

Despite the development of highly effective SARS-CoV-2 vaccines, there remains a great need for effective treatment of COVID-19. Through a public–private partnership in the United States, a system has been created to efficiently test a variety of therapeutic agents across the full spectrum of this disease. In addition to lessening the morbidity and mortality of COVID-19, this approach may also be useful in combating future epidemics.
Abstract
Working in an unprecedented time frame, the Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) public–private partnership developed and launched 9 master protocols between 14 April 2020 and 31 May 2021 to allow for the coordinated and efficient evaluation of multiple investigational therapeutic agents for COVID-19. The ACTIV master protocols were designed with a portfolio approach to serve the following patient populations with COVID-19: mild to moderately ill outpatients, moderately ill inpatients, and critically ill inpatients. To facilitate the execution of these studies and minimize start-up time, ACTIV selected several existing networks to launch the master protocols. The master protocols were also designed to test several agent classes prioritized by ACTIV that covered the spectrum of the disease pathophysiology. Each protocol, either adaptive or pragmatic, was designed to efficiently select those treatments that provide benefit to patients while rapidly eliminating those that were either ineffective or unsafe. The ACTIV Therapeutics-Clinical Working Group members describe the process by which these master protocols were designed, developed, and launched. Lessons learned that may be useful in meeting the challenges of a future pandemic are also described.
Just 100 years after the devastating 1918 influenza pandemic, which left an estimated 50 million dead worldwide, the SARS-CoV-2 pandemic has infected more than 163 million and killed more than 3.3 million people globally in just over 16 months. Advances in science, however, now enable implementation of biomedical interventions—diagnostics, vaccines, and therapeutics—alongside public health interventions to combat pandemics. In April 2020, to harness the collective scientific power of both public and private sectors, the U.S. National Institutes of Health (NIH) established the public–private partnership Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV). ACTIV leverages the scientific innovation, knowledge, and biomedical resources of the U.S. government and the private sector to address a shared research agenda. Its goal is to accelerate the development of vaccines and therapeutics to mitigate COVID-19 morbidity and mortality and to hasten an end to the pandemic (Figure 1) (1). Bringing together biomedical resources from 18 pharmaceutical companies, the NIH, the Biomedical Advanced Research and Development Authority, the Centers for Disease Control and Prevention, other U.S. government agencies, and academics, the ACTIV partnership was organized into the following 4 working groups: Preclinical, Therapeutics-Clinical, Vaccines, and Clinical Trial Capacity.
Figure 1. Organization of the ACTIV partner leadership and working groups.
![Figure 1. Organization of the ACTIV partner leadership and working groups. ACTIV = Accelerating COVID-19 Therapeutic Interventions and Vaccines; FDA = Food and Drug Administration; NIH = National Institutes of Health. (Reproduced from Collins and Stoffels [1] with permission of the Journal of the American Medical Association.).](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/d168/8244665/484fbe87ba63/aim-olf-M211269-M211269ff1.jpg)
ACTIV = Accelerating COVID-19 Therapeutic Interventions and Vaccines; FDA = Food and Drug Administration; NIH = National Institutes of Health. (Reproduced from Collins and Stoffels [1] with permission of the Journal of the American Medical Association.).
The Therapeutics-Clinical Working Group (TX-Clinical WG) had 2 charges: first, develop a systematic review process for identification and prioritization of therapeutic candidates, and second, create master protocols for efficient, flexible, rigorous assessment of safety and efficacy of selected candidates. The initial working group membership included 32 experts from 22 organizations. To tackle both assigned tasks, the ACTIV TX-Clinical WG split into 2 subgroups, one to develop a process for prioritizing agents (subject of a separate publication) and a second for master protocol development.
When the TX-Clinical WG launched, the clinical research landscape reflected the lack of a harmonized research agenda for COVID-19 therapeutics. Hundreds of trials were registered in ClinicalTrials.gov, but most lacked essential design features, such as randomization, controls, and adequate sample sizes to generate actionable evidence (2). In the context of increasing case counts, hospitals operating beyond surge capacity, growing numbers of small or poorly designed clinical trials, trials competing for identical patient populations, and limited understanding of disease pathogenesis, the TX-Clinical WG designed a series of rigorous master protocols aligned with the emerging stages of disease pathogenesis and available candidate treatments. The TX-Clinical WG also laid the groundwork for implementing the trials, which included interviewing and identifying the protocol leadership, regulatory sponsors, drug suppliers, and clinical trial networks necessary to fully develop the protocols, infrastructure, and governance to conduct the trials. Execution of the trials leveraged all resources of ACTIV, and later of Operation Warp Speed, to make them operational.
Rationale for the Use of Master Protocols in a Pandemic
Per the definition from the U.S. Food and Drug Administration, a master protocol uses a single trial infrastructure, trial design, and protocol to evaluate 1 or more drugs in 1 or more diseases for efficient and accelerated drug development (3). For ACTIV, master protocols were chosen as the primary vehicle for assessing selected therapeutics for several reasons. First, the ability to study multiple agents under a single, overarching protocol was essential, given the large number of agents anticipated for study. The Agent Prioritization subgroup of the TX-Clinical WG reviewed agents from multiple therapeutic classes targeting different aspects of disease pathogenesis and natural history (such as antiviral agents, immune modulators, and supportive therapies). Further, agents were selected for study in different populations (for example, hospitalized persons both in and out of the intensive care unit; infected but not yet hospitalized persons, both symptomatic and asymptomatic; and those at risk but not yet infected [ACTIV population priorities are shown in Appendix Figure 1]). Designing individual protocols to evaluate every agent in every relevant population was simply not feasible.
Appendix Figure 1. Priority populations for ACTIV.

To appropriately prioritize agents for the master protocols, the desired target populations needed to be agreed on by the ACTIV Therapeutics-Clinical Working Group. After much deliberation, the group decided given the high hospitalization and death rate early in the pandemic that the COVID-19 patient population would be prioritized in the following order for agent review: 1) hospitalized/moderately ill (non-ICU) and critically ill/ventilated (ICU), 2) outpatient/ambulatory ill, and 3) prophylaxis. ACTIV = Accelerating COVID-19 Therapeutic Interventions and Vaccines; ICU = intensive care unit.
Second, the ability to incorporate innovative design elements into a master protocol was seen as a significant advantage. The research objectives of screening numerous agents to identify the most promising candidates based on preliminary evidence and providing substantial evidence of safety and efficacy to support regulatory approval called for innovative trial designs to provide efficiency, flexibility, and power.
Finally, speed was of the utmost importance. Master protocols require time and resources for upfront planning exceeding those of a traditional, single-agent protocol. This early investment, however, was anticipated to realize and retain trial efficiency, as more agents became available for testing, by avoiding study start-up under a new protocol with additional agents.
Critical Design Decisions in Developing ACTIV Master Protocols
With the decision that master protocols would be the vehicle for agent evaluation, the next step was to agree on critical trial design features to ensure rapid initiation of and consistency across various protocols developed, beginning with the research objectives (Figure 2). Some selected agents would be approved for another indication with a well-characterized safety profile in the indicated population. The evaluation of repurposed agents could begin with a phase 3 investigation designed to provide evidence of efficacy and safety for expanded approval as a SARS-CoV-2 treatment. Other agents would be early in development with minimal clinical data available, requiring an exploratory trial design to evaluate tolerability and pharmacologic activity. Seamless phase 2/3 trial designs (4) would be ideal to screen these agents for graduation to a more rigorous investigation within the same protocol, where graduation rules could be based on the likelihood that the agent would prove successful in a phase 3 trial of reasonable size (5). For either exploratory or confirmatory research objectives, adaptive platform trial designs (6) can provide flexibility and speed of decision making—both essential during a pandemic. With an adaptive platform design, the trial infrastructure is established, master protocol developed, and study launched when at least 1 agent and appropriate comparator are available for study. Other agents are added to the platform as they become available, through amendments to the master protocol. Interim analyses of accumulating data are done throughout the study to determine if any agents demonstrate futility and should be discontinued, preserving resources for more promising agents, or if any agents demonstrate early evidence of efficacy, safety, and tolerability and could graduate for further study or proceed to regulatory submissions. Theoretically, once an adaptive platform design is established, the master protocol can run in perpetuity. Not knowing agent availability, criticality of timing for treatment in disease pathogenesis, or pandemic duration, ACTIV deemed the adaptive platform design the ideal choice.
Figure 2. Design decisions for ACTIV master protocols.
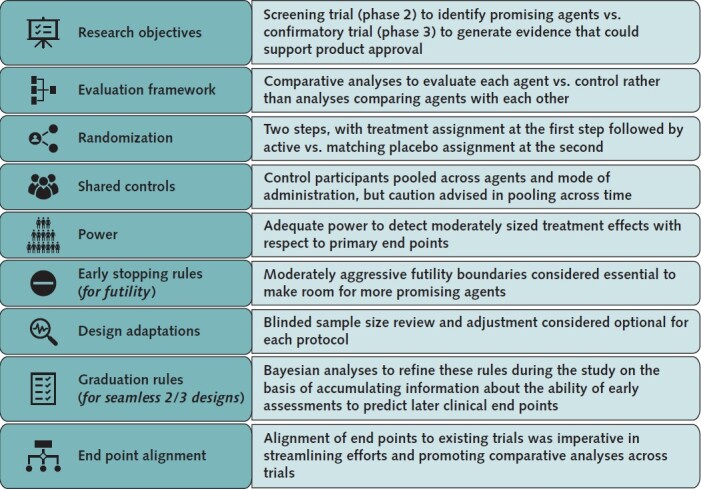
Each decision made for the master protocols was critical for tailoring them to the specific needs of the patients and the portfolio of studies that ACTIV was seeking to create to best address the therapeutic testing needs for the pandemic. ACTIV = Accelerating COVID-19 Therapeutic Interventions and Vaccines.
The framework for evaluating each agent was a second key decision in developing the ACTIV master protocols. Without proven SARS-CoV-2 therapies, our primary objective was to generate evidence that 1 or more selected agents was safe and effective. Comparison of agents or determination of best agent in class was, therefore, not a focus in designing the master protocols; each agent would be compared with a suitable control. The goal was to generate evidence to support regulatory approval of each agent independently of other agents. Focusing on this evaluation framework provided flexibility in starting and stopping the study of individual agents while the master protocol continued.
Understanding that standard of care would evolve throughout the pandemic and new treatment information and supportive care interventions would be obtained, agents would necessarily be evaluated in ACTIV as add-on therapies to current standard of care and could therefore enter the platform without stopping the trial. If matching placebos were available, the comparison control would be matching placebo plus standard of care, enabling double-blind implementation of the master protocols. This feature, combined with randomization, was considered important for primary end points requiring subjective assessment (for example, time to recovery as opposed to mortality).
Randomization was considered essential to generating reliable evidence from ACTIV master protocols, and nonrandomized studies were never a real consideration. ACTIV anticipated that some novel agents would be selected for evaluation, and ACTIV's ability to provide high-quality, comparative data on safety, tolerability, and pharmacologic activity of those agents, consistent with early-stage drug development, was critical.
Because each protocol was designed to evaluate multiple agents, it was decided during planning that whenever possible, a 2-step randomization procedure would be implemented, with agent assignment at stage 1 and active versus placebo assignment (for each agent with matching placebo available) at stage 2. Equal allocation would be used at the first step, with the possibility to adapt this ratio if needed. For example, 1 agent in a protocol might have more stringent safety exclusion criteria than other agents. Weighting the first step of randomization in its favor would facilitate recruitment for that agent. As agents entered and left the platform, this 2-step allocation algorithm could be easily adapted to the number of active interventions.
Given the desire to efficiently evaluate agents, a decision was made to share control participants among agents within a master protocol, thereby minimizing participants receiving placebo and reducing the overall sample size required for adequate power. Ability to share control participants is a key design innovation available in master protocols. The absence of a precision medicine objective, where patients are targeted for an intervention on the basis of their phenotype or genotype, allowed for broad sharing of control participants, in contrast to other master protocols (7). For implementation, allocation ratios at step 2 of randomization would reflect this sharing (for example, 3:1 for active vs. placebo with 3 agents).
Although early agreement was reached to share control participants among concurrently tested agents, even when method of administration differed (for example, injection vs. oral), concerns emerged about sharing control participants across time. Data from control participants generated before an agent entered the study might not be comparable to data from concurrent control participants given possible evolution of the disease or concurrent supportive care, and if so, potential for bias would be introduced into the primary analyses if nonconcurrent controls were used. Whether borrowing of control data across time would be allowed, and if so, how distant in time data could be, was determined for each protocol. Another limitation on control sharing resulted from some agents having more restrictive safety exclusions than other agents in a master protocol. Comparative analyses of such an agent would be limited to control data from participants who would be eligible to receive the agent. Careful monitoring would be required to ensure that any restrictions were not substantially affecting the power of the planned analyses.
The next critical design decision concerned the power available for statistical analyses of each agent and the potential effect on power of interim analyses or other design adaptations. Consistent with the urgency for identifying safe, effective treatments, agreement was reached for the first master protocols to provide adequate power to detect moderate treatment effects with respect to primary end points. At the same time, fairly aggressive futility stopping rules would be used to optimize resources for the most promising agents. Some later protocols focused on agents hypothesized to have very large effects. Statisticians from all stakeholders (pharmaceutical, government, and academic statisticians) worked together to conduct extensive simulations for each protocol and present benefits and risks of various analysis proposals to the ACTIV team. Decisions made for frequency of interim data reviews and stopping rules for both futility and efficacy reflected ACTIV's overarching goals, namely, to determine agent efficacy and safety most efficiently while ensuring a meaningful clinical effect of successful agents. Additional adaptations, such as blinded sample size re-estimation during a study, were proposed for some protocols with similar considerations.
For master protocols with research objectives spanning both exploratory and confirmatory phases (for example, seamless phase 2/3 designs), decisions about graduation rules from 1 phase to the other were needed. These rules typically depend on early phase assessments (such as symptoms or viral load for outpatients) that are believed, but not yet proven, to predict end points at a later phase (such as hospitalization and sustained recovery). For SARS-CoV-2, little was known about relationships between early and late phase end points for any patient populations, making a priori specification of graduation rules difficult. The master protocol design team of the TX-Clinical WG agreed that, where possible, Bayesian statistical methods would be used to pool accumulating data across agents in a master protocol to assess these relationships and adjust graduation rules accordingly.
The final design consideration that generated lengthy discussion was alignment of end points across ACTIV master protocols. The TX-Clinical WG agreed that end points could differ by patient population (for example, outpatient vs. inpatient) but believed that it was important to harmonize end points with existing (non-ACTIV) trials of the same population and to select simple, established measures that resonate with regulators, clinicians, and patients to streamline efforts and promote comparability across trials. Because ACTIV protocols were intended to generate evidence to support regulatory approval, input from regulatory partners was instrumental in guiding end point determination. Early ACTIV studies would rely on clinical end points for efficacy evaluation because surrogate end points (for example, virologic) were not yet established. The COVID-19 ordinal scale was identified as a reliable and meaningful clinical end point to support product approval (8, 9), and early ACTIV protocols incorporated variations of that scale as primary end points.
Processes to Develop and Launch the ACTIV Master Protocols
Having agreed on the critical design elements of each master protocol, the TX-Clinical WG next needed to determine how many master protocols should be developed and how they should be differentiated. In theory, designing 1 large, complex master protocol encompassing all agents, regardless of drug class or patient population, was possible; however, given the need for quick start-up and ease of interpretability of trial results to facilitate their translation to clinical practice, the group decided to simultaneously launch multiple master protocols by leveraging existing infrastructure where possible. Protocols would be differentiated by patient population and drug class. Developing separate master protocols for inpatient and outpatient populations would enable existing networks with experience in different populations to be used for ACTIV trials. Aligning master protocols to drug classes allowed trial designs to be tailored to the requirements of each class regarding safety data collection, definition and timing of end points for capturing predicted drug effects, and other considerations.
Protocols of existing COVID-19 trials (such as ACTT [Adaptive COVID-19 Treatment Trial], REMAP-CAP [Randomised, Embedded, Multi-factorial, Adaptive Platform Trial for Community-Acquired Pneumonia], and I-SPY COVID-19 TRIAL [An Adaptive Platform Trial for Critically Ill Patients]) were collected and reviewed by the working group to determine whether the networks for these protocols could be engaged to develop and launch an ACTIV master protocol. The ACTT-1 and ACTT-2 protocols were selected as the basis for a master protocol for immune modulators in hospitalized patients (ACTIV-1). Repurposing the ACTT protocols for ACTIV-1 meant that the team started with a well-designed, field-tested trial found to be successful in identifying an effective intervention, remdesivir. This made drafting the protocol more efficient and helped harmonize data collection and end points with existing trials. The team adapted the ACTT protocol to reflect critical ACTIV design decisions (Figure 2) and knowledge gained about the pandemic. Before the protocol was finalized, remdesivir gained emergency use authorization (10) and was incorporated as standard of care. ACTIV-1 was designed quickly to evaluate the first agents prioritized, but finding a network to house the protocol after the fact slowed the implementation process.
In contrast, the outpatient and inpatient master protocols to investigate neutralizing antibodies and other antiviral agents, ACTIV-2 and ACTIV-3, were developed after first identifying existing, NIH-funded clinical trial networks for implementation. INSIGHT (International Network for Strategic Initiatives in Global HIV Trials) (hospitalized patients), CTSN (Cardiothoracic Surgical Trials Network) and the PETAL (Prevention and Early Treatment of Acute Lung Injury) Network (critically ill patients), and ACTG (AIDS Clinical Trials Group) (outpatients) were engaged, taking advantage of existing clinical trial infrastructure and NIH support contracts. Advance network selection increased acceptance by network investigators, who helped accelerate overall protocol initiation. This hastened launch was critical for ACTIV-2 and ACTIV-3 because of the need to test SARS-CoV-2 neutralizing antibodies and other antiviral agents for which the protocols were designed. Another added advantage of selecting the networks before designing the protocol was the combined expertise (for example, infectious disease and critical care) available during protocol design from networks that had not previously collaborated (Figure 3).
Figure 3. Summary of ACTIV master protocols along disease progression and their current status.

The top illustration outlines the disease progression and how each ACTIV master protocol targets the individual patient population. Our understanding of viral and immunomodulatory responses throughout the disease progression continues to evolve as we learn from available clinical data. ACTIV-1 is a phase 3 master protocol that tests promising immune modulators. ACTIV-2 is designed as a phase 2 trial that can expand seamlessly to phase 3 to evaluate the efficacy and safety of various investigational agents, including monoclonal antibodies and antiviral agents. ACTIV-3 primarily aims to assess safety and efficacy of investigational agents to reduce time to sustained recovery. The sister protocol, ACTIV-3B, aims to evaluate the safety and efficacy of investigational agents at improving outcomes for hospitalized patients with acute respiratory distress syndrome related to COVID-19. ACTIV-4 master protocols evaluate the safety and efficacy of various antithrombotic agents that aim to prevent, treat, and address COVID-19–associated coagulopathy (CAC), or clotting, as well as help understand the effects of CAC across 3 patient populations: inpatient, outpatient, and convalescent. ACTIV-5 is designed as a proof-of-concept phase 2 study to rapidly evaluate proposed treatments and advance them to phase 3 trials if efficacy is demonstrated. Finally, ACTIV-6 tests existing prescription and over-the-counter medications for people to self-administer (orally or with an inhaler), with the aim of providing evidence-based treatment options for most adult patients with COVID-19 and mild to moderate symptoms. ACTG = AIDS Clinical Trials Group; ACTIV = Accelerating COVID-19 Therapeutic Interventions and Vaccines; ARDS = acute respiratory distress syndrome; CONNECTS = Collaborating Network of Networks for Evaluating COVID-19 and Therapeutic Strategies; CRO = contract research organization; CTSN = Cardiothoracic Surgical Trials Network; DCRI = Duke Clinical Research Institute; ICU = intensive care unit; IFN = interferon; IV = intravenous; INSIGHT = International Network for Strategic Initiatives in Global HIV Trials; LMWH = low-molecular-weight heparin; NCATS = National Center for Advancing Translational Sciences; NEJM = New England Journal of Medicine; NHLBI = National Heart, Lung, and Blood Institute; NIAID = National Institute of Allergy and Infectious Diseases; nMAB = neutralizing monoclonal antibody; nPAB = neutralizing polyclonal antibody; PCORnet = National Patient-Centered Clinical Research Network; PETAL = Prevention and Early Treatment of Acute Lung Injury; TIN = Trial Innovation Network; TRI = Technical Resources International; UFH = unfractionated heparin; VA = Department of Veterans Affairs.
With ACTIV-4, ACTIV-5, and ACTIV-6, ACTIV similarly aligned with existing networks and investigators to leverage ongoing efforts and infrastructure. The suite of ACTIV-4 protocols was launched by the National Heart, Lung, and Blood Institute's CONNECTS (Collaborating Network of Networks for Evaluating COVID-19 and Therapeutic Strategies) to test antithrombotic agents in all patient populations to address the rampant clotting conditions in patients with COVID-19. ACTIV-5 was launched to allow quick screening of agents that are ready only for phase 2 to determine if they should advance into one of the larger phase 3 master protocols. ACTIV-6, a light touch study, evaluates the efficacy of repurposed agents with solid safety records in COVID-19 outpatients. The need for ACTIV-6 arose from public and physician belief that agents (specifically ivermectin) had clinical benefit but that clinical study data were insufficient to inform clinical guidelines.
In all cases, factors considered in selecting a network included the need for global reach necessary for continued enrollment given geographic epidemiologic variability of the pandemic, clinical network capacity with site numbers to meet enrollment targets, network experience enrolling similar patient populations, determination of whether multiple networks should collaborate, institutional review board and data and safety monitoring board challenges from networks, and contracting mechanisms for each network affecting the ability to rapidly onboard and activate sites and vendors.
Key Aspects of Success and Considerations for Future Research About Pandemic Response
The need for high-quality clinical trials to evaluate candidate therapeutics for safety and efficacy in COVID-19 has been an urgent priority since the start of the pandemic. As of May 2021, ACTIV has launched 9 master protocols to create a portfolio of treatment trials to address the spectrum of COVID-19 disease (Figure 3); all were developed with input from all ACTIV stakeholders, including the Food and Drug Administration. In these efforts, ACTIV succeeded in bringing together experts from government, industry, and academia and experienced clinical trial networks to urgently address this need. The following 9 themes, which could inform future preparedness and response efforts, were interwoven into the project's success and ability to overcome challenges.
Singular goal. This team of experts representing government, industry, and academia were highly motivated by a common goal of quickly designing rigorous, controlled trials to produce clinically actionable data, and scores of ACTIV members volunteered countless hours to this endeavor. This model allowed input from diverse experts without concern about receiving “credit” for success. Further, there were no secondary agendas beyond accelerating evidence acquisition for safe, effective therapies. The parallel process for identifying agents for study through the Agent Prioritization subgroup also helped expedite the overall process. Groups outside the TX-Clinical WG were responsible for other tasks, such as advanced product commitments, supply chains, identification of sites, and contract support for the studies. Being part of a functional U.S. government system enabled the group to have a laser focus on their piece of the larger effort.
Broad spectrum of expertise. The TX-Clinical WG members collectively reflected expertise across multiple key areas, including regulatory processes, preclinical and clinical drug development, pharmacokinetics and pharmacodynamics, and biostatistics, enabling reliance on the group to accelerate the common goal.
Leadership. The ACTIV executive committee, which included senior leadership from the NIH, the Food and Drug Administration, Operation Warp Speed, industry, and others, was responsive to and supportive of the needs of the TX-Clinical WG when challenges were elevated. The TX-Clinical WG leadership, co-led by representatives from the NIH and industry, escalated needs and barriers to the ACTIV executive committee. Over time, ACTIV executive committee support solved challenges that might have otherwise hindered success. Administrative and organizational leadership from the Foundation for the National Institutes of Health was essential to the working group's success.
Speed, efficiency, and rigor as driving principles in master protocol design. The desire for speed, efficiency, and rigor led to an early decision to use adaptive platform trials done under overarching master protocols for the simultaneous testing of multiple agents and to allow interventions to be added as new data emerged and more was learned about COVID-19 disease pathogenesis. Various statistical approaches to trial design, adaptation, and analysis were adopted, including both frequentist and Bayesian approaches, taking into account the particular research objectives of each protocol. It was also agreed that scientific rigor and ability to assess both safety and efficacy should not be sacrificed for speed; therefore, the master protocols were appropriately powered to see clinically meaningful, definitive results. Although this requires larger sample sizes, the important benefits will be ease of interpretation and confidence in results. Another speed-enhancing decision was made to enable ACTIV trial teams to work with pharmaceutical companies during their phase 1 studies, enabling rapid onboarding of agents into the master protocols if phase 1 results were promising. Over time, commitment to speed, efficiency, and rigor ultimately resulted in a process and protocols in which pharmaceutical companies trusted that the trials would produce data that would support their clinical development plans.
Global clinical trials research capacity. Given anticipated and actual variability of the COVID-19 pandemic, the decision to create several clinical research networks with global research capacity has enabled global enrollment despite country shifts in COVID-19 incidence. Global sites are anticipated to contribute information around potential potency differences based on emergence of SARS-CoV-2 variants and mutations of concern.
Tradeoffs and challenges. Enrollment rates and site activation were initially slow despite rapid protocol development. ACTIV was launched in April 2020, more than 3 months into the pandemic, and clinical research capacities at the major research institutions were already engaged in investigator-initiated protocols or industry trials. Early outreach letters to sites by Francis Collins, the NIH director, conveyed that the ACTIV protocols were a priority of the U.S. government research agenda. In addition to trial competition, commitment to rigor enabling assessment of both safety and efficacy led to longer protocol initiation times at clinical trial sites due to regulatory and operational requirements. Sites worked through logistic hurdles and grappled with handling the additional burden of implementing a clinical trial on top of delivering clinical care. However, once the master protocols were implemented at clinical trial sites, the teams became efficient at adding and eliminating therapeutic candidates with no interruption to the trials. It would take far more time and resources to design and implement independent phase 2 and 3 trials for each individual candidate. Furthermore, multiple individual trials create competition for resources, as well as for participants, at each site. Differences in approaches within and across regulatory agencies also created challenges for country-level approvals. In some countries, regulators prioritized specific country trials, resulting in delays. Likewise, with different regulatory approaches to a given master protocol (such as inclusion of pregnant women), the master protocols are implemented differently in different countries.
Future research response agenda prioritized by the U.S. government for health emergencies. The ACTIV TX-Clinical WG initiated 9 master protocols within 13 months (with the ACTIV-2 and ACTIV-3 initiation within months; Figure 4 presents a timeline, and additional ACTIV trial timelines are shown in Appendix Figures 2 and 3). On the basis of this success, the team recommends that when the next pandemic strikes, a public–private partnership similar to ACTIV be quickly established, ensuring assembly of relevant expertise and resources to accelerate a prioritized research agenda that includes rigorous clinical trials. Repurposing existing, field-tested protocols; early involvement of federally funded investigators and clinical trial networks; and alignment with ongoing efforts can bring critically needed efficiency to identify safe, effective therapeutics to mitigate morbidity and mortality from a novel deadly pathogen (Figure 5). In addition, the ACTIV team would recommend that the U.S. government and the infectious disease community keep global clinical trial networks active and “trial-ready” for the next pandemic. Even more effective would be to bring together all clinical trial sites capable of performing trials in a pandemic, to unify the national research response in order to prioritize master protocols and eliminate competition from smaller, less clinically relevant clinical trials—similar to efforts by the U.K. National Institute for Health Research (11) in response to COVID-19. A final lesson that the ACTIV TX-Clinical WG would emphasize to any team desiring to design and execute master protocols in a future pandemic is not to wait for the perfect scenario or protocol design to emerge, because that can waste valuable time; move forward with the best strategy that can be executed under urgent timelines.
Figure 4. Timeline for ACTIV-2 and ACTIV-3 master protocol development.

The time to design, obtain approval, and launch is shown here. Overall, trial initiation completed in about 2.5 mo. Having a dedicated network and principal investigator champion during the trial design and setup resulted in rapid trial activation. ACTIV = Accelerating COVID-19 Therapeutic Interventions and Vaccines; CRO = contract research organization; FDA = Food and Drug Administration; IND = investigational new drug.
Appendix Figure 2. Timelines for ACTIV master protocol development.

Each ACTIV master protocol undergoes 3 main development stages: 1) protocol development and approval, which consists of designing and drafting the protocol, onboarding participating companies for the refinement of the protocol, submitting for Food and Drug Administration (FDA) pre–investigational new drug (IND) review, revising the protocol, and submitting for FDA IND review; 2) operational planning/setup, which consists of assembling required resources (e.g., sponsors, networks, and contract research organizations) and initiating the study at the site level (e.g., site registration and activation); and 3) ongoing operations, which consists of trial execution, protocol amendments, and additional site identification, registration, and activation. ACTIV = Accelerating COVID-19 Therapeutic Interventions and Vaccines.
Appendix Figure 3. Timelines for ACTIV-3B and ACTIV-6 master protocol development.

ACTIV = Accelerating COVID-19 Therapeutic Interventions and Vaccines.
Figure 5. Strategic decisions and considerations for the ACTIV master protocols.

ACTIV = Accelerating COVID-19 Therapeutic Interventions and Vaccines; EUA = emergency use authorization; FDA = Food and Drug Administration; PI = principal investigator.
Appendix: Members of the ACTIV Therapeutics-Clinical Working Group
Membership
Members of the ACTIV Therapeutics-Clinical Working Group who authored this work: Lisa LaVange (University of North Carolina, Chapel Hill); Stacey J. Adam (Foundation for the National Institutes of Health); Judith S. Currier (University of California, Los Angeles); Elizabeth S. Higgs, Lora A. Reineck, and Sarah W. Read (National Institutes of Health); and Eric A. Hughes (Novartis Pharma).
Members of the ACTIV Therapeutics-Clinical Working Group who contributed to this work but did not author it: Neil Aggarwal, John Beigel, Sam Bozzette, Christine Colvis, Michelle Culp, Josh Fessel, Ellen Gadbois, Keith W. Hoots, Andrei Kindzelski, Walter Koroshetz, Joanne Lumsden, Hilary Marston, Ashley Parker, Amy Patterson, Yves Rosenberg, and Michael Proschan (National Institutes of Health); Mark Eisner (Genentech); Timothy Buchman (U.S. Department of Health and Human Services); Timothy Burgess (Uniformed Services University of the Health Sciences); Joan Butterton (Merck & Co.); Sylva Collins, Angelo R. De Claro, Daniel Rubin, Yuan Li Shen, and Peter Stein (U.S. Food and Drug Administration); Ruxandra Draghia-Akli (Janssen Pharmaceutical Companies of Johnson & Johnson); Carl Garner (Eli Lilly and Company); Keith Gottesdiener (Prime Medicine); David M. Hone, Revell L. Phillips, and Ronald B. Reisler (Defense Threat Reduction Agency); Jacqueline Kirchner and Mike Poole (Bill and Melinda Gates Foundation); Elliot Levy (Amgen); John W. Mellors (University of Pittsburgh); Sandeep Menon (Pfizer); Linda Mollica, Naimish Patel, and Peter Wung (Sanofi); Amanda Peppercorn (GlaxoSmithKline); Martha Petrovick (Massachusetts Institute of Technology); David Wholley (Foundation for the National Institutes of Health); John Young (Roche); and Helen Chen, Alex Cwalina, and Hana Nasr (Deloitte Consulting).
Participating COVID-19 ACTIV Groups
Domestic and International Agencies
Biomedical Advanced Research and Development Authority
European Medicines Agency
National Institutes of Health
National Institutes of Health: National Cancer Institute
National Institutes of Health: National Center for Advancing Translational Sciences
National Institutes of Health: National Heart, Lung, and Blood Institute
National Institutes of Health: National Institute of Allergy and Infectious Diseases
National Institutes of Health: Office of the Director
U.S. Army Medical Research and Development Command
U.S. Centers for Disease Control and Prevention
U.S. Department of Veterans Affairs
U.S. Food and Drug Administration
U.S. Operation Warp Speed
White House COVID Response Team
Industry Partners
AbbVie
Amgen
AstraZeneca
Bristol Myers Squibb
Dewpoint Therapeutics
Eisai Co.
Eli Lilly and Company
Evotec
Gilead Sciences
GlaxoSmithKline
Johnson & Johnson
Karuna
KSQ Therapeutics
Merck & Co.
Moderna
Novartis International
Novavax
Pfizer
Rhythm Pharmaceuticals
Roche
Sanofi
Takeda Pharmaceutical Company
Vir Biotechnology
Nonprofit Organizations
Bill & Melinda Gates Foundation
Foundation for the National Institutes of Health
Fred Hutchinson Cancer Research Center
RTI International
Footnotes
This article was published at Annals.org on 29 June 2021.
* For members of the ACTIV Therapeutics-Clinical Working Group, see the Appendix.
References
- 1. Collins FS , Stoffels P . Accelerating COVID-19 therapeutic interventions and vaccines (ACTIV): an unprecedented partnership for unprecedented times. JAMA. 2020;323:2455-2457. [PMID: ] doi: 10.1001/jama.2020.8920 [DOI] [PubMed] [Google Scholar]
- 2. Bugin K , Woodcock J . Trends in COVID-19 therapeutic clinical trials. Nat Rev Drug Discov. 2021;20:254-255. [PMID: ] doi: 10.1038/d41573-021-00037-3 [DOI] [PubMed] [Google Scholar]
- 3. Woodcock J , LaVange LM . Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377:62-70. [PMID: ] doi: 10.1056/NEJMra1510062 [DOI] [PubMed] [Google Scholar]
- 4. Geiger MJ , Skrivanek Z , Gaydos B , et al. An adaptive, dose-finding, seamless phase 2/3 study of a long-acting glucagon-like peptide-1 analog (dulaglutide): trial design and baseline characteristics. J Diabetes Sci Technol. 2012;6:1319-27. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nanda R , Liu MC , Yau C , et al. Effect of pembrolizumab plus neoadjuvant chemotherapy on pathologic complete response in women with early-stage breast cancer: an analysis of the ongoing phase 2 adaptively randomized I-SPY2 trial. JAMA Oncol. 2020;6:676-684. [PMID: ] doi: 10.1001/jamaoncol.2019.6650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Saville BR , Berry SM . Efficiencies of platform clinical trials: a vision of the future. Clin Trials. 2016;13:358-66. [PMID: ] doi: 10.1177/1740774515626362 [DOI] [PubMed] [Google Scholar]
- 7. Redman MW , Papadimitrakopoulou VA , Minichiello K , et al. Biomarker-driven therapies for previously treated squamous non-small-cell lung cancer (Lung-MAP SWOG S1400): a biomarker-driven master protocol. Lancet Oncol. 2020;21:1589-1601. [PMID: ] doi: 10.1016/S1470-2045(20)30475-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection.. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect Dis. 2020;20:e192-e197. [PMID: ] doi: 10.1016/S1473-3099(20)30483-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adaptive COVID-19 Treatment Trial (ACTT) [clinical trial]. Accessed at https://clinicaltrials.gov/ct2/show/NCT04280705 on 15 April 2021.
- 10. Beigel JH , Tomashek KM , Dodd LE , et al; ACTT-1 Study Group Members. Remdesivir for the treatment of Covid-19 — final report. N Engl J Med. 2020;383:1813-1826. [PMID: ] doi: 10.1056/NEJMoa2007764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.RECOVERY Trial. Nuffield Department of Population Health. 2021. Accessed at www.recoverytrial.net on 15 April 2021.