

ABSTRACT
One of the longest human microRNA (miRNA) clusters is located on chromosome 19 (C19MC), containing 46 miRNA genes, which were considered to be expressed simultaneously and at similar levels from a common long noncoding transcript. Investigating the two tissue types where C19MC is exclusively expressed, we could show that there is a tissue-specific and chromosomal position-dependent decrease in mature miRNA levels towards the 3ʹ end of the cluster in embryonic stem cells but not in placenta. Although C19MC transcription level is significantly lower in stem cells, this gradual decrease is not present at the primary miRNA levels, indicating that a difference in posttranscriptional processing could explain this observation. By depleting Drosha, the nuclease component of the Microprocessor complex, we could further enhance the positional decrease in stem cells, demonstrating that a tissue-specific, local availability of the Microprocessor complex could lie behind the phenomenon. Moreover, we could describe a tissue-specific promoter being exclusively active in placenta, and the epigenetic mark analysis suggested the presence of several putative enhancer sequences in this region. Performing specific chromatin immunoprecipitation followed by quantitative real-time PCR experiments we could show a strong association of Drosha with selected enhancer regions in placenta, but not in embryonic stem cells. These enhancers could provide explanation for a more efficient co-transcriptional recruitment of the Microprocessor, and therefore a more efficient processing of pri-miRNAs throughout the cluster in placenta. Our results point towards a new model where tissue-specific, posttranscriptional ‘fine-tuning’ can differentiate among miRNAs that are expressed simultaneously from a common precursor.
KEYWORDS: C19MC, miRNA, Drosha, DGCR8, enhancer, ChIP-qPCR
Introduction
MicroRNAs (miRNAs) are short non-coding RNAs that form ribonucleoprotein complexes with Argonaute (AGO) proteins to fine-tune the expression of their target mRNA molecules. These approximately 22-nucleotide-long single-stranded nucleic acids are formed via consecutive cleavage and maturation steps from long primary transcripts (pri-miRNAs): during the canonical pathway in animal cells, the imperfect secondary structured hairpins are cleaved from the transcript by the Drosha/Dgcr8 Microprocessor complex, and the so-formed precursor miRNAs (pre-miRNAs) are transported out from the nucleus by the Exportin-5 system. In the cytoplasm, another RNaseIII type enzyme, Dicer removes the apical loop of the hairpin, forming short, double-stranded RNA molecules with 3ʹ overhangs of two nucleotides. Several subsequent maturation events result in a formation of an RNA-induced silencing complex (RISC) where one strand of the Dicer cleavage product becomes associated with an AGO protein and matures to an effector complex. The so-formed RISC starts scanning the mRNA population in the cell to find its target, typically a short sequence in the 3ʹ untranslated region complementary to the ‘seed sequence’ (two to eight nucleotides at the 5ʹ part) of the miRNA; the target mRNA is then either degraded or its translation is inhibited by various mechanisms [1,2].
miRNA genes can be located in introns (or even in exons) of protein-coding and non-coding transcripts (even in the coding region of DGCR8 itself, see [3]), where their expression is linked to the host gene transcription, but not necessarily coupled to splicing and maturation of the host transcript [4]. On the other hand, miRNA genes can be regulated by their own promoters, even if positioned in an intron [5]. Previous studies revealed that miRNAs are often clustered in the human genome [6], and such clustered miRNAs are functionally linked [7]. During evolution, several long miRNA clusters have been formed, and in primates, they are typically involved in stem cell regulation and placenta physiology [8,9]. Two exceptionally long miRNA clusters (MC) in the human genome are located on chromosome 14 (the C14MC, with 52 miRNA genes) and on chromosome 19 (the C19MC, with 46 miRNA genes), and there is emerging evidence that at least C19MC controls migration and invasion of human trophoblasts and take part in cell-to-cell communication during pregnancy [8–10]. The genomic loci of both clusters are complex: the C14MC is interrupted by a C/D snoRNA cluster and it is currently unknown if they are regulated independently [8,9], whereas the C19MC is located nearby the short miR-371-3 cluster which seems to have a distinct regulation [11]. Another common aspect of these long clusters is that their expression is regulated by imprinting: in the placenta, the C14MC is expressed from the maternally inherited allele, whereas the C19MC is expressed exclusively from the paternally inherited chromosome [8,9,11].
Although previous reports indicated that all miRNAs in a cluster share a common regulation, several recent studies found individual miRNAs showing distinct expression patterns, indicating more elaborate regulatory mechanisms (for a recent review, see [12]). As a prominent example, components of the let-7 cluster mature differently from the same polycistronic transcript, and due to the high variability of individual processing, pri-miRNA levels were found to be a better indicator for mature miRNA expression than primary transcription [13]. Moreover, RNA binding proteins such hnRNPA1 can bind to the stem-loop structure of distinct pre-miRNAs in a cluster, providing separate regulation for individual miRNAs, as shown for the hsa-miR-18a in the miR-17-92 (oncomiR-1) cluster [14–16]. Clearly, common transcriptional regulation is a necessary but may not be a sufficient condition for similar expression levels of miRNAs within the same cluster.
The C19MC has an additional unique feature: there are several Alu elements dispersed throughout the cluster, located typically flanking the pre-miRNA sequences [17–19]. This genomic arrangement and the fact that many of the miRNAs in the cluster share similar seed sequences indicate that the C19MC were formed by gene duplication events mediated by the Alu elements during primate evolution, and the common ancestor sequence was most likely an ancient miRNA similar to the miR-371 species [17,20]. The cluster is expressed predominantly in embryonic stem cells (ESCs) and in the reproductive system including the placenta [21,22], but some individual miRNAs from the cluster (such as hsa-miR-498) were found to be expressed in the foetal brain [23]. Moreover, previous studies of C19MC regulation remained controversial: earlier investigators claimed that due to the presence of Alu sequences, the cluster is transcribed by RNA polymerase III [18]. In a later study, however, it was revealed that this is not the case but the C19MC is transcribed by RNA polymerase II as a primate-specific, long non-protein-coding transcript with a complex splicing pattern, and the miRNA genes are intron-encoded [19]. Following that, Bellemer et al. provided evidence that during miRNA maturation, the Microprocessor complex recognizes the intron-coding long transcript near the site of transcription, and after Drosha cleavage, the DGCR8 remains attached to the precursors for longer [24]. However, the functional role of this latter association remains unclear, as well as the elaborate, potentially distinct regulatory processes of individual miRNAs from this large transcript.
In this study, we aimed to understand the tissue-specific transcriptional and posttranscriptional regulation of miRNAs expressed from the C19MC cluster. By examining and comparing human ESC lines with placenta-derived cells, we provided evidence that there is a strong correlation between the position and the expression level of a given miRNA from the cluster: significantly lower miRNA steady-state levels could be detected towards the 3ʹ region of the cluster in ESCs. We could reveal that the major source of this difference is originated at the posttranscriptional level of miRNA maturation, showing a decreasing pri-miRNA processing efficiency towards the 3ʹ region of C19MC, mediated by the lowered local availability of Drosha. Moreover, we could show that there is a placenta-specific promoter that contributes to tissue specificity, and by performing chromatin immunoprecipitation with antibody against Drosha, followed by quantitative real-time PCR (ChIP-qPCR), we could provide evidence that in placenta, Drosha is associated with certain enhancer regions located upstream of C19MC. These data point towards a potential enhancer-mediated, cotranscriptional recruitment of the Microprocessor to the transcription complex, providing the basis for the more efficient processing of miRNAs throughout the entire cluster in placenta. Our results can contribute to a new model of miRNA expression from long clusters, where tissue-specific promoters or enhancers could influence the processing of miRNAs from a common long non-coding transcript.
Results
C19MC miRNA expression levels show a position-dependent profile
Previous studies indicated that C19MC is expressed predominantly in embryonic stem cells and placenta [21,22] so we started analysing small RNA sequencing data from these tissue types (see Methods). Examining the abundance of miRNAs expressed from this cluster, we found that in placenta samples, these miRNAs contribute to a very high fraction, up to 40% of the total cellular miRNA population (Fig. 1A). This high expression level was in contrast to that in ESCs, where all C19MC species were detected but their overall expression was around 1% of the total miRNA pool (Fig. 1A and 1B). Individual miRNAs of the cluster showed heterogeneous expression levels in both tissue types, being more abundant in placenta samples (Fig. 1B). When the miRNA expression levels were compared with their genomic positions two different trends were revealed: in placenta, the expression of miRNAs slightly increases towards the 3ʹ end of the cluster, while in ESCs, the expression levels show a decreasing tendency (Fig. 2A). Moreover, if data were normalized to the corresponding placenta expression level, this position-dependent relative decrease in miRNA levels was further enhanced (Fig. 2B). Thorough analysis of the data reveals that the expression difference between the two tissue sample sets doubles at approximately every 12th miRNA, so the ~10-fold difference at the beginning of the cluster emerges to a ~ 135-fold difference at the 3ʹ end. Similar tendency could also be observed when independent datasets from Okae et al. [25] or from Mong et al. [10] were analysed (Suppl. Fig. 1). The data represents steady-state expression levels which are the combined results of transcription efficiency and RNA decay processes. Therefore, we aimed to investigate which level of regulation is responsible for this tissue-specific, position-dependent miRNA expression profile.
Figure 1.
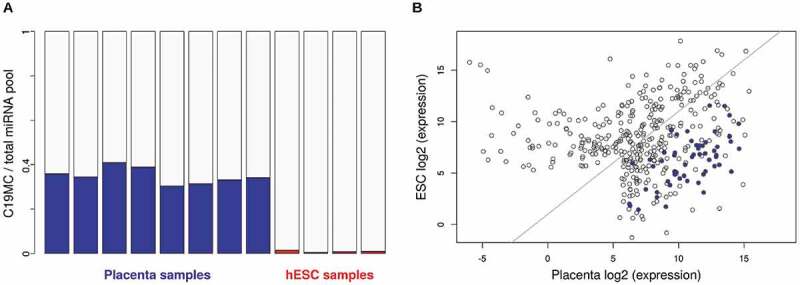
Investigation of mature C19MC miRNA expression. (A) Abundance of C19MC miRNAs in the total cellular miRNA population of placenta (n = 8; blue bars) and ESC (n = 4; red bars) samples (see Materials and Methods for dataset identifiers). (B) Expression levels of individual C19MC miRNAs. Averages of log2-transformed CPMs are shown in placenta (x-axis) and in ESC (y-axis). C19MC miRNAs are marked by blue dots
Figure 2.
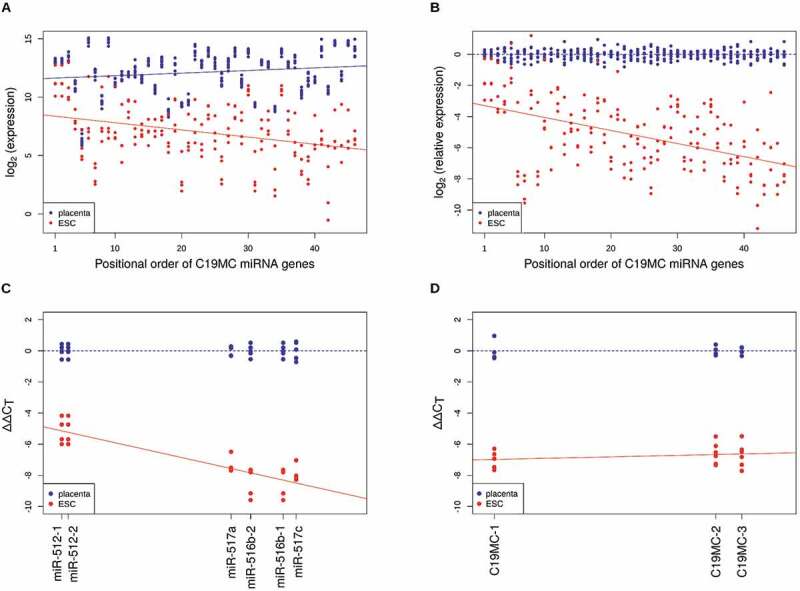
C19MC miRNA levels are genomic position-dependent. MiRNAs encoded by the same pre-miRNA (5p and 3p arms) were summed and ordered by their genomic position from first to 46th (x-axis). Y-axis shows log2-transformed (A) CPMs and (B) CPMs normalized to average placenta levels for each sample (placenta (n = 8; blue dots); ESC (n = 4; red dots)). Blue and red lines illustrate fitted linear models for placenta and ESC samples, respectively. For panel (B), the β-coefficient of regression is −0.08401, with p value of 3.5e-12. (C) Small RNA-seq results were reproduced by qRT-PCR on selected miRNA targets (β-coefficient: −0.08134, p value: 2.53e-06; blue dots are placenta samples (n = 5), red dots are ESC samples (n = 4)). (D) Pri-miRNA expression levels were measured by qRT-PCR and were found to be position-independent (β-coefficient: 0.00928, p value: 0.321). Primers for detecting the indicated PCR amplicons are listed in Suppl. Table 1
The position effect is attributed to tissue-specific pri-miRNA processing efficiency
To validate the expression profiles in the two tissues, we measured the steady-state levels of selected miRNA species by quantitative real-time PCR in the placenta-derived JAR and in the human embryonic stem cell originated HuES9 cell lines. The position-dependent profile was indeed confirmed (Fig. 2C): expression levels in hESCs were lower in general but when compared to their corresponding levels in placenta samples, miRNAs located at the 5ʹ regions of the cluster had higher abundance than those from the 3ʹ regions (e.g. hsa-miR-512 versus hsa-miR-517c). This tendency of gradual decrease towards the 3ʹ end in stem cells was statistically significant when linear regression models were fitted on the data (Fig. 2B and 2C). However, when pri-miRNA transcript levels were examined, this position-dependent decrease in abundance was not detected, although the steady-state expression level difference between the two cell types could still be revealed (Fig. 2D).
In order to further characterize this phenomenon, we quantified the processing efficiency by measuring the ratios of the unprocessed and the total pri-miRNAs in the two tissue types (Fig. 3A, and see also Materials and Methods). Interestingly, the processing was more efficient in hESCs but showed a position-dependent decrease towards the end of the cluster when compared to placenta cells (Fig. 3B). These results indicated that there is a tissue-specific, posttranscriptional process acting on the pri-miRNA species.
Figure 3.
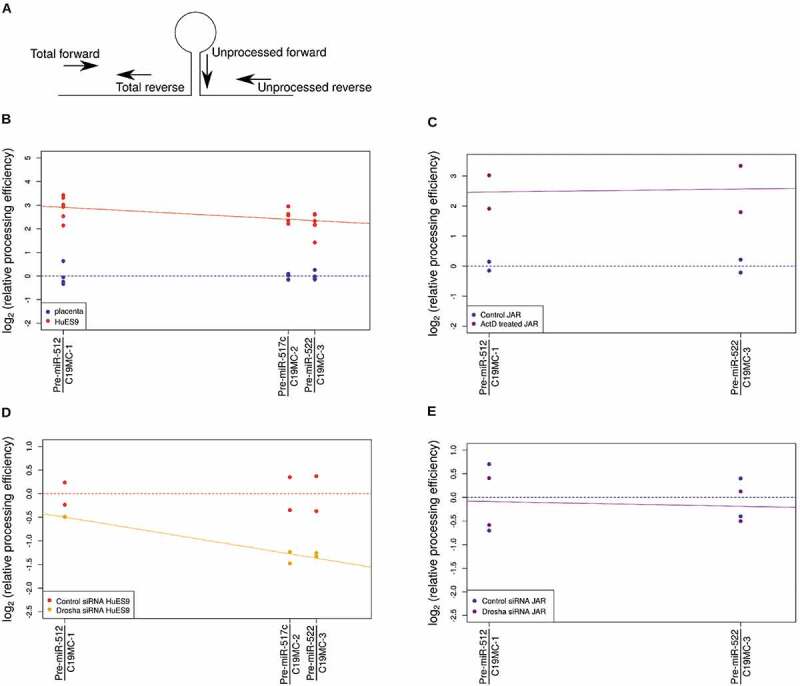
Position dependence of pri-miRNA processing efficiency. (A) Processing efficiency was measured at multiple positions of the cluster. At each position, two pairs of PCR primers were used: for the total pri-miRNA level measurements, both primers are located outside the pre-miRNA stem-loop; for the unprocessed pri-miRNA levels, a forward primer on the pre-miRNA stem-loop and a reverse primer downstream from the pre-miRNA were used. (B) Processing efficiency shows a position-dependent difference between placenta (n = 4; blue dots) and ESC samples (n = 6, red dots) (β-coefficient: −0.014825, p value: 0.0184). (C) Processing efficiency difference between control (n = 2; blue dots) and ActD treated JAR cells (n = 2; purple dots) was not affected by chromosomal position (β-coefficient: 0.002638, p value: 0.9256). (D) Drosha depletion by siRNA (n = 2; orange dots) reduced processing efficiency in a position-dependent manner in HuES9 cells (β-coefficient: −0.0228201, p value: 0.00131). (E) In JAR cells, Drosha depletion (n = 2; purple dots) had no effect compared to the control siRNA treatment (n = 2, blue dots) (β-coefficient: −0.002625, p value: 0.881)
Position-dependent processing is influenced by Microprocessor recruitment
We first hypothesized that pri-miRNA processing could be related to the transcript levels: in placenta, the much higher transcript level might saturate the miRNA processing machinery, causing a lower processing efficiency. To test this theory in the JAR cell line, we performed actinomycin D treatment to inhibit transcription and quantified the processing efficiency of pri-miRNAs in the C19M cluster. The transcripts in JAR cells had half-lives of less than 1 hour, and after 8 hours of treatment, the levels decreased to what was comparable to that in hESCs (Suppl. Fig. 2). At that time point, when measured at the two ends of the cluster, the processing efficiency did not show the position-dependent difference detectable in hESCs, although the processing efficiency increased because of the lower pri-miRNA input due to transcription inhibition (Fig. 3C). These results demonstrated that transcription level per se could not explain the position-dependent decrease in processing efficiency.
There are several RNA binding proteins involved in miRNA stability and processing, and a significant portion of them have been shown to bind the stem-loop structure of various pri-miRNAs, influencing their further maturation steps [12]. We searched for such factors that could potentially regulate miRNAs of the C19MC and found that hnRNPA1 and SRSF1 could be predicted to bind to several sequences in the cluster. However, when the scores of these binding sites were examined, they showed no correlation with the miRNA expression levels in the cluster (Suppl. Fig. 3). This indicated that although these (and potentially other) factors could regulate miRNA members of the cluster, their activity could not explain the tissue-specific, position-dependent processing difference.
To investigate whether the Microprocessor level or its recruitment is connected to processing efficiency, we examined the levels of Drosha in the cells. The mRNA levels were comparable in the two tissues so we knocked down Drosha by siRNA treatment (Suppl. Fig. 4) to see if this influences the processing. It was intriguing that in contrast to JAR cells (Fig. 3E), Drosha depletion in ESCs caused a strong decrease in processing efficiency towards the 3ʹ end of the cluster and it further enhanced the already present position-dependent processing decline within the cluster (Fig. 3D). When performing Drosha overexpression in JAR cells, no changes in the overall processing could be detected. On the other hand, when Drosha was expressed at higher levels in ESCs, the processing efficiency showed an increase towards the 3ʹ end of C19MC (Suppl. Fig. 5). These results supported the idea that during miRNA maturation from this cluster, tissue-specific processes regulate the local concentration of Drosha and thereby the Microprocessor, significantly influencing its access to the RNA template and therefore its processivity.
A placenta-specific promoter could cause more efficient co-transcriptional Microprocessor recruitment
To further study the tissue-specific aspect of the position-dependent processing regulation, we analysed epigenetic datasets from the NIH Roadmap Epigenomics Project. By examining data from human embryonic stem cell and placenta samples, we could reveal that there are distinct promoter activities in the two cell types. CAGE-seq data analysis [26] points towards 5 different transcription start sites, 4 of which are placenta-specific (TSS1-4), whereas TSS5 is active in ESCs (Fig. 4A). Moreover, a previously identified CpG island overlaps TSS1, and its methylated status in several tissues but placenta [11] indicates that this promoter region is exclusively active in the placenta, initiating the transcription of a longer RNA species. In contrast to that, in ESCs transcription is initiated 3ʹ downstream of this region which is also supported by the validated binding site of the stem cell-specific Nanog transcription factor, as well as the TATA-Box-Binding protein (TBP) site at TSS5 (Fig. 4A, see also the H3K4me3 marks of active transcription start sites [27]). By investigating our placenta and ESCs samples, we could indeed show that a longer transcript is present only in placenta samples (Fig. 4B). The two putative promoters show distinct epigenetic marks (different patterns of modified histones and DNase hypersensitive sites, Fig. 4A) which could well explain the tissue-specific differences in transcription intensity. In addition, the extensive regions with H3K27 acetylation and H3K4 tri-methylation in placenta samples not only indicate active enhancers and strong transcription start sites, but their presence in broad regions could also boost Microprocessor recruitment [28]. In order to investigate this potential aspect of regulation, we carried out ChIP-qPCR experiments using antibody against Drosha to analyse its binding to selected regions upstream of C19MC. We used the DNase I hypersensitive (DHS) sites to predict putative enhancers and designed real-time PCR primers to amplify seven selected DNA segments (DHS 1–7, Fig. 4A). For background normalization, we used two irrelevant promoter sequences, the elongation factor 1-alpha (EF1α) promoter being constitutively active, and the cardiac-specific alpha-actin (ACTC1) promoter being inactive in both tissues, to exclude potential binding signals related to Drosha’s non-canonical functions (e.g. in DNA repair, see [29]). By performing the experiments we could reveal Drosha association with selected sequences in placenta, showing a highest peak on the DHS 6 putative enhancer region; in ESCs, however, only very weak Drosha signals could be obtained (Fig. 4C and Suppl. Fig. 6). These findings were in line with the previous results on the distinct, tissue-specific promoters. In addition, an independent support for the tissue-specific promoter effect was revealed when we analysed the datasets from Mong et al.: the authors used the CRISPR/dCas9 Synergistic Activation Mediator system to activate the stem cell-specific C19MC promoter in HEK293 cells where it is normally transcriptionally silent. [10] By analysing their miRNA expression data, we could indeed provide evidence that the miRNA expression profiles show the similar, genomic position-dependent gradual decrease that were detected in other ESCs (Suppl. Fig. 1B). As a conclusion of our various analyses, the strong association of Drosha with the promoter/enhancer regions in placenta cells could explain the more efficient co-transcriptional recruitment of Drosha/DGCR8 in these cell types which ensures that the Microprocessor could efficiently process the entire region of the long non-coding transcripts from the miRNA cluster on the human chromosome 19.
Figure 4.
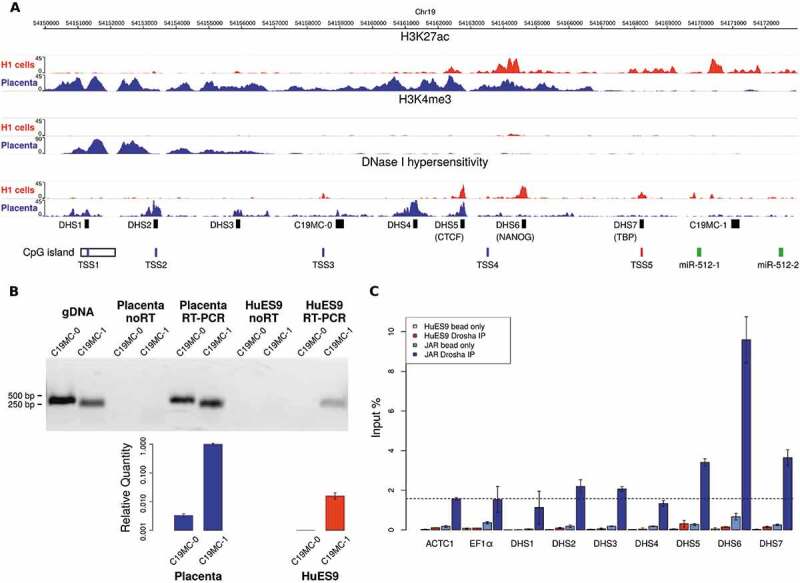
Promoter usage analysis in placenta tissues and in embryonic stem cells (H1 cell line as example) by investigating the epigenetic marks on a 22 kb DNA segment upstream of C19MC. (A) H3K4me3 modifications indicate two putative tissue-specific promoters with distinct active enhancer regions (H3K27ac modifications and DNase hypersensitive regions). The upper promoter is active in placenta, with four identified transcription start sites (TSS1-4 by CAGE-Seq data [26], shown in blue); while a more downstream alternative promoter initiates transcription in ESC (TSS5, shown in red). The TSS1 is located in a longer CpG island shown to be methylated in non-placental tissues [11]. PCR primer targets are marked by black boxes: the seven putative enhancers (DNase I hypersensitive sites, DHS1-7) and the two pri-miRNA transcripts (C19MC-0 for the longer, and C19MC-1 for the shorter form). The first two miRNAs of the cluster (hsa-miR-512-1 and −2) are marked by green boxes (B) The pri-miRNA with a longer 5ʹ end (C19MC-0) was detected only in the placenta sample (RT-PCR end point detection, gel image on upper panel). JAR genomic DNA (gDNA) was used for primer testing; the ‘noRT’ samples serve as negative controls. For the placenta and the HuES9 samples, C19MC-0 and C19MC-1 transcripts were also quantified by real-time PCR, using PolR2A as endogenous control; error bars show S.E.M. values. (C) ChIP-qPCR results analysing Drosha binding to the DHS sites and two unrelated promoter regions (ACTC1 and EF1α), the latter two being used for background control (indicated with a dashed line). Significant enrichment for DHS5-7 regions were detected in JAR cells (blue bars), but only weak binding in HuES9 cells (red bars, see separately also in Suppl. Fig. 6). Beads without antibody were used as ChIP negative controls (light blue and light red bars for JAR and HuES9, respectively), error bars indicate standard deviations
Discussion
In this study, we investigated the regulation of the miRNA cluster on human chromosome 19, especially focusing on the tissue-specific differences between placenta and human embryonic stem cells. The C19MC is one of the longest human miRNA clusters and the presence of scattered Alu sequences among the pre-miRNAs indicates that retrotransposon elements-mediated gene duplication events could have been responsible for the formation of the cluster [17–19]. For this reason, originally it had been proposed that the cluster is transcribed entirely by RNA polymerase III [18], however, it was later proved that RNA polymerase II transcribes this heavily spliced long RNA molecule [19]. Those studies all suggested that the numerous encoded miRNA species are processed simultaneously and at similar levels after transcription, however, we could prove that this is in fact not the case: apart from differences in individual miRNA levels, in human ESCs, there is a tissue-specific, chromosomal position-dependent miRNA expression profile which is different from what can be detected in placenta. We could provide evidence that there is a gradual decrease in the steady-state levels of miRNAs when moving towards the 3ʹ end of the cluster in ESCs, indicating tissue-specific regulatory differences (Fig. 2A-C). Investigating the potential mechanism(s) behind this observation we could show that such positional difference could not be detected on the pri-miRNA levels, demonstrating that at least on the transcript level, the 5ʹ and 3ʹ regions of this long noncoding RNA are present at similar levels in the examined tissues (Fig. 2D). Moreover, when transcription was hindered in a placenta cell line and when it was comparable to the lower level found in hESCs, the pri-miRNA processing from the 5ʹ and the 3ʹ end of C19MC did not show the gradual decrease present in stem cells (Fig. 3B-C). These results clearly proved that transcription level per se cannot explain the observed phenomenon and posttranscriptional mechanism(s) are responsible for the tissue-specific positional effect.
There are examples of other mammalian miRNA clusters where individual miRNAs are regulated distinctly from their neighbours: the hsa-miR-18a, for instance, shows different expression in its cluster, and posttranscriptional regulation by the hnRNPA1 protein has been found to be responsible for that [14–16]. We have tested some RNA regulatory proteins for the C19MC and although pri-miRNA binding was clearly predicted throughout the cluster, an overall effect on the miRNAs expression could not be detected (Suppl. Fig. 3). When testing the pri-miRNA processing efficiency, a clear difference was revealed between the two examined tissues: the efficiency showed a position-dependent gradual decrease in hESCs as compared to placenta (Fig. 3B). A previous study revealed that the DGCR8 protein component of the Microprocessor seems to stay longer on the pre-miRNA after the Drosha cleavage but the authors could not explain the significance or the consequence of this observation [24]. Based on this we hypothesized that the Microprocessor availability or the difference in its local activity might be responsible for the positional decrease in miRNA processing in hESCs. To investigate the mechanism, we tested the miRNA maturation by knocking down or overexpressing Drosha in the examined tissues. It was intriguing to see that changes in the level of Drosha could strongly influence the positional effect in hESCs but did not affect the miRNA processing efficiency in placental cells (Fig. 3 and Suppl. Fig. 5). The results indicated that in stem cells, the availability of the active Microprocessor complex could be limited which would explain the lower releasing efficiency of pre-miRNAs towards the 3ʹ end of this particularly long primary noncoding transcript.
In a recent study [30], Donayo and colleagues revealed that apart from previous findings of the selective regulation by hnRNPA1 [14], an additional hierarchical processing of pre-miRNAs in the miR-17-92 cluster results in different expression levels of the mature miRNA species. Moreover, they showed that the oncogenic amplification of this cluster leads to the sequestration of Microprocessor complexes in the cells, lowering the processing efficiency of other miRNA clusters. Such a mechanism could be in line with our findings but would not explain the tissue-specific differences in processing of C19MC. So, what could cause the decrease in availability of the Drosha/DGCR8 complex in embryonic stem cells as compared to placenta cells? One explanation could lie in the transcriptional regulation of the cluster. Here we described a placenta-specific promoter that overlaps with the previously described promoter of the cluster [11] and it is very active in placenta but silent in hESCs (Fig. 4A and 4B). This clearly explains the much stronger transcription activity observed in placenta cells but the detected epigenetic histone marks also point to the idea that this region can work as a potential enhancer: as described earlier for certain ‘super-enhancer’ regions [31,32], it can more efficiently recruit proteins and regulators to the RNA polymerase II apparatus, carrying out a more robust co-transcriptional processing of the transcript. Such a mechanism was shown to exist for long noncoding RNAs [33], including co-transcriptional recruitment of the Drosha/DGCR8 complex [13,28,34]. As a similar regulation could well explain our results showing tissue-specific differences in formation of miRNAs from C19MC, we performed ChIP-qPCR experiments using an antibody against Drosha, and could provide evidence that Drosha is indeed associated with several potential enhancer sequences located in the region upstream of the C19MC locus (Fig. 4C). This result fits well into the model of Microprocessor recruitment to the transcription apparatus already at the initiation site of transcription [28,34], and the level and the local availability of Drosha could also contribute to explaining the efficient processing of miRNA precursors throughout the C19MC region in placenta (Fig. 5). By investigating similar, super-enhancer mediated recruitment mechanisms it seems that it is not limited to the more efficient co- or posttranscriptional processing of the transcripts, such as splicing or pre-miRNA cleavage, but could also contribute to the more efficient recruitment of RNA export factors [35]. In addition, such tissue-specific sequestration of the miRNA processing apparatus could help in understanding why the misregulation of C19MC could be connected to tumour formation, as elevated level of C19MC transcription was found to be an associated marker of triple-negative breast cancers [36], or when a genomic rearrangement mediated fusion of C19MC with the TTYH1 gene resulted in the formation of embryonal brain tumours [37]. On the other hand, it still needs to be addressed how the previously described imprinting regulation of C19MC [11] is connected to the identified enhancer region and Microprocessor recruitment.
Figure 5.
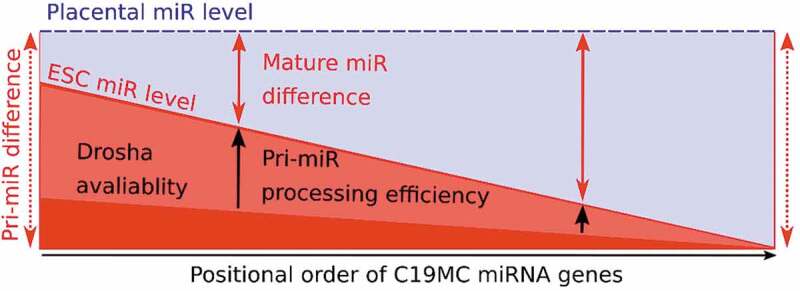
Cumulative model of the position-dependent miRNA maturation at C19MC. Normalized mature miRNA levels of C19MC show a position-dependent difference between placenta and ESCs (indicated with a blue and a red line, respectively). The constant difference of primary transcript levels (pri-miRNAs levels, represented by vertical dotted red arrows) could not explain this phenomenon, however, pri-miRNA processing efficiencies (represented by vertical black arrows) significantly contribute to this positionally increasing difference. Based on the results of the present study, tissue-specific difference in processing efficiency of C19MC is regulated by Drosha/Microprocessor level and local availability
The Drosha recruitment model supported by our ChIP-qPCR data could explain the local Drosha availability and the efficient processing of C19MC but fails to explain why the ESCs are more sensitive to changes in Drosha levels whereas placenta cells are not (Fig. 3 and Suppl. Fig. 5). When considering expression levels, the transcription of C19MC in placenta is very high, and miRNAs from the cluster make up a huge amount (40%) of the total miRNA pool in the cell (Fig. 1A). Together with the efficient recruitment of Drosha to the promoter in placenta, this massive amount of miRNA could ‘titrate out’ available Drosha (e.g. Microprocessor complexes) from other, less transcriptionally active miRNA loci, similarly to what was shown for the highly expressed miR-17 ~ 92 cluster [30]. In stem cells, however, the C19MC is expressed in a much lower level (approx. 1% of the total cellular miRNA pool, see Fig. 1A), and other strongly expressed miRNA loci could ‘titrate out’ Drosha locally, making the Microprocessor complexes less available for the C19MC in stem cells, therefore being more sensitive to the overall Drosha level. Our data analyses indicate that the mentioned miR-17 ~ 92 cluster (making up 10–30% of the total miRNA pool) or the miR-302 ~ 367 cluster (also making up a significant, 10–20% of the total miRNA pool) could act such local “recruiters“ in stem cells. Nevertheless, to gain a more detailed picture on the molecular mechanisms, further studies should also address the level and potential recruitment of DGCR8, or other associated factors of the Microprocessor complex in relation to C19MC processing. Activating the placenta-specific promoter by the already described CRISPR/dCas9 Synergistic Activation Mediator system [10] could also provide further details about the connection among transcription-recruitment-processing; however, these experiments are beyond the scope of the current investigation.
In conclusion, our results could reveal a new transcription-coupled, tissue-specific regulatory mechanism of miRNA maturation from long clusters. Although we could provide evidence that the rate-limiting step in the processing of the 3ʹ region of C19MC is the local availability of the effector Drosha/DGCR8, further studies are required to elucidate the exact details of this mechanism. Nevertheless, together with the described regulation of other oncogenic clusters, our results can well contribute to a novel model of a tissue-specific regulation of miRNA maturation from long genomic clusters.
Materials and methods
Cell culture maintenance and treatments
The JAR placental choriocarcinoma cell line was maintained under standard conditions in 5% CO2 incubator at 37°C, in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin-streptomycin (Gibco). Transcription inhibition was achieved by 5 μg/mL of actinomycin D (Sigma-Aldrich) treatment, and samples were taken at 1, 2, 4 and 8 h after treatment.
The HuES9 embryonic stem cell line was originally provided by Dr. Douglas Melton (HHMI). The cells were cultured on Matrigel (Corning) coated six-well plates in mTeSR medium (Stemcell Technologies) and were grown to 70% density before transfection. For nucleofection, cells were treated overnight with the rho-associated protein kinase (ROCK) inhibitor Y-27,632 (Selleckchem), then detached with Accutase (Thermo Fisher Scientific) and washed with 1× PBS at 37°C.
106 cells per reaction were used for electroporation using the A-023 program with the Amaxa Human Stem Cell Nucleofector Kit 1 (cat. #: VPH-5012, Lonza) for HuES9 cells, or the X-005 program with the Amaxa Cell Line Nucleofector Kit V (cat. #: VVCA-1003, Lonza) for JAR cells, according to the manufacturer’s protocol; the cells were seeded on six-well plates and harvested 24 hrs after transfection. For knock-down experiments, 25 nM of siRNA targeting Drosha (catalogue #4,390824) and a negative control (cat. #4,390843) were used as recommended by the manufacturer (Thermo Fisher Scientific). For Drosha overexpression experiments, GFP-tagged Drosha expressing plasmid was used (Addgene #62520 plasmid, [38]) and transfection efficiency was verified using fluorescence microscopy. In case of HuES9 cells, the similar nucleofection method was applied as described above. For efficient plasmid transfection into JAR cells, the FuGENE® HD reagent (Promega) was used according to the manufacturer’s instruction.
Study participants, placenta sample collection and handling
Study participants had been recruited during routine prenatal care or following hospital admission during the third trimester of pregnancy at 1st Department of Obstetrics and Gynaecology, Semmelweis University, Budapest, Hungary. In this study, five placenta samples were collected right after C-sections at term pregnancies without any indications of gestational complications. The study protocol was approved by the Scientific and Research Ethics Committee of the Medical Research Council (ETT TUKEB) [No: 24387–2/2016] and written informed consent was obtained from each patient. The research was conducted in accordance with the Declaration of Helsinki.
Placenta samples were collected according to the protocol described by Pasupathy et al. [39]. Four areas suitable for sampling were located on the maternal surface; damaged areas (calcification, haematoma, etc.) were excluded. About 1–2 mm from the basal membrane was removed and pea-sized tissue samples were taken from the placental cotyledons. The samples were washed twice in 1× PBS solution at 4°C and placed in RNAlater™ stabilizing solution (Thermo Fisher Scientific) to avoid RNA degradation.
RNA isolation
Total RNA isolation was done by using TRIzol™ Reagent (Thermo Fisher Scientific) as described in the user guide. RNA integrity was analysed by agarose gel electrophoresis, sample purity and concentration were measured by a Nanodrop spectrophotometer (Thermo Fisher Scientific).
cDNA preparations and real-time PCR quantifications
For mRNA or pri-miRNA analysis, 1 μg total RNA was reverse transcribed by random oligomers using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific); cDNA samples were diluted 1:10 before subsequent amplifications. Drosha mRNA level was measured by using TaqMan® Gene Expression Master Mix (Thermo Fisher Scientific) and pre-designed Drosha TaqMan® assay (cat. #4331182). In the case of C19MC pri-miRNA, RT-PCR was done by using SYBR Green PCR Master Mix with custom-made PCR primers (Suppl. Table 1). Real-time PCR measurements were done on a StepOnePlus™ platform (Thermo Fisher Scientific) according to the manufacturer’s instructions. The ΔΔCt method was applied for relative quantifications, using a set of endogenous control mRNAs for normalization: for TaqMan® analyses, the PolR2A (assay Hs00172187_m1), and the RPLP0 (assay Hs9999902_m1); for SYBR® Green assays, custom-made primers for PolR2A and RPLP0 (for details, see Suppl. Table 1).
For mature miRNA quantification, the expression analysis was performed using the miRCURY LNA™ Universal RT miRNA PCR Assay (Qiagen), according to the manufacturer’s instructions. Briefly, RNA samples (5 ng/µl) were reverse-transcribed and the UniSp6 RNA spike-in template was added to each reaction for controlling the quality of cDNA synthesis. cDNA samples were diluted 1:80 before subsequent amplifications. RT-PCR was done by using miRCURY SYBR® Green master mix (Qiagen) and real-time PCR reactions were run on a StepOnePlus™ platform (Thermo Fisher Scientific) according to the manufacturer’s protocol. Pre-designed assays were used to measure the levels of hsa-miR-512, hsa-miR-517a, hsa-miR-516b and hsa-miR-517 c. In these cases, the hsa-miR-103a internal control miRNA was used for normalization during the relative quantifications by the ΔΔCt method.
Pri-miRNA processing efficiency was calculated with normalization of uncleaved pri-miRNA level to the total amount of pri-miRNA (see also Fig. 3A); primers are listed in Suppl. Table 1.
Chromatin immunoprecipitation (ChIP) qPCR
ChIP-qPCR measurements were done using the ChIP Kit (cat. #ab500, Abcam) according to the manufacturer’s instructions. Briefly, 106 cells were collected per ChIP, chromatin was cross-linked by formaldehyde (Sigma), and cells were lysed and sonicated for 10 minutes. Sheared DNA fragment length was analysed by gel electrophoresis. Immunoprecipitations were done by anti-Drosha antibody (cat. #ab12286, Abcam), and for positive control, the anti-H3 antibody (cat. #ab1791, Abcam); as a negative control, only Protein A beads were used. For normalization purposes, input chromatin DNA was used. qPCR was done by using SYBR Green PCR Master Mix with custom-made PCR primers (Suppl. Table 1) on a StepOnePlus™ platform (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Western blot
Cell lysates were collected 24 h after transfection with siRNAs. Samples were briefly sonicated and protein concentration was measured by Lowry method. About 30 μg of protein samples per lane were run on 8% acrylamide gels and electroblotted onto PVDF membranes (BioRad). After washing with TBS-Tween, membranes were blocked by 5% milk/TBS-Tween, and subsequently incubated with Anti-Drosha antibody (cat. #ab12286, Abcam) on 4°C overnight. Membranes were washed three times with TBS-Tween and then incubated in Anti-Rabbit IgG secondary antibody (Jackson ImmunoResearch) solution for 1 h at room temperature. Membranes were washed twice with TBS-Tween, and for signal detection ECL reagent (Thermo Fisher Scientific) was used, and the membranes were exposed to Agfa films. Anti-beta Actin antibody (cat. #ab20272, Abcam) was used as a loading control and to normalize Drosha expression. Expression levels were determined by densitometry of the scanned images using the ImageJ software [40].
Next-generation sequencing data analyses, visualization and statistics
Publicly available sequencing data were reanalysed from the PRJNA187509 NCBI BioProject (placenta samples) [41] and from SRA data with accession name: SRR026761, SRR1616134, SRR1616135, SRR1203788 (hESC samples) [42–44]. Histone modification data (H3K27ac and H3K4me3) from hESC and placenta samples (E003 H1, E008 H9, E014 HuES48, E015 HuES6, E016 HuES64 and E091 Placenta) were downloaded from NIH Roadmap Epigenomics [45].
Raw data were trimmed by CutAdapt (1.10) [46] and reads were mapped to hg38 assembly of the human genome by BWA aln (0.7.12) algorithm [47]. Read counting was carried out by FeatureCounts from the Rsubread package (1.26.1) [48] using miRBase (v.21) [49] annotations. Normalization was done by edgeR package (3.18.1) [50] and ‘count per million’ (CPM) values were used. For quantification of miRNA genes, the two arms of each miRNA were summed. Logarithmic transformed relative expression levels were calculated similarly to the ΔΔCt method: logarithmic transformed values of the target samples were normalized to the mean of logarithmic transformed values of the control samples.
Statistics and visualization were done in R(3.4.4) [51] software environment. Simple linear regression models were fitted by the lm function [52] and associated p-values were used to decide whether the explanatory variable (e.g. genomic position) has a significant influence on the response variable (e.g. relative miRNA expression). The relationship between the response and the explanatory variable was described by the slope (β-coefficient) of the fitted linear model.
Epigenomics data was displayed with the WashU Epigenome Browser [53].
Supplementary Material
Acknowledgments
The authors are grateful to Kornélia Némethy and Gerda Wachtl for excellent technical help.
Funding Statement
This study was supported by the grants VEKOP-2.1.1-15-2016-00156 and VEKOP-2.3.3-15-2017-00014, and project no. 2018-1.2.1-NKP-2018-00005 from the National Research, Development and Innovation Fund of Hungary.
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Bartel DP, Metazoan MicroRNAs. Cell. 2018;173:20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gebert LFR, MacRae IJ.. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20:21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Guo WT, Wang Y. Dgcr8 knockout approaches to understand microRNA functions in vitro and in vivo. Cell Mol Life Sci. 2019;76:1697–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kim YK, Kim VN. Processing of intronic microRNAs. Embo J. 2007;26:775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ramalingam P, Palanichamy JK, Singh A, et al. Biogenesis of intronic miRNAs located in clusters by independent transcription and alternative splicing. RNA. 2014;20(1):76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Altuvia Y, Landgraf P, Lithwick G, et al. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005;33:2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kim YK, Yu J, Han TS, et al. Functional links between clustered microRNAs: suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res. 2009;37:1672–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Morales-Prieto DM, Ospina-Prieto S, Chaiwangyen W, et al. Pregnancy-associated miRNA-clusters. J Reprod Immunol. 2013;97(1):51–61. [DOI] [PubMed] [Google Scholar]
- [9].Malnou EC, Umlauf D, Mouysset M, et al. Imprinted MicroRNA gene clusters in the evolution, development, and functions of mammalian placenta. Front Genet. 2019;9:706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mong EF, Yang Y, Akat KM, et al. Chromosome 19 microRNA cluster enhances cell reprogramming by inhibiting epithelial-to-mesenchymal transition. Sci Rep. 2020;10(1):3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Noguer-Dance M, Abu-Amero S, Al-Khtib M, et al. The primate-specific microRNA gene cluster (C19MC) is imprinted in the placenta. Hum Mol Genet. 2010;19(18):3566–3582. [DOI] [PubMed] [Google Scholar]
- [12].Michlewski G, Caceres JF. Post-transcriptional control of miRNA biogenesis. RNA. 2019;25(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Conrad T, Marsico A, Gehre M, et al. Microprocessor activity controls differential miRNA biogenesis in vivo. Cell Rep. 2014;9(2):542–554. [DOI] [PubMed] [Google Scholar]
- [14].Concepcion CP, Bonetti C, Ventura A. The microRNA-17-92 family of microRNA clusters in development and disease. Cancer J. 2012;18(3):262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Michlewski G, Guil S, Semple CA, et al. Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol Cell. 2008;32(3):383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Michlewski G, Caceres JF. Antagonistic role of hnRNP A1 and KSRP in the regulation of let-7a biogenesis. Nat Struct Mol Biol. 2010;17(8):1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lehnert S, Van Loo P, Thilakarathne PJ, et al. Evidence for co-evolution between human microRNAs and Alu-repeats. PLoS One. 2009;4(2):e4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006;13(12):1097–1101. [DOI] [PubMed] [Google Scholar]
- [19].Bortolin-Cavaille M-L, Dance M, Weber M, et al. C19MC microRNAs are processed from introns of large Pol-II, non-protein-coding transcripts. Nucleic Acids Res. 2009;37(10):3464–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhang R, Wang Y-Q, Su B. Molecular evolution of a primate-specific microRNA family. Mol Biol Evol. 2008;25(7):1493–1502. [DOI] [PubMed] [Google Scholar]
- [21].Bar M, Wyman SK, Fritz BR, et al. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells. 2008;26(10):2496–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ren J, Jin P, Wang E, et al. MicroRNA and gene expression patterns in the differentiation of human embryonic stem cells. J Transl Med. 2009;7(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Flor I, Bullerdiek J. The dark side of a success story: microRNAs of the C19MC cluster in human tumours. J Pathol. 2012;227(3):270–274. [DOI] [PubMed] [Google Scholar]
- [24].Bellemer C, Bortolin-Cavaille ML, Schmidt U, et al. Microprocessor dynamics and interactions at endogenous imprinted C19MC microRNA genes. J Cell Sci. 2012;125:2709–2720. [DOI] [PubMed] [Google Scholar]
- [25].Okae H, Toh H, Sato T, et al. Derivation of Human Trophoblast Stem Cells. Cell Stem Cell. 2018;22(1):50–63 e6. [DOI] [PubMed] [Google Scholar]
- [26].Noguchi S, Arakawa T, Fukuda S, et al. FANTOM5 CAGE profiles of human and mouse samples. Sci Data. 2017;4(1):170112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liang G, Lin JC, Wei V, et al. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc Natl Acad Sci U S A. 2004;101(19):7357–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Suzuki HI, Young RA, Sharp PA. Super-enhancer-mediated RNA processing revealed by integrative microrna network analysis. Cell. 2017;168(6):1000–14 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lu WT, Hawley BR, Skalka GL, et al. Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair. Nat Commun. 2018;9:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Donayo AO, Johnson RM, Tseng HW, et al. Oncogenic biogenesis of pri-miR-17 approximately 92 reveals hierarchy and competition among polycistronic MicroRNAs. Mol Cell. 2019;75:340–56 e10. [DOI] [PubMed] [Google Scholar]
- [31].Pott S, Lieb JD. What are super-enhancers? Nat Genet. 2015;47(1):8–12. [DOI] [PubMed] [Google Scholar]
- [32].Sengupta S, George RE. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer. 2017;3(4):269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schlackow M, Nojima T, Gomes T, et al. Distinctive patterns of transcription and RNA processing for human lincRNAs. Mol Cell. 2017;65(1):25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Church VA, Pressman S, Isaji M, et al. Microprocessor recruitment to elongating RNA polymerase II is required for differential expression of MicroRNAs. Cell Rep. 2017;20(13):3123–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Viphakone N, Sudbery I, Griffith L, et al. Co-transcriptional loading of RNA export factors shapes the human transcriptome. Mol Cell. 2019;75(2):310–23 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jinesh GG, Flores ER, Brohl AS. Chromosome 19 miRNA cluster and CEBPB expression specifically mark and potentially drive triple negative breast cancers. PLoS One. 2018;13(10):e0206008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kleinman CL, Gerges N, Papillon-Cavanagh S, et al. Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet. 2014;46(1):39–44. [DOI] [PubMed] [Google Scholar]
- [38].Tang X, Zhang Y, Tucker L, et al. Phosphorylation of the RNase III enzyme Drosha at Serine300 or Serine302 is required for its nuclear localization. Nucleic Acids Res. 2010;38(19):6610–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pasupathy D, Dacey A, Cook E, et al. Study protocolA prospective cohort study of unselected primiparous women: the pregnancy outcome prediction study. BMC Preg Childbirth. 2008;8:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Williams Z, Ben-Dov IZ, Elias R, et al. Comprehensive profiling of circulating microRNA via small RNA sequencing of cDNA libraries reveals biomarker potential and limitations. Proc Natl Acad Sci U S A. 2013;110(11):4255–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Morin RD, O’Connor MD, Griffith M, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Asikainen S, Heikkinen L, Juhila J, et al. Selective microRNA-Offset RNA expression in human embryonic stem cells. PLoS One. 2015;10(3):e0116668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen T, Xiang JF, Zhu S, et al. ADAR1 is required for differentiation and neural induction by regulating microRNA processing in a catalytically independent manner. Cell Res. 2015;25:459–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Roadmap Epigenomics C, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12. [Google Scholar]
- [47].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liao Y, Smyth GK, Shi W. featurecounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. [DOI] [PubMed] [Google Scholar]
- [49].Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–D62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314. [Google Scholar]
- [52].Chambers JM, Hastie TJ. Chapter 4:Linear models. Chambers JM, Hastie TJ, eds. Statistical models in S.California: Wadsworth & Brooks/Cole; 1992. [Google Scholar]
- [53].Zhou X, Maricque B, Xie M, et al. The human epigenome browser at Washington University. Nat Methods. 2011;8(12):989–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.