

ABSTRACT
RNA helicases contribute to diverse aspects of RNA metabolism through their functions in re-arranging RNA structures. Identification of the remodelling targets of RNA helicases is a critical step in elucidating their cellular functions. Here, we show that, in contrast to many other ribosome biogenesis factors, the DExD box ATPase DDX55 predominantly localizes to the nucleoplasm and we identify a nuclear localization signal within the C-terminal region of the protein. DDX55 associates with pre-ribosomal subunits in human cells and is required for maturation of large subunit pre-rRNAs. Interestingly, in vitro analyses show that DDX55 selectively associates with double-stranded RNA substrates, which also stimulate its ATPase activity, and our data suggest that the C-terminal region of DDX55 contributes to this substrate specificity. The C-terminal region of DDX55 is also necessary for recruitment of the helicase to pre-ribosomes and, using in vivo crosslinking, we reveal a binding site for DDX55 in helix H62 of the 28S ribosomal RNA. Taken together, these data highlight the importance of the C-terminal region of DDX55 in substrate specificity and recruitment, and identify domain IV as a potential remodelling target of DDX55 during LSU biogenesis.
KEYWORDS: RNA helicase, ribosome, ribonucleoprotein complex (RNP), ATPase, RNA-protein interaction
Introduction
A hallmark of RNA is its ability to form intricate secondary and tertiary structures, and NTP-dependent RNA helicases are key regulators of RNA folding and rearrangement [1,2]. These ubiquitously expressed enzymes are characterized by a helicase core domain composed of tandem RecA-like domains containing conserved sequence motifs involved in RNA substrate and/or NTP binding and/or NTP hydrolysis. While the eIF4A-like sub-family of DExD box RNA helicases are considered minimal helicases as they comprise only this core domain, most other RNA helicases carry N- or C-terminal extensions that contribute to the function, interactions, or regulation of these proteins [3]. The N-/C-terminal extensions of some RNA helicases include defined domains, e.g. the oligosaccharide binding (OB) fold present in DExH box proteins. However, this is often not the case and the precise functions of many such extensions remain elusive. Mechanistically, conformational changes within DExD box helicases upon ATP and substrate binding cause transition from an inactive, open state to an active, closed state in which the conserved sequence motifs within the tandem RecA-like domains are in close proximity. This induces displacement of a single RNA strand leading to local unwinding, and rearrangement of the catalytic site then triggers ATP hydrolysis and disassembly of the helicase-RNA complex. Interestingly, the portfolio of functions attributed to DExD box ATPases extends beyond unwinding as some of these proteins have also been shown to be capable of RNA strand annealing, displacing proteins from RNA substrates and/or acting as RNA clamps within ribonucleoprotein complexes (RNPs) [4]. As a result of these molecular functions, DExD box helicases contribute to many different aspects of RNA metabolism. Most notably, the assembly and function of large ribonucleoprotein complexes (RNPs), such as ribosomes and spliceosomes, require the action of numerous DExD box RNA helicases. Although an increasing number of RNA helicases have been shown to be multifunctional, many DExD box proteins appear to be dedicated for particular substrates. Alongside modulation of the catalytic activity of RNA helicases by cofactor proteins, the recruitment of RNA helicases to their target RNAs/RNPs represents an important level of RNA helicase regulation [2].
Eukaryotic ribosomes are large ribonucleoprotein complexes (RNPs), composed of four ribosomal RNAs (rRNA) and 79 (Saccharomyces cerevisiae (yeast))/80 (human) ribosomal proteins (RPs) [5,6], that are responsible for the translation of mRNAs into proteins. As a consequence of this essential function, ribosome synthesis has emerged as a central hub, co-ordinating protein synthesis with cellular growth and proliferation [7]. Moreover, perturbation of ribosome production underlies various genetic disorders collectively termed ribosomopathies [8]. Ribosome synthesis is well characterized in the yeast model system and while fundamental aspects of the pathway appear to be conserved in higher eukaryotes, numerous factors involved in human ribosome production have been found to have different or additional functions compared to their yeast counterparts, highlighting the need for deeper understanding of the human ribosome assembly pathway [7]. Assembly of the large (LSU, 60S) and small (SSU, 40S) ribosomal subunits is a process that spans several cellular compartments; transcription of rRNA precursors and initial assembly of particles in the nucleolus is followed by further maturation of ribosomal subunits in the nucleoplasm, nuclear export, and final quality control in the cytoplasm [7,9,10]. This pathway requires the coordinated action of several hundred biogenesis factors that associate transiently with the pre-ribosomal subunits to scaffold rRNA folding and protein assembly or to mediate specific processes such as pre-rRNA maturation, rRNA modification, and nuclear export of pre-ribosomal subunits [11,12,13]. Among these assembly factors, are numerous RNA helicases, which have been implicated in diverse aspects of the maturation process [14]. For example, Dhr1/DHX37, Prp43, Rok1, and Has1 are implicated in regulating the dynamics of small nucleolar RNAs (snoRNAs) on pre-ribosomes [15,16,17,18,19,20,21,22,23]. Other RNA helicases, including Mak5, Has1, Rok1, and Spb4, are proposed to function in catalysing rRNA rearrangements that promote either the recruitment or release of particular RPs or biogenesis factors [22–24]. For example, in yeast, Spb4 is thought to remodel a hinge region at the base of a large eukaryotic expansion segment to facilitate recruitment of the pre-60S export factor Arx1 [22].
Here, we show that the putative DExD box RNA helicase, DDX55 is an active ATPase that localizes to the nucleoplasm of HEK293 cells. Our data reveal that DDX55 associates with pre-ribosomes and we identify a binding site of this protein within domain IV of the 28S rRNA of the pre-LSU. We further show that the C-terminal extension of DDX55 contains a nuclear localization signal (NLS) and is required for recruitment of the helicase to pre-ribosomal complexes.
Results
DDX55 localizes in the nucleoplasm and co-migrates with pre-ribosomes in sucrose gradients
The putative RNA helicase DDX55 is the human homologue of the yeast nucleolar RNA helicase Spb4, which is required for assembly of the LSU [22]. To explore if DDX55 may fulfil a similar function, the sub-cellular localization of DDX55 was first determined. A stably transfected HEK293 cell line for the inducible expression of C-terminally His-PreScission protease cleavage site-2xFlag (Flag) tagged DDX55 was generated. Cells expressing DDX55-Flag were fixed and immunofluorescence was performed using antibodies against the Flag tag and, as a nucleolar marker, the human ribosome biogenesis factor UTP14A [25]. Nuclear material was visualized by DAPI staining. This revealed that DDX55-Flag predominantly localizes to the nucleoplasm and is largely, but not completely, excluded from nucleoli (Fig. 1A). Next, to gain insight into whether DDX55 may contribute to the assembly of ribosomal subunits, the potential association of DDX55 with pre-ribosomal complexes was analyzed. Whole cell extracts prepared from cells expressing DDX55-Flag were separated by sucrose density gradient centrifugation. An absorbance profile of the gradient at 260 nm was generated to identify the fractions containing the (pre-)ribosomal 40S, 60S, and 80S complexes (Fig. 1B; upper panel). Proteins present in each gradient fraction were precipitated and the distribution of DDX55-Flag was analysed by western blotting. DDX55-Flag was distributed throughout the gradient but was enriched in fractions containing free proteins or small complexes and those containing (pre-)60S complexes (Fig. 1B; lower panel), implying that like its yeast counterpart, DDX55 may act in ribosome biogenesis.
Figure 1.

DDX55 is present in the nucleoplasm and associates with late pre-LSU complexes. (A) The sub-cellular localization of Flag-tagged DDX55 was determined by immunofluorescence (DDX55) using antibodies against the Flag tag. Co-immunofluorescence using an α-UTP14A served as a nucleolar marker and nuclear material was visualized using DAPI staining. Scale bar represents 10 μm. (B) Whole cell extracts prepared from HEK293 cells expressing DDX55-Flag were separated by sucrose density gradient centrifugation. During fractionation, a profile of absorbance at 260 nm was generated and the peaks corresponding to free proteins/small complexes, 80S mature/90S pre-ribosomes, and (pre-)40S and (pre-)60 particles are indicated (upper panel). Proteins in each fraction were separated by SDS-PAGE and analysed by western blotting using an α-Flag antibody (lower panel)
DDX55 crosslinks to helices H62-63 of the 28S rRNA sequence
The finding that DDX55 co-migrates with pre-ribosomal complexes in sucrose density gradients suggests that this helicase physically associates with such particles. The crosslinking and analysis of cDNA (CRAC) approach [26,27] was therefore employed to identify the pre-rRNA binding site(s) of DDX55. HEK293 cells expressing either DDX55-Flag or the Flag tag alone were crosslinked using light at 254 nm to covalently link proteins to their associated RNAs. Complexes were first affinity purified via their Flag tag under native conditions. A partial RNase digest was then performed to degrade RNA sequences not protected by association with a protein. Complexes were immobilized on NiNTA via their His tag, and the RNA fragments were labelled with [32P] and ligated to sequencing adaptors. Protein-RNA complexes were separated by denaturing polyacrylamide gel electrophoresis (PAGE), transferred to a nitrocellulose membrane and detected using autoradiography. Signals migrating at a position corresponding to the molecular weight of DDX55-Flag (+RNA fragments) were detected in the samples derived from cells expressing DDX55-Flag (A and B) but not the Flag tag alone, demonstrating that DDX55 directly associates with RNA in vivo (Fig. 2A). These regions of the membrane, and a corresponding area from the lane containing the control sample, were excised, proteins were degraded and RNA fragments isolated. After reverse transcription, the cDNA library was amplified by PCR and subjected to Illumina next-generation sequencing. The obtained sequencing reads were mapped to the human genome and the relative proportions of reads derived from different types of RNA were determined (Fig. 2B). Compared to the Flag tag control, both DDX55-Flag samples showed an enrichment of reads derived from rRNAs (Flag – 5%, DDX55-Flag A – 12%, DDX55-Flag B 13%), tRNAs (Flag – 4%, DDX55-Flag A – 22%, DDX55-Flag B 22%) and snoRNAs (Flag – 0.5%, DDX55-Flag A – 2%, DDX55-Flag B – 2%). Unspecific co-purification of tRNA/snoRNA sequences with non-tRNA/snoRNA-binding proteins has been observed in previous CRAC experiments (see, for example [28]), likely occurring due to the high abundance and accessibility of these RNAs for crosslinking. As no particular (subset of) tRNA species were enriched with either of the DDX55 samples compared to the control and the snoRNAs enriched with DDX55 do not belong to a particular class (box C/D, box H/ACA) or guide modifications in a specific rRNA region (data not shown), it is unlikely that these types of RNA are bona fide substrates of this helicase. However, given the co-migration of DDX55 with pre-ribosomal subunits (Fig. 1B), the enrichment of rRNA sequences with DDX55-Flag strongly supports the direct association of DDX55 with pre-rRNAs in vivo and, therefore, the distribution of the sequencing reads on the rDNA sequence encoding the full-length 47S pre-rRNA transcript was examined. In the two DDX55-Flag samples, but not the Flag control, the majority of reads mapped to a specific region of the 28S rRNA sequence, indicating that this represents a binding site of the protein (Fig. 2C). Depiction of the numbers of sequencing reads mapping to each nucleotide of the 28S rRNA sequence on the 25S rRNA secondary structure [29] revealed the enrichment of sequences derived from helices H62-63 at the base of ES27 with DDX55 (Fig. 2D). As no structural information on human nucleoplasmic pre-LSU complexes is currently available, the identified DDX55 binding site was mapped onto the tertiary structure of the mature ribosome [6], revealing that this region of domain IV is non-protein-bound in mature LSU complexes (Fig. 2E), but forms part of the intersubunit interface within assembled 80S ribosomes.
Figure 2.

DDX55 crosslinks to helices H62-63 of the 28S ribosomal RNA sequence in vivo. (A) HEK293 cells expressing DDX55-Flag or the Flag tag alone were crosslinked using light at 254 nm. The tagged proteins and crosslinked RNAs were retrieved on α-Flag beads under native conditions and subjected to a partial RNase digest. Complexes were then immobilized on NiNTA in denaturing conditions and co-purified RNA fragments were [32P] labelled, ligated to sequencing adaptors. After elution, protein-RNA complexes were separated by denaturing PAGE, transferred to a nitrocellulose membrane and radiolabelled RNAs were detected by autoradiography. (B) The regions of the membranes containing crosslinked DDX55-Flag complexes were excised, as well as a corresponding region of the lane containing the Flag sample. RNAs were released by proteinase treatment, purified and converted to a cDNA library that was subjected to deep sequencing. The obtained sequence reads were mapped to the human genome and the relative proportions of reads mapped to gene features encoding different classes of RNA was determined. Abbreviations – ribosomal RNA (rRNA), transfer RNA (tRNA), small nucleolar RNA (snoRNA), non-coding RNA (ncRNA), small nuclear RNA (snRNA), long non-coding RNA (lncRNA), microRNA (miRNA), mitochondrial RNA (mtRNA). (C) The number of reads mapping to each nucleotide of the rDNA sequence encoding the 47S pre-rRNA is shown above a schematic view of the transcript (upper panel). The number of mutations, arising during reverse transcription due to the presence of amino acid-crosslinked nucleotides, mapping to each nucleotide is also shown (lower panel). (D) The number of sequencing reads in the DDX55-His6-Prc-Flag CRAC data mapping to each nucleotide of the 28S rRNA sequence is shown on the secondary structure of the mature 28S rRNA using the indicated colour scale. A magnified view of the region crosslinked by DDX55 is shown. (E) The number of sequencing reads in the DDX55-His6-Prc-Flag CRAC data mapping to each nucleotide of the tertiary structure of the 28S rRNA (grey) within the mature LSU (PBD: 4V6X) is shown using a colour scale as in (D). The ribosomal proteins of the LSU are shown in surface view in pale cyan. The positions of key ribosomal features are indicated. CP – central protuberance
RNAi-mediated depletion of DDX55 affects pre-rRNA processing
The crosslinking of DDX55 to H62-63 of the 28S rRNA sequence suggests that this protein is involved in maturation of the LSU. The requirement of a biogenesis factor for a particular step of ribosome assembly often manifests in changes in the levels of pre-rRNA intermediates and defects in the production of the mature rRNAs (Fig. 3A). An RNAi-based rescue system was therefore established in which endogenous DDX55 could be depleted using siRNAs and expression of siRNA-insensitive, Flag tagged DDX55 (DDX55siins-Flag) could be induced. Proteins and RNAs were extracted from HEK293 cells expressing the Flag tag, transfected with non-target siRNAs (siNT) or siRNAs against DDX55 (siDDX55) as well as from siDDX55-treated cells expressing DDX55siins-Flag. Western blot analysis of the levels of DDX55 (anti-DDX55) and, as a control, tubulin, confirmed efficient depletion of DDX55 in the cells transfected with siDDX55 and expression of DDX55-Flag close to the endogenous level (Fig. 3B). Total RNA extracted from these cells was separated by denaturing agarose gel electrophoresis and analysed by northern blotting using probes hybridizing to different regions of the pre-rRNA transcript, which detect all major pre-rRNA species present in human cells (Fig. 3A). Compared to the cells treated with siNT, a mild accumulation of the 32S pre-rRNA was observed in cells depleted of DDX55 (Fig. 3C, D). The increased level of this pre-rRNA species suggests that DDX55 is required for LSU biogenesis and likely functions downstream of the pre-rRNA cleavage in ITS1 that generates the 32S pre-rRNA. To demonstrate the importance of this mild pre-rRNA processing defect for LSU biogenesis, pulse-chase metabolic labelling was used to monitor the production of nascent rRNAs. This confirmed the accumulation of the 32S pre-rRNA in cells treated with siRNAs against DDX55 compared to those treated with non-target siRNAs and also revealed that production of the mature 28S rRNA is reduced in cells lacking DDX55 (Fig. 3E, F).
Figure 3.
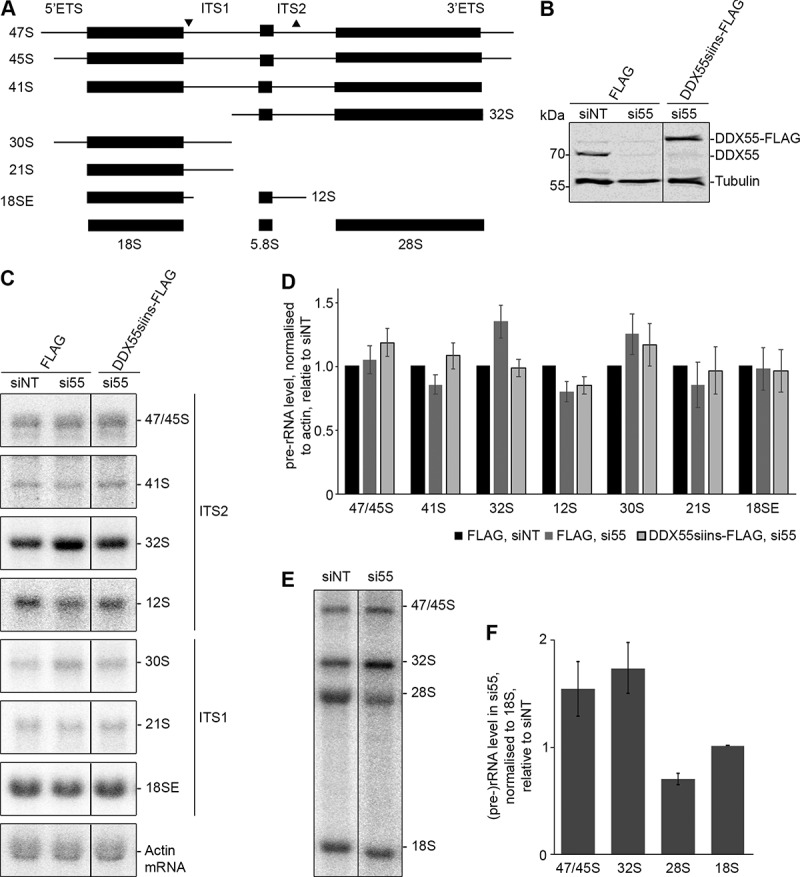
Pre-rRNA processing in cells depleted of DDX55. (A) Simplified processing scheme showing the major pre-rRNA intermediates present in human cells. Mature rRNA sequences are shown as black rectangles, and internal and external transcribed spacers (ITS and ETS respectively) are represented by black lines. The hybridization positions of probes used for northern blotting are indicated by triangles on the initial 47S pre-rRNA transcript. (B) HEK293 cells expressing the Flag tag or siRNA-insensitive, Flag-tagged DDX55 (DDX55siins-Flag) were treated with siRNAs targeting the firefly luciferase (siNT) or DDX55 (si55). Cells were harvested 90 h after transfection, and proteins were separated by SDS-PAGE and analysed by western blot using antibodies against DDX55 (α-DDX55) or, as a loading control tubulin (α-tubulin). (C) Total RNA prepared from siRNA-treated cells as in (B) was separated by denaturing agarose gel electrophoresis and transferred to a nylon membrane. Pre-rRNA species and the actin mRNA were detected by northern blotting using the probes indicated to the right of the panel. (D) The levels of pre-rRNA species in three independent experiments were quantified, normalized according to the actin mRNA, and are presented relative to the Flag, siNT sample as mean ± standard error. (E) HeLa cells treated with siNT or siDDX55 were subjected to pulse-chase metabolic labelling. Total RNAs were separated by denaturing agarose gel electrophoresis and transferred to a nylon membrane. Abundant labelled RNAs were detected using a phosphorimager. (F) The levels of (pre-)rRNA species in three independent pulse-chase experiments were quantified, normalized according to the 18S rRNA, and are presented relative to the siNT sample as mean ± standard error
A nuclear localization signal is present within the C-terminal region of DDX55
The identification of a binding site of DDX55 on pre-ribosomes raised the question of how the helicase associates with these complexes. After its production in the cytoplasm, DDX55 requires import into the nucleoplasm and recruitment to its pre-rRNA target site. Many DExD box helicases, including DDX55, carry N- and/or C-terminal extensions that can contribute to helicase interactions and function (Fig. 4A). To investigate whether the C-terminal tail of DDX55 contributes to association of the helicase with its substrate, the amino acid sequences of the C-terminal region of DDX55 and several of its homologues were aligned to identify evolutionarily conserved features. This revealed a putative nuclear localization signal (NLS [30]) and the presence of numerous basic amino acids (Fig. 4B).
Figure 4.

The C-terminal region of DDX55 contains a nuclear localization signal. (A) Schematic overview of DDX55. Amino acid numbers corresponding to specific domain boundaries (www.uniprot.org) are given above. (B) The amino acid sequences of human DDX55 and its homologues from the species indicated were aligned. Basic amino acids are highlighted in red, evolutionarily conserved amino acids are marked with a black background, and conserved basic amino acids are shown in white with a red background. A putative bipartite nuclear localization signal (NLS) identified in the human DDX55 sequence is indicated by a green box. (C) The subcellular localization of Flag tagged, full-length DDX55 (DDX55), DDX55 lacking the C-terminal tail (DDX551-403), DDX55 lacking the C-terminal tail but coupled to the NLS of the SV40 large T-antigen (DDX551-403+NLS) and DDX55 carrying amino acid substitutions (K537A, R538A, K539A, R540A) within the predicted NLS sequence (DDX55NLSmut-Flag) were determined by immunofluorescence using an α-Flag antibody. Co-immunofluorescence using α-UTP14A served as a nucleolar marker and nuclear material was visualized using DAPI staining. Scale bar represents 10 μm. (D) The subcellular localization of a GFP-GFP-GST reporter (GGG) and the reporter coupled to the predicted DDX55 NLS (GGG-DDX55533-563) was determined by fluorescence microscopy. DAPI staining was used to visualize nuclear material. Scale bar represents 10 μm
HEK293 cell lines for the expression of Flag tagged, C-terminally truncated DDX55 (DDX551-403) and truncated DDX55 coupled to the NLS of the SV40 large T-antigen (DDX551-403+NLS) were generated to determine whether the C-terminal extension is required for import of DDX55 to the nucleoplasm. Analysis of the sub-cellular localizations of these proteins by immunofluorescence revealed that, in contrast to the full-length protein, DDX551-403 is cytoplasmic but that addition of an NLS restores the normally nucleoplasmic localization (Fig. 4C). To explore the functionality of the identified NLS sequence, four basic amino acids expected to mediate import receptor interaction (K537, R538, K539, and R540) were substituted with alanine, and the localization of the mutated protein (DDX55NLSmut) was determined by immunofluorescence (Fig. 4C, right panel). The cytoplasmic localization of DDX55NLSmut demonstrated the requirement of this sequence for nuclear import of DDX55. Moreover, coupling of the predicted NLS sequence (DDX55533-562) to a normally cytoplasmic GFP-GFP-GST reporter [31] lead to its nuclear import, confirming that this sequence acts as an import signal for DDX55 (Fig. 4D).
The ATPase activity of DDX55 is stimulated by double-stranded RNA and the C-terminal region of DDX55 contributes to the specificity of RNA binding
Clusters of basic amino acids within the C-terminal extensions of some RNA helicases, e.g. Mss116 [32] have been shown to contribute to RNA substrate binding. As the C-terminal region of DDX55 contains numerous basic residues (Fig. 4B), this raised the possibility that this sequence could contribute to the interaction of DDX55 with the pre-rRNA. To directly analyse RNA binding, full-length DDX55 and C-terminally truncated DDX55 (DDX551-403) carrying N-terminal ZZ and C-terminal His tags were recombinantly expressed in Escherichia coli (E. coli) and purified via their His tags (Fig. 5A). Anisotropy experiments were then performed to monitor the association of these proteins with fluorescently-labelled single- and double-stranded RNA (ssRNA and dsRNA, respectively) substrates in vitro. Full-length DDX55 associated only weakly with the ssRNA (K.d. = 3.70 ± 0.50 μM) but bound with high affinity to the dsRNA substrate (K.d. = 0.99 ± 0.09 μM; Fig. 5B, C). In contrast, DDX551-403 interacted poorly with the dsRNA (K.d. = 3.70 ± 0.78 μM) but could readily bind the ssRNA substrate (K.d. = 0.42 ± 0.03 μM; Fig. 5B, C). These results indicate that DDX55 preferentially binds dsRNA substrates and suggest that the C-terminal region contributes to maintaining this specificity. It is likely that the strong binding of DDX551-403 to ssRNA reflects non-specific interactions of the core region.
Figure 5.

The ATPase DDX55 preferentially binds and is stimulated by double-stranded RNAs. (A) ZZ-DDX55-His7, ZZ-DDX551-403-His7 and ZZ-DDX55T60A-His7 were recombinantly expressed in E. coli and purified via their His tags on a nickel matrix. Purified proteins were separated by SDS-PAGE and visualized by Coomassie staining. (B,C) Fluorescence anisotropy experiments were performed using fluorescently labelled, single-stranded (B) or double-stranded (C) RNAs and different amounts of purified ZZ-DDX55-His7 and ZZ-DDX551-403-His7. Data from three independent experiments are presented as mean ± standard deviation. (D) In vitro NADH-coupled ATPase assays were performed using no protein (no prot.), DDX55, DDX55T60A or DDX551-403. Samples contained no RNA (-), single- or double-stranded RNA (ss and ds respectively) or a mimic of the cellular RNA sequence crosslinked by DDX55 (H62)
The preferential binding of DDX55 to duplex RNA is in line with previous observations that DExD-box RNA helicases typically bind dsRNA substrates to mediate local strand unwinding [4]. To determine whether the C-terminal region and the substrate specificity it promotes is important for the catalytic activity of DDX55, the RNA-dependent ATPase activity of DDX55 was investigated. In addition to the purified full-length and C-terminally truncated versions of DDX55, DDX55 carrying a threonine to alanine substitution within a conserved ‘GKT’ motif (DDX55T60A) that, in other RNA helicases, has been implicated in ATP binding and hydrolysis, was overexpressed with N-terminal ZZ and C-terminal His tags and purified from E. coli (Fig. 5A). In vitro NADH-coupled ATPase assays performed using these proteins showed that, compared to a control sample containing no protein, full-length DDX55 hydrolysed ATP in the absence of an RNA substrate. The specificity of this catalytic activity is supported by the finding that DDX55T60A did not hydrolyse ATP above the background level observed in the control. DDX551-403, however, showed only very weak ATP hydrolysis activity, suggesting that the C-terminal region likely helps maintain the tandem RecA-like domains in an appropriate conformation for ATP hydrolysis. Addition of an ssRNA substrate only mildly stimulated ATP hydrolysis by DDX55, consistent with the weak binding of the full-length protein to ssRNA. A strong increase in the ATPase activity DDX55 was, however, observed in the presence of dsRNA (Fig. 5D). The RNA sequence crosslinked by DDX55 in vivo (28S-H62) contains a short RNA duplex that is likely unwound by endogenous DDX55. Addition of an RNA fragment containing this sequence (H62) only weakly stimulated the ATPase activity of DDX55 (Fig. 5D), however, this likely reflects the instability of this short duplex outside the context of the pre-ribosome and under the conditions used in the in vitro ATPase assay.
The C-terminal region of DDX55 is required for recruitment of the helicase to pre-ribosomes
We next analysed the influence of the C-terminal region of DDX55 on binding to cellular RNAs. HEK293 cells expressing DDX55-Flag, DDX551-403-Flag, DDX551-403+NLS-Flag or the Flag tag were exposed to UV light to covalently link the proteins to associated RNAs. Protein-RNA complexes were tandem affinity purified and co-purified RNA fragments were labelled with [32P]. Complexes were separated by denaturing PAGE and after transfer to a nitrocellulose membrane, radiolabelled protein-RNA complexes were detected by autoradiography. As previously (Fig. 2A), the full-length protein robustly crosslinked to RNA in vivo and very little RNA was co-precipitated by the cytoplasmic, C-terminally truncated DDX551-403, which would not encounter its pre-ribosomal substrate. Interestingly, however, even though imported into the nucleoplasm, DDX551-403+NLS also did not associate with cellular RNAs (Fig. 6A). As prior analysis of the RNA sequences crosslinked to DDX55 showed that they are mostly derived from (pre-)rRNAs (Fig. 2), this suggests that the C-terminal tail of DDX55 may be required for recruitment of the helicase to pre-ribosomal complexes. The small amount of RNA co-purified with DDX551-403 likely reflects unspecific interactions with non-substrate, cytoplasmic RNAs, as the in vitro analysis showed that DDX551-403 readily binds to single-stranded RNAs (Fig. 5B).
Figure 6.
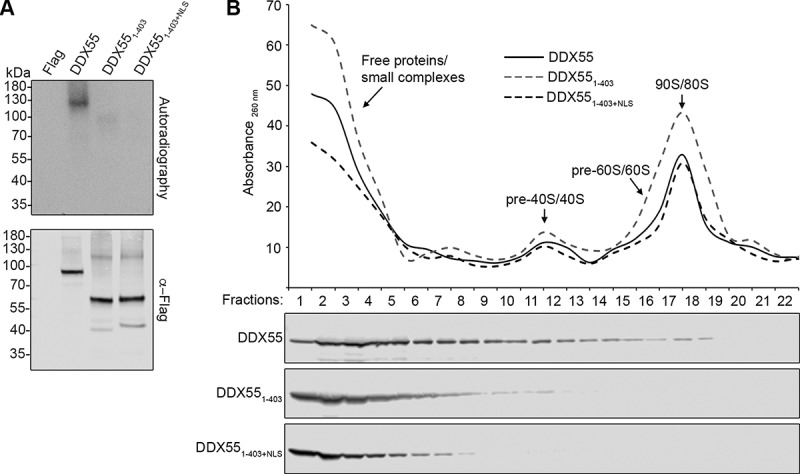
The C-terminal region of DDX55 is required for pre-ribosome association. (A) HEK293 cells expressing the Flag tag alone, DDX55-Flag, DDX551-403-Flag or DDX551-403+NLS-Flag were crosslinked using light at 254 nm. The tagged protein and crosslinked RNAs were retrieved on α-Flag beads under native conditions and subjected to a partial RNase digest. Complexes were then immobilized on NiNTA in denaturing conditions and co-purified RNA fragments were [32P] labelled. After elution, protein-RNA complexes were separated by denaturing PAGE, transferred to a nitrocellulose membrane and radiolabelled RNAs were detected by autoradiography. Protein samples were separated by SDS-PAGE and subjected to western blotting using an α-Flag antibody. (B) Whole cell extracts prepared from HEK293 cells expressing HEK293 cells expressing DDX55-Flag, DDX551-403-Flag or DDX551-403+NLS-Flag were separated by sucrose density gradient centrifugation. During fractionation, an absorbance profile at 260 nm was generated and the peaks corresponding to 80S mature/90S pre-ribosomes, and (pre-)40S and (pre-)60 particles are indicated (upper panel). Proteins in each fraction were separated by SDS-PAGE and analysed by western blotting using an α-Flag antibody (lower panel)
To confirm the role of the C-terminal region of DDX55 in recruitment of the protein to pre-ribosomes, extracts prepared from cells expressing DDX55-Flag, DDX551-403-Flag or DDX551-403+NLS-Flag were separated by sucrose density gradient centrifugation. In contrast to DDX55-Flag, neither DDX551-403-Flag or DDX551-403+NLS-Flag co-migrated with pre-ribosomal subunits further supporting the requirement for the C-terminal region for association of DDX55 with its pre-ribosomal substrates (Fig. 6B).
Discussion
Elucidating the functions of RNA helicases necessitates identification of their substrate RNAs and here, we have discovered that the DExD box ATPase DDX55 associates with the 28S rRNA within pre-ribosomal particles and is necessary for efficient LSU biogenesis. Interestingly, our data show that the C-terminal region of the protein beyond the helicase core domain is required for pre-ribosome association and nuclear import of DDX55. Amino acid sequence analysis uncovered a bipartite NLS, characteristic of cargoes of importin alpha, a protein bound by DDX55 (data not shown). A truncated form of DDX55 lacking this sequence localizes exclusively to the cytoplasm and substitution of basic amino acids within this motif prevents nuclear import of the full-length protein whereas coupling of the N-terminal region and helicase core domain to an alternative NLS restores the normally nucleoplasmic localization of the protein, confirming the functionality of the identified NLS.
As DDX55 is a predicted RNA-dependent ATPase and RNA helicase, which contains evolutionarily conserved stretches of basic amino acids within its C-terminal region, we investigated binding of the helicase core and C-terminal region to different RNA substrates. Interestingly, in vitro DDX55 displayed a preference for binding dsRNA, rather than ssRNA and, consistent with this, the ATPase activity of DDX55 was more strongly stimulated by dsRNA than ssRNA. Notably, DDX55 lacking the C-terminal region had a much higher affinity for ssRNA than the full-length protein, suggesting that in the absence of the C-terminal region, the helicase core non-specifically interacts with non-substrate RNAs. The lower affinity of DDX551-403 for dsRNA and the fact that this truncated protein had little ATPase activity, suggest that the C-terminal region also helps maintain the helicase core region in an appropriate conformation for specific RNA binding and catalytic activity. The binding of DDX55 to dsRNA is consistent with its classification as a DExD box helicase as these proteins typically perform local strand unwinding of target duplexes. The selective activation of DDX55 by this type of RNA likely represents a mechanism via which the inherent promiscuity of the helicase can be minimized until its pre-ribosomal substrate is encountered.
The identification of a crosslinking site of DDX55 in 28S-H62 suggests that this could represent the endogenous helicase substrate. Consistent with this model, RNA structure probing analysis revealed that an equivalent region of domain IV of the yeast 25S rRNA is remodelled by the S. cerevisiae homologue of DDX55, Spb4 [22]. Interestingly, a second pre-rRNA contact site in domain VI of the 25S rRNA, which is likely to act as a binding platform for the helicase, was identified for Spb4. The finding that, even when imported into the nucleoplasm via the SV40 large T antigen NLS, DDX55 lacking the C-terminal region does not associate with pre-ribosomes, implicates this part of the protein in recruitment of DDX55 to its pre-rRNA target site. As the helicase core region (DDX551-403) binds dsRNA, but with lower affinity than the full-length protein, this supports direct RNA interactions of both the catalytic core and residues within the C-terminal region. The absence of a second in vivo crosslinking site for DDX55 could reflect subtle differences in the conformation of domain VI of the yeast and human pre-ribosomes leading to greater (yeast) or lesser (human) accessibility of the pre-rRNA for UV-induced crosslinking. It is also possible, however, that for DDX55, the C-terminal region rather contributes to pre-ribosome recruitment by maintaining the protein in a conformation necessary for the formation of interactions with components of pre-ribosomal particles.
The identification of 28S-H62 as a pre-rRNA contact of the helicase core region of DDX55 raises the question of what consequences remodelling of this pre-rRNA region could be. Often defects in ribosomal subunit maturation indirectly cause defects in pre-rRNA processing. However, in contrast to yeast Spb4, whose depletion causes strong accumulation of the initial 35S pre-rRNA transcript and the LSU pre-rRNA 27SB [22], RNAi-mediated depletion of DDX55 leads to only mild accumulation of the 32S pre-rRNA species. This difference may, on the one hand, suggest that the residual protein present after siRNA treatment is sufficient to largely fulfil the cellular requirement for DDX55 but, on the other hand, could reflect differences in the kinetics of LSU pre-rRNA processing and the stability of these pre-RNA species between yeast and human. The nucleoplasmic localization of DDX55 suggests that it acts at a relatively late stage of pre-60S biogenesis and the accumulation of the early 32S pre-rRNA therefore likely reflects feedback arising due to impaired downstream maturation events. In a complex, multifaceted process like ribosome assembly, it is challenging to define the precise functions of particular biogenesis factors and to differentiate these from indirect consequences of lack of a particular protein. This is especially the case for RNA helicases where their action in rRNA rearrangements can influence diverse downstream events. While the precise role of DDX55 during LSU maturation therefore still remains elusive, the identification of 28S-H62 of domain IV as a direct RNA contact site provides insight into the likely target of the remodelling activity of the helicase.
Materials and methods
Molecular cloning
The coding sequence (CDS) of DDX55 (NM_020936.3) and a truncated version comprising only amino acids 1–403 (DDX551-403) were cloned into pQE80-derived plasmids for the expression of N-terminally ZZ- and C-terminally His7-tagged proteins in E. coli. Site-directed mutagenesis was used to modify the coding sequence to induce substitutions of threonine 60 for alanine (DDX55T60A). The complete DDX55 CDS and that encoding amino acids 1–403 were also cloned into pcDNA5-based plasmids for the expression of C-terminally His6-PreScission protease cleavage site-2xFlag (Flag)-tagged proteins in human cells. The coding sequence was modified using site-directed mutagenesis to substitute lysine 537, arginine 538, lysine 539 and arginine 540 for alanine (DDX55NLSmut). Alternatively, five mutations that do not induce amino acid substitutions were introduced within the target site of siDDX55 to enable expression of siRNA-insensitive DDX55. An additional pcDNA5-based construct for the expression of DDX551-403 coupled to the NLS of the SV40 large T antigen was also generated. The sequence encoding DDX55533-562 was also cloned into a plasmid for the expression of an N-terminally GFP-GFP-GST-tagged peptide in human cells [31].
Cell culture and RNAi
HEK293 Flp-In T-Rex and HeLa cells were cultured in 1x Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% foetal calf serum (FCS) at 37°C with 5% CO2. To generate stably transfected cell lines expressing DDX55-Flag or its derivatives from a defined genomic locus, HEK293 Flp-In T-Rex cells were transfected with the appropriate pcDNA5-based plasmids and a construct for expression of the Flp recombinase. Transfected cells were selected using hygromycin B and blasticidin and population cell lines were expanded. Expression of the tagged protein from the transgene was induced by addition of 1 μg/ml or 5 ng/ml tetracycline for 24 h. For RNAi, HeLa cells were transfected with 50 nmol of siRNA (siNT – 5ʹ-UCGUAAGUAAGCGCAACCCdTdT-3ʹ; siDDX55 – 5ʹ- AGAGGGUUCUGAUAUUGAAdTdT-3ʹ) using lipofectamine RNAiMAX (Thermo Scientific) according to the manufacturer’s instructions and cells were harvested after 90 h.
Immunofluorescence and fluorescence microscopy
Immunofluorescence was performed as previously described [33]. In brief, HEK293 cell lines were grown on coverslips and expression of Flag-tagged proteins was induced by addition of tetracycline before fixation using 4% paraformaldehyde. Treatment with 0.1% Triton-X-100 in PBS for 20 at room temperature (RT) was used to permeabilise the cells before blocking with PBS supplemented with 10% FCS and 0.1% Triton-X-100 for 1 h at RT. Cells were then incubated at RT for 2 h with primary antibodies (α-Flag – Sigma-Aldrich (F3165); α-UTP14A – Proteintech (11,474-1-AP)) diluted in PBS containing 10% FCS. After thorough washing of the coverslips, cells were incubated at RT for 1 h with Alexa Fluor 488- and Alexa Fluor-543-conjugated antibodies. Washing steps were repeated and coverslips were mounted using Vectashield containing DAPI. Confocal microscopy was then performed to visualize cells. Alternatively, HeLa P4 cells grown on coverslips were transiently transfected with constructs for the expression of GFP-tagged proteins using calcium phosphate and cells were fixed using 4% paraformaldehyde before mounting and visualization as described above.
Sucrose density gradient centrifugation
HEK293 cells lines for the expression of DDX55-Flag, DDX551-403-Flag or DDX551-403+NLS-Flag were treated with tetracycline for 24 h to induce protein expression before harvesting. Cells were lysed by sonication in a buffer containing 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 5 mM MgCl2 and 1 mM DTT and cellular debris were pelleted by centrifugation. The resultant whole cell lysates were separated by 10–45% sucrose gradients by centrifugation for 16 h at 23,500 rpm in an SW-40Ti rotor as previously [34]. Gradients were fractionated and an absorbance profile at 260 nm was generated. Proteins in each fraction were precipitated with 20% trichloroacetic acid (TCA) before separation by SDS-PAGE and analysis by western blotting using an α-Flag antibody.
Crosslinking and analysis of cDNA (CRAC)
CRAC was performed as previously described [26,28,34]. HEK293 cells expressing DDX55-Flag or the Flag tag were exposed to UV light at 254 nm. The cells were lysed and protein-RNA complexes were enriched on α-FlagM2-magnetic beads. After elution from the beads with Flag-peptide, exposed RNA sequences were digested using RNase-IT. Complexes were then immobilized on NiNTA beads via their His tag under denaturing conditions. The RNA was radioactively labelled and 5ʹ and 3ʹ adaptors were ligated. The 5ʹ adaptor contains a unique molecular identifier (UMI) sequence (NNNNNAGC) to allow consolidation of multiple sequencing reads PCR amplified from the same template RNA. The radioactively labelled complexes were separated on 4–12% Bis-Tris NuPAGE gradient gels and transferred onto nitrocellulose membrane. Radioactive signals were visualized by autoradiography and appropriate regions of the membrane were excised. RNA was eluted from the membrane using Proteinase K, and after phenol-chloroform extraction was used as a template for reverse transcription to generate a cDNA library that was amplified by PCR and subjected to Illumina Next-generation sequencing.
Bioinformatics analysis of the obtained sequencing data involved collapsing of identical sequences containing the same UMI into one sequence as well as removal of low-quality sequences, trimming of UMIs, and discarding of sequences shorter than 21 nt. STAR aligner was used to align the remaining sequences to a modified version of the human genome version GRCh38.p13 primary assembly, including the 47S and 5S rDNA sequences on two additional chromosomes. The 18S and 28S rRNA sequences were adapted according to Krogh et al. 2016 [35]. Annotated 5S sequences within the primary assembly were exchanged for ‘N’ to enable mapping to only the single 5S sequence. A modified version of the gencode annotation release version 29 was used for genome annotation. Double-annotated genes were deleted and miRNA annotation was modified according to the miRNA database. Hit counting was performed using featureCounts and summary plots were created using python scripts. For mapping onto the rRNA secondary [29] and tertiary (PDB 4V6X) [6] structures, the maximum number of reads mapping to a nucleotide was set to 100% and the relative number of reads mapping to all other nucleotides was depicted using a colour scale.
RNA-IP
HEK293 cells expressing DDX55-Flag, DDX551-403-Flag or DDX551-403+NLS-Flag were UV crosslinked and protein-RNA complexes were enriched on α-FlagM2 beads as described above. After immobilization on NiNTA, RNase-treatment was performed and RNA fragments were 5ʹ radiolabelled with [32P]. Complexes were eluted by Proteinase K-mediated digestion before separation by denaturing PAGE and transfer to nitrocellulose membrane. Radiolabelled complexes were detected by autoradiography.
Pre-rRNA analysis by northern blotting and pulse-chase metabolic labelling
Total RNA was extracted from siRNA-treated cells using TRI reagent (Sigma) according to the manufacturer’s instructions. RNAs were separated on a 1.2% denaturing (glyoxal) agarose gel and transferred to a nylon membrane by vacuum blotting. Pre-rRNAs were detected using [32P]-labelled DNA oligonucleotide probes complementary to different regions of the transcript (ITS1 – 5ʹ-CCTCGCCCTCCGGGCTCCGTTAATGATC-3ʹ; ITS2 – 5ʹ-GAGTCCGCGGTGGAG-3ʹ) [36,37]. Pulse-chase metabolic labelling was performed as previously [36]. In brief, siRNA-treated cells were grown in phosphate-free DMEM for 1 h, phosphate-free DMEM supplemented with 15 μCi/mL [32P]-labelled orthophosphate for 1 h before washing and growth in unlabelled DMEM for a further 3 h. Total RNA was extracted, separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane by capillary blotting and labelled RNAs were detected using a phosphorimager. Quantification of northern blot signals was done using ImageQuant software (GE Healthcare).
Recombinant protein expression in E. coli
E. coli BL21-codon plus cells were transformed with plasmids for the expression of N-terminally ZZ- and C-terminally His7-tagged DDX55, DDX55T60A, or DDX551-403 and protein expression was induced with 1 µM IPTG at 18°C for 16 h. Cells were harvested and lysed by sonication in Lysis buffer (50 mM Tris-HCl pH 7, 500 mM NaCl, 1 mM MgCl2, 10 mM Imidazole, 1 mM PMSF, 10% glycerol) and insoluble material was pelleted by centrifugation at 20,000 rcf for 20 min at 4°C. The supernatant was incubated with 10% polyethyleneimine (PEI) on a rotating wheel for 15 min at 4°C. The solution was cleared by centrifugation at 33,000 rcf for 30 min at 4°C. His-tagged proteins in the supernatant were immobilized on cOmplete His-Tag purification resin (Roche). After sequential washing steps with Wash buffer I (50 mM Tris-HCl pH 7, 500 mM NaCl, 1 mM MgCl2, 30 mM imidazole, 10% glycerol) and Wash buffer II (50 mM Tris-HCl pH 7, 1 M NaCl, 1 mM MgCl2, 30 mM imidazole, 10% glycerol), bound proteins were eluted using Elution buffer (50 mM Tris-HCl pH 7, 500 mM NaCl, 1 mM MgCl2, 300 mM imidazole, 10% glycerol). The eluate was dialysed against a buffer containing 50 mM Tris-HCl pH 7, 120 mM NaCl, 2 mM MgCl2, 20% glycerol overnight and stored at −80°C.
NADH-couple ATPase assays
Hydrolysis of ATP was monitored using NADH-coupled ATPase assays as previously described [38]. Briefly, reactions containing 45 mM Tris-HCl pH 7.4, 2 mM MgCl2, 25 mM NaCl, 450 μM NADH,1.5 mM phosphoenolpyruvate (PEP, Sigma-Aldrich), 4 mM ATP and 20 U/ml pyruvate kinase/lactate dehydrogenase (Sigma-Aldrich) were supplemented with 1.5 μM recombinant protein and 1.5 μM RNA (Single-stranded – 5ʹ-GUAAUGUAAGUGAACGUAAAACAAAACAAAAC-3ʹ; Double-stranded – 5ʹ- UCGUAAGUAAGCGCAACCCTT-3ʹ and 5ʹ-TTGGGUUGCGCUUACUUACGA) as appropriate. The decrease in absorbance of NADH at 340 nm was measured using a BioTEK Synergy HT microplate spectrophotometer. The rate of ATP hydrolysis was calculated using the following equation:
where Kpath is the molar absorption co-efficient for a defined optical path length which is defined as reaction volume (150 μl/well) and background NADH decomposition.
Anisotropy measurements
Anisotropy measurements were performed as previously described [39]. Recombinant proteins were re-dialysed for 16 h against a buffer containing 50 mM Tris-HCl pH 7.5, 120 mM NaCl, 2 mM MgCl2. Anisotropy of 20 nM fluorescently-labelled RNA (single-stranded – 5ʹ-GUAAUGAAAGU-Atto647-3ʹ; double-stranded 5ʹ-Flu-GACAUACUGCGCCUUCAAAUA-3ʹ and 5ʹ-AUUUGAAGGCGCAGUAUGUCT-3ʹ) was measured in the presence of 0–7 µM of protein. The measurements were performed at room temperature with a FluoroMax-4 spectrofluorometer (Horiba Scientific). The fluorophore was excited at 640 nm and emission was measured at 660 nm (single-stranded RNA) or excited at 647 nm and emission was measured at 517 nm (double-stranded RNA). The data were analysed using the following equation:
where r0 is the anisotropy of free RNA, Δrmax is the amplitude, and [protein]tot and [RNA]tot are the total protein and total RNA concentrations, respectively.
Acknowledgments
We thank Aikaterini Vrentzou for molecular cloning, Prof. Detlef Doenecke for providing plasmids and Christina James for help with microscopy. We also thank Prof. Claudia Höbartner and her group for providing fluorescently labelled RNAs.
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft (SFB1190 to M.T.B and K.E.B) and the University Medical Centre Göttingen (to K.E.B. and M.T.B.).
Disclosure statement
The authors declare no conflict of interest.
References
- [1].Jarmoskaite I, Russell R.. RNA helicase proteins as chaperones and remodelers. Annu Rev Biochem. 2014;83:697–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sloan KE, Bohnsack MT. Unravelling the mechanisms of RNA helicase regulation. Trends Biochem Sci. 2018;43:237–250. [DOI] [PubMed] [Google Scholar]
- [3].Ozgur S, Buchwald G, Falk S, et al. The conformational plasticity of eukaryotic RNA-dependent ATPases. Febs J. 2015;282:850–863. [DOI] [PubMed] [Google Scholar]
- [4].Linder P, Jankowsky E. From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol. 2011;12:505–516. [DOI] [PubMed] [Google Scholar]
- [5].Ben-Shem A, Garreau de Loubresse N, Melnikov S, et al. SOM: the structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 2011;334:1524–1529. [DOI] [PubMed] [Google Scholar]
- [6].Anger AM, Armache J-P, Berninghausen O, et al. Structures of the human and Drosophila 80S ribosome. Nature. 2013;497:80–85. [DOI] [PubMed] [Google Scholar]
- [7].Bohnsack KE, Bohnsack MT. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. Embo J. 2019;38:e100278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mills EW, Green R. Ribosomopathies: there’s strength in numbers. Science. 2017;358. DOI: 10.1126/science.aan2755 [DOI] [PubMed] [Google Scholar]
- [9].Klinge S, Woolford JLJ. Ribosome assembly coming into focus. Nat Rev Mol Cell Biol. 2019;20:116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gerhardy S, Menet AM, Pena C, et al. Assembly and nuclear export of pre-ribosomal particles in budding yeast. Chromosoma. 2014;123:327–344. [DOI] [PubMed] [Google Scholar]
- [11].Henras AK, Plisson-Chastang C, O’Donohue MF, et al. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA. 2015;6:225–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sloan KE, Warda AS, Sharma S, et al. Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017;14:1138–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sloan KE, Gleizes P-E, Bohnsack MT. Nucleocytoplasmic transport of RNAs and RNA-protein complexes. J Mol Biol. 2016;428:2040–2059. [DOI] [PubMed] [Google Scholar]
- [14].Martin R, Straub AU, Doebele C, et al. DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol. 2013;10:4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Watkins NJ, Bohnsack MT. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA. 2012;3:397–414. [DOI] [PubMed] [Google Scholar]
- [16].Sardana R, Liu X, Granneman S, et al. The DEAH-box helicase Dhr1 dissociates U3 from the pre-rRNA to promote formation of the central pseudoknot. PLoS Biol. 2015;13:e1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Choudhury P, Hackert P, Memet I, et al. The human RNA helicase DHX37 is required for release of the U3 snoRNP from pre-ribosomal particles. RNA Biol. 2019;16:54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bohnsack MT, Martin R, Granneman S, et al. Prp43 bound at different sites on the Pre-rRNA performs distinct functions in ribosome synthesis. Mol Cell. 2009;36:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bohnsack MT, Kos M, Tollervey D. Quantitative analysis of snoRNA association with pre-ribosomes and release of snR30 by Rok1 helicase. EMBO Rep. 2008;9:1230–1236. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Martin R, Hackert P, Ruprecht M, et al. A pre-ribosomal RNA interaction network involving snoRNAs and the A pre-ribosomal RNA interaction network involving snoRNAs and the Rok1 helicase. Rna. 2014;20:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liang X-H, Fournier MJ. The helicase Has1p is required for snoRNA release from pre-rRNA. Mol Cell Biol. 2006;26:7437–7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bruning L, Hackert P, Martin R, et al. RNA helicases mediate structural transitions and compositional changes in pre-ribosomal complexes. Nat Commun. 2018;9:5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Khoshnevis S, Askenasy I, Johnson MC, et al. The DEAD-box protein Rok1 orchestrates 40S and 60S ribosome assembly by promoting the release of Rrp5 from Pre-40S ribosomes to allow for 60S maturation. PLoS Biol. 2016;14:e1002480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dembowski JA, Kuo B, Woolford JL. Has1 regulates consecutive maturation and processing steps for assembly of 60S ribosomal subunits. Nucleic Acids Res. 2013;41:7889–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Warda AS, Freytag B, Haag S, et al. Effects of the Bowen-Conradi syndrome mutation in EMG1 on its nuclear import, stability and nucleolar recruitment. Hum Mol Genet. 2016;25:5353–5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bohnsack MT, Tollervey D, Granneman S. Identification of RNA helicase target sites by UV cross-linking and analysis of cDNA. Methods Enzymol. 2012;511:275–288. [DOI] [PubMed] [Google Scholar]
- [27].Haag S, Kretschmer J, Sloan KE, et al. Crosslinking methods to identify RNA methyltransferase targets in vivo. Methods Mol Biol. 2017;1562:269–281. [DOI] [PubMed] [Google Scholar]
- [28].Memet I, Doebele C, Sloan KE, et al. The G-patch protein NF-kappaB-repressing factor mediates the recruitment of the exonuclease XRN2 and activation of the RNA helicase DHX15 in human ribosome biogenesis. Nucleic Acids Res. 2017;45:5359–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Petrov AS, Bernier CR, Gulen B, et al. Secondary structures of rRNAs from all three domains of life. PLoS One. 2014;9:e88222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kosugi S, Hasebe M, Tomita M, et al. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A. 2009;106:10171–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Baade I, Spillner C, Schmitt K, et al. Extensive identification and in-depth validation of importin 13 cargoes. Mol Cell Proteomics. 2018;17:1337–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mallam AL, Jarmoskaite I, Tijerina P, et al. Solution structures of DEAD-box RNA chaperones reveal conformational changes and nucleic acid tethering by a basic tail. Proc Natl Acad Sci U S A. 2011;108:12254–12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Haag S, Warda AS, Kretschmer J, et al. NSUN6 is a human RNA methyltransferase that catalyzes formation of m5C72 in specific tRNAs. RNA. 2015;21:1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sloan KE, Leisegang MS, Doebele C, et al. The association of late-acting snoRNPs with human pre-ribosomal complexes requires the RNA helicase DDX21. Nucleic Acids Res. 2015;43:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Krogh N, Jansson MD, Hafner SJ, et al. Profiling of 2ʹ-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 2016;44:7884–7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sloan KE, Mattijssen S, Lebaron S, et al. Both endonucleolytic and exonucleolytic cleavage mediate ITS1 removal during human ribosomal RNA processing. J Cell Biol. 2013;200:577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Haag S, Kretschmer J, Bohnsack MT. WBSCR22/Merm1 is required for late nuclear pre-ribosomal RNA processing and mediates N7-methylation of G1639 in human 18S rRNA. RNA. 2015;21:180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kiianitsa K, Solinger JA, Heyer W-D. NADH-coupled microplate photometric assay for kinetic studies of ATP-hydrolyzing enzymes with low and high specific activities. Anal Biochem. 2003;321:266–271. [DOI] [PubMed] [Google Scholar]
- [39].Kretschmer J, Rao H, Hackert P, et al. The m(6)A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5ʹ-3ʹ exoribonuclease XRN1. RNA. 2018;24:1339–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]