

SUMMARY
Responding to different dynamic levels of stress is critical for mammalian survival. Disruption of mineralocorticoid receptor (MR) and glucocorticoid receptor (GR) signaling is proposed to underlie hypothalamic-pituitary-adrenal (HPA) axis dysregulation observed in stress-related psychiatric disorders. In this study, we show that FK506-binding protein 51 (FKBP5) plays a critical role in fine-tuning MR:GR balance in the hippocampus. Biotinylated-oligonucleotide immunoprecipitation in primary hippocampal neurons reveals that MR binding, rather than GR binding, to the Fkbp5 gene regulates FKBP5 expression during baseline activity of glucocorticoids. Notably, FKBP5 and MR exhibit similar hippocampal expression patterns in mice and humans, which are distinct from that of the GR. Pharmacological inhibition and region- and cell type-specific receptor deletion in mice further demonstrate that lack of MR decreases hippocampal Fkbp5 levels and dampens the stress-induced increase in glucocorticoid levels. Overall, our findings demonstrate that MR-dependent changes in baseline Fkbp5 expression modify GR sensitivity to glucocorticoids, providing insight into mechanisms of stress homeostasis.
Graphical abstract
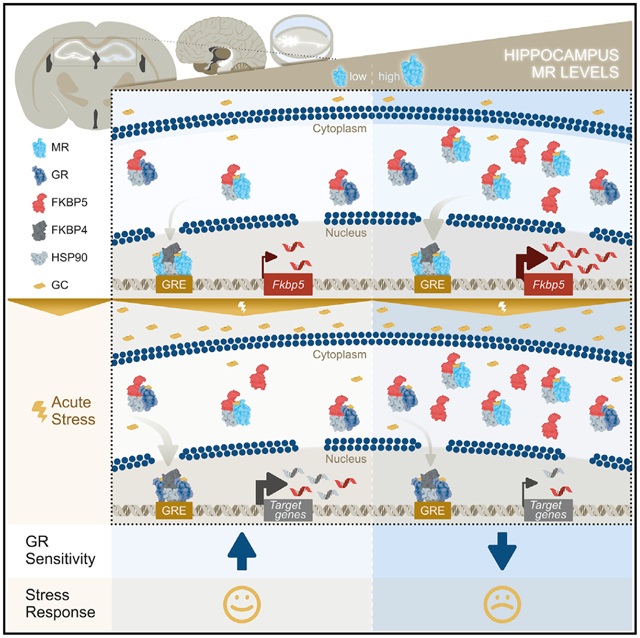
In brief
Hartmann et al. demonstrate that MRs regulate baseline FKBP5 expression in the hippocampus. This regulation leads to a modification of GR sensitivity to glucocorticoids during acute stress. The results suggest that FKBP5 acts as a key modulator of HPA axis activity by mediating the fine-tuning of hippocampal MR:GR balance.
INTRODUCTION
Stress-related psychiatric disorders, including major depression disorder (MDD) and posttraumatic stress disorder (PTSD), represent significant global disease and social burden, but the underlying molecular mechanisms are still poorly understood (Fenster et al., 2018; Maddox et al., 2019). In addition to the autonomic nervous system, the primary control module of the stress response in mammals is the hypothalamic-pituitary-adrenal (HPA) axis, which regulates the circadian and stress-induced release of glucocorticoids (cortisol in humans, corticosterone in mice). It is well established that coordinated secretion of glucocorticoids, in response to acute stress, is beneficial for the individual (de Kloet et al., 2005). Alternatively, aberrant glucocorticoid release as a result of chronic stress or traumatic experiences can be damaging for the brain and increase the susceptibility to develop mental disorders, including MDD and PTSD (Lupien et al., 2009). Therefore, disturbed activation or regulation of the body’s stress response through the HPA axis represents a common pathophysiological aspect of multiple stress-related diseases (de Kloet et al., 2005; Lupien et al., 2009).
Glucocorticoids orchestrate the activity of the HPA axis and neuronal circuits via the glucocorticoid receptor (GR, encoded by the Nr3c1 gene) and the mineralocorticoid receptor (MR, encoded by the Nr3c2 gene). These two nuclear receptors belong to the ligand-dependent transcription factor family (De Kloet et al., 1998; Ulrich-Lai and Herman, 2009). MRs have a 10-fold higher affinity for glucocorticoids than do GRs, suggesting different roles for each receptor in the regulation of HPA axis activity (Reul and de Kloet, 1985; Reul et al., 2014). MRs are largely occupied under basal glucocorticoid conditions (circadian trough), whereas GR occupancy is increased when glucocorticoid levels rise during the circadian peak or following stress. Thus, while MRs are involved in basal activity and onset of stress-induced HPA axis activity, GRs primarily drive its termination (de Kloet et al., 2018).
The Hsp90-associated co-chaperone FK506-binding protein 51 (FKBP5), which is encoded by the Fkbp5 gene, is a negative regulator of GR activity and plays a key role in the termination of the stress response by GRs (Binder, 2009). FKBP5 limits GR function by decreasing ligand-binding sensitivity, thereby delaying nuclear translocation and ultimately reducing GR-dependent transcriptional activity. Notably, the expression of Fkbp5 is stimulated by glucocorticoids as part of an intracellular ultra-short negative feedback loop for GR activity (Hubler and Scammell, 2004; Vermeer et al., 2003). Hence, augmented transcription and translation of FKBP5 following GR activation is associated with increased levels of circulating cortisol/corticosterone and altered negative feedback inhibition of the stress response (Binder et al., 2008; Denny et al., 2000; Hartmann et al., 2012; Häusl et al., 2021; Hoeijmakers et al., 2014; Ising et al., 2008; O’Leary et al., 2011; Touma et al., 2011; Westberry et al., 2006). Interestingly, recent evidence also points to potential regulation of FKBP5 expression via MR signaling (McCann et al., 2021; van Weert et al., 2019). In addition, previous genetic and epigenetic evidence in humans has implicated the NR3C1, NR3C2, and FKBP5 genes as associated with stress-related disorders (i.e., MDD and PTSD) (Binder et al., 2004; Criado-Marrero et al., 2018; Hardeveld et al., 2015; Keller et al., 2017; Klengel et al., 2013; Klok et al., 2011; van Rossum et al., 2006).
Regulation of the HPA axis occurs at numerous central nervous system (CNS) nodes, including rapid effects at the paraventricular nucleus of the hypothalamus (PVN), which abundantly expresses GR, but little to no MR (Arnett et al., 2016; Häusl et al., 2021; de Kloet et al., 1988, 2018; Laryea et al., 2013). The hippocampus also exerts strong regulatory control of the HPA axis. This has been observed in hippocampal lesion studies, as well as in forebrain-specific GR knockout mice, which showed impairments in HPA axis feedback inhibition (Arnett et al., 2016; Boyle et al., 2006; Dedovic et al., 2009; Fanselow and Dong, 2010; Furay et al., 2008; Herman et al., 2016; Jacobson and Sapolsky, 1991).
In recent years, an imbalance between central MR and GR signaling has been proposed to underlie HPA axis dysregulation associated with the susceptibility to psychopathology such as MDD and PTSD; however, the underlying molecular mechanisms are far from clear (Harris et al., 2013; De Kloet and Derijk, 2004; de Kloet and Joëls, 2017; Medina et al., 2013). Interestingly, the MR, GR, and FKBP5 are all strongly expressed in the hippocampus (Patel et al., 2000; Scharf et al., 2011). Thus, FKBP5 is ideally positioned to regulate MR:GR balance in the hippocampus, not only through its well established, inhibitory actions on GRs, but possibly also through an interplay with the MR. In this study, we explored the underlying molecular mechanisms and the extent to which this imbalance may be driven by an FKBP5-mediated modulation of both the GR and MR. By utilizing a number of analytic and causal approaches across species—biotinylated oligonucleotide immunoprecipitation (oligoIP) in mouse primary hippocampal neurons, single-cell RNA sequencing data, human postmortem brain tissue expression analyses, pharmacological approaches, as well as region- and cell type-specific GR and MR knockout mice—we propose a model in which FKBP5 acts as a key modulator of the HPA axis by fine tuning the MR:GR balance in the hippocampus.
RESULTS
FKBP5 and MR exhibit similar expression patterns in the hippocampus, which is distinct from that of GR
Earlier work has shown that, even under baseline conditions, Fkbp5 mRNA expression is more pronounced in the hippocampus compared to other brain regions that regulate behavioral and neuroendocrine stress responsivity, including the PVN, the basolateral amygdala, or the prefrontal cortex (PFC) (Scharf et al., 2011). Given that MRs are largely occupied under basal glucocorticoid conditions (circadian trough), whereas GR occupancy primarily occurs during rising glucocorticoid levels (circadian peak or stress), we postulated that the hippocampal FKBP5 expression pattern at baseline would more closely resemble that of the MR than the GR. Using radioactive in situ hybridization (ISH) and fluorescent RNAscope, we initially confirmed that baseline (glucocorticoid trough) Fkbp5 mRNA levels exhibit a specific pattern in the hippocampus, with high expression in the CA2 and DG subregions and lower levels in the CA1 and CA3 (Figures 1A and 1B). Interestingly, the hippocampal expression patterns of Nr3c1 (GR) and Fkbp5 were fairly distinct, with particularly lower Nr3c1 than Fkbp5 expression in CA2, while those of Nr3c2 (MR) and Fkbp5 were more similar. In line with this observation, the Fkbp5 expression within all hippocampal subregions was positively correlated with the expression of Nr3c2, with no correlation observed between Fkbp5 and Nr3c1 levels (Figure 1C). Notably, human hippocampus postmortem samples of healthy control individuals (n = 6; see Table S1 for subject details) had mRNA expression profiles of FKBP5, NR3C1, and NR3C2 matching those of their murine equivalents (Figure 1D). Along these lines, FKBP5 mRNA levels were positively correlated with NR3C2 expression levels in human hippocampal tissue, while there was no correlation between FKBP5 and NR3C1 (Figure 1E).
Figure 1. Fkbp5, Nr3c1, and Nr3c2 mRNA expression patterns in the human and mouse hippocampus.
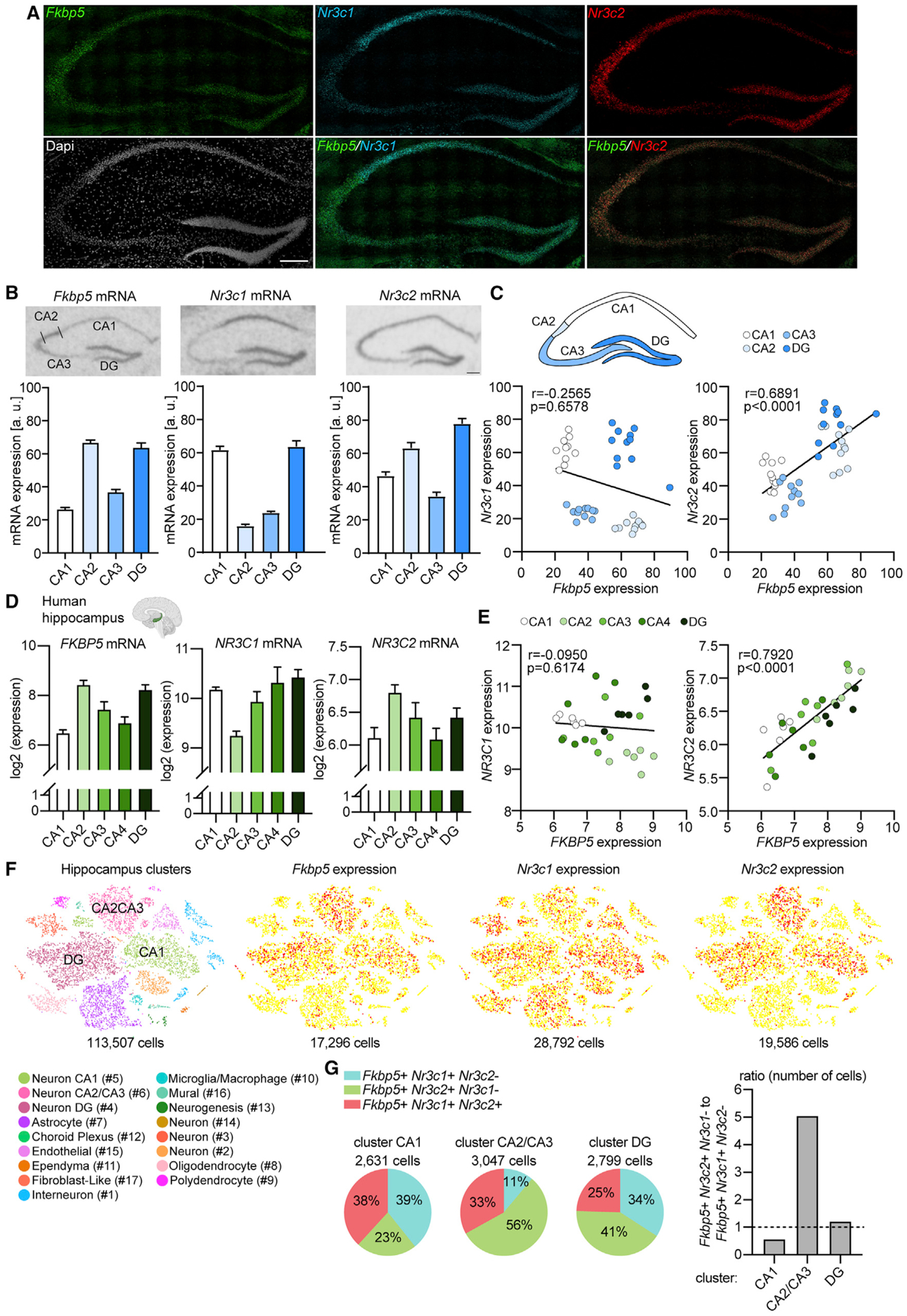
(A) Fkbp5, Nr3c1, and Nr3c2 mRNA expression in the mouse hippocampus determined by RNAscope. Fkbp5 mRNA (green) and Nr3c2 mRNA (red) are strongly expressed in CA2 and dentate gyrus (DG). Nr3c1 mRNA (cyan) is prominently expressed in CA1 and DG. DAPI stain (gray) shows area examined. Overlay of Fkbp5 and Nr3c2 reveals strong overlap in expression of Fkbp5 and Nr3c2 specifically in the CA2. Fkbp5 and Nr3c1 expression does not show a high a degree of overlap in the CA1, CA2, or CA3. n = 4 mice. Scale bar, 250 μm.
(B) Hippocampal Fkbp5, Nr3c1 (glucocorticoid receptor [GR]), and Nr3c2 (mineralocorticoid receptor [MR]) mRNA expression determined by in situ hybridization (ISH) in C57BL/6J mice. Fkbp5 and Nr3c2 exhibit similar expression patterns in the hippocampus, which is distinct from that of Nr3c1. (Top panel) Representative autoradiographs of hippocampal Fkbp5, Nr3c1, and Nr3c2 mRNA expression. (Lower panel) Quantified expression of Fkbp5, Nr3c1, and Nr3c2 mRNA. Areas of interest are CA1, CA2, CA3, and DG. n = 11 mice. Scale bar, 250 μm.
(C) Correlation of Fkbp5 and Nr3c1 (left; Pearson correlation coefficient, r = −0.2565, p = 0.6578) or Nr3c2 (right; r = 0.6891, p < 0.0001) mRNA levels in hippocampal subregions CA1, CA2, CA3, and DG. Each dot represents the levels of Fkbp5 and the respective receptor in the same mouse.
(D) Microarray data from the Allen Brain Institute (Hawrylycz et al., 2012) showing FKBP5, NR3C1, and NR3C2 mRNA expression in human hippocampal subregions CA1, CA2, CA3, CA4, and DG. n = 6 subjects (see also Table S1).
(E) Correlation of FKBP5 and NR3C1 (left; Pearson correlation coefficient, r = −0.0950, p = 0.6174) or NR3C2 (right; r = 0.7920, p < 0.0001) mRNA levels in human hippocampal subregions CA1, CA2, CA3, CA4, and DG. Each dot represents the levels of FKBP5 and the respective receptor in one individual.
(F) Single-cell RNA sequencing data (Saunders et al., 2018) of the mouse hippocampus (n = 113,507 cells) depicts several different cell types (left) and the expression plots for Fkbp5, Nr3c1, and Nr3c2.
(G) Percentage of Fkbp5-positive cells expressing either only Nr3c1, only Nr3c2, or both receptors in individual neuronal clusters CA1, CA2/CA3, and DG (left). (Right) Ratio of the number of cells expressing Fkbp5 and only Nr3c2 to cells expressing Fkbp5 and only Nr3c1 in neuronal clusters CA1, CA2/CA3, and DG (Yates’ chi-square = 854.177; p < 2.2e–16). Data are presented as mean + SEM.
To further investigate the relationship of the expression profiles of Fkbp5, Nr3c1, and Nr3c2, we took advantage of a publicly available single-cell RNA sequencing dataset consisting of 113,507 single cells isolated from the mouse hippocampus (Saunders et al., 2018). The single-cell expression data revealed a complex cellular composition, including among others, neurons, astrocytes, microglia/macrophages, and oligodendrocytes (Figure 1F). Fkbp5 was detected in 17,296 cells and was prominently expressed in neuronal clusters #4 (Fkbp5+ cells: 5,815), #5 (Fkbp5+ cells: 3,913), and #6 (Fkbp5+ cells: 4,461), which include neurons of the CA1 (#5), CA2/CA3 (#6), and DG (#4) (Figure 1F). In contrast, Nr3c1 was expressed in 28,792 cells. Although Nr3c1 was also strongly present in neuronal clusters CA1 (Nr3c1+ cells: 7,235), CA2/CA3 (Nr3c1+ cells: 2,959), and DG (Nr3c1+ cells: 5,957), it additionally showed a more widely distributed expression in other cell types such as astrocytes and oligodendrocytes. Similar to Fkbp5, Nr3c2 was detected in a total of 19,568 cells and showed a pronounced expression in clusters CA1 (Nr3c2+ cells: 5,048), CA2/CA3 (Nr3c2+ cells: 5,877), and DG (Nr3c2+ cells: 6,598), but less so in other clusters. Next, we focused on the number of Fkbp5-expressing cells that co-express either Nr3c1, Nr3c2, or both receptors specifically in cluster CA1 (2,631 cells), CA2/CA3 (3,047 cells), and DG (2,799 cells) (Figure 1G). Fkbp5-expressing cells showed a cluster-dependent co-expression pattern with Nr3c1 and Nr3c2, recapitulating the ISH and RNAscope results (number of cells in percent; cluster CA1: Fkbp5+ Nr3c1+ Nr3c2−, 39%; Fkbp5+ Nr3c2+ Nr3c1−, 23%; Fkbp5+ Nr3c1+ Nr3c2+, 38%; cluster CA2/CA3: Fkbp5+ Nr3c1+ Nr3c2−, 11%; Fkbp5+ Nr3c2+ Nr3c1−, 56%; Fkbp5+ Nr3c1+ Nr3c2+, 33%; cluster DG: Fkbp5+ Nr3c1+ Nr3c2−, 34%; Fkbp5+ Nr3c2+ Nr3c1−, 41%; Fkbp5+ Nr3c1+ Nr3c2+, 25%). Along these lines, analyses of the ratio of the number of cells expressing Fkbp5 and Nr3c2, but not Nr3c1, to the number of cells expressing Fkbp5 and Nr3c1, but not Nr3c2, further confirmed this cluster-dependent relationship of Fkbp5 and the two receptors (Yates’ chi-square = 854.177; p < 2.2e‒16), with cluster CA2/CA3 representing a primarily Fkbp5+ Nr3c2+ population.
Importantly, the specific hippocampal expression patterns of Fkbp5, Nr3c1, and Nr3c2 were also confirmed at the protein level via triple immunofluorescence (Figure 2; Figure S1). Notably, at this level, using high-resolution immunofluorescence, it is clear that the GR (Nr3c1) is most prevalent in CA1, whereas FKBP5 is most prominently overlapping with the MR (Nr3c2) in CA2 (Figure 2B).
Figure 2. FKBP5, GR, and MR protein expression patterns in the mouse hippocampus.
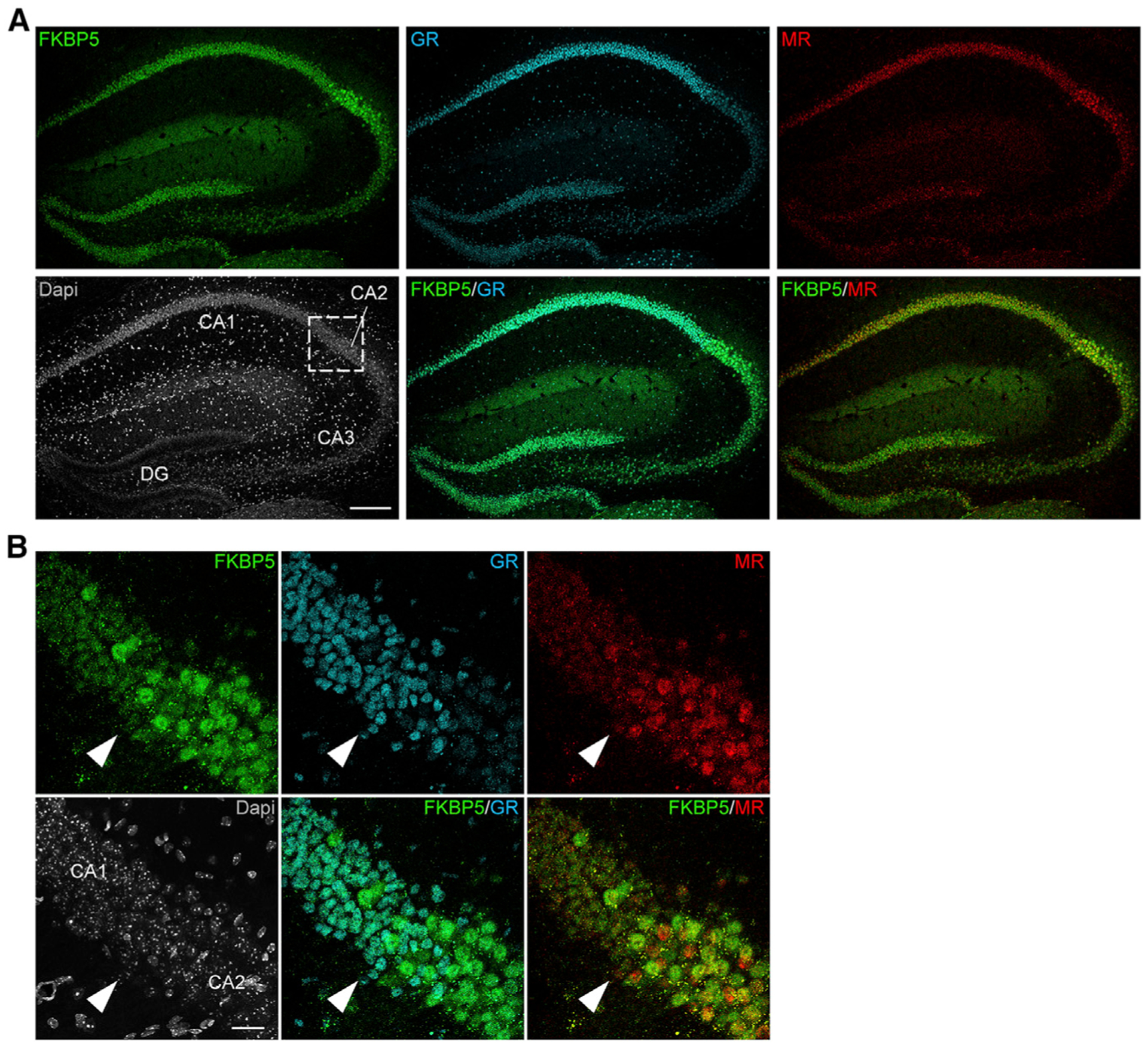
(A) Coronal sections of C57BL/6J mice (n = 5) were stained for FKBP5 (FK506-binding protein 51), GR, and MR protein as well as DAPI (4′,6-diamidino-2-phenylindole). FKBP5 and MR exhibit similar expression patterns in the hippocampus, which is distinct from that of the GR. Scale bar, 250 μm.
(B) Higher magnification images of the approximate CA1–CA2 boundary (white arrow) in the hippocampus. FKBP5 and MR expression is most prominent in hippocampal subregion CA2, whereas GR expression is strongly expressed in the CA1. Scale bar, 25 μm. See also Figure S1.
Overall, our mapping data of the hippocampus suggest that baseline FKBP5 expression might primarily be regulated by the MR rather than the GR.
The MR, not the GR, regulates FKBP5 expression under baseline conditions in primary hippocampal neurons
In order to further explore whether the MR is primarily regulating baseline FKBP5 expression, we utilized an oligoIP method (Zannas et al., 2019) to assess the dynamics of MR and GR binding to functional Fkbp5-glucocorticoid response elements (GREs) within the gene’s promoter region and determine the impact on FKBP5 levels in hippocampal primary neurons of C57BL/6J mice (Figure 3A). First, we confirmed that induction of FKBP5 expression by dexamethasone (Dex), a synthetic stress hormone and potent GR-selective agonist, is primarily mediated by the GR. This was demonstrated by increased GR binding and decreased MR binding to the Fkbp5-GRE oligonucleotide in response to increasing concentrations of Dex (Figures 3B–3E).
Figure 3. GRs regulate FKBP5 expression under Dex-stimulated, but not under baseline, conditions in mouse primary hippocampal neurons.
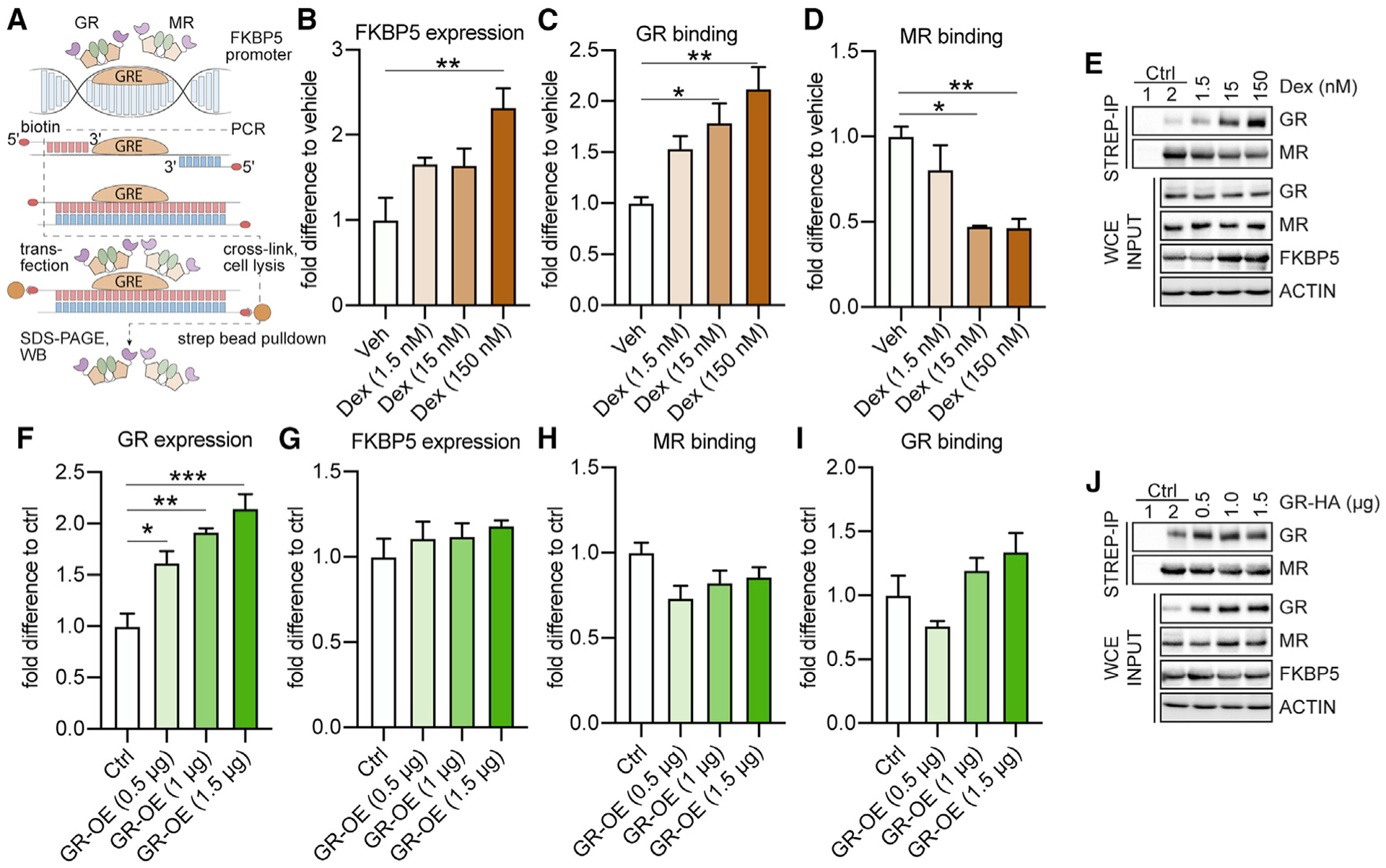
The effects of altered GR levels on FKBP5 expression and on MR and GR binding to two Fkbp5-glucocorticoid response elements (GREs) were examined under baseline conditions and following dexamethasone (Dex) stimulation in primary hippocampal neurons, using biotinylated oligonucleotide immunoprecipitation (oligoIP).
(A) Schematic summary of the experimental setup. After oligoIP, MR and GR binding to the Fkbp5-GRE oligonucleotide were quantified by western blotting using antibodies specific for the respective receptor. In addition, FKBP5, MR, or GR expression levels were quantified by western blotting from whole-cell extracts (WCEs).
(B) Dex treatment increased FKBP5 levels in a concentration-dependent manner (F3,8 = 7.278, p < 0.05).
(C and D) GR binding to the Fkbp5-GRE oligonucleotide was increased following Dex treatment (F3,8 = 8.9, p < 0.01), while MR binding was decreased (F3,8 = 10.35, p < 0.01).
(E) Example blots of (B)–(D). Ctrl (control) 1: magnetic beads lacking conjugated streptavidin. Ctrl 2: cells treated with vehicle (Veh).
(F) Transfection with a GR-expressing plasmid concentration dependently increased GR protein expression (F3,8 = 19.25, p < 0.001).
(G–I) FKBP5 expression and MR and GR binding to the Fkbp5-GRE oligonucleotide are not altered following GR overexpression (OE) under baseline conditions.
(J) Example blots of (F)–(I). Ctrl 1: magnetic beads lacking conjugated streptavidin. Ctrl 2: cells transfected with empty Ctrl vector.
One-way analysis of variance (ANOVA) + Bonferroni post hoc test: *p < 0.05, **p < 0.01, ***p < 0.001. Data are presented as mean + SEM (n = mean derived from three independent experiments).
Next, we assessed FKBP5 levels as well as receptor binding following GR or MR manipulation. Interestingly, dose-dependent overexpression (OE) of the GR under baseline conditions did not alter FKBP5 expression, nor were there any significant changes in MR or GR binding to the Fkbp5-GRE oligonucleotide (Figures 3F–3J). In contrast, MR overexpression led to a significant, dose-dependent increase in FKBP5 expression and enhanced MR binding to the Fkbp5-GRE oligonucleotide. GR binding was significantly decreased following MR overexpression (Figures 4A–4E).
Figure 4. MR drives FKBP5 expression under baseline conditions, which fine-tunes GR stress responsiveness in mouse primary hippocampal neurons.
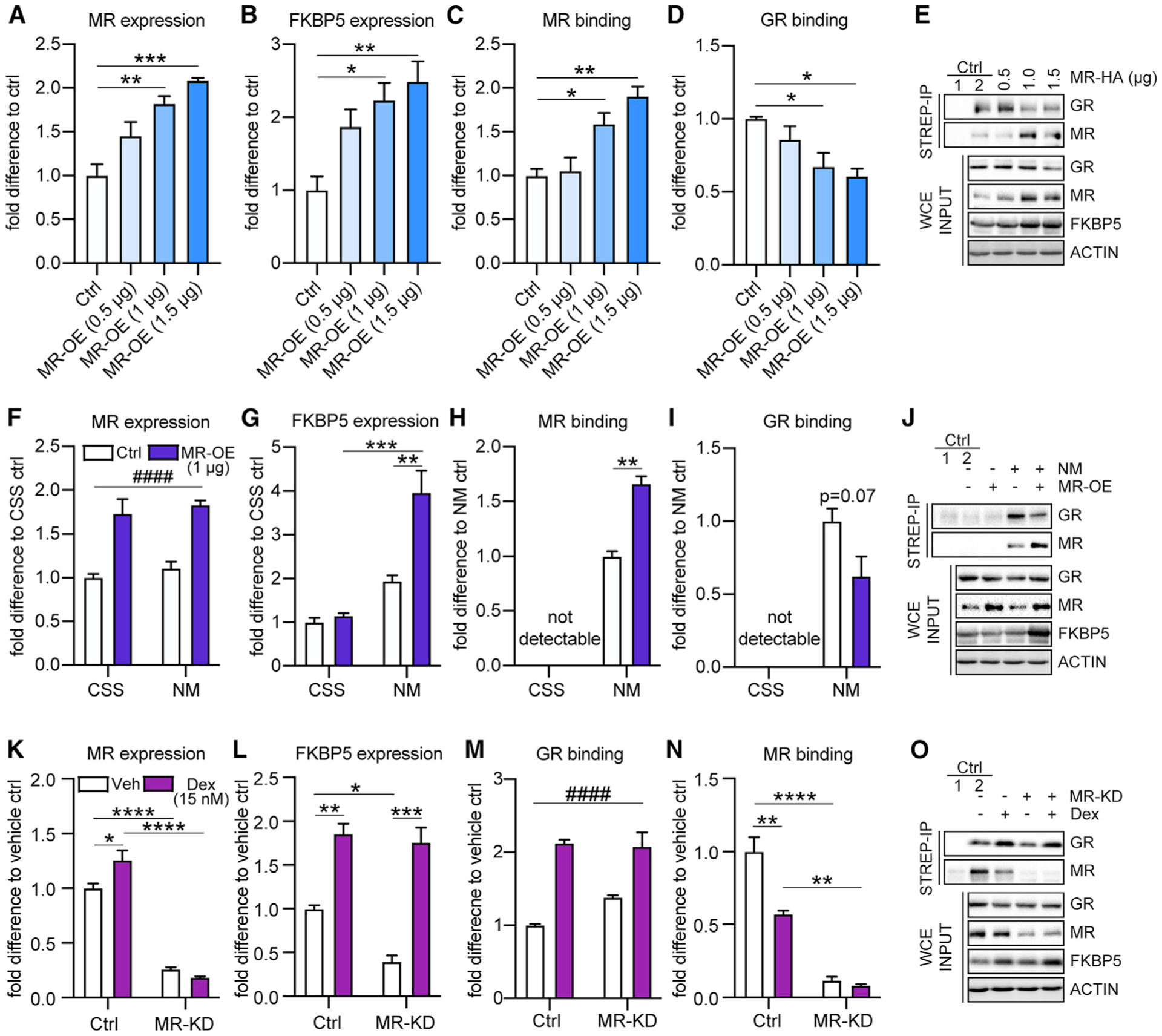
The effects of altered MR levels on FKBP5 expression as well as on receptor binding to GREs within Fkbp5’s promoter region were examined under baseline conditions, in medium supplemented with charcoal-stripped serum (CSS, steroid-free; resulting in no receptor activation) and following Dex stimulation using biotinylated oligoIP in primary hippocampal neurons.
(A) Transfection of a MR-expressing plasmid concentration dependently increased MR protein expression (F3,8 = 18.30, p < 0.001).
(B–D) Under baseline conditions, OE of MR significantly increased FKBP5 expression (F3,8 = 7.578, p < 0.01) as well as MR binding to the Fkbp5-GRE oligonucleotide (F3,8 = 12.98, p < 0.01), while GR binding was decreased (F3,8 = 6.461, p < 0.05).
(E) Example blots of (A)–(D). Ctrl 1: magnetic beads lacking conjugated streptavidin. Ctrl 2: cells transfected with empty Ctrl vector.
(F–I) Only under normal media (NM) conditions, OE of MR (main treatment effect F1,8 = 57.60, p < 0.0001) significantly increased FKBP5 expression (treatment-by-condition interaction F1,8 = 12.71, p < 0.01) as well as MR binding to the Fkbp5-GRE oligonucleotide (t4 = 8.241, p < 0.01), while GR binding was decreased (t4 = 2.351, p = 0.07). These effects were abolished in neurons cultured in medium supplemented with CSS.
(J) Example blots of (F)–(I). Ctrl 1: magnetic beads lacking conjugated streptavidin. Ctrl 2: cells transfected with empty Ctrl vector.
(K) Knockdown (KD) of MR led to significantly reduced MR expression under vehicle (Veh) and Dex conditions. In addition, Dex treatment increased MR expression under control conditions (treatment-by-condition interaction F1,8 = 11.71, p < 0.01).
(L) MR KD significantly reduced FKBP5 expression under vehicle conditions. In contrast, Dex treatment significantly increased FKBP5 expression, which was even more pronounced under MR KD conditions (treatment-by-condition interaction F1,8 = 5.168, p < 0.05).
(M) Dex treatment significantly increased GR binding to the Fkbp5-GRE oligonucleotide independent of MR expression (main treatment effect F1,8 = 83.18, p < 0.0001).
(N) KD of MR significantly decreased MR binding to the Fkbp5-GRE oligonucleotide. MR binding was significantly decreased following Dex treatment under control conditions (treatment-by-condition interaction F1,8 = 14.34, p < 0.01).
(O) Example blots of (K)–(N). Ctrl 1: magnetic beads lacking conjugated streptavidin. Ctrl 2: vehicle treated cells transfected with scrambled small interfering RNA (scr-siRNA) Ctrl vector.
One-way ANOVA + Bonferroni post hoc test, two-way ANOVA + Bonferroni post hoc test, and unpaired, two-tailed Student’s t test for simple comparisons: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ####p < 0.0001 (two-way ANOVA main treatment effect). Data are presented as mean + SEM; n = mean of three independent experiments.
In addition, we assessed FKBP5 levels and receptor binding following MR overexpression under control (normal medium) and steroid-free, non-receptor-activating conditions (medium supplemented with charcoal-stripped serum [CSS]) (Figures 4F–4J). While MR overexpression significantly increased FKBP5 levels in neurons cultured in normal medium, this effect was absent in cells supplemented with CSS (Figure 4G). FKBP5 expression was also significantly higher when MR was overexpressed under normal versus CSS conditions. Along these lines, MR binding to the Fkbp5-GRE oligonucleotide was not detectable in CSS medium. In contrast, MR binding was significantly increased following MR overexpression in normal medium conditions (Figure 4H). Moreover, GR binding was not detectable under CSS conditions. In addition, there was a trend toward decreased GR binding in the MR overexpressing group compared to the controls under normal medium conditions (Figure 4I). Taken together, these data further emphasize that regulation of baseline FKBP5 levels not only depends on MR expression, but also its activation.
Next, we assessed the impact of a MR knockdown on FKBP5 levels as well as on receptor binding to the Fkbp5-GRE oligonucleotide under control conditions and following Dex treatment. MR knockdown significantly reduced FKBP5 levels under vehicle conditions without impairing Dex-induced enhancement of FKBP5 expression (Figures 4K–4O). In fact, compared to vehicle, the Dex-mediated induction of FKBP5 was even more pronounced under MR knockdown conditions (Figure 4L). In addition, GR binding to the Fkbp5-GRE oligonucleotide was significantly increased following Dex treatment while the opposite effect was observed for MR binding under vehicle conditions (Figures 4M and 4N). As expected, MR knockdown significantly decreased MR binding to the Fkbp5-GRE oligonucleotide, independent of treatment. Taken together, these data suggest that the MR, rather than the GR, regulates hippocampal FKBP5 levels at baseline and thereby fine-tunes GR stress responsivity.
GR activation increases Fkbp5 mRNA levels in vivo, while GR deletion does not alter Fkbp5 expression
It is well established that stress and GR activity induce Fkbp5 expression in the mouse brain (Lee et al., 2010; Scharf et al., 2011; Wagner et al., 2012). Consistent with this, and our above results in primary hippocampal neurons, we found that Fkbp5 mRNA expression was significantly increased in the hippocampus of C57BL/6J mice, 4 h after injection with the potent GR-selective agonist Dex (Figure 5A), as well as following overnight treatment with corticosterone via drinking water (Figure S2A). However, in support of the hypothesis that baseline Fkbp5 levels are primarily regulated by the MR, pharmacological blockade of GR, administering the GR antagonist RU486 overnight via drinking water, induced no significant changes in Fkbp5, Nr3c1, or Nr3c2 mRNA expression in the hippocampus of C57BL/6J mice (Figure S2B). Likewise, mice lacking the GR in forebrain glutamatergic neurons (GRNex-CKO) showed no significant differences in hippocampal Fkbp5 mRNA expression compared to littermate controls (Figure 5B). In addition, hippocampal Nr3c2 mRNA expression was also not altered in GRNex-CKO mice (Figure 5C). Thus, while Dex- and corticosterone-mediated GR activation enhances Fkbp5 expression, pharmacological inhibition or absence of the GR does not appear to alter baseline Fkbp5 levels.
Figure 5. Basal Fkpb5 mRNA levels in the hippocampus are regulated by the MR.
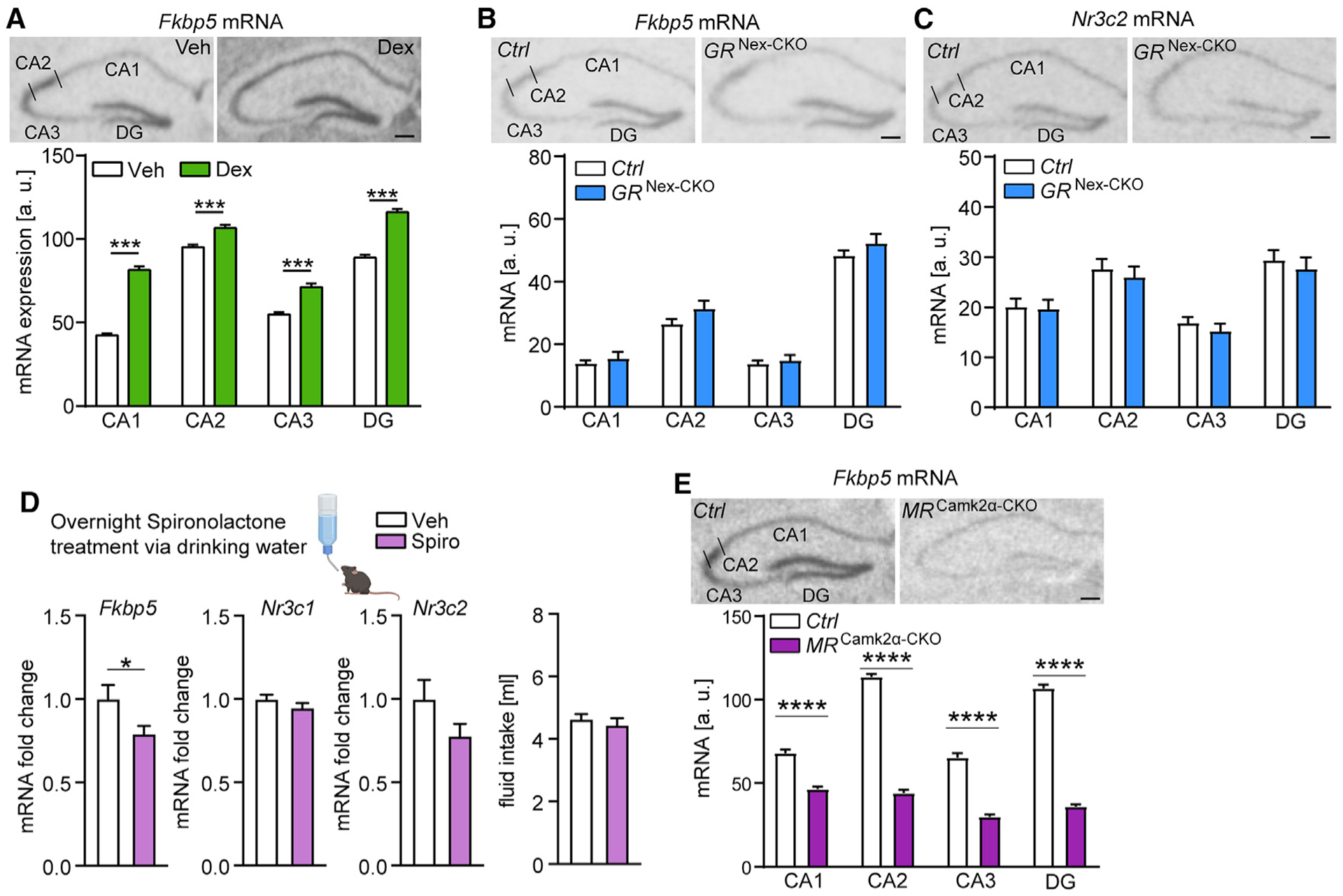
(A) GR activation (Dex injection) leads to increased hippocampal Fkbp5 mRNA expression in C57BL/6J mice determined by ISH. (Top panel) Representative autoradiographs of hippocampal Fkbp5 mRNA expression. (Lower panel) Quantified expression of Fkbp5 mRNA (treatment-by-subregion interaction F3,180 = 25.72, p < 0.0001; n = 23–24 mice per group).
(B and C) No alterations in hippocampal Fkbp5 (B) and Nr3c2 (C) mRNA expression in glutamatergic GR knockout mice (n = 9–11 mice per group). (Top panel) Representative autoradiographs of hippocampal Fkbp5 or Nr3c2 mRNA expression determined by ISH. (Lower panel) Quantified expression of Fkbp5 or Nr3c2 mRNA.
(D) Fkbp5 mRNA expression is decreased in the hippocampus of C57BL/6J mice following overnight treatment with the MR antagonist spironolactone (Fkbp5, t19 = 2.108, p < 0.05; n = 10–11 mice per group), while Nr3c1 and Nr3c2 mRNA levels are not altered. Overnight fluid intake did not differ between vehicle- and spironolactone-treated mice.
(E) MR deletion in forebrain neurons (MRCamk2α-CKO) leads to lower hippocampal Fkbp5 mRNA expression determined by ISH (genotype-by-hippocampal subregion interaction F3,88 = 77.2, p < 0.0001; n = 10–14 mice per group). (Top panel) Representative autoradiographs of hippocampal Fkbp5 mRNA expression. (Lower panel) Quantified expression of Fkbp5 mRNA.
Areas of interest are CA1, CA2, CA3, and DG. Two-way ANOVA + Bonferroni post hoc test and unpaired, two-tailed Student’s t test for simple comparisons: *p < 0.05, ***p < 0.001, ****p < 0.0001. Data are presented as mean + SEM. Scale bars, 250 μm. See also Figures S2 and S3.
Pharmacological inhibition and conditional deletion of MR decrease hippocampal Fkbp5 mRNA levels
Given the potentially distinct GR- and MR-specific roles in HPA axis regulation under baseline versus stress conditions, we aimed to further dissect the contribution of MR in the regulation of hippocampal Fkbp5 expression in vivo. Thus, we pharmacologically blocked the MR in wild-type animals and generated different conditional MR knockout mouse lines to investigate the impact of receptor depletion on hippocampal Fkbp5 and Nr3c1 mRNA expression. C57BL/6J mice treated with the MR antagonist, spironolactone, via drinking water exhibited a significant downregulation of Fkbp5 mRNA expression in the hippocampus compared to vehicle controls, while Nr3c1 and Nr3c2 mRNA levels remained unaffected (Figure 5D). Along these lines, forebrain-specific MR knockout mice showed significantly decreased hippocampal Fkbp5 mRNA levels compared to control littermates (Figure 5E). Similarly, Fkbp5 mRNA levels were significantly decreased in mice lacking MR specifically in the CA2 region of the hippocampus (MRAmigo2-CKO mice; Figure S3A). In addition, Nr3c1 mRNA levels were significantly increased in MR-CKOAmigo2-CKO mice (Figure S3B). These data further support the observation that Fkbp5 expression is particularly sensitive to MR regulation during baseline activity of glucocorticoids.
Forebrain MR deletion leads to GR hypersensitivity during the acute stress response
Given the distinct Fkbp5 and Nr3c1 mRNA expression patterns in the conditional MR knockout mouse lines, we explored the impact of acute stress on hippocampal gene expression and peripheral corticosterone levels in MRCamk2α-CKO mice. Fkbp5 mRNA expression is robustly increased 4 h after exposure to an acute Dex treatment or restraint stress (30 min) (Scharf et al., 2011). In order to obtain a similarly strong Fkbp5 induction, while also ensuring that the brain and plasma collection occurs during the HPA axis response and not recovery phase, we applied a prolonged, 4-h restraint stressor. Inaccordance withourearlier results, hippocampal Fkbp5 mRNA levels were decreased in MRCamk2α-CKO mice compared to controls. Moreover, 4 h of acute restraint stress led to increased Fkbp5 expression in both genotypes (Figure 6A). However, the stress-induced induction of Fkbp5 mRNA levels (delta to baseline) in the CA1, CA2, and DG of MRCamk2α-CKO mice was significantly larger compared to littermate controls (Figure 6B).
Figure 6. Forebrain-specific MR deletion leads to GR hypersensitivity during the acute stress response.
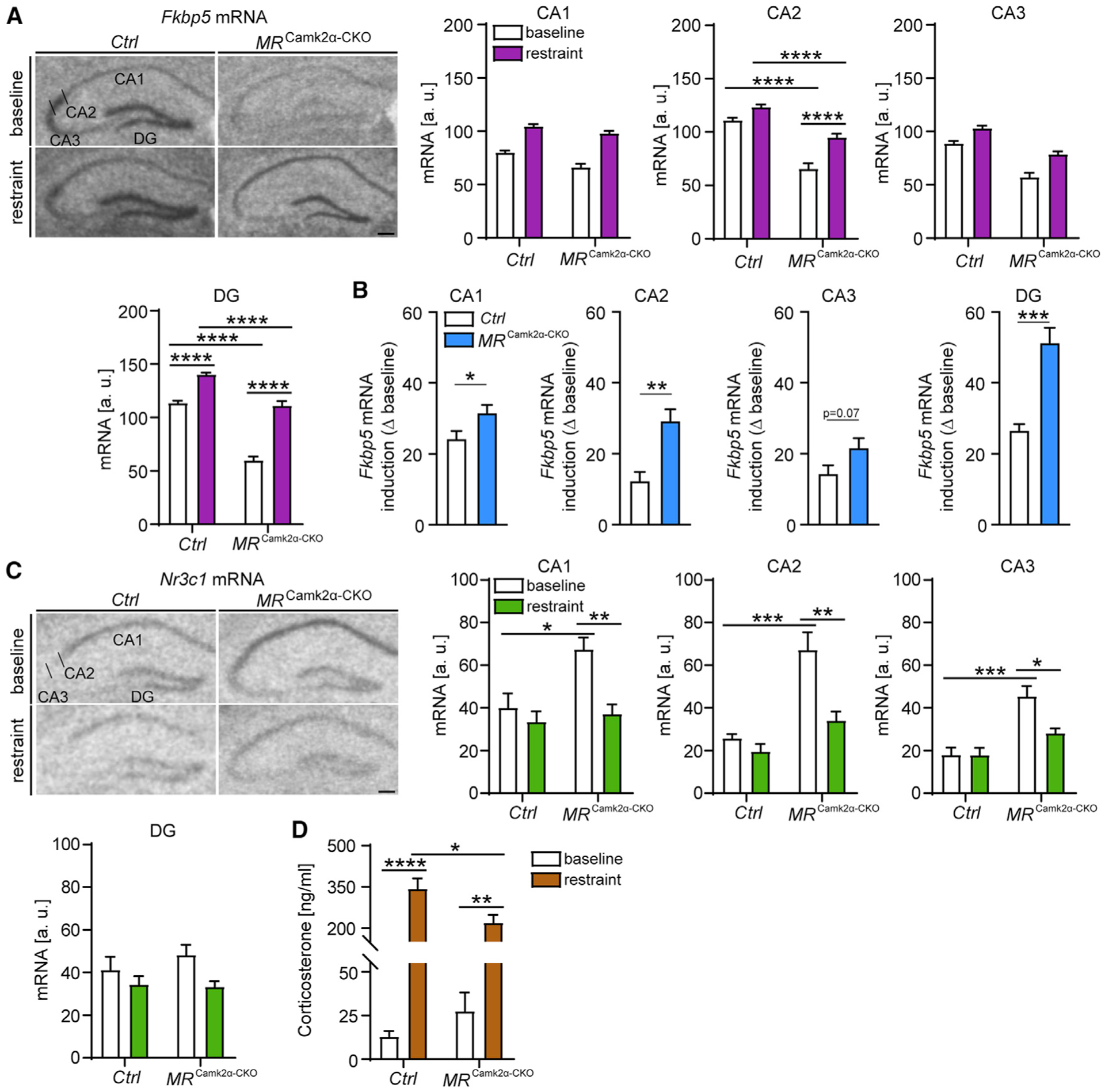
(A) (Left) Representative autoradiographs of hippocampal Fkbp5 mRNA expression in MRCamk2α-CKO mice. CA1, CA2, CA3, and DG show quantified expression of Fkbp5 mRNA. Fkbp5 levels are decreased in the CA1, CA2, CA3, and DG of conditional forebrain MR knockout mice. 4 h of restraint stress increases Fkbp5 expression in the hippocampus, which is even more pronounced in MRCamk2α-CKO mice (CA1: main condition effect, F1,21 = 137.8, p < 0.0001; main genotype effect, F1,21 = 18.03, p < 0.001; CA2: genotype-by-condition interaction, F1,21 = 6.183, p < 0.05; CA3: main condition effect, F1,21 = 36.71, p < 0.0001; main genotype effect, F1,21 = 88.06, p < 0.0001; DG: genotype-by-condition interaction, F1,21 = 16.5, p < 0.001).
(B) The induction of Fkbp5 mRNA by 4-h restraint stress (delta to baseline) in CA1, CA2, and DG of MRCamk2α-CKO mice is increased compared to littermate controls (CA1, t11 = 2.315, p < 0.05; CA2, t11 = 4.179, p < 0.01; CA3, t11 = 2.01, p = 0.07; DG, t11 = 5.717, p < 0.0001).
(C) (Left) Representative autoradiographs of hippocampal Nr3c1 mRNA expression in MRCamk2α-CKO mice. CA1, CA2, CA3, and DG show quantified expression of Nr3c1 mRNA. Nr3c1 levels are increased in the CA1, CA2, and CA3 of conditional forebrain MR knockout mice under baseline conditions. In contrast, 4 h of restraint stress decreases Nr3c1 expression in the hippocampus of MRCamk2α-CKO mice, while no changes are observed in littermate controls (CA1: genotype-by-condition interaction, F1,19 = 4.794, p < 0.05; CA2: genotype-by-condition interaction, F1,19 = 6.144, p < 0.05; CA3: genotype-by-condition interaction, F1,19 = 5.399, p < 0.05). No significant changes in Nr3c1 mRNA expression were observed in the DG.
(D) 4 h of restraint stress leads to increased corticosterone levels, an effect that is significantly blunted in in MRCamk2α-CKO mice (genotype-by-condition interaction, F1,19 = 6.228, p < 0.05).
Two-way ANOVA + Bonferroni post hoc test and unpaired, two-tailed Student’s t test for simple comparisons: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are presented as mean + SEM; n = 4–7 mice per group. Scale bars, 250 μm.
MRCamk2α-CKO mice showed significantly increased Nr3c1 mRNA levels under baseline conditions compared to control littermates across all hippocampal subregions. Remarkably, acute restraint stress significantly decreased Nr3c1 mRNA expression in the CA1, CA2, and CA3 of MRCamk2α-CKO mice, without producing an effect in control animals (Figure 6C). No stress or genotype-dependent changes in Nr3c1 mRNA expression were observed in the DG.
Given the involvement of the hippocampus in HPA axis regulation and the striking, stress-induced changes in Fkbp5 and Nr3c1 mRNA levels observed in MRCamk2α-CKO mice, we assessed whether forebrain-specific MR deletion would also alter peripheral corticosterone levels under control and/or stress conditions. Notably, MRCamk2α-CKO mice demonstrated a significantly lower stress-induced increase in glucocorticoid levels compared to control littermates (Figure 6D). Overall, our results suggest that MR-dependent changes in baseline Fkbp5 expression may modify GR sensitivity to ultimately alter GR-dependent stress responses and HPA axis regulation.
DISCUSSION
It is well established that MRs are involved in basal activity and onset of stress-induced HPA axis activity, whereas GRs primarily drive its termination. An imbalance between MR- and GR-mediated actions may lead to an exaggerated or inadequate HPA axis response to stress, impaired containment, delayed recovery, and compromised adaptation (Harris et al., 2013). Consequently, such changes may lead to a condition of neuroendocrine dysregulation and impaired behavioral adaptation, which can potentially aggravate stress-induced deterioration and promote susceptibility to mood and anxiety disorders (De Kloet et al., 1998, 2018). However, the underlying molecular mechanisms of how a shift in the balanced actions of these two receptors is produced are still poorly understood. Our data illustrate that MR-mediated regulation of baseline Fkbp5 expression alters GR sensitivity to glucocorticoids during stress. Thus, FKBP5 acts as a key regulator of HPA axis activity by fine-tuning the MR:GR balance in the hippocampus.
GRs are widely distributed throughout the brain, with highest expression levels found in stress-regulating centers such as the PVN as well as in the prefrontal cortex-hippocampal-amygdala circuitry. Conversely, MRs show a more distinct expression pattern, most prominently in the hippocampus, amygdala, and the lateral septum (Ahima et al., 1991; Arriza et al., 1988; van Eekelen et al., 1991; Hartmann et al., 2017; Patel et al., 2000). Alternatively, Fkbp5 is ubiquitously expressed throughout the adult mouse brain under basal conditions, and it can show a pronounced increase in expression in response to various stressors, Dex, and cortisol treatment (Lee et al., 2010; Scharf et al., 2011; Wagner et al., 2012).
Although GR expression is high in the PVN (acting as key mediator of the negative feedback), basal Fkbp5 mRNA levels are very low (Haüsl et al., 2021; Scharf et al., 2011). In contrast, the most pronounced expression of Fkbp5 under baseline conditions has been found in the hippocampus. Intriguingly, regions with low basal Fkbp5 expression showed a higher stress-mediated Fkbp5 mRNA induction than did regions with high basal expression. Such region-specific expression differences of Fkbp5 under baseline and stress conditions may be explained by the expression and activity of different transcription factors. It is well established that GR activity, especially after stress, is able to induce Fkbp5 expression. Recent evidence also points to a potential regulation of FKBP5 via MR signaling. Our results consistently illustrate that Nr3c2 (MR) and Fkbp5 share the same mRNA and protein expression profiles in all hippocampal subregions. In contrast, the expression pattern of Nr3c1 (GR) and Fkbp5 is more distinct. In addition, we found a strong positive correlation between hippocampal Fkbp5 and Nr3c2 expression under baseline conditions, while there was no correlation between Nr3c1 and Fkbp5. Importantly, we were able to recapitulate these expression profiles, and confirmed a positive correlation of FKBP5 and NR3C2 expression, in human hippocampus from postmortem samples of healthy individuals.
OligoIP experiments in mouse primary hippocampal neurons further elucidated the dynamics and binding characteristics of the MR and GR to GREs within the promoter region of the Fkbp5 gene. In addition, the impact of the MR and GR on FKBP5 expression was assessed under baseline and stress-like conditions (induced by treatment with the GR agonist Dex). Under baseline conditions, FKBP5 expression was strongly regulated by changes in MR levels and dependent on MR binding to the Fkbp5-GREs. Accordingly, overexpression of MR resulted in increased FKBP5 expression and MR binding to the Fkbp5-GRE oligonucleotide only under normal media conditions, but not when the medium was supplemented with CSS to inhibit receptor activation. At the same time alterations in GR levels had no impact on GR binding to the Fkbp5-GRE oligonucleotide or on FKBP5 expression. Alternatively, Dex treatment enhanced GR binding to the Fkbp5-GREs as well as FKBP5 expression, while MR binding was decreased. Similar observations of increased GR binding to a different Fkbp5-GRE (within intron 5, GRE2) has been reported in the hippocampus of rats following acute challenges, including forced swim stress (Mifsud and Reul, 2016). Interestingly, acute stress also enhances heterodimerization of the GR and MR as well as binding of the MR to GRE2 up to 3 h after stress onset. In contrast, we observed a dose-dependent decrease of MR binding to the Fkbp5-GRE oligonucleotide following GR activation (Dex treatment). These discrepancies might be due to the different GREs within the Fkbp5 gene, type of receptor activation (stress versus Dex treatment), timing of the analyses (24 versus up to 3 h after GR activation), and/or differences in experimental conditions and techniques. Of note, the 70-bp-long biotinylated oligonucleotide probes that were used in our experiments do not contain the same chromatin structure or epigenetic signature as the endogenous Fkbp5 gene. In addition, there are more GREs within the Fkbp5 gene (including those in introns). Thus, despite being a valuable tool, this method only represents an estimate for studying the interactions between transcription factors such as MRs and GRs and their specific DNA binding sites.
In addition to our oligoIP analyses, GR activation (using Dex) elevated hippocampal Fkbp5 mRNA expression in C57BL/6J mice, confirming previous findings (Scharf et al., 2011). Interestingly the induction of Fkbp5 expression was even more pronounced in hippocampal subregions CA1 and DG, which express lower basal Fkbp5 mRNA levels (compared to CA2 and CA3) as well as Nr3c2 levels, but high levels of Nr3c1 mRNA. Notably, pharmacological inhibition of the GR in adult C57BL/6J mice as well as deletion of the GR in glutamatergic forebrain neurons (GRNex-CKO mice) did not alter Fkbp5 or Nr3c2 mRNA expression in the hippocampus. In contrast, deletion of the MR in the forebrain (MRCamk2α-CKO mice) or hippocampal CA2 region (MRAmigo2-CKO mice) resulted in lower basal Fkbp5 mRNA levels, which is consistent with recent reports (McCann et al., 2021; van Weert et al., 2019). These effects were not likely due to compensatory mechanisms during development since pharmacological inhibition of the MR in adult C57BL/6J mice also resulted in decreased hippocampal Fkbp5 mRNA levels. However, morphological changes in the CA2 have previously been reported in MRAmigo2-CKO mice (McCann et al., 2021). Thus, manipulation of the MR and consequently the lack of FKBP5 and increased GR sensitivity in the hippocampus may not only contribute to changes in gene expression/regulation and HPA axis feedback, but also result in profound structural and cellular alterations. In addition, although the recombination pattern of the Camk2α-Cre largely resembles that of the Nex-Cre (i.e., primarily confined to forebrain glutamatergic neurons of the cortex and hippocampus), subtle expression is also observed in the caudate putamen, central amygdala, septum, and bed nucleus of the stria terminalis (BNST) (Hartmann et al., 2017). Thus, we cannot completely rule out a potential contribution of these brain regions to the observed phenotype and comparative findings shown in this study. Taken together, these findings support the hypothesis that MRs primarily drive FKBP5 expression in the hippocampus under basal conditions.
Confirming previous studies, basal Nr3c1 levels in the hippocampus were increased in both conditional MR knockout mouse lines (ter Horst et al., 2012; McCann et al., 2021), which is likely due to adaptive/compensatory changes following early onset of MR deletion. An interactive regulation of the two receptors has also been demonstrated in global GR overexpressing mice, where increased GR coincides with lower hippocampal Nr3c2 mRNA levels (Reichardt et al., 2000). Likewise, MR forebrain overexpressing mice show lower Nr3c1 mRNA levels in the hippocampus (Rozeboom et al., 2007).
The acute restraint stress experiment in MRCamk2α-CKO mice further demonstrates the complex interplay and dynamic regulation of the MR:GR balance, as well as the extent to which these interactions can be modulated by FKBP5. Under baseline conditions MRCamk2α-CKO mice expressed high levels of hippocampal Nr3c1, which enhanced their feedback sensitivity during the acute stress response. In addition, Fkbp5 levels were low, most likely due to the lack of MRs in the hippocampus. It is well established that FKBP5 protein reduces the sensitivity of the GR toward glucocorticoids (Binder, 2009; Denny et al., 2000; Hartmann et al., 2012; Klengel et al., 2013; Scammell et al., 2001; Schülke et al., 2010; Touma et al., 2011; Westberry et al., 2006; Wochnik et al., 2005). Thus, increased Nr3c1 levels, together with the low expression of Fkbp5 in the hippocampus of MRCamk2α-CKO mice under baseline conditions, shift the hippocampal GR into a state of “hypersensitivity” during the acute stress response. Indeed, Nr3c1 levels in MRCamk2α-CKO mice were reduced in response to acute stress, likely in order to counteract the GR hypersensitivity (an effect that was not observed in littermate controls). At the same time, restraint stress resulted in increased hippocampal Fkbp5 mRNA levels, which was even more pronounced in MRCamk2α-CKO mice most likely due to the GR hypersensitivity during the acute stress response. Importantly, note that in addition to Fkbp5 regulation, altered MR expression can have multiple other effects on gene expression and cell signaling, all of which might ultimately regulate GR sensitivity.
The importance of a balanced action of corticosterone via MRs and GRs has repeatedly been suggested (de Kloet et al., 2005; Oitzl et al., 2010). Previous studies report that MRCamk2α-CKO mice demonstrate a distinct and dynamic pattern of circulating corticosterone depending on the type and severity of a stimulus as well as its duration. MR deletion in forebrain neurons resulted in increased basal corticosterone levels (ter Horst et al., 2012). The fact that we did not observe significantly increased basal corticosterone levels in MRCamk2α-CKO mice might be due to the relatively small sample size. MRCamk2α-CKO mice also demonstrate an initial higher corticosterone response to a short period (5 or 10 min) of restraint stress (ter Horst et al., 2012; Ter Horst et al., 2014).
This effect is abolished after a 40-min restraint stress (Berger et al., 2006). Longer lasting stimuli (90–120 min), such as exposure to a novel environment, result in significantly lower corticosterone levels in MRCamk2α-CKO mice compared to littermate controls, which is independent of prior restraint stress (ter Horst et al., 2012). Along these lines, we were able to demonstrate that MRCamk2α-CKO mice showed a reduced corticosterone response to 4 h of restraint stress. Collectively, these results point toward a more efficient negative feedback mediated by strengthened GR actions on neuroendocrine control, and they are consistent with the hippocampal Nr3c1 and Fkbp5 mRNA expression profiles. Interestingly, global GR overexpressing mice demonstrate an enhanced glucocorticoid feedback (Reichardt et al., 2000; Ridder et al., 2005).
In summary, our current data reveal another crucial role of FKBP5 in regulating HPA axis activity by acting as a mediator of the MR:GR balance in the hippocampus. Our findings demonstrate that FKBP5 levels under baseline conditions are dependent on MR levels, whereas the GR is primarily involved in driving FKBP5 induction following its activation (i.e., Dex treatment). Within the hippocampus of mice and humans, Nr3c2 expression is much more closely correlated with Fkbp5 than with Nr3c1 levels. In addition, MR signaling regulates GR sensitivity to stress-induced glucocorticoid release by modulating baseline Fkbp5 expression in the hippocampus (Figure 7). This provides additional insights into the molecular mechanisms underlying the MR:GR balance hypothesis. Future studies should address whether (basal and high glucocorticoid-induced) Fkbp5 levels might possibly also alter MR sensitivity. Taken together, our findings suggest that therapeutic targeting of MR, GR, and FKBP5 may be complementary in manipulating CNS and peripheral regulation of stress homeostasis. Our data further underline the important, but largely unappreciated role of MR signaling in stress-related psychiatric disorders.
Figure 7. Working model of MR-dependent changes in baseline FKBP5 expression that modify GR sensitivity to glucocorticoids and subsequent HPA axis activity.
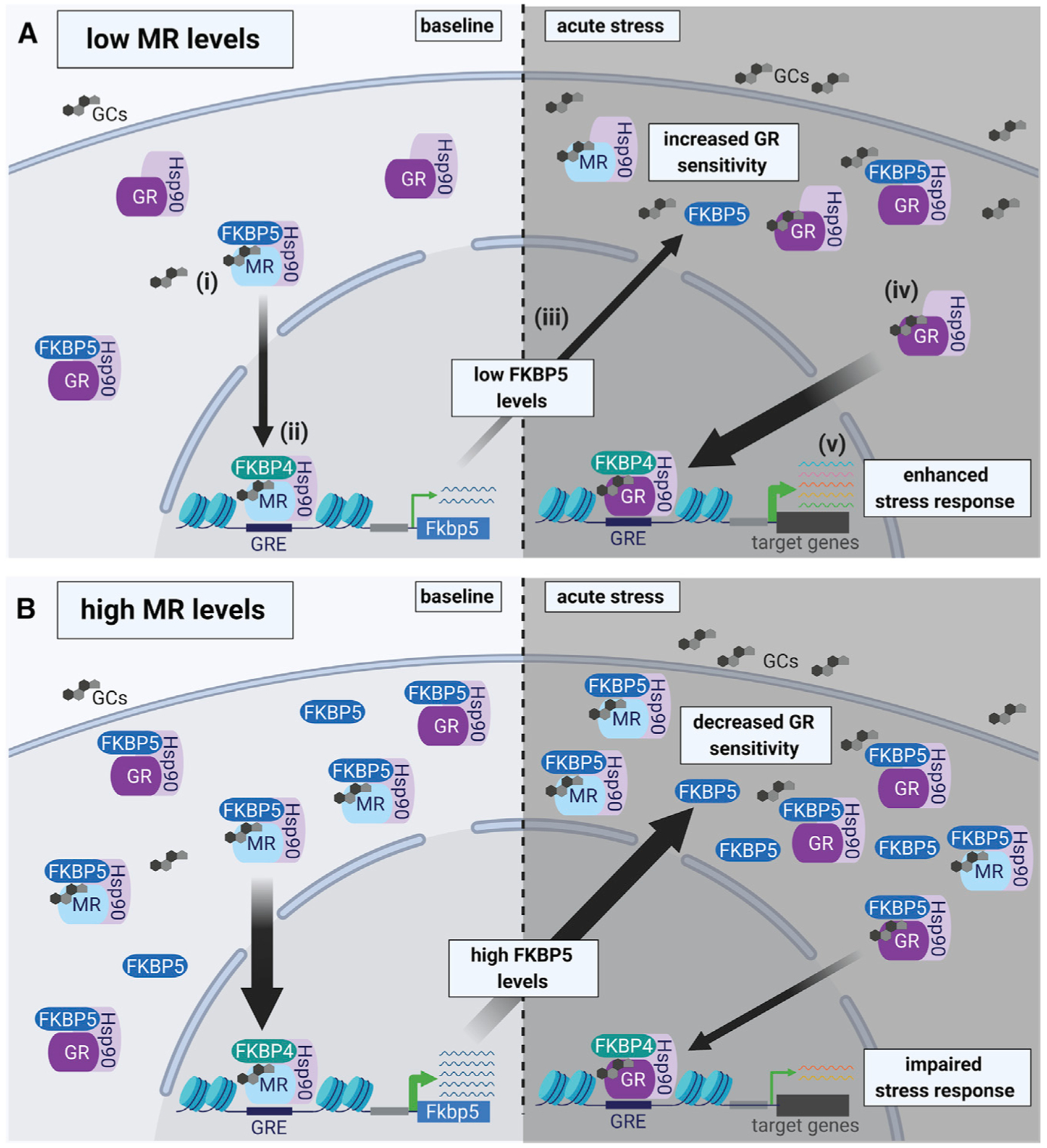
In the hippocampus MRs are largely occupied under basal glucocorticoid conditions, whereas GR occupancy is increased when glucocorticoid levels rise following acute stress. Upon glucocorticoid binding to the MR under baseline conditions (i), FKBP5 is replaced by FKBP4, which promotes the translocation of the MR-Hsp90 complex into the nucleus and subsequent DNA binding (to FKBP5 GREs) (ii). Thereby, the MR increases FKBP5 transcription and translation (iii), which can impact GR sensitivity (iv) and the subsequent stress response during acute stress (v).
(A) Low MR levels result in low FKBP5 expression under baseline conditions. In turn, low FKBP5 levels lead to increased GR sensitivity during acute stress, resulting in an enhanced stress response.
(B) In contrast, high levels of MR promote increased FKBP5 expression under baseline conditions. High levels of FKBP5 result in decreased GR sensitivity during acute stress, which leads to an impaired stress response. Of note, MRs and GRs can function either as homodimers or heterodimers (de Kloet et al., 2005; Mifsud and Reul, 2016). For simplicity, we did not include homodimers and heterodimers of corticosteroid receptors in this illustration. GCs, glucocorticoids; Hsp90, heat shock protein 90; FKBP4, FK506-binding protein 4.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jakob Hartmann (jhartmann@mclean.harvard.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Original/source data for Figures 1D and E (Human postmortem microarray analysis) were publicly available from the Allen Brain Institute and can be downloaded at https://human.brain-map.org/static/download. Original/source data for Figures 1F and 1G (Single-cell RNA sequencing analysis) were publicly available from the McCarroll Lab (Department of Genetics, Harvard Medical School) and can be downloaded at http://dropviz.org.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary hippocampal neuronal cell culture
Primary hippocampal neurons were obtained from C57BL/6J mouse embryos (E17.5–18.5) and maintained in Neurobasal-A medium with 2% B27 and 0.5 mM GlutaMAX-I (GIBCO) at 37°C and 5% CO2 (Dotti et al., 1988). For oligoIP experiments, neurons were transfected at DIV17-19.
Animals and animal housing
Male mice, aged 2 to 4 months, were used for all experiments. Deletion of the GR from forebrain glutamatergic neurons was achieved by breeding GRlox/lox mice (Tronche et al., 1999) to Nex-Cre mice (Goebbels et al., 2006) to obtain GRGlu-Ctrl (GRlox/lox) and GRGlu-CKO (GRlox/lox:Nex-Cre) mice (Hartmann et al., 2017). Conditional MR mutant mice were obtained by breeding MRlox/lox mice to Camk2α–Cre mice (Berger et al., 2006; Minichiello et al., 1999) or Amigo2-Cre mice (McCann et al., 2021), respectively, to obtain Ctrl (MRlox/lox) and MRCamk2α-CKO (MRlox/lox:Camk2α-Cre) or Ctrl (MRlox/lox) and MRAmigo2-CKO (MRlox/lox:Amigo2-Cre) mice. For experiments in wild-type animals, C57BL/6J male mice were obtained from The Jackson Laboratory. All animals were kept under standard laboratory conditions and were maintained on a 12 h light–dark cycle (lights on from 07:00 am to 07:00 pm), with food and water provided ad libitum. All experiments conformed to National Institutes of Health guidelines and were carried out in accordance with the European Communities’ Council Directive 2010/63/EU and the McLean Hospital Institutional Animal Care and Use Committee. All efforts were made to minimize animal suffering during the experiments. The protocols were approved by the committee for the Care and Use of Laboratory animals of the Government of Upper Bavaria, Germany or by the local Institutional Animal Care and Use Committee, respectively.
Human postmortem microarray analysis
Human microarray data were publicly available from the Allen Brain Institute (Hawrylycz et al., 2012). Log2 expression levels from donors (n = 6) were collected for FKBP5, NR3C1 and NR3C2 from each of the hippocampal subregions, CA1, CA2, CA3, CA4 and dentate gyrus (DG). See Table S1 for subject details.
METHOD DETAILS
In situ hybridization
Mice were sacrificed by decapitation following quick anesthesia by isoflurane. Brains were removed, snap-frozen in isopentane at −40°C, and stored at −80°C. Frozen brains were sectioned at −20°C in a cryostat microtome at 18 μm, thaw mounted on Super Frost Plus slides, dried and stored at −80°C. In situ hybridization using 35S UTP labeled ribonucleotide probes (Fkbp5, Nr3c1 and Nr3c2) was performed as described previously (Schmidt et al., 2007). The slides were exposed to Kodak Biomax MR films (Eastman Kodak Co., Rochester, NY) and developed. Autoradiographs were digitized, and expression (i.e., signal intensity in arbitrary units) was determined by optical densitometry utilizing the freely available NIH ImageJ software. Each region of interest (left and right hemisphere) was manually outlined. The mean of two measurements of one brain slice was calculated for each animal. The data were analyzed blindly, always subtracting the background signal of a nearby structure not expressing the gene of interest from the measurements.
RNAscope
RNAscope technology provides a more precise method for multiplex fluorescent cellular level in situ hybridization. Mice were sacrificed by decapitation following quick anesthesia by isoflurane. Brains were removed, snap-frozen in isopentane at −40°C, and stored at −80°C. Frozen brains were sectioned in the coronal plane at −20°C in a cryostat microtome at 18 μm, mounted on Super Frost Plus slides, and stored at −80°C. The RNA Scope Fluorescent Multiplex Reagent kit (cat. no. 320850, Advanced Cell Diagnostics, Newark, CA, USA) was used for mRNA staining. Probes used for staining were: mm-Nr3c1-C1, mm-Fkbp5-C2 and mm-Nr3c2-C3. The staining procedure was performed according to manufacturer’s specifications as described previously (McCullough et al., 2018). Briefly, sections were fixed in 4% paraformaldehyde for 15 min at 4°C. Subsequently, brain sections were dehydrated in increasing concentrations of ethanol. Next, tissue sections were incubated with protease IV for 30 min at room temperature. Probes were hybridized for 2 h at 40°C followed by 4 hybridization steps of the amplification reagents 1 to 4. Next, sections were counterstained with DAPI (4′,6-diamidino-2-phenylindole), coverslipped and stored at 4°C until image acquisition. Sixteen-bit images of the dorsal hippocampus were acquired on a Leica SP8 confocal microscope using a 40x objective (n = 4 mice). For every individual marker, all images were acquired using identical settings for laser power, detector gain, and amplifier offset.
Immunohistochemistry
Mice were deeply anesthetized with isoflurane and perfused intracardially with 4% paraformaldehyde. Brains were removed, post-fixed overnight in 4% paraformaldehyde following overnight incubation in 30% sucrose solution at 4°C, and then stored at −80°C. Frozen brains were coronally sectioned in a cryostat microtome at 35 μm. Triple-immunofluorescence was performed on free-floating sections as described previously (Hartmann et al., 2017). Sections were incubated with primary antibodies (goat anti-FKBP5 (F-14, Santa Cruz, 1:500), rabbit anti-GR (M-20, Santa Cruz, 1:1000) and mouse anti-MR (MABS496, clone 6G1, Millipore-Sigma, 1:100)) overnight at 4°C and labeled with AlexaFluor-conjugated secondary antibodies (1:1000)). Sections were mounted on Super Frost Plus slides and covered with Vectashield mounting medium (Vector Laboratories, Burlingame, USA) containing DAPI. Sixteen-bit images of the dorsal hippocampus were acquired on a Leica SP8 confocal microscope using 10x or 63x objectives (n = 5 mice). For every individual marker, all images were acquired using identical settings for laser power, detector gain, and amplifier offset.
Acute stress paradigm
Mice were restrained in a 50 mL falcon tube. Each tube had 2 holes drilled into the bottom, as well as in the lid to allow the animals to breathe normally and move their tail. After 4 h, animals were removed from the tube, deeply anesthetized with isoflurane and sacrificed by decapitation. Control animals were kept undisturbed in their home cage until sacrifice. Trunk blood was collected in labeled 1.5 mL EDTA-coated microcentrifuge tubes (Sarstedt, Germany) and kept on ice until centrifugation. After centrifugation (4°C, 8000 rpm for 15 min) plasma was removed and transferred to new, labeled tubes and stored at −20°C until corticosterone quantification. For mRNA analysis, brains were removed, snap-frozen in isopentane at −40°C, and stored at −80°C for in situ hybridization.
Corticosterone assessment
Corticosterone (CORT) concentrations were determined by radioimmunoassay using a Corticosterone double antibody 125I RIA kit (sensitivity: 12.5 ng/ml, MP Biomedicals Inc) and were used according to the manufacturers’ instructions. Radioactivity of the pellet was measured with a gamma counter (Packard Cobra II Auto Gamma; Perkin-Elmer). Final CORT levels were derived from the standard curve.
Dexamethasone treatment
Male C57BL/6J mice were administered dexamethasone (Dex, Sigma, St Louis, MO, USA, catalog no. D1159) intraperitoneally (i.p.) at a dose of 10 mg/kg dissolved in saline. The injection volume was 10 μl/g body weight. Vehicle treated mice were injected with the same amount of saline. Injections were performed between 08:00 am and 09:00 am. 4 h after the injection, all mice were sacrificed by decapitation following quick anesthesia by isoflurane. Brains were removed, snap-frozen in isopentane at −40°C, and stored at −80°C until further processing.
CORT, RU486, and spironolactone treatment
Male C57BL/6J mice were single housed 4 days prior to the experiment and their daily water intake was monitored. On the experimental day, mice were treated overnight (14 h) with either CORT (0.1 mg/ml in drinking water resulting in a ~25 mg/kg average dose based on the initially determined fluid intake; 4-pregnen-11β 21-DIOL-3 20-DIONE 21-hemisuccinate, #Q1562-000, Steraloids), RU486 (0.05 mg/ml in 0.5% EtOH resulting in a ~10 mg/kg average dose based on the initially determined fluid intake; Mifepristone, #475838, Sigma-Aldrich) or Spironolactone (0.124 mg/ml in 0.4% EtOH resulting in a ~20 mg/kg average dose based on the initially determined fluid intake; #S3378, Sigma-Aldrich). Control animals received their respective vehicle solutions. The next morning all mice were sacrificed by decapitation following quick anesthesia by isoflurane. Brains were removed, snap-frozen in isopentane at −40°C, and stored at −80°C until further processing. Overnight fluid intake did not differ between treatment groups.
qPCR
Tissue punches of the dorsal hippocampus were collected, total RNA was isolated and purified using the Quick-RNA Miniprep kit (Zymo research, Irvine, CA, USA, catalog no. R1054) according to the manufacturer’s protocol. RNA templates were reverse transcribed into cDNA with the Superscript IV kit (Thermo Scientific, Waltham, MA, USA, catalog no. 18091200) and random hexamer primers. cDNA was amplified on an Applied Biosystems ViiA7 Real-Time PCR System with POWRUP SYBR Green Master Mix (Thermo Scientific, Waltham, MA, USA, catalog no. 4368706). Primer sequences for Fkbp5, Nr3c1, Nr3c2 and Gapdh (housekeeper) can be found in the key resources table. Ct values were normalized using the established delta-delta Ct method (2–ΔΔCt) and normalized to Gapdh Cts.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-FKBP5 (1:500) | Santa Cruz | Cat#sc-11518; RRID: AB_2246889 |
| Rabbit anti-GR (1:1000) | Santa Cruz | Cat#sc-1004; RRID: AB_2155786 |
| Mouse anti-MR (1:100) | Millipore-Sigma | Cat#MABS496; RRID: AB_2811270 |
| Donkey anti-Rabbit 647 (1:1000) | Abcam | Cat#ab150075; RRID: AB_2752244 |
| Goat-anti-Mouse 594 (1:1000) | Abcam | Cat#ab150116; RRID: AB_2650601 |
| Donkey anti-Goat 488 (1:1000) | Invitrogen | Cat#A-11055; RRID: AB_2534102 |
| Rabbit anti-FKBP5 (1:1000) | Bethyl | Cat#A301-430A; RRID: AB_961006 |
| Goat anti-MR (1:800) | Santa Cruz | Cat# sc-6860; RRID: AB_2298883 |
| Rabbit anti-GR (1:800) | Cell Signaling | Cat#12041; RRID: AB_2631286 |
| Mouse anit-FLAG (1:5000) | Sigma | Cat#F3165; RRID: AB_259529 |
| Rat anti-Ha (1:8000) | Roche | Cat#11867423001; RRID: AB_390918 |
| Goat anti-Actin (1:5000) | Santa Cruz | Cat#sc-1616; RRID: AB_630836 |
| Chemicals, peptides, and recombinant proteins | ||
| Mm-Nr3c1-C1 | ACDBio | Cat#475261 |
| Mm-Fkbp5-C2 | ACDBio | Cat#457241-C2 |
| Mm-Nr3c2-C3 | ACDBio | Cat#456331-C3 |
| Dexamethasone | Sigma | Cat#D1159 |
| 4-pregnen-11β 21-DIOL-3 20-DIONE 21-hemisuccinate | Steraloids | Cat#Q1562-000 |
| RU486 | Sigma-Aldrich | Cat#475838 |
| Spironolactone | Sigma-Aldrich | Cat#S3378 |
| POWRUP SYBR Green Master Mix | Thermo Scientific | Cat#4368706 |
| RIPA buffer | Merck | Cat#20-188 |
| Protease Inhibitor cocktail | Sigma | Cat#04693132001 |
| Dynabeads M-280 | Thermo Scientific | Cat#11205D |
| Protein G Dynabeads | Thermo Scientific | Cat#10007D |
| Critical commercial assays | ||
| RNAscope kit | ACDBio | Cat#320850 |
| Corticosterone Double Antibody RIA kit | MP Biomedicals | Cat#0712010-CF |
| Quick-RNA Miniprep kit | Zymo Research | Cat#R0154 |
| Superscript IV kit | Thermo Scientific | Cat#18091200 |
| Experimental models: Cell lines | ||
| Primary hippocampal neurons | This paper | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6J male mice | Jackson Laboratory | Cat#000664 |
| GRNex-CKO male mice | Hartmann et al., 2017 | N/A |
| MRCamk2α-CKO male mice | Berger et al., 2006 | N/A |
| MRAmigo2-CKO male mice | McCann et al., 2021 | N/A |
| Oligonucleotides | ||
| Fkbp5-fwd 5′ CGGCGAC AGGTCTTCTACTT 3′ | Life Technologies | N/A |
| Fkbp5-rev 5′ TCTTCACCC TGCTCAGTCAT 3′ | Life Technologies | N/A |
| Nr3c1-fwd 5′ TGCTGTT TATCTCCACTGAATTACA 3′ | Life Technologies | N/A |
| Nr3c1-rev 5′ TCCTTAGGA ACTGAGGAGAGAAGC 3′ | Life Technologies | N/A |
| Nr3c2-fwd 5′ ATGGGTACC CGGTCCTAGAG 3′ | Life Technologies | N/A |
| Nr3c2-rev 5′ AAGCCTCATCT CCACACACC 3′ | Life Technologies | N/A |
| Gapdh-fwd 5′ TATGACT CCACTCACGGCAA 3′ | Life Technologies | N/A |
| Gapdh-rev 5′ ACATACTC AGCACCGGCCT 3′ | Life Technologies | N/A |
| Biotinylated-oligonucleotide probe 5′ GACTTGGTGAGAGAAAAACAG TCCCTAAGAATGGCGCCAAGCAT AAATATCTGTTGAATCAAAAATCAAG 3′ | IDT | N/A |
| Nr3c2-siRNA sequence: 5′ GTGAAGT GGGCCAAGGTACTTCCAGGAT TTAAAAACTTGCC 3′ | IDT | N/A |
| Deposited data | ||
| Human postmortem microarray data | Hawrylycz et al., 2012 | https://human.brain-map.org/static/download |
| Single-cell RNA sequencing | Saunders et al., 2018 | http://dropviz.org |
| Software and Algorithms | ||
| Seurat | Stuart et al., 2019 | https://satijalab.org/seurat/ |
| ImageJ | NIH | https://imagej.nih.gov/ij |
| Prism 7 | GraphPad | https://www.graphpad.com |
Biotinylated oligoIP
OligoIP was performed in mouse primary hippocampal neurons using a previously established method (Ibrahim et al., 2013) (schematically outlined in Figure 3A). In short, single-stranded complementary biotinylated-oligonucleotide probes (length 70 bp) spanning part of Fkbp5’s promotor region including two GREs (underlined) (5′ GACTTGGTGAGAGAAAAACAGTCCCTAAGAATGGCGCCAAG CATAAATATCTGTTGAATCAAAAATCAAG 3′, Integrated DNA technologies (IDT), Leuven, Belgium) were annealed. Subsequently, 0.5 pmol/106 cells of probes were transfected into neuronal cells (3–3,5 × 106 cells per replicate). After 24 h, the cells were incubated for 24 hours at 37°C with either vehicle (DMSO) or dexamethasone (1.5 nM, 15 nM and 150 nM in Figures 3B–3E; 15nM in Figures 4K–4O). For HA-GR or HA-MR overexpression (pRK7-HA-GR, pRK7-HA-MR, pRK7 empty vector as CTRL (Schülke et al., 2010) or MR knockdown (Nr3c2-siRNA 5′ GTGAAGTGGGCCAAGGTACTTCCAGGATTTAAAAACTTGCC 3′, IDT, Leuven, Belgium) experiments cells were transfected in parallel to oligonucleotide probes. In this case cells were harvested 48 h after transfection. Subsequently cells were cross-linked with 1% formaldehyde/ PBS at room temperature for 15 min and were lysed in RIPA buffer (Merck, 20-188, completed with protease inhibitor cocktail, Sigma, 04693132001). Lysates were precipitated using streptavidin-coupled magnetic beads (Dynabeads M-280, Thermo Scientific, 11205D) or control beads lacking conjugated streptavidin (Protein G Dynabeads, Thermo Scientific, 10007D), and both input and eluates were quantified for MR and GR by western blotting.
Western blot analysis
Protein extracts were obtained by lysing cells in RIPA buffer (Merck, 20-188, completed with protease inhibitor cocktail, Sigma, 04693132001). Proteins were separated by SDS-PAGE and electro-transferred onto PVDF membranes. Western Blots were placed for blocking in Tris-buffered saline (TBST; 50 mM Tris-Cl, pH 7.6; 150 mM NaCl, 0.05% Tween 20) and 5% non-fat milk for 1 h at room temperature and subsequently incubated with primary antibody TBST overnight at 4°C. The following primary antibodies were used: FKBP5/FKBP51 (1:1,000, Bethyl, A301-430A), MR (1:800, Santa Cruz, N-17), GR (1:800, Cell Signal, #12041), FLAG (1:5,000, Sigma, F3165), HA (1:8,000, 11867423001) and Actin (1:5,000, Santa Cruz, I-19). Subsequently, the blots were washed with TBST and probed with the respective horseradish-peroxidase or fluorophore-conjugated secondary antibody for 2 h at room temperature.
The immuno-reactive bands were detected either by using ECL detection reagent (Millipore, WBKL0500) or directly by excitation of the respective fluorophore. Recording of the band intensities was performed with the ChemiDoc MP from Bio-Rad. Protein data were normalized to Actin, which was detected on the same blot in the same lane (multiplexing).
Single-cell RNA sequencing analysis
Mouse hippocampus single-cell RNA sequencing data were publicly available from the McCarroll Lab (Department of Genetics, Harvard Medical School) (Saunders et al., 2018). t-distributed stochastic neighbor embedding (t-SNE) plots for hippocampus global clustering of cell-types, as well as for Fkbp5, Nr3c1 and Nr3c2 expression profiles were generated using the Seurat 3.0 package in R (Stuart et al., 2019). The cluster-specific number of Fkpb5, Nr3c1 and Nr3c2 expressing cells were extracted for further analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
The data presented are shown as means + standard error of the mean (SEM). All data were analyzed by the commercially available software GraphPad 7.0. When two groups were compared, the unpaired, two-tailed Student’s t test was applied. For four or more group comparisons, one-way or two-way analysis of variance (ANOVA) was performed, followed by the Bonferroni posthoc test, as appropriate. mRNA expression associations were evaluated with Pearson correlation. Yate’s chi-square test was performed for the single cell data. P values of < 0.05 were considered statistically significant.
Supplementary Material
Highlights.
FKBP5 and MRs, but not GRs, exhibit similar hippocampal expression patterns
MRs, rather than GRs, regulate FKBP5 expression in hippocampal neurons at baseline
Inhibition and deletion of MRs decrease hippocampal Fkbp5 mRNA levels in vivo
Forebrain MR deletion leads to GR hypersensitivity during acute stress
ACKNOWLEDGMENTS
We thank Daniela Harbich, Andrea Reßle, and Bianca Schmid (Max Planck Institute of Psychiatry, Munich, Germany) for excellent technical assistance and support. We thank the scientific illustrator Dr. Matteo Oliverio for excellent design of the graphical abstract. This study was supported by a NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation (awarded to J.H., grant no. 24774), the National Institutes of Health (R01MH108665, P50MH115874), and the Intramural Research Program of the National Institute of Environmental Health Sciences, National Institutes of Health (Z01ES100221, to S.M.D.). T.K. was supported by research grants from NICHD (R21HD088931, R21HD097524), NIMH (R21MH117609), and ERA-Net Neuron (01EW2003). Figure 7 was created with BioRender.
DECLARATION OF INTERESTS
N.D. is currently an employee of Sunovion Pharmaceuticals. K.M.M. is currently an employee of Encoded Therapeutics Inc. N.P.D. has served as a paid consultant for Sunovion Pharmaceuticals and is on the scientific advisory board for Sentio Solutions, Inc. for unrelated work. K.J.R. has received consulting income from Alkermes and Bio X Cell and is on scientific advisory boards for Janssen and Verily for unrelated work. He has also received a sponsored research grant support from Takeda, Alto Neuroscience, and Brainsway for unrelated work. T.K. has received consulting income from Alkermes for unrelated work. The remaining authors declare no competing interests.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109185.
REFERENCES
- Ahima R, Krozowski Z, and Harlan R (1991). Type I corticosteroid receptor-like immunoreactivity in the rat CNS: Distribution and regulation by corticosteroids. J. Comp. Neurol 313, 522–538. [DOI] [PubMed] [Google Scholar]
- Arnett MG, Muglia LM, Laryea G, and Muglia LJ (2016). Genetic approaches to hypothalamic-pituitary-adrenal axis regulation. Neuropsychopharmacology 41, 245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Simerly RB, Swanson LW, and Evans RM (1988). The neuronal mineralocorticoid receptor as a mediator of glucocorticoid response. Neuron 1, 887–900. [DOI] [PubMed] [Google Scholar]
- Berger S, Wolfer DP, Selbach O, Alter H, Erdmann G, Reichardt HM, Chepkova AN, Welzl H, Haas HL, Lipp H-P, and Schütz G (2006). Loss of the limbic mineralocorticoid receptor impairs behavioral plasticity. Proc. Natl. Acad. Sci. USA 103, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder EB (2009). The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 34 (Suppl 1), S186–S195. [DOI] [PubMed] [Google Scholar]
- Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Pütz B, Papiol S, Seaman S, Lucae S, Kohli MA, et al. (2004). Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat. Genet 36, 1319–1325. [DOI] [PubMed] [Google Scholar]
- Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, Tang Y, Gillespie CF, Heim CM, Nemeroff CB, et al. (2008). Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 299, 1291–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle MP, Kolber BJ, Vogt SK, Wozniak DF, and Muglia LJ (2006). Forebrain glucocorticoid receptors modulate anxiety-associated locomotor activation and adrenal responsiveness. J. Neurosci 26, 1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criado-Marrero M, Rein T, Binder EB, Porter JT, Koren J 3rd, and Blair LJ (2018). Hsp90 and FKBP51: Complex regulators of psychiatric diseases. Philos. Trans. R. Soc. Lond. B Biol. Sci 373, 20160532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kloet ER, and Derijk R (2004). Signaling pathways in brain involved in predisposition and pathogenesis of stress-related disease: Genetic and kinetic factors affecting the MR/GR balance. Ann. NY Acad. Sci 1032, 14–34. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, and Joëls M (2017). Brain mineralocorticoid receptor function in control of salt balance and stress-adaptation. Physiol. Behav 178, 13–20. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, De Kock S, Schild V, and Veldhuis HD (1988). Antiglucocorticoid RU 38486 attenuates retention of a behaviour and disinhibits the hypothalamic-pituitary adrenal axis at different brain sites. Neuroendocrinology 47, 109–115. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Vreugdenhil E, Oitzl MS, and Joëls M (1998). Brain corticosteroid receptor balance in health and disease. Endocr. Rev 19, 269–301. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Joëls M, and Holsboer F (2005). Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci 6, 463–475. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Meijer OC, de Nicola AF, de Rijk RH, and Joëls M (2018). Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front. Neuroendocrinol 49, 124–145. [DOI] [PubMed] [Google Scholar]
- Dedovic K, Duchesne A, Andrews J, Engert V, and Pruessner JC (2009). The brain and the stress axis: The neural correlates of cortisol regulation in response to stress. Neuroimage 47, 864–871. [DOI] [PubMed] [Google Scholar]
- Denny WB, Valentine DL, Reynolds PD, Smith DF, and Scammell JG (2000). Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology 141, 4107–4113. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, and Banker GA (1988). The establishment of polarity by hippocampal neurons in culture. J. Neurosci 8, 1454–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, and Dong HW (2010). Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65, 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenster RJ, Lebois LAM, Ressler KJ, and Suh J (2018). Brain circuit dysfunction in post-traumatic stress disorder: From mouse to man. Nat. Rev. Neurosci 19, 535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furay AR, Bruestle AE, and Herman JP (2008). The role of the forebrain glucocorticoid receptor in acute and chronic stress. Endocrinology 149, 5482–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, and Nave K-A (2006). Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 44, 611–621. [DOI] [PubMed] [Google Scholar]
- Hardeveld F, Spijker J, Peyrot WJ, de Graaf R, Hendriks SM, Nolen WA, Penninx BWJH, and Beekman ATF (2015). Glucocorticoid and mineralocorticoid receptor polymorphisms and recurrence of major depressive disorder. Psychoneuroendocrinology 55, 154–163. [DOI] [PubMed] [Google Scholar]
- Harris AP, Holmes MC, de Kloet ER, Chapman KE, and Seckl JR (2013). Mineralocorticoid and glucocorticoid receptor balance in control of HPA axis and behaviour. Psychoneuroendocrinology 38, 648–658. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Wagner KV, Liebl C, Scharf SH, Wang XD, Wolf M, Hausch F, Rein T, Schmidt U, Touma C, et al. (2012). The involvement of FK506-binding protein 51 (FKBP5) in the behavioral and neuroendocrine effects of chronic social defeat stress. Neuropharmacology 62, 332–339. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Dedic N, Pöhlmann ML, Häusl A, Karst H, Engelhardt C, Westerholz S, Wagner KV, Labermaier C, Hoeijmakers L, et al. (2017). Forebrain glutamatergic, but not GABAergic, neurons mediate anxiogenic effects of the glucocorticoid receptor. Mol. Psychiatry 22, 466–475. [DOI] [PubMed] [Google Scholar]
- Häusl AS, Brix LM, Hartmann J, Pöhlmann ML, Lopez J-P, Menegaz D, Brivio E, Engelhardt C, Roeh S, Bajaj T, et al. (2021). The co-chaperone Fkbp5 shapes the acute stress response in the paraventricular nucleus of the hypothalamus of male mice. Mol. Psychiatry, Published online March 1, 2021. 10.1038/s41380-021-01044-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, et al. (2012). An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, McKlveen JM, Ghosal S, Kopp B, Wulsin A, Makinson R, Scheimann J, and Myers B (2016). Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr. Physiol 6, 603–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers L, Harbich D, Schmid B, Lucassen PJ, Wagner KV, Schmidt MV, and Hartmann J (2014). Depletion of FKBP51 in female mice shapes HPA axis activity. PLoS ONE 9, e95796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubler TR, and Scammell JG (2004). Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones 9, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim EE, Babaei-Jadidi R, and Nateri AS (2013). The streptavidin/biotinylated DNA/protein bound complex protocol for determining the association of c-JUN protein with NANOG promoter. Curr. Protoc. Stem Cell Biol Chapter 1, 18.10, Unit. [DOI] [PubMed] [Google Scholar]
- Ising M, Depping A-M, Siebertz A, Lucae S, Unschuld PG, Kloiber S, Horstmann S, Uhr M, Müller-Myhsok B, and Holsboer F (2008). Polymorphisms in the FKBP5 gene region modulate recovery from psychosocial stress in healthy controls. Eur. J. Neurosci 28, 389–398. [DOI] [PubMed] [Google Scholar]
- Jacobson L, and Sapolsky R (1991). The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr. Rev 12, 118–134. [DOI] [PubMed] [Google Scholar]
- Keller J, Gomez R, Williams G, Lembke A, Lazzeroni L, Murphy GM Jr., and Schatzberg AF (2017). HPA axis in major depression: cortisol, clinical symptomatology and genetic variation predict cognition. Mol. Psychiatry 22, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, Pace TWW, Mercer KB, Mayberg HS, Bradley B, et al. (2013). Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci 16, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klok MD, Giltay EJ, Van der Does AJW, Geleijnse JM, Antypa N, Penninx BWJH, de Geus EJC, Willemsen G, Boomsma DI, van Leeuwen N, et al. (2011). A common and functional mineralocorticoid receptor haplotype enhances optimism and protects against depression in females. Transl. Psychiatry 1, e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laryea G, Schütz G, and Muglia LJ (2013). Disrupting hypothalamic glucocorticoid receptors causes HPA axis hyperactivity and excess adiposity. Mol. Endocrinol 27, 1655–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RS, Tamashiro KLK, Yang X, Purcell RH, Harvey A, Willour VL, Huo Y, Rongione M, Wand GS, and Potash JB (2010). Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology 151, 4332–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, and Heim C (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci 10, 434–445. [DOI] [PubMed] [Google Scholar]
- Maddox SA, Hartmann J, Ross RA, and Ressler KJ (2019). Deconstructing the gestalt: Mechanisms of fear, threat, and trauma memory encoding. Neuron 102, 60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann KE, Lustberg DJ, Shaughnessy EK, Carstens KE, Farris S, Alexander GM, Radzicki D, Zhao M, and Dudek SM (2021). Novel role for mineralocorticoid receptors in control of a neuronal phenotype. Mol. Psychiatry 36, 350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough KM, Morrison FG, Hartmann J, Carlezon WA Jr., and Ressler KJ (2018). Quantified coexpression analysis of central amygdala sub-populations. eNeuro 5, ENEURO.0010-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina A, Seasholtz AF, Sharma V, Burke S, Bunney W Jr., Myers RM, Schatzberg A, Akil H, and Watson SJ (2013). Glucocorticoid and mineralocorticoid receptor expression in the human hippocampus in major depressive disorder. J. Psychiatr. Res 47, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifsud KR, and Reul JMHM (2016). Acute stress enhances heterodimerization and binding of corticosteroid receptors at glucocorticoid target genes in the hippocampus. Proc. Natl. Acad. Sci. USA 113, 11336–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, and Klein R (1999). Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24, 401–414. [DOI] [PubMed] [Google Scholar]
- O’Leary JC 3rd, Dharia S, Blair LJ, Brady S, Johnson AG, Peters M, Cheung-Flynn J, Cox MB, de Erausquin G, Weeber EJ, et al. (2011). A new anti-depressive strategy for the elderly: Ablation of FKBP5/FKBP51. PLoS ONE 6, e24840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oitzl MS, Champagne DL, van der Veen R, and de Kloet ER (2010). Brain development under stress: Hypotheses of glucocorticoid actions revisited. Neurosci. Biobehav. Rev 34, 853–866. [DOI] [PubMed] [Google Scholar]
- Patel PD, Lopez JF, Lyons DM, Burke S, Wallace M, and Schatzberg AF (2000). Glucocorticoid and mineralocorticoid receptor mRNA expression in squirrel monkey brain. J. Psychiatr. Res 34, 383–392. [DOI] [PubMed] [Google Scholar]
- Reichardt HM, Umland T, Bauer A, Kretz O, and Schütz G (2000). Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol. Cell. Biol 20, 9009–9017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reul JMHM, and de Kloet ER (1985). Two receptor systems for corticosterone in rat brain: Microdistribution and differential occupation. Endocrinology 117, 2505–2511. [DOI] [PubMed] [Google Scholar]
- Reul JMHM, Collins A, Saliba RS, Mifsud KR, Carter SD, Gutierrez-Mecinas M, Qian X, and Linthorst ACE (2014). Glucocorticoids, epigenetic control and stress resilience. Neurobiol. Stress 1, 44–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridder S, Chourbaji S, Hellweg R, Urani A, Zacher C, Schmid W, Zink M, Hörtnagl H, Flor H, Henn FA, et al. (2005). Mice with genetically altered glucocorticoid receptor expression show altered sensitivity for stress-induced depressive reactions. J. Neurosci 25, 6243–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozeboom AM, Akil H, and Seasholtz AF (2007). Mineralocorticoid receptor overexpression in forebrain decreases anxiety-like behavior and alters the stress response in mice. Proc. Natl. Acad. Sci. USA 104, 4688–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scammell JG, Denny WB, Valentine DL, and Smith DF (2001). Overexpression of the FK506-binding immunophilin FKBP51 is the common cause of glucocorticoid resistance in three New World primates. Gen. Comp. Endocrinol 124, 152–165. [DOI] [PubMed] [Google Scholar]
- Scharf SH, Liebl C, Binder EB, Schmidt MV, and Müller MB (2011). Expression and regulation of the Fkbp5 gene in the adult mouse brain. PLoS ONE 6, e16883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MV, Sterlemann V, Ganea K, Liebl C, Alam S, Harbich D, Greetfeld M, Uhr M, Holsboer F, and Müller MB (2007). Persistent neuroendocrine and behavioral effects of a novel, etiologically relevant mouse paradigm for chronic social stress during adolescence. Psychoneuroendocrinology 32, 417–429. [DOI] [PubMed] [Google Scholar]
- Schülke JP, Wochnik GM, Lang-Rollin I, Gassen NC, Knapp RT, Berning B, Yassouridis A, and Rein T (2010). Differential impact of tetratri-copeptide repeat proteins on the steroid hormone receptors. PLoS ONE 5, e11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Horst JP, van der Mark MH, Arp M, Berger S, de Kloet ER, and Oitzl MS (2012). Stress or no stress: Mineralocorticoid receptors in the forebrain regulate behavioral adaptation. Neurobiol. Learn. Mem 98, 33–40. [DOI] [PubMed] [Google Scholar]
- Ter Horst JP, van der Mark M, Kentrop J, Arp M, van der Veen R, de Kloet ER, and Oitzl MS (2014). Deletion of the forebrain mineralocorticoid receptor impairs social discrimination and decision-making in male, but not in female mice. Front. Behav. Neurosci 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touma C, Gassen NC, Herrmann L, Cheung-Flynn J, Büll DR, Ionescu IA, Heinzmann JM, Knapman A, Siebertz A, Depping AM, et al. (2011). FK506 binding protein 5 shapes stress responsiveness: Modulation of neuroendocrine reactivity and coping behavior. Biol. Psychiatry 70, 928–936. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, and Schütz G (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet 23, 99–103. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, and Herman JP (2009). Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci 10, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eekelen JA, Bohn MC, and de Kloet ER (1991). Postnatal ontogeny of mineralocorticoid and glucocorticoid receptor gene expression in regions of the rat tel- and diencephalon. Brain Res. Dev. Brain Res 61, 33–43. [DOI] [PubMed] [Google Scholar]
- van Rossum EFC, Binder EB, Majer M, Koper JW, Ising M, Modell S, Salyakina D, Lamberts SWJ, and Holsboer F (2006). Polymorphisms of the glucocorticoid receptor gene and major depression. Biol. Psychiatry 59, 681–688. [DOI] [PubMed] [Google Scholar]
- van Weert LTCM, Buurstede JC, Sips HCM, Vettorazzi S, Mol IM, Hartmann J, Prekovic S, Zwart W, Schmidt MV, Roozendaal B, et al. (2019). Identification of mineralocorticoid receptor target genes in the mouse hippocampus. J. Neuroendocrinol 31, e12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeer H, Hendriks-Stegeman BI, van der Burg B, van Buul-Offers SC, and Jansen M (2003). Glucocorticoid-induced increase in lymphocytic FKBP51 messenger ribonucleic acid expression: A potential marker for glucocorticoid sensitivity, potency, and bioavailability. J. Clin. Endocrinol. Metab 88, 277–284. [DOI] [PubMed] [Google Scholar]
- Wagner KV, Marinescu D, Hartmann J, Wang X-D, Labermaier C, Scharf SH, Liebl C, Uhr M, Holsboer F, Müller MB, and Schmidt MV (2012). Differences in FKBP51 regulation following chronic social defeat stress correlate with individual stress sensitivity: Influence of paroxetine treatment. Neuropsychopharmacology 37, 2797–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westberry JM, Sadosky PW, Hubler TR, Gross KL, and Scammell JG (2006). Glucocorticoid resistance in squirrel monkeys results from a combination of a transcriptionally incompetent glucocorticoid receptor and overexpression of the glucocorticoid receptor co-chaperone FKBP51. J. Steroid Bio-chem. Mol. Biol 100, 34–41. [DOI] [PubMed] [Google Scholar]
- Wochnik GM, Rüegg J, Abel GA, Schmidt U, Holsboer F, and Rein T (2005). FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J. Biol. Chem 280, 4609–4616. [DOI] [PubMed] [Google Scholar]
- Zannas AS, Jia M, Hafner K, Baumert J, Wiechmann T, Pape JC, Arloth J, Ködel M, Martinelli S, Roitman M, et al. (2019). Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-κB-driven inflammation and cardiovascular risk. Proc. Natl. Acad. Sci. USA 116, 11370–11379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original/source data for Figures 1D and E (Human postmortem microarray analysis) were publicly available from the Allen Brain Institute and can be downloaded at https://human.brain-map.org/static/download. Original/source data for Figures 1F and 1G (Single-cell RNA sequencing analysis) were publicly available from the McCarroll Lab (Department of Genetics, Harvard Medical School) and can be downloaded at http://dropviz.org.