

Abstract
Characterisation of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) genetic diversity through space and time can reveal trends in virus importation and domestic circulation and permit the exploration of questions regarding the early transmission dynamics. Here, we present a detailed description of SARS-CoV-2 genomic epidemiology in Ecuador, one of the hardest hit countries during the early stages of the coronavirus-19 pandemic. We generated and analysed 160 whole genome sequences sampled from all provinces of Ecuador in 2020. Molecular clock and phylogeographic analysis of these sequences in the context of global SARS-CoV-2 diversity enable us to identify and characterise individual transmission lineages within Ecuador, explore their spatiotemporal distributions, and consider their introduction and domestic circulation. Our results reveal a pattern of multiple international importations across the country, with apparent differences between key provinces. Transmission lineages were mostly introduced before the implementation of non-pharmaceutical interventions, with differential degrees of persistence and national dissemination.
Keywords: SARS-CoV-2, phylogenetics, molecular epidemiology, phylogeography, transmission lineages
1. Introduction
The rapid generation of substantial numbers of virus genomic sequences during the coronavirus-19 (COVID-19) pandemic is without precedent. Laboratories and institutes around the world produced and shared over 300,000 whole severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) genome sequences in the GISAID repository during 2020 and features a total of 1,428,296 sequence entries by 10 May 2021 (Shu and McCauley 2017), providing an unparalleled data set that permits detailed analyses of virus transmission and dissemination. These achievements have provided insights into the sources of SARS-CoV-2 importation and early transmission dynamics in individual countries and geographical regions (Candido et al. 2020; Geoghegan et al. 2020; Lu et al. 2020; Alteri et al. 2021) and have enabled the exploration of viral transmission history at a global scale (Worobey et al. 2020). Phylogenetic methods, including molecular clock models and phylogeographic and phylodynamic methods, are now used routinely to analyse such genomic data from emerging outbreaks (Grubaugh et al. 2019a). The resolution level of evolutionary and transmission history obtained using these methods is contingent on the virus’ evolutionary rate and the depth and representativeness of sampling of cases across space and time (Duchene et al. 2020). While heterogeneous sampling and sequencing among countries can bias and affect the output of some phylogeographic methods (Lemey et al. 2020; Kalkauskas et al. 2021), general trends in the transmission of viral lineages can still be inferred from smaller samples of genomic sequences from individual locations.
The utility of pathogen genomic surveillance during outbreaks has developed during various past emerging epidemics (Rambaut and Holmes 2009; Park et al. 2015; Faria et al. 2017) and has gained further momentum during the current global health crisis. Information about epidemiological trends can be effectively complemented with genomic analyses in order to understand case-specific (Meredith et al. 2020) and general transmission patterns (Du Plessis et al. 2021). This framework can be extended to account for other factors that affect the spread of pathogens, ranging from human mobility on a global scale (Lemey et al. 2014) to particular social networks (Vasylyeva et al. 2016). The analysis of local- and national-scale data sets during the current pandemic has provided insights into the processes affecting the introduction and circulation of the virus into new locations (Moreno et al. 2020) and provided genomic context for other data sources (Gudbjartsson et al. 2020; Popa et al. 2020; Sekizuka et al. 2020). Indeed, the integration and analysis of multiple data sources about an emerging epidemic has the potential to compensate for surveillance blind spots and better understand poorly sampled outbreaks (Grubaugh et al. 2019b).
The COVID-19 epidemic in Ecuador was marked by a dramatic and widely publicised early phase (Long 2020), with an estimated basic reproductive number (R0) of 3.54 (Ortiz-Prado et al. 2021). Ecuador is a small middle-income South American country with the seventh largest population in the continent; half of the country’s population lives in Guayas province (host of the country’s most populated city, Guayaquil) and Pichincha province (host of the country’s second most populated city, the capital Quito; Fig. 1A). The first case was reported in the country on 27 February 2020 (a patient who returned from abroad through Guayaquil with date of symptom onset of February 15) and was followed by the declaration of a National Health Emergency on 11 March 2020. Public health interventions were implemented shortly thereafter: mass gatherings were restricted on 13 March, and a partial lockdown that included the closure of international borders was implemented on 17 March. Finally, a full lockdown that included a curfew and the limitation of domestic mobility in private and public vehicles came into effect on 25 March (Ortiz-Prado et al. 2021). The country’s port city of Guayaquil was the first epicentre of the epidemic, facing a severe increase in the numbers of cases between late February and early April. The province of Guayas reached its highest effective reproductive number Rt (defined as the average number of secondary cases caused by a primary cases at a point in time t; Nishiura and Chowell 2009) on 14 March (Rt estimates vary between 3.96 and 4.91), with 1,462 cases reported that day (Fernández-Naranjo et al. 2021), and reported a cumulative incidence of 146.94 cases per 100,000 people by 18 April (Ortiz-Prado et al. 2021). The actual number of cases are likely to have been much higher when evaluated through the lens of excess mortality data (as obtained from the National Institute of Statistics and Census; Instituto Nacional de Estadística y Censos) and would explain why local diagnostic and healthcare services became rapidly overwhelmed (Long 2020; Ortiz-Prado et al. 2021). After the peak and decline of the epidemic’s first wave in Ecuador, restrictions were maintained during April and May and progressively relaxed over the following months, as the epicentre of Ecuador’s epidemic moved to the capital city of Quito, located in Pichincha province (which on 23 July 2020 overtook Guayaquil as the city with the greatest number of COVID-19 confirmed cases). The last restrictions were finally lifted on 13 September, although use of personal protective equipment and physical distancing guidelines remained in place.
Figure 1.
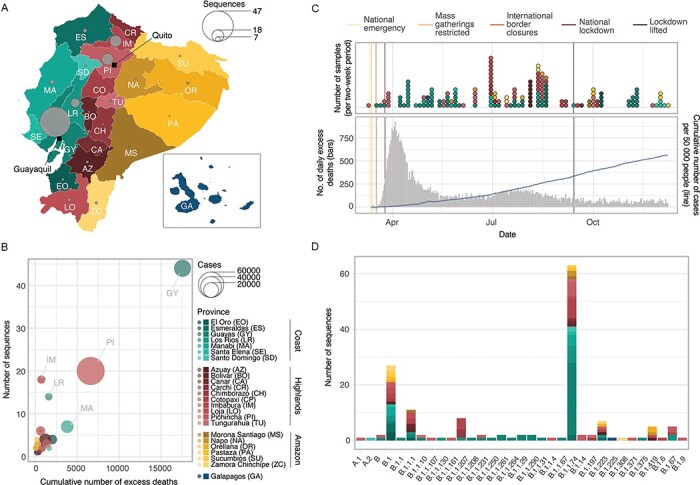
Overview of genomic sampling and SARS-CoV-2 genetic diversity in Ecuador. (A) Number of sequences from Ecuador analysed in this study per province across the four main geographic regions: the coast (shades of green), the highlands (shades of yellow), the Amazon (shades of orange), and the Galápagos (blue). (B) Number of deaths (attributed to laboratory-confirmed COVID-19 cases) versus number of whole genome sequences available per province. Circle radius shows the number of cases per province. (C) Timelines showing collection dates of sequences from the four geographic regions across time (upper panel) and the COVID-19 epidemiological curves in Ecuador during 2020 (cumulative number of laboratory-confirmed cases as reported by the Ministry of Health in the blue line, number of daily excess deaths compared to the same dates in 2019 as reported by the National Institute of Statistics and Census in grey; lower panel). (D) Geographic distribution of SARS-CoV-2 lineages identified in Ecuador.
To date, the source and diversity of circulating transmission lineages in Ecuador and their reach across the country remain unexplored. International importations are expected to have played an important role in seeding transmission chains in Ecuador, as observed in other countries in Latin America (Candido et al. 2020; Laiton-Donato et al. 2020; Franco et al. 2021) and elsewhere (Du Plessis et al. 2021). We undertake phylogenetic analyses of 160 SARS-CoV-2 whole genome sequences sampled from Ecuadorian cases and place them within the context of global viral genetic diversity in order to characterise the genomic epidemiology of SARS-CoV-2 in the country. We identify introduction events and transmission lineages within Ecuador and investigate their spatiotemporal distribution, and we hypothesise about the role of domestic seeding on viral transmission dynamics.
2. Methods
2.1. Genomic sequencing of SARS-CoV-2 samples from Ecuador
Clinical samples were collected from patients with a laboratory-confirmed SARS-CoV-2 infection in Ecuador during 2020 (Fig. 1A–C). Samples collected by the Microbiology Institute at Universidad San Francisco de Quito (IM-USFQ) were obtained from third-level hospitals (i.e. specialised tertiary referral hospitals) across all 24 provinces in the country without unified selection criteria (Marquez et al. 2020). Samples collected by the Omics Sciences Laboratory at Universidad de Especialidades Espiritu Santo (UEES) were obtained from samples collected from the laboratory’s diagnostic service and selected at random for sequencing. Samples collected by the National Institute of Investigations in Public Health (Instituto Nacional de Investigación en Salud Pública, INSPI) were obtained from the national epidemiological SARS-CoV-2 surveillance system. Samples collected by the Biomedical Research Unit at Zurita & Zurita Laboratories were obtained from community patients from Quito who presented clinical signs of reinfection. This complete Ecuadorian sample set was collected between 9 March and 9 December 2020, with limited representation during the early months of the epidemic when compared to excess mortality data (Fig. 1C).
From these samples, we generated 160 complete SARS-CoV-2 genomic sequences using different methodologies. From these, 121 genomes represent samples collected between the implementation of the national lockdown (25 March) and the lifting of restrictions (13 September). IM-USFQ generated 108 whole genome sequences using Oxford Nanopore MinION sequencing and the ARTIC Network primer scheme approach as previously described (Marquez et al. 2020). UEES generated 33 whole genome sequences through Illumina sequencing on a MiniSeq platform (Illumina, San Diego, CA). INSPI generated 15 sequences either in collaboration with Charité—Berlin University of Medicine (through Illumina sequencing) or on site at the Centre for Multidisciplinary Research of the Direction of Research, Development and Innovation (through Oxford Nanopore MinION sequencing as described in Lopez-Alvarez, Parra, and Cuellar 2020). Zurita & Zurita Laboratories generated four sequences through Illumina sequencing on a MiSeq platform (Illumina, San Diego, CA). Details of sample collection, sequencing, and genome assembly are summarised in Supplementary Table S1. Sample collection dates and the province-level geographical location of residence of the patient were included as metadata for all sequences in the country.
To determine the viral genetic diversity circulating in the country during the sampling period, all sequences from Ecuador were phylogenetically assigned under the global Pango lineage system using the Pangolin v2.2.2 tool (https://virological.org/t/pangolin-web-application-release/482).
2.2. Global SARS-CoV-2 data sets
The Ecuadorian virus sequences were analysed in the context of global SARS-CoV-2 genomic diversity by including all high-quality SARS-CoV-2 genome sequences and their accompanying metadata available in GISAID (Shu and McCauley 2017) on 1 January 2021 (sequences were retained if they were >29,000 nucleotides long and <5 per cent of the sequence was missing). Sequences without a complete sample collection date or not attributed to human hosts were excluded, yielding a total of 218,771 sequences from samples collected from 1 December 2019 up until 10 December 2020.
The large number of SARS-CoV-2 genomes generated during 2020 makes full-scale phylogenomic analyses computationally prohibitive. We therefore subsampled sequences from the abovementioned full data set (i.e. all GISAID sequences included in our analyses, excluding the complete set of Ecuadorian sequences) using two approaches. First, we randomly sampled one sequence per country per day from the full data set over the complete sampling period, to create a ‘systematically-subsampled data set’ (comprised of 8,606 sequences). In parallel, we arbitrarily generated three ‘randomly-subsampled data sets’ consisting of 8,606 randomly chosen sequences from the full data set, to match the size of the systematically subsampled data set. These randomly subsampled data sets were used to evaluate the performance of the background SARS-CoV-2 sequences as the genomic context for the identification of transmission lineages within Ecuador (see Section 2.3). Finally, we added the sequences from Ecuador to each data set, resulting in a total of 8,766 sequences per data set.
Each data set was aligned to the Wuhan-Hu-1 (GenBank accession: MN908947.3) reference genome sequence (Wu et al. 2020) using Minimap 2.17 (Li 2018) to generate multiple sequence alignments. Sites containing >90 per cent gaps relative to the sequences in their respective alignment were masked, whilst the untranscribed terminal regions were trimmed. After masking and trimming, the resulting alignments had a final length of 29,409 nucleotides, with the shortest partial genome sequences being cut down to 28,955 nucleotides long.
2.3. Phylogenetic identification of transmission lineages
We followed a similar rationale and methodology to that described in Du Plessis et al. (2021) to identify local transmission lineages. Phylogenetically linked sequences were inferred to have descended from a common ancestor if they were associated with a single inferred introduction event into Ecuador from an international location (Candido et al. 2020; Du Plessis et al. 2021). Ecuadorian transmission lineages therefore correspond to lineages of sequences sampled within the country that descend from a node inferred to have also occurred in Ecuador, which must in turn have descended from outside of the country. Given the unstructured sampling of the Ecuadorian sequences, some transmission lineages will likely correspond to epidemiologically linked cases (i.e. targeted investigation of epidemiological clusters); these have been identified as such in the text whenever the information was available.
Maximum likelihood (ML) phylogenetic trees were estimated from the systematically subsampled data set and the randomly subsampled data sets using IQtree 2.1.1 (Minh et al. 2020) under a GTR + Γ substitution model. Node support was estimated through an SH-like approximate Likelihood Ratio Test using 1,000 replicates (Guindon et al. 2010). While the randomly subsampled data sets were not analysed further, the tree for the systematically subsampled data set was re-rooted by heuristically searching for the root placement that minimises the mean squared residual of a regression of sequence sample date against root-to-tip genetic distance, calculated using TempEst v1.5.3 (Rambaut et al. 2016), to maximise the temporal signal of the data set. The same regression was used to assess the clock-like behaviour of the data set.
Subsequent analyses in our pipeline require an evolutionary rate estimation. We performed an exploratory analysis on a random selection of 866 genomes from the systematically subsampled data set (∼10 per cent of the sequences, ensuring that representatives of the earliest and latest collection dates were included) in order to estimate the evolutionary rate of the data set over the sampling period. We used BEAST v.1.10.4 (Suchard et al. 2018) to obtain a clock rate estimate using the Hasegawa-Kishino-Yano (HKY) substitution model and a strict molecular clock with a continuous-time Markov chain prior (Ferreira and Suchard 2008). We employed a Skygrid coalescent tree prior (Gill et al. 2013) that accounts for the 50 epidemiological weeks over which the genomes were sampled, plus a cut-off period that precedes the earliest collected SARS-CoV-2 sequences. Independent Markov Chain Monte Carlo (MCMC) chains were run for 40 million steps and subsequently combined after discarding the initial 10 per cent of each run as a burn-in. Convergence of relevant parameters was assessed by visually inspecting the MCMC trace plots and posterior probability distributions of parameters from independent chains and using effective sample size (ESS) estimates approaching 100 (for this analysis: median ESS = 87.9; interquartile range (IQR) = 50.5–276.2) from the combined chains using Tracer v1.7.1 (Rambaut et al. 2018). While these ESS values are normally considered low for standard Bayesian phylogenetic analyses, the high node density of the SARS-CoV-2 phylogeny and the currently available models in BEAST appeared to affect the MCMC mixing.
The systematically subsampled data set was analysed with BEAST v1.10.5 (https://github.com/beast-dev/beast-mcmc/releases/tag/v1.10.5pre_thorney) using a newly implemented method that significantly reduces analysis time by using a simple model to estimate a time-calibrated tree (see Didelot, Siveroni, and Volz 2021). This approach takes a previously estimated rooted phylogenetic tree (henceforth called the data tree) instead of an alignment and rescales branches in this tree into time. Under this model, the likelihood of each branch length (in mutations) is defined as a function of a Poisson distribution with a mean directly proportional to the clock rate (Volz and Frost 2017; Didelot, Siveroni, and Volz 2021); we therefore used a rate of 6.28 × 10−4 substitutions/site/year, based on the median clock rate estimate obtained from our exploratory analysis. We defined a coalescent Skygrid prior, similar to the one described for the exploratory analysis, and used the previously mentioned re-rooted ML tree as a starting data tree. Independent MCMC chains were run for 100 million steps and combined (after discarding 10 per cent of each run as burn-in) to produce a posterior tree distribution. Convergence was assessed through the examination of trace plots and ESS estimates as previously described.
To identify nodes associated with transmission lineages in Ecuador, we used a discrete phylogeographic model consisting of a two-state discrete trait analysis (DTA; Lemey et al. 2009) implemented in BEAST v1.10.4 (Suchard et al. 2018). The posterior distribution of trees generated in the previous step was resampled down to 500 time-calibrated trees using LogCombiner v1.10.4, and BEAST was used to sample this tree space. Tips were assigned to one of two possible states (Ecuador vs non-Ecuador), and reconstruction of ancestral node states was undertaken using an asymmetric substitution model (Lemey et al. 2009). The expected number of DTA transitions between international locations and Ecuador were estimated using a robust counting approach (Minin and Suchard 2008). Two independent MCMC chains of 5 million steps each were combined for this analysis, after discarding the first 500,000 steps of each run as burn-in. A Maximum Clade Credibility (MCC) tree was generated from the DTA posterior tree distribution by sampling 1,000 trees from the combined MCMC runs in TreeAnnotator v.1.10. Each internal node was assigned a posterior probability for its inferred location, and these were used to evaluate uncertainty regarding the assignment of potential transmission lineages in Ecuador.
2.4. Transmission lineages and transmission lineage groups
All phylogenetic clusters of sequences from Ecuador were inspected visually on the MCC tree to assign individual transmission lineages. The nomenclature of these country-specific transmission lineages followed a one-letter code in alphabetical order, defined by the earliest sample collection date in each transmission lineage. General features of each transmission lineage were summarised, such as the earliest and latest samples in each lineage, the number of provinces in which the transmission lineage had been identified, and the number of sequences belonging to said lineage (used as a proxy of transmission lineage size). The consistency with which sequences were grouped into these transmission lineages was evaluated by visually inspecting the ML trees for the randomly subsampled data sets and comparing the clusters of Ecuadorian sequences to those from the DTA analysis.
3. Results
3.1. SARS-CoV-2 genetic diversity in Ecuador
The samples from laboratory-confirmed individuals obtained across mainland Ecuador (and one sample from the Galápagos Islands) were collected as they became available through different hospitals and laboratories and yielded representative genomes from all provinces in the country (Fig. 1A). The number of sequences per province correlates with the cumulative number of excess deaths per province during 2020 (compared to the mean number of deaths per province between 2015 and 2019; Spearman’s ρ = 0.556, P = 0.005), suggesting that the number of sequences per province is approximately proportional to the number of infections (Fig. 1B). Despite the testing limitations in the country throughout 2020, the number of sequences also correlate to the cumulative number of patients with a positive or suspected positive COVID-19 PCR test over the sampling period (Spearman’s ρ = 0.603, P = 0.002). Despite the limited number of sequences from Ecuador, the representativeness of our sample is similar to that of other countries in the region. We estimate Ecuador produced 8 sequences for every 10,000 reported cases or 12 sequences for every 1,000 officially reported COVID-19 deaths. This is more representative than Peru (4 sequences per 10,000 cases/10 sequences per 1,000 deaths) or Brazil (3 sequences per 10,000 cases/10 sequences per 1,000 deaths) but less representative than Uruguay (159 sequences per 10,000 cases, and a higher number of SARS-CoV-2 genome sequences than reported deaths) (Supplementary Fig. S1, File S1). It should be noted however that these estimates rely on the testing intensities between provinces in Ecuador and between different countries; limited testing in Ecuador could mean that the overall representation is lower than estimable from official reports.
Wide variation in the total numbers of cases across provinces in Ecuador is reflected in the variation in the number of sequences obtained (Supplementary File S2). While heavily affected provinces such as Pichincha (72,305 confirmed cases until 10 December) and Guayas (26,080 confirmed cases) account for larger numbers of sequences (47 and 18 for Guayas and Pichincha, respectively), less affected provinces in the southern Highlands (Azuay—12,670 confirmed cases, and Loja—7,252) and the Amazon (Morona Santiago—3,422, Napo—1,605, Orellana—2,100, Pastaza—2,360, and Zamora Chinchipe—1,628) are represented by few sequences (28 in total). Only a single sequence was obtained from the Galápagos because of the extremely low number of cases in this province. Manabí province appears to be underrepresented (14,061 confirmed cases), while Imbabura (5,695 confirmed cases) and Los Ríos (4,707 confirmed cases) are represented by higher numbers of genomes per death (Fig. 1B). Sequence sampling rates for each province (excluding Galápagos) varied between 3 and 86 sequences per 1,000 deaths (2). The temporal distribution of samples collected in Ecuador during 2020 does not strongly match trends in reported excess deaths. More samples were collected in July and August, but fewer genomes were sampled in the early epidemic months (March to May) despite the high number of excess deaths reported then (Fig. 1C). Sequence representation is greater for the coastal provinces during the early months of the epidemic (March to June), when the epicentre of the epidemic was based in the port city of Guayaquil (in the Guayas province; Long 2020), and shifted towards higher sampling in the highlands and Amazon provinces, as the epicentre of the epidemic shifted towards the capital city of Quito (in Pichincha province) and as more cases were reported in the Amazon.
Virus genomes from Ecuador were assigned to specific Pango lineages (Rambaut et al. 2020) using the pangolin tool (https://virological.org/t/pangolin-web-application-release/482). The genomes were assigned to 33 different global lineages and predominantly B.1.1.74 (39.4 per cent of all Ecuadorian sequences; Fig. 1D), one of the lineages descended from B.1.1, which became one of the most dominant lineages during the early phase of the pandemic in Europe and North America (after the virus was introduced from Asia; Rambaut et al. 2020; Alteri et al. 2021). The geographic distribution of the SARS-CoV-2 lineage diversity in the country shows distinctive patterns: the most prevalent Pango lineages (B.1.1.74, B.1, and B.1.1.1) are found in multiple provinces, while the majority of the lineages observed at low frequencies were found in more heavily affected (and therefore better sampled) provinces, particularly Guayas and Pichincha. The heavily affected (and highly populated) province of Guayas (where Guayaquil is located) exhibits a predominance of the B.1.1.74 lineage (59.1 per cent of all sequences from this province; Fig. 1D). B.1.1.74 is also abundant in the provinces of Los Ríos, which neighbours Guayas (49 per cent), and Imbabura, which neighbours Pichincha (38.9 per cent). Other common lineages, such as B.1 (16.9 per cent of all sequences from Ecuador) and B.1.1.1 (6.9 per cent of all sequences from Ecuador), are distributed across various geographical regions.
3.2. Identification of Ecuadorian transmission lineages
We undertook exploratory phylogenetic analyses using different sequence subsampling schemes, as the exceptionally large number of available SARS-CoV-2 sequences prevents full analysis of the complete global data set. We estimated ML trees of the Ecuadorian sequences in the context of different background data sets and performed Bayesian phylogenetic inference on a systematically subsampled data set. The clustering patterns of Ecuadorian sequences in the ML trees showed some variation between data sets but in the majority of cases remained consistent (Supplementary File S3). We therefore derive our results from the systematically subsampled data set and discuss these in light of the randomly subsampled data sets.
We consistently found that a sizeable proportion of sequences from Ecuador do not cluster with other sequences from the country (54/160 sequences for the systematically subsampled data set, 48–51/160 for the randomly subsampled data sets; Supplementary File S3) and were therefore assigned as singletons and not associated with further virus spread within Ecuador detectable through genomic analysis. These singletons could in fact represent introduction events that produced no forward transmission or cases where forward transmission did occur but was not captured in this study due to the small sample size. We note that the majority of the singleton sequences were collected before mid-July (Supplementary Fig. S2); we speculate that they could represent predominantly early introduction events that occurred before the implementation of a national lockdown on 16 March. While it is possible to establish a possible limit of dates on which each singleton was introduced, based on the last ancestral node inferred to have occurred outside of Ecuador, the precise importation date will fall somewhere between the inferred age of this preceding node and the collection date of the singleton sequence. The low sampling density in Ecuador and our subsampling schemes are likely to introduce uncertainty in estimating the age of these nodes and we therefore excluded these analyses from our results.
The remaining sequences (106/160 sequences for the systematically subsampled data set) fall into two distinct categories. Firstly, 20 monophyletic clusters of Ecuadorian sequences were identified, capturing multiple introduction events and some local viral circulation patterns. These clusters were assigned to be separate Ecuadorian transmission lineages, named A through V (with exceptions detailed in the paragraph below; Fig. 2A, Supplementary Figs S3–S9). Each represents a single introduction event of the virus from an international destination, followed by local forward transmission in Ecuador (Du Plessis et al. 2021).
Figure 2.
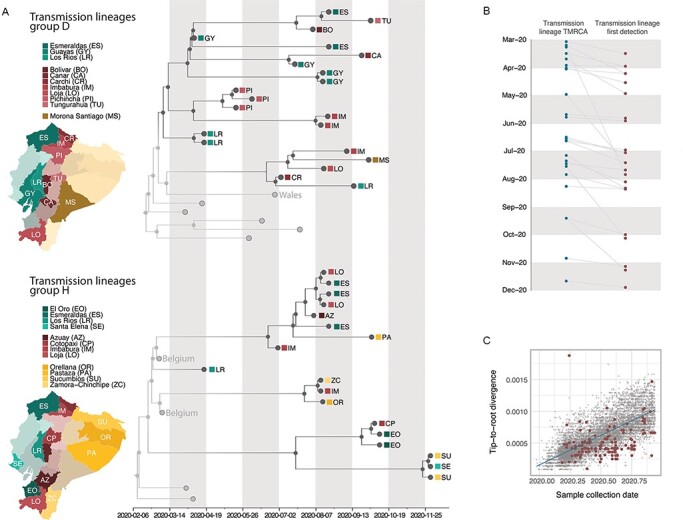
Time calibrated phylogenetic trees for the major transmission lineages in Ecuador. (A) Subtrees extracted from a time-calibrated Maximum Clade Credibility (MCC) tree of SARS-CoV-2 whole genome sequences, corresponding to the two largest clusters of sequences from Ecuador. Tree tips are coloured by sampling location (in Ecuador, red, versus outside of Ecuador, grey); nodes and branches are coloured by inferred location through a two-state DTA analysis. The province where each sequence was sampled is annotated on the tips, and maps highlight these provinces. Tips that correspond to sequences that cluster together within the major Ecuadorian clusters are also annotated with the region where the samples were collected. (B). Detection lag of individual transmission lineages in Ecuador, showing the median TMRCA of each transmission lineage from our data set (blue) connected by a grey line to the date of the earliest sequence in that transmission lineage (red). (C) Root-to-tip genetic distances (based on a heuristically rooted maximum likelihood tree) versus sample collection dates for the SARS-CoV-2 data set used in this analysis. Data points corresponding to sequences collected in Ecuador are highlighted in red, and the linear regression trendline is shown in blue.
Secondly, we identified two large monophyletic clusters that include sequences from international locations and Ecuador. These were not identified strictly as individual transmission lineages through our DTA approach but rather as genetically similar groups of individual transmission lineages. While these may represent clusters of independent introductions from shared sources, there is also a possibility that these groups of transmission lineages in fact correspond to single introduction events misidentified by our DTA analysis, given the variation in the intensity of SARS-CoV-2 sampling across countries (Lemey et al. 2020; Worobey et al. 2020), including Ecuador, and the limited genetic divergence observed in SARS-CoV-2 over the time span being analysed (Villabona-Arenas, Hanage, and Tully 2020). This likely resulted in the ancestral nodes being inferred to have existed outside of Ecuador due to the phylogenetic placement of the Ecuadorian sequences (Fig. 2A). While there is a possibility that these in fact represent multiple closely related yet independently introduced transmission lineages, we here identify them as transmission lineage groups (labelled with a single letter and highlighted with an asterisk, D* and H*) for summary purposes. The remaining unambiguous transmission lineages were each identified with their own letters and are shown in Supplementary Figs S3–S9.
Table 1 provides details for each Ecuador transmission lineage (named sequentially according to the collection date of the earliest sequence in each lineage). We identify 82 (95 per cent HPD: 81–84) SARS-CoV-2 introduction events from other countries into Ecuador through a robust counting approach (Minin and Suchard 2008). This estimate assumes that transmission lineage groups D* and H* are comprised of two and three individual transmission lineages, respectively (with an additional singleton inferred as part of H*; Fig. 2A). The detection lag (defined as the number of days between the inferred transmission lineage time to the most recent common ancestor (TMRCA) and its earliest sampled sequence) ranged between 1 and 140 days (Table 1), with a median of 16 days (IQR: 7–31 days; Fig. 2B, Supplementary Fig. S10).
Table 1.
Summary of transmission lineages identified in Ecuador.
| Transmission lineage | Number of sequences (% of total sequences) | Earliest sample collection date | Latest sample collection date | Median TMRCA (95% HPD) | Detection lag (days)c | Provinces where it has been sampled |
|---|---|---|---|---|---|---|
| A | 3 (1.875) | 16/3/2020 | 23/3/2020 | 3/3/2020 (25/2/2020–9/3/2020) | 13 | Guayas |
| B | 5 (3.125) | 30/3/2020 | 30/6/2020 | 16/3/2020 (2/3/2020–30/3/2020) | 14 | Imbabura, Los Rios, Pichincha |
| C | 2 (1.25) | 30/3/2020 | 30/3/20 | 29/3/2020 (26/3/2020–30/3/2020) | 1 | Pichincha |
| Da | 21 (13.125) | 7/4/2020 | 1/10/2020 | 2/3/2020b (19/2/2020–14/3/2020) | 36b | Bolivar, Cañar, Carchi, Esmeraldas, Guayas, Imbabura, Loja, Los Rios, Morona Santiago, Pichincha, Tungurahua |
| E | 3 (1.875) | 7/4/2020 | 30/6/2020 | 2/4/2020 (10/3/2020–7/4/2020) | 5 | Guayas, Manabi |
| F | 6 (3.75) | 13/4/2020 | 10/10/2020 | 12/7/2020 (2/6/2020–28/7/2020) | 23 | Azuay, Bolivar, El Oro, Guayas, Loja |
| G | 8 (5) | 17/4/2020 | 7/1/2020 | 1/4/2020 (25/2/2020–9/4/2020) | 16 | Chimborazo, Imbabura, Los Rios |
| Ha | 18 (11.25) | 17/4/2020 | 30/11/2020 | 21/2/2020b (11/2/2020–28/2/2020) | 56b | Azuay, Cotopaxi, El Oro, Esmeraldas, Imbabura, Loja, Orellana, Pastaza, Santa Elena, Sucumbios, Zamora Chinchipe |
| I | 3 (1.875) | 29/4/2020 | 7/8/2020 | 22/3/2020 (24/2/2020–8/4/2020) | 38 | Guayas |
| J | 2 (1.25) | 26/5/2020 | 15/6/2020 | 22/5/2020 (3/5/2020–26/5/2020) | 4 | Napo |
| K | 2 (1.25) | 29/5/2020 | 20/7/2020 | 25/5/2020 (13/5/2020–29/5/2020) | 4 | Guayas |
| L | 5 (3.125) | 3/7/2020 | 19/8/2020 | 16/6/2020 (8/6/2020–2/7/2020) | 17 | Azuay, Loja, Napo, Orellana |
| M | 3 (1.875) | 14/7/2020 | 13/8/2020 | 30/4/2020 (26/3/2020–18/6/2020) | 75 | Los Rios, Zamora Chinchipe |
| N | 7 (4.375) | 14/7/2020 | 20/8/2020 | 20/6/2020 (19/5/2020–12/7/2020) | 24 | Azuay, Esmeraldas, Imbabura, Los Rios, Orellana, Pichincha, Zamora Chinchipe |
| O | 3 (1.875) | 22/7/2020 | 7/9/2020 | 15/7/2020 (2/7/2020–22/7/2020) | 7 | Imbabura |
| P | 3 (1.875) | 27/7/2020 | 11/4/2020 | 9/3/2020 (24/2/2020–8/4/2020) | 140 | Guayas |
| Q | 2 (1.25) | 11/8/2020 | 26/11/2020 | 6/7/2020 (22/5/2020–24/7/2020) | 36 | Guayas, Pichincha |
| R | 2 (1.25) | 12/8/2020 | 12/8/2020 | 9/8/2020 (20/7/2020–12/8/2020) | 3 | Imbabura |
| S | 2 (1.25) | 1/10/2020 | 5/10/2020 | 13/9/2020 (16/8/2020–1/10/2020) | 18 | Cotopaxi, Tungurahua |
| T | 2 (1.25) | 5/11/2020 | 9/11/2020 | 27/10/2020 (5/10/2020–4/11/2020) | 9 | Guayas |
| U | 2 (1.25) | 9/11/2020 | 9/11/2020 | 13/9/2020 (16/8/2020–1/10/2020) | 57 | Guayas |
| V | 3 (1.875) | 28/11/2020 | 9/12/2020 | 21/11/2020 (1/11/2020−27/11/2020) | 7 | Santo Domingo de los Tsachilas, Sucumbios |
Transmission complex, composed of two or more potential introductions of very genetically similar viruses.
TMRCA of various transmission lineages and sequences outside of Ecuador; node not inferred in Ecuador.
Number of days between the inferred TMRCA and the earliest collection date in the transmission lineage.
3.3. Size and persistence of transmission lineages
Initial molecular clock analyses showed that our data set contains strong temporal signal overall, although many sequences from Ecuador showed lower than average genetic divergence from the root (Fig. 2C). The inferred TMRCAs of Ecuadorian transmission lineages ranged from February to November 2020 (Table 1); from this list, transmission lineages C and S are composed of pairs of sequences that share an epidemiological link.
The TMRCAs estimated for the two large transmission lineage groups D* and H* are the earliest in our data; however, these might not represent true lineage ancestors within Ecuador, because each group could represent more than one introduction from other countries. After excluding these larger groups, we still identified six transmission lineages for which the 95 per cent HPDs of the TMRCA include the date of implementation of the national lockdown, 16 March. Therefore, these transmission lineages likely correspond to introduction events that occurred before restrictions on incoming international flights were adopted in Ecuador. An additional four transmission lineages have TMRCA estimates between late March and November 2020. These may correspond to more recent introductions (following the progressive relaxation of the lockdown in Ecuador between May and September). The most likely exceptions to this are transmission lineages C (TMRCA: 2020.2412, 95 per cent HPD: 2020.2328-2020.2446) and M (TMRCA: 2020.3278, 95 per cent HPD: 2020.1864-2020.4187). Incomplete sampling of these lineages and detection lags could result in the date of introduction being substantially earlier than the date of the TMRCA (Duchêne, Duchêne, and Ho 2015), which would place the introduction date for these transmission lineages prior to the implementation of the lockdown.
Transmission lineages vary in size from sequence pairs (transmission lineages C, J, K, Q, R, S, T, and U) to larger clusters of 16–21 sequences (transmission lineage group D*, depending on whether D* is considered as a single lineage or as multiple lineages). The number of sequences in each transmission lineage is correlated with the number of days between the earliest and most recent sampling dates within the lineage (assuming that D* and H* are composed of multiple individual transmission lineages each). However, this result could be driven by the single largest transmission lineage in the data set. A similar pattern is observed when comparing the time between the inferred TMRCA and the most recent sequence of each transmission lineage (equivalent to the persistence time plus the detection lag; Supplementary Fig. S11). We also observed that lineages that were detected earlier tend to be larger (contain more sequences) and persist longer but are not more geographically widespread (Fig. 3A; Supplementary Figs S12–S14). However, transmission lineages first detected or with a TMRCA between June and August are found on average in a greater number of provinces (from earliest detection of transmission lineages: mean = 3.27 provinces, median = 3 provinces; from TMRCAs of transmission lineages: mean = 3.7 provinces, median = 4 provinces) compared to transmission lineages first detected at any other time of the year (from earliest detection of transmission lineages: mean = 2.21 provinces, median = 1.5 provinces; from TMRCAs of transmission lineages: mean = 2 provinces, median = 1 province) (Fig. 3A). Overall, transmission lineages with earlier TMRCAs and that were first detected earlier in the year persisted for longer timespans (i.e. there is a greater number of days between the first detection and the most recent detection of a transmission lineage; Supplementary Fig. S13). It should be noted however that these patterns also resemble the increased sampling that occurred over the months of July and September (seen in Fig. 1C), suggesting that these patterns could be explained by sampling intensity across time.
Figure 3.
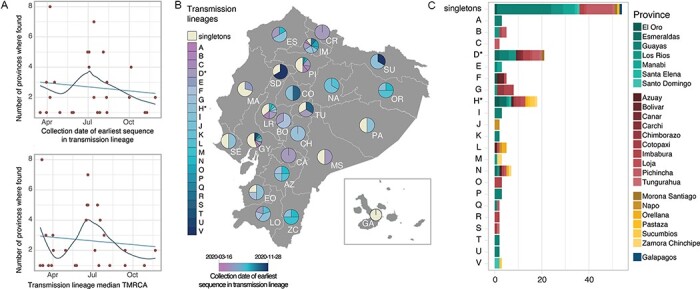
SARS-CoV-2 transmission lineages in Ecuador. (A) Summary of the geographic spread of transmission lineages in Ecuador, showing the number of provinces where each transmission lineage is found compared to the collection date of the earliest sequence in each transmission lineage (upper panel) or the inferred median TMRCA for each transmission lineage (lower panel). The trend lines show a linear regression in light blue and a fitted local polynomial regression in dark blue. (B) Contribution of individual transmission lineages and singleton sequences in each province. Transmission lineages (shades of blue) are ordered based on the earliest sample collection date in the group from earliest (darker) to more recent (lighter). (C) Bar plot summarising the provinces where each transmission lineage was sampled over the study sampling period.
3.4. Geographical distribution of transmission lineages
Singletons and transmission lineages are found across multiple provinces and regions of Ecuador (Fig. 3B–C). Singletons represent an important proportion of the sequences in various provinces across central Ecuador ranging between 33.3 per cent in Tungurahua and 52.3 per cent in Guayas (Fig. 3B), an observation that is particularly important for provinces with large numbers of sequences (Supplementary Fig. S14). On the other hand, different transmission lineages are found either in single provinces or across multiple regions (Fig. 3C). The large transmission lineage groups D* and H* include sequences from provinces across three geographical regions each (the coastal region, the highlands, and the Amazon region) and potentially show internal seeding events of the virus across provincial boundaries (Figs 2A, 3C). Even when accounting for the possibility that these lineage groups are comprised of multiple transmission lineages, sequences from different provinces and regions clustered together (Fig. 2A).
Consistent with the spatiotemporal sampling patterns (Fig. 1C), the older transmission lineages (shown in darker blue in Fig. 3B) were identified predominantly in provinces on the coast and highland regions, while younger transmission lineages (shown in lighter blue in Fig. 3B) were identified in specific provinces in the north and more broadly in the south. The first epicentre of the COVID-19 epidemic in Ecuador, the province of Guayas, is represented by a high frequency of singleton lineages, with a high diversity of individual transmission lineages first identified at different times during 2020. A similar pattern is observed for the second epicentre of the epidemic, the province of Pichincha, but with fewer different transmission lineages and less representation of the youngest transmission lineages (Fig. 3A). We note that these patterns could be affected by differences in the number of sequences available for each province (Fig. 1A; Supplementary Fig. S15).
4. Discussion
The early weeks of the COVID-19 epidemic in Ecuador were characterised by a severe spike in the number of cases in the city of Guayaquil, the largest in the country located in the province of Guayas, and by high attack rates (i.e. new cases in a population at risk divided by the size of that population at risk) across various coastal provinces (Ortiz-Prado et al. 2021). The outbreak overwhelmed local healthcare systems, resulting in one of the highest excess death rates in the world during early 2020 (Long 2020). Information about the importation of SARS-CoV-2 into Ecuador and the domestic spread of the virus is needed to explain the drastic effects of the pandemic in the country during March and April 2020 and to explain the large difference in disease burden between Guayaquil and the capital, Quito.
An important determinant of the early dynamics of COVID-19 outbreaks has been human mobility and the number of introduction events of the virus into a new location with an immunologically naïve population, as was observed during the early stages of the emergence of SARS-CoV-2 in China (Kraemer et al. 2020). The large proportion of singletons observed in Guayas and various other coastal provinces, particularly given their tendency to occur early in the epidemic (Supplementary Fig. S2), could be suggestive of multiple independent introduction events with limited forward transmission. This would also explain why the earliest sequences in the local transmission lineages and the sequences assigned to the most common Pango lineage in Ecuador (B.1.1.74) were predominantly sampled in coastal provinces during the early weeks of the epidemic (Fig. 2A, Supplementary Fig. S16).
Two additional factors support the hypothesis that Guayas played an important role in seeding of viral transmission to other regions in Ecuador: (i) the city of Guayaquil hosts the second busiest international airport in the country and one of only two in the coastal region (the second international airport located in the province of Manabí hosts limited flights to a few international destinations; Hidalgo et al. 2020) and (ii) the overall timing of the seeding events (which necessarily have to predate the inferred TMRCA of a lineage) corresponds to the school holiday period in the coastal region (February to April), when international travel and large social gatherings are more likely to occur. The inferred TMRCAs, which can serve as an upper bound for the true introduction dates of the largest lineages (assuming these are better represented with the available sequences), show that these transmission lineages most likely arrived in Ecuador before the date when non-pharmaceutical interventions (NPIs) were implemented and before travel restrictions came into place.
The genetic diversity of SARS-CoV-2 can be quantified using the Pango dynamic nomenclature system. Pango lineages reflect the history of significant events in the epidemic and geographic spread of the virus (Fountain-Jones et al. 2020) and can be used to explore likely source locations of virus importations, for example, the high representation of lineages descended from B.1.1 in the province of Guayas (Fig. 1D). The emergence of B.1.1 in Europe and North America around February 2020 (Rambaut et al. 2020) might suggest these regions contributed to seeding the epidemic in this province of Ecuador (despite the limitations on identifying exact source locations due to poor surveillance in many countries). B.1.1.74, the most prevalent lineage in Ecuador, descended from B.1.1 and was more frequently sampled in Guayas during the early months of the epidemic (Supplementary Fig. S16). This further reveals that locations where B.1.1 was prevalent during the epidemic’s first wave likely played a role as importation sources. However, the high proportion of singletons observed in Guayas also suggests that onward transmission of introduced virus was less common. Insights from regions sampled at very high intensities, such as the United Kingdom, show that the majority of introductions lead to small, transient, dead-end transmission lineages, whereas a smaller number of introductions lead to larger and longer-lasting transmission lineages (Du Plessis et al. 2021). If this phenomenon is a general property of the first wave of SARS-CoV-2 transmission, as appears to be the case given similar early observations in Brazil (Candido et al. 2020), Panama (Franco et al. 2021), Uruguay (Elizondo et al. 2021), Spain (Díez-Fuertes et al. 2021), and the Netherlands (Oude Munnink et al. 2020), we can expect that many of the introductions to Guayas led to few new cases and that most of the ongoing transmission was derived from only a few introductions. This pattern of transmission heterogeneity would emerge due to the over-dispersion of SARS-CoV-2 transmission (Lloyd-Smith et al. 2005; Li, Grassly, and Fraser 2017), a phenomenon that could play an important role in the viral epidemic dynamics (Adam et al. 2020; Du Plessis et al. 2021; Geidelberg et al. 2021).
The larger transmission lineages identified here suggest that virus transmission was high between neighbouring and well-connected provinces. This might have been an important determinant of the transmission dynamics between the main cities in the country. In contrast to Guayaquil, Quito is the second-largest city in the country and presented a much less severe first epidemic wave, despite hosting the busiest international airport in the country. The city is located in the province of Pichincha, which exhibits a large proportion of singletons but fewer distinct global and transmission lineages overall. Importantly, the transmission lineages observed in Pichincha are mostly not shared with Guayas. This suggests that either independent international introductions or domestic seeding events likely drove the early epidemic in Pichincha. Our phylogenetic analyses suggest that some transmission lineages in Pichincha were introduced from other provinces (Fig. 2A, e.g., the monophyletic Pichincha clade in lineage group D*; Supplementary Fig. S6), suggesting that domestic travel might have played an important role in the establishment of SARS-CoV-2 transmission in this region. It is also possible that a later burst of international introductions of new lineages occurred after air travel and lockdown measures were lifted; however, the sampling dates of singleton genomes from Pichincha (between March and July) suggest that introductions into this province likely occurred before the lockdown and not during the relaxation of NPIs. The Pichincha singletons account for the earliest sequences of this kind in our data set but could represent instances where limited or no additional spread occurred following their introduction.
Ultimately, more comprehensive analyses on the sources and drivers of transmission would require a deeper sampling of key locations where transmission was high, and the inclusion of complementary data sources such as real-time mobility and transportation data could provide a better overview of the forces shaping the observed viral genetic diversity in Ecuador. Provinces such as Azuay, Guayas, and Pichincha represent the main air travel entry points but the role of land mobility across the northern border with Colombia and the southern border with Peru should also be considered to further understand the role of other Latin American countries in regional viral transmission.
Our analysis highlights some important patterns but is limited by various factors. Most notably, the number of genome sequences in our study, although large by historical standards, is small compared to the current trends for virus genome sequencing during the COVID-19 pandemic. The sample size restricts our ability to infer further details about local virus transmission dynamics from sequence data alone. The trajectories of individual lineages, from their international sources to their spread across the country and their subsequent local circulation, are best analysed from larger data sets, or in conjunction with additional data sources to manage and ameliorate the potential consequences of sampling biases (De Maio et al. 2015; Kalkauskas et al. 2021). Incorporating data from self-reported travel histories and human mobility can help to maximise the utility of smaller samples, collected in settings where the sequencing of large numbers of genomes lies beyond local technological or financial capacity, or where high sampling densities are unfeasible (e.g. in remote locations). Moreover, the broad range of lag times between the inferred TMRCAs and the earliest sampled sequence per transmission lineage suggest that a considerable number of transmission lineages are not detected immediately after being introduced to the country, reducing our capacity to identify a potential port of entry and delaying the possibility of responding rapidly to new seeding events. This can be particularly relevant for the identification of variants of concern, where their potential for higher transmissibility (Faria et al. 2021; Tegally et al. 2021; Volz et al. 2021) could require faster response times to identify the sources of importation and establish effective contact tracing to contain further spread.
The first year of the COVID-19 pandemic has shown how global connectivity plays a key role in the development of national epidemics caused by respiratory viruses, reminiscent of other pathogens such as influenza viruses (Lemey et al. 2014). Our results from Ecuador showcase the relevance of importations in establishing local viral circulation and the potential consequences of interprovincial mobility for highly connected locations. In particular, it shows that importations have been a common occurrence even after the implementation of lockdown measures and travel restrictions and that seeding events across provinces can occur frequently. While air travel is limited between provinces, the connectivity provided by land travel can serve as a means for pathogen spread, highlighting the vulnerability of highly connected and remote locations alike. The notion that two large cities with busy international airports can manifest such different transmission dynamics and viral genetic diversity is also relevant, as it shows that a combination of multiple factors determines the outcome of an epidemic within a specific location. Ultimately, the type of interventions chosen to mitigate this high degree of connectivity, the necessity of an early implementation of these interventions, and the adherence to these by the general population are paramount in determining their efficacy.
Supplementary Material
Acknowledgements
We would like to thank all the clinical personnel from numerous public and private healthcare institutions who provided access to laboratory-confirmed samples to generate SARS-CoV-2 genomic sequences. We also thank the Centre for Arbovirus Discovery, Diagnosis, Genomics and Epidemiology (CADDE); Eva Harris from the A2CARES network; and Maria de Lourdes Torres, Diego Quiroga, and Stella de la Torre for their methodological, logistical, and financial support (through the USFQ Emergency Grants) towards the sequencing of viral genomes. We are grateful to all laboratories and institutions worldwide involved in the generation of virus genome data shared on GISAID.
Contributor Information
Bernardo Gutierrez, Department of Zoology, University of Oxford, Oxford, Oxfordshire OX1 3SY, UK; Colegio de Ciencias Biológicas y Ambientales, Universidad San Francisco de Quito, Quito 170901, Ecuador.
Sully Márquez, Instituto de Microbiología, Universidad San Francisco de Quito, Quito 170901, Ecuador.
Belén Prado-Vivar, Instituto de Microbiología, Universidad San Francisco de Quito, Quito 170901, Ecuador.
Mónica Becerra-Wong, Instituto de Microbiología, Universidad San Francisco de Quito, Quito 170901, Ecuador.
Juan José Guadalupe, Laboratorio de Biotecnología Vegetal, Universidad San Francisco de Quito, Quito 170901, Ecuador.
Darlan Da Silva Candido, Department of Zoology, University of Oxford, Oxford, Oxfordshire OX1 3SY, UK.
Juan Carlos Fernandez-Cadena, Omics Sciences Laboratory, Faculty of Medical Sciences, Universidad de Especialidades Espíritu Santo, Samborondón 092301, Ecuador.
Gabriel Morey-Leon, Faculty of Medical Sciences, Universidad de Guayaquil, Guayaquil 090613, Ecuador.
Rubén Armas-Gonzalez, Faculty of Sciences, Escuela Superior Politécnica del Litoral, Guayaquil 090112, Ecuador.
Derly Madeleiny Andrade-Molina, Omics Sciences Laboratory, Faculty of Medical Sciences, Universidad de Especialidades Espíritu Santo, Samborondón 092301, Ecuador.
Alfredo Bruno, Instituto Nacional de Investigación en Salud Pública, Guayaquil 3961, Ecuador; Universidad Agraria del Ecuador, Guayaquil 090104, Ecuador.
Domenica De Mora, Instituto Nacional de Investigación en Salud Pública, Guayaquil 3961, Ecuador.
Maritza Olmedo, Instituto Nacional de Investigación en Salud Pública, Guayaquil 3961, Ecuador.
Denisse Portugal, Instituto Nacional de Investigación en Salud Pública, Guayaquil 3961, Ecuador.
Manuel Gonzalez, Instituto Nacional de Investigación en Salud Pública, Guayaquil 3961, Ecuador.
Alberto Orlando, Instituto Nacional de Investigación en Salud Pública, Guayaquil 3961, Ecuador.
Jan Felix Drexler, Institute of Virology, Charité-Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin 10117, Germany.
Andres Moreira-Soto, Institute of Virology, Charité-Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin 10117, Germany.
Anna-Lena Sander, Institute of Virology, Charité-Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin 10117, Germany.
Sebastian Brünink, Institute of Virology, Charité-Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin 10117, Germany.
Arne Kühne, Institute of Virology, Charité-Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin 10117, Germany.
Leandro Patiño, Instituto Nacional de Investigación en Salud Pública, Guayaquil 3961, Ecuador.
Andrés Carrazco-Montalvo, Instituto Nacional de Investigación en Salud Pública, Quito 170403, Ecuador.
Orson Mestanza, Servicio de Genética, Instituto Nacional de Salud del Niño San Borja, Lima 15037, Perú.
Jeannete Zurita, Facultad de Medicina, Pontificia Universidad Católica del Ecuador, Quito 170143, Ecuador; Unidad de Investigaciones en Biomedicina, Zurita & Zurita Laboratorios, Quito 170104, Ecuador.
Gabriela Sevillano, Unidad de Investigaciones en Biomedicina, Zurita & Zurita Laboratorios, Quito 170104, Ecuador.
Louis Du Plessis, Department of Zoology, University of Oxford, Oxford, Oxfordshire OX1 3SY, UK.
John T McCrone, Institute of Evolutionary Biology, University of Edinburgh, Edinburgh EH9 3JW, UK.
Josefina Coloma, School of Public Health, University of California, Berkeley CA 94704, USA.
Gabriel Trueba, Instituto de Microbiología, Universidad San Francisco de Quito, Quito 170901, Ecuador.
Verónica Barragán, Instituto de Microbiología, Universidad San Francisco de Quito, Quito 170901, Ecuador.
Patricio Rojas-Silva, Instituto de Microbiología, Universidad San Francisco de Quito, Quito 170901, Ecuador.
Michelle Grunauer, Escuela de Medicina, Universidad San Francisco de Quito, Quito 170901, Ecuador.
Moritz U G Kraemer, Department of Zoology, University of Oxford, Oxford, Oxfordshire OX1 3SY, UK.
Nuno R Faria, Department of Zoology, University of Oxford, Oxford, Oxfordshire OX1 3SY, UK; MRC Centre for Global Infectious Disease Analysis, J-IDEA, Imperial College London, London SW7 2AZ, UK.
Marina Escalera-Zamudio, Department of Zoology, University of Oxford, Oxford, Oxfordshire OX1 3SY, UK.
Oliver G Pybus, Department of Zoology, University of Oxford, Oxford, Oxfordshire OX1 3SY, UK; Department of Pathobiology and Population Sciences, Royal Veterinary College London, London NW1 0TU, UK.
Paúl Cárdenas, Instituto de Microbiología, Universidad San Francisco de Quito, Quito 170901, Ecuador.
Data availability
The newly generated sequences have been publicly shared through the GISAID platform. The data sets (including Ministry of Health and National Institute of Statistics and Census data) and code used to generate the analyses presented in this study, as well as a list of accession numbers for the Ecuador SARS-CoV-2 genome sequences analysed here are available through GitHub at https://github.com/BernardoGG/SARS-CoV-2_Genomic_lineages_Ecuador. We thank Andrés N. Robalino and Carlos Oporto for their contribution to the open licence Ecuacovid repository (https://github.com/andrab/ecuacovid) used to extract epidemiological data for this study.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
Financial support was provided by the Clarendon Fund and the Department of Zoology of the University of Oxford (D.S.C.); WT fellowship 204311/Z/16/Z and MRC-FAPESP awards MR/S0195/1 and 18/14389-0 (N.R.F.); Branco Weiss Fellowship and EU grant 874850 MOOD (M.U.G.K.); WT Collaborators Award 206298/Z/17/Z (J.T.M.); Leverhulme Trust ECR Fellowship ECF-2019-542 (M.E.Z.); the Oxford Martin School (O.G.P., M.U.G.K. and L.d.P.); and the NIH Global Health Equity Scholars award FIC D43TW010540 (P.C.) For the purpose of open access, the authors have applied a CC BY-NC public copyright licence to this manuscript.
Conflict of interest:
The authors report no conflicts of interests.
References
- Adam D. C. et al. (2020) ‘Clustering and Superspreading Potential of SARS-CoV-2 Infections in Hong Kong’, Nature Medicine, 26: 1714–9.doi: 10.1038/s41591-020-1092-0. [DOI] [PubMed] [Google Scholar]
- Alteri C. et al. (2021) ‘Genomic Epidemiology of SARS-CoV-2 Reveals Multiple Lineages and Early Spread of SARS-CoV-2 Infections in Lombardy, Italy’, Nature Communications, 12: 434.doi: 10.1038/s41467-020-20688-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candido D. S. et al. (2020) ‘Evolution and Epidemic Spread of SARS-CoV-2 in Brazil’, Science, 369: 1255–60.doi: 10.1126/science.abd2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maio N. et al. (2015) ‘New Routes to Phylogeography: A Bayesian Structured Coalescent Approximation’, PLOS Genetics, 11: e1005421.doi: 10.1371/journal.pgen.1005421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X., Siveroni I., and Volz E. M. (2021) ‘Additive Uncorrelated Relaxed Clock Models for the Dating of Genomic Epidemiology Phylogenies’, Molecular Biology and Evolution, 38: 307–17.doi: 10.1093/molbev/msaa193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díez-Fuertes F. et al. (2021) ‘A Founder Effect Led Early SARS-CoV-2 Transmission in Spain’, Journal of Virology, 95: e01583-20.doi: 10.1128/JVI.01583-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Plessis L. et al. (2021) ‘Establishment and Lineage Dynamics of the SARS-CoV-2 Epidemic in the UK’, Science, 371: 708–12.doi: 10.1126/science.abf2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchêne D., Duchêne S., and Ho S. Y. (2015) ‘Tree Imbalance Causes a Bias in Phylogenetic Estimation of Evolutionary Timescales Using Heterochronous Sequences’, Molecular Ecology Resources, 15: 785–94.doi: 10.1111/1755-0998.12352. [DOI] [PubMed] [Google Scholar]
- Duchene S. et al. (2020) ‘Temporal Signal and the Phylodynamic Threshold of SARS-CoV-2’, Virus Evolution, 6: veaa061.doi: 10.1093/ve/veaa061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elizondo V. et al. (2021) ‘SARS-CoV-2 Genomic Characterization and Clinical Manifestation of the COVID-19 Outbreak in Uruguay’, EmergingMicrobes and Infections, 10: 51–65.doi: 10.1080/22221751.2020.1863747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria N. R. et al. (2021) ‘Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil’, Science, 372: 815–21.doi: 10.1126/science.abh2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— (2017) ‘Establishment and Cryptic Transmission of Zika Virus in Brazil and the Americas’, Nature, 546: 406–10.doi: 10.1038/nature22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Naranjo R. P. et al. (2021) ‘Statistical Data Driven Approach of COVID-19 in Ecuador’, Infectious Disease Modelling, 6: 232–43.doi: 10.1016/j.idm.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira M. A., and Suchard M. A. (2008) ‘Bayesian Analysis of Elapsed Times in Continuous‐time Markov Chains’, Canadian Journal of Statistics, 36: 355–68. [Google Scholar]
- Fountain-Jones N. M. et al. (2020) ‘Emerging Phylogenetic Structure of the SARS-CoV-2 Pandemic’, Virus Evolution, 6: veaa082.doi: 10.1093/ve/veaa082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco D. et al. (2021) ‘Early Transmission Dynamics, Spread, and Genomic Characterization of SARS-CoV-2 in Panama’, Emerging Infectious Diseases, 27: 612–5.doi: 10.3201/eid2702.203767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geidelberg L. et al. (2021) ‘Genomic Epidemiology of a Densely Sampled COVID-19 Outbreak in China’, Virus Evolution, 7: veaa102.doi: 10.1093/ve/veaa102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J. L. et al. (2020) ‘Genomic Epidemiology Reveals Transmission Patterns and Dynamics of SARS-CoV-2 in Aotearoa New Zealand’, Nature Communications, 11: 6351.doi: 10.1038/s41467-020-20235-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill M. S. et al. (2013) ‘Improving Bayesian Population Dynamics Inference: A Coalescent-based Model for Multiple Loci’, Molecular Biology and Evolution, 30: 713–24.doi: 10.1093/molbev/mss265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh N. D. et al. (2019a) ‘Tracking Virus Outbreaks in the Twenty-First Century’, Nature Microbiology, 4: 10–9.doi: 10.1038/s41564-018-0296-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— (2019b) ‘Travel Surveillance and Genomics Uncover a Hidden Zika Outbreak during the Waning Epidemic’, Cell, 178: 1057–71.e11.doi: 10.1016/j.cell.2019.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson D. F. et al. (2020) ‘Spread of SARS-CoV-2 in the Icelandic Population’, New England Journal of Medicine, 382: 2302–15.doi: 10.1056/NEJMoa2006100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S. et al. (2010) ‘New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0’, Systematic Biology, 59: 307–21.doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hidalgo G. et al. (2020) Anuario De Estadísticas De Transporte 2019. Quito: Instituto Nacional de Estadísticas y Censos. [Google Scholar]
- Kalkauskas A. et al. (2021) ‘Sampling Bias and Model Choice in Continuous Phylogeography: Getting Lost on a Random Walk’, PLOS Computational Biology, 17: e1008561.doi: 10.1371/journal.pcbi.1008561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer M. U. G. et al. (2020) ‘The Effect of Human Mobility and Control Measures on the COVID-19 Epidemic in China’, Science, 368: 493–7.doi: 10.1126/science.abb4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiton-Donato K. et al. (2020) ‘Genomic Epidemiology of Severe Acute Respiratory Syndrome Coronavirus 2, Colombia’, Emerging Infectious Diseases, 26: 2854–62.doi: 10.3201/eid2612.202969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P. et al. (2020) ‘Accommodating Individual Travel History and Unsampled Diversity in Bayesian Phylogeographic Inference of SARS-CoV-2’, Nature Communications, 11: 5110.doi: 10.1038/s41467-020-18877-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— (2014) ‘Unifying Viral Genetics and Human Transportation Data to Predict the Global Transmission Dynamics of Human Influenza H3N2’, PLoS Pathogens, 10: e1003932.doi: 10.1371/journal.ppat.1003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— (2009) ‘Bayesian Phylogeography Finds Its Roots’, PLoS Computational Biology, 5: e1000520.doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2018) ‘Minimap2: Pairwise Alignment for Nucleotide Sequences’, Bioinformatics, 34: 3094–100.doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L. M., Grassly N. C., and Fraser C. (2017) ‘Quantifying Transmission Heterogeneity Using Both Pathogen Phylogenies and Incidence Time Series’, Molecular Biology and Evolution, 34: 2982–95.doi: 10.1093/molbev/msx195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Smith J. O. et al. (2005) ‘Superspreading and the Effect of Individual Variation on Disease Emergence’, Nature, 438: 355–9.doi: 10.1038/nature04153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long G. (2020), Ecuador’s Virus-hit Guayaquil is Grim Warning for Region. Financial Times <https://www.ft.com/content/5e970473-0710-44f6-bfae-2a830b78a3a1> (accessed 27 May 2021.)
- Lopez-Alvarez D., Parra B., and Cuellar W. J. (2020) ‘Genome Sequence of SARS-CoV-2 Isolate Cali-01, from Colombia, Obtained Using Oxford Nanopore MinION Sequencing’, Microbiology Resource Announcements, 9: e00573-20.doi: 10.1128/MRA.00573-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J. et al. (2020) ‘Genomic Epidemiology of SARS-CoV-2 in Guangdong Province, China’, Cell, 181: 997–1003.e9.doi: 10.1016/j.cell.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez S. et al. (2020) ‘Genome Sequencing of the First SARS-CoV-2 Reported from Patients with COVID-19 in Ecuador’, medRxiv.doi: 10.1101/2020.06.11.20128330. [DOI] [Google Scholar]
- Meredith L. W. et al. (2020) ‘Rapid Implementation of SARS-CoV-2 Sequencing to Investigate Cases of Health-care Associated COVID-19: A Prospective Genomic Surveillance Study’, TheLancet Infectious Diseases, 20: 1263–71.doi: 10.1016/S1473-3099(20)30562-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B. Q. et al. (2020) ‘IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era’, Molecular Biology and Evolution, 37: 1530–4.doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin V. N., and Suchard M. A. (2008) ‘Counting Labeled Transitions in Continuous-time Markov Models of Evolution’, Journal of Mathematical Biology, 56: 391–412.doi: 10.1007/s00285-007-0120-8. [DOI] [PubMed] [Google Scholar]
- Moreno G. K. et al. (2020) ‘Revealing Fine-scale Spatiotemporal Differences in SARS-CoV-2 Introduction and Spread’, Nature Communications, 11: 5558.doi: 10.1038/s41467-020-19346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiura H., and Chowell G. (2009) ‘The Effective Reproduction Number as a Prelude to Statistical Estimation of Time-Dependent Epidemic Trends’. In: Chowell, G.et al. (eds) Mathematical and Statistical Estimation Approaches in Epidemiology, pp. 103–21. Springer Netherlands: Dordrecht. [Google Scholar]
- Ortiz-Prado E. et al. (2021) ‘Epidemiological, Socio-demographic and Clinical Features of the Early Phase of the COVID-19 Epidemic in Ecuador’, PLOS Neglected Tropical Diseases, 15: e0008958.doi: 10.1371/journal.pntd.0008958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oude Munnink B. B. et al. (2020) ‘Rapid SARS-CoV-2 Whole-genome Sequencing and Analysis for Informed Public Health Decision-making in the Netherlands’, Nature Medicine, 26: 1405–10.doi: 10.1038/s41591-020-0997-y. [DOI] [PubMed] [Google Scholar]
- Park D. J. et al. (2015) ‘Ebola Virus Epidemiology, Transmission, and Evolution during Seven Months in Sierra Leone’, Cell, 161: 1516–26.doi: 10.1016/j.cell.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa A. et al. (2020) ‘Genomic Epidemiology of Superspreading Events in Austria Reveals Mutational Dynamics and Transmission Properties of SARS-CoV-2’, Science Translational Medicine, 12: eabe2555.doi: 10.1126/scitranslmed.abe2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2018) ‘Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7’, Systematic Biology, 67: 901–4.doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., and Holmes E. (2009) ‘The Early Molecular Epidemiology of the Swine-origin A/H1N1 Human Influenza Pandemic’, PLoS Currents, 1: RRN1003.doi: 10.1371/currents.rrn1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2020) ‘A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology’, Nature Microbiology, 5: 1403–7.doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— (2016) ‘Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen)’, Virus Evolution, 2: vew007.doi: 10.1093/ve/vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekizuka T. et al. (2020) ‘Haplotype Networks of SARS-CoV-2 Infections in the Diamond Princess Cruise Ship Outbreak’, Proceedings of the National Academy of Sciences of the United states of America, 117: 20198–201.doi: 10.1073/pnas.2006824117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y., and McCauley J. (2017) ‘GISAID: Global Initiative on Sharing All Influenza Data - from Vision to Reality’, Eurosurveillance, 22: 30494.doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard M. A. et al. (2018) ‘Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10’, Virus Evolution, 4: vey016.doi: 10.1093/ve/vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H. et al. (2021) ‘Detection of a SARS-CoV-2 Variant of Concern in South Africa’, Nature, 592: 438–43.doi: 10.1038/s41586-021-03402-9. [DOI] [PubMed] [Google Scholar]
- Vasylyeva T. I. et al. (2016) ‘Integrating Molecular Epidemiology and Social Network Analysis to Study Infectious Diseases: Towards a Socio-molecular Era for Public Health’, Infection, Genetics and Evolution, 46: 248–55.doi: 10.1016/j.meegid.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villabona-Arenas C. J., Hanage W. P., and Tully D. C. (2020) ‘Phylogenetic Interpretation during Outbreaks Requires Caution’, Nature Microbiology, 5: 876–7.doi: 10.1038/s41564-020-0738-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz E. et al. (2021) ‘Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 In England’, Nature, 593: 266–9.doi: 10.1038/s41586-021-03470-x. [DOI] [PubMed] [Google Scholar]
- Volz E. M., and Frost S. D. W. (2017) ‘Scalable Relaxed Clock Phylogenetic Dating’, Virus Evolution, 3: vex025.doi: 10.1093/ve/vex025. [DOI] [Google Scholar]
- Worobey M. et al. (2020) ‘The Emergence of SARS-CoV-2 in Europe and North America’, Science, 370: 564–70.doi: 10.1126/science.abc8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F. et al. (2020) ‘A New Coronavirus Associated with Human Respiratory Disease in China’, Nature, 579: 265–9.doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The newly generated sequences have been publicly shared through the GISAID platform. The data sets (including Ministry of Health and National Institute of Statistics and Census data) and code used to generate the analyses presented in this study, as well as a list of accession numbers for the Ecuador SARS-CoV-2 genome sequences analysed here are available through GitHub at https://github.com/BernardoGG/SARS-CoV-2_Genomic_lineages_Ecuador. We thank Andrés N. Robalino and Carlos Oporto for their contribution to the open licence Ecuacovid repository (https://github.com/andrab/ecuacovid) used to extract epidemiological data for this study.