

Abstract
Aims
Inflammation plays an important role in cardiovascular disease (CVD) development. The NOD-like receptor protein-3 (NLRP3) inflammasome contributes to the development of atherosclerosis in animal models. Components of the NLRP3 inflammasome pathway such as interleukin-1β can therapeutically be targeted. Associations of genetically determined inflammasome-mediated systemic inflammation with CVD and mortality in humans are unknown.
Methods and results
We explored the association of genetic NLRP3 variants with prevalent CVD and cardiovascular mortality in 538 167 subjects on the individual participant level in an explorative gene-centric approach without performing multiple testing. Functional relevance of single-nucleotide polymorphisms on NLRP3 inflammasome activation has been evaluated in monocyte-enriched peripheral blood mononuclear cells (PBMCs). Genetic analyses identified the highly prevalent (minor allele frequency 39.9%) intronic NLRP3 variant rs10754555 to affect NLRP3 gene expression. rs10754555 carriers showed significantly higher C-reactive protein and serum amyloid A plasma levels. Carriers of the G allele showed higher NLRP3 inflammasome activation in isolated human PBMCs. In carriers of the rs10754555 variant, the prevalence of coronary artery disease was significantly higher as compared to non-carriers with a significant interaction between rs10754555 and age. Importantly, rs10754555 carriers had significantly higher risk for cardiovascular mortality during follow-up. Inflammasome inducers (e.g. urate, triglycerides, apolipoprotein C3) modulated the association between rs10754555 and mortality.
Conclusion
The NLRP3 intronic variant rs10754555 is associated with increased systemic inflammation, inflammasome activation, prevalent coronary artery disease, and mortality. This study provides evidence for a substantial role of genetically driven systemic inflammation in CVD and highlights the NLRP3 inflammasome as a therapeutic target.
Keywords: Cardiovascular diseases, Coronary artery disease, Inflammation, Inflammasome, NLRP3
Graphical Abstract
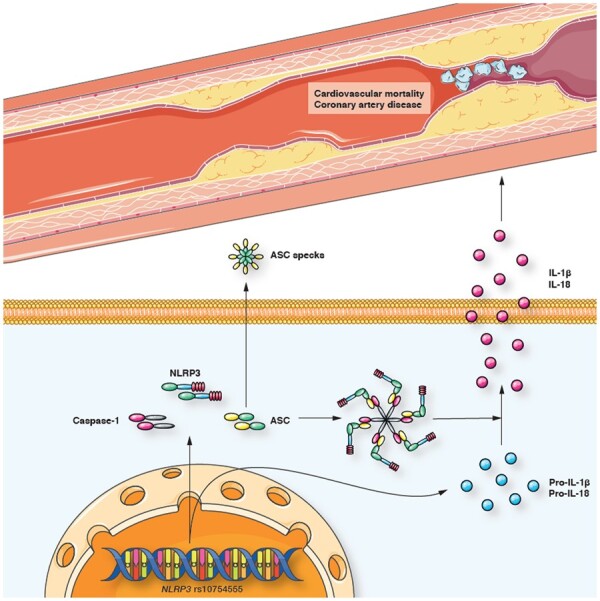
rs10754555 associates with higher NLRP3 mRNA expression and NLRP3 inflammasome activation, which induces the release of IL-1β and IL-18. This leads to systemic inflammation, higher risk for coronary artery disease, and cardiovascular mortality.
 See page 1757 for the editorial comment on this article (doi: 10.1093/eurheartj/ehab201)
See page 1757 for the editorial comment on this article (doi: 10.1093/eurheartj/ehab201)
Introduction
Vascular inflammation is important in the initiation and progression of atherosclerotic vascular diseases.1 Inflammatory markers such as high-sensitivity C-reactive protein (hsCRP) and serum amyloid A (SAA) are associated with increased mortality in patients with manifest cardiovascular disease (CVD)2 and healthy subjects with elevated inflammatory markers are at increased risk for the development of CVD.3,4 Inflammation in patients with CVD is characterized by the activation of monocytes, which adhere to the endothelium and migrate into the sub-endothelial layer, where they are activated by endogenous mediators such as modified lipoproteins triggering an innate immune response.1,5 These monocytes differentiate into tissue macrophages, acquire lipids and lipoproteins, and transform into foam cells contributing to atherosclerotic plaque formation.6,7
Interleukin (IL)-1β represents one of the key cytokines released by activated monocytes and macrophages leading to vascular (micro)inflammation.1 The processing of pro-IL-1β into mature IL-1β is tightly regulated by a multimeric intracellular protein complex, the NOD-like receptor protein 3 (NLRP3) inflammasome.8 In addition to exogenous triggers, the NLRP3 inflammasome is activated by a variety of endogenous mediators such as urate, cholesterol crystals, and oxidized low-density lipoprotein.9 Moreover, we recently observed that lipoproteins such as the triglyceride-associated apolipoprotein C3 (ApoC3) directly mediate alternative NLRP3 activation in human monocytes leading to vascular injury in vivo.10 The CANTOS trial demonstrated that inhibition of IL-1β, the effector cytokine of the NLRP3 inflammasome, with the monoclonal antibody canakinumab reduced recurrent cardiovascular (CV) events in patients with previous myocardial infarction (MI) and elevated hsCRP >2 mg/L on top of maximally tolerated statin therapy.11 Recently, the COLCOT trial reported that colchicine, an anti-inflammatory agent for the treatment of conditions such as gout, reduced a composite CV endpoint after MI by 23%.12 Importantly, modulating NLRP3 inflammasome activity represents one mechanism by which colchicine reduces inflammation.12
Despite growing experimental evidence for the NLRP3 inflammasome being a key driver of CVD and increased understanding of its molecular regulation, the clinical relevance of inflammasome activation in patients at risk for or with prevalent CVD is incompletely understood. In the present study, we assessed the association of a gene variant affecting NLRP3 gene expression and function with the prevalence of coronary artery disease (CAD) and CV mortality in 538 167 subjects.
Methods
Detailed description of the methods can be found in the Supplementary material online.
Genetic association validation studies
The association between single-nucleotide polymorphisms (SNPs) and all-cause as well as CV mortality was studied by genotype or in an additive genetic model. Since the current study is a gene-centric and not a genome-wide association study (GWAS), it did not require genome-wide significance. Due to the explorative nature of the study, we did not account for the issue of multiple testing and thus report unadjusted P-values. Findings were validated in participants of 10 studies comprising 526 091 participants. Study details are described in the Supplementary material online.
Statistical analyses
Continuous variables are presented as mean ± standard deviation or mean ± 95% confidence intervals (CIs) for normally distributed variables, or as median and interquartile range for variables with skewed distributions. Categorical variables are presented as frequencies. Differences between continuous variables were assessed using one-way analysis of variance (ANOVA) or Kruskal–Wallis test where appropriate. Differences between categorical variables were determined using the χ2 test. Generalized linear models were used to estimate age- and sex-adjusted marginal means of hsCRP or SAA according to rs10754555 SNP carrier status. In LURIC and GerMIFS, the association between rs10754555 genotype, CAD and severe CAD (only in LURIC) as well as mortality was assessed by logistic and Cox regression analyses. Severe CAD was defined as angiographically visualized ≥50% stenosis. To study the effect of age, an interaction term between rs10754555 and age was added to the respective models. Moreover, patients were divided into two groups at the age of 60 years corresponding to the first tertile of age in LURIC. Univariate and multivariable adjusted analyses were performed with adjustment for age, sex, diabetes mellitus, systolic blood pressure, body mass index, smoking status, estimated glomerular filtration rate, low-density lipoprotein cholesterol, hsCRP, presence of CAD, and previous MI. In the experimental studies, one-way ANOVA followed by Dunnett’s post hoc tests were used to assess significant differences across rs10754555 genotype. Genotype distributions were tested for Hardy–Weinberg equilibrium using exact tests (https://ihg.helmholtz-muenchen.de/cgi-bin/hw/hwa1.pl). Meta-analyses on the association between rs10754555 SNP carrier status and CV mortality were performed by using hazard ratios (HRs) and standard errors derived from multivariable adjusted Cox regression models at individual participant level provided by each study. Standard normal random-effects weighted meta-analysis was performed using the STATA package ‘metan’. Between-study heterogeneity I2 was determined as described previously.13 Small-study effects were excluded by using the Egger test provided within the STATA package ‘metabias’. To study the effect of ApoC3, triglycerides, and urate, an interaction term with rs10754555 was introduced in the respective models. ApoC3, triglycerides, and urate were divided into two categories (Quartiles 1–3 vs. Quartile 4). All other analyses were performed using SPSS version 25 and R version 3.3.3. The significance level was set at 0.05.
Results
NLRP3 genetic variants
We used GWAS data from the LURIC study comprising 3061 patients referred for coronary angiography as cohort for SNP pre-selection. Prioritization of an SNP with effects on the expression of NLRP3 is shown in Figure 1A and identified rs10754555 as significant eQTL in the ‘Blood eQTL browser’ (P = 2.32 × 10−6) and the ‘GTEX database’ (P = 9.80 × 10−10, Supplementary material online, Tables S1 and S2). To validate rs10754555 as an eQTL of NLRP3, the association between rs10754555 and NLRP3 mRNA expression in whole blood and peripheral blood mononuclear cells (PBMCs) was assessed in 36 cohorts comprising 31 556 samples included in the eQTLGen consortium14 (Figure 1B). In these analyses, rs10754555 qualified as a significant eQTL of NLRP3 (Z-score: 11.03, false discovery rate <0.05, P = 2.73 × 10−28). The allele and genotype frequencies of rs10754555 are consistent with Hardy–Weinberg equilibrium as shown in Supplementary material online, Table S3. Data from the Roadmap Epigenomics project indicate that rs10754555 maps with promoter and enhancer histone marks and DNase hypersensitivity (Supplementary material online, Figure S1). Importantly, heterozygous and homozygous rs10754555 carriers showed significantly higher levels of hsCRP (Figure 1C) and SAA (Figure 1D) as compared to non-carriers indicating that this variant is associated with a systemic pro-inflammatory state.
Figure 1.

Identification of single-nucleotide polymorphisms regulating NLRP3 expression. (A) Variant prioritization approach. (B) Expression quantitative trait locus meta-analysis for rs10754555 in whole blood or peripheral blood mononuclear cells in the eQTLGen consortium comprising 31 556 samples from 36 cohorts. (C) Age and sex-adjusted least square means of high-sensitivity C-reactive protein and (D) serum amyloid A in 3061 participants of the LURIC study (mean ± 95% confidence interval).
Biological relevance of rs10754555
The biological relevance of the rs10754555 variant was tested in monocyte-enriched PBMCs (Figure 2A, Supplementary material online, Table S4, and Supplementary material online, Figure S2A), which revealed higher NLRP3 mRNA expression in heterozygous and homozygous carriers of the G allele as compared to PBMCs from non-carriers (Figure 2B). Importantly, the plasma levels of IL-18 and IL-1β as NLRP3-dependent cytokines were also significantly higher in G allele carriers (Figure 2C and D). To directly assess NLRP3 inflammasome activation according to the rs10754555 variant carrier status, we quantified apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) specks in plasma. Notably, the rs10754555 G allele was associated with plasma ASC specks (Figure 2E–G). These findings confirm that carriers of the rs10754555 NLRP3 G allele are characterized by greater inflammasome activation.
Figure 2.

Functional effects of rs10754555 on expression of NLRP3 and inflammasome activation in freshly isolated human peripheral blood mononuclear cells. (A) Experimental work-flow. (B) mRNA expression of NLRP3 in freshly isolated peripheral blood mononuclear cells. (C) Plasma levels of interleukin-18 and (D) and interleukin-1β according to rs10754555 genotype. (E) Representative fluorescence microscopy of Alexa Fluor-488-labeled ASC specks from plasma and GFP-ASC in the supernatant of THP-1 cells (representative of three independent experiments). (F) Mean fluorescence intensity of ASC specks in plasma samples according to rs10754555 genotype. (G) Representative flow cytometry images of ASC speck quantification in plasma. Each dot represents an individual patient, and whiskers of the box plots represent 5 and 95 percentiles. LPS, lipopolysaccharide; qPCR, quantitative polymerase chain reaction; TNF, tumour necrosis factor.
To corroborate these results, activation of the NLRP3 inflammasome was modelled by stimulating the isolated PBMCs with known inflammasome activators [i.e. lipopolysaccharide (LPS), adenosine triphosphate (ATP), and nigericin] and measuring the release of IL-1β into the cell culture supernatant. Upon stimulation with LPS, LPS + ATP, and LPS + nigericin, PBMCs from heterozygous and homozygous NLRP3 rs10754555 G allele carriers released significantly more IL-1β compared to cells from non-carriers (Figure 3A–C). Unstimulated monocytes did not release detectable concentrations of IL-1β. To determine the specificity of these findings, release of IL-6 and tumour necrosis factor (TNF) into cell culture supernatants was quantified (Supplementary material online, Figure S2B–G), which did not differ according to rs10754555 variant carrier status. To prove the relevance of rs10754555 in vivo, we transplanted NOD-SCID mice with human PBMCs from non-carriers and homozygous rs10754555 carriers and subjected them to perivascular carotid injury, a mouse model for re-endothelialization, which we have recently shown to be NLRP3 dependent10 (Figure 3D and E). Re-endothelialization was significantly impaired in humanized mice receiving PBMCs from homozygous rs10754555 carriers, in which NLRP3 protein expression was higher as compared to non-carriers (Figure 3F).
Figure 3.

Modulation of NLRP3 inflammasome response by rs10754555 in freshly isolated human peripheral blood mononuclear cells and humanized mice. (A–C) Concentration of interleukin-1β in the supernatant of freshly isolated peripheral blood mononuclear cells stimulated with lipopolysaccharide (10 ng/mL, 3 h), lipopolysaccharide (3 h) and ATP (5 mM, 1 h), lipopolysaccharide (3 h) and nigericin (1 µM, 1 h). Each dot represents an individual patient, and whiskers of the box plots represent 5 and 95 percentiles. (D) Experimental outline of the murine perivascular carotid injury model in NOD-SCID mice transplanted with human peripheral blood mononuclear cells (i.e. humanized mice). (E) Re-endothelialized area 72 h after carotid injury in humanized mice and representative microphotographs. (F) Western blot of NLRP3 protein expression in transplanted peripheral blood mononuclear cells from nine individual donors. Mean ± 95% confidence interval.
Association between rs10754555 and the risk of coronary artery disease
Supplementary material online, Tables S5 and S6 summarize the baseline characteristics of participants of the LURIC study population separated by rs10754555 genotype as well divided at age of 60 years. Minor allele frequency (MAF) (G) for rs10754555 was 39.9%. The prevalence of traditional CV risk factors such as age, sex, body mass index, smoking, hypertension, and lipid parameters did not differ between non-carriers and carriers of the rs10754555 NLRP3 G allele. Moreover, there was no significant difference in the medication across different rs10754555 genotypes (Supplementary material online, Table S7). Since there was a trend towards lower prevalence of hypertension and diabetes in homozygous rs10754555 carriers in LURIC, we assessed the association between rs10754555, blood pressure, and presence of hypertension in UKBiobank, which did not differ significantly between the groups (Supplementary material online, Table S8), whereas the prevalence of diabetes was higher in rs10754555 G allele carriers. In homozygous rs10754555 G allele carriers, the risk for CAD and severe CAD was significantly higher as compared to non-carriers (Figure 4A and Supplementary material online, Table S9). This association was present in participants below 60 years of age [odds ratio (OR) for prevalent CAD: 2.04, 95% CI 1.15–3.61; OR for severe CAD: 2.28, 95% CI 1.29–4.01], but not in those above 60 years (OR for prevalent CAD: 0.83, 95% CI 0.55–1.25; OR for severe CAD: 0.73, 95% CI 0.49–1.03) revealing an age-dependent association of rs10754555 with the development of atherosclerotic CVD. We confirmed these findings in the GerMIFS studies II–VII with individual patient data available. Importantly, also in GerMIFS, rs10754555 was associated with a higher risk for CAD in subjects aged below 60 years (OR 1.12, 95% CI 1.02–1.22, Figure 4B and Supplementary material online, Table S10).
Figure 4.

Risk of coronary artery disease as a function of rs10754555 genotype. (A) Odds ratios for prevalent coronary artery disease and severe coronary artery disease (visual stenosis ≥50% on coronary angiography) according to rs10754555 genotype in 3061 participants of the LURIC study divided into two groups at age of 60 years (first tertile of age) and (B) in a meta-analysis of 12 076 participants with individual patient data available included in GerMIFS. Results are adjusted for age and sex.
Association between rs10754555 and CV mortality
In LURIC, all-cause mortality and CV mortality were significantly higher in heterozygous (HR 1.26, 95% CI 1.08–1.45 and 1.22, 95% CI 1.01–1.47) and homozygous (HR 1.31, 95% CI 1.08–1.59 and 1.35, 95% CI 1.07–1.72) rs10754555 variant carriers (Supplementary material online, Table S11). There was no association between rs10754555 and other clinical endpoints such as fatal cancer or fatal infection (Supplementary material online, Table S12). Interestingly, the percentage of rs10754555 G allele carriers decreased with increasing age (Supplementary material online, Table S13). Supplementary material online, Figure S3 compares the effect of the rs10754555 genotype with other CV risk factors. Furthermore, we assessed the association between rs10754555 genotypes and CV mortality in 10 prospective clinical trials enrolling 526 091 subjects with or without pre-existing CAD. Baseline characteristics for each individual study are shown in Supplementary material online, Tables S14–S22. Analyses were performed at an individual participant level. Additive genetic models show that the rs10754555 genotype is associated with significantly higher CV mortality in subjects from secondary prevention studies (HR 1.14, 95% CI 1.07–1.21) and in subjects from primary prevention studies (HR 1.06, 95% CI 1.01–1.11), without significant heterogeneity (I2 = 22.2%, P = 0.253 for secondary prevention studies and I2 = 0.0%, P = 0.999 for primary prevention studies, Figure 5A and B). Small-study effects were excluded using the Egger test (P = 0.341 for meta-analysis on CV mortality in secondary prevention studies).
Figure 5.

Risk of cardiovascular mortality as a function of rs10754555 genotype. Random-effects meta-analysis on cardiovascular mortality associated with rs10754555 genotype in (A) secondary and (B) primary prevention cohorts/studies. Shown are the hazard ratios for cardiovascular mortality associated with rs10754555 NLRP3 variant in 33 488 participants from eight studies comprising patients with prevalent coronary artery disease (i.e. secondary prevention) and in 492 603 participants from two studies from the general population. Analyses from each individual study were adjusted for age and gender.
Known NLRP3 inflammasome activators and the association between rs10754555 and mortality
Several endogenous NLRP3 inflammasome activators have been identified, of which ApoC3, triglycerides, and urate are of particular importance in CVD. The release of IL-1β from PBMCs stratified according to the rs10754555 genotype was modulated by baseline triglyceride or urate concentrations (Supplementary material online, Figure S4A–F and Supplementary material online, Tables S23 and S24). Furthermore, PBMCs from heterozygous or homozygous rs10754555 carriers released significantly higher concentrations of IL-1β after stimulation of ApoC3 or monosodium urate (Supplementary material online, Figure S5). Therefore, we assessed the association between rs10754555 and CV mortality with respect to ApoC3, triglyceride, or urate plasma levels. In LURIC, rs10754555 was only associated with CV mortality in subjects with high ApoC3 and triglyceride plasma levels (i.e. in the 4th quartile, Figure 6A and B and Supplementary material online, Tables S25 and S26). This was confirmed in subjects of the UKBiobank and was independent of age and also present in subjects with elevated triglycerides due to SNPs in the APOC3 gene locus (Figure 6B and Supplementary material online, Tables S27–S30). Vice versa, in UKBiobank, triglyceride plasma levels were associated with higher CV mortality (HR 1.17, 95% CI 1.08–1.26) in the total population, with the strongest effect in homozygous rs10754555 carriers (HR 1.57, 95% CI 1.30–1.90; Supplementary material online, Table S31). Similar results were obtained when participants of LURIC and UKBiobank were dichotomized according to urate plasma levels or carriers of SNPs associated with higher urate (Figure 6C and Supplementary material online, Tables S32–S37).
Figure 6.

Apolipoprotein C3, triglycerides, and urate modulate the association between rs10754555 and cardiovascular mortality. (A) Association between rs10754555 genotype and cardiovascular mortality in 3061 participants of the LURIC study divided in subjects with low (≤17.3 mg/dL, Quartiles 1–3) and high (>17.3 mg/dL, Quartile 4) apolipoprotein C3 plasma levels. (B) Association between rs10754555 genotype and cardiovascular mortality in the LURIC study and in 483 258 participants of UKBiobank divided into subjects with low (≤201 mg/dL, Quartiles 1–3) and high (>201 mg/dL, Quartile 4) triglyceride plasma levels. (C) Association between rs10754555 genotype and cardiovascular mortality in LURIC and UKBiobank divided into subjects with low (≤5.1 mg/dL, Quartiles 1–3) and high (>5.1 mg/dL, Quartile 4) urate plasma levels. Interaction refers to the interaction term between apolipoprotein C3, triglycerides, or urate and rs10754555 included in the Cox regression models.
Discussion
The main and novel finding of this study is that genetically determined sterile inflammation mediated by a specific cellular pathway (i.e. NLRP3) associates with higher prevalence of CAD and higher CV mortality. These associations are particularly prominent in the younger population, in which the influence of genetic predisposition likely predominates over lifestyle and environmental risk factors for (premature) CVD. Moreover, these findings highlight the NLRP3 inflammasome as a pathophysiologically important pathway and a potential therapeutic target.
Sterile inflammation is a hallmark of patients with atherosclerotic CVD,1 with experimental data showing a pivotal role of the NLRP3 inflammasome. In NLRP3- and IL-1β-deficient mice, atherosclerotic lesion formation was markedly reduced.9,15 Nevertheless, the effect of NLRP3 on atherosclerosis is dependent on the experimental atherosclerosis model, the type of atherogenic diet, and the gender of the mice.16 NLRP3 inflammasome activation and subsequently enhanced IL-1β production have been linked to maladaptive vascular remodelling after injury and adverse endothelial activation.17,18 Acceleration of atherosclerosis by clonal haematopoiesis is partially mediated by NLRP3-dependent IL-1β secretion19,20 and attenuated in subjects with genetic IL-6 signalling deficiency due to missense mutations of the IL-6 receptor.21
Our study links genetically driven inflammation with CVD prevalence and outcomes. An intronic variant within the NLRP3 locus has been identified, which is not associated with other CV risk factors or alterations in lipids, but appears to specifically increase systemic (micro)inflammation. rs10754555 represents an intronic NLRP3 variant, which is scored as NLRP3 eQTL by the provided evidence. Moreover, rs10754555 maps with promoter and enhancer histone marks, and with DNAse I-sensitive regions. This indicates that rs10754555 might indeed associate with increased NLRP3 mRNA transcription. Accordingly, rs10754555 was identified as NLRP3 eQTL in whole blood and PBMCs in the eQTLGen consortium. Importantly, our experimental studies show that the rs10754555 genotype is associated with higher NLRP3 mRNA expression, higher IL-18 plasma levels, increased ASC speck formation and inflammasome activation in human monocyte-enriched PBMCs, which represent a major inflammatory effector cell type in blood.1 Moreover, we have shown that PBMCs from rs10754555 G allele carriers suppressed re-endothelialization in humanized mice. The release of IL-6 and TNF from monocytes treated with known NLRP3 activators was not linked to the rs10754555 carrier status. This indicates that this genetic variant is not associated with unspecific pro-inflammatory cell activation, but specifically with NLRP3 inflammasome activation. rs10754555 was only associated with higher risk for CAD in subjects aged <60 years. This SNP–age interaction was confirmed in the GerMIFS studies and by applying the same age cut-off. SNP–environment interactions and in particular SNP–age interactions were reported for CVD-relevant SNPs but also for SNPs in genes involved in inflammation such as IL1R1.22–25 This observation points to an interaction between age and NLRP3 activation. Although the NLRP3 inflammasome is associated with a functional decline in aging,26,27NLRP3 gene expression and NLRP3 inflammasome activation have been reported to decrease with age.28,29 Moreover, we found that the percentage of heterozygous and homozygous rs10754555 carriers decreased with increasing age, which could explain the lack of association between rs10754555 and CAD in the elderly.
Importantly, rs10754555 is associated with CV mortality but not mortality related to infection or cancer. Our validation cohorts comprise a wide range of different patient populations including patients with prevalent CAD as well as subjects from the general population. Across these studies, the rs10754555 NLRP3 variant was consistently associated with increased CV mortality. Moreover, the high frequency of the risk allele (MAF 39.9%) indicates that increased NLRP3 inflammasome activity might contribute substantially to CV mortality on the population level. In agreement with the pre-clinical data on the association between NLRP3 inflammasome activation and atherosclerosis,9 our study shows that the rs10754555-mediated increase in all-cause mortality is mainly driven by CV deaths. These data highlight an important role of the innate immune system in the pathophysiology of CVD. Similar to NLRP3, gain-of-function mutations within the IL-6 receptor locus were found to be associated with increased risk for CAD.30,31
Indeed, in animal studies, inhibition of the NLRP3 inflammasome by the selective, small-molecule inhibitor MCC950 reduced experimental autoimmune encephalomyelitis and myocardial infarction.32,33 Compelling evidence for the benefit of therapeutically targeting NLRP3-dependent pathways is provided by studies using the monoclonal, IL-1β-targeting antibody canakinumab. In patients after MI with persistently elevated hsCRP, canakinumab lowered the rate of recurrent CV events by 15%, when 150 mg of canakinumab was administered.11Post hoc analyses of the CANTOS trial revealed persistently elevated levels of the NLRP3-dependent cytokine IL-18, which are unaffected by canakinumab treatment and still associated with future CV events.34 Therefore, inhibition of the NLRP3 inflammasome or its assembly in contrast to the specific inhibition of one effector cytokine could potentially provide a stronger reduction in CV events. Nevertheless, IL-1β release can also be induced by other inflammasome sensors such as absent in melanoma 2 (AIM2), which is activated by double-stranded DNA by exogenous pathogens and also during tissue damage.35 In addition to canakinumab, treatment with colchicine, which modulates the NLRP3 inflammasome, reduces CV events in patients post-MI with a stronger effect as compared to canakinumab.12 Since treatment with canakinumab or colchicine is associated with potential serious adverse events, strategies to select patients for targeted treatment are necessary. Screening for genetic variants associated with NLRP3 activation and subsequently elevated IL-1β and IL-18 may help to identify subjects with increased risk for CV events—especially at young age—as a result of sustained (micro)inflammation particularly when plasma levels of known inflammasome activators such as ApoC3, triglycerides, or urate are elevated.
Some limitations of our study should be considered. Although our results highlight the NLRP3 inflammasome as a potential factor promoting CV mortality, further studies are needed to prove that in particular carriers of the rs10754555 NLRP3 variant benefit from a specific anti-inflammatory treatment. In the present study, the NLRP3 variant rs10754555 is linked to mortality. Based on the study designs, we cannot show an association between the mutant carrier status and non-fatal CV events. The age-dependent association of rs10754555 with CAD risk could not have been validated in CARDIOGRAM due to limited access to individual patient data. Therefore, this interaction was validated in the GerMIFS studies (N = 6389 CAD cases and N = 5687 controls), which are part of the CARDIOGRAM consortium.
In conclusion, this is the first study to demonstrate the association between genetically driven inflammation and CVD by engaging a specific pro-inflammatory pathway (i.e. the NLRP3 inflammasome). These findings set the stage for individualized treatments in subjects with inflammation-driven high CV risk.
Supplementary material
Supplementary material is available at European Heart Journal online.
Data availability
All data are available from the corresponding author upon request.
Supplementary Material
Acknowledgements
The authors thank the staff and participants of the ARIC study and UKBiobank for their important contributions.
Funding
This work was supported by grants of Deutsche Forschungsgemeinschaft (DFG, SFB TRR 219, Project-ID 322900939) for Stephen Zewinger, Stefan Wagenpfeil, Michael Böhm, Rafael Kramann, Barbara Niemeyer, Danilo Fliser, and Thimoteus Speer, CORONA Stiftung (Ulrich Laufs), Universität Leipzig (Ulrich Laufs), as well as European Uremic Toxin (EUTox) Work Group of the ERA-EDTA (Danilo Fliser, Thimoteus Speer). Support for genotyping in the LURIC cohort was provided by the seventh framework program of the European commission (AtheroRemo, grant 201668). The measurement of DNA methylation in LURIC was supported by the seventh framework program of the European commission (RiskyCAD, grant 305739) and the Competence Cluster of Nutrition and Cardiovascular Health (nutriCARD), which is funded by the German Federal Ministry of Education and Research (grant 01EA1801A). The Atherosclerosis Risk in Communities study has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services (contract numbers HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I, and HHSN268201700005I), R01HL087641, R01HL59367, and R01HL086694; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. Infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research. GeneBank was supported, in part, by grants P01HL076491, R01HL103931, R01HL113452, R01HL103866, R01DK106000, R01HL126827, R01HL133169, and R01HL148110 from the National Institutes for Health (NIH). INVEST was supported by the University of Florida and grants from BASF Pharma and Abbott Laboratories and was supported by NIH grants R01HL074730 and U01GM074492. The INVEST GWAS genotyping was supported by RIKEN. Additional support for this project comes from NIH grant KL2 TR001429 (C.W.M.). LIFE-Heart was funded by the Leipzig Research Center for Civilization Diseases (LIFE). LIFE is an organizational unit affiliated to the Medical Faculty of the University of Leipzig. LIFE is funded by means of the European Union, by the European Regional Development Fund and by funds of the Free State of Saxony within the framework of the excellence initiative. The PROSPER study was supported by an investigator-initiated grant obtained from Bristol-Myers Squibb. J.W.J. is an Established Clinical Investigator of the Netherlands Heart Foundation (grant 2001 D 032). Support for genotyping was provided by the seventh framework programme of the European commission (grant 223004) and by the Netherlands Genomics Initiative (Netherlands Consortium for Healthy Aging grant 050-060-810). S.C.’s effort is supported, in part, by the National Institutes of Health (R01 NR013396). The Coronary Disease Cohort Study was funded by the New Zealand Health Research Council and the Heart Foundation of New Zealand, with funding for genotyping from the Christchurch Heart Institute Trust. TRIUMPH was sponsored by the National Institutes of Health: Washington University School of Medicine SCCOR Grant P50 HL077113. The PLATO trial was supported by Uppsala Clinical Research Center, AstraZeneca, and the Swedish Heart-Lung Foundation.
Conflict of interest: W.M. reports other from Synlab Holding Deutschland GmbH, during the conduct of the study, grants from Siemens Healthineers, grants and personal fees from Aegerion Pharmaceuticals, grants and personal fees from AMGEN, grants from AstraZeneca, grants and personal fees from Sanofi, grants and personal fees from Amryt Pharmaceuticals, grants and personal fees from BASF, grants and personal fees from Abbott Diagnostics, grants and personal fees from Numares AG, grants and personal fees from Berlin-Chemie, grants and personal fees from Akzea Therapeutics, grants from Bayer Vital GmbH, grants from bestbion dx GmbH, grants from Boehringer Ingelheim Pharma GmbH Co KG, grants from Immundiagnostik GmbH, grants from Merck Chemicals GmbH, grants from Novartis Pharma GmbH, grants from Olink Proteomics, grants and personal fees from AMGEN, personal fees from Novartis Pharma, and personal fees from Vifor Pharma, outside the submitted work. R.M.C.-D. reports grants from NIH, during the conduct of the study. W.H.W.T. reports grants from National Institutes of Health, personal fees from Sequana Medical AG, personal fees from Owkin Inc, personal fees from Relypsa Inc, personal fees from American Board of Internal Medicine, and personal fees from Springer Nature AG, outside the submitted work. S.L.H. reports other from Procter & Gamble, other from Roche Diagnostics, and grants from NIH, outside the submitted work. In addition, S.L.H. has a patent Cleveland Heart Lab/Quest Diagnostics with royalties paid and a patent Procter & Gamble with royalties paid. L.W. reports grants from GlaxoSmithKline, during the conduct of the study, grants from AstraZeneca, grants from Boehringer Ingelheim, grants from Bristol-Myers Squibb/Pfizer, grants from Merck & Co, grants from Roche Diagnostics, and personal fees from Abbott, outside the submitted work. N.S. reports personal fees from Amgen, personal fees from AstraZeneca, grants and personal fees from Boehringer Ingelheim, personal fees from Eli Lilly, personal fees from Merck Sharp & Dohme, personal fees from Novartis, personal fees from Novo Nordisk, personal fees from Pfizer, and personal fees from Sanofi, outside the submitted work. D.J.S. reports grant Bristol-Myers Squibb, during the conduct of the study. W.K. reports personal fees from AstraZeneca, personal fees from Novartis, personal fees from Pfizer, personal fees from The Medicines Company, personal fees from DalCor, personal fees from Kowa, personal fees from Amgen, personal fees from Corvidia, personal fees from Daiichi-Sankyo, personal fees from Genentech, personal fees from Genentech, personal fees from Novo Nordisk, personal fees from Esperion, personal fees from Berlin-Chemie, personal fees from Sanofi, personal fees from Bristol-Myers Squibb, grants and non-financial support from Abbott, grants and non-financial support from Roche Diagnostics, grants and non-financial support from Beckmann, and grants and non-financial support from Singulex, outside the submitted work. E.B. reports grants from NIH, during the conduct of the study. M.S. receives funding from Pfizer Inc. for a project not related to the present work. M.E.K. reports other from SYNLAB Holding Deutschland GmbH, outside the submitted work. R.N.D. reports grants from New Zealand Heart Foundation and grants from Health Research Council of New Zealand, during the conduct of the study. M.-P.D. reports personal fees and other from Dalcor, personal fees and other from GlaxoSmithKline, other from AstraZeneca, other from Pfizer, other from Servier, and other from Sanofi, outside the submitted work. In addition, M.-P.D. has a patent Methods for Treating or Preventing Cardiovascular Disorders and Lowering Risk of Cardiovascular Events issued to Dalcor, no royalties received, a patent Genetic Markers for Predicting Responsiveness to Therapy with HDL-Raising or HDL Mimicking Agent issued to Dalcor, no royalties received, and a patent Methods for using low-dose colchicine after myocardial infarction with royalties paid to Invention assigned to the Montreal Heart Institute. H.W. reports grants and personal fees from Eli Lilly and Company, other from AstraZeneca, grants and personal fees from Omthera Pharmaceuticals, grants and personal fees from Eisai Inc., grants and personal fees from DalCor Pharma UK Inc., grants and personal fees from CSL Behring LLC, grants and personal fees from American Regent, grants and personal fees from Sanofi-Aventis Australia Pty Ltd, grants and personal fees from Esperion Therapeutics Inc., personal fees from Genentech, Inc., and grants, personal fees and other from Sanofi-Aventis, outside the submitted work. S.K.J. reports grants from AstraZeneca, outside the submitted work. V.T. reports employment from deCODE genetics/Amgen, outside the submitted work. C.H. reports grants and personal fees from GlaxoSmithKline and grants and personal fees from AstraZeneca, during the conduct of the study. H.S. reports personal fees from MSD Sharp & Dohme, personal fees from Amgen, personal fees from Bayer Vital GmbH, personal fees from Boehringer Ingelheim, personal fees from Daiichi-Sankyo, personal fees from Novartis, personal fees from Servier, personal fees from Brahms, personal fees from Bristol-Myers Squibb, personal fees from Medtronic, personal fees from Sanofi-Aventis, personal fees from Synlab, grants and personal fees from AstraZeneca, and personal fees from Pfizer and Vifor, outside the submitted work. R.K. reports grants from Chugai and personal fees from Bayer, outside the submitted work. T.S. reports personal fees from Amgen, personal fees from Sanofi-Aventis, personal fees from Bayer, and personal fees from Astellas, outside the submitted work.
Translational perspective
Inflammation plays a crucial role in the development of atherosclerotic cardiovascular disease (CVD). Interventional studies highlight the NOD-like receptor protein 3 (NLRP3) inflammasome as an important mediator of CVD. Here, we report that a genetic variant within the NLRP3 gene locus refers to a systemic pro-inflammatory state. This variant is associated with coronary artery disease risk and cardiovascular mortality predominately in younger subjects. Therefore, genetically determined inflammation represents an important driver of atherosclerotic CVD. Identification of subjects at high inflammation-driven cardiovascular risk sets the stage for individualized treatments.
References
- 1. Hansson GK, Hermansson A.. The immune system in atherosclerosis. Nat Immunol 2011;12:204–212. [DOI] [PubMed] [Google Scholar]
- 2. Ridker PM, Hennekens CH, Buring JE, Rifai N.. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000;342:836–843. [DOI] [PubMed] [Google Scholar]
- 3. Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh JC; Emerging Risk Factors Collaboration. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet 2010;375:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ridker PM, Rifai N, Stampfer MJ, Hennekens CH.. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 2000;101:1767–1772. [DOI] [PubMed] [Google Scholar]
- 5. Libby P, Ridker PM, Hansson GK.. Progress and challenges in translating the biology of atherosclerosis. Nature 2011;473:317–325. [DOI] [PubMed] [Google Scholar]
- 6. Libby P. Inflammation in atherosclerosis. Nature 2002;420:868–874. [DOI] [PubMed] [Google Scholar]
- 7. Speer T, Ridker PM, von Eckardstein A, Schunk SJ, Fliser D.. Lipoproteins in chronic kidney disease: from bench to bedside. Eur Heart J 2021;doi:10.193/eurheartj/ehaa1050. [DOI] [PubMed] [Google Scholar]
- 8. Latz E, Xiao TS, Stutz A.. Activation and regulation of the inflammasomes. Nat Rev Immunol 2013;13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E.. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, Klug M, Schunk SJ, Schmit D, Kramann R, Körbel C, Ampofo E, Laschke MW, Selejan SR, Paschen A, Herter T, Schuster S, Silbernagel G, Sester M, Sester U, Aßmann G, Bals R, Kostner G, Jahnen-Dechent W, Menger MD, Rohrer L, März W, Böhm M, Jankowski J, Kopf M, Latz E, Niemeyer B, Fliser D, Laufs U, Speer T.. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol 2020;21:30–41. [DOI] [PubMed] [Google Scholar]
- 11. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Trial Group C. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 12. Tardif J-C, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, Berry C, López-Sendón J, Ostadal P, Koenig W, Angoulvant D, Grégoire JC, Lavoie M-A, Dubé M-P, Rhainds D, Provencher M, Blondeau L, Orfanos A, L’Allier PL, Guertin M-C, Roubille F.. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med 2019;381:2497–2505. [DOI] [PubMed] [Google Scholar]
- 13. Higgins JP, Thompson SG, Deeks JJ, Altman DG.. Measuring inconsistency in meta-analyses. BMJ 2003;327:557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Võsa U, Claringbould A, Westra HJ, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Kasela S, Pervjakova N, Alvaes I, Fave MJ, Agbessi M, Christiansen M, Jansen R, Seppälä I, Tong L, Teumer A, Schramm K, Hemani G, Verlouw J, Yaghootkar H, Sönmez R, Brown A, Kukushkina V, Kalnapenkis A, Rüeger S, Porcu E, Kronberg-Guzman J, Kettunen J, Powell J, Lee B, Zhang F, Arindrarto W, Beutner F, Brugge H, Dmitreva J, Elansary M, Fairfax BP, Georges M, Heijmans Bt Kähönen M, Kim Y, Knight Jc Kovacs P, Krohn K, Li S, Loeffler M, Marigorta Um Mei H, Momozawa Y, Müller-Nurasyid M, Nauck M, Nivard M, Penninx B, Pritchard J, Raitakari O, Rotzchke O, Slagboom Ep Stehouwer CDA, Stumvoll M, Sullivan P, Hoen PACt, Thiery J, Tönjes A, van Dongen J, van Iterson M, Veldink J, Völker U, Wijmenga C, Swertz M, Andiappan A, Montgomery Gw Ripatti S, Perola M, Kutalik Z, Dermitzakis E, Bergmann S, Frayling T, van Meurs J, Prokisch H, Ahsan H, Pierce B, Lehtimäki T, Boomsma D, Psaty Bm Gharib Sa Awadalla P, Milani L, Ouwehand W, Downes K, Stegle O, Battle A, Yang J, Visscher PM, Scholz M, Gibson G, Esko T, Franke L.. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv 2018;447367. [Google Scholar]
- 15. Alexander MR, Moehle CW, Johnson JL, Yang Z, Lee JK, Jackson CL, Owens GK.. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J Clin Invest 2012;122:70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grebe A, Hoss F, Latz E.. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res 2018;122:1722–1740. [DOI] [PubMed] [Google Scholar]
- 17. Xiao H, Lu M, Lin TY, Chen Z, Chen G, Wang WC, Marin T, Shentu TP, Wen L, Gongol B, Sun W, Liang X, Chen J, Huang HD, Pedra JH, Johnson DA, Shyy JY.. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation 2013;128:632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, Weissleder R, Libby P, Swirski FK, Nahrendorf M.. Targeting interleukin-1beta reduces leukocyte production after acute myocardial infarction. Circulation 2015;132:1880–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, Robertson AA, Cooper MA, Andres V, Hirschi KK, Martin KA, Walsh K.. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017;355:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, Danesh J, Frossard P, Saleheen D, Melander O, Sukhova GK, Neuberg D, Libby P, Kathiresan S, Ebert BL.. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, Libby P, Kathiresan S, Natarajan P.. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation 2020;141:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Portelli MA, Dijk FN, Ketelaar ME, Shrine N, Hankinson J, Bhaker S, Grotenboer NS, Obeidat M, Henry AP, Billington CK, Shaw D, Johnson SR, Pogson ZEK, Fogarty A, McKeever TM, Nickle DC, Bossé Y, van den Berge M, Faiz A, Brouwer S, Vonk JM, de Vos P, Brandsma C-A, Vermeulen CJ, Singapuri A, Heaney LG, Mansur AH, Chaudhuri R, Thomson NC, Holloway JW, Lockett GA, Howarth PH, Niven R, Simpson A, Blakey JD, Tobin MD, Postma DS, Hall IP, Wain LV, Nawijn MC, Brightling CE, Koppelman GH, Sayers I.. Phenotypic and functional translation of IL1RL1 locus polymorphisms in lung tissue and asthmatic airway epithelium. JCI Insight 2020;5:e132446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi G, Gu CC, Kraja AT, Arnett DK, Myers RH, Pankow JS, Hunt SC, Rao DC.. Genetic effect on blood pressure is modulated by age: the Hypertension Genetic Epidemiology Network Study. Hypertension 2009;53:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simino J, Shi G, Bis JC, Chasman DI, Ehret GB, Gu X, Guo X, Hwang S-J, Sijbrands E, Smith AV, Verwoert GC, Bragg-Gresham JL, Cadby G, Chen P, Cheng C-Y, Corre T, de Boer RA, Goel A, Johnson T, Khor C-C, Lluís-Ganella C, Luan J, Lyytikäinen L-P, Nolte IM, Sim X, Sõber S, van der Most PJ, Verweij N, Zhao JH, Amin N, Boerwinkle E, Bouchard C, Dehghan A, Eiriksdottir G, Elosua R, Franco OH, Gieger C, Harris TB, Hercberg S, Hofman A, James AL, Johnson AD, Kähönen M, Khaw K-T, Kutalik Z, Larson MG, Launer LJ, Li G, Liu J, Liu K, Morrison AC, Navis G, Ong RT-H, Papanicolau GJ, Penninx BW, Psaty BM, Raffel LJ, Raitakari OT, Rice K, Rivadeneira F, Rose LM, Sanna S, Scott RA, Siscovick DS, Stolk RP, Uitterlinden AG, Vaidya D, van der Klauw MM, Vasan RS, Vithana EN, Völker U, Völzke H, Watkins H, Young TL, Aung T, Bochud M, Farrall M, Hartman CA, Laan M, Lakatta EG, Lehtimäki T, Loos RJF, Lucas G, Meneton P, Palmer LJ, Rettig R, Snieder H, Tai ES, Teo Y-Y, van der Harst P, Wareham NJ, Wijmenga C, Wong TY, Fornage M, Gudnason V, Levy D, Palmas W, Ridker PM, Rotter JI, van Duijn CM, Witteman JCM, Chakravarti A, Rao DC, Alizadeh BZ, de Boer RA, Boezen HM, Bruinenberg M, Franke L, van der Harst P, Hillege HL, van der Klauw MM, Navis G, Ormel J, Postma DS, Rosmalen JGM, Slaets JP, Snieder H, Stolk RP, Wolffenbuttel BHR, Wijmenga C.. Gene-age interactions in blood pressure regulation: a large-scale investigation with the CHARGE, Global BPgen, and ICBP Consortia. Am J Hum Genet 2014;95:24–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takeuchi F, Isono M, Katsuya T, Yamamoto K, Yokota M, Sugiyama T, Nabika T, Fujioka A, Ohnaka K, Asano H, Yamori Y, Yamaguchi S, Kobayashi S, Takayanagi R, Ogihara T, Kato N.. Blood pressure and hypertension are associated with 7 loci in the Japanese population. Circulation 2010;121:2302–2309. [DOI] [PubMed] [Google Scholar]
- 26. Youm Y-H, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, Münzberg H, Rosen CJ, Ingram DK, Salbaum JM, Dixit VD.. Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab 2013;18:519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spadaro O, Goldberg EL, Camell CD, Youm YH, Kopchick JJ, Nguyen KY, Bartke A, Sun LY, Dixit VD.. Growth hormone receptor deficiency protects against age-related NLRP3 inflammasome activation and immune senescence. Cell Rep 2016;14:1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cho SJ, Rooney K, Choi AMK, Stout-Delgado HW.. NLRP3 inflammasome activation in aged macrophages is diminished during Streptococcus pneumoniae infection. Am J Physiol Lung Cell Mol Physiol 2018;314:L372–L387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Connat JL, Dumont A, Rialland M, Faivre B, Sorci G.. Nlrp3 gene expression in circulating leukocytes declines during healthy aging. J Gerontol A: Biol Sci Med Sci 2018;73:1045–1049. [DOI] [PubMed] [Google Scholar]
- 30. Interleukin 6, Receptor Mendelian Randomisation Analysis C, Swerdlow DI, Holmes MV, Kuchenbaecker KB, Engmann JE, Shah T, Sofat R, Guo Y, Chung C, Peasey A, Pfister R, Mooijaart SP, Ireland HA, Leusink M, Langenberg C, Li KW, Palmen J, Howard P, Cooper JA, Drenos F, Hardy J, Nalls MA, Li YR, Lowe G, Stewart M, Bielinski SJ, Peto J, Timpson NJ, Gallacher J, Dunlop M, Houlston R, Tomlinson I, Tzoulaki I, Luan J, Boer JM, Forouhi NG, Onland-Moret NC, van der Schouw YT, Schnabel RB, Hubacek JA, Kubinova R, Baceviciene M, Tamosiunas A, Pajak A, Topor-Madry R, Malyutina S, Baldassarre D, Sennblad B, Tremoli E, de Faire U, Ferrucci L, Bandenelli S, Tanaka T, Meschia JF, Singleton A, Navis G, Mateo Leach I, Bakker SJ, Gansevoort RT, Ford I, Epstein SE, Burnett MS, Devaney JM, Jukema JW, Westendorp RG, Jan DBG, van der Graaf Y, de Jong PA, Mailand-van der Zee AH, Klungel OH, de Boer A, Doevendans PA, Stephens JW, Eaton CB, Robinson JG, Manson JE, Fowkes FG, Frayling TM, Price JF, Whincup PH, Morris RW, Lawlor DA, Smith GD, Ben-Shlomo Y, Redline S, Lange LA, Kumari M, Wareham NJ, Verschuren WM, Benjamin EJ, Whittaker JC, Hamsten A, Dudbridge F, Delaney JA, Wong A, Kuh D, Hardy R, Castillo BA, Connolly JJ, van der Harst P, Brunner EJ, Marmot MG, Wassel CL, Humphries SE, Talmud PJ, Kivimaki M, Asselbergs FW, Voevoda M, Bobak M, Pikhart H, Wilson JG, Hakonarson H, Reiner AP, Keating BJ, Sattar N, Hingorani AD, Casas JP; Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet 2012;379:1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sarwar N, Butterworth AS, Freitag DF, Gregson J, Willeit P, Gorman DN, Gao P, Saleheen D, Rendon A, Nelson CP, Braund PS, Hall AS, Chasman DI, Tybjærg-Hansen A, Chambers JC, Benjamin EJ, Franks PW, Clarke R, Wilde AAM, Trip MD, Steri M, Witteman JCM, Qi L, van der Schoot CE, de Faire U, Erdmann J, Stringham HM, Koenig W, Rader DJ, Melzer D, Reich D, Psaty BM, Kleber ME, Panagiotakos DB, Willeit J, Wennberg P, Woodward M, Adamovic S, Rimm EB, Meade TW, Gillum RF, Shaffer JA, Hofman A, Onat A, Sundström J, Wassertheil-Smoller S, Mellström D, Gallacher J, Cushman M, Tracy RP, Kauhanen J, Karlsson M, Salonen JT, Wilhelmsen L, Amouyel P, Cantin B, Best LG, Ben-Shlomo Y, Manson JE, Davey-Smith G, de Bakker PIW, O'Donnell CJ, Wilson JF, Wilson AG, Assimes TL, Jansson J-O, Ohlsson C, Tivesten Å, Ljunggren Ö, Reilly MP, Hamsten A, Ingelsson E, Cambien F, Hung J, Thomas GN, Boehnke M, Schunkert H, Asselbergs FW, Kastelein JJP, Gudnason V, Salomaa V, Harris TB, Kooner JS, Allin KH, Nordestgaard BG, Hopewell JC, Goodall AH, Ridker PM, Hólm H, Watkins H, Ouwehand WH, Samani NJ, Kaptoge S, Di Angelantonio E, Harari O, Danesh J; IL6R Genetics Consortium Emerging Risk Factors Collaboration. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet 2012;379:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Nunez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O'Neill LA.. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015;21:248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Hout GP, Bosch L, Ellenbroek GH, de Haan JJ, van Solinge WW, Cooper MA, Arslan F, de Jager SC, Robertson AA, Pasterkamp G, Hoefer IE.. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J 2017;38:828–836. [DOI] [PubMed] [Google Scholar]
- 34. Ridker PM, MacFadyen JG, Thuren T, Libby P.. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1beta inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J 2020;41:2153–2163. [DOI] [PubMed] [Google Scholar]
- 35. Paulin N, Viola JR, Maas SL, de Jong R, Fernandes-Alnemri T, Weber C, Drechsler M, Döring Y, Soehnlein O.. Double-strand DNA sensing Aim2 inflammasome regulates atherosclerotic plaque vulnerability. Circulation 2018;138:321–323. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the corresponding author upon request.