

Abstract
Purpose of Review
Current therapies for autoimmune disorders often employ broad suppression of the immune system. Antigen-specific immunotherapy (ASI) seeks to overcome the side-effects of immunosuppressive therapy by specifically targeting only disease-related autoreactive T and B cells. Although it has been in development for several decades, antigen-specific immunotherapy (ASI) still is not in use clinically to treat autoimmunity. Novel ways to deliver antigen may be effective in inducing ASI. Here we review recent innovations in antigen delivery
Recent Findings
New ways to deliver antigen include particle and non-particle approaches. One main focus has been the targeting of antigen presenting cells in a tolerogenic context. This technique often results in the induction and/or expansion of regulatory T cells (Tregs), which has the potential to be effective against a complex, polyclonal immune response.
Summary
Whether novel delivery approaches can help bring ASI into general clinical use for therapy of autoimmune diseases remains to be seen. However, pre-clinical work and early results from clinical trials using these new techniques show promising signs.
Keywords: Autoimmunity, antigen-specific immunotherapy, microparticles, nanoparticles, Tregs
Introduction
Type 1 diabetes (T1D) is a disease caused by autoimmune destruction of the insulin-secreting beta cells found in the pancreas. An alternative to insulin replacement is to re-establish a tolerant state characterized by a balance between a regulatory and effector response, particularly during the time window in which re-establishment might leave remaining beta cells [1]. Although the target of the immune response in T1D is a single cell type, the importance of different antigens in the various stages of disease progression means the most effective treatment is broad immune suppression, leading to potentially unwanted side effects, such as increased susceptibility to infection or cancer [2,3]. Yet another alternative is antigen-specific immunotherapy (ASI). While it avoids the safety issues of broad-based immunosuppression, ASI requires knowledge of the initiating antigen and/or the other antigens to which autoimmunity is spread as disease progresses, although the addition of each new antigen to an ASI formulation potentially contributes to new safety concerns. It should be noted, however, that this problem may be overcome by induction of bystander suppression [2,3]. While T cells are likely the most important autoreactive cells in T1D [4], their simple deletion or inactivation would not produce such bystander suppression [2]. In light of this, many efforts to induce antigen-specific tolerance have included a focus on the generation and/or expansion of suppressive regulatory T cells (Tregs) [4]. Here, we briefly describe ASI as treatment for T1D, note the difficulties associated with it, and review recent innovations for tolerogenic antigen delivery. We also discuss the development of antigen delivery techniques for other autoimmune disorders, which may eventually be applied to T1D.
Antigen-specific Immunotherapy: Background
Numerous diabetogenic autoantigens have been identified in mice and humans [5–7], including the recently described post-translationally modified hybrid insulin peptides (HIPs) [8–10]. Delivering insulin as a tolerogenic antigen marked one of the earliest attempts at T1D tolerization [3], but others have also made use of peptides such as glutamic acid decarboxylase 65 (GAD65) [11–14], or a mimotope for the diabetogenic T cell clone BDC2.5 [15]. ASI approaches have been successful in preclinical studies [4,16,17]. However, while ASI works well in allergy treatment and protein replacement therapy, it has not yet been approved for treatment of autoimmunity [2,17]. Although clinical trials of ASI have been taking place for many years, they have mostly been unsuccessful [2,3,11,14,17–19]. Nevertheless, several clinical trials are currently being undertaken, and their frequency is increasing [17]. One reason for the lack of success is the failure to combat complex, polyclonal immune responses, which could potentially be accomplished by the infectious, bystander tolerance mediated by Tregs [2]. Whether innovative ways to deliver antigen can help remedy this shortcoming by promoting specific regulatory immune responses remains to be seen.
Novel Delivery: Non-particle
Table 1 summarizes some recent innovative ways to deliver antigen in ASI. As can be seen, there is considerable diversity among the approaches.
Table 1.
Recent Non-particle Antigen Delivery Approaches.
| Antigen Delivery Approach | Antigens | Route of Administration | Result | Reference |
|---|---|---|---|---|
| Microneedle | Proinsulin | i.d. | Stimulates adoptively transferred T cells | [20] |
| C19-A3 (nanoparticle) | i.d. | Uptake by Langerhans cells in ex vivo human skin | [21] | |
| Peptide-loaded tolDCs | C19-A3 | i.d. | Safety and stability in human patients | [26] |
| Myelin peptides +/− AQ4 | i.v. |
|
[27] | |
| mRNA-electroporated DCs |
|
|
|
[28] |
| Bacteria |
|
Oral gavage |
|
[30–31] |
Abbreviations: DC = dendrictic cell, tolDC = tolerogenic dendritic cell, i.d. = intradermal, i.v. = intravenous, i.p. = intraperitoneal, AQ4 = aquaporin-4, InsB = insulin B chain, GAD65 = glutamic acid decarboxylase, IGRP = islet-specific glucose-6-phosphatase catalytic subunit-related protein, T1D = type 1 diabetes, Tregs = regulatory T cells
Microneedles have been used to increase the safety of antigen delivery, either as full protein or peptide [20,21]. A proinsulin-coated array was used to stimulate adoptively transferred T cells in draining lymph nodes (LNs), presumably following uptake and processing by skin-resident antigen presenting cells (APCs) [20]. In ex vivo human skin, it was found that microneedles were able to inject gold nanoparticles coated with proinsulin peptide C19-A3 into the epidermis [21]. In both these cases, the use of microneedles, as opposed to traditional transdermal injection which causes skin trauma, reduced the likelihood of an inflammatory response and made it more likely that APCs presented peptide to T cells in a tolerogenic context.
Apoptotic cells coupled with peptide(s) have also been explored for their ability to induce tolerance. Our laboratory has investigated the use of ethylenecarbodiimide to both fix self-protein/peptide antigens to splenocytes and induce apoptosis [7,22–24]. This proved protective in non-obese diabetic (NOD) mice as well as in experimental autoimmune encephalomyelitis (EAE), the murine model of multiple sclerosis (MS), through both T cell anergy and Tregs. In addition, this tolerogenic approach was shown to be both safe and efficacious in a phase 1 clinical trial [25]. Two other recent studies [26,27] found that tolerogenic dendritic cells (tolDCs) loaded with antigenic peptides/proteins are safe in human patients. In the first [26], tolerogenic cells were obtained by leukapheresis, followed by selection for CD14+ monocytes. They were then loaded with C19-A3 and administered intradermally (i.d.) to T1D patients. The treatment was found to be safe and the patients maintained stable β cell function over a 6 month follow-up. In the second [27], a mix of patients with either MS or neuromyelitis optica spectrum disorders (NMOSDs) were treated with TolDCs that were loaded with 7 myelin peptides (with or without the dominant NMOSD antigen, aquaporin-4) prior to intravenous (i.v.) administration. This study demonstrated the safety of the approach and noted an increase in IL-10 production by peripheral blood mononuclear cells (PBMCs), as well as an increase in regulatory Tr1 cells. An alternative to loading dendritic cells (DCs) with peptide/protein takes advantage of the low cost and high transfection efficiency of mRNA. In one recent study, ex vivo DCs were electroporated with a construct that includes CD4+ and CD8+ T1D epitopes [28]. Systemic or local administration of the DCs lead to enhanced proliferation of adoptively transferred transgenic cells and upregulation of their CD25 expression.
Tolerogenic antigen can also be delivered by bacteria. Nonpathogenic Salmonella typhimurium and Lactococcus lactis have been used to deliver proinsulin, pre-proinsulin, and GAD65 as part of a combination therapy in three different investigations [29–31]. Plasmids for the peptide antigens, as well as IL-10 (and TGF-β in two studies), were transfected into cells, which were delivered orally to NOD mice along with anti-CD3, leading to protection from T1D and an increase in FoxP3+ Tregs (and Tr1 cells in one study).
Novel Delivery: Nano- or micro-particle
A popular approach to antigen delivery employs micro- or nano-particle carriers. Several groups have used these particulate systems as treatment for autoimmunity generally and for T1D in particular [32]. Particles in this section can be divided into three categories: encapsulating liposomes/lipoplexes [28,33–35] and encapsulating non-liposomes [36–39], decorated metal particles [21,40–45], and negatively charged PLG nanoparticles [46–52]. Here, we only highlight recent, novel antigen delivery micro/nanoparticles (summarized in Figure 1 and Table 2). Reviews of older studies can be found elsewhere [2,3,32,53–55].
Figure 1. Illustration of Nano- and Micro-particle Approaches Under Investigation for Induction of Antigen-Specific Immunotherapy (ASI).
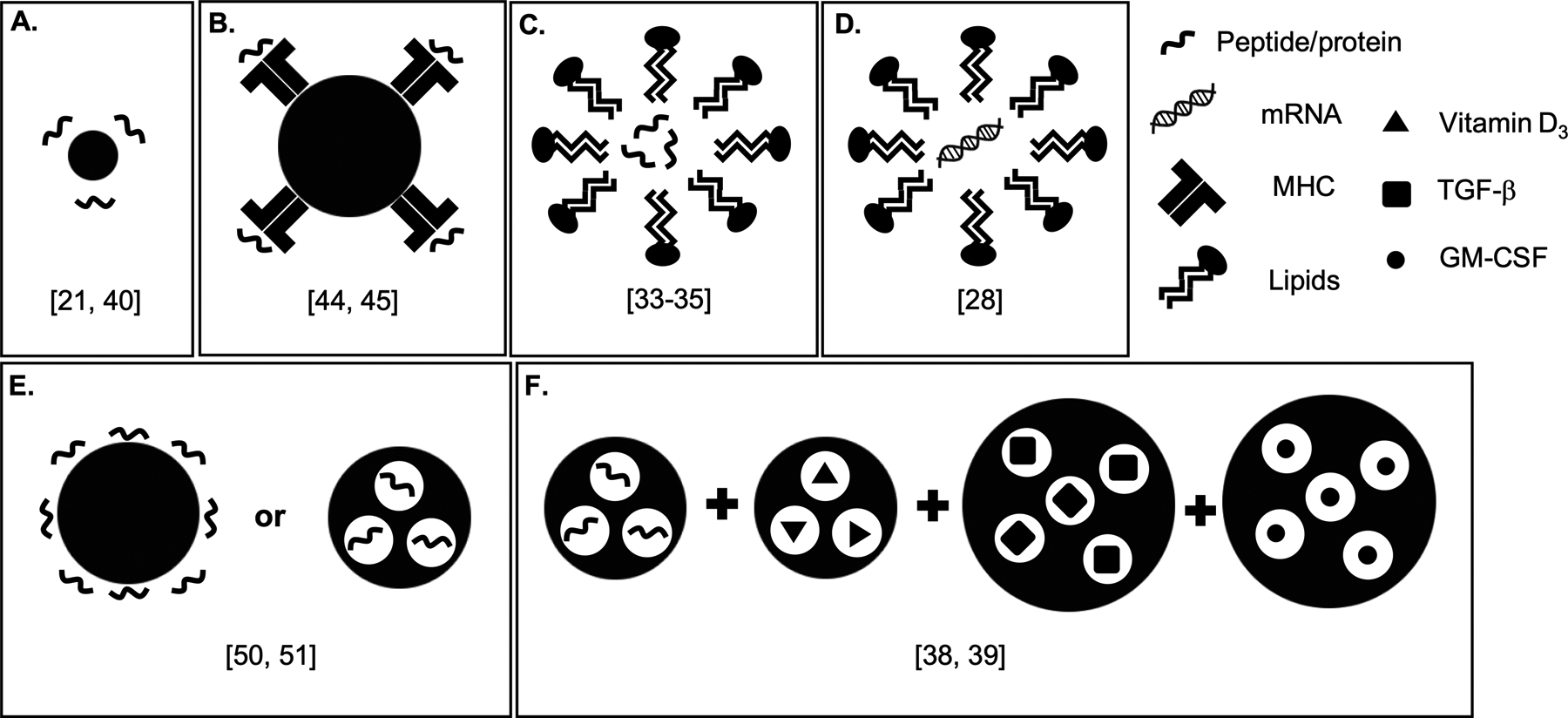
A) Gold nanoparticles coated with peptide [21, 40]. B) Iron oxide nanoparticles coated with pMHC [44, 45]. C) Liposome incorporating α-galactosylceramide or phosphatidylserine, encapsulating peptide [33–35]. D) Lipoplex encapsulating mRNA [28]. E) PLG nanoparticles coupled with or encapsulating peptide [46–52]. F) Dual-sized microparticles encapsulating peptides/proteins and other factors [38, 39].
Table 2.
Recent Micro- and Nano-particle Antigen Delivery Approaches.
| Antigen Delivery Approach | Antigens | Route of Administration | Result | Reference |
|---|---|---|---|---|
| Encapsulating liposome / lipolex | InsB9–23 | i.p. |
|
[33] |
| InsA90–110 InsB25–54 |
i.p. |
|
[34,35] | |
|
|
|
[28] | |
| Encapsulating non-liposome |
|
s.c. |
|
[38, 39] |
| Coated metal nanoparticles |
|
i.d. |
|
[21, 40] |
| pMHC-coated metal nanoparticles |
|
i.v. |
|
[44, 45] |
| Negatively charged PLG nanoparticles |
|
i.v. |
|
[50, 51] |
Abbreviations: DC = , i.p. = intraperitoneal, i.d. = intradermal, s.c. = sub-cutaneous, i.v. = intravenous, InsA/B = insulin A or B chain, GAD65 = glutamic acid decarboxylase, IGRP = islet-specific glucose-6-phosphatase catalytic subunit-related protein, MOG = myelin oligodendrocyte glycoprotein, PDC = pyruvate dehydrogenase complex, CYPD = cytochrome P450, FTCD = formimidoyltransferase cyclodeaminas, T1D = type 1 diabetes, EAE = experimental autoimmune encephalomyelitis, Treg = regulatory T cell, Teff = effector T cell
Regarding liposomes/lipoplexes, all three examples encapsulate biological molecules and are meant to target APCs. Both Akimoto et al. [33] and Villalba et al. [34,35] used liposomes to encapsulate insulin peptides. The former incorporated the lipid α-galactosylceramide (α-GalCer), a ligand for iNKT cells, and encapsulated insulin B chain 9–23 (InsB9–23), resulting in a synergistic augmentation of tolerance, including transferrable prevention of T1D in young NOD mice, reduction of insulitis, and increase of pancreatic FoxP3+ Tregs. The latter used the lipid phosphatidylserine to mimic apoptotic cells, while the cores contained peptides from insulin A and B chains. A single injection in NOD mice at 8 weeks of age (WOA) lowered disease incidence, decreased insulitis, and was safe and well tolerated [34]. A second study [35] determined that liposomes combined with the β cell regenerating agent liraglutide maintained low blood glucose for longer than either therapy alone and that there are early signs this may be effective in humans. Another study discussed above [28] used lipoplexes to encapsulate mRNA and protect it from degradation, comparing the product to electroporated DCs. Unlike the liposomes described above [34,35], the lipoplexes carried a positive electrical charge. Although this is sometimes associated with toxicity [56,57], none was observed. After intraperitoneal (i.p.) injection, lipoplexes were taken up by more lymphoid cells than electroporated DCs, while i.v. injection localized lipoplexes predominantly to the spleen, and DCs were brought mostly to the lungs. Both i.p. and local i.d. injection following transfer of transgenic CD4+ BDC2.5 cells lead to increased proliferation and upregulation of CD25 as opposed to control mRNA. Inclusion of mRNA for IL-27 enhanced the tolerogenic potential of the technique, as measured by IL-10 secretion from T cells following i.p. administration. The wider tissue distribution of the lipoplexes versus DCs, their ability to target tolerogenic LN stromal cells, and the comparative ease of working with them as compared to ex vivo cells, may make them more suited for translation to clinical use.
Non-lipid particles can also encapsulate peptide antigens. One group has developed a system using four varieties of poly(lactic-co-glycolic acid) (PLGA or PLG) microparticles, two small (1 μm) loaded with antigen and vitamin D3 (VD3), and two large (30 μm) loaded with TGF-β1 and GM-CSF. DCs attracted to the s.c. injection site by GM-CSF take up the antigen and are rendered tolerogenic by TGF-β1 and VD3. Initially, the small particles encapsulated InsB9–23 and protected young NOD mice from T1D development [36], but failed to protect when administered at 8 weeks or age (WOA) [38]. The same system was used with the peptide myelin oligodendrocyte glycoprotein 35–55 (MOG35–55) to protect against EAE in C57BL/6 mice [37]. Reformulating the particles to encapsulate full-length insulin and more VD3, TGF-β1, and GM-CSF was protective in NOD mice when administered at 8 WOA [38]. When diabetic NOD mice were given insulin pellets, then treated, the microparticles prolonged remission to hyperglycemia. Compared to unloaded controls, the small particles of the fully loaded formulation were found to be taken up more by DCs than macrophages, a key goal of this group’s strategy. The protective treatment coincided with an increase in FoxP3+ Tregs in the spleen and pancreatic LNs, and an upregulation of PD-1 on both CD4+ and CD8+ T cells in draining LNs, potentially an important result, as β cell destruction is carried out by cytotoxic CD8+ cells. A recent follow-up study [39] seeking to extend the age of effective dual microparticle treatment to 12 WOA by combining low or ultra-low dose i.p. anti-CD3 administration failed to induce protection.
Metal nanoparticles may also be loaded with peptide antigens. The particles from the previously discussed study [21] used small gold particles coated with carbohydrates to enhance solubility, glutathione to facilitate intracellular activation, and C19-A3 peptide. It was found that delivery through microneedles did not greatly affect their physical properties, which allowed their diffusion into the dermis and epidermis following injection into ex vivo human skin, the latter significant as the dermis is the location of potentially tolerogenic Langerhans cells. A more recent study [40] made use of the same particles, but decorated them with BDC2.5 mimotope peptide P31. Possibly due to its relatively low solubility, coupling to particles resulted in response from transferred BDC2.5 T cells in a greater number of lymphoid tissues than the peptide alone. A similar effect was seen when particles were injected via microneedles. Finally, transferred BDC2.5 cells also responded to increasing doses of particles decorated with hybrid insulin peptide in terms of both proliferation and activation, with fewer T cells expressing IFN-γ.
Iron oxide nanoparticles coated with peptide-loaded major histocompatibility complex molecules (pMHC) [41–45] are unique among the particle approaches described here, as these particles directly interact with T cells instead of APCs. Both MHC class I and II molecules have been used for the expansion of T cells with regulatory properties, and, in some cases [42,44] even regulatory B cells have been described. An advantage of this system is that, unlike those approaches that only convert already present cells into Tregs, the expansion seen here is not reliant on the initial number of peptide-specific T cells [2]. The pMHC-nanoparticle approach has not shown any safety issues or hazardous suppression of immunity [42–44] and has previously proven successful in models of EAE, T1D, and collagen-induced arthritis (CIA) [41,42]. In two recent studies [44,45], attention has been turned to models of liver autoimmunity. Autoreactive CD4+ T cells specific for peptide antigens enriched in the liver, but not found in the liver alone, were re-programmed and expanded as disease-suppressing Tr1-like cells [44]. Remarkably, pMHC-nanoparticles loaded with antigens associated with one model of liver autoimmunity were able to suppress autoimmunity in others, leading the authors to characterize this approach as organ- rather than disease-specific. The tolerance did not impact normal immunity against infection and tumor, even within the liver. The ubiquitous distribution of the antigens prompted the question of whether T cells specific for them would be activated by non-liver autoimmunity and, if so, whether or not those cells could also be utilized by pMHC-nanoparticles. In mice with EAE, that strategy ended up being effective in reducing disease [45]. However, in mice with both EAE and liver autoimmunity, not only did the Tr1-like cells fail to suppress EAE, they were found to traffic primarily to the liver and liver-draining LNs, and not the central nervous system (CNS) or CNS draining LNs. This may be due to a number of possible reasons, including size and vasculature of the liver versus the CNS, differences in antigen availability following damage from the two different autoimmune pathologies, and/or expression of chemokine receptors induced by pMHC-nanoparticles.
In each of the approaches described above, a complex biomolecule was used to either target a specific cell type, enhance tolerogenic capability, or promote the mechanism of tolerance induction. By contrast, our laboratory and collaborators have developed highly negatively charged PLG nanoparticles that are able to either encapsulate or be coupled with protein/peptide antigens and are taken up by tolerogenic macrophages through the MARCO scavenger receptor. Tolerance induction involves multiple mechanisms, including T cell anergy and the production of regulatory cells. We have previously used these particles to prevent or treat disease in EAE [46–48] or adoptive transfer models of T1D [49]. We view it as a strong advantage that our nanoparticles rely solely on their physical characteristics for uptake by tolerogenic APCs in the spleen and liver for subsequent induction of regulatory T cell subsets which induce and maintain long-term tolerance. We expect that this will facilitate their production under good manufacturing practices (GMP) and aid their translation to clinical use.
A recent study coupled a hybrid insulin peptide (HIP) fusion peptide composed of fragments of insulin and chromogranin A (2.5HIP) to negatively charged PLG nanoparticles and administered them i.v. to immunodeficient NOD.SCID mice that had received ex vivo activated diabetogenic BDC2.5 T cells [50]. This prompted anergy in inflammatory cytokine-secreting T cells and altered the ratio between effectors and FoxP3+ Tregs in favor of the latter, ultimately leading to protection from T1D development. As 2.5HIP is a major neoepitope in NODs, these results are likely the first step in a series of tolerance induction accomplishments in mice and ultimately in humans.
Apart from T1D studies, another direction for tolerance induction is celiac disease (CD). PLG nanoparticles encapsulating gliadin protein were shown to be efficacious for treating three separate CD mouse models reducing clinical signs of inflammation and disease [51]. In one of the models (transgenic mice expressing human CD4 and HLA-DQ8), gliadin-encapsulating nanoparticles prior to disease induction increased FoxP3 mRNA in spleen cells re-stimulated with gliadin. This aligned the treatment with our other nanoparticle-based tolerance induction strategies and provided hope that it may be translatable into clinical practice. Remarkedly, we have recently reported results of a recent double-blind, placebo-controlled phase 1/2a clinical trial showing specific prevention of gliadin-specific IFN-γ T cell responses and protection from gut histopathological changes following oral gluten challenge in CD patients tolerized with gliadin-encapsulating PLG nanoparticles [52].
Conclusion
The difficulties of lifelong insulin dependency, as well as the risks of broad immunosuppression, indicate that clinically successful ASI is urgently needed. New delivery approaches, both particle and non-particle based, may make ASI more safe and feasible in clinical trials. An overarching theme of the strategies discussed here is an increased targeting to APCs as a way to make antigens more tolerogenic. When that increased tolerogenicity results in induction or expansion of regulatory cells, infectious bystander tolerance might enable ASI to address polyclonal autoimmunity, thereby increasing the feasibility and, potentially, the safety as well.
Key Points.
Antigen-specific immunotherapy (ASI) seeks to overcome the problems associated with broad immunosuppression, but has not yet achieved clinical acceptance for the treatment of autoimmunity, such as type 1 diabetes (T1D).
New ways to deliver antigen include particle and non-particle based approaches, many of them leading to increased uptake by antigen presenting cells (APCs).
Novel mechanisms for antigen delivery may make ASI more effective, safe, even against a complex, polyclonal immune response.
Financial Support and Sponsorship
Supported in part by Juvenile Diabetes Research Foundation research grant SRA 2018-566-S-B (to SDM). TN was supported by JDRF Postdoctoral Fellowship 3-PDF-2018-582-A-N.
Footnotes
Conflict of Interest
TN declares no conflict of interest. SDM is a co-founder, member of the Scientific Advisory Board, grantee, paid consultant, and holds stock options in COUR Pharmaceutical Development Co.
References and Recommended Reading
Papers of particular interest, published within the past 10 years have been highlighted as:
* of special interest
** of outstanding interest
- 1.In’t Veld P. Insulitis in human type 1 diabetes: a comparison between patients and animal models. Semin Immunopathol 2014, 36:569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. *.Serra P, Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nat Biotechnol 2019, 37:238–251. [DOI] [PubMed] [Google Scholar]; Review of antigen-specific strategies, including a summary of clinical trials and a thorough discussion of the reasons for clinical trial failure.
- 3. *.Loaiza Naranjo JD, Bergot AS, Buckle I, Hamilton-Williams EE. A question of tolerance-antigen-specific immunotherapy for type 1 diabetes. Curr Diab Rep 2020, 20:70. [DOI] [PubMed] [Google Scholar]; Review of antigen-specific treatment for T1D, including many recent studies, organized by category.
- 4. *.Roep BO, Wheeler DCS, Peakman M. Antigen-based immune modulation therapy for type 1 diabetes: the era of precision medicine. Lancet Diabetes Endocrinol 2019, 7:65–74. [DOI] [PubMed] [Google Scholar]; Review of antigen-specific treatment for T1D, including a summary of clinical trials.
- 5.Babon JA, DeNicola ME, Blodgett DM, Crèvecoeur I, Buttrick TS, Maehr R, Bottino R, Naji A, Kaddis J, Elyaman W, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med 2016, 22:1482–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearson JA, Wong FS, Wen L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J Autoimmun 2016, 66:76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prasad S, Kohm AP, McMahon JS, Luo X, Miller SD. Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9–23 epitope and involves functional epitope spreading. J Autoimmun 2012, 39:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, Armstrong M, Powell RL, Reisdorph N, Kumar N, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016, 351:711–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. *.Baker RL, Rihanek M, Hohenstein AC, Nakayama M, Michels A, Gottlieb PA, Haskins K, Delong T. Hybrid insulin peptides are autoantigens in type 1 diabetes. Diabetes 2019, 68:1830–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]; Recent study demonstrating PBMCs from human T1D patients respond to HIPs.
- 10. *.Wiles TA, Powell R, Michel R, Beard KS, Hohenstein A, Bradley B, Reisdorph N, Haskins K, Delong T. Identification of hybrid insulin peptides (HIPs) in mouse and human islets by mass spectrometry. J Proteome Res 2019, 18:814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]; Recent study confirming the presence of HIPs in mouse and human iselts.
- 11.Ludvigsson J, Faresjö M, Hjorth M, Axelsson S, Chéramy M, Pihl M, Vaarala O, Forsander G, Ivarsson S, Johansson C, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. New Eng J Med 2008, 359:1909–1920. [DOI] [PubMed] [Google Scholar]
- 12.Ludvigsson J, Hjorth M, Chéramy M, Axelsson S, Pihl M, Forsander G, Nilsson N, Samuelsson BO, Wood T, Aman J, et al. Extended evaluation of the safety and efficacy of GAD treatment of children and adolescents with recent-onset type 1 diabetes: a randomised controlled trial. Diabetologia 2011, 54:634–640. [DOI] [PubMed] [Google Scholar]
- 13.Chéramy M, Skoglund C, Johansson I, Ludvigsson J, Hampe CS, Casas R. GAD-alum treatment in patients with type 1 diabetes and the subsequent effect on GADA IgG subclass distribution, GAD65 enzyme activity and humoral response. Clin Immunol 2010, 137:31–40. [DOI] [PubMed] [Google Scholar]
- 14.Wherrett DK, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Herold KC, Marks JB, et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet 2011, 378:319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Judkowski V, Rodriguez E, Pinilla C, Masteller E, Bluestone JA, Sarvetnick N, Wilson DB. Peptide specific amelioration of T cell mediated pathogenesis in murine type 1 diabetes. Clin Immunol 2004, 113:29–37. [DOI] [PubMed] [Google Scholar]
- 16.Xu D, Prasad S, Miller SD. Inducing immune tolerance: a focus on Type 1 diabetes mellitus. Diabetes Manag (Lond) 2013, 3:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinman L, Ho PP, Robinson WH, Utz PJ, Villoslada P. Antigen-specific tolerance to self-antigens in protein replacement therapy, gene therapy and autoimmunity. Curr Opin Immunol 2019, 61:46–53. [DOI] [PubMed] [Google Scholar]
- 18.Pozzilli P, Pitocco D, Visalli N, Cavallo MG, Buzzetti R, Crinò A, Spera S, Suraci C, Multari G, Cervoni M, et al. No effect of oral insulin on residual beta-cell function in recent-onset type I diabetes (the IMDIAB VII). IMDIAB Group. Diabetologia 2000, 43:1000–1004. [DOI] [PubMed] [Google Scholar]
- 19.Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C, Cuthbertson D, Rafkin-Mervis LE, Chase HP, Leschek E. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial--Type 1. Diabetes Care 2005, 28:1068–1076. [DOI] [PubMed] [Google Scholar]
- 20.Arikat F, Hanna SJ, Singh RK, Vilela L, Wong FS, Dayan CM, Coulman SA, Birchall JC. Targeting proinsulin to local immune cells using an intradermal microneedle delivery system; a potential antigen-specific immunotherapy for type 1 diabetes. J Control Release 2020, 322:593–601. [DOI] [PubMed] [Google Scholar]
- 21.Dul M, Nikolic T, Stefanidou M, McAteer MA, Williams P, Mous J, Roep BO, Kochba E, Levin Y, Peakman M, et al. Conjugation of a peptide autoantigen to gold nanoparticles for intradermally administered antigen specific immunotherapy. Int J Pharm 2019, 562:303–312. [DOI] [PubMed] [Google Scholar]
- 22.Turley DM, Miller SD. Peripheral tolerance induction using ethylenecarbodiimide-fixed APCs uses both direct and indirect mechanisms of antigen presentation for prevention of experimental autoimmune encephalomyelitis. J Immunol 2007, 178:2212–2220. [DOI] [PubMed] [Google Scholar]
- 23.Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, Getts MT, Martin AJ, Luo X, Terry RL, et al. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J Immunol 2011, 187:2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Getts DR, McCarthy DP, Miller SD. Exploiting apoptosis for therapeutic tolerance induction. J Immunol 2013, 191:5341–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lutterotti A, Yousef S, Sputtek A, Stürner KH, Stellmann JP, Breiden P, Reinhardt S, Schulze C, Bester M, Heesen C, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med 2013, 5:188ra175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikolic T, Zwaginga JJ, Uitbeijerse BS, Woittiez NJ, de Koning EJ, Aanstoot HJ, Roep BO. Safety and feasibility of intradermal injection with tolerogenic dendritic cells pulsed with proinsulin peptide-for type 1 diabetes. Lancet Diabetes Endocrinol 2020, 8:470–472. [DOI] [PubMed] [Google Scholar]
- 27.Zubizarreta I, Flórez-Grau G, Vila G, Cabezón R, España C, Andorra M, Saiz A, Llufriu S, Sepulveda M, Sola-Valls N, et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc Natl Acad Sci U S A 2019, 116:8463–8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol Ther Methods Clin Dev 2020, 16:50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Husseiny MI, Du W, Mbongue J, Lenz A, Rawson J, Kandeel F, Ferreri K. Factors affecting Salmonella-based combination immunotherapy for prevention of type 1 diabetes in non-obese diabetic mice. Vaccine 2018, 36:8008–8018. [DOI] [PubMed] [Google Scholar]
- 30.Mbongue JC, Rawson J, Garcia PA, Gonzalez N, Cobb J, Kandeel F, Ferreri K, Husseiny MI. Reversal of new onset type 1 diabetes by oral Salmonella-based combination therapy and mediated by regulatory T-cells in NOD mice. Front Immunol 2019, 10:320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cook DP, Cunha J, Martens PJ, Sassi G, Mancarella F, Ventriglia G, Sebastiani G, Vanherwegen AS, Atkinson MA, Van Huynegem K, et al. Intestinal delivery of proinsulin and IL-10 via Lactococcus lactis combined with low-dose anti-CD3 restores tolerance outside the window of acute type 1 diabetes diagnosis. Front Immunol 2020, 11:1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neef T, Miller SD. Tolerogenic nanoparticles to treat islet autoimmunity. Curr Diab Rep 2017, 17:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akimoto H, Fukuda-Kawaguchi E, Duramad O, Ishii Y, Tanabe K. A novel liposome formulation carrying both an Iisulin peptide and a ligand for invariant natural killer T cells induces accumulation of regulatory T cells to islets in nonobese diabetic mice. J Diabetes Res 2019, 2019:9430473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villalba A, Rodriguez-Fernandez S, Ampudia RM, Cano-Sarabia M, Perna-Barrull D, Bertran-Cobo C, Ehrenberg C, Maspoch D, Vives-Pi M. Preclinical evaluation of antigen-specific nanotherapy based on phosphatidylserine-liposomes for type 1 diabetes. Artif Cells Nanomed Biotechnol 2020, 48:77–83. [DOI] [PubMed] [Google Scholar]
- 35.Villalba A, Rodriguez-Fernandez S, Perna-Barrull D, Ampudia RM, Gomez-Muñoz L, Pujol-Autonell I, Aguilera E, Risueño RM, Cano-Sarabia M, Maspoch D, et al. Antigen-specific immunotherapy combined with a regenerative drug in the treatment of experimental type 1 diabetes. Sci Rep 2020, 10:18927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis JS, Dolgova NV, Zhang Y, Xia CQ, Wasserfall CH, Atkinson MA, Clare-Salzler MJ, Keselowsky BG. A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice. Clin Immunol 2015, 160:90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho JJ, Stewart JM, Drashansky TT, Brusko MA, Zuniga AN, Lorentsen KJ, Keselowsky BG, Avram D. An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis. Biomaterials 2017, 143:79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis JS, Stewart JM, Marshall GP, Carstens MR, Zhang Y, Dolgova NV, Xia C, Brusko TM, Wasserfall CH, Clare-Salzler MJ, et al. Dual-sized microparticle system for generating suppressive dendritic cells prevents and reverses type 1 diabetes in the nonobese diabetic mouse model. ACS Biomater Sci Eng 2019, 5:2631–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stewart JM, Posgai AL, Leon JJ, Haller MJ, Keselowsky BG. Combination treatment with antigen-specific dual-sized microparticle system plus anti-CD3 immunotherapy fails to synergize to improve late-stage type 1 diabetes prevention in nonobese diabetic mice. ACS Biomater Sci Eng 2020, 6:5941–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh RK, Malosse C, Davies J, Malissen B, Kochba E, Levin Y, Birchall JC, Coulman SA, Mous J, McAteer MA, et al. Using gold nanoparticles for enhanced intradermal delivery of poorly soluble auto-antigenic peptides. Nanomed 2020, 32:102321. [DOI] [PubMed] [Google Scholar]
- 41.Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, Yang Y, Medarova Z, Moore A, Santamaria P. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity 2010, 32:568–580. [DOI] [PubMed] [Google Scholar]
- 42.Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, Tsai S, Wang J, Garabatos N, Izquierdo C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 2016, 530:434–440. [DOI] [PubMed] [Google Scholar]
- 43.Singha S, Shao K, Yang Y, Clemente-Casares X, Solé P, Clemente A, Blanco J, Dai Q, Song F, Liu SW, et al. Peptide–MHC-based nanomedicines for autoimmunity function as T-cell receptor microclustering devices. Nature Nanotechnol 2017, 12:701–710. [DOI] [PubMed] [Google Scholar]
- 44. **.Umeshappa CS, Singha S, Blanco J, Shao K, Nanjundappa RH, Yamanouchi J, Parés A, Serra P, Yang Y, Santamaria P. Suppression of a broad spectrum of liver autoimmune pathologies by single peptide-MHC-based nanomedicines. Nat Commun 2019, 10:2150. [DOI] [PMC free article] [PubMed] [Google Scholar]; Recent study using iron oxide nanoparticles coated with peptide-MHC to treat several mouse models of liver autoimmunity, also confirming lack of impact on general immune system function.
- 45. **.Umeshappa CS, Mbongue J, Singha S, Mohapatra S, Yamanouchi J, Lee JA, Nanjundappa RH, Shao K, Christen U, Yang Y, et al. Ubiquitous antigen-specific T regulatory type 1 cells variably suppress hepatic and extrahepatic autoimmunity. J Clin Invest 2020, 130:1823–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]; Recent study investigating ubiquitous nature of particles and antigens from previous study, determining that they are capable of suppressing non-liver autoimmunity and that liver inflammation sequesters expanded regulatory T cells.
- 46. *.Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol 2012, 30:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]; First demonstration that i.v. injection of carboxylated PLG nanoparticles can induce antigen-specific toleranc
- 47.Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, Miller SD. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano 2014, 8:2148–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCarthy DP, Yap JW, Harp CT, Song WK, Chen J, Pearson RM, Miller SD, Shea LD. An antigen-encapsulating nanoparticle platform for T(H)1/17 immune tolerance therapy. Nanomed 2017, 13:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. *.Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Shea LD, Miller SD. Tolerogenic Ag-PLG nanoparticles induce Tregs to suppress activated diabetogenic CD4 and CD8 T cells. J Autoimmun 2018, 89:112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstration that PLG nanoparticles bearing diabetogenic peptide epitopes can inhibit the effector function of both activated autoreactive CD4 and CD8 T cells
- 50. *.Jamison BL, Neef T, Goodspeed A, Bradley B, Baker RL, Miller SD, Haskins K. Nanoparticles containing an insulin-ChgA hybrid peptide protect from Ttansfer of autoimmune diabetes by shifting the balance between effector T cells and regulatory T cells. J Immunol 2019, 203:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstration that PLG nanoparticles bearing HIP antigen protect against adoptive transfer T1D, working both through induction of T cell anergy and a change in the relative proportion of FoxP3+ Tregs.
- 51 *.Freitag TL, Podojil JR, Pearson RM, Fokta FJ, Sahl C, Messing M, Andersson LC, Leskinen K, Saavalainen P, Hoover LI, et al. Gliadin nanoparticles Induce immune tolerance to gliadin in mouse models of celiac disease. Gastroenterol 2020, 158:1667–1681.e1612. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstration that gliadin-loaded PLG nanoparticles show signs of effectiveness in celiac models, including the increase of FoxP3 Tregs in a transgenic human-like model.
- 52. *.Kelly CP, Murray JA, Leffler DA, Getts DR, Bledso AC, Smithson G, First MR, Morris A, Boyne M, Elhofy A, et al. TAK-101 nanoparticles induced gluten-specific tolerance in celiac disease. Gastroenterol. 2021, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstration of efficacy of gliadin-loaded PLG nanoparticles for tolerance induction in celiac disease patients.
- 53.McCarthy DP, Hunter ZN, Chackerian B, Shea LD, Miller SD. Targeted immunomodulation using antigen-conjugated nanoparticles. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2014, 6:298–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Getts DR, Shea LD, Miller SD, King NJ. Harnessing nanoparticles for immune modulation. Trends Immunol 2015, 36:419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serra P, Santamaria P. Nanoparticle-based autoimmune disease therapy. Clin Immunol 2015, 160:3–13. [DOI] [PubMed] [Google Scholar]
- 56.Lockman PR, Koziara JM, Mumper RJ, Allen DD. Nanoparticle surface charges alter blood-brain barrier integrity and permeability. J Drug Target 2004, 12:635–641. [DOI] [PubMed] [Google Scholar]
- 57.Malik N, Wiwattanapatapee R, Klopsch R, Lorenz K, Frey H, Weener JW, Meijer EW, Paulus W, Duncan R. Dendrimers: relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125I-labelled polyamidoamine dendrimers in vivo. J Control Release 2000, 65:133–148. [DOI] [PubMed] [Google Scholar]