to the infection.
(G) Expression of Ifnb1 in WT, IFNAR KO, or STING KO BMDMs either mock infected (open symbols and dashed line) or infected with an MOI of 10 live H37Rv (filled symbols and solid line) for 4 h. WT and STING KO BMDMs were either left untreated or treated with 500 U/mL IFNβ. Ifnb1 expression was quantified by qRT-PCR. Each point represents a technical replicate and the bars are the mean from two independent experiments from each KO genotype. Statistics shown for comparisons of Ifnb1 expression after H37Rv infection.
Repeating the previous metabolic analyses in STING KO BMDMs confirms the predictions of our model. Live H37Rv infection in STING KO BMDMs does not restrain glycolysis and only slightly decreases mitochondrial respiration (Figures 6B, 6C, S6A, and S6B). Furthermore, STING KO BMDMs accumulate less mROS than do WT BMDMs after infection (Figure 6D). Consistently, the addition of exogenous IFNβ to infected STING KO BMDMs restores the metabolic dysfunction seen in each assay (Figures 6B-6D, S6B, and S6C), further validating the causal link between type I IFN and mitochondrial damage in macrophages. We confirmed that these metabolic changes track with IFNβ protein concentration: STING KO BMDMs fail to secrete IFNβ 24 h after infection, and the concentration of exogenous IFNβ used throughout this study approximates the concentration measured in supernatants of WT BMDMs infected with H37Rv (Figure S6A).
We next performed RNA-seq on STING KO BMDMs infected with live and HK H37Rv in the absence and presence of exogenous IFNβ. As predicted by our metabolic data, the STING KO response to live H37Rv clusters with the WT or STING KO response to HK H37Rv (Figure 6E), and fewer genes are differentially expressed when directly comparing infection conditions in STING KO BMDMs (Figure S6D). In addition, STING KO BMDMs maintain higher expression of genes encoded on mtDNA than do WT BMDMs upon infection with live H37Rv (Figure 6F). Exogenous IFNβ added to STING KO BMDMs infected with live H37Rv makes the transcriptional response much more closely resemble the WT response to live H37Rv (Figures 6E and 6F), confirming that the lack of induction of type I IFN is the main difference in the Mtb response of STING KO BMDMs.
An Mtb-initiated, type I IFN-mitochondrial positive feedback loop would be predicted to drive increased Ifnb1 expression in BMDMs with intact type I IFN signaling (Figure 6A). Indeed, WT BMDMs expressed greater than 3-fold more Ifnb1 than did IFNAR KO BMDMs after 4 h of infection with live H37Rv (Figure 6G), while expressing similar amounts of Tnf and Il1b (Figure S6E). Exogenous IFNβ further increases Ifnb1 expression in WT BMDMs. We confirmed that live H37Rv infection of STING KO BMDMs does not induce Ifnb1 expression at this time point (Collins et al., 2015; Wassermann et al., 2015; Watson et al., 2015; Wiens and Ernst, 2016) (Figure 6G). Consistent with the positive feedback loop operating through mitochondrial damage and STING activation, exogenous IFNβ in addition to live H37Rv does not rescue full Ifnb1 expression in STING KO BMDMs compared to infected WT BMDMs (Figure 6G).
DISCUSSION
In this study, we describe a role for type I IFN as a master regulator of macrophage metabolism and identify this as the main mechanism modulating metabolism during Mtb infection. Live, but not HK, Mtb restrains macrophage glycolysis and induces hallmarks of mitochondrial stress. We show that both the transcriptional and metabolic responses of WT macrophages to live Mtb resemble the responses to type I IFN alone. We then demonstrate the causal role of type I IFN in the metabolic changes initiated by Mtb infection both in vitro and in vivo; macrophages lacking type I IFN signaling maintain mitochondrial function and glycolytic capacity after infection with live Mtb. Our conclusions are strengthened by experiments showing that exogenous type I IFN in addition to HK Mtb recapitulates the metabolic restraint observed after live Mtb infection.
The utility of type I IFN signatures for the prediction of tuberculosis (TB) progression in humans (Berry et al., 2010; Zak et al., 2016) reinvigorated interest in mechanisms linking type I IFN and Mtb pathogenesis. Although the consensus remains that type I IFN worsens disease, inconsistences in this simple interpretation abound—type I IFN treatment benefits patients with multiple drug-resistant Mtb (Giosué et al., 1998; Palmero et al., 1999), type I IFN improves Mtb vaccination responses (Giacomini et al., 2009; Grode et al., 2005; Gröschel et al., 2017), and most mouse models of impaired type I IFN signaling have only minor differences in disease progression or none at all (the present study) (Moreira-Teixeira et al., 2018). The role that we report herein for type I IFN in regulating the macrophage response to live Mtb might help explain how type I IFN augments vaccination strategies; type I IFN administration with an attenuated vaccine strain might improve anamnestic responses by making the transcriptional response to the vaccine more closely resemble a virulent infection. It is tempting to use the pleiotropic nature of type I IFN to explain the inconsistencies in disease progression during manipulation of this pathway: type I IFN mediates both beneficial and harmful effects in the host, and their relative contributions can lead to a wide spectrum of outcomes. Our incomplete understanding of the specific effects of type I IFN on the macrophage-bacterial interface limits our ability to modulate this pathway in the laboratory or the clinic. The IFNβ-mediated restraint of glycolysis shown in this study might be a key harmful effect for this cytokine, since glycolysis is crucial for macrophage control of Mtb (Braverman et al., 2016; Huang et al., 2018; Marín Franco et al., 2020).
The connections between immunometabolic changes and proliferation are often neglected, especially in macrophages. Cell division places a substantial metabolic and biosynthetic burden on cells, and bulk measurements can hide effects secondary to changes in cell number. In this study, we find evidence that Mtb infection and type I IFN decrease macrophage metabolism through both direct cell-intrinsic metabolic rewiring and indirect inhibition of macrophage proliferation; the normalized bulk measurements and single-cell assays confirm the cell-intrinsic effects, and the transcriptional changes and cessation of EdU incorporation clearly support cell-cycle arrest. Interestingly, there is a growing appreciation that proliferation is crucial for tissue macrophage function (Jenkins et al., 2011; Rosas et al., 2014) and that Mtb alters host cell proliferation (Cumming et al., 2017; Huang et al., 2018; Pisu et al., 2020), including modulation of bone marrow stem cell differentiation in a type I IFN-dependent manner (Khan et al., 2020). The connection between type I IFN, metabolic modulation, and proliferation shown in this study provides another avenue to explore the multifaceted role of type I IFN in Mtb pathogenesis.
The study of the modulation of metabolism by type I IFN is in its infancy, but it is clear that this cytokine family can have drastically different metabolic consequences in different cell types (Burke et al., 2014; Kissig et al., 2017; Pantel et al., 2014; Wu et al., 2016). The effects of type I IFN on macrophage metabolism have not been explored thoroughly, although intriguing interactions exist between type I IFN and macrophage cholesterol homeostasis (Blanc et al., 2011, 2013; York et al., 2015). Although a recent report showed that macrophages have divergent metabolic responses to different TLR ligands that are known to differentially induce type I IFN, the authors did not explore the direct contributions of type I IFN (Ahmed et al., 2019). Additional studies implicate type I IFN in macrophage metabolic changes during intracellular bacterial infections (Hos et al., 2017; Howard et al., 2018), but they rely on indirect evidence. Since the metabolic profile we observed during live Mtb infection has not been reported previously as an outcome of type I IFN signaling in macrophages as far as we know, we characterized the metabolic alterations in macrophages after type I IFN treatment. We describe a metabolic response to type I IFN itself that mirrors that to live H37Rv; IFNβ treatment impairs mitochondrial function and restrains the shift toward aerobic glycolysis in LPS-stimulated macrophages. This restraint highlights the need to extend current immunometabolic models derived from studies with single purified stimuli to include how multiple signals are integrated to coordinate metabolic responses across the wide spectrum of diseases that include robust type I IFN signaling.
The metabolic changes effected by IFNβ itself would predict that both infected macrophages and bystander macrophages (themselves uninfected but exposed to soluble type I IFN) decrease their energy metabolism upon infection. Indeed, the type I IFN-mediated metabolic restraint observed in bulk pulmonary macrophages supports this conclusion, since only ~2% of AMs and <1% of MDMs are infected 2 weeks after a high-dose aerosol infection similar to the one used in this study (Rothchild et al., 2019). Future studies should compare the metabolic profiles of infected and bystander cells to unravel the relative contributions of soluble type I IFN, infection-specific processes, and their interactions (Banks et al., 2019).
We show that the metabolic restraint of macrophages during Mtb infection requires STING signaling and that treatment of Mtb-infected STING KO macrophages with exogenous type I IFN recapitulates the mitochondrial damage seen in WT macrophages. Current models place type I IFN downstream of mitochondrial damage and subsequent cGAS-STING signaling (Hopfner and Hornung, 2020; Weinberg et al., 2015; West et al., 2015). We suggest that the additional placement of type I IFN upstream of mitochondrial dysfunction described in this study creates a positive feedback loop in macrophages. Although we show evidence that type I IFN signaling potentiates type I IFN expression in a STING-dependent manner consistent with a mitochondrial-driven loop, further study is required to rule out other established IFN feedback mechanisms.
Does mitochondrial damage or type I IFN induction initiate the loop during Mtb infection? Studies using recombinant mycobacterial proteins suggest that Mtb effector molecules target the mitochondria (Cadieux et al., 2011; Shin et al., 2010; Sohn et al., 2011), but further studies are needed to characterize direct Mtb-mitochondrial interactions in more physiologic settings. It is noteworthy, however, that the lack of robust mitochondrial damage in IFNAR and STING KO BMDMs argues against widely held assumptions that direct pathogen effectors are the primary explanation for metabolic changes during infections with intracellular bacteria. Although STING is required for robust type I IFN expression during Mtb infection (Collins et al., 2015; Wassermann et al., 2015; Watson et al., 2015), recent studies suggest that the ligands are mitochondrial rather than mycobacterial (Lienard et al., 2020; Wiens and Ernst, 2016). The model proposed in the current study might help resolve this perceived conflict: early minor activation by mycobacterial ligands might initiate an IFN-mitochondrial-STING loop that is required for robust induction. The minor induction of type I IFN by other recognition receptors (Cheng and Schorey, 2018; Leber et al., 2008; Pandey et al., 2009; Troegeler et al., 2017) or mitochondrial damage by other host pathways (Weindel et al., 2020) might also contribute to initiation. Consistent with this, STING KO BMDMs infected with live H37Rv and WT BMDMs only treated with IFNβ had intermediate levels of mROS (i.e., higher than in mock-treated cells but lower than in WT BMDMs infected with live H37Rv).
In summary, we think that this study provides important mechanistic insight into the metabolic response of macrophages to a clinically relevant pathogen. Furthermore, we think that investigating the metabolic effects mediated by type I IFN on macrophages offers many opportunities to disentangle the complex beneficial and detrimental roles that this fundamental cytokine family plays in immunity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Elizabeth Gold (Elizabeth.Gold@seattlechildrens.org).
Materials availability
There are restrictions to the availability of the MyD88-TRIF double KO mouse line used in this study due to the requirement for a material transfer agreement for mouse distribution.
Data and code availability
The RNA sequencing data generated during this study are available in GEO: GSE162620. Additional data supporting the current study are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
WT (C57BL6/J; RRID:IMSR_JAX:000664), IFNAR KO (B6.129S2-Ifnar1tm1Agt/Mmjax, RRID:MGI:3703445), STING KO (B6(Cg)-Sting1tm1.2Camb/J, RRID:IMSR_JAX:025805), and IL10 KO (B6.129P2-Il10tm1Cgn/J, RRID:IMSR_JAX:002251) strains of Mus musculus were obtained from The Jackson Laboratory. MyD88-TRIF double KO mice were generated from the two strains MyD88 KO (B6.129P2(SJL)-Myd88tm1.1Defr/J, RRID:IMSR_JAX:009088) and TRIF KO (C57BL/6J-Ticam1Lps2/J, RRID:IMSR_JAX:005037). All KO mouse experiments used only homozygous animals. All mice were housed and maintained in specific pathogen–free conditions at the Seattle Children’s Research Institute (SCRI). All experiments were approved by the Institutional Animal Care and Use Committee and then performed in compliance with the relevant protocols. Healthy eight- to 14-week-old female mice without any previous procedure history were used for all experiments and were age-matched within each experiment.
Bone marrow-derived macrophages
Female mice (see section on Mice for further details) were euthanized with carbon dioxide asphyxiation. Bone marrow was harvested from the femur and tibia by flushing with BMDM media (RPMI 1640 supplemented with recombinant M-CSF (50 ng/mL), L-glutamine (final concentration 4 mM), and 10% fetal bovine serum (FBS)) plus penicillin and streptomycin (p/s). The flushed marrow was homogenized by pipetting and then filtered through a 70 μm filter before being placed at 37°C, 5% CO2 in four 15 cm plates. Fresh BMDM media was added on day 3 or 4 and then on day 6 the adherent cells were washed twice and then lifted with phosphate buffered saline (PBS) + 2mM EDTA. Cells were replated in BMDM media without p/s and rested overnight at 37°C, 5% CO2 before treatments or infections were added.
Pulmonary macrophage subsets
WT or IFNAR KO female mice were either left uninfected or infected with Mtb by aerosol (see below for infection details) and then group housed, segregated by genotype, in a biosafety level 3 facility in an animal biohazard containment suite.
Bronchoalveolar lavage (BAL)
Uninfected mice and mice infected for 15 days were euthanized (cervical dislocation) and cells present in the pulmonary alveoli were collected by BAL using four consecutive rounds of three washes with an intratracheal instillation of 0.7mL sterile PBS. The BAL fluid was kept on ice and then cells were collected by centrifugation (350 g for 10 minutes at 4°C) and resuspended in ice-cold RPMI + 10% FBS + penicillin and streptomycin. The alveolar macrophages (AM) were then counted with a hemocytometer (excluding red blood cells) and plated at 40,000 cells per well of an Agilent XFp miniplate. Five hours after plating, the non-adherent cells were removed by gentle but thorough washing to produce a purified population of AM which were then analyzed using the XFp Seahorse. To account for the variations in AM numbers introduced by the increased washing, 20x micrographs were taken immediately before loading onto the machine and all seahorse measurements were normalized to the # of cells counted in a predefined area within each well.
Fluorescence-activated cell sorting (FACS)
Infected mice were euthanized (cervical dislocation) and the superior, middle, and inferior lobes of the right lung were isolated by dissection. Single-cell suspensions of lung mononuclear cells were prepared by Liberase TM (70 μg/ml) digestion in 1 μM HEPES buffer containing deoxyribonuclease I (DNase I) (30 μg/ml) for 30 min at 37°C and mechanical disruption using a gentleMACS dissociator (Miltenyi Biotec), followed by filtering through a 70 μm cell strainer. Red blood cells were removed by incubating with ACK lysis buffer for 2 minutes before neutralization with FBS. Cells were stained with LIVE/DEAD Fixable Violet Dead Cell kit and anti-CD16/CD32 was added to block Fc receptors to reduce non-specific binding before addition of the fluorescently-conjugated antibodies listed in the Key Resources Table. Stained cells were resuspended in PBS + 5% FBS + 10 mM HEPES before sorting with a FACSAria II (BD Biosciences). AM were identified as live singlets that were CD45+CD3−CD19−Ly6G−CD11c+SiglecF+ and Monocyte Derived Macrophages (MDM) were identified as live singlets that were CD45+CD3−CD19−Ly6G−SiglecF−MHC-II+CD64+ (and see Figure S5 for gating). Sorted AM and MDM were kept on ice in RPMI 1640 + 10% FBS before plating in Agilent XFp mini-plates and subsequent XFp Seahorse analysis.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Brilliant violet 570 rat monoclonal anti-mouse CD11b (clone M1/70) | Biolegend | Cat# 101233; RRID:AB_10896949 |
| Brilliant violet 605 Armenian hamster monoclonal anti-mouse CD11c (clone N418) | Biolegend | Cat# 117333;RRID:AB_11204262 |
| Brilliant violet 711 rat monoclonal anti-mouse Ly-6G (clone 1A8) | Biolegend | Cat# 127643; RRID:AB_2565971 |
| Brilliant violet 785 rat monoclonal anti-mouse Ly-6C (clone HK1.4) | Biolegend | Cat# 128041; RRID:AB_2565852 |
| Alexa fluor 488 rat monoclonal anti-mouse MERTK (clone DS5MMER) | Thermo Fisher Scientific | Cat# 53-5751-82; RRID:AB_2784751 |
| PerCP cyanine5.5 mouse monoclonal anti-mouse CD45.2 (clone 104) | Biolegend | Cat# 109828; RRID:AB_893350 |
| Phycoerythrin rat monoclonal anti-mouse Siglec-F (clone E50-2440) | BD Biosciences | Cat# 552126; RRID:AB_394341 |
| PE cyanine7 mouse monoclonal anti-mouse CD64 (clone X54-5/7.1) | Biolegend | Cat# 139313; RRID:AB_2563903 |
| APC rat monoclonal anti-mouse I-A/I-E (MHC class II) clone M5/114.15.2) | Biolegend | Cat# 107614; RRID:AB_313329 |
| APC-eFluor 780 Rat monoclonal anti-mouse CD19 (clone eBio1D3(1D3)) | Thermo Fisher Scientific | Cat# 47-0193-82; RRID:AB_10853189 |
| APC-eFluor 780 rat monoclonal anti-mouse CD3 (clone 17A2) | Thermo Fisher Scientific | Cat# 47-0032-82; RRID:AB_1272181 |
| Unconjugated rat monoclonal anti-mouse CD16/CD32 (clone 2.4G2) | BD Biosciences | Cat# 553141; RRID:AB_394656 |
| Functional-grade mouse monoclonal anti-mouse IFNAR (cloneMAR1-5A3) | Thermo Fisher Scientific | Cat# 16-5945-85; RRID:AB_1210688 |
| Unconjugated rabbit polyclonal antiGlucose-6-phosphate isomerase (GPI) | Thermo Fisher Scientific | Cat# PA5-26787; RRID:AB_2544287 |
| Unconjugated rabbit polyclonal antiAldolase C | Proteintech | Cat# 14884-1-AP; RRID:AB_2226691 |
| HRP mouse monoclonal anti-beta actin (clone mAbcam 8226) | Abcam | Cat# ab20272; RRID:AB_445482 |
| HRP goat polyclonal anti-Rabbit IgG Fc | Jackson ImmunoResearch Labs | Cat# 111-035-008; RRID:AB_2337937 |
| Bacterial and virus strains | ||
| Mycobacterium tuberculosis, H37Rv | BEI Resources | Cat#NR-123 |
| Mycobacterium tuberculosis, H37Rv ΔRD1 | David Sherman, Rothchild et al., 2019 | N/A |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Lipopolysaccharide from Salmonella Minnesota | List Labs | Cat#R595 |
| Synthetic triacylated lipopeptide (Pam3CSK4) | Invivogen | Cat#vac-pms |
| Interferon-β Protein, Recombinant mouse | Millipore Sigma | Cat#IF011 |
| Recombinant human M-CSF | PeproTech | Cat#300-25 |
| MitoTEMPO | Enzo Biochem | Cat#ALX-430-150 |
| Liberase TM sesearch Grade | Millipore Sigma | Cat#5401127001 |
| DNase I grade II | Millipore Sigma | Cat#10104159001 |
| ACK lysing buffer | Thermo Fisher Scientific | Cat#A1049201 |
| 20% paraformaldehyde aqueous solution | Electron Microscopy Sciences | Cat#15713-S, CAS#50-00-0 |
| TRIzol reagent | Thermo Fisher Scientific | Cat#15596026 |
| RIPA buffer (10X) | Cell Signaling Technology | Cat#9806 |
| Halt protease inhibitor Cocktail (100X) | Thermo Fisher Scientific | Cat#78430 |
| Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) | Millipore Sigma | Cat#C2920, CAS#370-86-5 |
| Oligomycin A | Millipore Sigma | Cat#75351, CAS#579-13-5 |
| Antimycin A from streptomyces sp. | Millipore Sigma | Cat#A8674, CAS#1397-94-0 |
| Rotenone | Millipore Sigma | Cat#R8875, CAS#83-79-4 |
| 2-deoxy-D-glucose | Millipore Sigma | Cat#D8375, CAS#154-17-6 |
| D-(+)-glucose | Millipore Sigma | Cat#G7528, CAS#50-99-7 |
| Tetramethylrhodamine, methyl ester, perchlorate (TMRM) | Thermo Fisher Scientific | Cat#T668, CAS#115532-50-8 |
| Critical commercial assays | ||
| CyQUANT LDH cytotoxicity assay | Thermo Fisher Scientific | Cat#C20301 |
| Pierce BCA protein assay kit | Thermo Fisher Scientific | Cat#23225 |
| VeriKine mouse interferon Beta ELISA | PBL Assay Science | Cat#42400 |
| ADP/ATP ratio assay kit (bioluminescent) | abcam | Cat#ab65313 |
| Click-iT Plus EdU alexa fluor 647 flow cytometry assay kit | Thermo Fisher Scientific | Cat#C10635 |
| FxCycle PI/RNase staining solution | Thermo Fisher Scientific | Cat#F10797 |
| Direct-zol-96 RNA kits | Zymo Research | Cat#R2056 |
| RNA to cDNA ecoDry premix (Oligo dT) | Takara Bio | Cat#639542 |
| Taqman fast universal PCR master mix (2x), no AmpErase UNG | Thermo Fisher Scientific | Cat#4364103 |
| LIVE/DEAD fixable violet dead cell stain kit | Thermo Fisher Scientific | Cat#L34964 |
| MitoSOX red mitochondrial superoxide indicator | Thermo Fisher Scientific | Cat#M36008 |
| MitoTracker green FM | Thermo Fisher Scientific | Cat#M7514 |
| Seahorse XFp cell glycolysis Stress Test Kit | Agilent | Cat#103017-100 |
| Seahorse XFp cell mito stress test kit | Agilent | Cat#103010-100 |
| Seahorse XFp real-time ATP rate assay kit | Agilent | Cat#103591-100 |
| Deposited data | ||
| RNA sequencing data | This paper | GEO: GSE162620 |
| Experimental models: organisms/strains | ||
| Mouse: STING KO: B6(Cg)-sting1tm1.2Camb/J Mus musculus | The Jackson Laboratory | Cat#025805, RRID:IMSR_JAX:025805 |
| Mouse: IFNAR KO: B6. 129S2-Ifnar1tm1Agt/Mmjax Mus musculus | The Jackson Laboratory (MMRRC) | Cat#32045-JAX, RRID:MGI:3703445 |
| Mouse: IL10 KO: B6.129P2-Il10tm1Cgn/J Mus musculus | The Jackson Laboratory | Cat#002251, RRID:IMSR_JAX:002251 |
| Mouse: MyD88-Trif double KO: B6.129P2(SJL)-Myd88tm1.1Defr/J x C57BL/6J-Ticam1Lps2/J | Aderem lab; this paper. Parental strains: The Jackson Laboratory | Parental strains: Cat#009088 (RRID:IMSR_JAX:009088) and Cat#005037 (RRID:IMSR_JAX:005037) |
| Mouse: WT: C57BL/6J Mus musculus | The Jackson Laboratory | Cat#000664, RRID:IMSR_JAX:000664 |
| Oligonucleotides | ||
| See Table S1 for full listing of oligonucleotides used | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| RStudio 1.1.463 | RStudio | https://www.rstudio.com, RRID:SCR_000432 |
| edgeR 3.26.8 | Robinson et al., 2010 | http://bioconductor.org/packages/release/bioc/html/edgeR.html, RRID:SCR_012802 |
| R 3.6.1 | Cran | https://www.r-project.org/, RRID:SCR_001905 |
| FastQC | Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/, RRID:SCR_014583 |
| GSNAP 2018-07-04 | Wu and Nacu, 2010 | http://research-pub.gene.com/gmap/, RRID:SCR_005483 |
| featureCounts v1.5.2 | Liao et al., 2014 | http://bioinf.wehi.edu.au/featureCounts/, RRID:SCR_012919 |
| CellProfiler image analysis Software 3.1.9 | McQuin et al., 2018 | https://cellprofiler.org/, RRID:SCR_007358 |
| (Fiji is just) ImageJ 1.53c | Schindelin et al., 2012 | https://fiji.sc/, RRID:SCR_002285 |
| Molecular signatures database (MSigDB) v7.2 | Subramanian et al., 2005 | https://software.broadinstitute.org/gsea/msigdb/index.jsp, RRID:SCR_016863 |
| Seahorse Wave 2.6.1.53 | Agilent | https://www.agilent.com/en-us/products/cell-analysis-(seahorse)/software-download-for-wave-desktop, RRID:SCR_014526 |
| FlowJo 10.5.3 | Becton, Dickinson and Company | https://www.flowjo.com/solutions/flowjo, RRID:SCR_008520 |
| Pathview 1.24.0 | Luo and Brouwer, 2013 | http://bioconductor.org/packages/release/bioc/html/pathview.html, RRID:SCR_002732 |
| HypeR 1.3.1 | Federico and Monti, 2020 | https://www.bioconductor.org/packages/release/bioc/html/hypeR.html |
| Other |
METHOD DETAILS
Infections and stimulations
BMDM infection in vitro with and without IFNβ treatment
An aliquot of H37Rv (a virulent strain of lineage 4 Mtb) was thawed and cultured at 37°C in 7H9 media supplemented with 0.2% glycerol, 0.1% tween-80, and 10% OADC at 37°C and ambient CO2. Cultures were used during log-phase growth, (i.e., when the optical density at 600nm (OD600) was between 0.1 and 0.3). The final concentration of bacteria was calculated using the OD600 and the appropriate number of bacteria was separated from the stock culture and centrifuged at 3000-4000 g for 10 minutes before resuspension in BMDM media at the appropriate concentration for the desired MOI. For some experiments, the resuspended H37Rv was heated to 99°C for 10 minutes and then cooled to RT for at least 5 minutes to produce heat killed (HK) H37Rv. Mock infections were performed with fresh BMDM media with no bacteria added. Some infections used an H37Rv strain with a deletion of the virulence determinant RD1 region (ΔRD1), provided by D. Sherman (Univ. of Washington, Seattle, WA) (Rothchild et al., 2019). The bacteria were added to the macrophages for 3-4 hours at 37°C, 5% CO2 before the macrophages were washed three times with pre-warmed BMDM media to remove extracellular bacteria and fresh media was added. Antibiotics were not used at any step of this process. In some experiments, 500 U/mL IFNβ was added to BMDM cultures 4 hours before infection and included with the bacteria during the 3-4 hour infection. No IFNβ was added after the wash at the end of the infection incubation. For CFU measurement, cells were lysed with 1% Triton X-100 in PBS and lysates were plated in serial dilutions in 0.05% Tween-80 in PBS on Middlebrook 7H10 agar plates and cultured at 37°C for 21-28 days.
Additional treatments
Frozen stocks of LPS or PAM3Csk4 were thawed and sonicated for 5 minutes before dilution in BMDM media. The diluted stimuli were again sonicated for 5 minutes before addition to BMDM cultures for a final concentration of 10 ng/mL for LPS and 300 ng/mL for PAM3Csk4. In some experiments, 500 U/mL IFNβ was added at the same time as LPS stimulation. Oligomycin A was diluted in BMDM media before addition to BMDM cultures at a final concentration of 1 μM. MitoTEMPO (Trnka et al., 2008) was added at a final concentration of 200uM for 1 hour to allow accumulation within mitochondria before subsequent infection or stimulations were added.
Mouse aerosol infection
For high-dose (~1000 to 2000 CFU) infections, mice were enclosed in an aerosol infection chamber (Glas-Col) and frozen stocks of bacteria were thawed and placed inside the associated nebulizer. To determine the infectious dose, three mice in each infection were sacrificed after the aerosolization was complete. The whole lung was homogenized in 0.05% Tween-80 in PBS with a gentleMACS Tissue Dissociator (Miltenyi Biotec) and serial dilutions were plated onto 7H10 plates for CFU enumeration, as described previously (Rothchild et al., 2020). Bacterial growth and dissemination was assessed as above for the right lung lobe and the entire spleen at day 14-15 after aerosol infection.
Metabolic flux analysis
XFp
An XFp analyzer (Agilent) was used to determine OCR and ECAR of macrophages in experiments including live H37Rv Mtb. BMDM were plated at 25,000 cells/well of an XFp miniplate (Agilent) on day 6 of differentiation and rested overnight. After sorting, MDM and AM were plated at 60,000 cells/well and rested overnight. The XFp Mito Stress Test Kit,XFp Glycolysis Stress Test Kit, or Real-time ATP Rate Assay Kit (Agilent) was run according to the manufacturer’s instructions. Briefly, the XFp cartridge was hydrated in sterile H2O overnight and then the H2O was replaced with XF Calibrant (Agilent) for 1 hour before calibration on the XFp analyzer. Macrophage culture media was removed and replaced with XF RPMI pH 7.4 (Agilent) supplemented with just 2 mM L-glutamine (Glycolysis Stress Test) or an additional 1mM sodium pyruvate and 10 mM glucose (Mito Stress Test and ATP Rate Assay) for 1 hour before a second wash with the same media immediately before loading in the analyzer. The kit compounds were resuspended in supplemented XF media and loaded into the cartridge injection ports such that the final concentrations of the compounds in each well were oligomycin A 5 μM, FCCP 3 μM (BMDM and sorted cells) or 5 μM (BAL AM), and rotenone and antimycin both at 1 μM (Mito Stress Test), oligomycin 5 μM and rotenone/antimycin both at 1 μM (ATP Rate), or glucose 10 mM, oligomycin 5 μM, 2-deoxyglucose 50 mM (Glycolysis Stress Test). FCCP concentrations were determined by a dedicated titration assay done separately on BMDM and BAL AMs.
XFe24
An Agilent XFe24 analyzer was used to compare the effects of IFNβ and/or LPS treatment on BMDM. BMDM were plated at 100,000 cells/well of an XFe24 miniplate (Agilent) on day 6 of differentiation and rested overnight. The XFe24 cartridge was hydrated in Agilent XF Calibrant overnight. BMDM culture media was removed and the cells were washed twice with appropriately supplemented XF RPMI pH 7.4 (Agilent) media 1 hour before loading in the analyzer. The same final concentrations of compounds as for the XFp analyzer were used, although the compounds were obtained independently rather than from the pre-formulated Agilent kits.
Mitochondrial flow cytometry
Mitochondrial membrane potential
After 6 days of differentiation, BMDM were replated at 400,000 cells/well of a non-TC treated 24 well dish in BMDM media without p/s. The culture media was removed, and macrophages were washed once with pre-warmed staining media (RPMI 1640 without phenol red + 10 mM HEPES buffer) before incubation with 100 nM TMRM and 100 nM MitoTracker Green (MTG) in staining media for 30 minutes at 37°C, 5% CO2. The staining media was removed, and macrophages were lifted off the culture plates using gentle pipetting after a 5-minute incubation at 4°C in ice-cold PBS + 2 mM EDTA. The macrophages were centrifuged in 96-well U-bottom plates at 300 g for 6 minutes at 4°C before staining with the LIVE/DEAD Fixable Violet Dead cell kit for 8 minutes. This was washed off and the cells were resuspended in PBS + 5% FBS before analysis on an LSR II cytometer (BD biosciences) using the following details (excitation laser; emission bandwidth filter): LIVE/DEAD (405 nm; 450/50), MTG (488 nm; 515/20), TMRM (532 nm; 575/25). Events were first gated for constant flow rate using time and then for singlets and FSC/SSC to gate out debris before selecting only live cells based on LIVE/DEAD exclusion. The ratio of TMRM/MTG was calculated for each live cell to account for total mitochondrial content and reported as the mitochondrial membrane potential (Δψm).
Mitochondrial reactive oxygen species
Analysis of mitochondrial reactive oxygen species (mROS) used the above protocol with the following adjustments. Staining was done with 2.5 mM MitoSOX Red (MSR) in staining media for 10 minutes at 37°C, 5% CO2. After washing off the LIVE/DEAD stain, the macrophages were fixed with 2% paraformaldehyde in PBS for 1 hour at room temperature before resuspension in PBS + 5% FBS and analysis on an LSR II (BD biosciences) using the following laser;filter combinations: LIVE/DEAD (405 nm; 450/50), MSR (405 nm; 585/42). Live non-debris singlets were analyzed for fluorescence in the MSR channel to quantify mROS. Excitation of MSR at 405 nm more specifically measures mitochondrial superoxide production by reducing the influence of cytosolic hydrogen peroxide (Robinson et al., 2006).
Cell count
Since all mitochondrial flow cytometry was acquired at a constant flow rate using a 96-well high-throughput system, relative cell numbers in each sample could be quantified. The number of live singlet cells collected during a stable 10 s flow rate (gating on time) was calculated for each sample and normalized to untreated, mock-infected conditions for each experiment.
Mitochondrial microscopy
After 6 days of differentiation, BMDM were replated at 200,000 cells/well of a glass-bottom TC-treated24 well dish (Greiner Bio-One). After infection, culture media was removed, and the cells were washed with pre-warmed staining media before adding 100 nM TMRM and 100 nM MTG diluted in staining media. After 30 minutes at 37°C, 5% CO2, micrographs were taken with a Ti-E inverted microscope (Nikon Instruments Inc.) using a 20X objective and the following filter cubes (excitation bandwidth filter; dichroic mirror; emission filter): TMRM (545/30; 570; 620/60) and MTG (470/40; 500; 535/50). For quantification of TMRM fluorescence, raw images in both channels first had background subtraction done in Fiji (ImageJ) (Schindelin et al., 2012) using the rolling ball algorithm (radius 40 pixels). Background subtracted images were then analyzed using CellProfiler (McQuin et al., 2018): Images were masked based on mitochondria identified using global RobustBackground thresholding in both the TMRM and MTG channels. TMRM fluorescence was quantified in the masked image to assess Δψm and MTG fluorescence quantified to assess mitochondrial mass.
Protein quantification
Western blot
After 6 days of differentiation, BMDM were replated at 106 cells/well of a TC-treated 6 well dish. After 24 hours of treatment with or without 500 U/mL IFNβ, culture media was removed, and the cells were washed once with PBS before protein collection with RIPA buffer and HALT protease inhibitor. Western blotting analyses were performed using standard techniques and transblotted onto nitrocellulose membranes. After blocking with 5% milk for 1 hour, membranes were probed with relevant primary antibodies anti-GPI (1:1000 dilution) or anti-ALDOC (1:5000 dilution) overnight at 4°C. Primary antibodies were detected by an HRP-conjugated secondary goat anti-rabbit antibody (1:10000). Membranes were then stripped and then re-blocked and probed with HRP-conjugated anti-β-actin (1:50000). Images were quantified using ImageJ densitometry analysis.
IFNβ enzyme-linked immunosorbent assay (ELISA)
BMDM cell culture supernatants were collected and frozen at −20°C. If the experiment included live Mtb, the supernatants were filtered twice through 0.2 μm filters (Pall Corporation) before removal from the BSL-3 suite. Supernatants were thawed at 4°C and IFNβ concentrations were determined with a mouse IFNβ ELISA assay according to the manufacturer’s specifications. Absorbance was recorded at 450 nm with wavelength correction performed by subtracting absorbance at 570 nm using a SpectraMax M2 plate reader (Molecular Devices).
Gene expression
RNA preparation
After 6 days of differentiation, BMDM were replated at 400,000 cells/well of a TC-treated 24 well dish. After the indicated amount of time of infection or stimulation, culture media was removed, and the cells were washed once with PBS before being lysed in TRIzol for 5 minutes at room temperature and frozen at −80°C. TRIzol was thawed and total RNA extracted using Direct-zol-96 extraction kits according to manufacturer’s instruction, including an on-column DNase treatment and elution with 25 μL H2O. RNA integrity was confirmed using 260/280 and 260/230 nm ratios measured by a Nanodrop One Microvolume UV-Vis Spectrophotometer (Thermo Fisher Scientific).
Quantitative reverse transcription PCR
RNA (500 – 1000 ng) was converted to cDNA using the RNA to cDNA EcoDry Premix (Oligo dT) according to manufacturer instructions. RT-qPCR reactions were carried out with 15-20 ng cDNA using TaqMan primer probes and the TaqMan Fast Universal PCR Master Mix (Thermo Fisher Scientific) in the QuantStudio 5 Real-Time PCR System (Applied Biosystems). Data were normalized to Eef1a1 expression in individual samples.
RNA-seq
Two different batches of RNA sequencing were done. The first batch was sent to BGI (bgi.com) for library prep and sequencing. The BV01 protocol was used for library prep and then the libraries were sequenced on a DNBSEQ-G400. The second batch was sent to Psomagen (Psomagen.com) for library preparation and sequencing. The Illumina TruSeq stranded kit was used for library prep and then the libraries were sequenced on an Illumina NovaSeq6000 S4. Both sequencing runs produced 100 bp paired end reads. Fastq files from both batches were processed in parallel (described in section on Quantification).
Cell-cycle analysis
Differentiated BMDM were plated at 4*105 cells/well of a non-TC treated 24 well plate and then infected with an MOI 10 of H37Rv, treated with 500 U/mL IFNβ, or left uninfected and untreated as controls. After 24 hours, a Click-iT Plus EdU Assay Kit and FxCycle PI/RNase were used according to manufacturer’s instructions to assess the cell-cycle status of the cells. Briefly, 5-ethynyl-2′-deoxyuridine (EdU) was added to a final concentration of 10 μM in experimental wells and the cells incubated for 1.5 hours at 37°C to allow incorporation. The macrophages were then washed with 1% bovine serum albumin (BSA) in PBS and were lifted off the culture plates using gentle pipetting after a 5-minute incubation at 4°C in ice-cold PBS + 2mM EDTA. The macrophages were centrifuged in 96-well U-bottom plates at 300 g for 6 minutes at 4°C before staining with the LIVE/DEAD Fixable Violet Dead cell kit for 8 minutes at 4°C before another wash and fixation with Component D (4% paraformaldehyde). The cells were washed and resuspended in 1X Permeabilization/Wash reagent and incubated at room temperature for 15 minutes before adding the reaction cocktail with Alexa Fluor 647 picolyl azide. After a 30-minute incubation at room temperature, the cells were washed and resuspended in FXCycle PI/RNase staining solution and incubated for 15 minutes before acquisition with an LSR II flow cytometer (BD biosciences). Cells were identified as being in S phase if they were EdU-Alexa Fluor 647 positive by flow, with medium to high intensity staining for PI.
ATP concentration
Differentiated BMDM were plated at 105 cells/well in a 96 well plate and then infected with an MOI 10 H37Rv or treated with 500 U/mL IFNβ. After 24 hours, ATP concentrations were measured using a bioluminescent ATP assay kit according to the manufacturer’s instructions. Briefly, the culture media was removed, and Nucleotide Releasing Buffer was added for 5 minutes with gentle agitation. These samples were transferred to a new plate and frozen at −80°C. Thawed samples were then added to the ATP monitoring enzyme working reagent and bioluminescence was measured after 2 minutes. A standard curve with ATP disodium was used to quantify ATP concentrations, and these were normalized to control BMDM to calculate fold change.
Lactate dehydrogenase assays
Concentrations of lactate dehydrogenase (LDH) present in the supernatants collected from BMDM cultures was one method used to monitor cell death. If the experiment included live Mtb, the supernatants were filtered twice through 0.2 μm filters (Pall Corporation) before removal from the BSL-3 suite. LDH concentrations were determined with the CyQUANT LDH Cytotoxicity Assay (Thermo Fisher Scientific) according to manufacturer’s instructions. Briefly, samples were mixed with the reconstituted reaction mixture for 30 minutes in the dark at room temperature before the stop solution was added. The absorbance at 750 nm (background) was subtracted from the absorbance at 490 nm (signal) and normalized to the background-subtracted 490 absorbance of dedicated untreated wells within each experiment that had been lysed at each time point with the provided 10X lysis buffer.
Bicinchoninic acid assays
The total protein present in adherent cultures was measured using bicinchoninic acid assays for Seahorse experiments to control for changes in cell number. After the Seahorse analyzer experiment finished, the plate was immediately removed from the machine, the media was removed from the cells, and ice-cold 1X RIPA buffer (10X buffer diluted in H2O) + 1X Halt protease inhibitor cocktail was added to the cells. Cells were kept on ice for at least 5 minutes and then the lysate was homogenized by pipetting and diluted 1:4 with PBS before quantifying total protein with the Pierce BCA Protein Assay Kit according to manufacturer’s instructions. Briefly, the working reagent was added to diluted samples in duplicate and albumin standard curves in triplicate for 30 minutes at 37°C and the protein concentration was calculated according to a 4-parameter logistic regression based on absorbance at 540 nm.
Quantification and statistical analysis
RNA-seq data
Initial processing and differential expression.
Initial quality assessment was performed with FastQC version 0.11.9 before read ends consisting of 50 or more of the same nucleotide were removed. Reads were aligned to the mouse + Mtb genome (NCBI GRCm38 mm10 + H37Rv Mtb) using GSNAP version 2018-07-04 (Wu and Nacu, 2010). Feature counts were extracted using the featureCounts software (Liao et al., 2014), distributed as part of the Subread package v1.5.2. Genes were filtered to only include those detected in at least 2 samples. Normalization (to find counts-per-million (CPM) for each gene) and differential analysis of gene expression was calculated using edgeR version 3.26.8. The threshold for significance for differential expression was set at as ∣log2FC∣ > 1 and FDR <0.001.
Principal component analysis
Input genes were restricted to those that were differentially expressed when directly comparing the conditions included in the analysis. Normalized CPM values for these genes were input into the prcomp function in R for visualization and identification of the proportion of variance explained by each principal component.
Clustering
The union of the sets of differentially expressed genes in any of the 3 comparisons (HK H37Rv versus Mock, Live H37Rv versus Mock, and IFNβ versus untreated) across the two time points (4 and 24 hours post infection) in WT BMDM (a total of 6337 genes) were clustered using the K-means clustering algorithm described in Hartigan and Wong (1979) and implemented in R.
Gene set enrichment and pathway visualization
The 1411 genes in cluster 4 were tested for enrichment in the Hallmark gene sets of the Molecular Signatures Database (MSigDB) (Liberzon et al., 2015) using a hypergeometric test implemented in the hypeR R package (Federico and Monti, 2020) with the number of protein coding genes (22,519) in the GRCm38 genome build as the background set. A self-contained rotational gene set tests (Wu et al., 2010), implemented in the edgeR package in R (McCarthy et al., 2012; Robinson et al., 2010), was used to directly test the directional regulation of the Hallmark Glycolysis pathway. The visualization of the log2FC (IFNβ treated versus untreated at 28 hours after treatment) of enzymes in the KEGG Glycolysis/Gluconeogenesis pathway (mmu00010) was adapted from the pathview R package (Luo and Brouwer, 2013). Because multiple genes can catalyze each enzymatic step, the labels on each set were generalized to represent the gene family. The method for summarizing expression changes across multiple genes was set to sum (node.sum = “sum”).
Statistical information
Data were analyzed using R. Data are either presented as individual technical replicates with a horizonal bar representing the mean, or as a point representing the mean with SEM error bars. Statistical significance for more than two groups was calculated with a one-way ANOVA followed by Tukey’s honestly significant difference (HSD) tests on pairwise comparisons. Comparisons between two groups were assessed with Student’s t test. (Adjusted) p values are indicated on graphs as appropriate: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Precise values of n and numbers of independent experiments are indicated in figure legends.
Supplementary Material
Highlights.
Live, but not killed, Mtb induces type I IFN-mediated metabolic changes in macrophages
IFNβ restrains glycolysis and drives mitochondrial stress in activated macrophages
Type I IFN signaling in vivo limits pulmonary macrophage metabolic responses to Mtb
STING signaling is upstream of mitochondrial damage during mycobacterial infection
ACKNOWLEDGMENTS
We would like to thank the Office of Animal Care at the Center for Global Infectious Disease Research at Seattle Children’s Research Institute, including especially Jessica Spaulding and Roberto Guirnalda for taking care of the mice; Rong Tian, Toni Hsu, Bo Zhou, Dennis Wang, and Mingyue Zhao for critical assistance in designing and interpreting metabolic assays; Alissa Rothchild and Johannes Nemeth for extensive scientific discussions and critical reading of the manuscript; Irina Podolskaia for technical assistance; and members of the Urdahl lab for fruitful discussions and sharing in vivo expertise. This study was supported by NIH grants U19AI135976, 75N93019C00070, R01AI032972, and U19AI100627.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109195.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahmed D, Roy D, Jaworski A, Edwards A, Abizaid A, Kumar A, Golshani A, and Cassol E (2019). Differential remodeling of the electron transport chain is required to support TLR3 and TLR4 signaling and cytokine production in macrophages. Sci. Rep 9, 18801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli LRV, Gigliotti Rothfuchs A, Gonçalves R, Roffê E, Cheever AW, Bafica A, Salazar AM, Feng CG, and Sher A (2010). Intranasal poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J. Clin. Invest 120, 1674–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres JS (2020). A metabolic handbook for the COVID-19 pandemic. Nat. Metab 2, 572–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks DA, Ahlbrand SE, Hughitt VK, Shah S, Mayer-Barber KD, Vogel SN, El-Sayed NM, and Briken V (2019). Mycobacterium tuberculosis inhibits autocrine type I IFN signaling to increase intracellular survival. J. Immunol 202, 2348–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, et al. (2010).An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, Hsieh WY, Robertson KA, Watterson S, Shui G, Lacaze P, Khondoker M, Dickinson P, Sing G, Rodríguez-Martín S, et al. (2011). Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol. 9, e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ, et al. (2013). The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman J, Sogi KM, Benjamin D, Nomura DK, and Stanley SA (2016). HIF-1α is an essential mediator of IFN-γ-dependent immunity to Mycobacterium tuberculosis. J. Immunol 197, 1287–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, Sowell RT, Kaech SM, and Pearce EL (2017). Metabolic instruction of immunity. Cell 169, 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JD, Platanias LC, and Fish EN (2014). Beta interferon regulation of glucose metabolism is PI3K/Akt dependent and important for antiviral activity against coxsackievirus B3. J. Virol 88, 3485–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadieux N, Parra M, Cohen H, Maric D, Morris SL, and Brennan MJ (2011). Induction of cell death after localization to the host cell mitochondria by the Mycobacterium tuberculosis PE_PGRS33 protein. Microbiology (Reading) 157, 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Gan H, and Remold HG (2006). A mechanism of virulence: Virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J. Immunol 176, 3707–3716. [DOI] [PubMed] [Google Scholar]
- Cheng Y, and Schorey JS (2018). Mycobacterium tuberculosis-induced IFN-β production requires cytosolic DNA and RNA sensing pathways. J. Exp. Med 215, 2919–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JHA, Rao NA, Aghajanirefah A, et al. (2014). mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345, 1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, Sherman DR, Gerner MY, and Urdahl KB (2018). Alveolar macrophages provide an early mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe 24, 439–446.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, Scharn CR, Stamm CE, Levine B, Chen ZJ, and Shiloh MU (2015). Cyclic GMP-AMP synthase is an innate immune DNA sensor for Mycobacterium tuberculosis. Cell Host Microbe 17, 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming BM, Rahman MA, Lamprecht DA, Rohde KH, Saini V, Adamson JH, Russell DG, and Steyn AJC (2017). Mycobacterium tuberculosis arrests host cycle at the G1/S transition to establish long term infection. PLoS Pathog. 13, e1006389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming BM, Addicott KW, Adamson JH, and Steyn AJC (2018). Mycobacterium tuberculosis induces decelerated bioenergetic metabolism in human macrophages. eLife 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming BM, Pacl HT, and Steyn AJC (2020). Relevance of the Warburg effect in tuberculosis for host-directed therapy. Front. Cell. Infect. Microbiol 10, 576596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker T, Müller M, and Stockinger S (2005). The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol 5, 675–687. [DOI] [PubMed] [Google Scholar]
- Diamond MS, and Farzan M (2013). The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol 13, 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorhoi A, Yeremeev V, Nouailles G, Weiner J 3rd, Jörg S, Heinemann E, Oberbeck-Müller D, Knaul JK, Vogelzang A, Reece ST, et al. (2014). Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur. J. Immunol 44, 2380–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenreich W, Rudel T, Heesemann J, and Goebel W (2019). How viral and intracellular bacterial pathogens reprogram the metabolism of host cells to allow their intracellular replication. Front. Cell. Infect. Microbiol 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SCC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJW, et al. (2014). TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat. Immunol 15, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federico A, and Monti S (2020). hypeR: An R package for geneset enrichment workflows. Bioinformatics 36, 1307–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini E, Remoli ME, Gafa V, Pardini M, Fattorini L, and Coccia EM (2009). IFN-β improves BCG immunogenicity by acting on DC maturation. J. Leukoc. Biol 85, 462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giosué S, Casarini M, Alemanno L, Galluccio G, Mattia P, Pedicelli G, Rebek L, Bisetti A, and Ameglio F (1998). Effects of aerosolized interferon-α in patients with pulmonary tuberculosis. Am. J. Respir. Crit. Care Med 158, 1156–1162. [DOI] [PubMed] [Google Scholar]
- Gleeson LE, and Sheedy FJ (2016). Metabolic reprogramming & inflammation: Fuelling the host response to pathogens. Semin. Immunol 28, 450–468. [DOI] [PubMed] [Google Scholar]
- Gleeson LE, Sheedy FJ, Palsson-McDermott EM, Triglia D, O’Leary SM, O’Sullivan MP, O’Neill LAJ, and Keane J (2016). Cutting edge: Mycobacterium tuberculosis induces aerobic glycolysis in human alveolar macrophages that is required for control of intracellular bacillary replication. J. Immunol 196, 2444–2449. [DOI] [PubMed] [Google Scholar]
- Grode L, Seiler P, Baumann S, Hess J, Brinkmann V, Nasser Eddine A, Mann P, Goosmann C, Bandermann S, Smith D, et al. (2005). Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guérin mutants that secrete listeriolysin. J. Clin. Invest 115, 2472–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gröschel MI, Sayes F, Shin SJ, Frigui W, Pawlik A, Orgeur M, Canetti R, Honoré N, Simeone R, van der Werf TS, et al. (2017). Recombinant BCG expressing ESX-1 of Mycobacterium marinum combines low virulence with cytosolic immune signaling and improved TB protection. Cell Rep. 18, 2752–2765. [DOI] [PubMed] [Google Scholar]
- Hackett EE, Charles-Messance H, O’Leary SM, Gleeson LE, Muøoz-Wolf N, Case S, Wedderburn A, Johnston DGW, Williams MA, Smyth A,et al. (2020). Mycobacterium tuberculosis limits host glycolysis and IL-1β by restriction of PFK-M via microRNA-21. Cell Rep. 30, 124–136.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartigan JA, and Wong MA (1979). Algorithm AS 136: A K-means clustering algorithm. Appl. Stat 28, 100–108. [Google Scholar]
- Hopfner KP, and Hornung V (2020). Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol 21, 501–521. [DOI] [PubMed] [Google Scholar]
- Hos NJ, Ganesan R, Gutiérrez S, Hos D, Klimek J, Abdullah Z, Krönke M, and Robinson N (2017). Type I interferon enhances necroptosis of Salmonella Typhimurium-infected macrophages by impairing antioxidative stress responses. J. Cell Biol 216, 4107–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard NC, and Khader SA (2020). Immunometabolism during Mycobacterium tuberculosis Infection. Trends Microbiol. 28, 832–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard NC, Marin ND, Ahmed M, Rosa BA, Martin J, Bambouskova M, Sergushichev A, Loginicheva E, Kurepina N, Rangel-Moreno J, et al. (2018). Mycobacterium tuberculosis carrying a rifampicin drug resistance mutation reprograms macrophage metabolism through cell wall lipid changes. Nat. Microbiol 3, 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Nazarova EV, Tan S, Liu Y, and Russell DG (2018). Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J. Exp. Med 215, 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip WKE, Hoshi N, Shouval DS, Snapper S, and Medzhitov R (2017). Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, Van Rooijen N, MacDonald AS, and Allen JE (2011). Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332, 1284–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji DX, Yamashiro LH, Chen KJ, Mukaida N, Kramnik I, Darwin KH, and Vance RE (2019). Type I interferon-driven susceptibility to Mycobacterium tuberculosis is mediated by IL-1Ra. Nat. Microbiol 4, 2128–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Downey J, Sanz J, Kaufmann E, Blankenhaus B, Pacis A, Pernet E, Ahmed E, Cardoso S, Nijnik A, et al. (2020). M. tuberculosis reprograms hematopoietic stem cells to limit myelopoiesis and impair trained immunity. Cell 183, 752–770.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissig M, Ishibashi J, Harms MJ, Lim H-W, Stine RR, Won K-J, and Seale P (2017). PRDM16 represses the type I interferon response in adipocytes to promote mitochondrial and thermogenic programing. EMBO J. 36, 1528–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmandas E, Boutens L, Ratter JM, Hijmans A, Hooiveld GJ, Joosten LAB, Rodenburg RJ, Fransen JAM, Houtkooper RH, van Crevel R, et al. (2016a). Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat. Microbiol 2, 16246. [DOI] [PubMed] [Google Scholar]
- Lachmandas E, Beigier-Bompadre M, Cheng SC, Kumar V, van Laarhoven A, Wang X, Ammerdorffer A, Boutens L, de Jong D, Kanneganti TD, et al. (2016b). Rewiring cellular metabolism via the AKT/mTOR pathway contributes to host defence against Mycobacterium tuberculosis in human and murine cells. Eur. J. Immunol 46, 2574–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber JH, Crimmins GT, Raghavan S, Meyer-Morse NP, Cox JS, and Portnoy DA (2008). Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog. 4, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienard J, Nobs E, Lovins V, Movert E, Valfridsson C, and Carlsson F (2020). The Mycobacterium marinum ESX-1 system mediates phagosomal permeabilization and type I interferon production via separable mechanisms. Proc. Natl. Acad. Sci. USA 117, 1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus RM, and Finlay DK (2016). Immunometabolism: Cellular metabolism turns immune regulator. J. Biol. Chem 291, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, and Brouwer C (2013). Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 29, 1830–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanillo PS, Shiloh MU, Portnoy DA, and Cox JS (2012). Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11, 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín Franco JL, Genoula M, Corral D, Duette G, Ferreyra M, Maio M, Dolotowicz MB, Aparicio-Trejo OE, Patiøo-Martínez E, Charton A, et al. (2020). Host-derived lipids from tuberculous pleurisy impair macrophage microbicidal-associated metabolic activity. Cell Rep. 33, 108547. [DOI] [PubMed] [Google Scholar]
- Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, and Sher A (2011). Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35, 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, et al. (2014). Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, and Smyth GK (2012). Differential expression analysis of multifactor RNA-seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, and O’Neill LAJ (2017). Mitochondria are the power-houses of immunity. Nat. Immunol 18, 488–498. [DOI] [PubMed] [Google Scholar]
- Moreira-Teixeira L, Mayer-Barber K, Sher A, and O’Garra A (2018). Type I interferons in tuberculosis: Foe and occasionally friend. J. Exp. Med 215, 1273–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortaz E, Adcock IM, Tabarsi P, Masjedi MR, Mansouri D, Velayati AA, Casanova JL, and Barnes PJ (2015). Interaction of pattern recognition receptors with Mycobacterium tuberculosis. J. Clin. Immunol 35, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LAJ, Kishton RJ, and Rathmell J (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmero D, Eiguchi K, Rendo P, Castro Zorrilla L, Abbate E, and González Montaner LJ (1999). Phase II trial of recombinant interferon-α2b in patients with advanced intractable multidrug-resistant pulmonary tuberculosis: long-term follow-up. Int. J. Tuberc. Lung Dis 3, 214–218. [PubMed] [Google Scholar]
- Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, Behr MA, Fitzgerald KA, Sassetti CM, and Kelliher MA (2009). NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog. 5, e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, and Longhi MP (2014). Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol. 12, e1001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, and Pearce EJ (2013). Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisu D, Huang L, Grenier JK, and Russell DG (2020). Dual RNA-seq of Mtb-infected macrophages in vivo reveals ontologically distinct host-pathogen interactions. Cell Rep. 30, 335–350.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MA, Cumming BM, Addicott KW, Pacl HT, Russell SL, Nargan K, Naidoo T, Ramdial PK, Adamson JH, Wang R, and Steyn AJC (2020). Hydrogen sulfide dysregulates the immune response by suppressing central carbon metabolism to promote tuberculosis. Proc. Natl. Acad. Sci. USA 117, 6663–6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, and Beckman JS (2006). Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. USA 103, 15038–15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas M, Davies LC, Giles PJ, Liao C-T, Kharfan B, Stone TC, O’Donnell VB, Fraser DJ, Jones SA, and Taylor PR (2014). The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science 344, 645–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothchild AC, Olson GS, Nemeth J, Amon LM, Mai D, Gold ES, Diercks AH, and Aderem A (2019). Alveolar macrophages generate a noncanonical NRF2-driven transcriptional response to Mycobacterium tuberculosis in vivo. Sci. Immunol 4, eaaw6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothchild AC, Mai D, Aderem A, and Diercks AH (2020). Flow cytometry analysis and fluorescence-activated cell sorting of myeloid cells from lung and bronchoalveolar lavage samples from Mycobacterium tuberculosis-infected mice. Bio Protoc. 10, e3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DG, Huang L, and VanderVen BC (2019). Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol 19, 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ, and Divangahi M (2021). Targeting immunometabolism in host defence against Mycobacterium tuberculosis. Immunology 162, 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DM, Jeon BY, Lee HM, Jin HS, Yuk JM, Song CH, Lee SH, Lee ZW, Cho SN, Kim JM, et al. (2010). Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 6, e1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn H, Kim JS, Shin SJ, Kim K, Won CJ, Kim WS, Min KN, Choi HG, Lee JC, Park JK, and Kim HJ (2011). Targeting of Mycobacterium tuberculosis heparin-binding hemagglutinin to mitochondria in macrophages. PLoS Pathog. 7, e1002435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trnka J, Blaikie FH, Smith RAJ, and Murphy MP (2008). A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic. Biol. Med 44, 1406–1419. [DOI] [PubMed] [Google Scholar]
- Troegeler A, Mercier I, Cougoule C, Pietretti D, Colom A, Duval C, Vu Manh T-P, Capilla F, Poincloux R, Pingris K, et al. (2017). C-type lectin receptor DCIR modulates immunity to tuberculosis by sustaining type I interferon signaling in dendritic cells. Proc. Natl. Acad. Sci. USA 114, E540–E549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid-Burgk JL, Schmidt T, Hornung V, Cole ST, and Ablasser A (2015). Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe 17, 799–810. [DOI] [PubMed] [Google Scholar]
- Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, and Cox JS (2015). The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 17, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, Sena LA, and Chandel NS (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindel CG, Bell SL, Vail KJ, West KO, Patrick KL, and Watson RO (2020). LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. eLife 9, e51071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, and Ghosh S (2011). TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens KE, and Ernst JD (2016). The mechanism for type I interferon induction by Mycobacterium tuberculosis is bacterial strain-dependent. PLoS Pathog. 12, e1005809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2020). Global tuberculosis report 2020 (World Health Organization; ). https://www.who.int/publications/i/item/9789240013131. [Google Scholar]
- Wu TD, and Nacu S (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Lim E, Vaillant F, Asselin-Labat ML, Visvader JE, and Smyth GK (2010). ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics 26, 2176–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, Patterson A, Smith AM, Chang CH, Liu Z, et al. (2016). Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 44, 1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, Gray EE, Zhen A, Wu NC, Yamada DH, et al. (2015). Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell 163, 1716–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, Mahomed H, Erasmus M, Whatney W, Hussey GD, et al. ; ACS and GC6-74 cohort study groups (2016). A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet 387, 2312–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data generated during this study are available in GEO: GSE162620. Additional data supporting the current study are available from the corresponding author on request.