

Abstract
Previous literature suggests that acute opioid use results in the functional impairment of the immune response, thereby decreasing resistance to viral infection. Here, we assessed if innate and adaptive immune responses are compromised ex vivo in persons who inject drugs (PWID) and whether long-term injection drug use may impact host susceptibility to in vitro HIV infection. We measured the frequency, activation state, and functional profile of NK cells, dendritic cells, CD4+ and CD8+ T cells in low-risk PWID who do not share needles, high-risk needle-sharing PWID, and control donors who did not inject drugs. We also assessed plasma levels of inflammatory markers and CD4+ T cell susceptibility to HIV infection. We observed a significant increase in the amount of sCD14 (p=0.0023, n=16) and sCD163 (p=0.0001, n=16) in the plasma of PWID compared to controls. Evidence of constitutive activation was noted in PWID as compared to controls with increased CD69 expression in CD56dim NK cells (p=0.0103, n=26) and increased CD38 and HLA-DR expression in CD4+ T cells (p=0.0355, n=23). However, no innate or adaptive functional differences were detected between PWID and controls including: NK cell direct or ADCC poly-functional response, TLR-stimulated dendritic cell/NK cross-talk, CD8+ T cell response to SEB or CMV/EBV/FLU peptides, or constitutive or anti-CD3/CD28 stimulated CD4+ T cell infectivity with CCR5-tropic or CXCR4-tropic HIV-1 isolates. Our data indicate that PWID who utilize opioids over as prolonged time frame can retain a functional ex vivo immune response without a measurable increase in CD4+ T cell infectivity suggesting that leukocytes from PWID are not intrinsically more susceptibility to infection with HIV than non-PWID controls.
Keywords: People Who Inject Drugs (PWID), Opioids, activation, HIV infection, memory response, HIV
Summary Sentence.
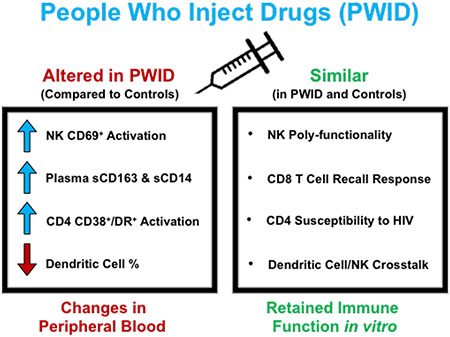
Long-term opioid injection-drug users have evidence of increased immune activation, but no detectable immune dysfunction in NK, DC and T cell nor increased susceptibility to in vitro HIV infection.
Graphical Abstract
Introduction.
Use of opioids such as morphine, heroin, and fentanyl is thought to induce a functional impairment of the innate and adaptive immune response (1–3), which in turn may reduce the systemic host intrinsic resistance to HIV infection. This conclusion is based largely on animal studies and in vitro data where opioids were administered in an acute setting without sufficient time to induce long-term use and tolerance (4–6). Likewise, opioid withdrawal in animal models of addiction has been observed to induce immune dysfunction, which may not relate to steady-state users (6, 7). In contrast, studies focusing on chronic opioid use yield conflicting results, showing evidence of either increased or decreased host immune functional responses (8, 9). Further research is necessary to address if persons who inject drugs (PWID) long-term can establish a homeostatic acclimation to opioids over time, allowing immune function (including activation and resistance to acute viral infection) to be retained.
NK cells represent a critical component of the host innate immune response against acute viral infection and serve as a front-line defense against a diverse array of pathogens (10–12). Unlike antigen-specific T cells, NK cells use the coordinated interaction of both inhibitory and activating receptors to lyse targets cells exhibiting signs of stress or antibody-coated targets utilizing the direct cytotoxicity pathway or antibody-dependent cellular cytotoxicity (ADCC) pathway, respectively (13–25). NK activity is also augmented by accessory cells such as myeloid dendritic cells (mDC) and plasmacytoid dendritic cells (pDC) that secrete NK-stimulatory cytokines such as IFN-alpha, IL-12 and IL-15 following pathogen recognition by Toll-like Receptors (TLRs) (26–32). In addition, dendritic cell accessory function is also required for generating de novo adaptive CD4+ T cell and CD8+ T cell responses following acute infection (33–35). However, TLR stimulated cytokine secretion by DCs can also induce CD4+ T cell activation, which in turn may predispose targets for greater HIV infection following needle-sharing or other high-risk behaviors (36, 37).
Here, we assessed if a history of chronic injection drug use alters the intrinsic susceptibility of isolated CD4+ T cells to in vitro HIV infection in PWID compared to control donors who do not inject drugs. In parallel, we compared innate and adaptive (recall) immune responses by measuring the frequency, activation state, and functional profile of NK cells, dendritic cells, CD4+ and CD8+ T cells in PWID and non-PWID controls. Due to the future potential of prophylactic immuno-therapy approaches such as BNAbs in the prevention of HIV infection among PWID (38–40), we thoroughly investigated the functional capacity of the antibody-dependent cellular cytotoxicity (ADCC) response by NK cells from PWID and controls. To capture the full breadth of individuals from the current opioid epidemic, we recruited participants who inject drugs at a high frequency (daily) and included low-risk PWID who do not share needles and high-risk needles-sharing PWID. The goal of our study was to test the hypothesis that chronic immune activation among PWID contributes to a weakened host innate and adaptive immune response associated with higher susceptibility to in vitro HIV infection. While we did confirm that PWID possess increased inflammatory markers and constitutive immune activation, our data did not support our hypothesis. Rather, our results here point to the maintenance of a highly functional innate and adaptive immune response in long-term PWID.
Methods.
Study participants and Clinical Assessment.
We recruited 50 high-risk needle-sharing PWID from the city of Philadelphia via community-based street outreach in specific neighborhoods previously identified as “risk pockets” (41, 42). Risk pockets are defined as locations within neighborhoods with a high HIV-1 prevalence where injectable drugs are sold, used, and exchanged for sex as identified from the Philadelphia Department of Public Health HIV Surveillance Reports (2014-2018). We utilized the “Prognostic Model for Sero-conversion Among Injection Drug Users” (43) to identify high-risk PWID subjects for our study based upon their frequency of injection and needle sharing behavior. Briefly, individuals were identified as high-risk PWID if they remained HIV-1 IgG sero-negative despite a history of more than 2 years of daily injections and frequent (weekly or greater) needle sharing with partners of unknown HIV status. To control for the increased risk associated with needle-sharing, 35 low-risk, non-sharing PWID were recruited from local needle exchange centers in Philadelphia with similarly high rate of daily injection drug use (Table 1). A comprehensive summary of the behavioral risk practices, drug usage patterns, recent sexual history and demographic information of our cohort has been chronicled in our previous reports (44–47). As a comparison to high-and low-risk PWID, 40 control donors that did not inject drugs were also recruited from the greater Philadelphia area. The study protocol and Informed Consent documents were approved by the University of PA Institutional Review Board. All PWID individuals screened or enrolled in the study were offered referral to drug cessation programs, counseled to enter the local needle-exchange programs to reduce their risk of exposure to blood borne pathogens, and offered additional health services as needed.
Table 1:
Demographic Information of Non-sharing and Needle-sharing PWID
| Control Donors (n=40) | Low-risk Non-sharing PWID (n=35) | High-risk Needle-sharing PWID (n=50) | |
|---|---|---|---|
| Mean Age | μ= 52.4 years | μ=41.9 years | μ=41.8 years |
| Hepatitis B Status | N/A | 23% Sero-positive | 26% Sero-positive |
| Hepatitis C Status | N/A | 69% Sero-positive | 85% Sero-positive |
| Frequency of Opioid Injection (Last 6 months) | N/A | μ=179 events | μ=160 events |
| Frequency of Needle-Sharing (Last 6 months) | N/A | μ=1 event | μ=34 events |
| Frequency of Cocaine Injection (Last 6 months) | N/A | μ=24 events | μ=49 events |
| Frequency of Unprotected Intercourse (Last 6 months) | N/A | μ=43 events | μ=37 events |
Flow Cytometry.
All cell surface antibodies and isotype controls were used at the recommended dilution of 0.25 μg antibody per million cells. Peripheral Blood Mono-nuclear Cells (PBMC) were stained with antibodies to phenotypic and functional markers for 15 minutes at room temperature in the dark, washed twice and fixed for 5 minutes at 4°C with Cytofix Buffer (BD Biosciences, San Jose, CA). The following surface antibodies (with clones shown in parentheses) and their appropriate isotype controls were obtained from BD Biosciences unless otherwise noted: CD69 FITC (FN50), CD107a PE (H4A3), CD56 PERCP Cy5.5 (B159), CD57 APC (NK-1), CD16 APC-H7 (3G8), CD3 BV510 (UCHT1), CD4 APC-H7 (RPA-T4), CD8 PE (RPA-T8), PD-1 FITC (MIH4), HLA-DR PERCP Cy5.5 (L243), CD38 APC (HIT2), Lineage FITC (cocktail), CD83 PE (HB15e), CD11c BV421 (B-ly6), BDCA-4 APC (REA380, Miltenyi Corporation, Auburn CA), and CD40 APC-H7 (5C3). Intra-cellular staining for IFN-gamma BV421 (B27), TNF-alpha FITC (MAB11), MIP-1 beta APC-H7 (D21-1351) or p24 FITC (Kc57, Beckman Coulter, Pasadena CA) was carried out in 1X Perm/Wash Buffer (BD Biosciences) as described by the manufacturer. A minimum of two hundred thousand events were collected on a BD LSR-II Flow Cytometer and samples were subsequently analyzed with FlowJo v9 software (Tree Star Incorporated, Ashland OR).
NK Poly-functional Response to Direct or ADCC Targets.
1x106 freshly isolated PBMC were washed and incubated in the presence or absence of 2x105 target cells at a 5:1 effector/target ratio along with 20 μl anti-CD107a monoclonal antibody and 0.133 μl of Golgi-stop (BD Biosciences) in a 200 μl total volume. As targets, K562 tumor cells were used to measure the direct NK cytotoxicity response. For ADCC experiments, CEM NK resistant tumor targets were coated with 1 μg gp120 from the HIV-1 IIIB isolate (ProSpec Protein Specialists, East Brunswick, NJ) for 30 minutes, washed and then incubated with a 1/1000 dilution of heat inactivated plasma from an HIV-1 infected elite controller reference subject for 15 minutes as previously reported (48). Timepoints were collected every hour and samples were stained with antibodies to NK cell phenotypic and functional markers as described above. The percentage of CD56+/CD3− gated NK cells staining positive for CD107a degranulation and/or cytokine production following incubation with target cells was determined after subtraction of background levels of staining in the absence of target cells (No Target Control). A 5:1 effector to target ratio was utilized because it ensures a saturating amount of targets so that every NK cells has access to a target thereby normalizing for differing NK frequency per PBMC among donors.
DC-stimulated NK Response.
5x106/ml PBMC were stimulated for 18 hours with 10 μg/ml of either the Toll-like Receptor 9 agonist, CpG-ODN 2216 or the Toll-like Receptor 7/8 agonist, R848, to stimulate plasmacytoid and myeloid Dendritic cells, respectively (Invivogen, San Diego, CA). On day 2, PBMCs were stained by Flow Cytometry to access DC-induced NK CD69 activation or utilized in a CD107a assay against K562 cells to assess DC-stimulated NK function. When assessing dendritic cell stimulation of NK functional responses against K562, background levels of unstimulated NK responses are subtracted from TLR stimulated conditions.
CD8+ T Cell Stimulation.
5x105 PBMC were co-cultured with 5 μg/mL SEB (Staphylococcal Enterotoxin B, Sigma Aldrich) or a 1 μg/mL mixture of overlapping CEF peptide pool comprising 23 peptides consisting of sequences derived from the human Cytomegalovirus, Epstein-Barr and Influenza Viruses (AIDS Research and Reference Reagent Repository, NIH) in the presence of 2.5 μl CD28/CD49d co-stimulation (BD) and 5 μg/mL Brefeldin A for 18 hours in a 200 μl volume. Unstimulated PBMC were used as a negative control. PBMC were washed, stained with antibodies to T Cell phenotypic markers and intra-cellular staining for IFN-gamma was carried out as described above. CD8+ T cells were gated by CD8+/CD3+ staining and the percentage of cells staining positive for CD107a and/or IFN-gamma was determined after subtraction of background levels of staining in unstimulated control cells.
HIV-1 Infection.
CD4+ primary T cells were isolated to 99% purity by negative selection using CD4 magnetic bead isolation kit II as described by the manufacturer (Miltenyi Corporation). To trigger activation, CD4+ primary T cells were incubated in the presence of anti-CD3/CD28 microbeads (Miltenyi Corporation) and 100 IU/ml hIL-2 (PeproTech, Rocky Hill, NJ) for 48 hours as described by the manufacturer. Constitutive activation was maintained by CD4+ primary T cells cultured for 48 hours with media in the presence of 100 IU/ml hIL-2 alone. 5x106 activated or IL-2-only CD4+ T cells were spinfected at 1800 rpm for two hours with 150 ng of p24 containing supernatant of the CXCR4-tropic HIV-1 primary isolate TYBE or the CCR5-tropic HIV-1 lab isolate BAL as previously described (31). HIV-1 infection was determined four days later by measuring intra-cellular levels of p24 by Flow Cytometry as described above. All viral strains were generated and tittered by the University of Pennsylvania Centers for AIDS Research Center.
Cytokine Measurements.
Circulating sCD14 (Magnetic Bead Panel, Millipore Sigma) and sCD163 (ELISA, Thermo Scientific) were assessed in plasma specimens by the University of Pennsylvania Immunology Core Facility utilizing according to the manufacture’s recommendations.
Statistical Analysis.
All graphic presentations were derived using GraphPad Prism software (version 8, GraphPad Software, La Jolla, CA) and represent the median and interquartile range.
Comparisons between groups were performed using a two-sided non-parametric Wilcoxon rank sum (Mann-Whitney) test (alpha = 0.05). All missing data originated from technical issues with the assays performed: no data was purposefully excluded from the analysis. No adjustment for multiple testing was performed due to the exploratory nature of our hypothesis-driven research.
Results.
Similar magnitude of NK direct poly-functionality despite heightened NK activation and increased plasma inflammatory markers among PWID compared to control donors.
In order to assess the functional capacity of the immune response in PWID, we recruited individuals who inject opioids daily from the greater Philadelphia area including low-risk PWID who do not share needles and high-risk needle-sharing PWID (Table 1). First, we characterized the plasma from PWID and controls to investigate if chronic opioid usage was associated with measurable changes in any inflammatory markers associated with myeloid immune activation. As shown in Figure 1A and B, we observed that PWID had significantly higher levels of plasma sCD14 (p=0.0023, n=16) and sCD163 (p=0.0001, n=16) compared to control donors. Interestingly, we did not observe a difference in the level of sCD14 or sCD163 between high-risk, needle sharing PWID and low-risk, non-sharing PWID (Figure 1). These data suggest that myeloid activation is driven primarily by exposure to opioids and/or other drugs, rather than to potential pathogens introduced by needle sharing behavior alone. We next sought to determine if the increase in inflammatory markers in PWID was associated with other phenotypic changes or immune activation parameters. Utilizing multi-parameter flow cytometry, we assessed the frequency of the major subsets of NK cells. As shown in Figure 2A, levels of CD56dim/CD3− (p=0.4453, n=26), CD56bright/CD3− (data not shown), and CD56+/CD3+ NKT cells (data not shown) were similar in all study groups. In contrast, we observed a significantly (p=0.0103, n=26) increased level of NK activation, as measured by constitutive CD69 expression, in PWID compared to controls (Figure 2B). We have previously identified that the heightened NK activation among PWID was primarily due to increased CD69 expression among needle-sharing PWID rather than non-sharing PWID (47). Of note, there was no correlation between NK CD69 activation and plasma levels of sCD14 or sCD163.
Figure 1. Increased levels of sCD14 and sCD163 in the plasma of PWID compared to controls.
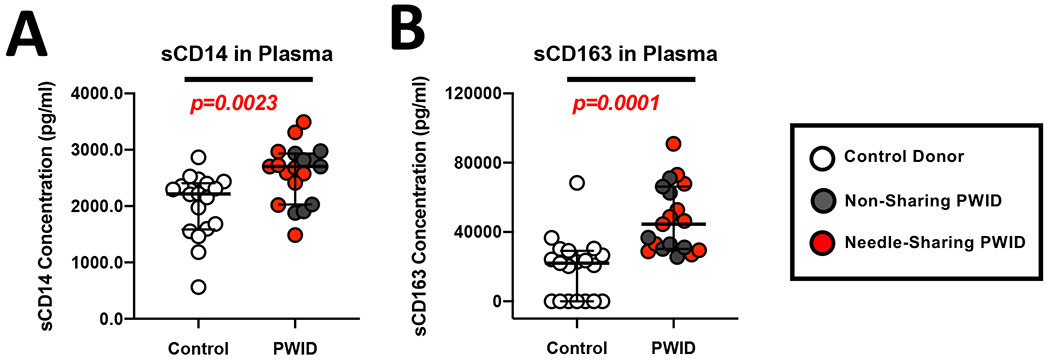
(A-B) Composite graph displaying the levels of sCD14 (A) and sCD163 (B) in the plasma from non-sharing (light gray circles) and needle-sharing (dark gray circles) PWID compared to control donors (white circles).
Figure 2. Similar magnitude and kinetics of NK direct poly-functionality despite heightened NK activation in PWID compared to control donors.
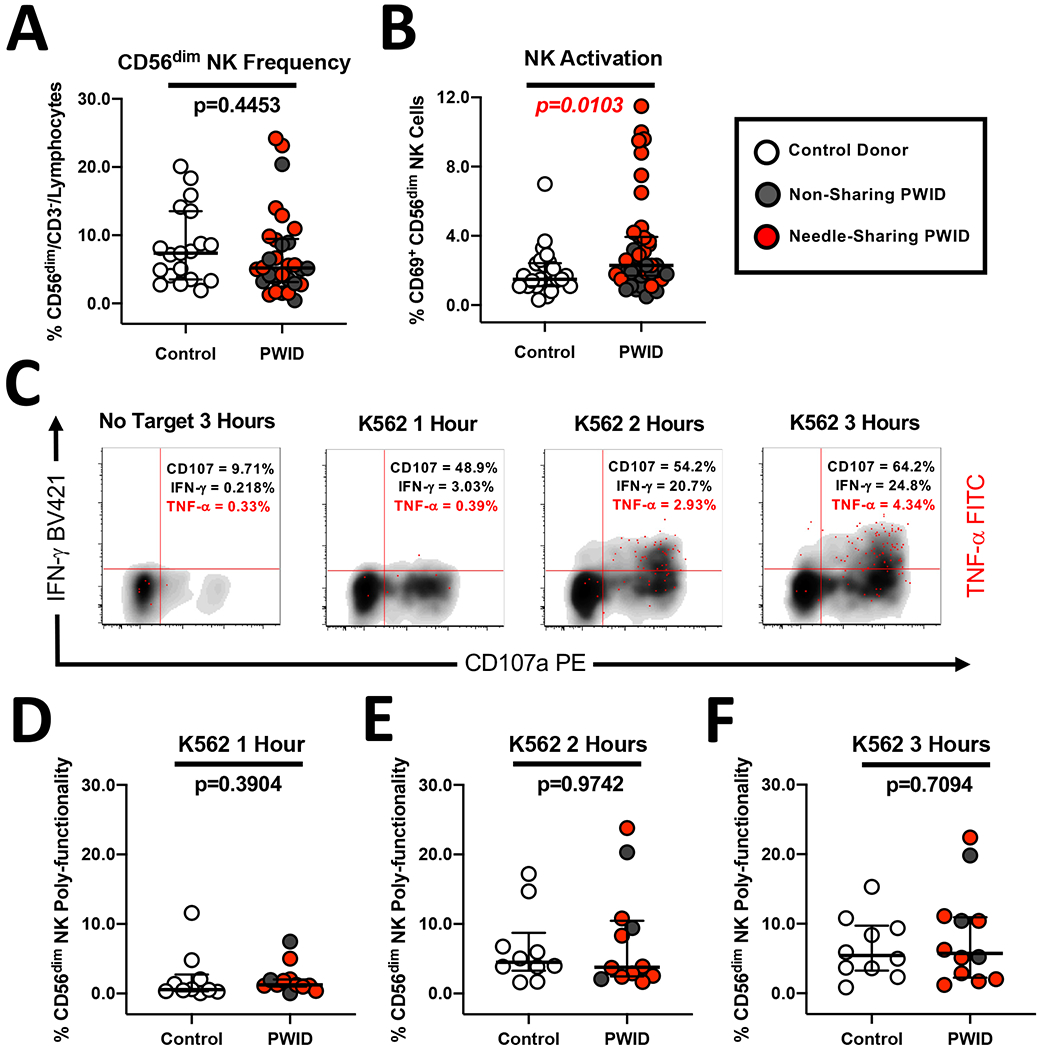
(A) Composite graph displaying the similar frequency of CD56dim/CD3− NK cells from non-sharing (light gray circles) and needle-sharing (dark gray circles) PWID compared to control donors (white circles). (B) Composite graph displaying the increased constitutive CD69 activation of CD56dim/CD3− NK cells from PWID compared to control uninfected donors. (C) Representative kinetic analysis of the NK poly-functional profile against K562 tumor targets recognized through the direct cytotoxicity pathway. PBMCs from a representative PWID were incubated for 3 hours in the presence or absence of MHC-devoid K562 cells at a 5:1 effector to target cell ratio. PBMCs were then stained with fluorescently conjugated antibodies to NK phenotypic markers and permeabilized to measure intra-cellular cytokine expression. The data is shown in a three-parameter poly-functional density plot with CD107a degranulating NK cells on the X-axis, IFN-gamma producing NK cells on the Y-axis, and NK cells producing TNF-alpha super-imposed on top as red dots. The percentage of CD56dim/CD3− gated NK cells staining positive for CD107a, IFN-gamma, and TNF-alpha is shown in the upper right-hand quadrant. (D-F) Composite graph of K562-induced NK poly-functionality from PWID and control donors was defined as the percentage of CD56dim/CD3− gated NK cells exhibiting two or more effector functions including degranulation and/or cytokine production at each timepoint after subtraction of background from the No Target control condition. Statistical analyses of two groups was performed using a non-parametric Mann-Whitney T-test with a two-tailed p-value.
We next investigated if the heightened NK activation observed among PWID translated to altered NK effector function against target cells lysed through the direct cytotoxicity pathway (such as K562 tumor cells). We utilized a NK poly-functionality assay (as shown for a representative PWID in Figure 2C) which can measure target induced CD107a degranulation, IFN-gamma production and TNF-alpha production over the course of a three-hour incubation with K562 cells. As shown in Figure 2D–F, there was no difference in the magnitude or kinetics of target-induced NK poly-functionality (defined as CD56dim NK cells exhibiting more than one effector function) among PWID or controls at any timepoint tested. There was also no significant difference among individual NK effector functions between PWID and controls at any timepoint tested. Together, these results indicate that while PWID have increased inflammatory markers in the plasma and heightened NK activation, the kinetics and magnitude of NK effector function elicited through the direct cytotoxicity pathway appear to be unaffected.
Similar magnitude of NK ADCC poly-functionality in PWID compared to controls.
Due to the critical role of Fc mediated effector function in preventative BNAb immuno-therapy approaches against HIV (49), we investigated strength of the antibody-dependent cellular cytotoxicity (ADCC) response by NK cells from PWID and controls. First, we characterized the expression of FcγR receptor, CD16, which mediates ADCC by CD56dim/CD3− NK cells. We observed that the geometric mean fluorescence intensity (GMFI) of CD16 (p=0.9121, n=26) was similar between controls, non-sharing PWID and needle-sharing PWID (Figure 3A). In addition, we determined that expression of the NK maturation marker CD57, which has been associated with enhanced ADCC activity on CD56dim/CD3− NK cells (50, 51), was also similar (p=0.7781, n=26) across study groups (Figure 3B). To assess the functional ADCC response, we utilized our kinetic assay of NK poly-functionality with CEM NKres target cells coated with the gp120 protein of HIV-1 and plasma from a representative HIV-1 infected subject to trigger ADCC as we described previously (48). As shown for a representative PWID individual in Figure 3C, there was a specific increase in the amount of CD107a degranulation, IFN-gamma production and to a lesser extent TNF-alpha production over the course of a three-hour incubation with HIV plasma-coated ADCC targets. However, we did not observe any difference in the magnitude of NK ADCC poly-functional response among PWID or controls at any timepoint tested (Figure 3D–F). Together, this data suggests that, like direct NK cytotoxic responses, NK ADCC is not affected in PWID, despite evidence of constitutive NK activation and increased soluble inflammatory markers.
Figure 3. Similar levels of NK CD16/CD57 expression and NK kinetics of ADCC poly-functionality in PWID and control donors.
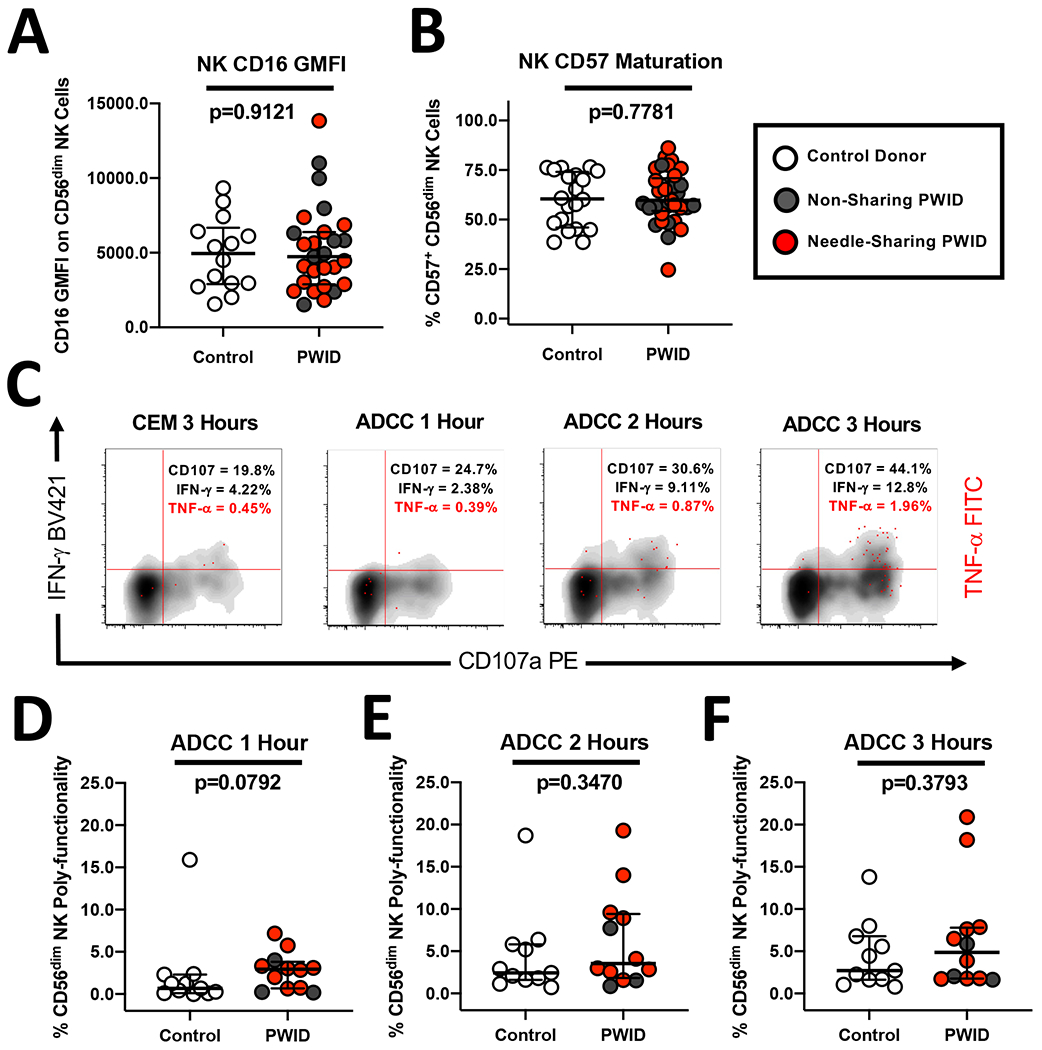
(A) Composite graph displaying the similar Geometric Mean Fluorescence Intensity (GMFI) of CD16 receptor on CD56dim/CD3− NK cells from non-sharing (light gray circles) and needle-sharing (dark gray circles) PWID compared to control uninfected donors (white circles). (B) Composite graph displaying the similar levels of CD57 maturation marker CD56dim/CD3− NK cells from PWID compared to control uninfected donors. (C) Representative kinetic analysis of the NK poly-functional profile against CEM targets coated with gp120/anti-HIV plasma and recognized through the ADCC pathway. PBMCs from a representative PWID were incubated for 3 hours in the presence or absence of ADCC coated targets at a 5:1 effector to target cell ratio. PBMCs were then stained with fluorescently conjugated antibodies to NK phenotypic markers and permeabilized to measure intra-cellular cytokine expression. The data is shown in a three-parameter poly-functional density plot with CD107a degranulating NK cells on the X-axis, IFN-gamma producing NK cells on the Y-axis, and NK cells producing TNF-alpha super-imposed on top as red dots. The percentage of CD56dim/CD3− gated NK cells staining positive for CD107a, IFN-gamma, and TNF-alpha is shown in the upper right-hand quadrant. (D-F) Composite graph of ADCC-induced NK poly-functionality from PWID and control uninfected donors as defined as the percentage of CD56dim/CD3− gated NK cells exhibiting two or more effector functions including degranulation and/or cytokine production at each timepoint after subtraction of background from the No Target control condition. Statistical analyses of two groups was performed using a non-parametric Mann-Whitney T-test with a two-tailed p-value.
Similar levels of DC/NK cross-talk despite reduced frequency of Plasmacytoid and Myeloid Dendritic cells in PWID compared to control donors.
Due to the critical role that dendritic cells play in augmenting NK responses (26–32), we assessed the phenotype and function of plasmacytoid (pDC) and myeloid dendritic cells (mDC) in PWID and controls. As shown in Figure 4A and B, we observed a significant reduction in the overall frequency of both mDC (p=0.0001, n=22) and pDC (p=0.0015, n=22) within the peripheral blood mono-nuclear cells (PBMC) compartment of PWID compared to controls. Despite the decreased amount of pDC and mDC, there was no detectable change in the activation profile of these subsets as the constitutive CD40 and CD83 expression on mDC and pDC was similar in PWID and controls (Figure 4C and D). During both acute and chronic infection, people living with HIV (PLWH) exhibit a profound loss of dendritic cells in the periphery due to a combination of factors including increased recruitment into the tissues as well as heightened activation induced apoptosis (52–64). While we did not measure levels of tissue homing receptors such as alpha 4 beta 7 on dendritic cells from individuals from our study, our results suggest that the observed decrease in dendritic cells among PWID is not due to factors associated with increased activation in peripheral blood subsets. Of note, there was no correlation between the levels of constitutive mDC and pDC activation and plasma levels of sCD14 or sCD163 (data not shown).
Figure 4. Similar levels of activation and DC/NK cross-talk despite reduced frequency of Plasmacytoid and Myeloid Dendritic cells in PWID compared to control donors.
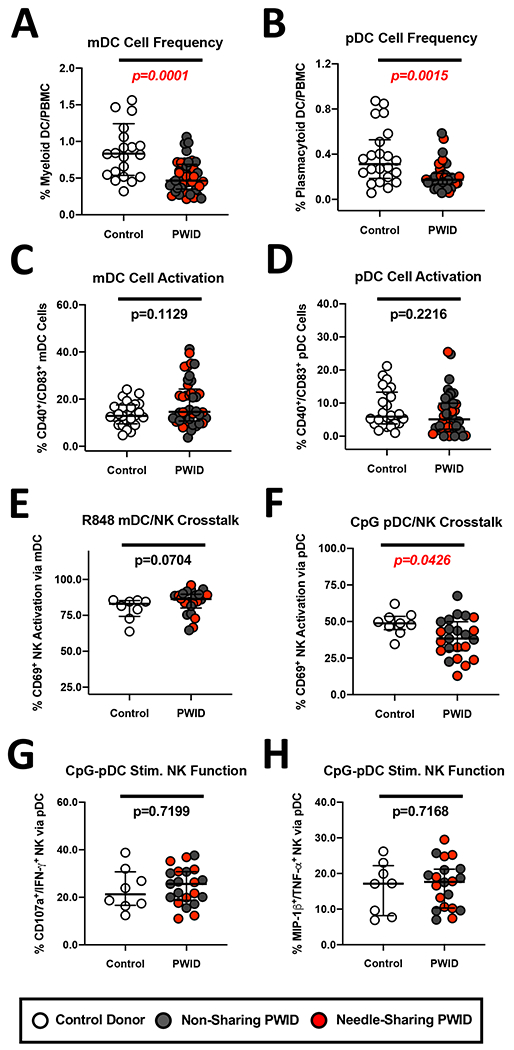
(A-B) Composite graph displaying the reduced frequency of (A) LIN−/HLA-DR+/CD11c+ Myeloid dendritic cells (mDC) cells and (B) LIN−/HLA-DR+/BDCA-4+ Plasmacytoid dendritic cells (pDC) from non-sharing (light gray circles) and needle-sharing (dark gray circles) PWID compared to control uninfected donors (white circles). (C-D) Composite graph displaying the similar levels of (C) mDC and (D) pDC constitutive activation as determined by CD40/CD83 expression from PWID compared to control uninfected donors. (E) Composite graph displaying the similar levels of mDC induced NK CD69 activation following R848 TLR 7/8 stimulation. (F) Composite graph displaying the reduced level of pDC induced NK CD69 activation following CpG-ODN 2216 TLR9 stimulation. (G-H) Functional analysis of pDC-stimulated NK poly-functional response against MHC-devoid K562 targets recognized through the direct cytotoxicity pathway. PBMCs were stimulated overnight with CpG-ODN 2216 TLR9 to induce pDC driven NK activation as described above and then incubated for 3 hours in the presence or absence of K562 coated targets at a 5:1 effector to target cell ratio. PBMCs were then stained with fluorescently conjugated antibodies to NK phenotypic markers and permeabilized to measure intra-cellular cytokine expression. The percentage of CD56dim/CD3− gated NK cells staining positive for (G) CD107a/IFN-gamma or (H) MIP-1beta/TNF-alpha was determined after subtraction of background from the unstimulated NK conditions. Statistical analyses of two groups was performed using a non-parametric Mann-Whitney T-test with a two-tailed p-value.
To test DC-NK crosstalk, we incubated PBMC with Toll-like Receptor (TLR) ligands to stimulate DC secretion of cytokines that could in turn activate NK cells. Despite the observed reduced frequency of mDC among PWID, R848 stimulation of the TLR7/8 pathway led to strong CD69 activation of upwards of 80% NK cells in both PWID and controls (Figure 4E). In contrast, stimulation of PDCs with the TLR9 agonist CpG-ODN 2216 resulted in significantly reduced NK CD69 activation (p=0.0426, n=21) in PWID compared to controls (Figure 4F), as anticipated due to observed reduced pDC frequency. We have previously reported that pDC/NK crosstalk in needle-sharing PWID can be impacted by the negative effect of Hepatitis C infection on production of IFN-alpha by pDC (46). However, when we measured the ability of CpG-ODN 2216stimulated pDC to augment NK poly-functionality against K562 cells as a measure of DC/NK cross-talk, we observed no reduction in the ability of pDC-stimulated NK cells from PWID to degranulate or produce cytokines compared to controls (Figure 4G and H). Together, these results suggested that while the frequency of pDC and mDC is reduced within the PBMC compartment of PWID compared to controls, sufficient dendritic cells stimulation is present to mediate NK activation and boost NK poly-functionality against K562 tumor targets.
Similar CD8+ T cell recall antigenic response and CD4 T cell HIV infectivity among PWID and controls despite an increased CD4+ T cell activation profile in PWID.
Due to the potential of opioids to negatively impact the adaptive immune compartment following acute exposure (4), we sought to explore the phenotype and functionality of CD8+ and CD4+ T cells in PWID who were exposed to opioids over a prolonged time period. As shown in Figure 5A, we observed a significant increase in the overall frequency of CD8+ T cells in PWID compared to controls (p=0.0424, n=18). In contrast, there was a minimal change in the activation profile (i.e.: CD38 and HLA-DR co-expression) of CD8+ T cells from PWID compared to control uninfected donors (Figure 5B). Among PWID, non-sharing and needle-sharing individuals showed similar CD8+ T cell frequency and activation. Using SEB as a superantigen and CEF peptide pool to measure the antigenic recall response to CMV, EBV and FLU, we measured the functional profile of CD8+ T cells to polyclonal and recall antigen stimuli. As shown in Figure 5C and D, we observed similar levels of CD107a degranulation and IFN-gamma production by CD8+ T cells from PWID and controls in response to all stimuli.
Figure 5. Similar levels of CD8 T cell activation and antigenic peptide response in PWID and control donors.
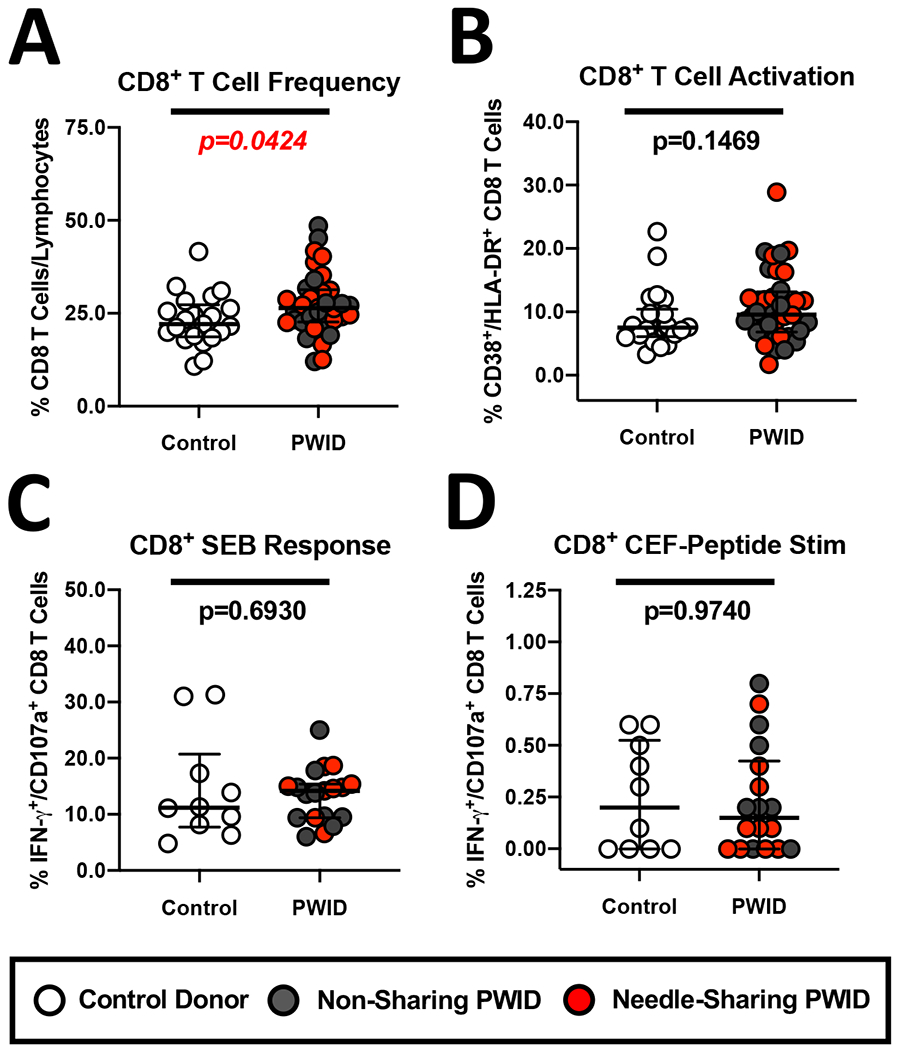
(A-B) Composite graph displaying the increased (A) frequency of CD8+/CD3+ T cells and (B) similar constitutive CD38+/HLA-DR+ activation of CD8 T cells from non-sharing (light gray circles) and needle-sharing (dark gray circles) PWID compared to control uninfected donors (white circles). (C-D) Functional analysis of the CD8 T cell response in PWID compared to control donors in response to (C) SEB or (D) a mixture of overlapping CEF peptide pool consisting of sequences derived from the human Cytomegalovirus, Epstein-Barr and Influenza Viruses (CEF) for 18 hours. Unstimulated or stimulated PBMC were stained with fluorescently conjugated antibodies to CD8 phenotypic markers and permeabilized to measure intra-cellular cytokine expression. The percentage of CD8+/CD3+ cells staining positive for CD107a and/or IFN-gamma was determined after subtraction of background levels of staining in unstimulated control cells. Statistical analyses of two groups was performed using a non-parametric Mann-Whitney T-test with a two-tailed p-value.
We next investigated if the observed increase in inflammatory markers and NK activation among PWID was accompanied by any changes in CD4+ T cell activation. As shown in Figure 6A, we observed significantly higher constitutive expression of the activation markers CD38 and HLA-DR on CD4+ T cells (p=0.0355, n=18) in PWID compared to controls. While the heightened activation profile of CD4+ T cells did not correlate with plasma levels of sCD14 or sCD163 (data not shown), our data suggest that injection drug use may increase the constitutive or induced susceptibility of CD4+ T cells to HIV-1 infection. This observation was of critical importance, particularly for PWID who engage in high-risk needle-sharing activity, as increased activation could lead to increased susceptibility to HIV infection through contaminated injection instruments. To test if CD4+ T cells from PWID are more susceptible to HIV infection, we assessed the constitutive or activation-induced in vitro susceptibility to HIV infection using CD4+ T cells derived from PWID and controls. To this end, we activated a portion of CD4+ T cells via anti-CD3/CD28 microbead stimulation and observed a comparable upregulation of CD69/PD-1 markers among both PWID and controls (Figure 6B). We also left a portion of CD4+ T cells under constitutive activation by being in culture with IL-2 alone. We then infected constitutive or anti-CD3/CD28 stimulated CD4+ T cells with CCR5-tropic (BAL) or CXCR4-tropic (TYBE) strains of HIV-1 as described previously (31). As shown in a representative PWID individual in Figure 6C, we observed detectable intra-cellular HIV-1 p24 expression and CD4 downregulation among CD4+ T cells infected with either CCR5-tropic or CXCR4-tropic strains of HIV. Across the cohorts, we observed no difference in the rate of CCR5-tropic BAL infection of constitutive or CD3/CD28 microbead stimulated CD4+ T cells between PWID and controls (Figure 6D and E). When assessing CXCR4-tropic TYBE strain infection of CD4+ T cells, we observed minimal to no infectivity in the absence of stimulation indicative that constitutive activation did not by itself affect infectivity (Figure 6F). In contract, after activation we observed strong infectivity of anti-CD3/CD28 bead stimulated CD4+ T cells with the CXCR4-tropic TYBE strain of HIV-1 (Figure 6G). However, we did not observe any significant difference in CXCR4-tropic HIV infection between non-sharing PWID, needle-sharing PWID, or controls (Figure 6G). Together, these results indicate that while CD4+ T cells from PWID exhibit evidence of heightened constitutive activation, this does not result in increased in vitro susceptibility to either CCR5-tropic or CXCR4-tropic strains of HIV-1.
Figure 6. Similar levels of CCR5-tropic and CXCR4-tropic infection despite heightened CD4 T cell activation in PWID and control donors.
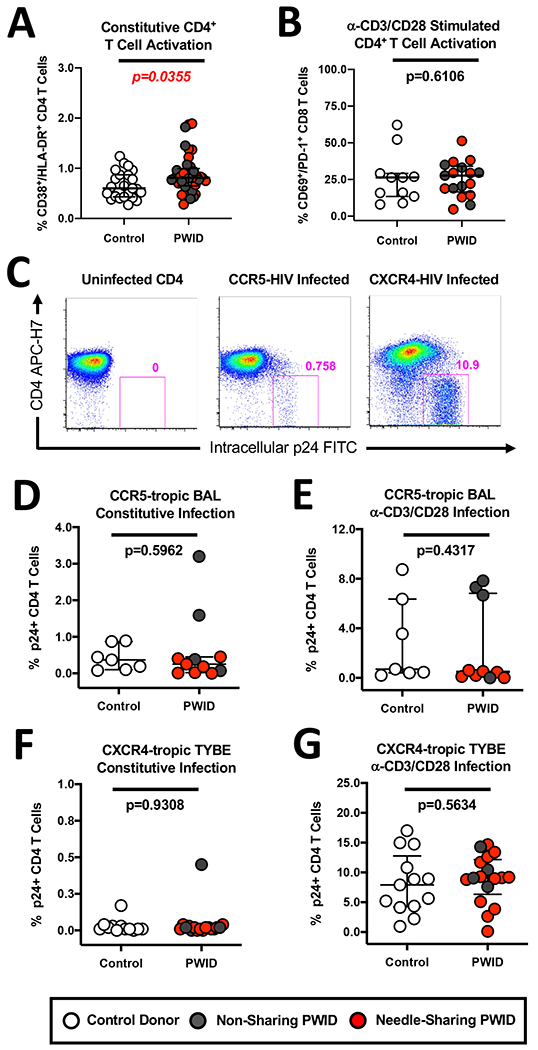
(A) Composite graph displaying the increased constitutive CD38+/HLA-DR+ activation of CD4+ T cells from non-sharing (light gray circles) and needle-sharing (dark gray circles) PWID compared to control uninfected donors (white circles). (B) Composite graph displaying similar levels of anti-CD3/CD28 bead induced CD69+/PD-1+ maturation of CD4+ T cells from PWID and control uninfected donors. (C) Representative infection of unstimulated and CD3/CD28-stimulated CD4+ T cells from a PWID individual with the CCR5-tropic BAL strain and the CXCR4-tropic TYBE strain of HIV-1, respectively. HIV p24 positive, CD4 downregulated CD4+ T cells that are productively infected are shown in the red gate (D-E) Composite graph displaying similar levels of infection of (D) constitutive and (E) CD3/CD28-stimulated CD4+ T cells from PWID and control uninfected donors with the CCR5-tropic BAL strain of HIV-1, respectively. (F-G) Composite graph displaying similar levels of infection of (F) constitutive and (G) CD3/CD28-stimulated CD4+ T cells from PWID and control uninfected donors with the CXCR4-tropic TYBE strain of HIV-1, respectively.
Discussion.
Here, we documented that long-term PWID have functional NK cells, dendritic cells, CD4+ T cells and CD8+ T cells when compared to individuals who do not inject drugs thereby refuting the commonly held view that chronic opioid use results in highly dysfunctional innate and adaptive immune responses (1–3). Importantly, our data support the rationale for using broadly neutralizing antibodies as a prophylactic HIV prevention strategy among PWID (38–40). Fc effector function has been shown to enhance the efficacy of BNAbs (49), and here we document the retention of strong ADCC activity among NK cells from PWID. While our study did not address if injection drug use results in altered expression of anti-viral restriction factors or miRNA molecules as has been previously described (3, 65, 66), we do report that CD4+ T cells from PWID are not intrinsically more susceptible to constitutive or activation-induced in vitro infection with CCR5-tropic or CXCR4-tropic strains of HIV than CD4+ T cells from control donors. Together, our results indicate that the increased incidence of HIV infection among PWID may be principally due to increased viral exposure from injection drug use activity (or other concurrent high-risk behaviors), rather than an intrinsically higher susceptibility within leukocytes leading to enhanced HIV infection.
To capture various types of individuals from the current injection drug use epidemic, we recruited both low-risk PWID who do not share needles and high-risk needles-sharing PWID. Non-sharing and needle-sharing PWID were similar in terms of their high frequency of prolonged opioid use and the frequency of co-injection with added drugs including cocaine (Table 1). While there was no measurable difference in the levels of plasma sCD14 and sCD163 between low-risk PWID and high-risk needles-sharing PWID (Figure 1), we did observe that high-risk needle-sharing PWID have a significantly higher constitutive NK CD69 expression profile compared to non-sharing PWID and controls (Figure 2B) supporting our previous findings (47). Here, we built upon that work by investigating if the increased NK activation among high-risk PWID led to a heightened CD57 maturation profile of the NK repertoire (Figure 3B). In contrast to a recent report showing that sero-negative (HESN) individuals exposed to HIV through unprotected sexual contact with a discordant partner show increased levels of NK CD57 maturation (67), we did not detect any difference in the levels of CD57 maturation among low-risk PWID and high-risk needle-sharing PWID. We speculate that NK differentiation may be better supported in the mucosa through chronic exposure to sexually transmitted pathogens rather than as the result of pathogens transmitted during needle-sharing. Importantly, we also determined that the increased constitutive NK activation profile in high-risk needle-sharing PWID did not translate to a difference in the magnitude of the NK poly-functional response to direct or ADCC targets at any time point tested compared to non-sharing PWID. Future studies will be needed to address whether the observed increase in constitutive NK cell and CD4+ T cell activation among PWID contributes to increased turnover in these cell populations due to long-term injection drug use.
The current opioid epidemic in the United States and the associated rise in HIV infections in PWID has highlighted the need to identify novel prophylactic (i.e., vaccine) or immuno-therapeutic strategies against HIV. Preventive approaches against HIV such as passive immunization with BNAbs may require the presence of a capable host innate ADCC immune functional response for optimal efficacy (49). In addition to confirming strong direct and ADCC NK function among PWID, we also determined that no other innate or adaptive functional differences were observed between needle-sharing and non-sharing PWID including dendritic cell TLR stimulated NK function, CD8+ T cell response to SEB or CMV/EBV/FLU peptides, and constitutive or anti-CD3/CD28 stimulated CD4+ T cell infectivity with CCR5-tropic or CXCR4-tropic HIV-1 isolates. While our results suggest that CD4+ T cells from PWID and controls exhibit similar levels of constitutive and activation induced infection with HIV, we cannot rule out that CD4+ T cells from PWID produce more infectious virus per cell due to heightened levels of constitutive activation. We posit that the length of exposure to opioids is a critical determinant in whether immune function is negatively impacted by drug exposure. In animal models, a profound defect in the innate and adaptive immune response is observed when opioids are administered in an acute setting without sufficient time to induce long-term use and tolerance (4–6). Our work showing that long-term injection drug use is associated with retained immune function potentially resolves the discrepancy between previous chronic opioid exposure studies which reported both increased and decreased host immune responses in people who inject drugs (8, 9).
Similar to acute exposure, opioid withdrawal in animal models of addiction also induces immune dysfunction, likely due to the stress of withdrawal overcoming tolerance mechanisms (6, 7). Further studies may help determine if medications for opioid use disorder (MOUD) could independently affect immune parameters measured here. Therefore, our study does not address any heightened chance of HIV infection during first opioid use or opioid relapse periods which can occur in a significant percentage of individuals participating in opioid cessation programs (68–71). In addition, our study does not address the impact of acute use, MOUD, or active comorbidities (HIV infection) on immune function. However, we do document that injection of opioids over a prolonged time period does not negatively affect ex vivo innate or adaptive recall immune functions. Future studies will be required to confirm if high-risk behavior in conjunction with active comorbidities provide for added HIV infection incidence in this population. Taken together, our data support the rationale for using strategies dependent on mobilizing innate or adaptive immune responses for the prevention of HIV-1 in long-term PWID.
Acknowledgements.
We would like to thank the University of Pennsylvania Immunology Core Facility for analyzing the plasma samples for inflammatory markers and the University of Pennsylvania Center For AIDS Research for the expansion and tittering of all HIV isolates utilized in this study. This work was supported by the following grants from the National Institutes of Health (NIDA R01DA028775, NIDA R21DA040554, T32AI007632, UM1AI126620, R01AI094603, R01 DA048728, R01 DA049666). Additional support was provided by Roberts and I Jacobs fund of The Philadelphia Foundation, Kean Family Professorship, AIDS funds from the Commonwealth of Pennsylvania and from the Commonwealth Universal Research Enhancement Program, Pennsylvania Department of Health, the Penn Center for AIDS Research (P30 AI 045008), and Cancer Center Grant (P30 CA10815). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations.
- PWID
Persons Who Inject Drugs
- NK cells
Natural Killer
- pDC
Plasmacytoid Dendritic Cells
- mDC
Myeloid Dendritic Cell
- IFN-γ
Interferon-Gamma
- TNF-α
Tumor Necrosis Factor-alpha
- ADCC
Antibody-Dependent Cellular Cytotoxicity
- TLR
Toll Like Receptor
- SEB
Staphylococcal enterotoxin B
- CEF Peptide Pool
Cytomegalovirus, Epstein Barr Virus and Influenza
Footnotes
The authors have no conflicts of interest.
Conflicts of Interest Disclosure.
The authors declare no scientific, commercial or financial conflicts of interest.
References.
- 1.Bhargava HN. Opioid peptides, receptors, and immune function. NIDA Res Monogr. 1990;96:220–33. [PubMed] [Google Scholar]
- 2.Boland JW, and Pockley AG. Influence of opioids on immune function in patients with cancer pain: from bench to bedside. Br J Pharmacol. 2018;175(14):2726–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rouveix B. Opiates and immune function. Consequences on infectious diseases with special reference to AIDS. Therapie. 1992;47(6):503–12. [PubMed] [Google Scholar]
- 4.Eisenstein TK, and Hilburger ME. Opioid modulation of immune responses: effects on phagocyte and lymphoid cell populations. J Neuroimmunol. 1998;83(1-2):36–44. [DOI] [PubMed] [Google Scholar]
- 5.Saurer TB, Carrigan KA, Ijames SG, and Lysle DT. Suppression of natural killer cell activity by morphine is mediated by the nucleus accumbens shell. J Neuroimmunol. 2006;173(1-2):3–11. [DOI] [PubMed] [Google Scholar]
- 6.Desjardins S, Belkai E, Crete D, Cordonnier L, Scherrmann JM, Noble F, et al. Effects of chronic morphine and morphine withdrawal on gene expression in rat peripheral blood mononuclear cells. Neuropharmacology. 2008;55(8):1347–54. [DOI] [PubMed] [Google Scholar]
- 7.Weed MR, Carruth LM, Adams RJ, Ator NA, and Hienz RD. Morphine withdrawal dramatically reduces lymphocytes in morphine-dependent macaques. J Neuroimmune Pharmacol. 2006;1(3):250–9. [DOI] [PubMed] [Google Scholar]
- 8.Diasso PDK, Birke H, Nielsen SD, Main KM, Hojsted J, Sjogren P, et al. The effects of long-term opioid treatment on the immune system in chronic non-cancer pain patients: A systematic review. Eur J Pain. 2020;24(3):481–96. [DOI] [PubMed] [Google Scholar]
- 9.Kreek MJ. Immune function in heroin addicts and former heroin addicts in treatment: pre- and post-AIDS epidemic. NIDA Res Monogr. 1990;96:192–219. [PubMed] [Google Scholar]
- 10.Caligiuri MA. Human natural killer cells. Blood. 2008;112(3):461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooper MA, Fehniger TA, and Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22(11):633–40. [DOI] [PubMed] [Google Scholar]
- 12.Della Chiesa M, Marcenaro E, Sivori S, Carlomagno S, Pesce S, and Moretta A. Human NK cell response to pathogens. Semin Immunol. 2014;26(2):152–60. [DOI] [PubMed] [Google Scholar]
- 13.Raulet DH, and Held W. Natural killer cell receptors: the offs and ons of NK cell recognition. Cell. 1995;82(5):697–700. [DOI] [PubMed] [Google Scholar]
- 14.Raghavan M, and Bjorkman PJ. Fc receptors and their interactions with immunoglobulins. Annu Rev Cell Dev Biol. 1996;12:181–220. [DOI] [PubMed] [Google Scholar]
- 15.Long EO, Burshtyn DN, Clark WP, Peruzzi M, Rajagopalan S, Rojo S, et al. Killer cell inhibitory receptors: diversity, specificity, and function. Immunol Rev. 1997;155:135–44. [DOI] [PubMed] [Google Scholar]
- 16.Lanier LL. NK cell receptors. Annu Rev Immunol. 1998;16:359–93. [DOI] [PubMed] [Google Scholar]
- 17.Bottino C, Biassoni R, Millo R, Moretta L, and Moretta A. The human natural cytotoxicity receptors (NCR) that induce HLA class I-independent NK cell triggering. Hum Immunol. 2000;61(1):1–6. [DOI] [PubMed] [Google Scholar]
- 18.De Maria A, and Moretta L. HLA-class I-specific inhibitory receptors in HIV-1 infection. Hum Immunol. 2000;61(1):74–81. [DOI] [PubMed] [Google Scholar]
- 19.Tomasello E, Blery M, Vely F, and Vivier E. Signaling pathways engaged by NK cell receptors: double concerto for activating receptors, inhibitory receptors and NK cells. Semin Immunol. 2000;12(2):139–47. [DOI] [PubMed] [Google Scholar]
- 20.Blery M, Olcese L, and Vivier E. Early signaling via inhibitory and activating NK receptors. Hum Immunol. 2000;61(1):51–64. [DOI] [PubMed] [Google Scholar]
- 21.Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol. 2001;19:197–223. [DOI] [PubMed] [Google Scholar]
- 22.Biassoni R, Cantoni C, Pende D, Sivori S, Parolini S, Vitale M, et al. Human natural killer cell receptors and co-receptors. Immunol Rev. 2001;181:203–14. [DOI] [PubMed] [Google Scholar]
- 23.Middleton D, Curran M, and Maxwell L. Natural killer cells and their receptors. Transpl Immunol. 2002;10(2-3):147–64. [DOI] [PubMed] [Google Scholar]
- 24.Djeu JY, Jiang K, and Wei S. A view to a kill: signals triggering cytotoxicity. Clin Cancer Res. 2002;8(3):636–40. [PubMed] [Google Scholar]
- 25.Biassoni R, Bottino C, Cantoni C, and Moretta A. Human natural killer receptors and their ligands. Curr Protoc Immunol. 2002;Chapter 14:Unit 14 0. [DOI] [PubMed] [Google Scholar]
- 26.Biron CA, Nguyen KB, Pien GC, Cousens LP, and Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. [DOI] [PubMed] [Google Scholar]
- 27.Chehimi J, and Trinchieri G. Interleukin-12: a bridge between innate resistance and adaptive immunity with a role in infection and acquired immunodeficiency. J Clin Immunol. 1994;14(3):149–61. [DOI] [PubMed] [Google Scholar]
- 28.Maghazachi AA, and Al-Aoukaty A. Chemokines activate natural killer cells through heterotrimeric G-proteins: implications for the treatment of AIDS and cancer. Faseb J. 1998;12(11):913–24. [DOI] [PubMed] [Google Scholar]
- 29.Robertson MJ. Role of chemokines in the biology of natural killer cells. J Leukoc Biol. 2002;71(2):173–83. [PubMed] [Google Scholar]
- 30.Waldmann T The contrasting roles of IL-2 and IL-15 in the life and death of lymphocytes: implications for the immunotherapy of rheumatological diseases. Arthritis Res. 2002;4 Suppl 3:S161–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomescu C, Chehimi J, Maino VC, and Montaner LJ. NK Cell Lysis of HIV-1-Infected Autologous CD4 Primary T Cells: Requirement for IFN-Mediated NK Activation by Plasmacytoid Dendritic Cells. J Immunol. 2007;179(4):2097–104. [DOI] [PubMed] [Google Scholar]
- 32.Tomescu C, Chehimi J, Maino VC, and Montaner LJ. Retention of viability, cytotoxicity, and response to IL-2, IL-15, or IFN-{alpha} by human NK cells after CD107a degranulation. J Leukoc Biol. 2009;85:871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanzavecchia A, and Sallusto F. The instructive role of dendritic cells on T cell responses: lineages, plasticity and kinetics. Curr Opin Immunol. 2001;13(3):291–8. [DOI] [PubMed] [Google Scholar]
- 34.Sallusto F, and Lanzavecchia A. The instructive role of dendritic cells on T-cell responses. Arthritis Res. 2002;4 Suppl 3:S127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zinkernagel RM. On the Role of Dendritic Cells Versus Other Cells in Inducing Protective CD8+ T Cell Responses. Front Immunol. 2014;5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Groot F, van Capel TM, Kapsenberg ML, Berkhout B, and de Jong EC. Opposing roles of blood myeloid and plasmacytoid dendritic cells in HIV-1 infection of T cells: transmission facilitation versus replication inhibition. Blood. 2006;108(6):1957–64. [DOI] [PubMed] [Google Scholar]
- 37.Fitzgerald-Bocarsly P, and Jacobs ES. Plasmacytoid dendritic cells in HIV infection: striking a delicate balance. J Leukoc Biol. 2010;87(4):609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barin F, and Braibant M. HIV-1 antibodies in prevention of transmission. Curr Opin HIV AIDS. 2019;14(4):273–8. [DOI] [PubMed] [Google Scholar]
- 39.Garber DA, Adams DR, Guenthner P, Mitchell J, Kelley K, Schoofs T, et al. Durable protection against repeated penile exposures to simian-human immunodeficiency virus by broadly neutralizing antibodies. Nat Commun. 2020;11(1):3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu ZJ, Bai J, Liu FL, Zhang XY, and Wang JZ. Focus on the therapeutic efficacy of 3BNC117 against HIV-1: In vitro studies, in vivo studies, clinical trials and challenges. Int Immunopharmacol. 2017;52:44–50. [DOI] [PubMed] [Google Scholar]
- 41.Williams CT, and Metzger DS. Race and distance effects on regular syringe exchange program use and injection risks: a geobehavioral analysis. Am J Public Health. 2010;100(6):1068–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.City of Philadelphia Department of Public Health. In: Department of Public Health ed Philadelphia: Office of HIV Planning; 2009. [Google Scholar]
- 43.Boileau C, Bruneau J, Al-Nachawati H, Lamothe F, and Vincelette J. A prognostic model for HIV seroconversion among injection drug users as a tool for stratification in clinical trials. J Acquir Immune Defic Syndr. 2005;39(4):489–95. [DOI] [PubMed] [Google Scholar]
- 44.Colon K, Speicher DW, Smith P, Taylor M, Metzger DS, Montaner LJ, et al. S100A14 Is Increased in Activated NK Cells and Plasma of HIV-Exposed Seronegative People Who Inject Drugs and Promotes Monocyte-NK Crosstalk. J Acquir Immune Defic Syndr. 2019;80(2):234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomescu C, Abdulhaqq S, and Montaner LJ. Evidence for the innate immune response as a correlate of protection in human immunodeficiency virus (HIV)-1 highly exposed seronegative subjects (HESN). Clin Exp Immunol. 2011;164(2):158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomescu C, Duh FM, Lanier MA, Kapalko A, Mounzer KC, Martin MP, et al. Increased plasmacytoid dendritic cell maturation and natural killer cell activation in HIV-1 exposed, uninfected intravenous drug users. AIDS. 2010;24(14):2151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomescu C, Seaton KE, Smith P, Taylor M, Tomaras GD, Metzger DS, et al. Innate activation of MDC and NK cells in high-risk HIV-1-exposed seronegative IV-drug users who share needles when compared with low-risk nonsharing IV-drug user controls. J Acquir Immune Defic Syndr. 2015;68(3):264–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tomescu C, Tebas P, and Montaner LJ. IFN-alpha augments natural killer-mediated antibody-dependent cellular cytotoxicity of HIV-1-infected autologous CD4+ T cells regardless of major histocompatibility complex class 1 downregulation. AIDS. 2017;31(5):613–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, and Ravetch JV. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell. 2014;158(6):1243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lopez-Verges S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H, et al. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood. 2010;116(19):3865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scheiter M, Lau U, van Ham M, Bulitta B, Grobe L, Garritsen H, et al. Proteome analysis of distinct developmental stages of human natural killer (NK) cells. Mol Cell Proteomics. 2013;12(5):1099–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lehmann C, Jung N, Forster K, Koch N, Leifeld L, Fischer J, et al. Longitudinal analysis of distribution and function of plasmacytoid dendritic cells in peripheral blood and gut mucosa of HIV infected patients. J Infect Dis. 2014;209(6):940–9. [DOI] [PubMed] [Google Scholar]
- 53.Reeves RK, Evans TI, Gillis J, Wong FE, Kang G, Li Q, et al. SIV infection induces accumulation of plasmacytoid dendritic cells in the gut mucosa. J Infect Dis. 2012;206(9):1462–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dave B, Kaplan J, Gautam S, and Bhargava P. Plasmacytoid dendritic cells in lymph nodes of patients with human immunodeficiency virus. Appl Immunohistochem Mol Morphol. 2012;20(6):566–72. [DOI] [PubMed] [Google Scholar]
- 55.Brown KN, Wijewardana V, Liu X, and Barratt-Boyes SM. Rapid influx and death of plasmacytoid dendritic cells in lymph nodes mediate depletion in acute simian immunodeficiency virus infection. PLoS Pathog. 2009;5(5):e1000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malleret B, Maneglier B, Karlsson I, Lebon P, Nascimbeni M, Perie L, et al. Primary infection with simian immunodeficiency virus: plasmacytoid dendritic cell homing to lymph nodes, type I interferon, and immune suppression. Blood. 2008;112(12):4598–608. [DOI] [PubMed] [Google Scholar]
- 57.Chehimi J, Azzoni L, Farabaugh M, Creer SA, Tomescu C, Hancock A, et al. Baseline Viral Load and Immune Activation Determine the Extent of Reconstitution of Innate Immune Effectors in HIV-1-Infected Subjects Undergoing Antiretroviral Treatment. J Immunol. 2007;179(4):2642–50. [DOI] [PubMed] [Google Scholar]
- 58.Brown KN, Trichel A, and Barratt-Boyes SM. Parallel loss of myeloid and plasmacytoid dendritic cells from blood and lymphoid tissue in simian AIDS. J Immunol. 2007;178(11):6958–67. [DOI] [PubMed] [Google Scholar]
- 59.Schmidt B, Fujimura SH, Martin JN, and Levy JA. Variations in plasmacytoid dendritic cell (PDC) and myeloid dendritic cell (MDC) levels in HIV-infected subjects on and off antiretroviral therapy. J Clin Immunol. 2006;26(1):55–64. [DOI] [PubMed] [Google Scholar]
- 60.Fantuzzi L, Purificato C, Donato K, Belardelli F, and Gessani S. Human immunodeficiency virus type 1 gp120 induces abnormal maturation and functional alterations of dendritic cells: a novel mechanism for AIDS pathogenesis. J Virol. 2004;78(18):9763–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pacanowski J, Develioglu L, Kamga I, Sinet M, Desvarieux M, Girard PM, et al. Early plasmacytoid dendritic cell changes predict plasma HIV load rebound during primary infection. J Infect Dis. 2004;190(10):1889–92. [DOI] [PubMed] [Google Scholar]
- 62.Chehimi J, Campbell DE, Azzoni L, Bacheller D, Papasavvas E, Jerandi G, et al. Persistent decreases in blood plasmacytoid dendritic cell number and function despite effective highly active antiretroviral therapy and increased blood myeloid dendritic cells in HIV-infected individuals. J Immunol. 2002;168(9):4796–801. [DOI] [PubMed] [Google Scholar]
- 63.Pacanowski J, Kahi S, Baillet M, Lebon P, Deveau C, Goujard C, et al. Reduced blood CD123+ (lymphoid) and CD11c+ (myeloid) dendritic cell numbers in primary HIV-1 infection. Blood. 2001;98(10):3016–21. [DOI] [PubMed] [Google Scholar]
- 64.Donaghy H, Pozniak A, Gazzard B, Qazi N, Gilmour J, Gotch F, et al. Loss of blood CD11c(+) myeloid and CD11c(−) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood. 2001;98(8):2574–6. [DOI] [PubMed] [Google Scholar]
- 65.Meijerink H, Indrati AR, Soedarmo S, Utami F, de Jong CA, Alisjahbana B, et al. Heroin use in Indonesia is associated with higher expression of CCR5 on CD4+ cells and lower ex-vivo production of CCR5 ligands. AIDS. 2015;29(3):385–8. [PubMed] [Google Scholar]
- 66.Purohit V, Rapaka RS, Rutter J, and Shurtleff D. Do opioids activate latent HIV-1 by down-regulating anti-HIV microRNAs? J Neuroimmune Pharmacol. 2012;7(3):519–23. [DOI] [PubMed] [Google Scholar]
- 67.Lima JF, Oliveira LM, Pereira NZ, Mitsunari GE, Duarte AJ, and Sato MN. Distinct natural killer cells in HIV-exposed seronegative subjects with effector cytotoxic CD56(dim) and CD56(bright) cells and memory-like CD57(+)NKG2C(+)CD56(dim) cells. J Acquir Immune Defic Syndr. 2014;67(5):463–71. [DOI] [PubMed] [Google Scholar]
- 68.Amato L, Davoli M, Ferri M, Gowing L, and Perucci CA. Effectiveness of interventions on opiate withdrawal treatment: an overview of systematic reviews. Drug Alcohol Depend. 2004;73(3):219–26. [DOI] [PubMed] [Google Scholar]
- 69.Maglione MA, Raaen L, Chen C, Azhar G, Shahidinia N, Shen M, et al. Effects of medication assisted treatment (MAT) for opioid use disorder on functional outcomes: A systematic review. J Subst Abuse Treat. 2018;89:28–51. [DOI] [PubMed] [Google Scholar]
- 70.Thomas I A Brief Overview of Identification and Management of Opiate Use Disorder in the Primary Care Setting. Nurs Clin North Am. 2019;54(4):495–501. [DOI] [PubMed] [Google Scholar]
- 71.Oesterle TS, Thusius NJ, Rummans TA, and Gold MS. Medication-Assisted Treatment for Opioid-Use Disorder. Mayo Clin Proc. 2019;94(10):2072–86. [DOI] [PubMed] [Google Scholar]