

Abstract
Although Epstein-Barr virus (EBV) is hypothesized to be a prerequisite for multiple sclerosis (MS), up to 15% of children with a diagnosis of MS were reported to be EBV-seronegative. When re-evaluating 25 EBV-seronegative children out of 189 pediatric patients with a diagnosis of clinically isolated syndrome/MS, we found anti-myelin oligodendrocyte glycoprotein (MOG) antibody in 11/25 (44%) EBV-seronegative, but only 9/164 (5.5%, p<0.001) EBV-seropositive patients. After critical review, MS remained a plausible diagnosis in only four of 14 EBV-seronegative/MOG antibody-negative patients. In children with an MS-like presentation, EBV seronegativity should alert clinicians to consider diagnoses other than MS, especially MOG-antibody disease.
Introduction
Among risk factors for MS, prior infection with Epstein-Barr virus (EBV) might be unique: almost all adult patients with MS have evidence of a prior infection, such that EBV infection is thought to be a requisite for the development of the disease.1,2 However, up to 15% of pediatric MS patients have been reported to be EBV-seronegative.3,4,5 These observations could be explained by 1) false-negative EBV serology, 2) presence of central inflammatory demyelinating diseases other than MS, or 3) prior EBV infection not being a requisite for MS onset.
To further explore these possibilities, we reanalyzed the EBV serology and reviewed the charts of pediatric patients who were diagnosed with the clinically isolated syndrome (CIS) or MS at a tertiary care pediatric MS center and had tested negative for EBV antibodies after disease onset.
Methods
Pediatric patients with a diagnosis of CIS or MS based on contemporary criteria6,7 seen between January 2006 and December 2018 at the University of California San Francisco (UCSF) Regional Pediatric MS Center were included in this Institutional Review Board-approved study. Informed consent and assent were obtained from the parents and patients before enrollment. We compared the demographics, genetic and serological markers of EBV-seronegative and -seropositive patients but only performed an in-depth clinical and MRI review of EBV-seronegative cases in this retrospective study of prospectively collected data.
Immunoglobulin G (IgG) antibodies against the EBV viral capsid antigen (VCA) were measured by standardized enzyme-linked immunosorbent assay (ELISA) (Wampole Laboratories, Princeton, NJ) as described previously.8 Stored serum samples from patients who were VCA IgG negative with the initial assay were tested for IgG to Epstein-Barr nuclear antigen-1 (EBNA-1) and VCA by Labor Berlin GmbH (Berlin, Germany) using Liaison® (DiaSorin, Saluggia, Italy) automated quantitative chemiluminescence immunoassays and an EBV-IgG immunoblot (recomLine EBV IgG, Mikrogen, Germany) according to the manufacturer’s instructions.2
Serum from EBV-seronegative patients was tested for myelin oligodendrocyte glycoprotein (MOG)-IgG and aquaporin-4 (AQP4)-IgG by live-cell based flow cytometry assays at the Mayo Clinic Neuroimmunology Laboratory, Rochester, MN, as previously described.9,10 DNA samples were tested by single-nucleotide polymorphisms (SNPs) for the presence of HLA-DRB1*15:01/15:03 as previously described.4
A neuroradiologist (CA) blinded to the EBV, MOG, and AQP4 serostatus reviewed all available clinical brain MRIs of EBV-seronegative patients. Two neurologists (BN, CC) blinded to MOG and AQP4-IgG serostatus reviewed all available clinical information.
We defined typical MS as a clinicoradiological presentation meeting all the following criteria: 1) unifocal or multifocal neurological deficit, including unilateral optic neuritis, a typical brainstem/cerebellar syndrome, a typical spinal cord syndrome, with acute/subacute onset, evolution over hours to days, persisting for at least 24 hours, 2) MRI evidence of CNS demyelination, including periventricular, juxtacortical, and infratentorial lesions or spinal cord lesions with clearly demarcated borders with a length less than two vertebral segments and not affecting the complete cross-section of the cord, 3) 2017 McDonald criteria for the diagnosis of relapsing-remitting MS.
Data were presented as frequencies (%), and median (interquartile range [IQR]). Statistical significance of differences of categorical variables was assessed by Fisher Exact test and of continuous variables by Wilcoxon rank-sum test.
Results
Of 189 patients with pediatric CIS/MS in the UCSF database, 25 (13%) were EBV-VCA IgG negative with initial testing. All 25 patients were also negative for EBNA-1 IgG and repeat VCA IgG, and by an EBV-IgG immunoblot tested in the second laboratory. The demographic characteristics of EBV-seronegative and -seropositive patients are summarized in Table 1.
Table 1.
Demographic characteristics, HLA-DR status, serum MOG-IgG and AQP4-IgG positivity according to EBV serostatus.
| EBV-VCA IgG negative N=25 | EBV-VCA IgG positive N=164 | p-value$ | |
|---|---|---|---|
| Age at first presentation, median (IQR), y | 6.7 (4.4–10.8) | 14.6 (12.5, 16.0) | <0.001 |
| Female, n (%) | 17 (68%) | 111 (67.7%) | 1.00 |
| Race, n (%) | 0.20 | ||
| White | 14 (56%) | 110 (67.1%) | |
| Black | 1 (4%) | 16 (9.8%) | |
| Asian | 4 (16%) | 15 (9.2%) | |
| Mixed | 5 (20%) | 20 (12.2%) | |
| Unknown | 1 (4%) | 1 (0.6%) | |
| Pacific Islander | 0 | 2 (1.2%) | |
| Ethnicity, n (%) | 0.003 | ||
| Hispanic | 5 (20%) | 77 (47.0%) | |
| Non-Hispanic | 19 (76%) | 87 (53.0%) | |
| Unknown | 1 (4%) | 0 | |
| Positive for DRB1*15:01 or 15:03 | 4 (16%) | 64 (39.0%) | 0.019 |
| MOG-IgG positive | 11 (44%) | 9 (5.5%) | <0.001 |
| AQP4-IgG positive | 0 | 0 | NA |
Fisher’s exact test for categorical variables and Wilcoxon rank-sum test for continuous variables.
Serum MOG-IgG and AQP4-IgG were tested in all EBV-seronegative and seropositive patients at some point after disease onset (median [IQR] time: 0.7 [0.3 – 1.5] years; with 20% having sample collection within three months of disease onset). Remarkably, 11/25 (44%) EBV-seronegative patients, but only nine of 164 (5.5%) EBV-seropositive patients tested positive for MOG-IgG. All EBV-seronegative, MOG-IgG positive patients met the criteria of Jarius et al. for a diagnosis of MOG-antibody disease (MOGAD).11 None of the patients tested positive for AQP4-IgG. HLA-DRB1*15:01/15:03 was less frequent in EBV-seronegative than in EBV-seropositive patients (Table 1).
Detailed demographic, genetic, and clinical findings for the 25 EBV-seronegative patients according to MOG-IgG status are listed in Table 2. MOG-IgG positive patients frequently had uni- or bilateral optic neuritis and/or spinal cord symptoms with a relapsing course, consistent with MOGAD. The presence of intrathecal IgG synthesis was low in both MOG-IgG positive and negative patients.
Table 2.
Demographic, and clinical characteristics of EBV-seronegative patients, and comparison by MOG-IgG status.
| Demographic and clinical data | Total N=25 | MOG-IgG positive N=11 | MOG-IgG negative N=14 |
|---|---|---|---|
| Retrospective follow-up time, median (IQR), months | 48 (20–70) | 48 (20–78) | 51 (24–70) |
| Age at first presentation, median (IQR), years | 6.7 (4.4–10.8) | 4.7 (4.2–9.4) | 8.2 (4.4–11.6) |
| Time from disease onset to blood sample collection, median (IQR), years | 0.7 (0.3–1.5) | 1.4 (0.3–2.8) | 0.7 (0.3–1.5) |
| Female, n (%) | 17 (68%) | 9 (82%) | 8 (57%) |
| Race, n (%) | |||
| White | 14 (56%) | 6 (55%) | 8 (57%) |
| Black | 1 (4%) | 0 | 1 (7%) |
| Asian | 4 (16%) | 1 (9%) | 3 (22%) |
| Mixed (White+Asian) | 5 (20%) | 4 (36%) | 1 (7%) |
| Unknown | 1 (4%) | 0 | 1 (7%) |
| Ethnicity, n (%) | |||
| Hispanic | 5 (20%) | 4 (36%) | 1 (7%) |
| Non-Hispanic | 19 (76%) | 7 (64%) | 12 (86%) |
| Unknown | 1 (4%) | 0 | 1 (7%) |
| Positive for DRB1*15:01 or 15:03 | 4 (16%) | 2 (18%) | 2 (14%) |
| Demyelinating phenotype at the onset, n (%) | |||
| Encephalopathy (ADEM-like) | 3 (12%) | 1 (9%) | 2 (14%) |
| ON | 6 (24%) | 4 (36%) | 2 (14%) |
| SC | 4 (16%) | 3 (27%) | 1 (7%) |
| Brainstem, cerebellar or hemispheric | 8 (32%) | 1 (9%) | 7 (50%) |
| Multifocal (ON + SC) | 3 (12%) | 2 (18%) | 1 (7%) |
| Unknown | 1 (4%) | 0 | 1 (7%) |
| History of bilateral ON, n (%) | 10 (40%) | 7 (64%) | 3 (21%) |
| History of recurrent ON, n (%) | 6 (24%) | 5 (45%) | 1 (7%) |
| History of bilateral or recurrent ON, n (%) | 11 (44%) | 7 (64%) | 4 (29%) |
| OCB or increased CSF IgG index, n (%) | |||
| Positive | 2 (8%) | 1 (9%) | 1 (7%) |
| Negative | 16 (64%) | 8 (73%) | 8 (57%) |
| Not tested or not available | 7 (28%) | 2 (18%) | 5 (36%) |
| Second clinical attack during follow-up, n (%) | 18 (72%) | 9 (82%) | 9 (64%) |
| Fulfill McDonald 2017 criteria for MS diagnosis, n (%) | 20 (80%) | 10 (91%) | 10 (71%) |
| MS disease-modifying treatments, n (%) | |||
| Received | 18 (72%) | 8 (73%) | 10 (71%) |
| Did not receive | 6 (24) | 2 (18%) | 4 (29%) |
| Unknown | 1 (4%) | 1 (9%) | 0 |
MOG: myelin oligodendrocyte glycoprotein, ADEM: acute disseminated encephalomyelitis, ON: optic neuritis, SC: spinal cord
Brain MRI scans of 21/25 EBV-seronegative patients were available for review by a neuroradiologist (Supplementary Materials). While 10 of 21 (48%) patients had corpus callosum lesions on at least one MRI scan, Dawson’s fingers were present in only 5 of 21 (24%). A relatively high proportion of patients (14 of 21, 67%) had tumefactive lesions. MRI findings were overall similar in MOG-IgG positive and negative patients.
A large proportion (10/11, 91%) of EBV-seronegative, MOG-IgG positive patients met the 2017 McDonald criteria. However, among the 14 EBV-seronegative, MOG-IgG negative patients, four did not meet the 2017 McDonald criteria.
We looked in-depth at the 10 MOG-IgG-negative EBV-seronegative patients who fulfilled the 2017 McDonald criteria for major “red flags” for the diagnosis of MS. Two of these had encephalopathy at disease onset, and one had bilateral optic neuritis and multiple cranial nerve palsies, which are unusual for MS. Three patients had insufficient information to confirm or rule out a diagnosis of MS (two did not have MRI images for review, and one did not have details regarding the initial clinical presentation). In the four remaining patients, MS was felt to be the most likely diagnosis. We provided MRI images and case descriptions of these four patients in Figure 1.
Figure 1.
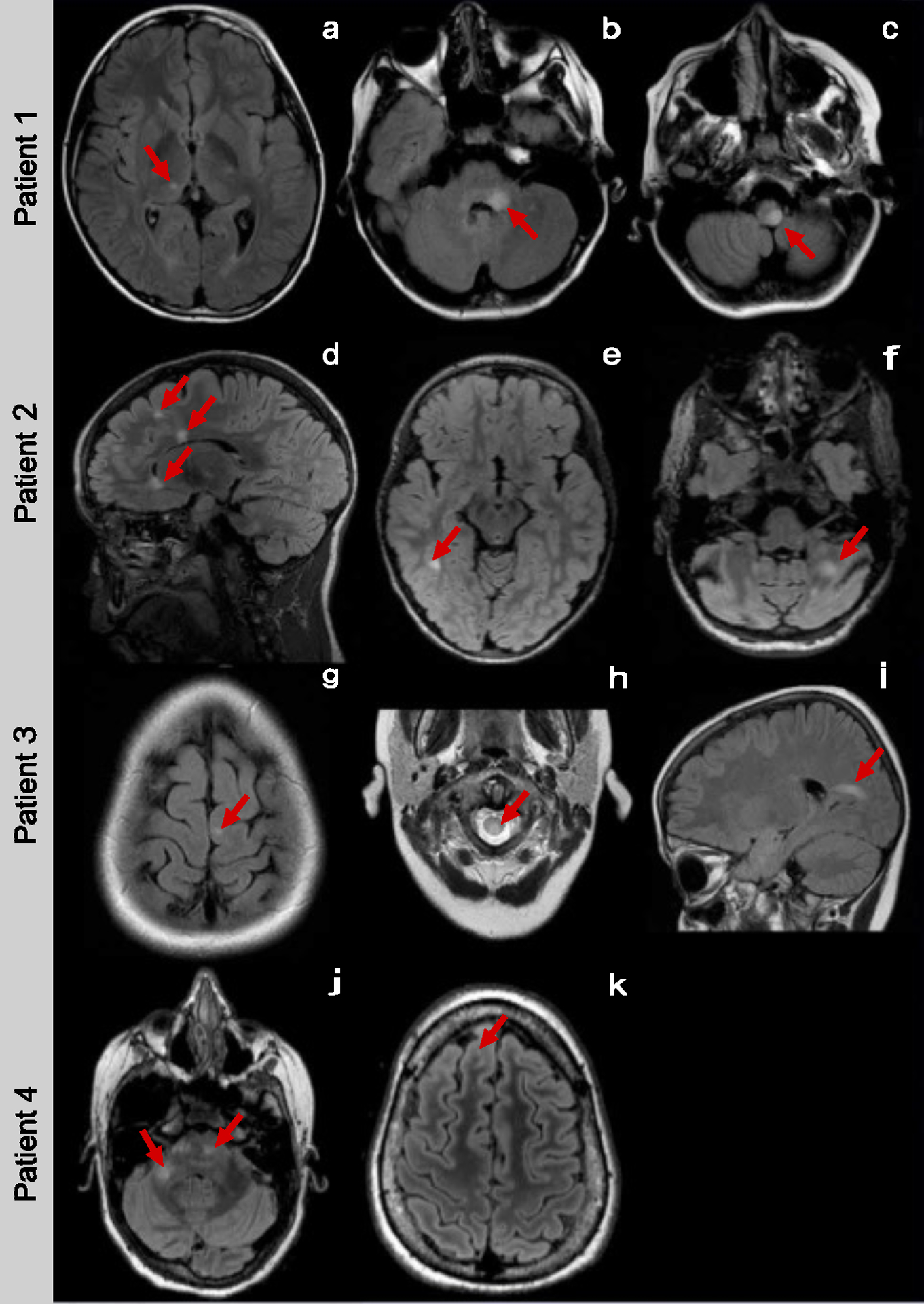
Case 1: An 11-year-old boy presented with facial weakness, double vision and hemiparesis. CSF analysis showed 7 white blood cells (WBCs) per μl, normal glucose, and protein, but no evidence of intrathecal IgG synthesis. His MRI showed deep grey matter, cerebellar peduncle, and partial medullary T2-bright lesions (Figure 1-a, b, c). Six months after the initial presentation, he developed right leg weakness and double vision. Disease activity stopped after initiating natalizumab.
Case 2: A 4-year old girl presented with poor coordination and falls. Her MRI images showed juxtacortical, periventricular, corpus callosum, and cerebellar T2-bright lesions suggestive of MS (Figure 1-d, e, f). CSF analysis showed no WBCs and normal protein and glucose. Intrathecal IgG synthesis was not evaluated. Five months after the initial presentation, she developed right facial weakness and dysarthria. The MRI images and clinical presentation were thought to be compatible with MS. Disease activity stopped after initiating natalizumab.
Case 3: A 10-year-old boy presented with bilateral leg weakness and sphincter dysfunction. CSF results were not available for review. His MRI showed juxtacortical, periventricular, infratentorial, and partial spinal cord T2-bright lesions (Figure 1-g, h, i). Six months after the initial presentation, he developed ataxia, and three months later, he presented with new-onset hand numbness. He started subcutaneous interferon beta-1a, but no further follow-up was available.
Case 4: A 14-year-old girl presented with slurred speech and extremity paresthesia. CSF analysis showed evidence of intrathecal IgG synthesis. Her MRI showed subcortical, pontine, and cerebellar peduncle T2-bright lesions (Figure 1-j, k). Although she had no further clinical attacks, subsequent MRI scans showed new T2-bright lesions. While she had no disease activity on natalizumab, she switched to glatiramer acetate due to safety concerns and remained clinically stable on that treatment.
Discussion
This study confirms that about 15% of children with a working diagnosis of CIS or MS are EBV-seronegative and suggests that a sizeable percentage of those patients, in fact, have an alternative diagnosis, in particular, MOGAD. While a negative EBV serology in children with an inflammatory demyelinating CNS disease may challenge a diagnosis of MS, it does not it rule out.
Numerous studies have consistently shown a practically universal EBV seropositivity in adult patients with MS5, as recently confirmed by a complete EBV-seropositivity in 901 patients with CIS or early MS in the German National MS Cohort.2 A large prospective study also reported the absence of incident adult-onset MS cases in EBV-seronegative individuals.12 These observations, in conjunction with the evidence of interaction of EBV infection with other MS risk factors,5 indicate a possible causal role of EBV infection in MS pathogenesis.13 The strong association of EBV and MS furthermore suggests that a negative EBV serology in patients with suspected inflammatory CNS disease may be a marker for the absence of MS. As about 30% of 10–14-year-olds in the general population of industrialized countries are still EBV-seronegative2, this may be particularly relevant in children with suspected inflammatory CNS disease.
Nevertheless, although a positive EBV serostatus is also associated with the risk of developing pediatric MS,3,4 up to 15% of patients with MS in the pediatric studies were reported to be EBV-seronegative.5 While this questions the hypothesis that EBV infection is a prerequisite for the development of MS, these previous studies did not review in detail clinical and paraclinical findings of EBV-seronegative patients and did not systematically search for serological markers of alternate diagnosis, such as MOG-IgG and AQP4-IgG.
In the present study, 11/25 (44%) EBV-seronegative children with a former diagnosis of CIS/MS tested positive for MOG-IgG measured by a cell-based assay, suggesting they had MOGAD rather than MS.
Four of 14 EBV-seronegative/MOG-IgG negative patients did not fulfill the 2017 McDonald criteria for MS, and three of the ten patients who fulfilled the 2017 McDonald criteria for MS had clinical features unusual for MS, again suggesting that they could have had an alternate diagnosis. Furthermore, the low frequencies of intrathecal IgG synthesis, HLA-DRB1*15:01/15:03, and Dawson’s fingers on MRI, and the frequent presence of tumefactive lesions on MRI in the EBV-seronegative/MOG-IgG negative children appear remarkable, as all of these findings are unusual in patients with classic MS. Accordingly, after critical review by two clinicians and a neuroradiologist, MS was felt to be the most plausible diagnosis on clinicoradiological grounds in only 4/25 (16%) EBV-seronegative children.
Limitations of this study include the retrospective analysis of clinical and paraclinical data, with some of these, such as intrathecal IgG synthesis, missing. Furthermore, only 20% of samples used for MOG-IgG testing were obtained within three months after disease onset. As in approximately half of children with demyelinating syndromes MOG-IgG is only transiently detectable14, MOG-IgG positivity in the 25 EBV-seronegative patients might have been even higher had we been able to test more sera obtained early during the disease course.
In summary, a sizeable proportion of EBV-seronegative patients with an MS-like presentation have MOGAD, and some of the remaining MOG-IgG negative cases may not have MS. While our work does not exclude that few children with MS may be EBV-seronegative, it should alert clinicians to carefully re-evaluate a diagnosis of MS in EBV-seronegative patients with suspected inflammatory CNS disease.
Supplementary Material
Acknowledgments
Source of Funding: National Institutes of Health (grant numbers: R01NS071463-04 and R01NS113828), Stiftung Charité (BIH Clinical Fellow Program to KR)
Footnotes
Potential Conflicts of Interest
The authors report no conflicts of interest relevant to this work.
References
- 1.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann. Neurol 2007;61(4):288–299. [DOI] [PubMed] [Google Scholar]
- 2.Abrahamyan S, Eberspächer B, Hoshi M-M, et al. Complete Epstein-Barr virus seropositivity in a large cohort of patients with early multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banwell B, Krupp L, Kennedy J, et al. Clinical features and viral serologies in children with multiple sclerosis: a multinational observational study. Lancet Neurol 2007;6(9):773–781. [DOI] [PubMed] [Google Scholar]
- 4.Waubant E, Mowry EM, Krupp L, et al. Common viruses associated with lower pediatric multiple sclerosis risk. Neurology 2011;76(23):1989–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobs BM, Giovannoni G, Cuzick J, Dobson R. Systematic review and meta-analysis of the association between Epstein–Barr virus, multiple sclerosis and other risk factors: [Internet]. Multiple Sclerosis Journal 2020;[cited 2020 Apr 3] Available from: https://journals.sagepub.com/doi/10.1177/1352458520907901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krupp LB, Tardieu M, Amato MP, et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult. Scler 2013;19(10):1261–1267. [DOI] [PubMed] [Google Scholar]
- 7.Krupp LB, Banwell B, Tenembaum S, International Pediatric MS Study Group. Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology 2007;68(16 Suppl 2):S7–12. [DOI] [PubMed] [Google Scholar]
- 8.James JA, Kaufman KM, Farris AD, et al. An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J. Clin. Invest 1997;100(12):3019–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waters PJ, Komorowski L, Woodhall M, et al. A multicenter comparison of MOG-IgG cell-based assays. Neurology 2019;92(11):e1250–e1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fryer JP, Lennon VA, Pittock SJ, et al. AQP4 autoantibody assay performance in clinical laboratory service. Neurol Neuroimmunol Neuroinflamm 2014;1(1):e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarius S, Paul F, Aktas O, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation 2018;15(1):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levin LI, Munger KL, O’Reilly EJ, et al. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann. Neurol 2010;67(6):824–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruprecht K. The role of Epstein-Barr virus in the etiology of multiple sclerosis: a current review. Expert Rev Clin Immunol 2020;16(12):1143–1157. [DOI] [PubMed] [Google Scholar]
- 14.Waters P, Fadda G, Woodhall M, et al. Serial Anti–Myelin Oligodendrocyte Glycoprotein Antibody Analyses and Outcomes in Children With Demyelinating Syndromes. JAMA Neurol 2020;77(1):82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.