

Summary paragraph
Strong connections exist between R-loops, genome instability, and human disease1–5. Indeed, R-loops are favoured in relevant genomic regions as regulators of certain physiological processes wherein homeostasis is typically maintained. For example, R-loop-associated transcription termination pause sites can induce the synthesis of antisense transcripts that enable the formation of local, RNA interference (RNAi)-driven heterochromation6. Pause sites are also protected against endogenous ssDNA breaks by BRCA17. Hypotheses about how DNA repair is enacted at pause sites includes a role for RNA, which is emerging as a normal, albeit unexplained, genome integrity regulator8.
Here we report a new species of single-stranded, DNA damage-associated sRNA (sdRNA) that is, surprisingly generated by a BRCA1/RNAi protein complex. sdRNAs promote PalB2/ Rad52 complex-driven DNA repair at R-loop, ssDNA break - rich transcriptional termination pause sites. sdRNA repair operates both in quiescent (G0) and proliferating cells. Thus, sdRNA repair can occur in intact tissue and/or stem cells, conceivably contributing to BRCA1 tumor suppression.
RNA is emerging as an important player in DNA repair. Small, double-stranded RNAs (diRNAs or DDRNAs), likely products of the RNAi, are produced at engineered double strand break (DSBs) and promote their repair9–13 by binding to Ago2 which guides the recruitment of these proteins to a suitable DNA break14,15. In yeast, endogenous RNA molecules mediate DSB16,17.
BRCA1 (p220), a driver of homologous recombination -driven DSB repair (HR-DSBR)18, also confronts ssDNA damage at R-loop termination sites7. RNAi factors participate in transcription termination, in part, through R loop-mediated heterochromatin formation6. Therefore, we asked whether BRCA1-mediated DNA repair at pause sites is RNAi-dependent.
BRCA1 and RNAi cooperate in DNA repair
To address this, we initially investigated whether BRCA1 and the RNAi machinery form physiological complexes that promote R-loop-mediated DNA damage repair, we co-immunoprecipitated (co-IPed) endogenous BRCA1, the helicase, Senataxin (SETX), Dicer, and Ago1 in HeLa cells (Fig. 1a and Extended Data Fig. 1a–d). These complexes were also detected in telomerase- immortalized primary mammary epithelial cells (HMEC) and fibroblasts (BJ-hTert) (Extended Data Fig. 1e). Results in RNAseH1-overexpressing cells implied that BRCA1/RNAi complex formation is, at least in part, R-loop-dependent (Extended Data Fig. 1f).
Figure 1 |. BRCA1 interacts with Dicer and Ago1/2 to prevent R-loop damage.
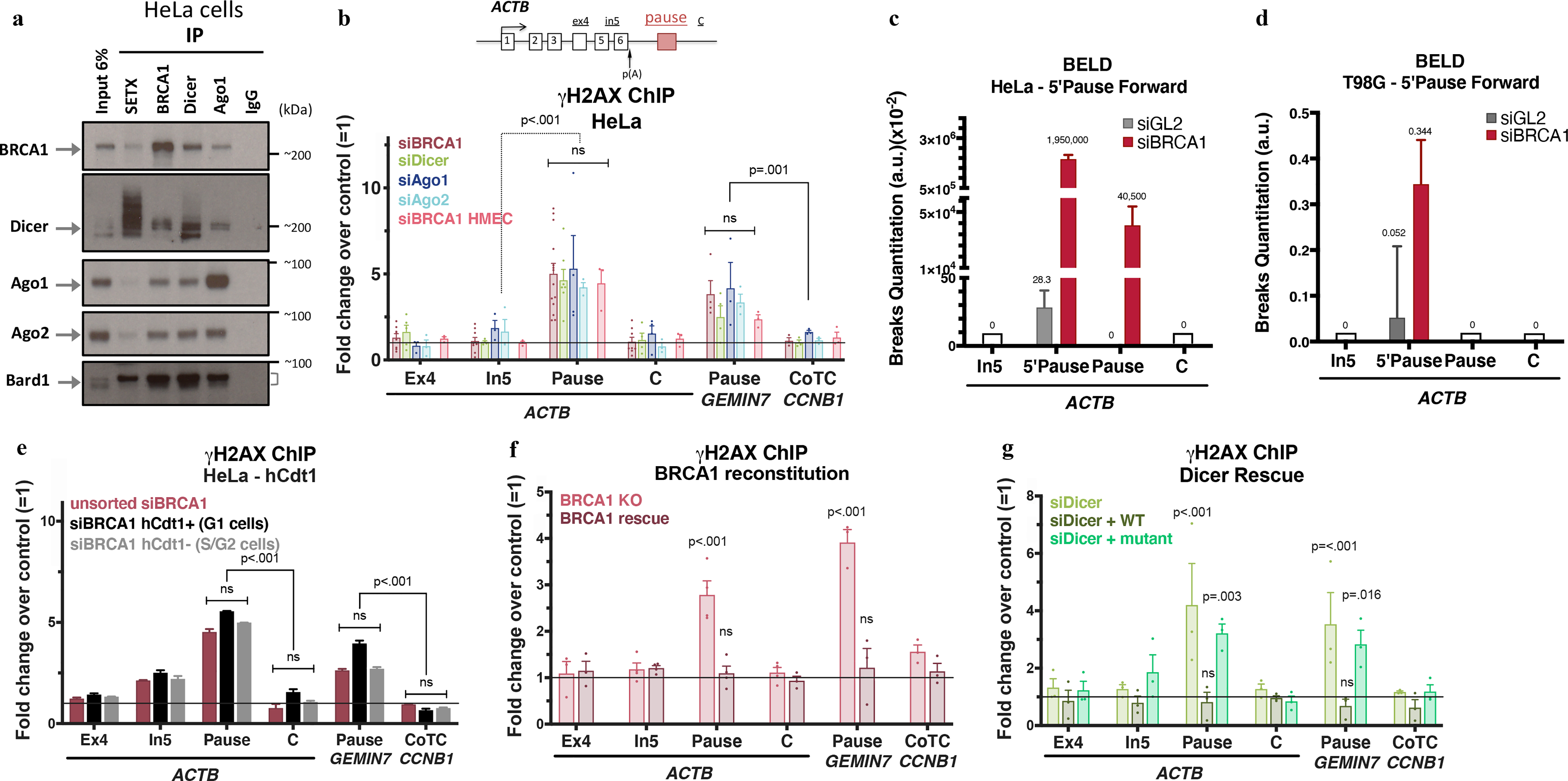
a, Immunoblots from co-IP of endogenous SETX, BRCA1, Dicer, or Ago1 present in nuclear extracts (NE) of HeLa cells. IgG, negative control; Cyt, cytoplasm; NS, Nuclear Soluble; Chrom, chromatin. n=7 b, DNA damage quantitation analysis using γ-H2AX ChIP qPCR analyses following the indicated depletion relative to mock siRNA. ACTB primer location is shown on the scheme. R-loop-positive pause sites (ACTB and GEMIN7) or R-loop-negative (CCNB1 CoTC). Error bars denote s.e.m. (n = 3–14 biological replicates). c-d, BELD quantitation on the ACTB pause site forward/coding strand in HeLa (c) and T98G cells (d). Average relative abundance of qPCR replicates ± s.d are shown. e, Cell cycle- dependent analysis of γ-H2AX ChIP signals in HeLa FUCCI cells. The histograms depict average fold changes of the qPCR replicates ± s.d. f-g, DNA damage analyses in BRCA1 (h) or Dicer (i) rescue experiments, showing average ChIP signals ± s.e.m. (n=3–4 biological replicates). Data were all analyzed by Two-way ANOVA with post-hoc Tukey HSD (except for f, one-way) and compared to data from the undamaged locus, or relevant control cells.
We have studied BRCA1 and RNAi factor recruitment to the human ACTB R-loop -forming transcription termination region, which spontaneously develops R loop-dependent ssDNA breaks7. RNAi factors binding to this region6 was BRCA1- dependent (Extended Data Fig. 1g).
To determine whether RNAi factors protect pause sites from DNA damage, we measured γ-H2AX ChIP signals at ACTB and two additional, R-loop-forming pause sites (GEMIN7 and ENSA). Depleting any of these RNAi factors led to a 5 fold increase in the γ-H2AX ChIP signal. By contrast, cotranscriptional cleavage (CoTC) terminators, which do not form R-loops6, e.g. CCNB1 and AKIRIN119, remained unaffected by BRCA1/RNAi depletion (Fig. 1b and Extended Data Fig. 1h–j).
The DNA damage at the ACTB pause site included ssDNA breaks (SSB) and a vicinal γH2AX signal7. To verify this claim, we developed a new SSB assay (BELD = Broken End Labeling and Detection; Methods Extended Data Fig. 1k–l). Here we employed Terminal deoxynucleotidyl transferase (TdT) labeling of broken DNA ends.
On the forward strand, a strong BELD signal was observed at the ACTB 5’Pause/Pause region, in BRCA1-depleted HeLa and T98G cells (Fig. 1c–d). The significant BELD signal decrease between 5’Pause and Pause sequences (87nt apart) implied that the breaks were enriched 5’ of the Pause site. BELD signal on the ACTB pause reverse strand was BRCA1-independent (Extended Data Fig. 1m). Two, independent assays confirmed that BRCA1 loss primarily induced ssDNA breaks at the forward strand of ACTB pause site.
BRCA1 HR function operates primarily during S/G218,20,21 where BRCA1 expression peaks22,23. To determine whether pause site BRCA1 DNA repair is also cell cycle- regulated, we performed DNA damage analysis in HeLa FUCCI and synchronized T98G cells (Fig. 1e and Extended Data Fig. 2a–e). Unexpectedly, γH2AX signals at the ACTB and GEMIN7 pause sites increased throughout interphase following BRCA1 depletion, possibly reflecting the existence of an unanticipated repair function for BRCA1 in G1 where BRCA1 levels, although low, remain detectable24.
We also undertook BRCA1 and Dicer rescue experiments, respectively using HMECs expressing a Doxycycline- inducible BRCA1 shRNA and Dicer-depleted HeLa cells that we reconstituted with either WT or an enzymatically-inactive Dicer mutant (Extended Data Fig. 2f). γ-H2AX ChIP analyses revealed that the DNA damage phenotype was clearly BRCA1 and Dicer depletion- dependent (Fig. 1f–g). Thus, BRCA1 and Dicer are active in pause site DNA damage repair, which likely involves a Dicer- processed RNA molecule, since catalytically inactive Dicer failed to rescue the damage induced by Dicer loss.
New sRNA species involved in DNA repair
In search of a role for sRNA in pause site repair, we performed genome-wide analyses, using NGS and a custom bioinformatics pipeline that allowed one to identify known and previously uncharacterized sRNA species. We searched for sRNAs whose abundance was differentially regulated following BRCA1 or RNAi depletion.
In HeLa cells, ~3500 sRNAs underwent a statistically significant decrease when compared to control conditions (mock and 53BP1 siRNA, no DNA damage) (Fig. 2a and Extended Data Fig. 3a). These results were confirmed in BRCA1-depleted HMECs (Extended Data Fig. 3b).
Figure 2 |. A new species of sRNA mediates locus- specific DNA damage repair.
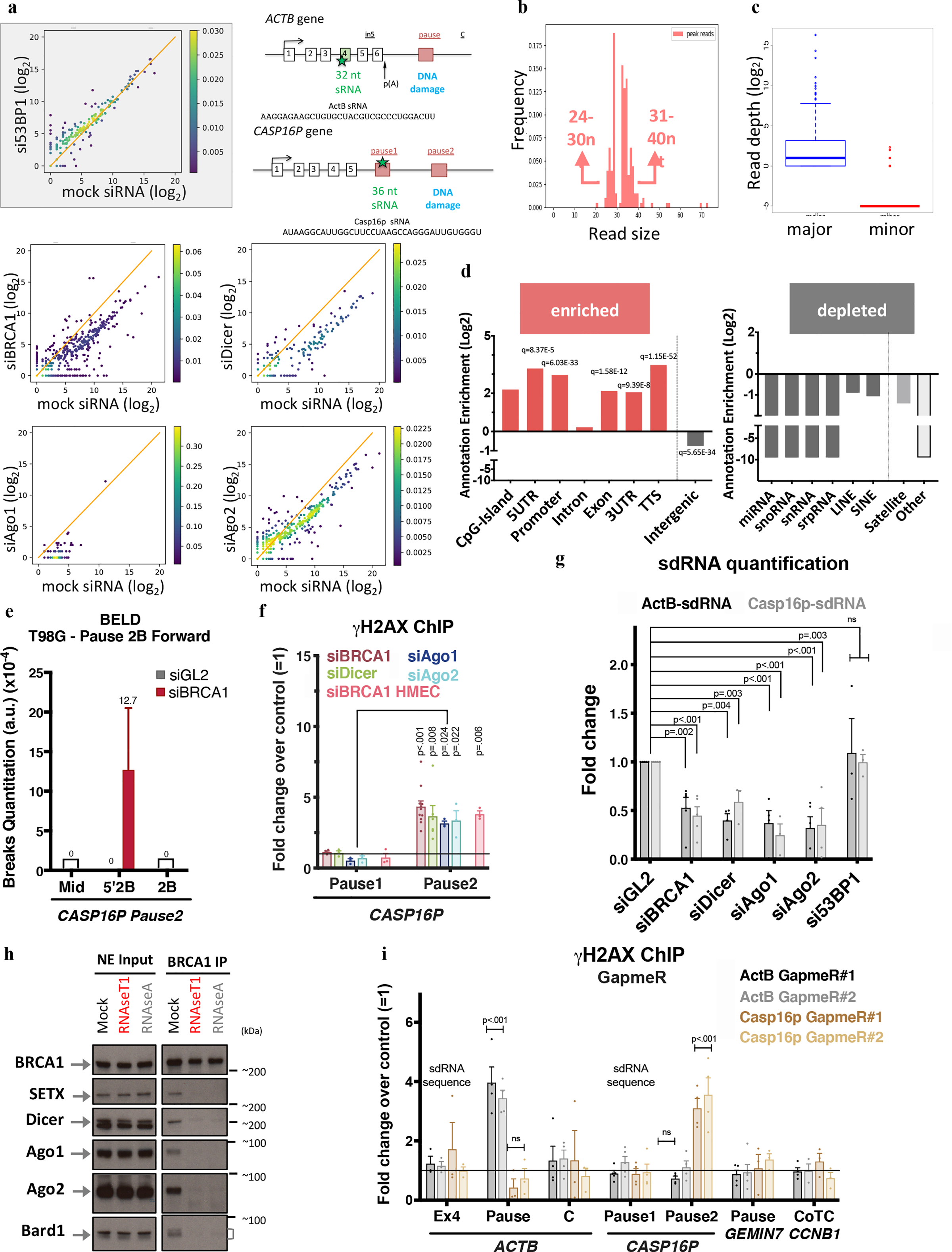
a, Density scattered plots comparing differential sRNA expression levels between mock siRNA and experimental samples. Diagonal line = no fold change in expression. b, New sRNAs size distribution frequency. c, Strand specificity analysis of the sequence reads obtained from the sRNA NGS. Boxplots depict read quantities by strand with the most (major) or the fewest (minor) reads. d, Annotation enrichment (Log2 scale) of genomic features that correspond to relevant sRNA sequences. q-values = multiple correction- adjusted p-values. e, A representative experiment depicting BELD quantitation of Forward/coding strand CASP16P pause2 site breaks in T98G cells. The average abundance of qPCR replicates ± s.d is shown. ActB- and Casp16p-sdRNA sequences and location are in the top right schematic. f, DNA damage quantitation at CASP16P pause sites. γ-H2AX ChIP qPCR analysis reflects the average fold change following a relevant depletion versus a mock siRNA condition (n =3–11 biological replicates). g, Taqman-based quantitative analysis of ActB- and Casp16p-sdRNA abundance in HeLa cells. Average qPCR values ± s.e.m. (n = 3–5 biological replicates). h, Immunoblotting of BRCA1 binding partners in NE pre- and post-RNaseA or RNaseT1 exposure, n=6. i, DNA damage analyses upon depletion of ActB- or Casp16p-sdRNA using two, individual GapmeRs for each target compared to a negative control GapmeR. Average γ-H2AX ChIP ± s.e.m. (n = 3–5 biological replicates). Data were analyzed by Two-way ANOVA with post-hoc Tukey HSD (except for g, multiple t-test) and compared to the undamaged locus or the relevant control cells.
Previous studies showed that sRNA, double-stranded and miRNA-like RNAs, engage in DNA damage repair12,25,26,11,15. Unexpectedly, the sRNAs we identified were 24 to 40nt long (Fig. 2b) and single -stranded (ss), with the NGS reads aligning to only one DNA strand (Fig. 2c). Furthermore, genomic annotation analysis revealed that the sRNA sequences were enriched for transcription- regulating elements prone to R-loop formation3,27 (Fig. 2d). Introns, intergenic regions, and sequences encoding other sRNAs (e.g. miRNAs) were under-represented in our catalog. Thus, this new class of BRCA1- and Dicer/Ago-dependent ss sRNAs is distinct from ds diRNAs/DDRNAs, which are shorter and appear after engineering a DSB10,15.
We also observed a genome-wide decrease in sRNA abundance after α-amanitin or DRB addition, suggesting that our sRNAs are RNA polymerase II products (Extended Data Fig. 3c–d). At the single gene level, we also observed a correlation between ACTB-derived nascent RNA transcripts and the corresponding ActB-sRNA following DRB treatment (Extended Data Fig. 3e), suggesting that nascent transcripts are precursor(s) of this new class of sRNAs.
sRNA function
We further analysed two of these sRNAs: 1) ActB sRNA, encoded within exon4 of ACTB and 2) Casp16p sRNA, encoded within the pause site of the CASP16P pseudogene.
BELD and γ-H2AX ChIP analyses revealed that BRCA1 depletion had induced cell cycle- independent damage accumulation (Pause2), as reflected by the presence of sense strand breaks (Fig. 2e and Extended Data Fig. 3f–h). RNAi factors also protected the CASP16P Pause2 site from DNA damage (Fig. 2f). Like ACTB, the phenotype was both BRCA1- and Dicer-dependent (Extended Data Fig. 3i–j). qPCR quantitation of ActB- and Casp16p-sRNAs, using custom-designed Taqman probes, confirmed the NGS data (Fig. 2g).
Importantly, depletion of Drosha, the dsRNA-specific Microprocessor endoribonuclease, revealed no DNA damage nor modulation of sRNA expression, implying that this new BRCA1/RNAi pathway is distinct from miRNA biogenesis (Extended Data Fig. 3k). Also, sRNA subcellular analyses showed that ~90% were nuclear, consistent with the hypothesis that most perform a DNA damage repair function (Extended Data Fig. 3l).
Surprisingly, BRCA1 co-IP with the RNAi factors and, more unexpectedly, with Bard1 was strongly compromised by single stranded RNA degradation (Fig. 2h and Extended Data Fig. 3m). These results suggest for the first time that single-stranded RNAs are required to maintain stability of this BRCA1-containing, multi- protein complex.
Next, we individually depleted the ActB- and Casp16p-sRNAs, using specific, single-stranded antisense DNA oligonucleotides (LNA GapmeRs). γ-H2AX ChIP analyses revealed that ActB-sRNA depletion led to increased DNA damage at the ACTB pause site but not the CASP16P gene and vice versa (Fig. 2i and Extended Fig. 3n–o).
These data strongly suggest that a new species of single-stranded sRNA, with characteristics that differ from what was previously reported11,26,28 is involved in DNA repair. Furthermore, these sRNAs, whose abundance is BRCA1- and RNAi-dependent, appear to act in a site-specific manner and “at a distance”, since their coding sequence lies upstream of the damage site (Fig. 1b and 2f, i). These two features of the single-stranded damage-associated sRNAs (sdRNA) suggest that they propagate a novel, sRNA-dependent DNA damage repair mechanism at R loop-forming termination pause sites.
sdRNAs can rescue BRCA1/RNAi depletion
We undertook complementation experiments by transfecting synthetic ActB- and Casp16p-sdRNAs into BRCA1-, Dicer-, or Ago1-depleted cells. The results showed that ActB- and Casp16p-sdRNAs, individually, led to damage repair in the same gene that encoded them (Fig. 3a). Thus, BRCA1-Dicer-Ago1 were no longer critical for repair once each sdRNA was provided in trans. Moreover, using BELD, we confirmed that BRCA1-depleted HeLa cells, complemented by either ActB- or Casp16p-sdRNA, manifested a specific sdRNA-driven reduction of free DNA ends at the relevant pause sites (Fig. 3b and Extended Data Fig. 4a).
Figure 3 |. BRCA1/Dicer/Ago promote sdRNA production; PalB2/Rad52 promote sdRNA-mediated repair.
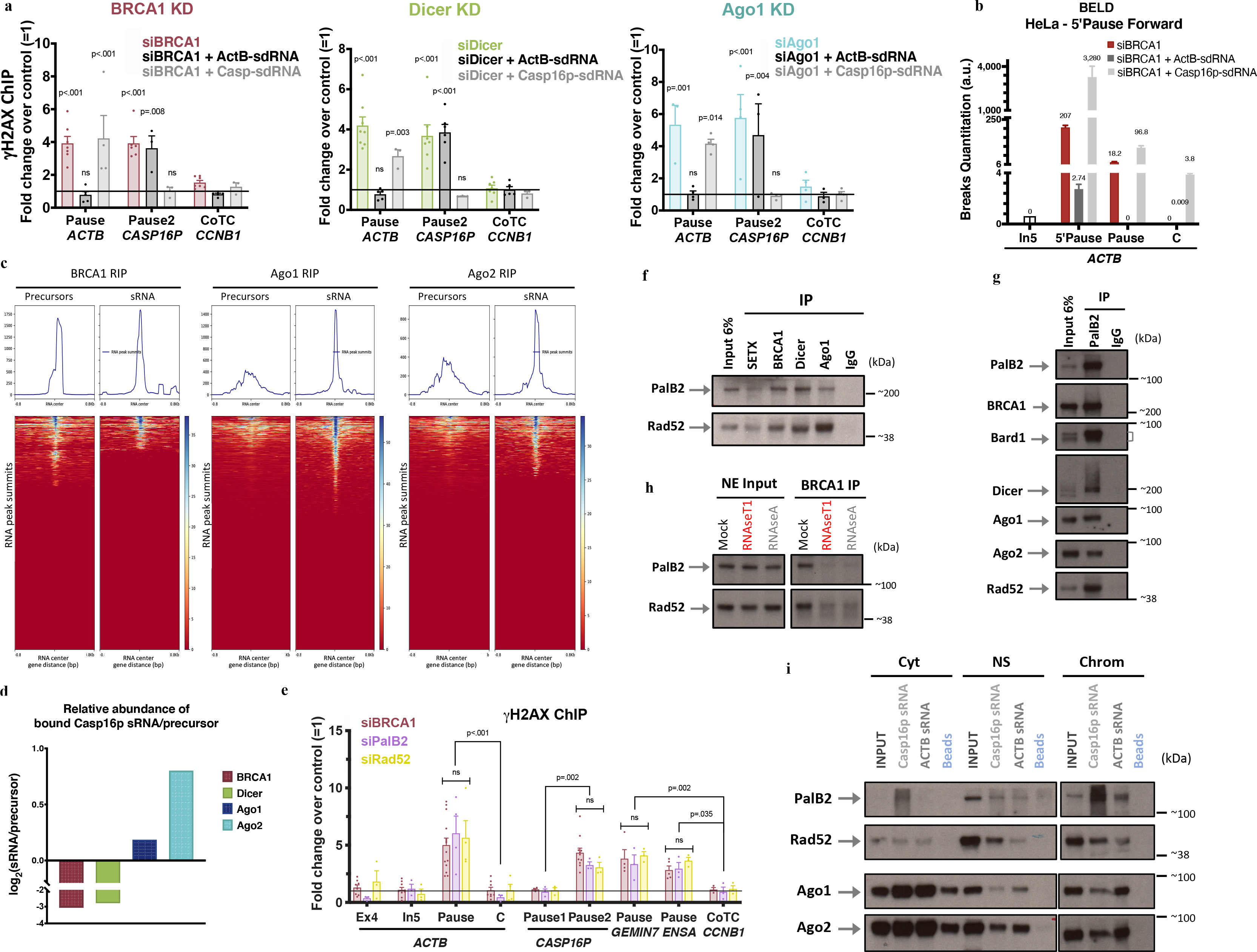
a, γ-H2AX ChIP analyses of ActB- and Casp16p-sdRNA complementation experiments in BRCA1-, Dicer- or Ago-depleted HeLa cells (n = 3–7; 3–7 and 3–4 biological replicates, respectively). b, Representative forward strand ACTB pause site BELD analysis in BRCA1-depleted HeLa cells, complemented with ActB- or Casp16p-sdRNA. Average fold change of qPCR replicates ± s.d. c, BRCA1, Ago1, and Ago2 genome-wide RNA RIP analyses of large (<300nt) and sRNAs, are shown as heat maps centered around the sdRNA peaks (+/− 0.8kb). d, HeLa WCE BRCA1, Dicer or Ago1/2 RIP analyses showing the binding ratio (Log2) between the Casp16p-sdRNA and its precursor. e, γ-H2AX ChIP DNA damage quantitation in PalB2- or Rad52-depleted HeLa cells. All ChIP analyses depict the average fold change ± s.e.m over the mock siRNA condition (n = 3–14 biological replicates). f-g, Immunoblots showing the proteins co-IPed from endogenous SETX, BRCA1, Dicer or Ago1 (f) and PalB2 (g) from HeLa NE, n=7. h, BRCA1 NE co-IP showing Rad52 and PalB2 immunoblots pre or post exposure to RNaseA or RNaseT1, n=6. i, RNA/protein binding assays, performed in HeLa subcellular fractions, using synthetic biotinylated Casp16p- and ActB-sdRNAs or streptavidin beads, were detected by immunoblotting (n=5). Cyt, cytoplasm; NS, Nuclear Soluble; Chrom, chromatin. ChIP data were all analyzed by Two-way ANOVA with post-hoc Tukey HSD and compared to the undamaged locus, or relevant control cells.
There was no repair following BRCA1 or Dicer complementation with the antisense of either sdRNA, the DNA version of ActB-sdRNA, indicating that site-specific repair occurred only in the presence of the cognate, strand-specific sdRNA (Extended Data Fig. 4b–c). Moreover, ActB-sdRNA, chemically blocked for polymerization in the 3’-direction, remained repair-competent, implying that this sdRNA doesn’t serve as a repair synthesis primer.
We then performed BRCA1, Ago1, and Ago2 RNA ImmunoPrecipitation (RIP) analyses. Two types of cDNA libraries (unfragmented RNAs (<300nt) and sdRNAs) were prepared, in parallel, for each RIP and analyzed by NGS. The relevant RIP reads were aligned to and centered around our catalog of ~3500 sdRNAs (Fig. 2a–d and Extended Data Fig. 5a–b). These data, represented as heat maps, showed that BRCA1 bound preferentially to putative sdRNA precursors (average length of ~120nt) while Ago1/Ago2 manifest greater avidity for the 24–40nt-long, repair-proficient sdRNAs (Fig. 3c).
Quantitation of the relative abundance of ActB- or Casp16p-sdRNAs and their candidate precursors bound to either BRCA1, Dicer, or Ago1/Ago2, supported the genome- wide results at the single gene level (Fig. 3d and Extended Data Fig. 5c), further suggesting that BRCA1/RNAi modulate the biogenesis/processing of the sdRNAs.
PalB2/Rad52/sdRNA repair complexes
The nature of sdRNAs is distinct from that of the RNAs previously known to participate in DNA repair. Nevertheless, in an effort to identify proteins contributing to sdRNA-mediated DNA repair, we tested several candidates based on the BRCA1/DNA repair literature25,29,30.
Two proteins scored positively, a) PalB2 -a known BRCA1-binding tumor and breast cancer suppressor31,32 and b) Rad52, a DNA repair protein. Of note, Rad52 was recently associated with RNA-mediated homologous DNA recombination, as well as R-loop processing in transcription- associated HR repair of DSBs16,17,33. Indeed, strong γ-H2AX ChIP signals were detected at the ACTB, CASP16P, GEMIN7 and ENSA termination pause sites after depleting PalB2 or Rad52 in HeLa cells (Fig. 3e and Extended Data Fig. 6a–b).
Additionnaly, PalB2 and Rad52 co-IP with BRCA1/RNAi complexes were detected in HeLa and HMECs and confirmed by reciprocal co-IP (Fig. 3f–g and Extended Data Fig. 6c–f). Moreover, similar to Dicer and Ago (Fig. 2h), BRCA1, PalB2 and Rad52 co-IP was lost when the nuclear extract was exposed to RNaseA or RNAseT1 (Fig. 3h), implying that ssRNA(s) is/are essential for the assembly of this new and larger DNA repair complex. Nuclear PalB2 and Rad52 RIP analyses also revealed that both proteins have a greater avidity for the repair-proficient Casp16p-sdRNA than for its precursor (Extended Data Fig. 6g).
To determine whether a 24–40nt long sdRNA is responsible for the assembly of this new repair complex, we performed RNA binding assays in pre-fractionated HeLa and T98G extracts, using synthetic, biotinylated Casp16p- and ActB-sdRNAs. Strikingly, each sdRNA interacted in the nucleus, and on chromatin where the repair takes place, with Ago and PalB2/Rad52, key components of the DNA repair complex, while the antisense sdRNA or their DNA analogs failed to do so (Fig. 3i and Extended Data Fig. 7a–c).
We next performed RNA pulldowns and γ-H2AX ChIP experiments, using two unrelated, single-stranded sRNA sequences of approximately the same base composition as endogenous ActB-sdRNA. These sRNAs mapped either upstream (−46bp from the 5’-end) or downstream (+31bp) from the 3’-end. The results indicated that only ActB-sdRNA (+31), located between sdRNA sequence and the DNA damage was able to bind BRCA1/RNAi complex and rescue ACTB pause site specific DNA damage upon BRCA1 or Dicer depletion (Extended Data Fig. 7d–f), suggesting that specific binding of a relevant sdRNA to Palb2, Rad52 and Ago proteins constitutes an important biochemical event during this new, sdRNA-driven, DNA damage repair process.
Interplay within the repair complex
To further address PalB2/Rad52 role, we performed sdRNAs complementation experiments in PalB2- or Rad52- depleted cells. In both conditions, neither sdRNA rescued the DNA damage that arose at either pause site (Fig. 4a).
Figure 4 |. Biological outcome of the interplay between sdRNA, PalB2 and Rad52 in pause site DNA damage repair.
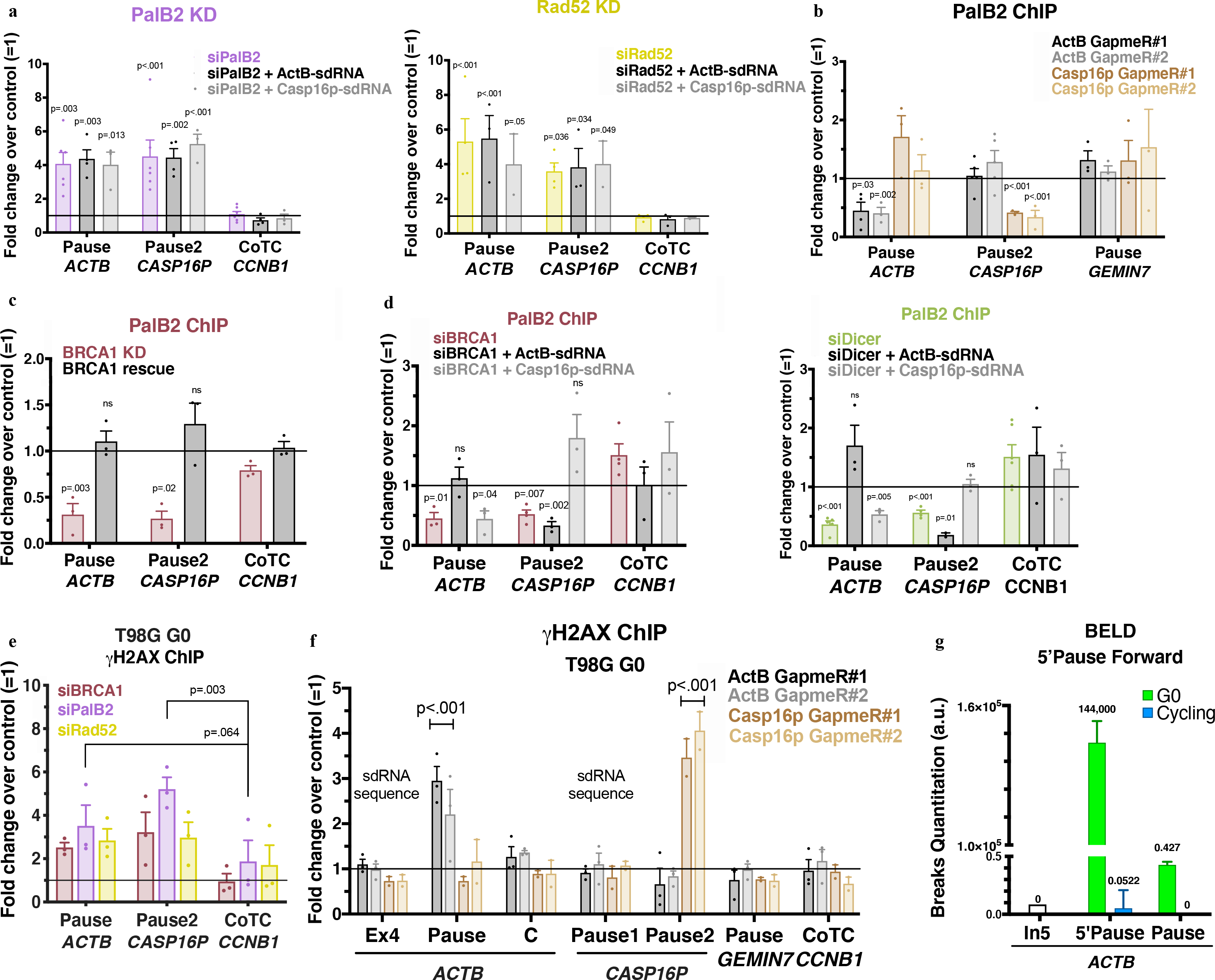
a, γ-H2AX ChIP analyses of ActB- and Casp16p-sdRNA complementation experiments in PalB2- or Rad52-depleted cells. b, PalB2 ChIP analyses with or without depletion of GapmeR-mediated ActB- or Casp16p-sdRNA. c, PalB2 ChIP analyses showing BRCA1-dependent PalB2 recruitment (n=3) d, PalB2 ChIP analyses of ActB- and Casp16p-sdRNA complementation experiments in BRCA1- or Dicer-depleted HeLa cells (n= 3–4 biological replicates). e, γ-H2AX ChIP quantitation analyzed in G0 BRCA1-, PalB2- or Rad52-depleted T98G cells (n= 3 biological replicates). f, γ-H2AX ChIP analyses in G0 T98G following ActB- or Casp16p-sdRNA GapmeR-mediated depletion (n = 3–4 biological replicates). g, ACTB forward strand BELD signal in quiescent (G0) and cycling T98G cells. Representative experiment showing the relative average abundance of the qPCR replicates ± s.d. ChIP analyses depict the average fold change ± s.e.m of the relevant depletion compared to the mock siRNA or negative GapmeR. Data were all analyzed by Two-way ANOVA with post-hoc Tukey HSD (panels c-e, multiple t-test) and compared to the undamaged locus, or relevant control cells.
NGS sRNAs RIP analyses confirmed that PalB2 and Rad52 are binding sdRNAs (Extended Data Fig. 8a). At the single gene level, the same RIP samples showed that, in both the NS and chromatin fraction, PalB2 bound ActB- and Casp16p-sdRNA inefficiently in the absence of Rad52, and vice versa, while the loss of Dicer or Ago1 had no effect in this sdRNA- replete setting (Extended Data Fig. 8b–c). Thus, both PalB2 and Rad52 must be present so that each protein can bind the sdRNA efficiently and thereby engage in PalB2/Rad52/sdRNA-mediated repair. Consistently, PalB2 loss prevented Rad52 recruitment to chromatin and vice versa (Extended Data Fig.8d–e).
Next, we tested for PalB2 and Rad52 recruitment to damaged pause sites in the absence of the sdRNAs. Both PalB2 and Rad52 failed to interact with the relevant pause site after GapmeR-mediated depletion of the ActB- or Casp16p-sdRNA (Fig. 4b and Extended Data Fig. 8f). Interestingly, PalB2 recruitment to a relevant pause site was BRCA1-dependent (Fig. 4c). In addition, while PalB2 pause site recruitment was impaired in the absence of BRCA1 or Dicer, complementation with the cognate sdRNA restored its binding (Fig. 4d). Thus, it appears that: 1) BRCA1 and Dicer engage in sdRNA homeostasis and 2) a relevant sdRNA must bind both PalB2 and Rad52 to execute cognate, site-specific DNA damage repair at R-loop positive pause sites.
sdRNA-mediated repair protects G0 cells
We also asked whether this new repair mechanism operates in quiescent cells. γ-H2AX ChIP analyses performed in BRCA1-, PalB2- or Rad52-depleted T98G cells arrested in G0 by serum starvation revealed similar DNA damage accumulation as cycling cells at the ACTB and CASP16P pause sites (Fig. 4e and Extended Data Fig. 9). BELD analysis showed that G0 cells harbored more DNA breaks than their cycling counterparts, nevertheless degradation of each sdRNA increased DNA damage at its cognate pause site only, like in cycling cells (Fig. 4f–g). These data predict that the BRCA1-sdRNA-PalB2/Rad52 repair pathway operates in G0, and, possibly, even in certain adult stem or stem-like cells.
Discussion
We have detected the molecular roots of an unanticipated process of sdRNA-mediated endogenous DNA repair. It targets ssDNA breaks at termination pause sites, using a novel and critical role for RNA in the assembly of BRCA1- repair complexes.
sdRNA-mediated repair requires BRCA1/RNAi, PalB2, and Rad52 and likely involves transcription regulation29,32,34–36. Our results suggest that BRCA1, also a transcription factor, and its partner, Bard1, which engages in RNA processing37–39 regulate both transcription and sdRNA synthesis. sdRNA in this model, can then be transferred to and activate a PalB2/Rad52 complex that promotes repair at single strand breaks that develop at pause sites in cycling and quiescent cells (Extended Data Fig. 10).
It is tempting to speculate that R-loops, themselves, affect sdRNA synthesis and repair. Indeed, they were detected at both ActB- and Casp16p-sdRNA coding sequences (data not shown).
Conceivably, the sdRNA-mediated repair mechanism prevents genome instability while allowing certain beneficial outcomes of R-loop formation. Our NGS analysis suggests that sdRNA abundance is directly linked to the rate of transcription. If so, and its abundance is metered, it might allow cells to adapt to an amount of pause site damage that occurs at a physiological transcription level8.
How sdRNA/PalB2/Rad52 repair complexes find their cognate DNA damage genomic site is unclear. If sdRNAs are synthesized in the vicinity of broken DNA, a property similar to what was described for DDRNA12, they might act as ‘scouts/pathfinders’ to efficiently transport appropriate repair factors to damaged DNA sequences40. Whether certain 3D DNA structures support an orderly, physiological transfer of key repair factors to damaged sites remains an interesting question for future investigation. Failure of such a mechanism might lead to genomic instability typical of BRCA1-deficient tumor cells.
For example, forced overexpression of BRCA1-regulated satellite RNAs41 in murine mammary gland tissue promoted tumor formation42, suggesting that excessive sdRNA expression might, paradoxically, elicit DNA damage at its coding site. Indeed, DNA damage accompanied ActB-sdRNA overexpression (Extended Data Fig. 10). Conceivably, it fosters persistent, local R-loop production. Collectively, these results support our model and suggest that the normal sdRNA regulation translates into DNA repair and tumor- suppression instead of chronic DNA damage, and, possibly, even tumorigenesis.
Methods
Cell Lines and culture conditions
HeLa cells were cultivated in DMEM supplemented with 10% Fetal Bovine Serum (FBS) and 100U/mL penicillin-streptomycin (Gibco). Human mammary epithelial cells (HMECs) were grown in Lonza Mammary Epithelial Cell Growth Medium (MEGM) supplemented with Lonza MEGM SingleQuots Supplement (Lonza, cc-3150). CD44low HMEC stably expressing a Dox-inducible shBRCA143 were maintained in Mammary Epithelial Cell Basal Medium + Supplement-Pack Mammary Epithelial Cell GM (PromoCell, C-21210). T98G cells were grown in DMEM supplemented with 10% FBS, 2mM L-Glutamine (Gibco), and 100U/mL penicillin-streptomycin. All cells were grown at 37°C in a 10% CO2 atmosphere, except for the CD44low HMEC which grew in a 5% CO2 atmosphere.
T98G cells were synchronized in G0 by serum deprivation (DMEM, 0% FBS, 2mM glutamine, 1% penicillin/streptomycin) for 72h. They were then released into the cell cycle by addition of complete cell culture medium (DMEM 10% FBS, 2mM glutamine, 1% penicillin/streptomycin). G1 cells were collected after 12h, S phase cells after 20h, G2 cells at 24h, and M phase cells at 30h post G0 release. HeLa FUCCI cells were sorted according to the expression of fluorescent cell cycle reporters (mKO2-hCdt1 as a G1 marker and mAG-hGeminin as a S/G2 marker)44. To assess the degree of synchronization, at each time point of all synchronization experiments, cells were stained with PI/RNase- staining buffer according to the manufacturer’s protocol (BD Pharmingen™, 550825). Flow cytometry analyses were performed using a BD Fortessa and analyzed using FlowJo software (Treestar). FACS analysis was confirmed by Western Blotting, using cell cycle specific antibodies. p27, Geminin/cyclin A, and phosphoH3 expression were respectively assessed for G0, S and M phase cells.
In BRCA1 rescue experiments, depletion of BRCA1 in CD44low HMEC-ShBRCA1 cells was achieved by treating the cells with 1.125ug/ml of doxycycline (Takara Bio, 631311) for 6 days (D6). The full restoration of BRCA1 expression was obtained in these cells by Dox removal (R) and culture for another 6 days in Dox-free media (D12). Cells were harvested for immunoblot, ChIP and RNA analysis at both D6 and D12.
Transfection
siRNAs (see sequences in Table 1) were transfected with Lipofectamine® RNAiMAX (Life Technologies, 13778–500) for 72h at a final concentration of 30nM. All siRNAs were obtained from Dharmacon. For complementation experiments, synthetic sdRNAs (Sigma, Custom DNA and RNA Oligos) were transfected with Lipofectamine® RNAiMAX 48h after siRNA transfection. sdRNAs (Table 1) were transfected for 24h. ActB sRNAs were transfected at a final concentration of 0.5nM; Casp16p sRNAs were transfected at a final concentration of 0.25nM. Antisense LNA® GapmeRs (Qiagen) were transfected for 30h with Lipofectamine® RNAiMAX according to the manufacturer’s instructions (see sequences in Table 1). ActB and Casp16p GapmeRs were transfected at a final concentration of 50nM and 30nM, respectively. A negative control GapmeR (Qiagen) was transfected at the relevant concentration for each sRNA GapmeR. Flag-tagged hDICER WT and enzymatic-dead mutant (110 ab) plasmids (gift of Sophia Francia10) were transfected into HeLa cells at 0.75μg/mL 24h after siRNA transfection with Lipofectamine® 2000 (Life Technologies, 11668500). Transfection was performed according to the manufacturer’s instructions.
Immunoblotting
Cell pellets were lysed for 15 min on ice with NETN-420 (420mM NaCl, 0.5mM EDTA, 20mM Tris-HCl (pH 8.0), 0.5% (v/v) Nonidet P-40 (NP-40)) supplemented with protease inhibitors. Whole cell extracts (WCE) were centrifuged at 14,200 rpm in a Sorvall Legend Micro21 centrifuge (Thermo Fisher Scientific) for 15min at 4 °C. Protein concentrations were measured using a BCA Protein Assay Kit (Pierce, PI21059). From each sample, 10ug of protein-containing extract were separated by gel electrophoresis in NuPAGE® 4–12% Bis-Tris protein precast gels (Life Technologies) using MES-SDS running buffer. Proteins were blotted onto 0.2 μm nitrocellulose membranes. Membranes were blocked with 5% milk in TBST (TBS + 0.1% Tween-20) for 1 h at room temperature and incubated with primary antibodies (See Table 2) diluted in 5% milk in TBST overnight at 4 °C. Membranes were then washed three times with TBST, incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature, washed again, and the relevant protein bands detected using Western Lightning plus ECL (Perkin Elmer, NEL10400).
Immunoprecipitation (IP):
Snap-frozen cells pellets (12 – 14×106 cells) were lysed on ice for 15 min in 15 mM NaCl, 60 mM KCl, 2 mM EDTA, 0.05% Triton X-100, 12% sucrose, 1 mM DTT, 0.65 mM spermidine and complete protease inhibitor cocktail (Roche) and then centrifuged at 13,000g for 15 min. Nuclei were washed three times in the same buffer and resuspended in nuclear lysis buffer (100mM Hepes Ph 7.5–8, 420mM NaCl, 0.5% NP40 in presence of protease inhibitors) for 10 min on ice. Chromatin was sheared using a cooled Bioruptor bath sonicator (Diagenode Bioruptor-Pico) for 2 min (30s ON - 45s OFF). Sonicated nuclear extracts (NE) were centrifuged at 14,000 rpm for 10 min at 4 °C in a Sorvall Legend Micro21 centrifuge (Thermo Fisher Scientific). Cytoplasmic (Cyt), nuclear soluble (NS), and chromatin (Chrom) fractions were extracted using a Subcellular Protein Fractionation kit for cultured cells (Thermo Fisher Scientific, 78840). The protein concentration was measured, using a BCA Protein Assay Kit. For RNase and other nuclease exposure experiments, cell extracts were exposed to 0.02ug/uL RNase A (Roche, 10109169001) for 10 min at 37 °C or 10U RNase T1 (Life Technologies, EN0541) for 15 min at 37 °C in the presence of 6mM EDTA or to 5U/uL Benzonase (Sigma, E1014) for 10 min at 37 °C. Aliquots of these treated extracts were analyzed for RNA and DNA content using a mock control (cell extracts incubated for 15 min at 37 °C) to assess the efficiency of RNA or genomic DNA digestion.
Aliquots representing 6% of the total amount of extract for each IP served as input samples. 120 μg of extract were diluted three-fold to reach 150mM NaCl in IP buffer (100mM Hepes Ph 7.5–8, 10mM NaCl, 1mM DTT, protease inhibitors) and incubated for 3 hours at 4 °C with the appropriate primary antibody (Table 2). Washed Protein G Dynabeads (20-μl slurry; Life Technologies, 10004) were added for an additional 15 minutes. The beads were then washed four times in washing buffer (10mM Hepes pH 7.5–8, 100mM NaCl, 10% glycerol, 0.2% Tween, and complete protease inhibitor cocktail) and eluted by boiling in 1× NuPAGE® LDS sample buffer (Life Technologies) with 1X NuPAGE® reducing agent. Proteins were electrophoresed in a 4–12% SDS polyacrylamide gel in MES-SDS buffer, and immunoblotting was performed as described above (see Immunoblotting section).
Chromatin immunoprecipitation
ChIP was performed as previously described7, using 15–25μg of chromatin incubated overnight at 4°C with the respective antibody prebound (Table 2) to Protein A and Protein G Dynabeads (Life Technologies). Real-time PCR was performed in a QuantStudio 6 Flex System (Applied Biosystems) with PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, 100029284) and the primers of interest (each present at 0.25μM) (see Table 3). ChIP-qPCR data were generated as relative quantities, using an input ChIP DNA standard curve with the assistance of Applied Biosystems QuantStudio software. Data were analyzed as percent of input, and results were presented as fold change over control samples.
DNA isolation
Genomic DNA (gDNA) was extracted by resuspending the relevant cells in lysis buffer (10mM Tris HCl pH 7.4, 10mM EDTA, 10mM NaCl, 1% N-Laurosyl-Sarcosine) and incubating the suspension at 63°C overnight in the presence of 0.66mg/mL proteinase K. The gDNA was precipitated with 0.3M Sodium Acetate and with two volumes of 100% EtOH and pelleted by centrifugation at 10,000rpm for 10 minutes at 4°C. The DNA pellet was washed twice with 70% EtOH and resuspended in water or TE.
Broken Ends Labeling and Detection (BELD) (adapted from 45–47)
a. In vitro DNA repair (Nick and Gap Repair reaction)46
2.5ug of gDNA with putative breaks were in vitro “sealed” before TdT labeling using 1000 U of T4 DNA ligase (NEB, M0202L) in presence of 0.1mM dNTP mix, 1X NEB buffer 2.1 and 1nM ATP for 10 min at 12°C. DNA samples were then incubated with 2.5 U of T4 polymerase (NEB, M0203L) for 15 min at 12°C. The enzymes were then heat-inactivated at 75°C for 20min. These samples are called “+NGR” samples. Mock samples prepared similarly but in the absence of enzymes were called “–NGR” samples.
b. 3’-ends labeling with TdT
5ug of gDNA were used to label 3’ free ends with 14 Biotin–dATP using 500U of Terminal deoxynucleotidyl Transferase (TdT; Roche, 03333566001). The no template-driven dATP/Biotinyl-dATP polymerization was performed in 1X TdT buffer (5mM CoCl2, 0.25uM 14-biot dATP (Life Technologies, 19524016), 250uM dATP at 37°C for one hour. The enzyme was inactivated for 10 min at 65°C.
c. Fragmentation of the labeled DNA and purification
Biotin-labeled DNA was fragmented into ~200–600 bp fragments using dsDNA Fragmentase (NEB, M0348A) in 1X Fragmentase buffer (NEB, B0349S), 1X BSA. The DNA-Fragmentase mixture was incubated for 5min on ice without enzyme, then at 37°C for 15min with the enzyme. The enzyme was inactivated by increasing the final EDTA concentration to 50 mM. Fragmented biotin-labeled DNA samples were purified using 5Prime Phase lock gel Heavy columns (Quantabio, 2302830) using an equal volume of Phenol-chloroform isoamyl alcohol 25:21:1 v/v mix followed by precipitation of the DNA in the upper phase with final concentrations of 2.5M NH4 Acetate and 70% EOH in the presence of Glycogen. The fragmented DNA was then resuspended in buffer EB (10 mM Tris-HCl, pH8) and quantified. 2% of the final volume was kept as INPUT.
DNA fragmentation was confirmed by running a duplicate of the labeled, fragmented, purified DNA in a 1.5% agarose gel.
d. Damaged DNA capture and elution
The biotinylated DNA fragments were captured using M-280 streptavidin Dynabeads® (Life Technologies, 11205D) using 30ul of initial bead volume for every 5ug of fragmented DNA. In summary, beads were washed four times in twice the volume of 2X no tween buffer (2M NaCl, 10mM Tris pH8, 1mM EDTA) and incubated overnight at 4°C in Blocking buffer (1M NaCl, 20mM Tris pH8, 1mM EDTA, 0.2% Triton X, 0.05% NP40, 2% BSA, 400ug tRNA ( Santa-Cruz, sc-240952)). Beads were then washed with 1X no tween buffer (1M NaCl, 5mM Tris pH8, 0.5mM EDTA) and resuspended in the same volume of the DNA sample with 2X no tween buffer (2M NaCl, 10mM Tris pH8, 1mM EDTA) and incubated with the fragmented DNA samples for 30 min at RT with rotation. The beads were then washed once with 2X no tween buffer, twice with 1X tween buffer (1M NaCl, 5mM Tris pH8, 0.5mM EDTA, 0.05% tween 20), once with 1X no tween buffer. Beads were washed with TE buffer (10mM Tris-HCl, pH8, 1mM EDTA), the dry beads were resuspended in 15ul TE.
e. Site Specific Enrichment of Biotin-dATP labelled DNA fragments
TE resuspended bead samples were used for primer extension (PE1) using custom tagged poly-dT primers (Sigma Aldrich; 100μM stock) and Phusion High Fidelity DNA Polymerase (NEB, M0530). The primer extension products containing tagged poly-dT DNA fragments were then purified using Illustra MicroSpin S-400 HR columns (GE Healthcare, 27–5140-01). A subsequent enrichment was performed for site-specific primer extension (PE2), using ACTB and CASP16P gene- specific primers (GSP) and the Phusion High Fidelity DNA Polymerase. The samples were then assessed for the presence of damaged sites using the relevant GSPs for the ACTB and CASP16P genes.
f. Assessment of the damaged sites using quantitative PCR
Real-time qPCR was performed on the enriched, ACTB- or CASP16P-specific tagged poly-dT DNA samples, using the QuantStudio 6 Flex System (Applied Biosystems) with PowerUp SYBR Green Master Mix (Thermo Fisher Scientific).
Two sets of qPCR were performed, the first using the enriched pool of ACTB and Casp16 specific labelled DNA fragments, with primers targeting for gene-specific tagged (GSP) amplicons. The GSP amplicons were then purified using a QIAquick Nucleotide Removal Kit (Qiagen, 28306). With appropriate dilutions, a second qPCR was performed on the purified samples and were amplified for primers targeting sites within or outside the GSP DNA fragments from the first qPCR. qPCR data were normalized over the input DNA and represented as relative quantities. Calculations and graphical representation of the data were performed using Microsoft Excel 2016 and Prism/GraphPad 8.
RNA isolation
Total RNA was isolated from cell pellets with TRIzol Reagent (Life Technologies, 15596026) according to the manufacturer’s instructions. For RNA isolation from subcellular fractions, cell pellets were fractioned using a Subcellular Protein Fractionation Kit for Cultured Cells (Thermo Fisher Scientific) according to the manufacturer’s instructions. TRIzol Reagent was added directly to the cytoplasmic and nuclear soluble fractions. For isolation of chromatin-associated RNA, the chromatin pellet was resuspended in TRIzol Reagent and disrupted using a 25-gauge needle and syringe. Fractionation efficiency was verified after cDNA synthesis by real-time qPCR detection of ribosomal RNAs as described in Conrad et al. (2017)48. Small RNAs were isolated using a mirVana miRNA Isolation Kit (Life Technologies, AM1561) according to the manufacturer’s instructions.
TaqMan® quantification of sdRNAs
cDNA was generated from TRIzol-extracted RNA using the TaqMan® Advanced miRNA cDNA Synthesis Kit with miR-Amp Universal Amplification (Life Technologies, A28007) according to the manufacturer’s instructions. sdRNAs were quantified by real-time PCR using TaqMan® Fast Advanced Master Mix (Life Technologies, 4444557) and custom assays designed by the TaqMan® Custom Assay and Oligo Service (Life Technologies). Samples were normalized to control assays using ΔΔCt analyses and results are presented as fold change over mock samples.
Reverse transcription
cDNA was generated using SuperScript IV VILO Master Mix with ezDNase Enzyme (Life Technologies, 11766050), according to the manufacturer’s instructions. Samples were quantified using real-time PCR with PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, 100029284) and the primers of interest at 0.25μM (see Table 3). Data was generated, using a cDNA standard curve with the assistance of Applied Biosystems QuantStudio software. Target loci were normalized to control genes, and all data were analyzed as fold change over control samples.
RNA immunoprecipitation
Cells were harvested in cold PBS and lysed in RSB-100 buffer (10mM Tris-HCl [pH 8], 100mM NaCl, 2.5mM MgCl2, 0.5% Nonidet P-40 (v/v), 0.5% Triton X-100 (v/v), protease inhibitors, 50U/mL RNaseOUT (Life Technologies, 10777–019 ) on ice for 10 minutes. Whole cell extracts (WCE) were sheared for 3 min, (30s ON - 30s OFF) (Bioruptor Pico, Diagenode). Debris was removed by centrifugation. Nuclear soluble (NS) and chromatin (Chrom) fractions were extracted using a Subcellular Protein Fractionation kit for cultured cells (Thermo Fisher Scientific). Relevant extracts were incubated with the primary antibody (see Table 2) at 4°C for two hours, followed by incubation with Protein G Dynabeads (Life Technologies) at 4°C for 1.5 hours. Beads were washed three times with RSB-100 buffer. TRIzol reagent was added directly to the beads, and RNA extraction proceeded as previously described. Where indicated, TaqMan cDNA synthesis and reverse transcription were followed by real-time PCR for assessment of the abundance of sRNA relative to its precursor.
RNA Pulldown
40uL of Dynabeads M-280 streptavidin (Life Technologies, 11206D) were pre-washed twice with 2 volumes of 0.1M NaOH, 50mM NaCl buffer for 2 min, washed once with 100mM NaCl and twice with 20mM Tris pH7.5. The beads were then resuspended in 50ul of RNA capture buffer (20mM Tris pH 7.5, 1M NaCl, 1mM EDTA, 1U/uL RNaseOUT™) containing 10pmol of biotinylated small RNA or unbiotinylated small RNA [See Table 1]. The beads were then incubated at room temperature with agitation and then washed twice with 20mM Tris pH 7.5 and once with 1X Protein-RNA Binding Buffer (20mM Tris pH 7.5, 50mM NaCl, 2 mM MgCl2, 0.1% tween 20 and 1U/uL RNaseOUT™). 120 μg of NE or cellular fractions were diluted three times in IP buffer (100mM Hepes Ph 7.5–8, 10mM NaCl, 1mM DTT, protease inhibitors) and further diluted in 10X Protein-RNA Binding Buffer. The extract was added to the beads and incubated for 2 hours at 40°C with agitation. Beads were then washed three times with 1X washing buffer (20mM Tris pH7.5, 10mM NaCl, 0.1% Tween) and resuspended in 1× NuPAGE® LDS sample buffer (Life Technologies) with 1X NuPAGE® reducing agent (Life Technologies). Samples were electrophoresed in a 4–12% SDS–polyacrylamide gel in MES-SDS buffer, and immunoblotting was performed as described above (see Immunoblotting section).
Library preparation
Small RNA samples extracted using the MirVana miRNA Isolation Kit (Life Technologies) to Enrich for small RNA (<200 nt) or using TRIZol reagents following’s the manufacturer’s instructions. Once isolated, the small RNAs were prepared for sequencing with an NEBNext Multiplex Small RNA Library Prep Kit for Illumina (NEB, E7300). Size selection was performed in a 6% polyacrylamide gel for RNAs in the 140–195bp range corresponding to RNA from 17 to 70bp. We sequenced the small RNA libraries in an Illumina NextSeq500 for 75bp single end reads. RIP samples were extracted using TRIzol reagents following’s the manufacturer’s instructions. The RIP samples were prepared for sequencing in two ways: 1) with a NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB, E7760) with no ribodepletion and no fragmentation (to capture RNA <300nt) and 2) with NEBNext Multiplex Small RNA Library Prep Kit (to capture sRNAs). We sequenced the RIP libraries in an Illumina NextSeq500 for paired end 150bp reads.
Bioinformatics Analysis
The small RNA library preparations had the adapters trimmed from the genuine sequences with FASTX-clipper49. The trimmed libraries were aligned to the hg19 genome50 with STAR aligner51 with the overhang parameter set to 74bp. We determined differential peak abundance quantification with MACS252 callpeak module enabling the parameter for bypassing the shifting model. We filtered the peaks for instances where at least 50% of the reads in the defined peak region overlapped the read distribution’s mode. Annotation of peaks was performed with HOMER53. The heatmaps were created using deeptools254.
Data availability
All the FASTQ sequence files from the small RNA, and RIP larger RNA libraries sequencing are available on the SRA database: BioProject PRJNA667516: Identification and characterization of novel small RNA species sdRNA: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA667516.
Code availibility
The code used for the identification of sdRNAs was developed in the Dana Farber Cancer Institutes Center for Computational Biology / Harvard School of Public Health’s Quantitative Biomedical Research Center, and the code repository is available at https://github.com/dkdeconti/sdRNAPeaksIdentification.
Extended Data
Extended Data Figure 1 |. BRCA1 interaction with RNAi factors.
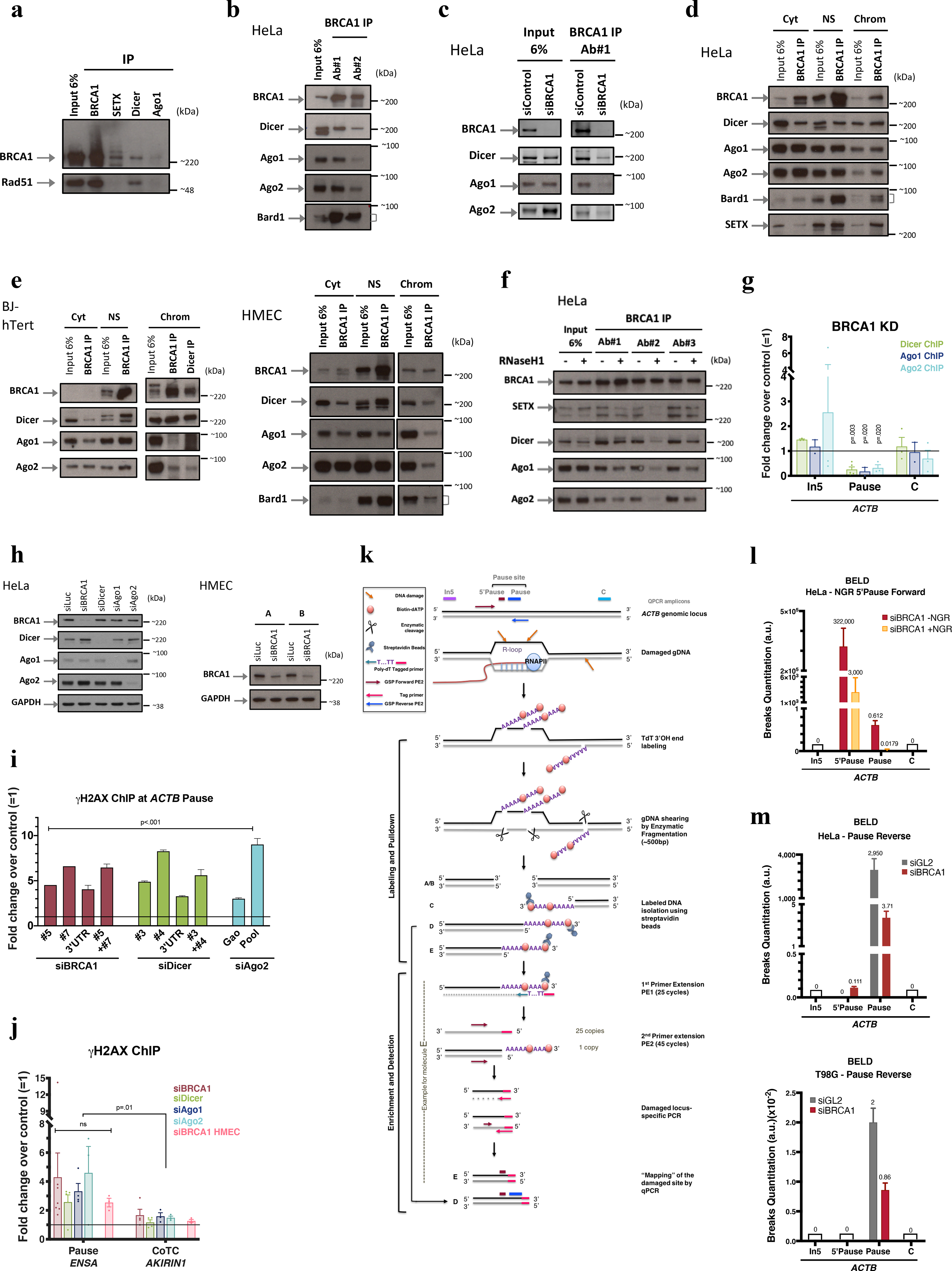
a, Co-IP of endogenous SETX, BRCA1, Dicer or Ago1 in HeLa NE showing that RAD51, an established BRCA1 binding partner involved in HR, failed to co-IP with SETX and co-IP’d very weakly, if at all, with Dicer and Ago1. n=3. b, Endogenous BRCA1 co-IP in HeLa NE, using two, BRCA1 Abs, each recognizing a different epitope (#1: N-terminus and #2: C-terminus). c, BRCA1 co-IP performed in siBRCA1-transfected HeLa NE; n=3 d-e, Co-IP of endogenous BRCA1 and RNAi factors present in subcellular HeLa fractions (d) and confirmed in HMEC and human fibroblast (BJ-hTert)(e) n=4. f, BRCA1 was co-IP’d, using three, monospecific Abs, in HeLa NE from cells overexpressing (+) or not (−) RNAseH1. Immunoblots showed with Ab#2 that a BRCA1/SETX/RNAi complex was R-loop sensitive (co-IP). IgG, negative control; Cyt, cytoplasm; NS, Nuclear Soluble; Chrom, chromatin. n=2 g, Dicer, Ago1, and Ago2 ChIP were performed in BRCA1-depleted HeLa,. Their ACTB pause site recruitment was BRCA1-dependent (n = 2–5 biological replicates). h, Representative immunoblots demonstrating siRNA-mediated silencing efficiency in HeLa cells and HMECs; n=10. i. Representative DNA damage analysis using individual siRNAs for each relevant target. j, DNA damage analyses at ENSA (R-loop-positive) and AKIRIN1 CoTC (R-loop-negative) transcription termination sites (n = 3–7 biological replicates). k, Broken End Labeling and Detection (BELD) strategy used to identify DNA breaks at the ACTB pause site. l-m, BELD quantitation of breaks at the ACTB pause site. l, BELD performed at the ACTB pause site in BRCA1- depleted cells with or without prior in vitro Nick and Gap Repair (+/− NGR) of breaks performed prior to TdT labeling. m, BELD performed on the ACTB pause site reverse strand. A representative example of each BELD analysis is shown, and the histograms depict the average values of qPCR replicates ± s.d. All ChIP data are represented as an average fold change ± s.e.m of the relevant KD condition compared to the mock. Data in g, i and j were respectively analyzed by multiple t-test, Two-way ANOVA with post-hoc Tukey HSD and unpaired t-test, and compared to results obtained with the undamaged locus or with relevant control cells.
Extended Data Figure 2 |. BRCA1 interacts with Dicer and Ago1/2 to prevent termination pause site damage throughout the cell cycle.
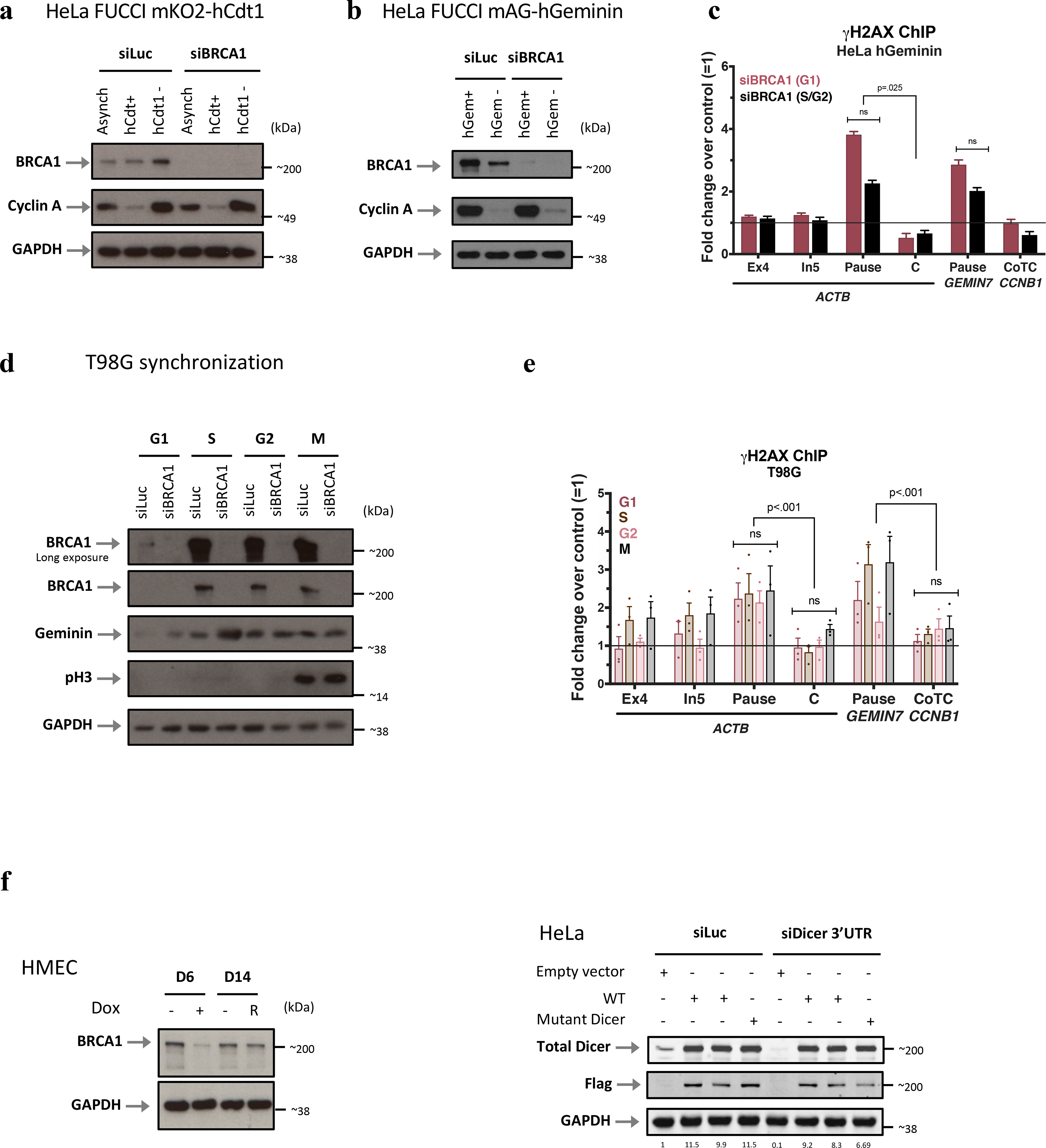
a-b, Immunoblots of Cyclin A and BRCA1 from WCE of FUCCI HeLa cells sorted according to the expression of the G1 marker, mKO2-hCdt1 (a) or the S/G2 phase mAG-hGeminin marker (b); n=4. c, Cell cycle- dependent analysis of DNA damage using γ-H2AX ChIP signals in HeLa FUCCI sorted according the S/G2 hGeminin marker indicating that G1 cells develop more damage than S/G2 cells. A representative experiment is shown, and the histograms depict the average fold change of qPCR replicates ± s.d. d, Immunoblots depicting the expression of BRCA1 during the various phases of the cell cycle upon release from serum starvation-induced G0 T98G cells; n=5. e, DNA damage analysis of synchronized T98G cells. f, Immunoblots validating the re-expression of BRCA1 (R) or Dicer (Flag-tagged WT and mutant Dicer, detected with LiCor and showing their quantitative expression) after BRCA1 or Dicer depletion, respectively in HMECs and HeLa cells; n=4. Data in c were analyzed by One-way Anova with post-hoc Tukey HSD and compared to an undamaged locus.
Extended Data Figure 3 |. DNA damage- induction after BRCA1, Dicer, and sdRNA.
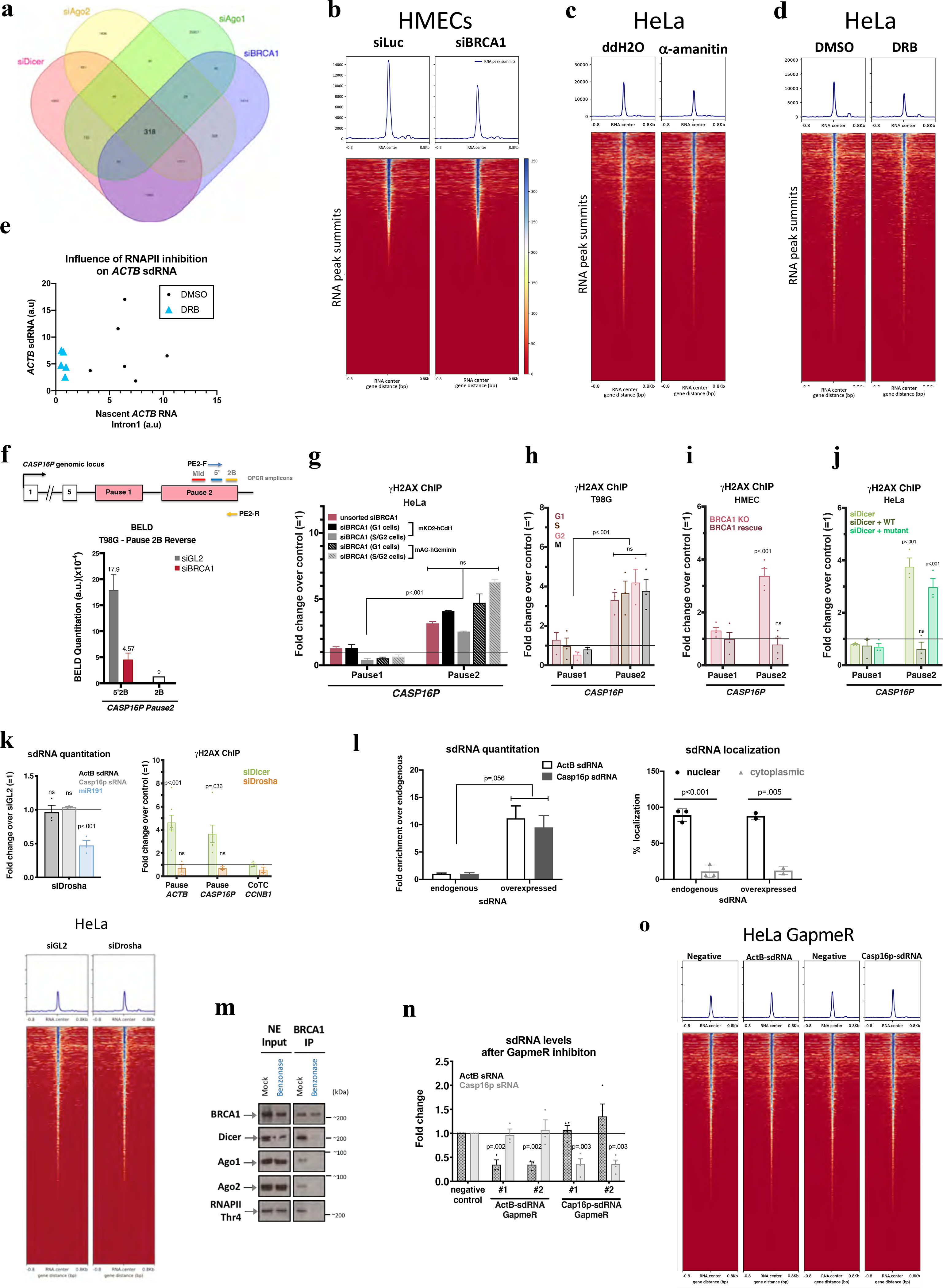
a, A Venn diagram showing a common set of deregulated sdRNAs following the indicated depletions. b-d, Heatmaps centered around the sdRNA peaks (+/− 0.8kb) showing effects of α-amanitin (c) and DRB (d) effects. e, Scatter plots suggesting a correlation between the abundance of ActB-sdRNA and the nascent ACTB RNA (detected using Intron1). f, (Top) Schematic of the CASP16P genomic locus. Primers used for BELD on the forward (PE2-F) and reverse (PE2-R) strands and the QPCR amplicons. (Bottom) BELD quantitation in T98G cells of the CASP16P Pause2 site reverse strand. Representative experiment showing the relative average abundance of the qPCR replicates ± s.d. g-j, DNA damage quantitation at the CASP16P pause site performed in G1 or S/G2 BRCA1-depleted HeLa FUCCI cells (representative graph)(g), or in BRCA1-depleted, synchronized T98G (n=3)(h) or in BRCA1 (n=4)(i) or Dicer (n=3)(j) rescue experiments. k, Quantitation of ActB- and Casp16p-sdRNA (top) and (right) genome-wide heatmaps centered around sdRNA peaks, showing that HeLa Drosha depletion didn’t affect sdRNA abundance (mir191=positive control). Average qPCR values ± s.e.m.. (bottom) Drosha depletion did not induce DNA damage at ACTB or CASP16P pause site CCNB1 CoTC is a negative control (n = 3 biological replicates for both graphs). l. Subcellular sdRNA quantitation, endogenously (n=2–3) or upon overexpression (representative). m. Benzonase effect on immunoblotted proteins co-IP’d with BRCA1, n=3. n, Efficacy and specificity of ActB- and Casp16p-sdRNA depletion using individual LNA GapmeRs (n=3–4). o. Heat maps centered around the sdRNA peaks showing no genome-wide effect upon ActB- or Casp16p-sdRNA depletion. γ-H2AX ChIP analyses shown as average fold change compared to relevant control cells. Data were analyzed by One-way (g) or Two-way (h-k, n) Anova with post-hoc Tukey HSD or multiple/unpaired t-test (l) and compared to an undamaged locus, or relevant control cells. ns= non significant.
Extended Data Figure 4 |. Complementation experiments performed with small DNA or antisense sRNA do not promote repair.

a, BELD quantitation of breaks on the forward (coding) strand of the CASP16P Pause2 termination site in BRCA1-depleted HeLa cells complemented with either ActB- or Casp16p-sdRNA, showing that only Casp16p-sdRNA can prevent accumulation of breaks at CASP16P Pause2. A representative experiment is shown, and the histograms depict the average values of the qPCR replicates ± s.d. b-c, DNA damage quantitation using γ-H2AX ChIP qPCR analyses performed in complementation experiments at the ACTB and CASP16P pause sites and at the CCNB1 CoTC. ChIP data are represented as the average fold change compared to an undepleted relevant control. Complementation experiments were performed in (b) BRCA1- or (c) Dicer-depleted HeLa cells reconstituted with the 3’ -non polymerizable sense sdRNA (3’-blocked ActB-sdRNA)(b-c), or ActB or Casp16p antisense (AS) sdRNA (b-c) or the corresponding sense or antisense (AS) sdRNA DNA sequence (b) (n = 2–6 biological replicates for b and n=2–6 for c). Data in b and c, were analyzed by Two-way Anova with post-hoc Tukey and compared to the relevant control cells.
Extended Data Figure 5 |. Relative Ago1 and Ago2 binding ratio of ActB-sdRNA compared to its precursor.
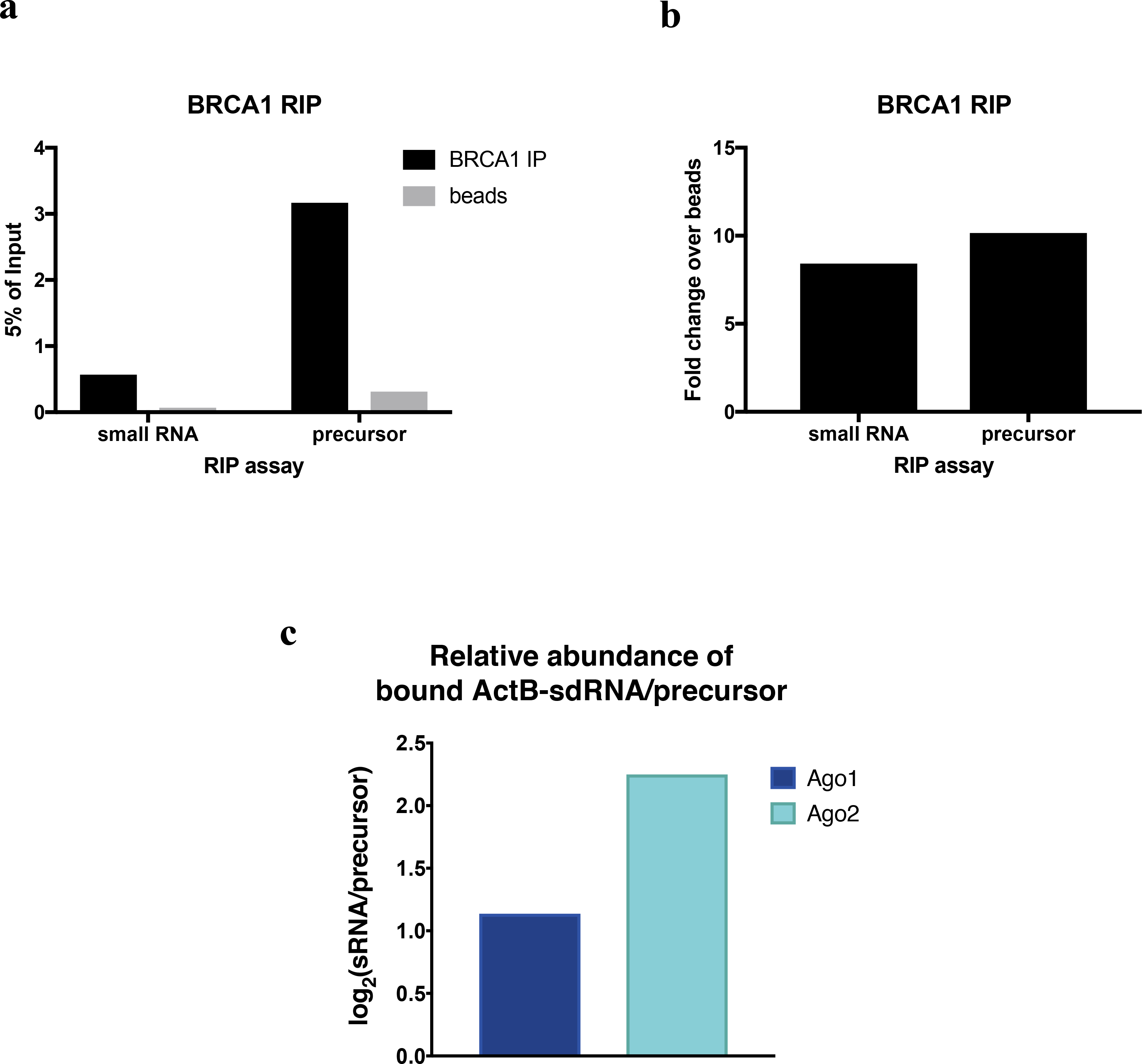
a-b, BRCA1 RIP analyses of the sdRNA or its precursor, showing the raw data represented as %input (a) or as the fold change of the signal obtained between the relevant Ab BRCA1 and a beads-only control (b). c, RIP analysis showing the binding ratio (Log2) between ActB-sdRNA and its precursor (quantified by Taqman and strand specific RT-qPCR) after Ago1 or Ago2 IP from HeLa whole cell extracts (WCE).
Extended Data Figure 6 |. Validation of siRNA- mediated depletion of PalB2 and Rad52 and specificity of PalB2 and Rad52 IP.
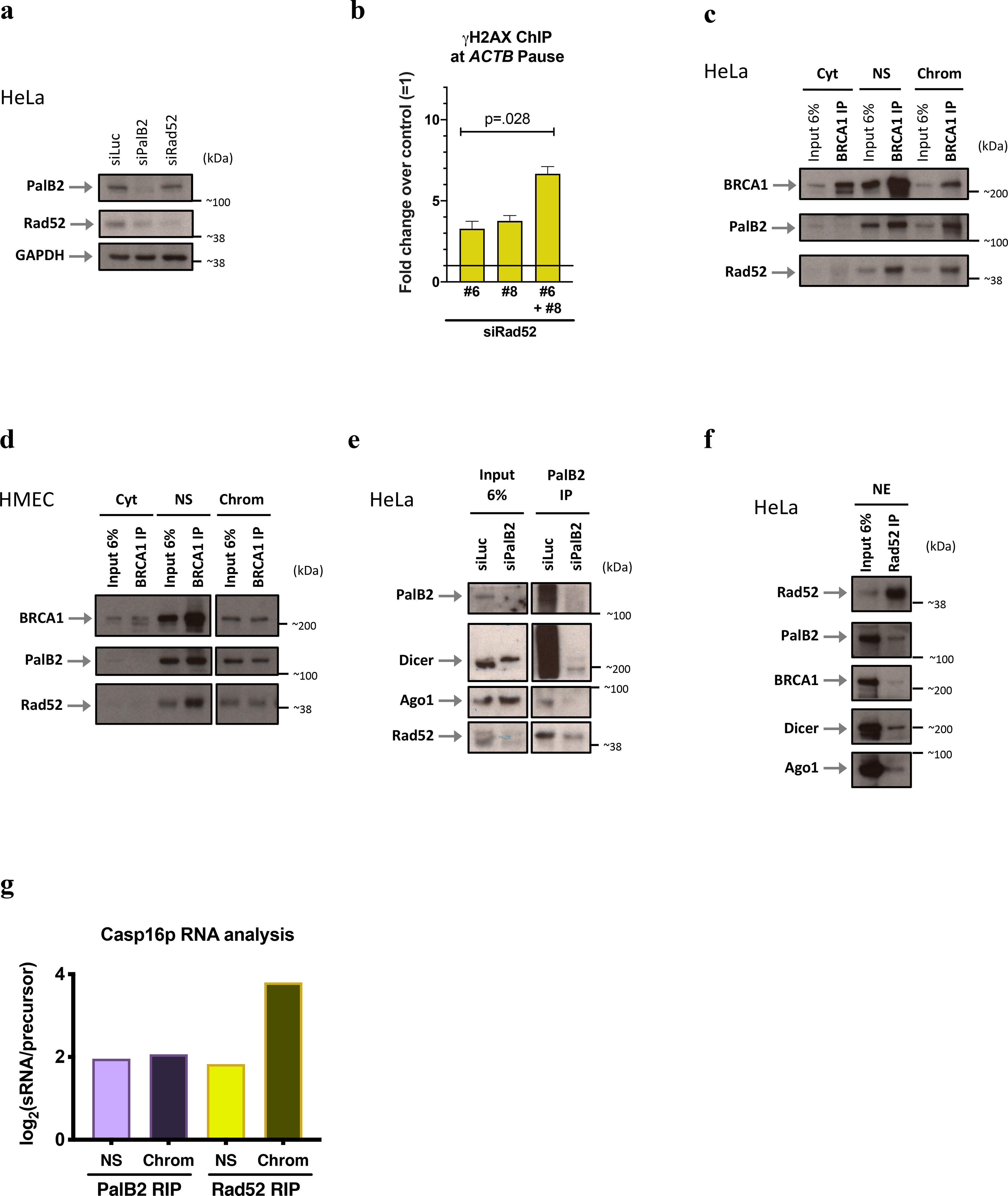
a, Representative immunoblots showing the efficiency of siRNA-mediated depletion of PalB2 and Rad52 in HeLa cells, n=7. b, Representative experiment showing γ-H2AX ChIP analyses upon Rad52 KD using 2 individual siRNAs. The histograms depict the average values of qPCR replicates ± s.d. Data were analyzed by unpaired t-test and compared to the relevant control cells. c-d, Co-immunoprecipitation (co-IP) of endogenous BRCA1 in subcellular fractions of HeLa cells (c) and HMECs (d); n=3. PalB2 and Rad52 proteins were detected by immunoblotting, using relevant antibodies. e, PalB2 co-IP validation in siPalB2 cells, n=2. f, Rad52 co-IP performed with NE from HeLa cells, n=3. g, Representative RIP analysis of Nuclear PalB2 and Rad52 binding affinity ratio (Log2) between Casp16p-sdRNA and its precursor (n=3). All immunoblots were developed using the indicated antibodies. Cyt, cytoplasm; NS, Nuclear Soluble; Chrom, chromatin.
Extended Data Figure 7 |. ActB and Casp16p antisense sdRNA or DNA sequences fail to attract the BRCA1/RNAi and the PalB2/Rad52 repair complexes.
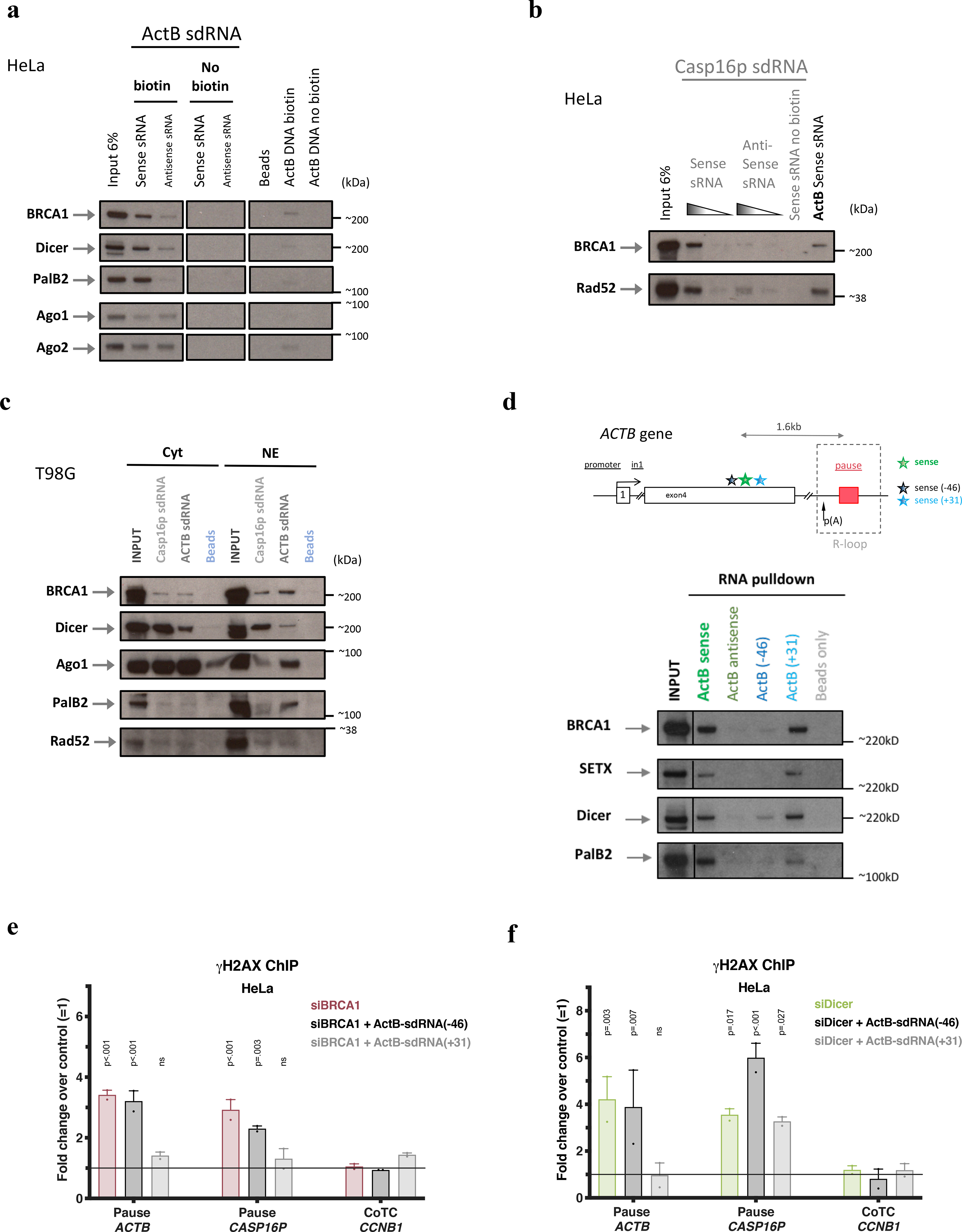
a, RNA/protein binding assay performed in HeLa NE using sense or antisense, synthetic biotinylated (or not) ActB-sdRNA [or its DNA sequence] or streptavidin beads, only, n=3. b, RNA/protein binding assay performed in HeLa NE, using sense or antisense synthetic biotinylated (or not) Casp16p-sdRNA. ActB-sdRNA was used as a positive control; n=3. c, RNA/protein binding assay performed in T98G subcellular fractions using synthetic, sense strand-derived, biotinylated ActB- or Casp16p-sdRNA; n=2. d, RNA/protein binding assay performed in HeLa NE using synthetic biotinylated sdRNAs with the sequence of ActB-sdRNA sense, antisense, ActB (−46) or ActB (+31) (See schematic of the ACTB locus depicting the location of the various sRNAs. ActB-sdRNA (−46) and ActB-sdRNA (+31) are respectively located 46nt upsteam of the 5’end of ActB-sdRNA and 31nt downstream of the 3’end of the ActB-sdRNA. n=3. The beads only were a negative control. All immunoblots were developed using the indicated antibodies. Cyt, cytoplasm; NE, Nuclear Extract. e-f, γ-H2AX ChIP analyses using the data collected from the complementation experiments using ActB-sdRNA sense, antisense, ActB-sdRNA (−46) or ActB-sdRNA (+31) performed in HeLa cells depleted for BRCA1 (e) or Dicer (f). ChIP results are shown as the average fold change ± s.e.m compared to the mock siRNA (n = 2–11 biological replicates for panel e and n=2–7 for panel f). Data were analyzed by Two-way Anova with post-hoc Tukey HSD and compared to the relevant control cells. ns= non significant.
Extended Data Figure 8 |. PalB2 binding affinity for sdRNA is independent of Dicer or Ago1/2, but the recruitment of PalB2 and Rad52 to pause sites was, respectively, Rad52- and PalB2-dependent.
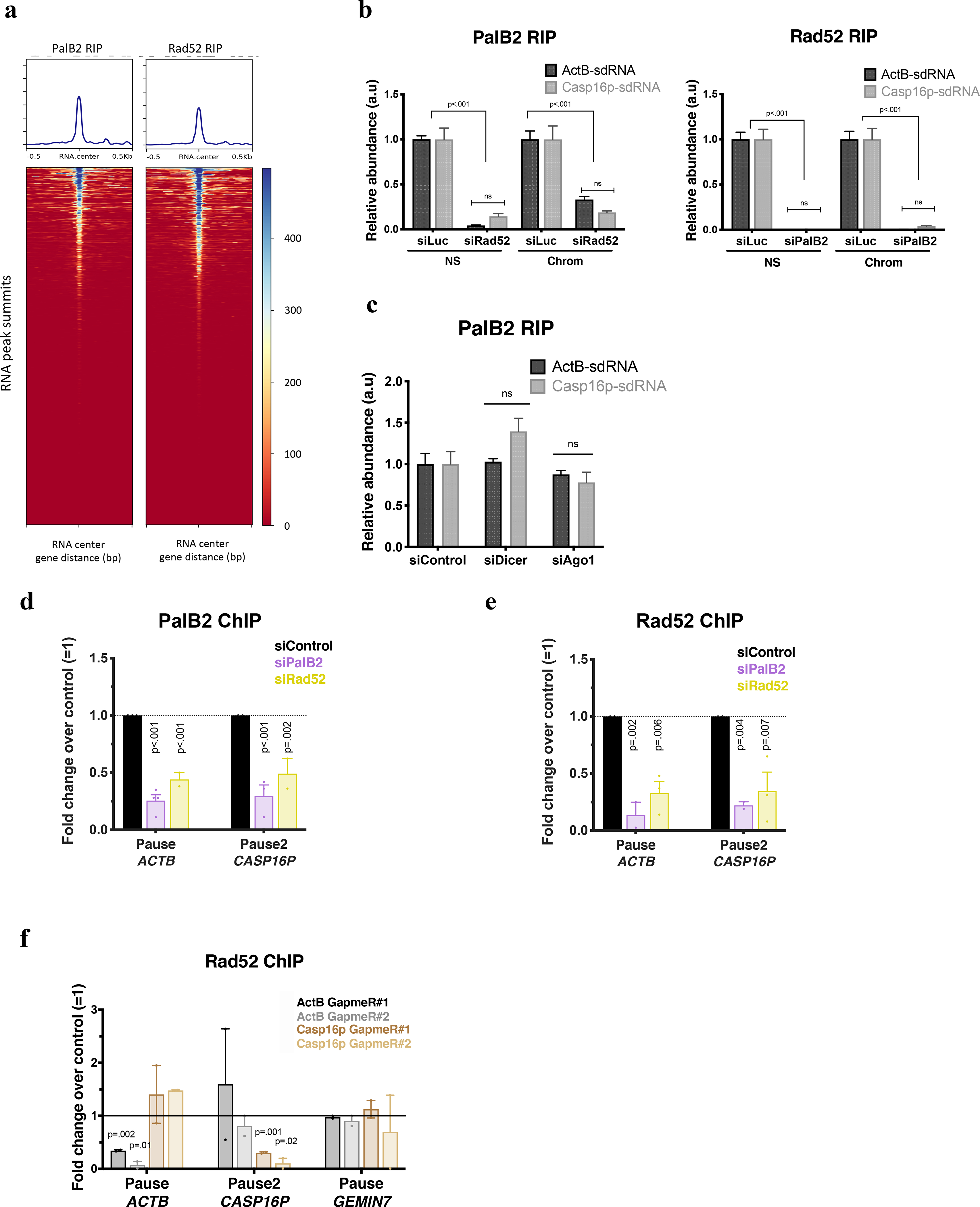
a, PalB2 and Rad52 genome wide RNA ImmunoPrecipitation (RIP) analyses of small RNAs, are shown as heat maps centered around the sdRNA peaks identified in Fig. 2. b, PalB2 and Rad52 RIP analyses of ActB- and Casp16p-sdRNA binding. Representative experiment showing qPCR replicates ± s.d. NS, Nuclear Soluble; Chrom, chromatin. c, Representative RIP analysis showing the relative abundance of PalB2-bound ActB- or Casp16p-sdRNAs (quantified by Taqman and RT-qPCR) after depletion of Dicer or Ago1. d-e, PalB2 (d) and Rad52 (e) ChIP analyses showing ChIP antibody specificity as well as how PalB2 or Rad52 recruitment is influenced following PalB2 or Rad52 depletion (n=2–4 and n=2–3 biological replicates, respectively for PalB2 and Rad52 ChIP). f, Rad52 ChIP analyses with or without depletion of GapmeR-directed ActB- or Casp16p-sdRNA. Data were analyzed by One-way (b-c) or Two-way (d-e) Anova with post-hoc Tukey HSD or multiple t-test (f) and compared to the relevant control cells. ns= non significant.
Extended Data Figure 9 |. Validation of PalB2 and Rad52 depletion efficiency and G0 arrest.

a, γ-H2AX ChIP quantitation analyzed in cycling or G0 BRCA1-, PalB2- or Rad52-depleted T98G cells (n= 3 biological replicates). b, Representative immunoblots showing the efficiency of siRNA-mediated depletion of PalB2 and Rad52 in both quiescent/G0 arrested and asynchronous T98G cells; n=4. p27 is a marker of quiescence in this setting.
Extended Data Figure 10 |. Overexpression of ActB- or Casp16p-sdRNA results in DNA damage at the pause site of the gene that encoded it.
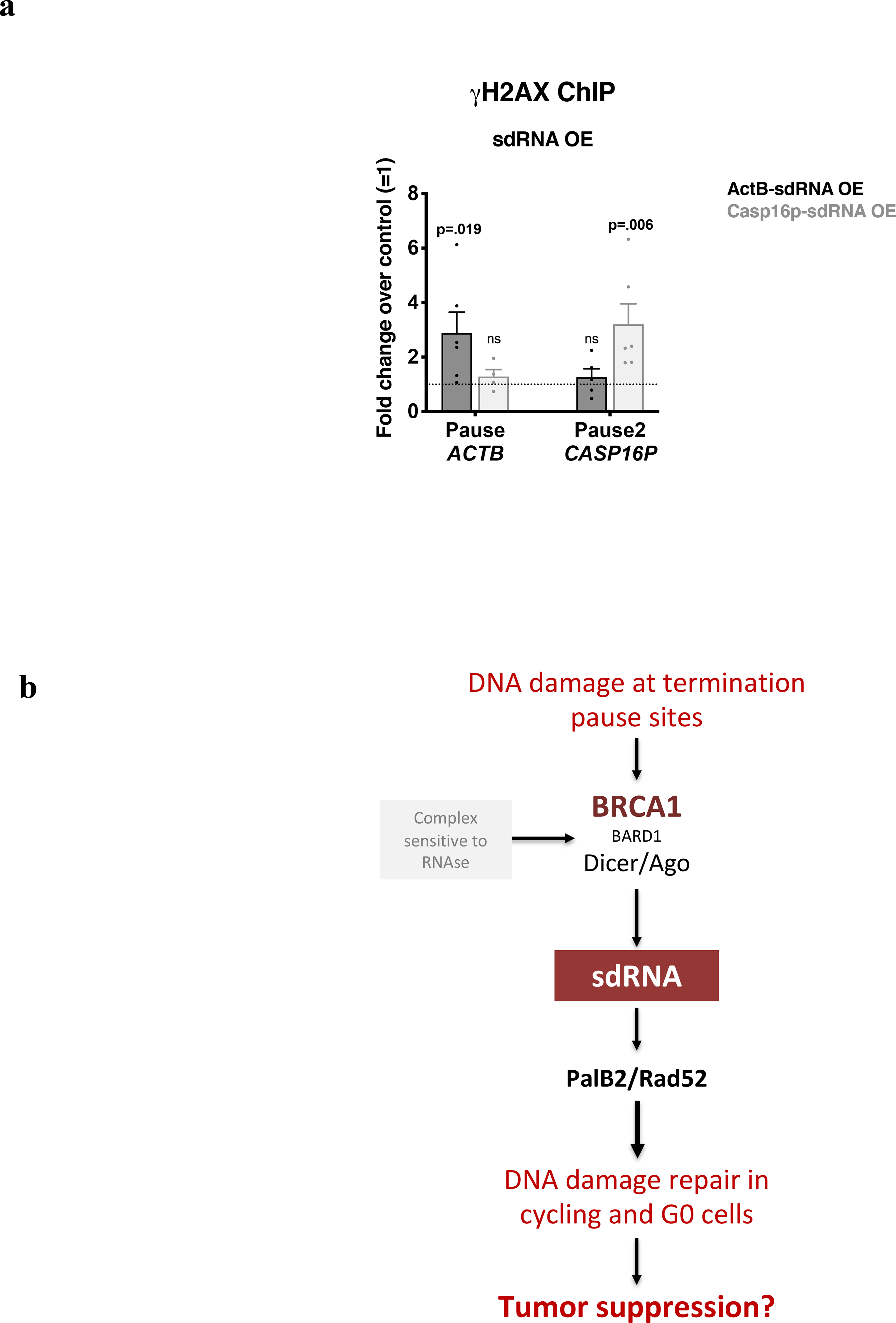
a, DNA damage quantitation at ACTB and CASP16P termination pause sites following ActB- or Casp16p-sdRNA overexpression (OE) using γ-H2AX ChIP qPCR analyses. The ChIP signal reflects the average fold change over the untransfected condition (control) (n = 4–6 biological replicates). Data were analyzed by Two-way Anova with post-hoc Tukey HSD and compared to the relevant control cells. b, Model of sdRNA-mediated damage repair at R-loop positive pause sites in cycling and quiescent cells.
Supplementary Material
Acknowledgements
We thank all Livingston laboratory members for support, technical advice, and helpful discussions. We also thank the members of the Center for Cancer Computational Biology (CCCB), and, in particular, Fieda O. Abderazzaq and Renee Rubio for their expertise in small RNA library preparation and sequencing. We are grateful for Drs David Pellman, and Myles Brown, for their helpful comments during manuscript preparation and to Dr. Will Foulkes for helpful discussions of PalB2 genetics. E.H. and other D.M.L. members were supported by grants from the National Cancer Institute (NCI) - Mechanisms of Breast Development and Carcinogenesis (2PO1CA80111–16) and, BRCA1 Function in Post Damage Foci (5R01CA136512–05), Deciphering the Mechanism Underlying BRCA1 Breast Cancer Development (5R35CA242143–02), as well as from the Susan B Komen Foundation for the Cure, The Breast Cancer Research Foundation, The Gray Foundation, The BRCA Foundation, and the Murray Winsten Foundation. E.H. was also supported by grants from Dana-Farber Cancer Institute (Barr Award - project 9618425 and Friend’s of Dana-Farber). L.G. was also supported by a Research Supplement to Promote Diversity in Health-Related Research Programs (3R01CA136512–08S1). K.S.-S. was supported by a Sir Henry Wellcome Fellowship (grant [101489/Z/13/Z]). D.DeC. and Y.E.W. were supported by grants from the National Cancer Institute (NCI) (5U24CA194354) and J.Q. by NCI grants1R35CA220523, 5U24CA194354 and 5U01CA190234.
Footnotes
Competing interests
Dr.Livingston is a member of the external advisory Boards of the Sidney Kimmel (Johns Hopkins) and the MIT Cancer Centers. He is also a member of the Oncode (the Netherlands) International Advisory Board and is a Science Partner of Nextech (Zurich, CH). Dr Livingston is also a scientific advisor to Constellation Pharma (Cambridge, MA) and to Cancer Research, UK. Neither Dr Livingston nor any other authors declare any competing interests.
References
- 1.Hamperl S & Cimprich KA The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair 19, 84–94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richard P & Manley JL R Loops and Links to Human Disease. Journal of Molecular Biology 429, 3168–3180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crossley MP, Bocek M & Cimprich KA R-Loops as Cellular Regulators and Genomic Threats. Molecular Cell 73, 398–411 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tubbs A & Nussenzweig A Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 168, 644–656 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skourti-Stathaki K & Proudfoot NJ A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 28, 1384–1396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skourti-Stathaki K, Kamieniarz-Gdula K & Proudfoot NJ R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 516, 436–439 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hatchi E et al. BRCA1 Recruitment to Transcriptional Pause Sites Is Required for R-Loop-Driven DNA Damage Repair. Molecular Cell 57, 636–647 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zong D, Oberdoerffer P, Batista PJ & Nussenzweig A RNA: a double-edged sword in genome maintenance. Nat Rev Genet 21, 651–670 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Zhang C & Peng G Non-coding RNAs: an emerging player in DNA damage response. Mutat Res Rev Mutat Res 763, 202–211 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Francia S et al. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 488, 231–235 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Francia S, Cabrini M, Matti V, Oldani A & d’Adda di Fagagna F DICER, DROSHA and DNA damage response RNAs are necessary for the secondary recruitment of DNA damage response factors. J. Cell. Sci. 129, 1468–1476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.d’Adda di Fagagna F A direct role for small non-coding RNAs in DNA damage response. Trends in Cell Biology 24, 171–178 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Sharma V & Misteli T Non-coding RNAs in DNA damage and repair. FEBS letters 587, 1832–1839 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao M et al. Ago2 facilitates Rad51 recruitment and DNA double-strand break repair by homologous recombination. Cell Res. 24, 532–541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei W et al. A role for small RNAs in DNA double-strand break repair. Cell 149, 101–112 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Keskin H et al. Transcript-RNA-templated DNA recombination and repair. Nature 515, 436–439 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazina OM, Keskin H, Hanamshet K, Storici F & Mazin AV Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair. Molecular Cell 67, 19–29.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen C-C, Feng W, Lim PX, Kass EM & Jasin M Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu Rev Cancer Biol 2, 313–336 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nojima T, Dienstbier M, Murphy S, Proudfoot NJ & Dye MJ Definition of RNA Polymerase II CoTC Terminator Elements in the Human Genome. Cell Reports 3, 1080–1092 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Escribano-Díaz C et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Molecular Cell 49, 872–883 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Wang B BRCA1 tumor suppressor network: focusing on its tail. Cell & Bioscience 2, 6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng C-X BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Research 34, 1416–1426 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durant ST & Nickoloff JA Good timing in the cell cycle for precise DNA repair by BRCA1. cc 4, 1216–1222 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Dimitrov SD et al. Physiological modulation of endogenous BRCA1 p220 abundance suppresses DNA damage during the cell cycle. Genes Dev. 27, 2274–2291 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meers C, Keskin H & Storici F DNA repair by RNA: Templated, or not templated, that is the question. DNA Repair 44, 17–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y-G & Qi Y RNA-directed repair of DNA double-strand breaks. DNA Repair 32, 82–85 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Bonnet A et al. Introns Protect Eukaryotic Genomes from Transcription-Associated Genetic Instability. Molecular Cell 67, 608–621.e6 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Miki D et al. Efficient Generation of diRNAs Requires Components in the Posttranscriptional Gene Silencing Pathway. Scientific Reports 7, 301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDevitt S, Rusanov T, Kent T, Chandramouly G & Pomerantz RT How RNA transcripts coordinate DNA recombination and repair. Nat Comms 9, 1091 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huen MSY, Sy SMH & Chen J BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 11, 138–148 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sy SMH, Huen MSY & Chen J PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proceedings of the National Academy of Sciences 106, 7155–7160 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ducy M et al. The Tumor Suppressor PALB2: Inside Out. Trends Biochem. Sci. 44, 226–240 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Yasuhara T et al. Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 175, 558–570.e11 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Gardini A, Baillat D, Cesaroni M & Shiekhattar R Genome-wide analysis reveals a role for BRCA1 and PALB2 in transcriptional co-activation. EMBO J 33, 890–905 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Meng X & Shen Z Association of human RAD52 protein with transcription factors. Biochem. Biophys. Res. Commun. 297, 1191–1196 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Lok BH & Powell SN Molecular pathways: understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin. Cancer Res. 18, 6400–6406 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleiman FE & Manley JL Functional interaction of BRCA1-associated BARD1 with polyadenylation factor CstF-50. Science 285, 1576–1579 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Kleiman FE & Manley JL The BARD1-CstF-50 interaction links mRNA 3’ end formation to DNA damage and tumor suppression. Cell 104, 743–753 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Kleiman FE et al. BRCA1/BARD1 inhibition of mRNA 3’ processing involves targeted degradation of RNA polymerase II. Genes Dev. 19, 1227–1237 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elf J Hypothesis: Homologous Recombination Depends on Parallel Search. Cell Syst 3, 325–327 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Zhu Q et al. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 477, 179–184 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu Q et al. Heterochromatin-Encoded Satellite RNAs Induce Breast Cancer. Molecular Cell 70, 842–853.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 43.Wang H et al. Inadequate DNA Damage Repair Promotes Mammary Transdifferentiation, Leading to BRCA1 Breast Cancer. Cell 178, 135–151.e19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakaue-Sawano A et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Leduc F et al. Genome-wide mapping of DNA strand breaks. PLoS ONE 6, e17353 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grégoire M-C et al. Quantification and genome-wide mapping of DNA double-strand breaks. DNA Repair 48, 63–68 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Grégoire M-C et al. The DNA double-strand ‘breakome’ of mouse spermatids. Cell. Mol. Life Sci. 75, 2859–2872 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conrad T & Ørom UA Cellular Fractionation and Isolation of Chromatin-Associated RNA. Methods in molecular biology (Clifton, N.J.) 1468, 1–9 (2017). [DOI] [PubMed] [Google Scholar]
- 49.hannonlab.cshl.edu. Available at: http://hannonlab.cshl.edu. (Accessed: 17 February 2019)
- 50.Lander ES et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 51.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biology 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramírez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research 44, W160–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the FASTQ sequence files from the small RNA, and RIP larger RNA libraries sequencing are available on the SRA database: BioProject PRJNA667516: Identification and characterization of novel small RNA species sdRNA: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA667516.