

Abstract
Highly efficient and straightforward synthetic routes toward the first total synthesis of 2-(p-hydroxybenzyl)-prodigiosins (2–5), isoheptylprodigiosin (6), and geometric isomers of tambjamine MYP1 ((E/Z)-7) have been developed. The crucial steps involved in these synthetic routes are the construction of methoxy-bipyrrole-carboxaldehydes (MBCs) and a 20-membered macrocyclic core and a regioselective demethylation of MBC analogues. These new synthetic routes enabled us to generate several natural prodiginines 24–27 in larger quantity. All of the synthesized natural products exhibited potent asexual blood-stage antiplasmodial activity at low nanomolar concentrations against a panel of Plasmodium falciparum parasites, with a great therapeutic index. Notably, prodiginines 6 and 24–27 provided curative in vivo efficacy against erythrocytic Plasmodium yoelii at 25 mg/kg × 4 days via oral route in a murine model. No overt clinical toxicity or behavioral change was observed in any mice treated with prodiginines and tambjamines.
Keywords: Antimalarials, natural products, prodiginines, prodigiosins, tambjamines, total synthesis
Graphical Abstract
INTRODUCTION
Natural products have been the most successful source of potential drug candidates to combat many infectious diseases.1–3 Many of the best antimalarials known to date have been derived from various natural products, including quinine4–5 and artemisinin,6 isolated from Cinchona species and Artemisia annua, respectively. Prodiginines or prodigiosins (PGs) and tambjamines (TAs) belong to a family of intriguing pyrrolylpyrromethane (PPM) alkaloid antibiotics isolated from various bacterial, marine, and terrestrial sources.7–13 4-Methoxy-2,2’-bipyrrole-5-carboxaldehyde (MBC, 1, Figure 1) is the common precursor in the biosynthetic pathway of both PGs and TAs.8, 14–15 The key structural difference between PGs and TAs is that TAs constitute an alkylamine moiety in the place of alkyl-pyrrole (ring-C) of PGs (Figure 1). Recently, there has been an increase of interest in these natural and synthetic products because they appear to have prominent therapeutic applications signified by their antimicrobial,16–18 anticancer,19–21 antitumor,16, 18, 22 antimalarial,23–26 and immunosuppressive27 activities with various modes of action.7, 28–30 Specifically, a synthetic PG analogue, obatoclax-3 (GX15-070; Figure 1), has completed phase-II clinical trials for the treatment of small cell lung cancer and is engaged in multiple clinical trials for the treatment of other cancer conditions.31–33
Figure 1.
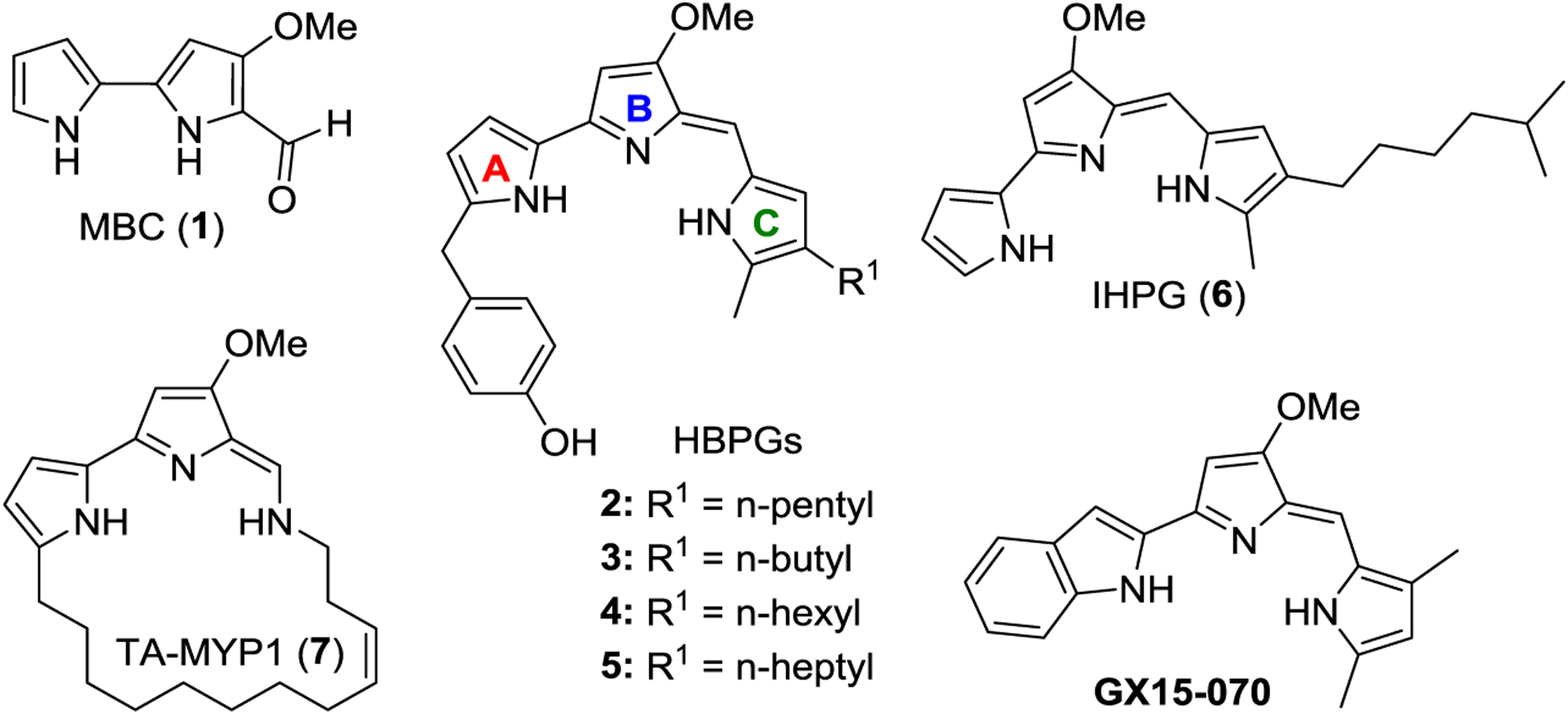
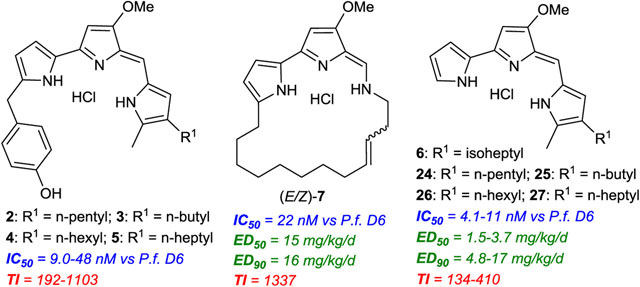
Naturally occurring prodiginines 2–6, tambjamine MYP1 (7), and their common biosynthetic precursor MBC (1) and synthetic prodiginine analogue GX15-070.
In 2008, Hemscheidt and co-workers reported the isolation of a novel 2-(p-hydroxybenzyl)-prodigiosin (HBPG, 2, Figure 1) from marine bacteria, Pseudoalteromonas rubra.34 HBPG 2 has been shown to have good antimicrobial and cytotoxicity activities. HBPG 2 was identified as the first naturally occurring PG containing a p-hydroxybenzyl substituent at the 2 position of ring-A. However, the gene product(s) responsible for the origin of p-hydroxybenzyl moiety on ring-A remains unknown. In addition, a liquid chromatography-mass spectrometry (LC-MS) analysis demonstrated the existence of several other HBPGs 3–5 (Figure 1), but the low quantity of these HBPGs precluded them from isolation and structural characterization. Recently, de Pascale et al. reported another new PG, isoheptylprodigiosin35 (IHPG, 6, Figure 1) from the marine Vibrio spartinae 3.6, with moderate antimicrobial activity. In 2019, Ross and the team reported a novel TA natural product, tambjamine MYP1 (TA-MYP1, 7, Figure 1) from Pseudoalteromonas citrea36 that was the first naturally occurring TA with a 20-membered macrocyclic ring. The supply of 2–7 from natural sources is rather limited. Therefore, the access to 2–7 and their analogues via the synthetic routes is highly desired for the investigation of extensive therapeutic applications, structure-activity relationships (SARs), and their mode(s) of action (MoA). To date, the total synthesis and further biological activities of 2–7 have not been reported.
As a part of an ongoing interest in discovery and development of new antiparasitic agents with novel MoA to overcome the emerging drug resistance, we have recently reported a large library of antimalarial PGs and TAs.24–26 A rigorous optimization process has produced several lead compounds with significantly improved oral efficacy and safety profiles. The promising biological activities and the intriguing structural features of these novel PGs and TAs merited for additional studies. Herein, we sought to design and execute the total synthesis and asexual blood-stage antimalarial evaluations of novel HBPGs 2–5, IHPG (6), geometric isomers of TA-MYP1 ((E/Z)-7), and several other natural PGs 24–27.
RESULTS AND DISCUSSION
Total Synthesis of 2-(p-Hydroxybenzyl)-prodigiosins (HBPGs, 2–5).
As shown in the retrosynthetic strategy (Scheme 1), HBPGs 2–5 could be constructed from two key precursors 8 and 9 by a condensation reaction. Compound 8 would be synthesized by Suzuki-cross coupling from pyrrole boronic acid pinacol ester 10 and bromo-pyrrole 11. Both intermediates 9 and 10 would be accessed from readily available pyrrole-carboxylates 14 and 12, respectively, via a sequence of Friedel-Crafts acylation, deoxygenative reduction, decarboxylation reactions, and an additional borylation toward 10. The synthesis of 11 has been well established in the literature starting from 4-methoxy-3-pyrrolin-2-one (13).37
Scheme 1.
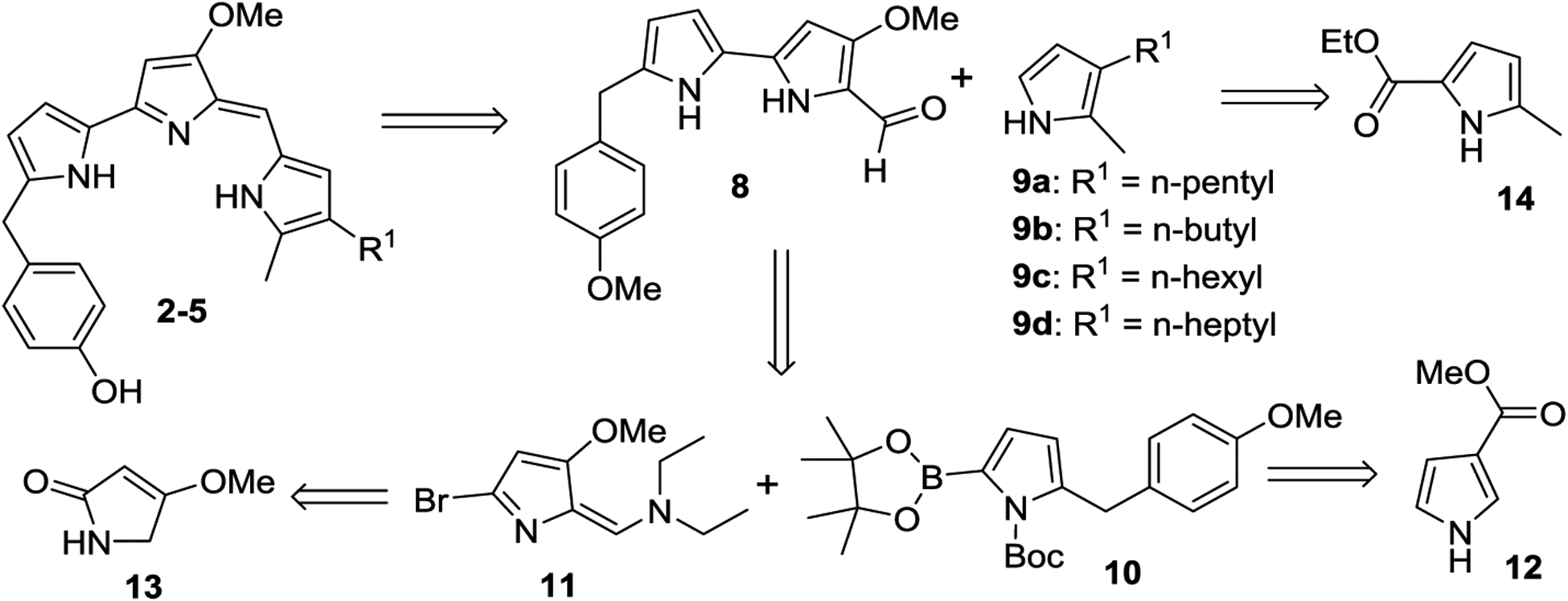
Retrosynthetic Strategy of HBPGs (2–5)
With the above retrosynthetic strategy (Scheme 1) in mind, our initial efforts focused on the construction of key MBC intermediate 8. Subjecting methyl-3-pyrrolecarboxylate (12) to Friedel-Crafts acylation24, 38 with 4-methoxybenzoyl chloride led to 5-acylated pyrrole 15, which when treated with NaBH4, in isopropyl alcohol (IPA) under reflux24, 39 furnished 5-benzylated-pyrrole 16 (Scheme 2). Interestingly, the spectral analysis of 16 demonstrated that the methyl ester on the pyrrole moiety was also converted to isopropyl ester, via a transesterification during the deoxygenative reduction with NaBH4/IPA. Upon treatment with NaOH in ethylene glycol under reflux, 16 delivered 2-(4-methoxybenzyl)-pyrrole (17) in a 77% yield.40 When compound 17 was treated with 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) in the presence of a catalytic amount of 2,2’-azobisisobutyronitrile (AIBN), followed by 4-(N,N-dimethylamino)pyridine (DMAP), triethylamine (Et3N), and di-tert-butyl dicarbonate (Boc2O), N-Boc-bromopyrrole 18 was obtained in excellent yield.40,41 Great care was required during the workup and purification of 18, which is unstable under solvent-free conditions (Experimental Section). Compound 18 was either dissolved in hexane and stored at 4 °C until further use or subjected to Pd-catalyzed borylation42–43 with pinacolborane to obtain the pyrrole boronic acid pinacol ester 10. Conversely, by use of minor modifications to the reported procedure,37 pyrrole-bromo-enamine 11 was prepared from 4-methoxy-3-pyrrolin-2-one (13) in good yield. A standard Suzuki cross-coupling reaction between compounds 10 and 11, and subsequent removal of the Boc group using LiOH in tetrahydrofuran (THF)/MeOH (1:1) led to the desired MBC intermediate 8 in a 73% yield (Scheme 2).40
Scheme 2.

Synthesis of the Key MBC Intermediate 8
Next, we focused on the construction of 2-methyl-3-alkyl-pyrroles 9a–d. By employing a similar strategy to the synthesis of 17, dialkyl pyrroles 9a–d were obtained in good yields from ethyl 5-methyl-pyrrole-2-carboxylate (14) and their corresponding acyl chlorides via the appropriate intermediates 19a–d and 20a–d (Scheme 3).
Scheme 3.

Synthesis of 2-Methy-3-alkyl-pyrroles (9a–d)
With substantial quantities of key synthons 8 and 9 in hand, an acid-catalyzed condensation24 smoothly provided 2-(p-methoxybenzyl)-prodigiosin (MBPG, 21) in an 84% yield (Scheme 4). We anticipated that the HBPG 2 could be obtained directly from 21 by a regioselective demethylation of OMe of the benzyl moiety. However, several demethylation conditions failed to provide the desired product 2, which resulted in the formation of several unwanted products. Then, our focus was directed on the demethylation of MBC intermediate 8 as the first step. To our delight, the treatment of 8 with BBr3 in DCM at −78 °C to room temperature led to the desired hydroxy-MBC intermediate 22 in a 91% yield. The structure of 22 was unambiguously established by the aid of 2D NMR data (supporting information), and the selected 1H→13C HMBC correlations are illustrated in Figure 2. Finally, an acid catalyzed condensation of 22 with an appropriate dialkyl- pyrrole 9 gave the corresponding HBPGs 2–5 in good yields (Scheme 4), and all of these final products were fully characterized with the aid of spectroscopic data. The spectral data of synthesized HBPG 2 are identical to those of the natural product 2.34
Scheme 4.

Synthesis of HBPGs (2–5) and MBPG (21)
Figure 2.

Selected 1H→13C HMBC correlations for 22
Total Synthesis of Isoheptylprodigiosin (IHPG, 6), Prodigiosin (24), 4”-Butylprodigiosin (25), 4”-Hexylprodigiosin (26) and 4”-Heptylprodigiosin (27).
With the success in synthesizing HBPGs 2–5 through the highly efficient synthetic route, we then focused on synthesizing IHPG (6). Consequently, Friedel-Crafts acylation, deoxygenative reduction, and decarboxylation reaction sequence provided 2-methyl-3-(isoheptyl)-pyrrole (23) in good yield from 14 and 5-methylhexanoic acid, via intermediates 19e and 20e (Scheme 5). Conversely, MBC (1) was obtained from 11 and N-Boc-pyrrole boronic acid, via Suzuki cross-coupling.37, 40 An acid-catalyzed condensation of 1 with 23 gave IHPG (6) in an 87% yield. The spectral data of synthesized and natural product 635 were compared and found to be identical in all respects. Adopting these newly developed synthetic routes, we synthesized several natural PGs, prodigiosin (24), 4”-butylprodigiosin (25), 4”-hexylprodigiosin (26), and 4”-heptylprodigiosin (27) in grams quantity for the first time (Scheme 5).
Scheme 5.

Synthesis of IHPG (6), Prodigiosin (24), 4”-Butylprodigiosin (25), 4”-Hexylprodigiosin (26), and 4”-Heptylprodigiosin (27).
Total Synthesis of Geometric Isomers of Tambjamine MYP1 ((E/Z)-7).
Next, our efforts shifted toward the total synthesis of geometric isomers of tambjamine MYP1 ((E/Z)-7). A similar retrosynthetic strategy to the construction of 2–5 was devised for the construction of (E/Z)-7. Using previously established methods, 1-(phenylsulfonyl)-2-pyrrolecarboxaldehyde (29) was prepared from pyrrole (28) in two steps.38, 44 Treatment of 29 with Grignard reagent 30, followed by one-pot deoxygenative reduction and deprotection25 of 31 with LiAlH4 in THF at 0 °C to reflux gave 2-alkyl-pyrrole 32 in a 71% yield (Scheme 6). Upon treating 32 with DBDMH, followed by Boc2O gave N-Boc-bromopyrrole 33 with an 86% yield. Since the intermediate 33 is highly unstable under solvent-free conditions, it was dissolved in hexane and stored at 4 °C until further use. However, subsequent Pd-catalyzed borylation of 33 with pinacolborane failed to deliver the desired product 34. Therefore, we took an alternative route that led to boronic acid ester 36. To achieve this, compound 32 was treated with Boc2O to give N-Boc-pyrrole 35, which was further treated with lithium diisopropylamide (LDA) and trimethyl borate resulting in 36 (Scheme 6). Great care was required during the workup of 36, which is highly unstable under solvent-free conditions. After completion, the reaction was quenched with NH4Cl solution, filtered through celite, and washed with THF. To avoid decomposition, the resulting filtrate containing 36 in THF was directly carried forward into the Suzuki coupling reaction with 11. However, the Suzuki coupling between 36 and 11 in THF/H2O (9:1) delivered 37 with only an ~10% yield. At this juncture, THF was swapped with 1,4-dioxane and carried forward into the Suzuki coupling with 11 followed by removal of the Boc group to provide 37 in an 81% yield. It is noteworthy that this newly developed synthetic strategy is highly efficient and feasible to generate multigram quantity of key MBC intermediate 37.
Scheme 6.

Synthesis of Key MBC Intermediate 37
With substantial quantity of key MBC precursor 37 readily available, we focused on the construction of the challenging complex macrocyclic ring. A standard aldehyde and amine coupling reaction was carried out between 37 and 38 to obtain Schiff base 39 (Scheme 7). However, several attempts to obtain 39 under a variety of acid catalysts/conditions were unsuccessful. Gratifyingly, upon treatment of 37 with 38 in the presence of anhydrous Na2SO4 in a sealed tube provided 39 in a 96% yield (Scheme 7). Attempts of ring-closure metathesis (RCM) of 39 using various Grubbs’ catalysts resulted in the formation of undesired products (not isolated and characterized). Since the amine functional groups appeared to react with Grubbs’catalyst,45 we decided to protect the NH group as in 40. RCM of 40 by Hoveyda-Grubbs’ catalyst, followed by removal of Boc group led to (E/Z)-7 with a poor yield (13%). Our alternative route via intermediate 42 led to (E/Z)-7 with good yield. A standard cross-metathesis reaction between 37 and N-Boc-amine 41 gave 42 in a 67% yield. Unfortunately, we were unable to convert 42 to the desired final product (E/Z)-7 under standard acid catalysts, including trifluoroacetic acid (TFA) and MeOH.HCl. To our delight, deprotection of N-Boc followed by an intramolecular cyclization of 42 with oxalyl chloride46 in one-pot delivered (E/Z)-7 in a 54% yield (Scheme 7).
Scheme 7.

Synthesis of Geometric Isomers of TA-MYP1 ((E/Z)-7)
The 1H-NMR analysis demonstrated that the synthetic compound (E/Z)-7 consists of a mixture of two geometric isomers in a ~14:1 ratio (Supporting Information). The major isomer of THE synthetic compound showed the following 1H-NMR pattern at the C10-C11 site, 5.75 (dt, J = 15.2, 6.8 Hz, C11-H), and 5.35 (dt, J = 15.2, 7.0 Hz, C10-H), while the natural product36 (Z- or cis-isomer) showed 5.55 (dt, J = 10.9, 7.7 Hz, C11-H), and 5.35 (dt, J = 10.9, 7.2 Hz, C10-H) (Table S1, Supporting Information). Notably, the coupling constant between CH-10 and CH-11 of the major isomer of the synthetic compound is larger (J =15.2 Hz) than that of the natural product (J = 10.9 Hz), demonstrating that the geometry at the C10-C11 of the major isomer of the synthetic compound is E or trans. All of these findings suggested that the mixture of E/Z isomers of synthetic TA (E/Z)-7 likely originated from a thermodynamically controlled cross-metathesis reaction between two olefins 37 and 41, with preferential formation of the thermodynamically favorable E isomer as major product (Scheme 7).
In Vitro Asexual Blood-Stage Antiplasmodial Activity and Structure-Activity Relationship (SAR) Studies.
In vitro blood-stage antiplasmodial activity of all of the synthesized compounds 2–6, (E/Z)-7, 21, 24–27, and 39 was evaluated against a panel of Plasmodium falciparum parasites with different geographic and genetic backgrounds using an SYBR Green-based assay.26, 47–48 Standard antimalarials chloroquine (CQ) and atovaquone (ATV) were used as reference drugs. Remarkably, all of these natural products exhibited potent activities at low nanomolar concentrations against CQ-sensitive and multidrug-resistant (MDR) parasites and were more effective than CQ. The results are summarized in Table 1. Natural HBPGs 2–4 with p-hydroxybenzyl group at the 2 position of ring-A exhibited great antiplasmodial activity at very low nanomolar concentrations against all P. falciparum strains (IC50 = 9.0 ± 0.52 nM–16 ± 0.92 nM against D6, Table 1), whereas HBPG 5 with p-hydroxybenzyl group at the 2 position of ring-A and n-heptyl moiety at the 4 position of ring-C exhibited slightly reduced potency (IC50 = 48 ± 2.8 nM against D6, Table 1) as compared to the corresponding HBPGs 2–4 containing the short-chain alkyl (C4–C6) substitutions at the 4 position of ring-C. Interestingly, MBPG 21 with p-methoxybenzyl group at the 2 position on ring-A and n-pentyl moiety at the 4 position of ring-C showed superior potency to that of the corresponding natural product HBPG 2 and the reference drug CQ (21 IC50 = 2.4 ± 0.14 nM vs 2 IC50 =16 ± 0.92 nM, and CQ IC50 = 15 ± 0.86 nM against D6, Table 1). The in vitro antiplasmodial activity and SARs of these PGs 2–5 and 21 demonstrated that the nature of substitutions on benzyl moiety at the 2 position of ring-A and the length of alkyl chain at the 4 position of ring-C play a critical role in altering the potency. Conversely, natural PGs 6 and 24–27 lacking the substitutions on ring-A and 4,5-di-alkyl substitutions on ring-C, exhibited the greatest antiplasmodial potency (IC50 = 4.1 ± 0.24 nM–11 ± 0.63 nM against D6). Taken together, these SAR results highlighted the impact of the substitutions on ring-A for optimal antiplasmodial potency. Similarly, both TA compounds (E/Z)-7 and 39 also exhibited great potency with low IC50 values across the testing panel, however, open-chained TA analogue 39 with two terminal olefins on ring-A and right-hand side amine group, exhibited slightly higher potency (~2-fold) than macrocyclic TA (E/Z)-7 (39 IC50 = 8.9 ± 0.51 nM vs (E/Z)-7 IC50 = 22 ± 1.3 nM against D6), and these results are comparable to the other synthetic TAs.26 It is noteworthy that the modes of action (MoA) are unknown for both PGs and TAs and they are equally effective against P. falciparum pan-sensitive and all MDR strains across the entire test panel (Table 1), suggesting potential to discover a new drug target to combat malarial parasites.
Table 1.
In Vitro Blood-Stage Antiplasmodial Activity and Cytotoxicity of PGs and TAs
| Compound | in vitro antiplasmodial activity IC50 (nM)a vs P. falciparum | cytotoxicity IC50 (nM)a vs HepG2 | therapeutic index vs HepG2 IC50/D6 IC50 | |||
|---|---|---|---|---|---|---|
| D6 | Dd2 | 7G8 | Tm90-C2B | |||
| 2 | 16 ± 0.92 | 28 ± 1.6 | 27 ± 1.6 | 29 ± 1.7 | 7023 ± 406 | 439 |
| 3 | 12 ± 0.69 | 12 ± 0.71 | 16 ± 0.91 | 17 ± 0.98 | 8880 ± 513 | 740 |
| 4 | 9.0 ± 0.52 | 11 ± 0.63 | 13 ± 0.75 | 12 ± 0.69 | 9927 ± 573 | 1103 |
| 5 | 48 ± 2.8 | 54 ± 3.1 | 44 ± 2.5 | 40 ± 2.3 | 9235 ± 533 | 192 |
| 6 | 4.1 ± 0.24 | 2.1 ± 0.12 | 2.1 ± 0.13 | 2.6 ± 0.15 | 1487 ± 86 | 363 |
| (E/Z)-7 | 22 ± 1.3 | 16 ± 0.92 | ntb | 13 ± 0.75 | 29412 ± 1700 | 1337 |
| 21 | 2.4 ± 0.14 | 3.2 ± 0.18 | 5.6 ± 0.32 | 2.3 ± 0.13 | 2639 ± 152 | 1099 |
| 24 | 8.0 ± 0.46 | 7.3 ± 0.42 | 9.2 ± 0.53 | 8.8 ± 0.51 | 1072 ± 62 | 134 |
| 25 | 11 ± 0.63 | 5.6 ± 0.31 | 8.7 ± 0.50 | 8.5 ± 0.49 | 3317 ± 192 | 301 |
| 26 | 5.9 ± 0.34 | 3.4 ± 0.20 | 3.6 ± 0.20 | 4.6 ± 0.27 | 2419 ± 140 | 410 |
| 27 | 4.2 ± 0.24 | 4.5 ± 0.26 | 4.1 ± 0.24 | 2.5 ± 0.14 | 1346 ± 78 | 320 |
| 39 | 8.9 ± 0.51 | 6.5 ± 0.38 | nt | 2.6 ± 0.15 | 1603 ± 92 | 180 |
| CQ | 15 ± 0.86 | 163 ± 9.4 | 171 ± 10 | 208 ± 12 | 37577 ± 2170 | 2505 |
| ATV | 0.10 ± 0.01 | 0.10 ± 0.01 | 0.20 ± 0.02 | 8256 ± 477 | 8496 ± 491 | 84960 |
Data are expressed as the mean ± SEM (n = 3); ATV, atovaquone; CQ, chloroquine; D6, P. falciparum CQ-sensitive strain; Dd2, MDR P. falciparum strain with Old World genetic background; 7G8, MDR P. falciparum strain with New World genetic background; and Tm90-C2B, MDR P. falciparum clinical isolate, ATV-resistant;
nt, not tested.
In Vitro Cytotoxicity.
In vitro general cytotoxicity was tested using human hepatic HepG2 cells.26, 48–49 There was no apparent mammalian cell cytotoxicity with IC50 values for these PPM natural products above 1 μM (Table 1), indicating a favorable therapeutic index (TI > 134).
In Vivo Asexual Blood-Stage Antimalarial Efficacy in Rodent Malaria Model.
In vivo antimalarial blood-stage efficacy of all of the synthesized PGs 2–6, 21, and 24–27 and TAs (E/Z)-7 and 39 was determined using a modified Thompson 4 day suppression model against Plasmodium yoelii in mice by oral route, and the results are summarized in Table 2.26, 48, 50 Seven- to eight week old female CF1 mice (Charles River Laboratories) were randomly placed in groups of four (n = 4; mean body weight of each group was ~30 g) and infected intravenously (iv) with 2.5 × 104 P. yoelli parasitized erythrocytes from a donor animal. Chloroquine (CQ) was used as a reference drug, and PEG-400 was used as a vehicle control. Test compounds 2–6, (E/Z)-7, 21, 24–27, and 39 were dissolved in PEG-400 and administered by oral gavage on 4 sequential days following the day of inoculation. The in vivo data were expressed as ED50 and ED90 values and reflect the dose required (estimated from dose-response curves) for suppression of parasitemia by 50 and 90%, respectively, related to vehicle-only controls as assessed on day 5 of each study. The animals were considered cured if they survive 28 days after the infection without detectable bloodstream parasites. Although HBPGs 2–3 with p-hydroxybenzyl group at the 2 position of ring-A exhibited great potency (Table 1), they did not show efficacy in the animal model with 5 mg/kg × 4 days of treatment. However, HBPGs 3 and 4 partially reduced parasitemia by 6 and 9%, respectively, with 25 mg/kg × 4 days of treatment. Interestingly, HBPG 5 with p-hydroxybenzyl group at the 2 position of ring-A and n-heptyl moiety at the 4 position of ring-C reduced parasitemia by 25 and 37% with 5 mg/kg × 4 days and 25 mg/kg × 4 days of treatments, respectively. MBPG 21 with p-methoxybenzyl group at the 2 position of ring-A significantly reduced parasitemia by 60 and 100% with 5 mg/kg × 4 days and 25 mg/kg × 4 days of treatments, respectively. However, MBPG 21 was not curative out to day 28 with the 25 mg/kg × 4 days treatment. These in vivo efficacy results demonstrated that the length of the alkyl chain at the 4 position of ring-C and the nature of substitutions on benzyl moiety at the 2 position of ring-A have substantial impact on the oral efficacy of HBPGs and MBPG. It is noteworthy that the crystalline nature of HBPGs 2–5 could be a reason for low aqueous solubility and/or poor oral bioavailability, as indicated by the inferior oral efficacy in the animal model. Pharmacology and solubility studies are underway and likely will offer further insight. Conversely, PGs 6 and 24–27 lacking the substitutions on ring-A each reduced parasitemia by > 60% with 5 mg/kg × 4 days of treatment, and all of these compounds exhibited excellent efficacy with parasitemia reduction by 100% on day 5 with 25 mg/kg × 4 days of treatment (ED50 and ED90 values are shown in Table 2). Notably, PGs 6 and 24–27 were curative out to day 28 in this model at 25 mg/kg × 4 days with good bioavailability. Most significantly, natural PGs 6, and 24–27 lacking the substitutions on ring-A demonstrated superior blood-stage efficacy as compared to the corresponding HBPGs 2–5 and 21 containing substitutions at position 2 on ring-A. Collectively, the in vivo results and the SAR observation of these PGs demonstrated that the substitutions on ring-A are not required for the optimal in vivo antimalarial efficacy. On the other hand, macrocyclic tambjamine (E/Z)-7 showed excellent efficacy with 100% parasitemia reduction with 25 mg/kg × 4 days of treatment, but it was not curative out to day 28. TA compound 39 (precursor of (E/Z)-7) containing both terminal olefin pendants on ring-A and right-hand-side amine group, showed moderate efficacy (42% parasitemia reduction) with 25 mg/kg × 4 days of treatment. Surprisingly, both TA compounds (E/Z)-7 and 39 were ineffective with 5 mg/kg × 4 days of treatment. The in vivo efficacy results of TAs demonstrated that terminal olefin groups on the TA scaffold were detrimental to antimalarial efficacy. Comprehensive SAR optimizations of a library of novel macrocyclic TA analogues will be studied to produce lead candidates for full preclinical studies.
Table 2.
In Vivo Blood-Stage Antimalarial Oral Efficacy of PGs and TAs
| compound | in vivo %parasitemia reduction on day 5 at | ED50 mg/kg/d | ED90 mg/kg/d | |
|---|---|---|---|---|
| 5 mg/kg/4d | 25 mg/kg/4d | |||
| PEG-400 | 0 | 0 | - | - |
| 2 | 0 | 0 | - | - |
| 3 | 0 | 6 | - | - |
| 4 | 0 | 9 | - | - |
| 5 | 25 | 37 | - | - |
| 6a | 65 | 100 | 2.7 | 5.9 |
| (E/Z)-7 | 0 | 100 | 15 | 16 |
| 21 | 60 | 100 | 3.6 | 18 |
| 24a | 63 | 100 | 3.7 | 17 |
| 25a | 67 | 100 | 2.5 | 8.5 |
| 26a | 70 | 100 | 1.5 | 4.8 |
| 27a | 89 | 100 | 2.6 | 9.2 |
| 39 | 0 | 42 | - | - |
| CQ | 100 | 100 | 1.5 | 3.3 |
Curative at 25 mg/kg/day
In Vivo Observations.
All mice were observed daily for mortality/morbidity and clinical signs of toxicity. Animal body weight, grooming, posture, and locomotion were monitored throughout the study. Animals with observable parasitemia following the experiment were euthanized, and animals cleared of parasites from their bloodstream were observed daily with assessment of parasitemia performed weekly until day 28, at which point, we scored the animal(s) as cured of infection. Body weights of the mice were not significantly affected by these PGs and TAs when compared to the control group, and there were no behavioral changes or clinical toxicity observed in any of the mice treated with PGs and TAs.
CONCLUSIONS
In summary, we have accomplished the first total synthesis of 2-(p-hydroxybenzyl)-prodigiosins (2–5), isoheptylprodigiosin (6), and geometric isomers of tambjamine-MYP1 ((E/Z)-7), via highly efficient, straightforward, and inexpensive chemical procedures using easily available building blocks in response to the demand for low-cost production of novel antiparasitic agents. The key features of our new strategies toward the total synthesis of intricate PPM natural products include (1) a robust protocol to construct the key MBC intermediate 8, via a stable pinacolborane intermediate 10; (2) a short and scalable approach to generate a variety of 2,3-dialkyl pyrroles; (3) a highly efficient and regioselective demethylation strategy for the hydroxy-MBC intermediate 22; (4) a high-yielding approach for the construction of key MBC intermediate 37, via a highly unstable boronic acid 36; and (5) a consecutive olefin cross metathesis followed by an intramolecular cyclization for the construction of challenging macrocyclic core of 7. Significantly, our new synthetic routes paved a way toward the generation of extensive libraries of novel PG and TA natural products for investigation of widespread therapeutic applications. Moreover, our new synthetic strategies are robust, allowing late-stage derivatization and potential to be carried out on larger scales. All of these compounds exhibited great potency at low nanomolar concentrations and they are equally effective against P. falciparum pan-sensitive D6 and MDR strains without any cross-resistance. These antimalarials likely operate by novel MoA. Particularly, PGs 6 and 24–27 are orally curative of malarial infection in the animal model. Extensive reports from our team will follow with future investigations on a large library of these natural product analogues, including antimalarial activities against multiple life stages of Plasmodium parasites, structural optimization, pharmacology, solubility, safety, and MoA.
EXPERIMENTAL SECTION
General.
NMR spectra were recorded on Bruker AMX-400 and AMX-600 spectrometers at 400 and 600 MHz, respectively. NMR experiments were recorded in CDCl3 and DMSO-d6 at 25 °C. Chemical shifts are given in parts per million (ppm) downfield from internal standard Me4Si (TMS). High-resolution mass spectra (HRMS) (electrospray ionization (ESI)) were recorded on a vanquish UHPLC/HPLC system coupled with a high resolution (35000) Q Exactive Orbitrap mass spectrometer. Melting points were recorded on an OptiMelt melting point apparatus and uncorrected. Unless otherwise stated, all reagents and solvents were purchased from commercial supplies and used without further purification. Reactions that required anhydrous conditions were carried out under an atmosphere of argon. Chromatography was executed on a CombiFlash instrument using silica gel (230–400 mesh) as stationary phase and mixtures of ethyl acetate and hexanes as eluents. Analytical HPLC analysis was performed on a C18 column (4.6 mm × 250 mm) with a linear elution gradient ranging from CH3OH/CH3CN/H2O (40%/10%/50%) to CH3OH (100%) in 0.15% trifluoroacetic acid at a flow rate of 0.9 mL/min. A purity of >95% has been established for all final compounds.
Synthesis of methyl 5-(4-methoxybenzoyl)-1H-pyrrole-3-carboxylate (15).
To a stirred suspension of anhydrous AlCl3 (21.28 g, 160 mmol) in 150 mL of anhydrous ethylene dichloride (EDC) was added 4-methoxybenzoyl chloride (16.32 g, 96 mmol) under an argon atmosphere at room temperature. After stirring for 15 min, a solution of methyl 1H-pyrrole-3-carboxylate (12) (10.0 g, 80 mmol) in EDC (50 mL) was added dropwise, and the reaction mixture was stirred at room temperature for 2 h. Then, the reaction was quenched with ice water (300 mL), and the product was extracted with dichloromethane (DCM; 3 × 100 mL). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was washed with hexane to afford the pure product 15 (19.5 g, 94%) as a white solid. mp 166–168 °C; 1H NMR (CDCl3, 600 MHz) δ 10.21 (br s, 1H), 7.98 (m, 2H), 7.71 (dd, J = 3.2, 1.1 Hz, 1H), 7.32 (dd, J = 3.2, 1.1 Hz, 1H), 7.02 (d, J = 8.8 Hz, 2H), 3.91 (s, 3H), 3.86 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ 183.8, 164.5, 163.3, 131.5, 131.4 (2C), 129.9, 128.3, 118.5, 118.2, 113.9 (2C), 55.5, 51.4; HRMS (ESI) calcd for C14H14NO4 (M + H)+ 260.0917, found 260.0912.
Synthesis of isopropyl 5-(4-methoxybenzyl)-1H-pyrrole-3-carboxylate (16).
To a stirred solution of 15 (15.0 g, 57.9 mmol) in 500 mL of anhydrous isopropyl alcohol (IPA) was added NaBH4 (15.3 g, 405 mmol) under an argon atmosphere at room temperature. The resulting suspension was heated at reflux for 18 h. The reaction mixture was cooled to room temperature and poured into 1000 mL of ice water and acidified with 2 N HCl while stirring, with the temperature maintained at below 10 °C. Then, the mixture was extracted with DCM (3 × 200 mL), and the combined organic extracts were washed with water and brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was chromatographed on silica gel, with ethyl acetate (10–20%)/hexanes (90–80%) as eluent, to afford the title compound 16 as a white amorphous solid (11.86 g, 75%). mp 73–75 °C; 1H NMR (CDCl3, 600 MHz) δ 8.50 (br s, 1H), 7.27 (dd, J = 2.7, 1.6 Hz, 1H), 7.12 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 6.38 (d, J = 0.9 Hz, 1H), 5.15 (m, 1H), 3.88 (s, 2H), 3.79 (s, 3H), 1.31 (d, J = 6.4 Hz, 6H); 13C NMR (CDCl3, 150 MHz) δ 164.9, 158.3, 132.3, 130.8, 129.7 (2C), 123.0, 116.9, 114.1 (2C), 107.3, 66.8, 55.3, 32.9, 22.1 (2C); HRMS (ESI) calcd for C16H20NO3 (M + H)+ 274.1438, found 274.1430.
Synthesis of 2-(4-methoxybenzyl)-1H-pyrrole (17).
Sodium hydroxide (3.22 g, 80.5 mmol) was added to a stirred solution of 16 (11.0 g, 40.3 mmol) in anhydrous ethylene glycol (100 mL) under an argon atmosphere at room temperature. The reaction mixture was heated to reflux and stirred at 160 °C for an hour. After cooling to room temperature, the reaction mixture was poured into water (300 mL), and the product was extracted with ethyl acetate (3 × 100 mL). The combined organic phases were dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was further chromatographed on silica gel, with ethyl acetate (5–10%)/hexanes (95–90%) as eluent, to afford the pure product 17 as a clear oil (5.8 g, 77%). 1H NMR (CDCl3, 600 MHz) δ 7.82 (br s, 1H), 7.16 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 6.68 (m, 1H), 6.17 (m, 1H), 6.00 (s, 1H), 3.95 (s, 2H), 3.82 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ 158.3, 131.5, 131.2, 129.7 (2C), 116.9, 114.0 (2C), 108.4, 106.2, 55.3, 33.2; HRMS (ESI) calcd for C12H14NO (M + H)+ 188.1070, found 188.1066.
Synthesis of tert-butyl 2-bromo-5-(4-methoxybenzyl)-1H-pyrrole-1-carboxylate (18).
To a stirred solution of 17 (5.0 g, 26.7 mmol) and 2,2’-azobisisobutyronitrile (AIBN, 0.22 g, 1.3 mmol) in THF (75 mL) was added portionwise 1,3-dibromo-5,5-dimethylhydatoin (DBDMH; 3.82 g, 13.3 mmol) over a period of 30 min under an argon atmosphere at −78 °C. After stirring for 2 h at −78 °C, triethylamine (1.35 g, 13.3 mmol), DMAP (0.33 g, 2.7 mmol) and Boc2O (5.83 g, 26.7 mmol) were added. The reaction mixture was stirred for 5 h while it was allowed to warm to room temperature, and then an additional amount of Boc2O (2.91 g, 13.3 mmol) was added and stirred for an additional hour at room temperature. The reaction mixture was quenched with water, and the product was extracted with hexane (3 × 100 mL). The combined organic layers were dried over anhydrous Na2SO4 and directly chromatographed on a short pad of silica gel, with ethyl acetate (5%)/hexanes (95%) as eluent, to afford the desired product 18 as a clear syrup (8.78 g, 90%). Since the compound 18 is unstable under solvent-free conditions, it was immediately dissolved in hexane and stored at 4 °C until further use. 1H NMR (DMSO-d6, 600 MHz) δ 6.98 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H), 6.28 (d, J = 3.5 Hz, 1H), 5.93 (d, J = 3.5 Hz, 1H), 4.02 (s, 2H), 3.70 (s, 3H), 1.41 (s, 9H); 13C NMR (DMSO-d6, 150 MHz) δ 158.2, 148.5, 136.3, 131.2, 129.7 (2C), 115.3, 114.2 (2C), 112.7, 99.7, 85.5, 55.5, 33.8, 27.6 (3C); HRMS (ESI) calcd for C17H20BrNaNO3 (M + Na)+ 388.0519, found 388.0510 and 390.0510 (+2, isotope).
Synthesis of tert-butyl 2-(4-methoxybenzyl)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrole-1-carboxylate (10).
To a stirred solution of 18 (6.0 g, 16.4 mmol) in 100 mL of anhydrous 1,4-dioxane were added [1,1’-bis(diphenylphosphino)ferrocene]-dichloropalladium(II).DCM complex (0.67 g, 0.82 mmol), triethylamine (8.3 g, 82.2 mmol) and pinacolborane (4.21 g, 32.6 mmol) under an argon atmosphere at room temperature. Then, the reaction mixture was stirred at 80 °C for 24 h. The cooled reaction mixture was filtered and the filtrate was diluted with ethyl acetate (300 mL) and washed with water, and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was further chromatographed on silica gel, with ethyl acetate (5–10%)/hexanes (95–90%) as eluent, to afford the pure product 10 as a clear syrup (4.41 g, 65%). 1H NMR (DMSO-d6, 400 MHz) δ 6.96 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.5 Hz, 2H), 6.47 (d, J = 3.2 Hz, 1H), 5.91 (d, J = 3.2 Hz, 1H), 4.04 (s, 2H), 3.69 (s, 3H), 1.38 (s, 9H), 1.24 (s, 12H); 13C NMR (CDCl3, 150 MHz) δ 157.9, 149.5, 135.0, 131.8, 129.8 (2C), 121.3, 113.7 (2C), 112.8, 109.9, 83.5, 83.1 (2C), 55.3, 34.2, 27.9 (3C), 24.6 (4C); HRMS (ESI) calcd for C23H32BNaNO5 (M + Na)+ 436.2270, found 436.2260.
Synthesis of N-((5-bromo-3-methoxy-2H-pyrrol-2-ylidene)methyl)-N-ethylethanamine (11).
To a stirred solution of diethylformamide (13.4 g, 132.7 mmol) in anhydrous DCM (50 mL) was added dropwise a solution of phosphorus oxybromide (POBr3; 31.6 g, 110.6 mmol) in DCM (50 mL) under an argon atmosphere at 0 °C. The resulting thick suspension was stirred at 0 °C for an hour to obtain the Vilsmeier complex as a white solid. Then, the solid was crushed with a spatula, and DCM (150 mL) was added, and the mixture was cooled to 0 °C. A solution of 4-methoxy-3-pyrroline-2-one (13, 5.0 g, 44.2 mmol) in DCM (50 mL) was added dropwise, and the mixture was warmed to room temperature and then heated at 50 °C for 6 h. The mixture was poured onto ice water, and the pH was adjusted to 8 by treating with a 5 N NaOH solution and then stirred for an additional 30 min. The organic layer was separated, and the aqueous layer was extracted with DCM (3 × 100 mL). The combined organic extracts were washed with water and brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was chromatographed on silica gel, with ethyl acetate (10–20%)/hexanes (90–80%) as eluent, to afford the title compound 11 as a tan solid (8.7 g, 76%). mp 37–39 °C; 1H NMR (CDCl3, 600 MHz) δ 7.01 (s, 1H), 5.61 (s, 1H), 4.14 (q, J = 7.4 Hz, 2H), 3.78 (s, 3H), 3.41 (q, J = 7.2 Hz, 2H), 1.31 (t, J = 7.4 Hz, 3H), 1.30 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 165.3, 138.6, 133.6, 120.7, 96.5, 58.0, 51.2, 44.6, 14.6, 12.5; HRMS (ESI) calcd for C10H16BrN2O (M + H)+ 259.0441, found 259.0434, and 261.0412 (+ 2, isotope).
Synthesis of 4-methoxy-5’-(4-methoxybenzyl)-1H,1’H-[2,2’-bipyrrole]-5-carbaldehyde (8).
To a degassed solution of 11 (1.50 g, 5.81 mmol) and 10 (3.60 g, 8.72 mmol) in 10% water/1,4-dioxane (100 mL) were added Pd(PPh3)4 (0.340 g, 0.29 mmol) and Na2CO3 (1.23 g, 11.62 mmol). The reaction mixture was stirred for 3 h at 100 °C and poured onto water (200 mL). The pH of the solution was lowered to pH 7 with 2 N HCl and extracted with ethyl acetate (3 × 100 mL). The combined organic extracts were washed with water and brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the obtained crude product was then dissolved in anhydrous THF (25 mL), followed by the dropwise addition of LiOH (1.39 g, 58.1 mmol) in anhydrous methanol (25 mL) under an argon atmosphere at room temperature. The resulting mixture was stirred for an additional 30 min at room temperature. On completion of the reaction, the solvent was evaporated under reduced pressure to obtain the crude product, which was next washed with water and the solid material was filtered. Upon washing with acetone (50 mL), the crude material gave the desired pure compound 8 as a pale yellow solid (1.31 g, 73%). mp > 200 °C; 1H NMR (DMSO-d6, 600 MHz) δ 11.27 (s, 1H), 10.99 (s, 1H), 9.26 (s, 1H), 7.15 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 6.62 (s, 1H), 6.22 (d, J = 2.4 Hz, 1H), 5.85 (s, 1H), 3.84 (s, 2H), 3.82 (s, 3H), 3.71 (s, 3H); 13C NMR (DMSO-d6, 150 MHz) δ 171.7, 159.2. 158.2, 134.7, 133.8, 132.3, 129.8 (2C), 123.0, 117.7, 114.2 (2C), 109.2, 108.3, 91.0, 58.2, 55.5, 33.0; HRMS (ESI) calcd for C18H19N2O3 (M + H)+ 311.1390, found 311.1381.
Synthesis of intermediates 19a-d.
Compounds 19a-d were synthesized and purified with excellent yields (92–95%) by the same procedure as described for 15 from ethyl 5-methyl-1H-pyrrole-2-carboxylate (14, 5.0 g, 32.6 mmol) and their corresponding acyl chlorides (39.2 mmol).
Ethyl 5-methyl-4-pentanoyl-1H-pyrrole-2-carboxylate (19a).
Yield: 7.36 g (95%) white amorphous solid; mp 122–124 °C; 1H NMR (CDCl3, 600 MHz) δ 9.52 (br s, 1H), 7.23 (d, J = 2.5 Hz, 1H), 4.36 (q, J = 7.2 Hz, 2H), 2.77 (t, J = 7.6 Hz, 2H), 2.61 (s, 3H), 1.69 (m, 2H), 1.43–1.37 (m, 2H), 1.39 (t, J = 7.2 Hz, 3H), 0.96 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 197.6, 161.4, 139.7, 122.2, 120.2, 116.8, 60.8, 40.0, 26.6, 22.6, 14.4, 14.1, 14.0; HRMS (ESI) calcd for C13H20NO3 (M + H)+ 238.1438, found 238.1430.
Ethyl 4-butyryl-5-methyl-1H-pyrrole-2-carboxylate (19b).
Yield: 6.70 g (92%) white amorphous solid; mp 129–131 °C; 1H NMR (CDCl3, 600 MHz) δ 10.09 (br s, 1H), 7.24 (d, J = 2.4 Hz, 1H), 4.36 (q, J = 7.1 Hz, 2H), 2.75 (t, J = 7.6 Hz, 2H), 2.61 (s, 3H), 1.73 (m, 2H), 1.39 (t, J = 7.1 Hz, 3H), 0.99 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 197.4, 161.4, 139.7, 122.2, 120.2, 116.8, 60.8, 42.2, 17.9, 14.4, 14.0, 13.9; HRMS (ESI) calcd for C12H18NO3 (M + H)+ 224.1281, found 224.1275.
Ethyl 4-hexanoyl-5-methyl-1H-pyrrole-2-carboxylate (19c).
Yield: 7.71 g (94%) white amorphous solid; mp 118–120 °C; 1H NMR (CDCl3, 600 MHz) δ 9.99 (br s, 1H), 7.23 (s, 1H), 4.36 (q, J = 7.1 Hz, 2H), 2.76 (t, J = 7.4 Hz, 2H), 2.61 (s, 3H), 1.70 (m, 2H), 1.39 (t, J = 7.1 Hz, 3H), 1.38 (m, 4H), 0.91 (t, J = 6.5 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 197.6, 161.3, 139.7, 122.2, 120.2, 116.8, 60.8, 40.2, 31.6, 24.2, 22.6, 14.4, 14.1, 14.0; HRMS (ESI) calcd for C14H22NO3 (M + H)+ 252.1594, found 252.1586.
Ethyl 4-heptanoyl-5-methyl-1H-pyrrole-2-carboxylate (19d).
Yield: 8.22 g (95%) white amorphous solid; mp 121–123 °C; 1H NMR (CDCl3, 600 MHz) δ 10.14 (br s, 1H), 7.23 (d, J = 2.4 Hz, 1H), 4.36 (q, J = 7.1 Hz, 2H), 2.76 (t, J = 7.5 Hz, 2H), 2.61 (s, 3H), 1.69 (m, 2H), 1.39 (t, J = 7.1 Hz, 3H), 1.37–1.30 (m, 6H), 0.89 (t, J = 6.7 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 197.6, 161.4, 139.8, 122.2, 120.2, 116.8, 60.8, 40.3, 31.7, 29.1, 24.5, 22.5, 14.4, 14.1, 14.0; HRMS (ESI) calcd for C15H24NO3 (M + H)+ 266.1751, found 266.1744.
Synthesis of intermediates 20a–d.
Compounds 20a–d were synthesized and purified with good yields (67–73%) by the same procedure as described for 16 from 19a–d.
Isopropyl 5-methyl-4-pentyl-1H-pyrrole-2-carboxylate (20a).
Yield: 4.97 g (71%) white amorphous solid; mp 70–72 °C; 1H NMR (CDCl3, 600 MHz) δ 8.78 (br s, 1H), 6.71 (d, J = 2.5 Hz, 1H), 5.18 (m, 1H), 2.37 (t, J = 7.7 Hz, 2H), 2.23 (s, 3H), 1.54 (m, 2H), 1.37–1.30 (m, 4H), 1.33 (d, J = 6.3 Hz, 6H), 0.91 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 160.9, 130.1, 122.8, 120.1, 115.5, 67.2, 31.6, 30.6, 25.7, 22.6, 22.1 (2C), 14.1, 11.3; HRMS (ESI) calcd for C14H24NO2 (M + H)+ 238.1802, found 238.1795.
Isopropyl 4-butyl-5-methyl-1H-pyrrole-2-carboxylate (20b).
Yield: 4.74 g (73%) white amorphous solid; mp 67–69 °C; 1H NMR (CDCl3, 600 MHz) δ 9.09 (br s, 1H), 6.71 (d, J = 1.8 Hz, 1H), 5.19 (s, 1H), 2.38 (t, J = 7.5 Hz, 2H), 2.23 (s, 3H), 1.53 (m, 2H), 1.37 (m, 2H), 1.33 (d, J = 6.3 Hz, 6H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 160.9, 130.1, 122.7, 120.1, 115.5, 67.2, 33.1, 25.4, 22.4, 22.1 (2C), 14.0, 11.3; HRMS (ESI) calcd for C13H21NaNO2 (M + Na)+ 246.1465, found 246.1458.
Isopropyl 4-hexyl-5-methyl-1H-pyrrole-2-carboxylate (20c).
Yield: 5.02 g (67%) white amorphous solid; mp 71–73 °C; 1H NMR (CDCl3, 600 MHz) δ 8.74 (br s, 1H), 6.71 (d, J = 2.5 Hz, 1H), 5.18 (s, 1H), 2.37 (t, J = 7.6 Hz, 2H), 2.23 (s, 3H), 1.53 (m, 2H), 1.32 (d, J = 6.2 Hz, 6H), 1.31 (m, 6H), 0.90 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 160.9, 130.1, 122.8, 120.1, 115.5, 67.2, 31.8, 30.9, 29.1, 25.7, 22.7, 22.1 (2C), 14.1, 11.3; HRMS (ESI) calcd for C15H25NaNO2 (M + Na)+ 274.1778, found 274.1767.
Isopropyl 4-heptyl-5-methyl-1H-pyrrole-2-carboxylate (20d).
Yield: 5.52 g (69%) white amorphous solid; mp 63–65 °C; 1H NMR (CDCl3, 600 MHz) δ 9.17 (br s, 1H), 6.72 (d, J = 2.5 Hz, 1H), 5.19 (s, 1H), 2.37 (t, J = 7.5 Hz, 2H), 2.23 (s, 3H), 1.54 (m, 2H), 1.34 (d, J = 6.3 Hz, 6H), 1.36–1.29 (m, 8H), 0.90 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 161.0, 130.2, 122.8, 120.1, 115.5, 67.2, 31.9, 30.9, 29.3, 29.2, 25.7, 22.7, 22.1 (2C), 14.1, 11.3; HRMS (ESI) calcd for C16H28NO2 (M + H)+ 266.2115, found 266.2107.
Synthesis of intermediates 9a–d.
Compounds 9a–d were synthesized and purified in good yields (75–82%) by the same procedure as described for 17 from 20a–d.
2-Methyl-3-pentyl-1H-pyrrole (9a).
Yield: 2.26 g (79%) clear oil; 1H NMR (CDCl3, 600 MHz) δ 7.73 (br s, 1H), 6.62 (t, J = 2.7 Hz, 1H), 6.04 (t, J = 2.7 Hz, 1H), 2.41 (t, J = 7.6 Hz, 2H), 2.21 (s, 3H), 1.56 (m, 2H), 1.39–1.34 (m, 4H), 0.93 (t, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 123.2, 119.8, 114.8, 108.9, 31.8, 31.1, 25.9, 22.7, 14.2, 11.0; HRMS (ESI) calcd for C10H18N (M + H)+ 152.1434, found 152.1431.
3-Butyl-2-methyl-1H-pyrrole (9b).
Yield: 2.28 g (82%) clear oil; 1H NMR (CDCl3, 600 MHz) δ 7.72 (br s, 1H), 6.63 (t, J = 2.6 Hz, 1H), 6.06 (t, J = 2.6 Hz, 1H), 2.44 (t, J = 7.7 Hz, 2H), 2.23 (s, 3H), 1.57 (m, 2H), 1.42 (m, 2H), 0.98 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 123.2, 119.7, 114.8, 108.9, 33.6, 25.6, 22.6, 14.1, 11.0; HRMS (ESI) calcd for C9H16N (M + H)+ 138.1277, found 138.1275.
3-Hexyl-2-methyl-1H-pyrrole (9c).
Yield: 2.55 g (77%) clear oil; 1H NMR (CDCl3, 600 MHz) δ 7.72 (br s, 1H), 6.62 (t, J = 2.7 Hz, 1H), 6.05 (t, J = 2.7 Hz, 1H), 2.41 (t, J = 7.8 Hz, 2H), 2.21 (s, 3H), 1.56 (m, 2H), 1.40–1.31 (m, 6H), 0.92 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 123.2, 119.8, 114.8, 108.9, 31.9, 31.4, 29.3, 25.9, 22.7, 14.2, 11.0; HRMS (ESI) calcd for C11H20N (M + H)+ 166.1590, found 166.1586.
3-Heptyl-2-methyl-1H-pyrrole (9d).
Yield: 2.55 g (75%) clear oil; 1H NMR (CDCl3, 600 MHz) δ 7.72 (br s, 1H), 6.62 (t, J = 2.5 Hz, 1H), 6.05 (t, J = 2.5 Hz, 1H), 2.42 (t, J = 7.6 Hz, 2H), 2.22 (s, 3H), 1.57 (m, 2H), 1.37–1.29 (m, 8H), 0.92 (t, J = 6.7 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 123.2, 119.8, 114.8, 108.9, 32.0, 31.4, 29.6, 29.3, 25.9, 22.7, 14.2, 11.0; HRMS (ESI) calcd for C12H22N (M + H)+ 180.1747, found 180.1742.
Synthesis of 4’-methoxy-5-(4-methoxybenzyl)-5’-((5-methyl-4-pentyl-1H-pyrrol-2-yl)methylene)-1H,5’H-2,2’-bipyrrole (21).
To a stirred solution of intermediates 8 (0.2 g, 0.64 mmol) and 9a (0.19 g, 1.29 mmol) in anhydrous methanol (50 mL) was added a few drops of methanolic HCl (3 N, catalytic amount) under an argon atmosphere at room temperature. The resulting bright red colored solution was stirred for 7 h at room temperature. On completion of the reaction, methanol was evaporated under reduced pressure and the obtained crude product was then chromatographed on silica gel, with ethyl acetate (30–40%)/hexanes (70–60%) as eluent, to afford the title compound 21 as a red solid (0.255 g, 84%). mp 130–132 °C; 1H NMR (CDCl3, 600 MHz) δ 12.66 (s, 1H), 12.60 (s, 1H), 12.50 (s, 1H), 7.30 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 6.86 (s, 1H), 6.80 (s, 1H), 6.62 (s, 1H), 6.02 (s, 1H), 5.99 (s, 1H), 4.04 (s, 2H), 3.96 (s, 3H), 3.78 (s, 3H), 2.54 (s, 3H), 2.39 (t, J = 7.3 Hz, 2H), 1.54 (m, 2H), 1.34 (m, 4H), 0.91 (t, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 165.6, 158.3, 147.6, 145.5, 143.1, 130.6, 129.9 (2C), 127.8, 127.4, 124.9, 121.3, 121.1, 118.5, 114.8, 114.1 (2C), 110.8, 92.5, 58.6, 55.2, 33.8, 31.4, 29.9, 25.3, 22.5, 14.1, 12.3; HRMS (ESI) calcd for C28H34N3O2 (M + H)+ 444.2646, found 444.2641.
Synthesis of 5’-(4-hydroxybenzyl)-4-methoxy-1H,1’H-[2,2’-bipyrrole]-5-carbaldehyde (22).
To a stirred suspension of 8 (1.0 g, 3.2 mmol) in anhydrous DCM (250 mL) was added dropwise BBr3 (1.0 M in DCM; 6.45 mL, 6.45 mmol) under an argon atmosphere at −78 °C. The resulting suspension was stirred for 8 h while it was allowed to warm up to room temperature. The reaction mixture was quenched with water (50 mL), and the DCM was evaporated under reduced pressure. Then, the crude material was filtered by a sintered funnel and washed with water and DCM (50 mL) to afford the pure compound 22 as a pale green solid (0.87 g, 91%). mp > 200 °C; 1H NMR (DMSO-d6, 600 MHz) δ 11.28 (s, 1H), 10.98 (s, 1H), 9.25 (s, 1H), 7.07 (d, J = 8.3 Hz, 2H), 6.69 (d, J = 8.3 Hz, 2H), 6.62 (s, 1H), 6.22 (d, J = 2.4 Hz, 1H), 5.84 (s, 1H), 3.82 (s, 3H), 3.79 (s, 2H); 13C NMR (DMSO-d6, 150 MHz) δ 171.5, 159.3. 156.1, 135.0, 134.0, 130.4, 129.7 (2C), 122.9, 117.7, 115.6 (2C), 109.3, 108.3, 91.0, 58.2, 33.1; HRMS (ESI) calcd for C17H17N2O3 (M + H)+ 297.1234, found 297.1225.
Synthesis of HBPGs 2–5.
Compounds 2–5 were synthesized and purified in excellent yields (82–85%) by the same procedure as described for 21 from 22, and their corresponding di-alkyl pyrroles 9a–d.
4-((4’-Methoxy-5’-((5-methyl-4-pentyl-1H-pyrrol-2-yl)methylene)-1H,5’H-[2,2’-bipyrrol]-5-yl)methyl)phenol (2).
Yield: 0.130 g (83%) red solid; mp 146–148 °C; 1H NMR (CDCl3, 400 MHz) δ 12.63 (br s, 1H), 12.58 (br s, 1H), 12.50 (br s, 1H), 7.24 (d, J = 8.3 Hz, 2H), 6.90 (s, 1H), 6.84 (s, 1H), 6.80 (d, J = 8.3 Hz, 2H), 6.65 (s, 1H), 6.05 (s, 1H), 6.03 (s, 1H), 4.95 (br s, 1H), 4.04 (s, 2H), 4.00 (s, 3H), 2.55 (s, 3H), 2.40 (t, J = 7.4 Hz, 2H), 1.55 (m, 2H), 1.34 (m, 4H), 0.92 (t, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 165.7, 154.6, 147.7, 145.7, 143.3, 130.5, 130.2 (2C), 128.0, 127.6, 125.0, 121.4, 121.1, 118.7, 115.7 (2C), 114.9, 111.0, 92.7, 58.8, 33.9, 31.6, 30.0, 25.5, 22.6, 14.2, 12.4; HRMS (ESI) calcd for C27H32N3O2 (M + H)+ 430.2489, found 430.2478.
4-((5’-((4-Butyl-5-methyl-1H-pyrrol-2-yl)methylene)-4’-methoxy-1H,5’H-[2,2’-bipyrrol]-5-yl)methyl)phenol (3).
Yield: 0.129 g (85%) red solid; mp > 200 °C; 1H NMR (CDCl3, 600 MHz) δ 12.67 (s, 1H), 12.62 (s, 1H), 12.55 (s, 1H), 7.25 (d, J = 8.4 Hz, 2H), 6.90 (s, 1H), 6.85 (t, J = 2.6 Hz, 1H), 6.80 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 2.0 Hz, 1H), 6.05 (t, J = 2.3 Hz, 1H), 6.03 (t, J = 1.7 Hz, 1H), 4.74 (br s, 1H), 4.05 (s, 2H), 4.00 (s, 3H), 2.55 (s, 3H), 2.41 (t, J = 7.6 Hz, 2H), 1.54 (m, 2H), 1.40–1.34 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 165.6, 154.5, 147.6, 145.6, 143.1, 130.1 (2C), 130.0, 127.9, 127.5, 124.9, 121.3, 121.0, 118.5, 115.5 (2C), 114.8, 110.8, 92.5, 58.6, 33.8, 32.4, 25.1, 22.3, 14.0, 12.3; HRMS (ESI) calcd for C26H30N3O2 (M + H)+ 416.2333, found 416.2320.
4-((5’-((4-Hexyl-5-methyl-1H-pyrrol-2-yl)methylene)-4’-methoxy-1H,5’H-[2,2’-bipyrrol]-5-yl)methyl)phenol (4).
Yield: 0.133 g (82%) red solid; mp 142–144 °C; 1H NMR (CDCl3, 600 MHz) δ 12.63 (s, 1H), 12.58 (s, 1H), 12.49 (s, 1H), 7.23 (d, J = 8.2 Hz, 2H), 6.89 (s, 1H), 6.84 (t, J = 3.1 Hz, 1H), 6.80 (d, J = 8.2 Hz, 2H), 6.65 (s, 1H), 6.05 (s, 1H), 6.02 (s, 1H), 4.99 (br s, 1H), 4.04 (s, 2H), 3.99 (s, 3H), 2.53 (s, 3H), 2.40 (t, J = 7.2 Hz, 2H), 1.54 (m, 2H), 1.35–1.27 (m, 6H), 0.91 (t, J = 6.3 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 165.7, 154.7, 147.7, 145.6, 143.3, 130.4, 130.2 (2C), 128.0, 127.6, 125.0, 121.4, 121.1, 118.8, 115.7 (2C), 114.9, 111.0, 92.7, 58.8, 33.9, 31.8, 30.3, 29.1, 255, 22.8, 14.2, 12.4; HRMS (ESI) calcd for C28H34N3O2 (M + H)+ 444.2646 found 444.2637.
4-((5’-((4-Heptyl-5-methyl-1H-pyrrol-2-yl)methylene)-4’-methoxy-1H,5’H-[2,2’-bipyrrol]-5-yl)methyl)phenol (5).
Yield: 0.138 g (83%) red solid; mp 157–159 °C; 1H NMR (CDCl3, 600 MHz) δ 12.61 (s, 1H), 12.56 (s, 1H), 12.47 (s, 1H), 7.22 (d, J = 8.0 Hz, 2H), 6.89 (s, 1H), 6.83 (s, 1H), 6.81 (d, J = 8.0 Hz, 2H), 6.64 (s, 1H), 6.04 (s, 1H), 6.02 (s, 1H), 5.12 (br s, 1H), 4.03 (s, 2H), 3.99 (s, 3H), 2.54 (s, 3H), 2.40 (t, J = 7.5 Hz, 2H), 1.54 (m, 2H), 1.33–1.29 (m, 8H), 0.90 (t, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 165.7, 154.6, 147.7, 145.7, 143.2, 130.5, 130.2 (2C), 128.0, 127.6, 125.0, 121.4, 121.1, 118.7, 115.7 (2C), 114.9, 111.0, 92.7, 58.8, 33.9, 32.0, 30.4, 29.4, 29.3, 25.5, 22.8, 14.2, 12.4; HRMS (ESI) calcd for C29H36N3O2 (M + H)+ 458.2802, found 458.2789.
Synthesis of ethyl 5-methyl-4-(5-methylhexanoyl)-1H-pyrrole-2-carboxylate (19e).
To a stirred solution of 5-methylhexanoic acid (5.1 g, 39.2 mmol) and DMF (few drops) in toluene (10 mL) was added SOCl2 (7.0 g, 58.8 mmol), and the mixture was heated at 80 °C for 5 h. After the solvents were removed in vacuo, the resulting clear oil was dissolved in EDC (25 mL) and transferred dropwise into a stirred suspension of 14 (3.0 g, 19.6 mmol) and AlCl3 (5.21 g, 39.2 mmol) in EDC (100 mL) under an argon atmosphere at room temperature. The reaction mixture was stirred at room temperature for 2 h, and then the reaction was quenched with ice water (100 mL), and the product was extracted with DCM (3 × 50 mL). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was then chromatographed on silica gel, with ethyl acetate (10–20%)/hexanes(90–80%) as eluent, to afford the title compound 19e as a white amorphous solid (4.26 g, 82%). mp 127–129 °C; 1H NMR (CDCl3, 600 MHz) δ 9.22 (br s, 1H), 7.23 (d, J = 2.5 Hz, 1H), 4.36 (q, J = 7.2 Hz, 2H), 2.75 (t, J = 7.6 Hz, 2H), 2.61 (s, 3H), 1.73–1.68 (m, 2H), 1.63–1.57 (m, 1H), 1.39 (t, J = 7.2 Hz, 2H), 1.28–1.21 (m, 3H), 0.91 (d, J = 6.6 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ 197.5, 161.3, 139.5, 122.2, 120.2, 116.7, 60.8, 40.5, 38,7, 27.9, 22.5 (2C), 22.3, 14.3, 14.1; HRMS (ESI) calcd for C15H24NO3 (M + H)+ 266.1751, found 266.1743.
Synthesis of isopropyl 5-methyl-4-(5-methylhexyl)-1H-pyrrole-2-carboxylate (20e).
Compound 20e (2.38 g, 70%, white amorphous solid) was synthesized and purified by the same procedure as described for 20a from 19e (3.4 g, 12.8 mmol). mp 47–49 °C; 1H NMR (CDCl3, 600 MHz) δ 8.70 (br s, 1H), 6.71 (d, J = 2.5 Hz, 1H), 5.18 (m, 1H), 2.37 (t, J = 7.5 Hz, 2H), 2.23 (s, 3H), 1.56–1.49 (m, 3H), 1.36–1.31 (m, 2H), 1.33 (d, J = 6.2 Hz, 6H), 1.20 (m, 2H), 0.88 (d, J = 6.6 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ 160.9, 130.0, 122.8, 120.1, 155.5, 67.2, 38.9, 31.2, 28.0, 27.2, 25.8, 22.7 (2C), 22.1 (2C), 11.4; HRMS (ESI) calcd for C16H28NO2 (M + H)+ 266.2115, found 266.2107.
Synthesis of 2-methyl-3-(5-methylhexyl)-1H-pyrrole (23).
Compound 23 (1.12 g, 72%, clear oil) was synthesized as described for 9a from 20e (2.3 g, 8.67 mmol), and carried forward into the next reaction without further purification. 1H NMR (CDCl3, 600 MHz) δ 7.73 (br s, 1H), 6.61 (s, 1H), 6.03 (s, 1H), 2.40 (t, J = 7.6 Hz, 2H), 2.20 (s, 3H), 1.57–1.50 (m, 3H), 1.38–1.33 (m, 2H), 1.23–1.20 (m, 2H), 0.88 (d, J = 6.6 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ 123.2, 119.8, 114.8, 108.8, 38.9, 31.7, 28.0, 27.4, 26.0, 22.7 (2C), 11.0; HRMS (ESI) calcd for C12H22N (M + H)+ 180.1747, found 180.1742.
Synthesis of 4-methoxy-1H,1’H-[2,2’-bipyrrole]-5-carbaldehyde (MBC, 1).
The MBC (1, 6.11 g, 83%) was synthesized and purified by the same procedure as described for 8 from 11 (10.0 g, 38.7 mmol) and N-Boc-2-pyrrole boronic acid (12.3 g, 58.1 mmol). mp > 200 °C; 1H NMR (DMSO-d6, 600 MHz) δ 11.41 (s, 1H), 11.23 (s, 1H), 9.31 (s, 1H), 6.90 (d, J = 1.2 Hz, 1H), 6.75 (br s, 1H), 6.27 (s, 1H), 6.12 (br s, 1H), 3.84 (s, 3H); 13C NMR (DMSO-d6, 150 MHz) δ 172.1, 159.1, 133.7, 123.8, 120.8, 117.8, 109.8, 108.0, 91.4, 58.2; HRMS (ESI) calcd for C10H11N2O2 (M + H)+ 191.0815, found 191.0810.
Synthesis of Isoheptylprodigiosin (IHPG, 6).
The IHPG (6, 0.443 g, 87%, red solid) was synthesized and purified in excellent yields by the same procedure as described for 21 from MBC (1, 0.25 g, 1.31 mmol) and 23 (0.473 g, 2.64 mmol). mp 118–120 °C; 1H NMR (CDCl3, 600 MHz) δ 12.75 (s, 2H), 12.61 (s, 1H), 7.25 (m, 1H), 6.99 (s, 1H), 6.94 (m, 1H), 6.70 (d, J = 2.4 Hz, 1H), 6.37 (m, 1H), 6.10 (d, J = 1.9 Hz, 1H), 4.02 (s, 3H), 2.56 (s, 3H), 2.41 (t, J = 7.5 Hz, 2H), 1.57–1.51 (m, 3H), 1.35–1.31 (m, 2H), 1.22 (m, 2H), 0.88 (d, J = 6.6 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ 165.7, 147.7, 146.9, 128.5, 128.4, 126.9, 125.1, 122.2, 120.7, 117.0, 116.0, 111.7, 92.8, 58.7, 38.8, 30.4, 28.0, 27.1, 25.4, 22.7 (2C), 12.5; HRMS (ESI) calcd for C22H30N3O (M + H)+ 352.2383, found 352.2373.
Synthesis of prodigiosin natural products 24–27.
Prodigiosins 24–27 were synthesized and purified in excellent yields (85–90%) by the same procedure as described for 21 from MBC (1, 0.7 g, 3.68 mmol) and their corresponding dialkyl pyrroles 9a-d (7.37 mmol).
4’-Methoxy-5’-((5-methyl-4-pentyl-1H-pyrrol-2-yl)methylene)-1H,5’H-2,2’-bipyrrole (24).
Yield: 1.15 g (87%) red solid; mp 151–153 °C; 1H NMR (CDCl3, 600 MHz) δ 12.74 (br s, 2H), 12.58 (br s, 1H), 7.24 (s, 1H), 6.96 (s, 1H), 6.93 (s, 1H), 6.70 (d, J = 2.2 Hz, 1H), 6.37 (m, 1H), 6.09 (d, J = 1.9 Hz, 1H), 4.02 (s, 3H), 2.56 (s, 3H), 2.41 (t, J = 7.6 Hz, 2H), 1.55 (m, 2H), 1.37–1.31 (m, 4H), 0.91 (t, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 165.7, 147.7, 146.9, 128.5, 128.4, 126.9, 125.1, 122.2, 120.7, 117.1, 116.0, 111.7, 92.8, 58.7, 31.4, 29.8, 25.3, 22.5, 14.1, 12.4; HRMS (ESI) calcd for C20H26N3O (M + H)+ 324.2070, found 324.2060.
5’-((4-Butyl-5-methyl-1H-pyrrol-2-yl)methylene)-4’-methoxy-1H,5’H-2,2’-bipyrrole (25).
Yield: 1.14 g (90%) red solid; mp 133–135 °C; 1H NMR (CDCl3, 600 MHz) δ 12.71 (br s, 2H), 12.57 (br s, 1H), 7.23 (s, 1H), 6.94 (s, 1H), 6.92 (s, 1H), 6.68 (d, J = 1.8 Hz, 1H), 6.35 (m, 1H), 6.08 (d, J = 1.6 Hz, 1H), 4.00 (s, 3H), 2.55 (s, 3H), 2.40 (t, J = 7.6 Hz, 2H), 1.53 (m, 2H), 1.36 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 165.8, 147.7, 146.9, 128.4 (2C), 126.9, 125.1, 122.3, 120.7, 117.0, 116.0, 111.7, 92.8, 58.7, 32.3, 25.1, 22.3, 13.9, 12.4; HRMS (ESI) calcd for C19H24N3O (M + H)+ 310.1914, found 310.1905.
5’-((4-Hexyl-5-methyl-1H-pyrrol-2-yl)methylene)-4’-methoxy-1H,5’H-2,2’-bipyrrole (26).
Yield: 1.17 g (85%) red solid; mp 135–137 °C; 1H NMR (CDCl3, 600 MHz) δ 12.74 (br s, 2H), 12.58 (br s, 1H), 7.24 (m, 1H), 6.96 (s, 1H), 6.93 (m, 1H), 6.69 (d, J = 2.3 Hz, 1H), 6.37 (m, 1H), 6.10 (d, J = 2.0 Hz, 1H), 4.02 (s, 3H), 2.56 (s, 3H), 2.41 (t, J = 7.6 Hz, 2H), 1.55 (m, 2H), 1.37–1.30 (m, 6H), 0.91 (t, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 165.8, 147.7, 146.9, 128.4, 128.3, 126.9, 125.1, 122.2, 120.7, 117.1, 116.0, 111.7, 92.8, 58.7, 31.7, 30.1, 28.9, 25.4, 22.6, 14.1, 12.5; HRMS (ESI) calcd for C21H28N3O (M + H)+ 338.2227, found 338.2216.
5’-((4-Heptyl-5-methyl-1H-pyrrol-2-yl)methylene)-4’-methoxy-1H,5’H-2,2’-bipyrrole (27).
Yield: 1.27 g (89%) red solid; mp 128–130 °C; 1H NMR (CDCl3, 600 MHz) δ 12.71 (br s, 1H), 12.70 (br s, 1H), 12.56 (br s, 1H), 7.23 (s, 1H), 6.94 (s, 1H), 6.90 (s, 1H), 6.68 (d, J = 1.9 Hz, 1H), 6.35 (m, 1H), 6.08 (d, J = 1.7 Hz, 1H), 4.00 (s, 3H), 2.55 (s, 3H), 2.39 (t, J = 7.6 Hz, 2H), 1.54 (m, 2H), 1.35–1.28 (m, 8H), 0.90 (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 165.8, 147.7, 146.9, 128.5, 128.4, 126.9, 125.1, 122.3, 120.7, 117.0, 116.0, 111.7, 92.8, 58.7, 31.8, 30.1, 29.2, 29.1, 25.4, 22.6, 14.1, 12.4; HRMS (ESI) calcd for C22H30N3O (M + H)+ 352.2383, found 352.2371.
Synthesis of 1-(phenylsulfonyl)-2-pyrrolecarboxaldehyde (29).
To a stirred suspension of pyrrole (28) (5.0 g, 74.6 mmol) and NaOH (8.95 g, 224 mmol) in 100 mL of EDC was added dropwise benzenesulfonyl chloride (15.8 g, 89.5 mmol) in 20 mL of EDC under an argon atmosphere at 0 °C. The resulting solution was allowed to react at ambient temperature overnight. The reaction was quenched by pouring the mixture into 200 mL of water. The organic layer was separated, and the aqueous layer was extracted with DCM (3 × 75 mL). The combined organic layers were washed with water and brine, and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the obtained crude material was washed with hot hexane to afford the pure N-phenylsulfonyl pyrrole (11.1 g), which was then dissolved in anhydrous EDC (150 mL) and added AlCl3 (14.3 g, 107.2 mmol). The reaction mixture was cooled to 0 °C, and 1,1-dichloromethyl methyl ether (9.25 g, 80.4 mmol) was added dropwise under an argon atmosphere. The resulting solution was stirred at 0 °C for 5 h and then poured into ice water (300 mL), and then the product was extracted with DCM (3 ×100 mL). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was then chromatographed on silica gel, with ethyl acetate (20–30%)/hexanes (80–70%) as eluent, to afford the title compound 29 as a white solid (10.5 g, 83%). mp 77–79 °C; 1H NMR (CDCl3, 600 MHz) δ 9.96 (s, 1H), 7.93 (d, J = 7.7 Hz, 2H), 7.67–7.64 (m, 2H), 7.56 (m, 2H), 7.18 (dd, J = 3.5, 1.3 Hz, 1H), 6.34 (t, J = 3.5 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ 178.8, 138.2, 134.6, 133.6, 129.6, 129.5 (2C), 127.5 (2C), 124.9, 112.6; HRMS (ESI) calcd for C11H10NO3S (M + H)+ 236.0376, found 236.0370.
Synthesis of 1-(1-(phenylsulfonyl)-1H-pyrrol-2-yl)dec-9-en-1-ol (31).
In a 500 mL Schlenk flask, small pieces of magnesium ribbon (2.04 g, 85.1 mmol) were heated with a heat gun under vacuum. The flask was allowed to cool to room temperature, and a small crystal of iodine (I2) and anhydrous THF (200 mL) was added. Then, 9-bromo-1-nonene (13.1 g, 63.8 mmol) was added to the reaction mixture and the resulting solution was stirred at 65 °C for 3 h. The reaction mixture (30) was cooled to room temperature, and cannulated into a stirred solution of 29 (10.0 g, 42.5 mmol) in THF (100 mL) under an argon atmosphere at 0 °C. The resulting solution was stirred at 0 °C for 2 h. Next, the reaction was quenched by the addition of 2 N HCl, with the temperature maintained below 10 °C, and then extracted with diethyl ether (3 × 100 mL). The combined organic layers were washed with water and brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was then chromatographed on silica gel, with ethyl acetate (10–20%)/hexanes (90–80%) as eluent, to afford the title compound 31 as a clear syrup (9.06 g, 59%). 1H NMR (CDCl3, 400 MHz) δ 7.80 (d, J = 7.6 Hz, 2H), 7.62 (t, J = 7.5 Hz, 1H), 7.52 (d, J = 7.6 Hz, 2H), 7.32 (dd, J = 2.7, 1.7 Hz, 1H), 6.31–6.27 (m, 2H), 5.82 (m, 1H), 5.01 (dd, J = 17.1, 1.6 Hz, 1H), 4.95 (dd, J = 10.2, 1.0 Hz, 1H), 4.82 (m, 1H), 2.74 (d, J = 4.0 Hz, 1H), 2.04 (q, J = 6.8 Hz, 2H), 1.81 (m, 2H), 1.38–1.24 (m, 10H); 13C NMR (CDCl3, 100 MHz) δ 139.3, 132.9, 138.4, 133.9, 129.5 (2C), 126.5 (2C), 123.5, 114.2, 112.4, 111.7, 65.2, 35.1, 33.8, 29.3, 29.2, 29.0, 28.9, 26.1; HRMS (ESI) calcd for C20H27NaNO3S (M + Na)+ 384.1604, found 384.1595.
Synthesis of 2-(dec-9-en-1-yl)-1H-pyrrole (32).
To a stirred suspension of LiAlH4 (6.63 g, 174.5 mmol) in anhydrous THF (300 mL) was added dropwise 31 (9.0 g, 24.9 mmol) in THF (50 mL) under an argon atmosphere at 0 °C. The reaction mixture was stirred for 3 h at the same temperature and then warmed to room temperature. Next, the resulting suspension was heated to reflux for 12 h. The reaction was quenched with the saturated solution of Na2SO4. The insoluble solid was filtered off and washed with DCM (200 mL). The filtrate was concentrated under reduced pressure to give the crude product, which was further chromatographed on silica gel, with ethyl acetate (5%)/hexanes (95%) as eluent, to afford 32 as a clear oil (3.63 g, 71%). 1H NMR (CDCl3, 600 MHz) δ 7.92 (br s, 1H), 6.69 (s, 1H), 6.17 (d, J = 2.6 Hz, 1H), 5.95 (s, 1H), 5.85 (m, 1H), 5.05 (d, J = 17.1 Hz, 1H), 4.98 (d, J = 10.1 Hz, 1H), 2.63 (t, J = 7.6 Hz, 2H), 2.08 (q, J = 6.8 Hz, 2H), 1.65 (m, 2H), 1.43–1.33 (m, 10H); 13C NMR (CDCl3, 150 MHz) δ 139.2, 132.9, 116.0, 114.2, 108.3, 104.9, 33.8, 29.7, 29.4 (3C), 29.1, 28.9, 27.7; HRMS (ESI) calcd for C14H24N (M + H)+ 206.1903, found 206.1899.
Synthesis of tert-butyl 2-bromo-5-(dec-9-en-1-yl)-1H-pyrrole-1-carboxylate (33).
Compound 33 (1.60 g, 86%, clear syrup) was synthesized and purified by the same procedure as described for 18 from 32 (1.0 g, 4.87 mmol). 1H NMR (CDCl3, 600 MHz) δ 6.24 (d, J = 3.5 Hz, 1H), 5.99 (d, J = 3.5 Hz, 1H), 5.78 (m, 1H), 4.98 (dd, J = 17.2, 1.6 Hz, 1H), 4.93 (dd, J = 10.1, 0.9 Hz, 1H), 2.67 (t, J = 7.5 Hz, 2H), 2.00 (q, J = 7.1 Hz, 2H), 1.57 (s, 9H), 1.30 (m, 2H), 1.25 (m, 10H); 13C NMR (DMSO-d6, 150 MHz) δ 148.7, 139.3, 137.7, 115.3, 115.1, 110.0, 98.9, 85.5, 33.6, 31.8, 29.1 (2C), 29.0, 28.9, 28.8, 28.7, 27.8 (3C); HRMS (ESI) calcd for C19H30BrNaNO2 (M + Na)+ 406.1352, found 406.1342, and 408.1321 (+2, isotope).
Synthesis of tert-butyl 2-(dec-9-en-1-yl)-1H-pyrrole-1-carboxylate (35).
To a stirred solution of 32 (5.0 g, 24.4 mmol) in anhydrous acetonitrile (100 mL) were added DMAP (0.30 g, 2.4 mmol) and Boc2O (7.97 g, 36.6 mmol) under an argon atmosphere at room temperature. The reaction mixture was stirred for 2 h at room temperature. DCM (200 mL) was added, and the solution was washed with water and brine and dried over anhydrous Na2SO4. Then, the solvent was evaporated under reduced pressure to give the crude product, which was further chromatographed on silica gel, with ethyl acetate (5%)/hexanes (95%) as eluent, to afford 35 as a clear oil (6.92 g, 93%). 1H NMR (CDCl3, 600 MHz) δ 7.20 (dd, J = 3.2, 1.8 Hz, 1H), 6.09 (t, J = 3.3 Hz, 1H), 5.96 (m, 1H), 5.83 (m, 1H), 5.02 (dd, J = 17.1, 1.5 Hz, 1H), 4.95 (dd, J = 10.2, 0.9 Hz, 1H), 2.84 (t, J = 7.7 Hz, 2H), 2.06 (q, J = 7.2 Hz, 2H), 1.65–1.59 (m, 2H), 1.61 (s, 9H), 1.39–1.32 (m, 10H); 13C NMR (CDCl3, 150 MHz) δ 149.6, 139.2, 136.6, 120.7, 114.1, 110.7, 109.8, 83.2, 33.8, 29.5, 29.4 (2C), 29.1, 28.9 (3C), 28.0 (3C); HRMS (ESI) calcd for C19H31NaNO2 (M + Na)+ 328.2247, found 328.2240.
Synthesis of tert-butyl 2-(dec-9-en-1-yl)-5-(dimethoxyboraneyl)-1H-pyrrole-1-carboxylate (36).
To a stirred solution of 35 (6.5 g, 21.3 mmol) in 100 mL of anhydrous THF was added dropwise lithium diisopropylamide (LDA, 2 M; 32.0 mL, 63.6 mmol) under an argon atmosphere at −78 °C. The reaction mixture was stirred for 2 h at −78 °C, then trimethyl borate (11.0 g, 106.5 mmol) was added, and the resulting solution was allowed to react at ambient temperature overnight. The reaction was quenched with saturated NH4Cl solution, filtered through celite, and washed with THF (50 mL). Note; decomposition of the product 36 was observed during the evaporation of solvent as it was highly unstable under solvent free conditions. Therefore, to avoid decomposition, THF was swapped with 1,4-dioxane (150 mL), and 36 was then carried forward into the Suzuki coupling reaction without further purification. HRMS (ESI) calcd for C21H36BNaNO4 (M + Na)+ 400.2630, found 400.2619.
Synthesis of 5’-(dec-9-en-1-yl)-4-methoxy-1H,1’H-[2,2’-bipyrrole]-5-carbaldehyde (37).
The MBC intermediate 37 was synthesized by the same procedure as described for 8 from 11, and 36. The pure compound 37 was obtained in an 81% yield (3.1 g, pale yellow solid) upon washing with hexane (200 mL). mp 143–145 °C; 1H NMR (DMSO-d6, 600 MHz) δ 11.23 (s, 1H), 10.90 (s, 1H), 9.25 (s, 1H), 6.60 (s, 1H), 6.21 (d, J = 2.5 Hz, 1H), 5.82 (t, J = 2.7 Hz, 1H), 5.77 (m, 1H), 4.98 (dd, J = 17.2, 1.9 Hz, 1H), 4.92 (dd, J = 10.2, 1.0 Hz, 1H), 3.82 (s, 3H), 2.53 (t, J = 7.6 Hz, 2H), 2.00 (q, J = 7.2 Hz, 2H), 1.58 (m, 2H), 1.33 (s, 2H), 1.29–1.26 (m, 8H); 13C NMR (DMSO-d6, 150 MHz) δ 171.5, 159.3, 139.3, 135.7, 134.0, 122.4, 117.6, 115.1, 109.2, 107.3, 90.8, 58.2, 33.6, 29.6, 29.3, 29.2, 29.1, 29.0, 28.7, 27.6; HRMS (ESI) calcd for C20H29N2O2 (M + H)+ 329.2224, found 329.2213.
Synthesis of N-((5-(dec-9-en-1-yl)-4’-methoxy-1H,5’H-[2,2’-bipyrrol]-5’-ylidene)methyl)but-3-en-1-amine (39).
MBC intermediate 37 (0.5 g, 1.52 mmol), but-3-en-1-amine (38, 0.43 g, 6.1 mmol), anhydrous Na2SO4 (50 g), and anhydrous DCM (100 mL) were placed in a sealed tube with a Teflon-lined cap under an argon atmosphere at room temperature. The reaction mixture was stirred and heated for 72 h at 60 °C. The mixture was filtered by a sintered funnel and washed with DCM (100 mL). The filtrate was concentrated under reduced pressure to give a crude residue, which was further chromatographed on silica gel, with ethyl acetate (40–60%)/hexanes (60–40%) as eluent, to afford the titled compound 39 in excellent yield (0.61 g, 96%) as a yellow amorphous solid. mp 91–93 °C; 1H NMR (CDCl3, 400 MHz) δ 13.53 (s, 1H), 10.54 (s, 1H), 9.18 (dd, J = 14.6, 7.1 Hz, 1H), 7.23 (d, J = 14.6 Hz, 1H), 6.67 (s, 1H), 6.00 (s, 1H), 5.88 (s, 1H), 5.82 (m, 2H), 5.20 (d, J = 17.1 Hz, 1H), 5.17 (d, J = 10.2 Hz, 1H), 4.99 (d, J = 17.1Hz, 1H), 4.93 (d, J = 10.2 Hz, 1H), 3.91 (s, 3H), 3.53 (q, J = 6.6 Hz, 2H), 2.70 (t, J = 7.6 Hz, 2H), 2.51 (q, J = 6.8 Hz, 2H), 2.03 (q, J = 6.9 Hz, 2H), 1.73 (m, 2H), 1.39–1.29 (m, 10H); 13C NMR (CDCl3, 100 MHz) δ 163.9, 142.9, 141.0, 139.4, 139.1, 133.4, 121.2, 118.9, 114.4, 114.2, 111.0, 108.7, 90.7, 58.5, 50.1, 34.6, 33.9, 29.5, 29.4 (2C), 29.2 (2C), 29.0, 28.1; HRMS (ESI) calcd for C24H36N3O (M + H)+ 382.2853, found 382.2842.
Synthesis of tert-butyl-but-3-en-1-yl((5-(dec-9-en-1-yl)-4’-methoxy-1H,5’H-[2,2’-bipyrrol]-5’-ylidene)methyl)carbamate (40).
Compound 40 (0.27 g, 94%, yellow amorphous solid) was synthesized by the same procedure as described for 35 from 39 (0.25 g, 0.61 mmol), and carried forward into the next reaction without further purification. mp 75–77 °C; 1H NMR (CDCl3, 400 MHz) δ 7.57 (br s, 1H), 6.52 (d, J = 3.4 Hz, 1H), 5.99 (d, J = 3.4 Hz, 1H), 5.95 (m, 1H), 5.84 (s, 1H), 5.82 (m, 1H), 5.21 (d, J = 17.2 Hz, 1H), 5.11 (d, J = 10.1 Hz, 1H), 5.01 (dd, J = 17.2, 1.5 Hz, 1H), 4.95 (dd, J = 10.1, 0.9 Hz, 1H), 4.51 (t, J = 7.3 Hz, 2H), 3.86 (s, 3H), 2.63 (t, J = 7.6 Hz, 2H), 2.53 (q, J = 6.8 Hz, 2H), 2.06 (q, J = 7.0 Hz, 2H), 1.66 (m, 2H), 1.57 (m, 9H), 1.39–1.28 (m, 10H); 13C NMR (CDCl3, 100 MHz) δ 169.7, 158.1, 153.1, 139.3, 136.6, 130.1, 127.9, 124.1, 117.3, 115.9, 114.3, 111.2, 107.7, 93.9, 82.9, 58.3, 45.6, 33.9, 32.7, 29.6, 29.5 (2C), 29.4, 29.2, 29.0, 28.2 (3C), 28.1; HRMS (ESI) calcd for C29H44N3O3 (M + H)+ 482.3377, found 482.3367.
Synthesis of tert-butyl 12-(5’-formyl-4’-methoxy-1H,1’H-[2,2’-bipyrrol]-5-yl)dodec-3-en-1-yl)carbamate (42).
Hoveyda-Grubbs 2nd generation catalyst (0.095 g, 0.152 mmol) was added to a stirred solution of 37 (0.250 g, 0.76 mmol) and tert-butyl but-3-en-1-ylcarbamate (41, 0.26 g, 1.52 mmol) in anhydrous DCM (100 mL) under an argon atmosphere at room temperature. The resulting mixture was stirred for 24 h at 50 °C. After completion of the reaction, the solvent was removed under reduced pressure to give the crude product, which was further chromatographed on silica gel, with ethyl acetate (20–30%)/hexanes (80–70%) as eluent, to afford 42 as a white amorphous solid (0.240 g, 67%). mp 114–116 °C; 1H NMR (DMSO-d6, 600 MHz) δ 11.23 (s, 1H), 10.90 (s, 1H), 9.25 (s, 1H), 6.71 (t, J = 5.2 Hz, 1H), 6.60 (s, 1H), 6.21 (d, J = 2.4 Hz, 1H), 5.82 (t, J = 2.5 Hz, 1H), 5.40 (m, 1H), 5.32 (m, 1H), 3.82 (s, 3H), 2.91 (t, J = 6.9 Hz, 2H), 2.53 (t, J = 7.6 Hz, 2H), 2.04 (q, J = 6.9 Hz, 2H), 1.93 (q, J = 6.1 Hz, 2H), 1.57 (m, 2H), 1.36 (s, 9H), 1.28 (m, 10H); 13C NMR (DMSO-d6, 150 MHz) δ 171.6, 159.3, 156.0, 135.7, 134.0, 132.1, 127.6, 122.4, 117.6, 109.2, 107.3, 90.8, 77.8, 58.2, 33.2, 32.5, 29.6, 29.5, 29.4, 29.3, 29.2, 29.1, 29.0, 28.7 (3C), 27.6; HRMS (ESI) calcd for C27H42N3O4 (M + H)+ 472.3170, found 472.3157.
Synthesis of geometric isomers of Tambjamine MYP1 ((E/Z)-7).
To a stirred solution of 42 (0.200 g, 0.42 mmol) in anhydrous methanol (50 mL) was added dropwise oxalyl chloride (180 uL, 2.13 mmol) under an argon atmosphere at room temperature. The resulting solution was stirred for 10 h at room temperature. On completion of the reaction, methanol was evaporated under reduced pressure and the obtained crude product was then extracted with DCM (3 × 50 mL). The combined organic phases were dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was further chromatographed on silica gel, with ethyl acetate (50–70%)/hexanes (50–30%) as eluent, to afford the pure product (E/Z)-7 in a ~14:1 ratio as a yellow amorphous solid (0.089 g, 54%). mp 92–94 °C; 1H NMR of major isomer (E) (CDCl3, 400 MHz) δ 13.66 (s, 1H), 10.92 (s, 1H), 9.11 (m, 1H), 7.20 (d, J = 14.9 Hz, 1H), 6.65 (t, J = 3.0 Hz, 1H), 5.97 (t, J = 3.0 Hz, 1H), 5.85 (d, J = 2.2 Hz, 1H), 5.74 (dt, J = 15.2, 6.8 Hz, 1H), 5.35 (dt, J = 15.2, 7.0 Hz, 1H), 3.91 (s, 3H), 3.53 (q, J = 5.8 Hz, 2H), 2.78 (t, J = 6.2 Hz, 2H), 2.39 (q, J = 6.3 Hz, 2H), 1.99 (q, J = 6.8 Hz, 2H), 1.75 (m, 2H), 1.40–1.11 (m, 10H); 13C NMR of major isomer (E) (CDCl3, 100 MHz) δ 163.7, 142.7, 140.5, 139.3, 137.5, 123.4, 120.9, 114.2, 111.4, 108.9, 90.5, 58.5, 49.2, 34.2, 32.6, 29.9, 29.7, 28.8, 28.3, 27.9, 27.5, 26.8; HRMS (ESI) calcd for C22H32N3O (M + H)+ 354.2540, found 354.2534.
Biological Experiments.
Culture Conditions.
P. falciparum strains D6, Dd2, 7G8, and TM90-C2B were cultured in human erythrocytes at 2% hematocrit in RPMI 1640 containing 0.5% Albumax II, 45 μg/mL hypoxanthine, and 5 μg/mL gentamicin.
In Vitro Antiplasmodial Activity.
In vitro antiplasmodial activity was determined by the Malaria SYBR Green I-based fluorescence (MSF) assay described previously47 with minor modifications. All synthesized PGs and TAs were evaluated for antiplasmodial activity with CQ and ATV as controls. Experiments were set up in triplicate in 96-well plates with a total of 100 μL total volume per well with 2% hematocrit, 0.2% parasitemia, and test compound dilutions in complete culture medium described above. Initial test compound dilutions were set between 2.5 and 2500 nM and subsequently adjusted to either a lower or higher range to achieve the most accurate IC50 (half- maximal inhibitory concentrations) values. The plates were incubated at 37 °C in low-oxygen conditions described previously for 72 h, at which point 100 μL of fluorescent dye-detergent mixture (0.2 μL SYBR Green I: 1.0 mL lysis buffer) was added and the plates were incubated in the dark at room temperature for an hour. After incubation with the fluorescent dye, a 96-well plate reader with an excitation wavelength set at 497 nm, and an emission at 520 nm was used to measure the fluorescence of each well. Fluorescence readings were plotted as a function of the test compound concentration, and the IC50 values for each test compound were calculated by nonlinear regression analysis (GraphPad Prism software) using the calculated test compound concentration that gave a 50% reduction in fluorescence compared to those of the drug-free controls.
In Vitro Cytotoxicity Assessment.
To identify toxicity to mammalian cells, an MTT assay using hepatic HepG2 cells was employed. Human hepatocarcinoma cells (HepG2) were cultured in complete Minimal Essential Medium (Gibco-Invitrogen, No. 11090-099) prepared by supplementing MEM with 0.19% sodium bicarbonate (Gibco-BRL no. 25080-094), 10% heat inactivated FBS (Gibco-Invitrogen No. 16000-036), 2 mM l-glutamine (Gibco-Invitrogen No. 25030-081), 0.1 mM MEM nonessential amino acids (Gibco-Invitrogen no. 11140-050), 0.009 mg/mL insulin (Sigma No. I1882), 1.76 mg/mL bovine serum albumin (Sigma no, A1470). Cells viability was assessed using trypan blue. Ninety-six-well plates were seeded with 2.5 × 104 cells in 170 μL of culture medium per well and incubated at 37 °C overnight in a humidified 5% CO2 atmosphere. Test compound plates were prepared with a Biomek 4000 automated laboratory station and 96-well plates containing 11 duplicate 1.6-fold serial dilutions of each test compound suspended in DMSO. A 30 μL sample of the diluted test compound was then added to 170 μL of medium per well. Cells were exposed to test compound concentrations ranging from 10 μg/mL to 0.15 μg/mL for 48 h. After incubation, 30 μL of a 1.5 mg/mL solution of MTT diluted in complete MEM media was added to each well, all plates were subsequently incubated in the dark for 1 h at room temperature. Next, the media and test compounds in each well were removed and plates were then left to dry in a hood for 15 min. 60 μL of acidified isopropyl alcohol was added to dissolve the formazan dye crystals created by the reduction of MTT. Plates were placed on a 3-D rotator for 15–30 min. Absorbance was determined with the Perkin Elmer EnSight plate reader. All experiments were run in triplicates and the IC50 values were determined using GraphPad Prism software using the nonlinear regression equation (sigmoidal dose-response, variable slope).
In Vivo Antimalarial Efficacy in a Rodent Malarial Model.
In vivo antimalarial efficacy was determined using a well-established modified Thompson 4 day suppression rodent model against P. yoelii. Seven to eight week old female CF1 mice (Charles River Laboratories) were randomly placed in groups of four (n = 4, mean body weight of each group was ~30 g) and infected intravenously with 2.5 × 104 P. yoelii (Kenya strain, MRA-428) parasitized erythrocytes from a donor animal. Test compounds’ administration commenced the day after the animals were inoculated (day 1). The test compounds were dissolved in PEG-400 and administered (5 and 25 mg/kg) by oral gavage once daily for 4 successive days; CQ phosphate was used as a positive control. Blood was collected via tail vein with the aid of a syringe needle on the day following the last dose and then at weekly intervals through day 28. Blood films were Giemsa-stained and examined microscopically to determine the levels of parasitemia. Animal body weight, grooming, posture and locomotion were monitored throughout the study. Animals with observable parasitemia following the experiment were euthanized; animals cleared of parasitemia from their bloodstream were observed daily with assessment of parasitemia performed weekly until day 28 at which point we score that the animal(s) are scored as cured of the infection and then euthanized. All treated mice with a negative smear on day 28 were considered cured (100% protection). ED50 and ED90 values (mg/kg/d) were derived graphically from the dose required to reduce parasite burden by 50 and 90%, respectively, relative to drug-free controls.
Animals and Ethic Statement at Portland VA Medical Center (PVAMC).
In vivo antimalarial testing was carried out at PVAMC, and the procedure was conducted under protocols approved by the PVAMC Institutional Animal Care and Use Committee (Protocol number 4475). Research was conducted in an AAALAC accredited facility in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adhered to principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 2011 edition.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases of the National Institute of Health under award number R01AI141972 and internal Portland State University funds. The National Science Foundation is acknowledged for support of the BioAnalytical Mass Spectrometry Facility at Portland State University (MRI1828573).
ABBREVIATIONS
- ATV
atovaquone
- AIBN
2,2’-azobisisobutyronitrile
- CQ
chloroquine
- DBDMH
1,3-dibromo-5,5-dimethylhydantoin
- DMAP
4-(N,N-dimethylamino)pyridine
- ED50
effective dose at 50% of the inhibition or reduction of the parasite load
- ED90
effective dose at 90% of the inhibition or reduction of the parasite load
- IC50
half-maximal inhibitory concentration
- HBPG
2-(p-hydroxybenzyl)-prodigiosin
- IHPG
isoheptylprodigiosin
- MBPG
2-(p-methoxybenzyl)-prodigiosin
- MBC
methoxy-bipyrrole-carboxaldehyde
- MoA
modes of action
- MDR
multiple drug-resistant
- PGs
prodiginines
- RCM
ring-closure metathesis
- SAR
structure-activity relationship
- TAs
tambjamines
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
1H, and 13C NMR spectra of all compounds, and HRMS and HPLC chromatograms of final compounds (PDF)
Molecular formula strings and in vitro data (CSV)
The authors declare no competing financial interest.
REFERENCES
- 1.Butler MS The role of natural product chemistry in drug discovery. J. Nat. Prod 2004, 67, 2141–2153. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ; Cragg GM Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod 2020, 83, 770–803. [DOI] [PubMed] [Google Scholar]
- 3.Butler MS Natural products to drugs: natural product derived compounds in clinical trials. Nat. Prod. Rep 2005, 22, 162–195. [DOI] [PubMed] [Google Scholar]
- 4.Singh SK; Singh S A brief history of quinoline as antimalarial agents. Int. J. Pharm. Sci. Rev. Res 2014, 25, 209–302. [Google Scholar]
- 5.Achan J; Talisuna AO; Erhart A; Yeka A; Tibenderana JK; Baliraine FN; Rosenthal PJ; D’Alessandro U Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria. Malaria J. 2011, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsu E The history of qing hao in the Chinese materia medica. Trans. R. Soc. Trop. Med. Hyg 2006, 100, 505–508. [DOI] [PubMed] [Google Scholar]
- 7.Fürstner A Chemistry and biology of roseophilin and the prodigiosin alkaloids: a survey of the last 2500 years. Angew. Chem. Int. Ed 2003, 42, 3582–3603. [DOI] [PubMed] [Google Scholar]
- 8.Hu DX; Withall DM; Challis GL; Thomson RJ Structure, chemical synthesis, and biosynthesis of prodiginine natural products. Chem. Rev 2016, 116, 7818–7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manderville RA Synthesis, proton-affinity and anti-cancer properties of the prodigiosin-group natural products. Curr. Med. Chem. Anticancer Agents 2001, 1, 195–218. [DOI] [PubMed] [Google Scholar]
- 10.Pinkerton DM; Banwell MG; Willis AC Total syntheses of tambjamines C, E, F, G, H, I and J, BE-18591, and a related alkaloid from the marine bacterium Pseudoalteromonas tunicata. Org. Lett 2007, 9, 5127–5130. [DOI] [PubMed] [Google Scholar]
- 11.Carté B; Faulkner DJ Defensive metabolites from three nembrothid nudibranchs. J. Org. Chem 1983, 48, 2314–2318. [Google Scholar]
- 12.Lindquist N; Fenical W New tambjamine class alkaloids from the marine ascidian atapozoa Sp and its nudibranch predators-origin of the tambjamines in atapozoa. Experientia 1991, 47, 504–506. [Google Scholar]
- 13.Franks A; Haywood P; Holmstrom C; Egan S; Kjelleberg S; Kumar N Isolation and structure elucidation of a novel yellow pigment from the marine bacterium Pseudoalteromonas tunicata. Molecules 2005, 10, 1286–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burke C; Thomas T; Egan S; Kjelleberg S The use of functional genomics for the identification of a gene cluster encoding for the biosynthesis of an antifungal tambjamine in the marine bacterium Pseudoalteromonas tunicata. Environ. Microbiol 2007, 9, 814–818. [DOI] [PubMed] [Google Scholar]
- 15.Lu W; Kancharla P; Reynolds KA MarH, a bifunctional enzyme involved in the condensation and hydroxylation steps of the marineosin biosynthetic pathway. Org. Lett 2017, 19, 1298–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boger DL; Patel M Total synthesis of prodigiosin, prodigiosene, and desmethoxy-prodigiosin: Diels-Alder reactions of heterocyclic azadienes and development of an effective palladium(II)-promoted 2,2’-bipyrrole coupling procedure. J. Org. Chem 1988, 53, 1405–1415. [Google Scholar]
- 17.Marchal E; Uddin MI; Smithen DA; Hawco CLA; Lanteigne M; Overy DP; Kerr RG; Thompson A Antimicrobial activity of non-natural prodigiosenes. RSC Advances 2013, 3, 22967–22971. [Google Scholar]
- 18.Kojiri K; Nakajima S; Suzuki H; Okura A; Suda H A new antitumor substance, BE-18591, produced by a streptomycete. I. Fermentation, isolation, physico-chemical and biological properties. J. Antibiot 1993, 46, 1799–1803. [DOI] [PubMed] [Google Scholar]
- 19.Regourd J; Ali AA; Thompson A Synthesis and anti-cancer activity of C-ring-functionalized prodigiosin analogues. J. Med. Chem 2007, 50, 1528–1536. [DOI] [PubMed] [Google Scholar]
- 20.Aldrich LN; Stoops SL; Crews BC; Marnett LJ; Lindsley CW Total synthesis and biological evaluation of tambjamine K and a library of unnatural analogs. Bioorg. Med. Chem. Lett 2010, 20, 5207–5211. [DOI] [PubMed] [Google Scholar]
- 21.Diaz de Grenu B; Iglesias Hernandez P; Espona M; Quinonero D; Light ME; Torroba T; Perez-Tomas R; Quesada R Synthetic prodiginine obatoclax (GX15-070) and related analogues: anion binding, transmembrane transport, and cytotoxicity properties. Chem. Eur. J 2011, 17, 14074–14083. [DOI] [PubMed] [Google Scholar]
- 22.Mukherjee PP; Goldschmidt ME; Williams RP Enzymic formation of prodigiosin analog by a cell-free preparation from Serratia marcescens. Biochim. Biophys. Acta 1967, 136, 182–184. [DOI] [PubMed] [Google Scholar]
- 23.Castro AJ Antimalarial activity of prodigiosin. Nature 1967, 213, 903–904. [DOI] [PubMed] [Google Scholar]
- 24.Papireddy K; Smilkstein M; Kelly JX; Shweta; Salem SM; Alhamadsheh M; Haynes SW; Challis GL; Reynolds KA Antimalarial activity of natural and synthetic prodiginines. J. Med. Chem 2011, 54, 5296–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kancharla P; Lu W; Salem SM; Kelly JX; Reynolds KA Stereospecific synthesis of 23-hydroxyundecylprodiginines and analogues and conversion to antimalarial premarineosins via a Rieske oxygenase catalyzed bicyclization. J. Org. Chem 2014, 79, 11674–11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kancharla P; Kelly JX; Reynolds KA Synthesis and structure-activity relationships of tambjamines and B-ring functionalized prodiginines as potent antimalarials. J. Med. Chem 2015, 58, 7286–7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Alessio R; Bargiotti A; Carlini O; Colotta F; Ferrari M; Gnocchi P; Isetta A; Mongelli N; Motta P; Rossi A; Rossi M; Tibolla M; Vanotti E Synthesis and immunosuppressive activity of novel prodigiosin derivatives. J. Med. Chem 2000, 43, 2557–2565. [DOI] [PubMed] [Google Scholar]
- 28.Rastogi S; Marchal E; Uddin I; Groves B; Colpitts J; McFarland SA; Davis JT; Thompson A Synthetic prodigiosenes and the influence of C-ring substitution on DNA cleavage, transmembrane chloride transport and basicity. Org. Biomol. Chem 2013, 11, 3834–3845. [DOI] [PubMed] [Google Scholar]
- 29.Melvin MS; Ferguson DC; Lindquist N; Manderville RA DNA binding by 4-methoxypyrrolic natural products. Preference for intercalation at AT sites by tambjamine E and prodigiosin. J. Org. Chem 1999, 64, 6861–6869. [DOI] [PubMed] [Google Scholar]
- 30.Cavalcanti BC; Junior HV; Seleghim MH; Berlinck RG; Cunha GM; Moraes MO; Pessoa C Cytotoxic and genotoxic effects of tambjamine D, an alkaloid isolated from the nudibranch Tambja eliora, on Chinese hamster lung fibroblasts. Chem. Biol. Interact 2008, 174, 155–162. [DOI] [PubMed] [Google Scholar]
- 31.Trudel S; Li ZH; Rauw J; Tiedemann RE; Wen XY; Stewart AK Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood 2007, 109, 5430–5438. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen M; Marcellus RC; Roulston A; Watson M; Serfass L; Murthy Madiraju SR; Goulet D; Viallet J; Belec L; Billot X; Acoca S; Purisima E; Wiegmans A; Cluse L; Johnstone RW; Beauparlant P; Shore GC Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 19512–19517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parikh SA; Kantarjian H; Schimmer A; Walsh W; Asatiani E; El-Shami K; Winton E; Verstovsek S Phase II study of obatoclax mesylate (GX15-070), a small-molecule BCL-2 family antagonist, for patients with myelofibrosis. Clin. Lymphoma Myeloma Leuk 2010, 10, 285–289. [DOI] [PubMed] [Google Scholar]
- 34.Feher D; Barlow RS; Lorenzo PS; Hemscheidt TK A 2-substituted prodiginine, 2-(p-hydroxybenzyl)prodigiosin, from Pseudoalteromonas rubra. J. Nat. Prod 2008, 71, 1970–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vitale GA; Sciarretta M; Palma Esposito F; January GG; Giaccio M; Bunk B; Sproer C; Bajerski F; Power D; Festa C; Monti MC; D’Auria MV; de Pascale D Genomics-metabolomics profiling disclosed marine Vibrio spartinae 3.6 as a producer of a new branched side chain prodigiosin. J. Nat. Prod 2020, 83, 1495–1504. [DOI] [PubMed] [Google Scholar]
- 36.Picott KJ; Deichert JA; deKemp EM; Schatte G; Sauriol F; Ross AC Isolation and characterization of tambjamine MYP1, a macrocyclic tambjamine analogue from marine bacterium Pseudoalteromonas citrea. Medchemcomm. 2019, 10, 478–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dairi K; Tripathy S; Attardo G; Lavallee JF Two-step synthesis of the bipyrrole precursor of prodigiosins. Tetrahedron Lett. 2006, 47, 2605–2606. [Google Scholar]
- 38.Kakushima M; Hamel P; Frenette R; Rokach J Regioselective synthesis of acylpyrroles. J. Org. Chem 1983, 48, 3214–3219. [Google Scholar]
- 39.Huggins MT; Lightner DA Intramolecular hydrogen bonding between remote termini. Tetrahedron 2001, 57, 2279–2287. [Google Scholar]
- 40.Kancharla P; Reynolds KA Synthesis of 2,2 ‘-bipyrrole-5-carboxaldehydes and their application in the synthesis of B-ring functionalized prodiginines and tambjamines. Tetrahedron 2013, 69, 8375–8385. [Google Scholar]
- 41.Chen W; Cava MP Convenient synthetic equivalents of 2-lithiopyrrole and 2,5-dilithiopyrrole. Tetrahedron Lett. 1987, 28, 6025–6026. [Google Scholar]
- 42.Murata M; Oyama T; Watanabe S; Masuda Y Palladium-catalyzed borylation of aryl halides or triflates with dialkoxyborane: A novel and facile synthetic route to arylboronates. J. Org. Chem 2000, 65, 164–168. [DOI] [PubMed] [Google Scholar]
- 43.Banwell MG; Goodwin TE; Ng S; Smith JA; Wong DJ Palladium-catalysed cross-coupling and related reactions involving pyrroles. Euro. J. Org. Chem 2006, 2006, 3043–3060. [Google Scholar]
- 44.Zelikin A; Shastri VR; Langer R Facile synthesis of 3-alkylpyrroles. J. Org. Chem 1999, 64, 3379–3380. [DOI] [PubMed] [Google Scholar]
- 45.Compain P Olerin metathesis of amine-containing systems: Beyond the current consensus. Adv. Synth. Catal 2007, 349, 1829–1846. [Google Scholar]
- 46.George N; Ofori S; Parkin S; Awuah SG Mild deprotection of the N-tert-butyloxycarbonyl (N-Boc) group using oxalyl chloride. RSC Adv. 2020, 10, 24017–24026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smilkstein M; Sriwilaijaroen N; Kelly JX; Wilairat P; Riscoe M Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents ChemotheR 2004, 48, 1803–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kancharla P; Dodean RA; Li Y; Pou S; Pybus B; Melendez V; Read L; Bane CE; Vesely B; Kreishman-Deitrick M; Black C; Li Q; Sciotti RJ; Olmeda R; Luong TL; Gaona H; Potter B; Sousa J; Marcsisin S; Caridha D; Xie L; Vuong C; Zeng Q; Zhang J; Zhang P; Lin H; Butler K; Roncal N; Gaynor-Ohnstad L; Leed SE; Nolan C; Ceja FG; Rasmussen SA; Tumwebaze PK; Rosenthal PJ; Mu J; Bayles BR; Cooper RA; Reynolds KA; Smilkstein MJ; Riscoe MK; Kelly JX Lead optimization of second-generation acridones as broad-spectrum antimalarials. J. Med. Chem 2020, 63, 6179–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferrari M; Fornasiero MC; Isetta AM MTT colorimetric assay for testing macrophage cytotoxic activity in vitro. J. Immunol. Methods 1990, 131, 165–172. [DOI] [PubMed] [Google Scholar]
- 50.Thompson PE; Bayles A; Olszewsk. B. PAM 1392 [2,4-diamino-6-(3,4-dichlorobenzylamino) quinazoline] as a chemotherapeutic agent: Plasmodium berghei, P. cynomolgi, P. knowlesi, and Trypanosoma cruzi. Exp. Parasitol 1969, 25, 32–49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.