

Abstract
The metal centres in metalloenzymes and molecular catalysts are responsible for the rearrangement of atoms and electrons during complex chemical reactions, and they enable selective pathways of charge and spin transfer, bond breaking/making and the formation of new molecules. Mapping the electronic structural changes at the metal sites during the reactions gives a unique mechanistic insight that has been difficult to obtain to date. The development of X-ray free-electron lasers (XFELs) enables powerful new probes of electronic structure dynamics to advance our understanding of metalloenzymes. The ultrashort, intense and tunable XFEL pulses enable X-ray spectroscopic studies of metalloenzymes, molecular catalysts and chemical reactions, under functional conditions and in real time. In this Technical Review, we describe the current state of the art of X-ray spectroscopy studies at XFELs and highlight some new techniques currently under development. With more XFEL facilities starting operation and more in the planning or construction phase, new capabilities are expected, including high repetition rate, better XFEL pulse control and advanced instrumentation. For the first time, it will be possible to make real-time molecular movies of metalloenzymes and catalysts in solution, while chemical reactions are taking place.
In 1895, Wilhelm Conrad Röntgen discovered X-rays, which have revolutionized imaging technology1,2. The power of X-rays led to one of the most profound discoveries of the twentieth century: the first high-resolution image of DNA, the molecule of life, revealing its double-helix molecular structure3. Whereas the penetrating power of X-rays benefits medical and large-scale imaging applications, it is the short wavelength and high energy of X-rays that enable the characterization of the atomic and electronic structure of matter, benefitting many areas of science. X-ray diffraction (XRD) techniques provide atomic-scale structural information of periodic systems (crystalline) and X-ray scattering provides atomic to mesoscale structural information of non-periodic systems (solutions, non-crystalline). Various forms of X-ray microscopy reveal detailed structural information of complex, non-periodic systems with better than 10-nm resolution, and can further provide elemental specificity. Numerous X-ray spectroscopy techniques provide detailed insights into the local atomic and electronic structure and bonding energetics of many systems in an element-specific manner (local chemical environment).
Ongoing efforts over the past 40 years have pushed these methods to ever better resolution and sensitivity with the development of increasingly brighter synchrotron radiation (SR) sources. In these sources, relativistic electrons circulate in a storage ring, consisting of a series of straight sections connected by bending magnets. The sideways acceleration of the electrons at the bending magnets creates an intense horizontal fan of SR, and periodic arrays of magnets (undulators, wigglers) in the straight sections force the electrons into slalom-like trajectories, creating even brighter SR. Several dozen such facilities now operate worldwide, serving tens of thousands of scientists annually. As the latest round of SR facility upgrades is underway, these powerful X-ray light sources are now approaching a physical limit of possible brightness, the diffraction limit. Another limit of SR sources is the duration of each X-ray pulse, typically 10–100 ps, which defines the fastest timescales accessible with SR (for instance, by stroboscopic probing of dynamics), analogous to the shutter time of an ultrafast camera.
Advancing beyond these limits was enabled by switching from a storage ring to a linear accelerator as the source of relativistic electrons; thus, the X-ray free-electron laser (XFEL) was born. The reduced electron beam emittance and higher energy, combined with electron bunch compression and a very long undulator (~100 m), causes the electrons to emit radiation no longer individually but cooperatively in the form of self-amplified spontaneous emission (SASE) (FIG. 1). The dramatically increased peak power of XFEL pulses (more than nine orders of magnitude over SR pulses) and the ability to produce extremely short pulses (typically 1–100 fs and, recently, down to the attosecond regime) have revolutionized X-ray science. Femtoseconds (10−15 s) are the fundamental timescale for nuclear motion. Combining this timescale with the atomic and electronic structure sensitivity of X-rays provides, for the first time, simultaneous spatial and temporal access to molecular systems during their various functions as molecules are transformed. How this groundbreaking task can be achieved in metalloenzymes and molecular transition-metal catalysts, using XFEL-based X-ray spectroscopy in combination with X-ray diffraction and scattering, is the subject of this Technical Review.
Fig. 1 |. X-ray free-electron laser scheme and experimental design for spectroscopy and diffraction and/or scattering experiments of metalloenzymes and molecular catalysts.

a | Coherent X-rays are generated using relativistic electrons from a linear accelerator propagating through a periodic array of magnets (undulator). The transverse undulating motion of the electrons in the magnetic field gives rise to (spontaneous) X-ray emission. Over a long propagation distance (~100 m), the X-ray field causes microbunching of the electrons at the X-ray wavelength, which, in turn, leads to stronger coherent emission, further microbunching and exponential growth of the coherent X-ray emission. At saturation, the X-ray pulses emitted from this self-amplified spontaneous emission (SASE) process have a relative bandwidth ΔE/E0 ≈ 0.2%, with a pulse duration of a few to several tens of femtoseconds (TABLE 1). Diagnostics and beam manipulation provide ways to characterize and change properties of the X-ray free-electron laser (XFEL) beam, such as intensity or photon flux, beam position or pointing, X-ray spectrum, polarization, repetition rate, pulse duration and arrival time. (Not all these properties can be easily changed at every beamline at all XFELs.) X-rays from the undulator are conditioned by beamline optics, which typically include a monochromator (based on ruled gratings or Bragg crystals) and focusing mirrors. Narrower bandwidth (and higher spectral brightness) is achievable using self-seeding, whereby monochromatization is done upstream (between undulator segments), with further amplification in subsequent undulator segments. Samples are introduced at the focus of the X-ray beam, for instance, by using a liquid injector to replace the sample volume at the repetition rate of the X-ray pulses. Numerous spectroscopy techniques are based on the detection of fluorescent X-rays, including fluorescence-detected X-ray absorption spectroscopy (XAS), X-ray emission spectroscopy (XES) and resonant inelastic X-ray scattering (RIXS). X-rays are collected (in the direction orthogonal to the beam propagation) and analysed by a spectrometer consisting of imaging optics and energy-selective elements (Bragg crystals for hard X-rays or dispersive ruled gratings for soft X-rays). b | Schematic of the XES and XAS spectral region (for a Mn compound170) showing the complementarity of the methods (left panel) and schematic of the RIXS spectrum (right panel). VtC, valence to core. Part a adapted with permission from REF.20. Part b adapted with permission from REF.170.
Diffraction limit.
The fundamental relationship between the size of a light source (D), the wavelength of the light (λ) and the divergence of the light beam (Δθ): Δθ ~ λ/πD.
Emittance.
Transverse emittance refers to the relationship between the angular spread (divergence) of an electron beam and the transverse size of the beam.
Bunch compression.
Condensing or squeezing the longitudinal (time) distribution of relativistic electrons in order to increase the peak current.
Self-amplified spontaneous emission (SASE).
Electrons propagating through periodic arrays of magnets exhibit transverse undulating motion, leading to X-ray photon emission. Over distances ~ 100 m, the X-ray field causes longitudinal microbunching of the electrons at the X-ray wavelength, causing positive feedback on the emission process.
Fluorescence yield.
The decay of core-excited states by emission of an X-ray fluorescence photon. X-ray fluorescence is a competitive process and its relative yield depends on the atomic number of the core-excited atom. In 3d transition metals, fluorescence decay dominates for K-edge excitation (31% for Mn, REF.68).
Background
The ability of transition metals to accept and donate electrons, at relatively low energy cost, puts them at the centre of many metalloenzymes and molecular catalysts. Among the 3d transition metals, those with unfilled 3d orbitals are of particular importance, as they are the most abundant in nature. (Of the 3d metals, Fe, Mn, Ni, Zn, Cu and Co are the most commonly used in catalysis. Only Zn has a full 3d shell and is usually not involved in redox processes.) Understanding the chemical function of 3d metals is essentially understanding their 3d electronic structure and bonding. X-ray spectroscopy is a powerful probe of the 3d metal electronic structure as it probes, directly or indirectly, the binding energetics and the charge and spin states with element specificity. Given the substantial differences in the experimental approaches, we distinguish between soft X-ray (<1 keV) and hard X-ray (>5 keV) spectroscopy.
The large difference in absorption cross section and fluorescence yield between soft and hard X-ray spectroscopy also has important practical consequences for the application of both approaches. Here, we will focus on photon-in photon-out spectroscopy methods for both hard and soft X-rays, as they are widely applicable and most commonly used for probing chemical reactions of metalloenzymes and molecular catalysts in solution (we note that soft X-ray spectroscopy is also performed in electron and ion yield modes).
Importantly, moving from a synchrotron to an XFEL requires rethinking experimental approaches, with new challenges and opportunities present. As described in a recent review, many of the pioneering experiments at the vacuum ultraviolet (VUV)/soft X-ray FEL, FLASH, which started user operation in 2005, addressed this issue and, thereby, laid the foundation for the methods discussed here4. An important issue that was identified and persists to date is that of X-ray-induced sample damage. A critical question that has to be addressed for each experiment is how to obtain useful information about the sample without the influence of damage.
The standard approach at a synchrotron (in which damage typically arises from average X-ray exposure5) is to cryogenically cool the sample to reduce the mobility of radicals created by ionization from X-rays. However, doing so precludes studies under functional conditions. Moreover, although the cryogenic technique with frozen samples works for hard X-rays, it is not suitable for soft X-ray spectroscopy studies on metalloenzymes. Even with cryo-cooling, soft X-rays induce radiation damage too rapidly, owing to their higher absorption cross section and the larger Auger electron yield. SR studies of metalloenzymes at room temperature are almost impossible in the soft X-ray energy range, although recent advances in sample delivery approaches allow some serial measurements at SR sources in the hard X-ray regime6–10.
Counter-intuitively, the very high peak power of XFEL pulses provides a critical advantage over synchrotrons, opening the door to studies of metalloenzyme samples at room temperature with both hard and soft X-rays. XFEL pulses are so short (~10–50 fs) that they can essentially outrun the sample damage caused by the diffusion of solvated electrons or radicals by probing the sample and getting useful information before the onset of damage. This concept is known as ‘probe-before-damage’ or ‘probe-before-destroy’. The limits above which damage-induced changes in a biological sample would compromise diffraction data have been estimated11. Subsequently, probe-before-destroy was first shown to work experimentally for diffraction12, for X-ray emission spectroscopy (XES) of solutions of manganese complexes13 and for soft X-ray spectroscopy of metalloenzymes14. It is now the standard approach for studying many samples at XFELs and for characterizing metalloenzymes and sensitive chemical materials, including molecular catalysts, under ambient and functional conditions in real time.
The work described in this Technical Review directly relies on this critically important probe-before-destroy approach. However, for successful execution, several additional experimental challenges must be addressed. First, because XFEL pulses often do destroy the sample, tailored methods of sample delivery and environment are required to replace the sample after each pulse15–19. Second, the stochastic nature of the SASE XFEL pulses causes complex spectral and temporal pulse profiles and significant shot-to-shot spectral, temporal and intensity fluctuations, requiring specialized diagnostics and spectroscopy instrumentation20–23. Third, it is possible to study the functional changes in metal centres in real time of the reaction with a pump–probe approach, in which a visible laser pump pulse or a trigger such as temperature or a chemical and/or substrate initiates the reaction, and an XFEL pulse probes the atomic and electronic structure as a function of the pump–probe delay time. This method requires shot-to-shot spatial and temporal pump–probe synchronization, spectral and spatial beam diagnostics, detection and data processing. Fourth, the interaction of intense XFEL pulses with matter may create new physical phenomena that need to be understood to properly interpret the data. We discuss examples of such nonlinear phenomena and their potential future applications in the last section of this Technical Review.
Photon-in photon-out spectroscopy.
X-ray spectroscopic methods that separately detect photons incident on the sample (photon-in) and photons leaving the sample (photon-out), to measure the spectroscopic observable. Examples include X-ray absorption spectroscopy in transmission and fluorescence yield mode, X-ray emission spectroscopy and resonant inelastic X-ray scattering.
Auger electron yield.
The decay of core-excited states by emission of an Auger electron. Auger decay is a competitive process and its relative yield depends on the atomic number of the core-excited atom. In 3d transition metals, Auger decay dominates for L-edge excitation with a minor fluorescence decay channel (0.5% for Mn, REF.68).
Stimulated emission.
The process by which an incident photon of a given energy (or wavelength) triggers an electronic transition in an excited atom or molecule to a lower electronic state, resulting in an emitted photon with the same wavevector, energy and phase as the incident photon.
Fourier transform limit.
The lower limit of the duration of a time pulse with a given frequency spectrum. This is based on the fundamental Fourier transform relationship between a time-dependent function and its frequency spectrum, and the lower limit implies a frequency-independent spectral phase.
Seeding.
In laser physics, a process in which a signal (typically from a weak laser) is injected into a gain medium (typically from a strong laser) to improve the output signal by stabilizing the wavelength and reducing variations in the output pulse energy and timing (jitter).
In this Technical Review, we describe how addressing these four challenges will create new opportunities at the current and the next generations of XFELs for spectroscopy of metalloenzymes and molecular catalysts. We first describe the basics of XFELs and their present-day capabilities. We then address hard X-ray and soft X-ray spectroscopy in turn, before discussing the capabilities that will be available in future facilities. Although, presently, there are only a few XFELs in operation, there is a steady worldwide growth of capacity. With new high-repetition-rate XFELs, such as the European XFEL and LCLS-II (Table 1), this capacity increase and the enhanced capabilities of XFELs will be further accelerated. We are optimistic that the explosive growth seen in synchrotron-based research over the last several decades is an indicator of what can be expected from XFEL-based research in the future, and X-ray spectroscopy of metalloenzymes and molecular catalysts will be a key research area and at the heart of this progress. We also want to direct the readers to several recent special issues of journals that provide a broad collection of specialized reviews on this topic24–28.
Table 1 |.
Characteristic parameters of the various X-ray free-electron laser facilities worldwide (adapted from REF.169)
| Location | Facility name | Photon energy (keV) | Pulse lengtha (fs) | Pulse energyb (mJ) | Repetition rate (Hz) | Flux (ph s−1)f | Start of operation |
|---|---|---|---|---|---|---|---|
| Japan | SACLA BL2,3 | 4–20 | 2–10 | 0.1–1 | 60 | 2–9 × 1013 | 2011 |
| SACLA BL1 | 0.04–0.15 | 60 | 0.1 | 60 | 3–9 × 1014 | 2015 | |
| Italy | FERMI FEL-1 | 0.01–0.06 | 40–90 | 0.08–0.2 | 10 (50) | 1–6 × 1015 | 2010 |
| FERMI FEL-2 | 0.06–0.3 | 20–50 | 0.01–0.1 | 10 (50) | 1–5 × 1014 | 2012 | |
| Germany | FLASH1 | 0.02–0.3 | 50–200 | 0.03–0.5 | (1–800) × 10c | 0.08–1 × 1018 | 2005 |
| FLASH2 | 0.01–0.3 | 50–200 | 0.03–0.3 | (1–800) × 10 | 0.05–2 × 1018 | 2015 | |
| Korea | PAL-XFEL | 2.5–15 | 5–50 | 0.8–1.5 | 60 | 0.4–2 × 1014 | 2016 |
| 0.25–1.2 | 5–50 | 0.2 | 60 | 0.6–3 × 1014 | 2016 | ||
| Switzerland | SwissFEL | 1.8–12.4 | 10–70 | 1 | 100 | 0.5–3 × 1014 | 2017 |
| 0.2–2 | 10–70 | 1 | 100 | 0.3–3 × 1015 | 2021 | ||
| Germany | European XFEL-SASE1,2 | 3–25 | 10–100 | 2 | 2,700 × 10d | 0.1–1 × 1017 | 2017 |
| European XFEL-SASE3 | 0.2–3 | 10–100 | 2 | 2,700 × 10 | 0.1–2 × 1018 | 2017 | |
| USA | LCLS | 0.3–12 | 2–500 | 2–4 | 120 | 0.03–1 × 1016 | 2009 |
| LCLS-II | 1–25 | 10–100 | 2–4 | 120 | 0.1–3 × 1015 | 2022e | |
| LCLS-II | 0.2–5 | 10–200 | 0.02–1 | 106 | 0.04–1 × 1019 | 2022e | |
| LCLS-II-HE | 0.2–13 | 10–200 | 0.02–1 | 106 | 0.01–1 × 1019 | 2026e |
Estimated pulse length (full width at half maximum) based on e-bunch length measurement or designed range.
Rough estimates of pulse energy, particularly for projects that are currently under construction.
Burst mode operation at 10 Hz, with each macropulse providing up to 800 bunches at 1 MHz.
Pulsed mode operation at 10 Hz, with each macropulse providing up to 2,700 bunches at 5 MHz.
Projected project completion dates.
Flux (photon s−1) is based on the combination of maximum pulse energy and repetition rate, and the range reflects the range of photon energies available from that source. Note that, for LCLS-II and LCLS-II-HE, the pulse energy (as a function of repetition rate) is constant up to ~300 kHz, and scales inversely with repetition rate above ~300 kHz.
Sources, properties and new capabilities
XFEL sources and properties.
Whereas optical lasers rely on the quantum process of stimulated emission in a gain medium, typically through multiple passes within a cavity, XFELs amplify coherent electromagnetic radiation using a single pass of relativistic GeV-scale electrons propagating through a very long undulator29,30 (FIG. 1). This SASE mode generates femtosecond coherent X-ray pulses, at multi-gigawatt peak powers and wavelengths spanning from tens of nanometres to below 1 Å (photon energies of hundreds of eV to above 10 keV). Thus, the peak brightness of XFEL pulses is ~109 times higher than that of current synchrotron X-ray pulses. The XFEL facilities FERMI31, LCLS20, SACLA32, PAL-XFEL33 and SwissFEL34 (Table 1) use normal conducting pulsed radio frequency accelerator technology operating at repetition rates in the 10–120-Hz range. FLASH35 and the European XFEL36 facilities use pulsed superconducting accelerator technology, creating a burst mode of pulse trains at 10 Hz, with each macropulse comprising up to 800 (FLASH) or 2,700 (European XFEL) pulses. The new XFELs now under development (LCLS-II37 and LCLS-II-HE38 in the USA, and SHINE in China) use continuous-wave superconducting radio frequency accelerator technology, providing high repetition rates with uniform or programmable time distributions. The first of these facilities (project name LCLS-II) is scheduled to start operation in 2022, delivering soft and tender X-ray pulses up to 5 keV (2.5 Å) at repetition rates up to 1 MHz (REF.37). In the second phase38, the X-ray range will be extended beyond 12 keV (1.0 Å). Other performance enhancements identified by the rapidly growing XFEL user community and demonstrated to date include ultrashort (<1 fs) X-ray pulse generation and characterization39,40, temporal coherence (near the Fourier transform limit) via seeding and related schemes41–48, and versatile operating modes such as two-colour multi-pulse sequences43,46,49–54, and large coherent bandwidth55. These enhancements, and the fact that the capacity and access to XFELs and lab-based femtosecond sources is growing worldwide56, will strongly benefit the science enabled by the X-ray spectroscopy and scattering techniques we review in the next section.
Time-dependent X-ray spectroscopy and scattering using an XFEL.
Using ultrabright femtosecond XFEL pulses for X-ray spectroscopy (the X-ray absorption spectroscopy (XAS), XES and resonant inelastic X-ray scattering (RIXS) methods described in BOXES 1 and 2) has created an enormous potential for time-dependent, operando studies of ultrafast dynamics and chemical reactions of metal complexes. The most common approach uses an optical laser pump pulse to initiate the chemical process and an XFEL pulse to probe it. The measurement is repeated at varying time delays between pump and probe to create a sequence of spectra or scattering patterns that reflect the temporal evolution of the system after excitation. Such sequences, sometimes called ‘molecular movies’, provide a way to follow reactions as they proceed. Critical to the success of this approach is the property of femtosecond XFEL pulses (described in the introduction to this Technical Review) to probe the system before destroying it. Femtosecond XFEL pulses are so short that they can outrun the diffusion of ions, radicals and electrons created by the interaction of X-rays and the sample, which is the main cause of the various forms of radiation damage observed at synchrotron facilities in molecular and biological catalysts5. Therefore, cryo-cooling is not required in XFEL experiments13,57. The combination of high peak power and ultrashort duration of XFEL pulses enables efficient probing of dilute species of catalysts in solution with sufficient signal-to-noise ratio. Radiation-sensitive molecular catalysts and metalloenzymes in solution are now routinely studied in XFELs to probe their steady states14 and to capture intermediates that occur on slow58,59 and ultrafast timescales60–62. However, at sufficiently high peak power, highly focused XFEL pulses start to create nonlinear effects that change the linear X-ray spectra63–67. These nonlinear phenomena and the magnitude of these effects on linear X-ray spectra critically depend on the experimental conditions, such as X-ray wavelength, pulse duration and sample concentration.
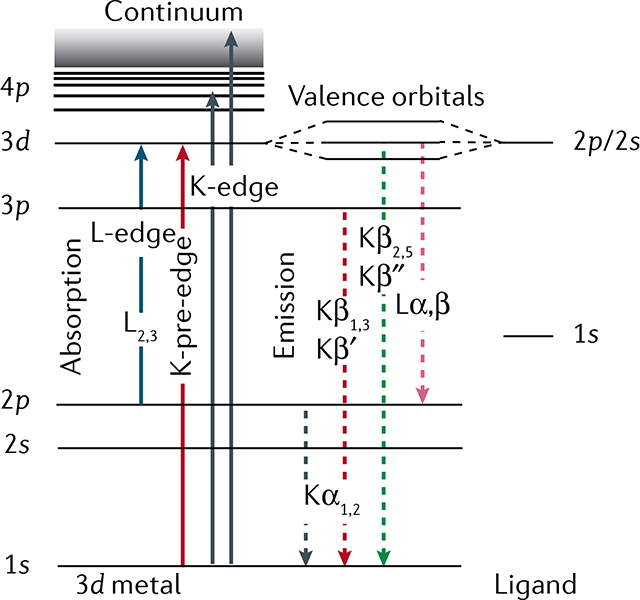
Box 1 |. Absorption and emission X-ray spectroscopy.
In general, X-ray spectroscopy is used to characterize electronic and geometric structures of elements in their chemical environment172, but in this Technical Review, we focus on 3d transition metals. X-ray spectroscopy is element-selective because the binding energies of core electrons are specific to each element173. It is sensitive to the chemical environment, which imposes small shifts on these binding energies174,175. By tuning the incident energy to a resonance, X-ray spectroscopy exhibits orbital specificity89,120,176,177. X-ray absorption spectroscopy (XAS) probes the electronic transitions from ground to core-excited states, whereas X-ray emission spectroscopy (XES) probes the fluorescence decay of core-excited states. The panel below shows a generalized energy-level diagram for 3d transition-metal systems for hard and soft X-ray spectroscopy. XAS and XES are complementary techniques providing information about local metal oxidation states, valence orbital populations and interactions, local metal spin states, ligand coordination, bond lengths and symmetry changes, and metal–ligand covalency.
XAS probes the transitions of a core electron to empty or partially unoccupied molecular orbitals, with symmetries given by the dipole selection rule176–180. K-edge XAS (hard X-rays) probes predominantly unoccupied molecular orbitals with p symmetry via 1s → 4p transitions. In the K-pre-edge region, weak quadrupolar 1s → 3d transitions can be observed, which gain intensity through mixing with orbitals with p symmetry. L-edge XAS (soft X-rays) predominantly probes the orbitals with d symmetry via 2p → 3d transitions. These transitions relate to the low-energy region of an XAS spectrum (first few eV in the soft X-ray range and first few tens of an eV in the hard X-ray range), also called the X-ray absorption near edge structure region. At higher energies (tens to hundreds of eV) is the extended X-ray absorption fine structure region, which is dominated by scattering processes. In this case, the energy of the X-ray photons liberate photoelectrons that propagate from the absorber atom and are backscattered by neighbouring atoms, producing interferences and the characteristic extended X-ray absorption fine structure oscillations181. This signal provides information about the atomic number, distance and coordination number of the atoms surrounding the metal absorber atom.
XES probes the transitions of an electron from an occupied orbital to an unoccupied or partially occupied core orbital182. These transitions result from spontaneous fluorescence decays of core-excited states reached by X-ray absorption of the system. Non-resonant K-edge XES (Kα, Kβ1,3, Kβ′, Kβ2,5 and Kβ˝), where the excitation energy is well above the core electron binding energy, provides information on the metal oxidation state, effective spin, metal bonding orbitals and nature of the ligand. L lines in XES of 3d transition metals and K lines in XES of ligand atoms such as N or O provide sensitivity to oxidation states, symmetry, energies and interactions of occupied orbitals, and metal–ligand covalency.
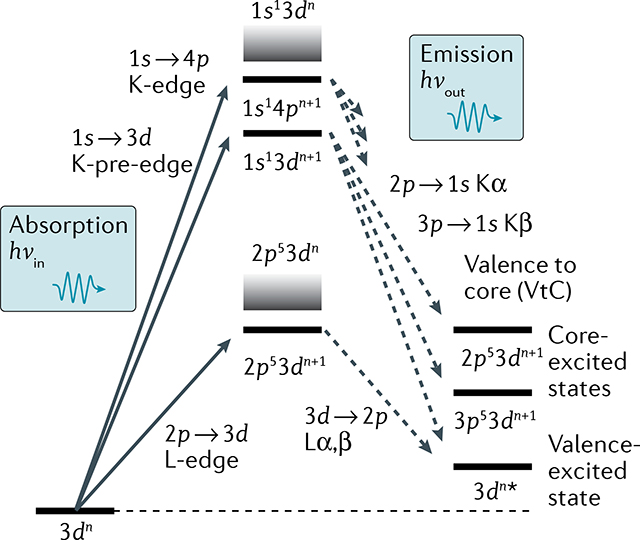
Box 2 |. Resonant inelastic X-ray scattering.
Resonant inelastic X-ray scattering (RIXS) is a multidimensional X-ray spectroscopy offering enhanced electronic structural information81,122,183,184. In RIXS, the energy of photons scattered/emitted by the sample is measured with high spectral resolution, while the incident photon energy is scanned across the absorption edge. The corresponding 2D spectrum shows the incident photon energy versus that transferred to the sample, defined as the difference in incident and emitted/scattered photon energies. This energy transfer is in the range of meV to eV for soft X-ray RIXS, corresponding to vibrational and valence-electronic d–d transitions, and up to several 100 eV for hard X-ray RIXS, corresponding to resonant core–electron transitions. RIXS is the X-ray analogue of resonance Raman scattering and provides the ability to probe the occupancy and interactions of both occupied and unoccupied orbitals in valence-excited and core-excited states.
In 1s RIXS, the incident hard X-ray energy is scanned across the K-pre-edge exciting 1s electrons to unoccupied 3d-derived valence orbitals, where 1s → 3d transitions lead to 1s13dn+1 intermediate states and 2p53dn+1, 3p53dn+1 or 3dn⋆ final states, respectively122 (see panel). Therefore, 1s RIXS probes 2p → 3d (similar to L-edge absorption), 3p → 3d (similar to M-edge absorption) or valence–electron transitions, with the advantages of hard X-rays.
In 2p3d RIXS, the incident soft X-ray energy is scanned across the L-edge, exciting 2p electrons to unoccupied 3d-derived valence orbitals, where 2p → 3d transitions lead to 2p53dn+1 intermediate states and 3dn⋆ final states122,185. The energy transfer corresponds to valence–electron transitions of ultraviolet–visible (UV/Vis) absorption, with the advantages of using an element-specific, chemical-site-specific and orbital-specific X-ray probe. 2p3d RIXS offers several additional advantages over UV/Vis absorption spectroscopy: it reveals d–d or ligand-field (LF) excitations over much wider energy ranges often hard to access with UV/Vis spectroscopy (down to below 1 eV and up to several 10 eV). LF or charge-transfer excitations can be selectively enhanced by resonant excitation, allowing to probe LF excitations unmasked from potentially overlapping charge-transfer transitions and solvent absorption that often dominate UV/Vis spectroscopy. Finally, due to spin–orbit interaction in the 2p-excited intermediate states, 2p3d RIXS allows probing transitions that are formally spin-forbidden in UV/Vis spectroscopy122,185,186.
Soft X-rays with typical absorption cross sections of ~10−17 to 10−18 cm2 and incident fluence of >1017 photons cm−2 within a core-hole lifetime (~4 fs for low-Z elements68,69) lead to an appreciable probability of instantaneous nonlinear effects. This corresponds to peak power densities >1015 W cm−2 at 500 eV. A hard X-ray study67 discussed the effects of high peak power and sample concentration on the X-ray emission spectra of Fe samples of varying concentrations. XFELs based on superconducting linacs, such as the existing European XFEL and the future LCLS-II, with kHz to MHz repetition rates with the same or smaller pulse energies compared with existing low-repetition-rate XFELs, are ideally suited for the traditional X-ray spectroscopy data collection. In some cases, however, nonlinear X-ray phenomena can be exploited to reveal information that is unavailable from linear X-ray spectroscopy, which is an area of active research.
Finally, the XFEL-based pump–probe approach has led to some important experimental changes in hard X-ray spectroscopy as compared with its long-standing use at synchrotron sources. To analyse the emitted or resonantly scattered (photon-out) spectrum at an XFEL, dispersive X-ray optics, where the whole spectrum is recorded in a shot-by-shot approach, are most commonly used13 (BOXES 1 and 2). In hard X-ray spectroscopy, non-resonant XES above the metal K-edge has proven very valuable to early XFEL applications60,70. The technique does not require monochromatic X-rays, which has the advantage that it can use the full SASE XFEL pulse, and it can be simultaneously applied with X-ray scattering and diffraction, two techniques that provide complimentary atomic structure information57,59,71.
Hard X-ray spectroscopy at XFELs
Hard X-ray spectroscopy techniques have been widely used for studying molecular catalysts and metalloenzymes at SR facilities. To avoid X-ray-induced changes, data are often collected at cryogenic temperature, meaning that only the chemical states that can be trapped by a freeze-quench method are accessible. There have also been various efforts in recent years to collect data at room temperature using jet and micro-fluidics systems6–10,72, and these often benefitted from sample delivery approaches initially developed for XFEL experiments. At XFELs, the ability to collect X-ray data at room temperature, that is, free from X-ray-induced changes, has advanced mechanistic studies of catalysis, by providing a way to follow a reaction as it proceeds. Among several reaction triggers, so far, photochemical pump–probe methods are most often carried out at XFELs, and have provided a way to capture chemical and structural changes at various time delays as short as tens of femtoseconds. The vast majority of catalytic reactions, however, proceed by substrate binding at catalytic sites. Such reactions proceed at much slower rates, due to diffusion-limited processes of substrates. In this case, it is the advantage of the probe-before-destroy nature of ultrashort XFEL pulses that allows capturing the population shift of reaction intermediates, thus, enabling the untangling of the sequence of events at reaction conditions without the requirement of cryo-cooling.
Oxidation states.
The oxidation state of an atom in a compound describes the degree to which the electron number of an atom has changed compared with the uncharged neutral form of the same atom. In case of redox reactions of first-row transition metals, these changes happen in the 3d shell; hence, the oxidation state is directly related to the number of 3d electrons present.
Spin states.
In transition-metal complexes, the spin state refers to the distribution of electrons in the valence shell. Often, there are two distributions possible for the same number of electrons: low-spin or high-spin configurations, having a low or a high number of unpaired electrons, respectively.
Although soft X-ray L-edge spectroscopy is a more direct probe of the 3d transition-metal electronic structure, the hard X-ray regime offers several practical advantages, as experiments under ambient pressure conditions and on bulk materials are facilitated, due to their larger penetration depth. Furthermore, the shorter hard X-ray wavelengths (~1–2 Å) are comparable to atomic distances, allowing simultaneous scattering/diffraction experiments that provide structural information at atomic resolution. Below, we describe examples of time-resolved XES and XAS experiments (FIG. 2) and multimodal experiments using combinations of spectroscopy and diffraction measurements with hard X-rays (FIG. 3).
Fig. 2 |. Hard X-ray spectroscopy at X-ray free-electron lasers.

a | Fe Kα X-ray emission spectroscopy (XES) of solutions of Fe/Mn containing ribonucleotide reductase R2c measured at room temperature prior to O2 incubation, and changes observed after 0.5, 2 and 8 s in situ O2 incubation. The change in spectral shape and full width at half maximum (FWHM) indicates, first, formation of a FeIV intermediate (within ~2 s), followed by the catalytically active FeIII state (schematic). b | Transient Fe Kβ XES difference spectra of Fe(CN)4(bpy) (bpy = 2,2′-bipyridine; structure in inset) at 50 fs and 1 ps after photoexcitation and the calculated difference involving either a doublet metal–ligand charge transfer (MLCT) or a quintet metal-centred excited state (top). The contour plot (bottom) shows light-induced changes of the XES up to 1.5 ps after excitation. The spectra indicate that, likely, only one excited state is present and the MLCT state fits the data best. c | Transient Fe valence-to-core (VtC) XES of Fe(CN)4(bpy) (top) and calculated spectra (bottom), showing light-induced changes in the picosecond time range due to photo-oxidation and ligand dissociation. d | Time-resolved X-ray absorption near edge structure (XANES) spectra of cytochrome c before and 600 fs after light excitation. The changes in the edge position and the shape resonances were interpreted as an out-of-plane motion of the haem Fe coupled with a loss of the Fe–S(Met) bond (inset). e | Extended X-ray absorption fine structure (EXAFS) spectra of Fe(terpy)2 (terpy = 2,2′:6′,2˝-terpyridine, structure in right inset, with equatorial (Neq) and axial (Nax) nitrogen atoms indicated) in the ground state and after excitation (“Laser ON”). The k2 weighted difference EXAFS modulation (k2Δχ(k)) as plotted in k-space (bottom inset) indicates light-induced changes in the Fe–ligand distances on the sub-picosecond timescale. k, photoelectron wavevector. Part a adapted with permission from REF.18. Part b adapted with permission from REF.75. Part c adapted with permission from REF.84. Part d adapted with permission from REF.60. Part e adapted with permission from REF.95.
Fig. 3 |. Multimodal detection methods in X-ray free-electron laser studies using hard X-rays.

a | Measurements of femtosecond time-resolved Kα X-ray emission spectroscopy (XES) (left) in parallel with X-ray diffuse scattering (XDS) (right) on a Ru–Co dyad (bottom). The kinetics of the XES show the conversion of the low-spin to the high-spin form of Co within 2 ps. At early times, the XDS data exhibit a strong dip in intensity (ΔS) at momentum transfer Q = 0.5 Å−1, indicating an expansion by 0.2 Å on the 500-fs timescale; a second feature at higher Q indicates thermal equilibration at ~12 ps. The schematic also contains information derived from transient optical absorption spectroscopy (TOAS) measurements, indicating ultrafast electron transfer from Ru to the bridge and transfer from the bridge to Co in under 500 fs. b | X-ray diffraction (XRD) in combination with Mn Kβ and Fe Kα XES of ribonucleotide reductase. The structure of the dinuclear metal centre found in crystals of oxidized ribonucleotide reductase is shown. Electron density is contoured at 1.2 s in blue. Omit density (green) indicates the position of the bridging oxygen atoms. The Mn Kβ spectrum obtained from crystals and solutions is shown, together with a calibration spectrum of MnIICl2 (bottom left); the Fe Kα spectrum of the crystals is shown on the bottom right. c | Combined XRD and XES studies on photosystem II. Left: the overall structure of the protein and the four-step catalytic cycle (Kok cycle), revealed by flashes 1F–4F. For each of the stable states S0, S1, S2 and S3, the XRD structure of the catalytic Mn4Ca cluster obtained from X-ray free-electron laser measurements is also shown, exhibiting a change in the distance between Mn atoms 1 and 4 (given in Å). Right: results from time-resolved XES and XRD measurements, together with kinetic simulations based on previous infrared (IR) or X-ray absorption (XAS) measurements. Bottom: XRD and XES data show concomitant Mn oxidation and insertion of a new oxygen (OX) in the Mn cluster on the 250-μs timescale during the S2 → S3 transition. The S1 state structure is shown in light grey and the different time point structures in various colours (yellow to olive). The electron density is contoured at 3, 4 and 5 σ around the O5 and OX atoms of the Mn cluster and Glu189, a critical mobile amino acid side chain. Part a adapted with permission from REF.99. Part b adapted with permission from REF.18. Part c adapted with permission from REFS58,59.
X-ray emission spectroscopy.
XES probes occupied states and can be performed off resonance with any incident photon energy above the absorption edge region of the absorbing metal (BOX 1), allowing the use of the full XFEL SASE pulse and combining the technique with scattering/diffraction. XFEL-based XES has been used widely to study changes in metal oxidation states and spin states of enzyme systems, including the haem Fe in cytochrome c (REFS60,61) and in myoglobin62, the Mn4Ca cluster in photosystem II (PS II) (REFS18,57–59,73), the dinuclear Mn–Fe site in ribonucleotide reductase (RNR)18 (FIGS 2a,3b) and the dinuclear Fe–Fe site in methane monooxygenase74. Numerous other photosensitive transition-metal complexes have been studied to understand the initial steps of photoexcitation and light-induced charge transfer, and the time-resolved XES provided detailed information about changes in the effective spin of the transition metal70,71,75–80 (FIG. 2b).
Although most XFEL-based XES studies have focused on the stronger Kα1,2 and Kβ1,3 XES lines, the weakest XES lines resulting from dipole-allowed valence-to-core transitions (VtC, also known as Kβ2,5 and Kβ”, see BOX 1 ) contain the most detailed information about the bonding and chemical environment of the absorbing atom81,82. Steady-state83 and time-resolved VtC-XES using an XFEL was reported84 (FIG. 2c). Comparing the observed spectral changes for three Fe(CN)-bipyridine compounds, recorded 0.2–50 ps after light excitation, with density functional theory calculations revealed the sensitivity of VtC-XES to bond length expansion. It was found that the integrated intensity of the VtC-XES spectrum is a measure of the metal–ligand bond length because it is primarily determined by the overlap of ligand valence orbitals with metal np orbitals. A decrease of intensity with increasing metal–ligand distance was observed. The spectra also showed signatures of photo-oxidation as measured by the energy position of the VtC-XES spectrum, where a blue shift was associated with increased oxidation state of the Fe centre. With further evidence of ligand dissociation for Fe(CN)6, and metal-centred triplet and quintet states for Fe(CN)4(bpy) and Fe(CN)2(bpy)2, where bpy = 2,2′-bipyridine, respectively, the study concluded that the metal–ligand bond length expanded by either 0.1 or 0.2 Å upon formation of the respective metal-centred triplet or quintet state on the sub-picosecond timescale84 (FIG. 2c).
X-ray absorption spectroscopy.
The X-ray absorption near edge structure region of XAS probes the unoccupied states via metal 1s → np transitions (see FIG. 1b and BOX 1 ) and, like XES, XAS can reveal details of ultrafast changes in the environment of transition metals in natural and synthetic molecules. Unlike XES, however, XAS measures changes of the absorption coefficient around the metal absorption edges and, therefore, requires monochromatized incoming X-rays to scan through a wide energy range (<50 eV for X-ray absorption near edge structure and >500 eV for X-ray absorption fine structure (EXAFS), see BOX 1). To observe small spectral changes, the normalization of data at each incoming X-ray energy, considering the fluctuation of both incoming X-ray intensity and probed sample volume during the shot-by-shot experiments at XFELs is crucial. Energy-dispersive XAS using a wide energy range has also been described85. When using monochromatized beams, the flux is reduced by a factor of about 100 for SASE pulses, but much less when using self-seeded pulses.
XAS was used at an XFEL to observe the formation of a high-spin state ~160 fs after light excitation in a Fe–tris-bipyridine compound, indicating a metal–ligand charge-transfer (MLCT) state at very short times86. Fe K-edge XAS was also used to study the ultrafast photodissociation process of CO bound to the haem group in myoglobin87. Changes of the Fe K-edge energy and intensity were fit by two processes with characteristic times of 70 and 400 fs. The faster process was interpreted as being connected to an elongation of the Fe–N bond length in the haem, coupled to a shortening of the Fe–His bond. The slower process was interpreted as an iron out-of-plane motion coupled to the motion of the helix F of the myoglobin, being the first step in the observed protein quake. These spectroscopic results agree well with independently conducted time-resolved crystallographic experiments on the sub-picosecond timescale88. Interestingly, an XES study on the photodissociation of NO-bound myoglobin showed that, within 100 fs, a ligand-free triplet state is formed and that it takes about 800 fs to form a domed high-spin quintet state62.
Metal–ligand charge-transfer (MLCT) state.
MLCT excitations are special cases of charge-transfer excitations in metal complexes. MLCT excited states result from one-electron transitions in which an electron is promoted from a metal-centred to a ligand-centred orbital.
XAS is sensitive to structure and ligand environment because the metal 1s → np transition energies and intensities change when metal–ligand orbitals overlap and ligand fields (LFs) change89. Examples for other ultrafast XAS studies include measurements on Cu–phenanthroline90 or on Ni porphyrins91. It is possible to extend the amount of information that can be obtained by XAS studies by using polarized photoexcitation. The idea is to exploit the fact that optical excitation in metal complexes is often anisotropic and, by using different relative polarizations of the optical pump and X-ray probe beams, it is possible to select different sub-ensembles of differently excited species. Polarized cobalt XAS studies on a cobalamin derivate, for example, showed that structural changes upon photoexcitation of the corrin ring occur mainly in the ring plane first along one direction at 19 fs and then in the perpendicular direction of the ring 50 fs later. These changes are followed by the elongation of the axial ligands on a 200–250-fs timescale, and the internal conversion to the ground state occurs on a 6.2-ps timescale. These results describe the features that influence the reactivity, stability and deactivation of electronically excited cobalamins92,93.
To obtain very precise measurements of distance changes of the ligands of transition-metal systems, EXAFS is an essential tool in SR studies. The EXAFS region of XAS was successfully collected at XFELs94,95. Precise signal normalization to the incoming beam intensity and the probed sample volume is critical when measuring the subtle changes in EXAFS spectra. This was achieved by recording the forward scattering signal from the solvent for each shot and using it for signal normalization. Time-resolved EXAFS with delay times of 0 to 600 fs and 10 ps on a solution of Fe(terpy)2, where terpy stands for 2,2′:6′,2″-terpyridine, showed a 0.2-Å change in the Fe–ligand distance between the ground state and the excited state, and revealed an intermediate spin state in the laser-induced low-spin (LS) to high-spin (HS) transition on the 100-fs timescale95 (FIG. 2e). In the HS final excited state, all Fe–N bonds were found to be expanded by ~0.2 Å. The intermediate state was best explained by expansion of the equatorial Fe–N bonds by 0.15 Å, while the axial Fe–N bonds did not yet expand95.
Multimodal measurements.
Combining complementary techniques leads to a more complete picture of the local changes of a transition metal site during a reaction. An example is the use of both XAS and XES to study the weak covalent bond between the haem Fe and an S-methionine (S-Met) ligand present in cytochrome c (REF.60). The presence of this bond ensures that the Fe stays in its LS state in both FeII and FeIII oxidation states. Loss of the S-Met bond changes the functionality of cytochrome c. The bond can be broken by laser excitation, after which it quickly reforms. 600 fs after excitation changes in the XAS spectrum indicate a change from a six-coordinate LS to a five-coordinate HS Fe configuration, including loss of the S-Met ligand and elongation of the His–Fe bond length (FIG. 2d). The sensitivity to structure and ligand environment with XAS was complemented with Kβ XES, which gives access to the metal spin state (number of unpaired electrons in the metal-derived 3d orbitals). This provided a 6.1-ps time constant for re-formation of the LS state and a 6.4-ps time constant for heat transfer out of the haem. The study shows that the Fe–(S-Met) bond enthalpy is stronger in the presence of protein constraints, demonstrating how biological systems use subtle differences in energetics to control chemical function of transition-metal systems60. An XES and XAS study on ferric cytochrome c found that haem-doming is also occurring in ferric haems upon photodissociation of bound ligands and results from populating HS states61.
It would be ideal to use multimodal techniques, simultaneously, to minimize systematic errors potentially introduced by varying conditions from each separate experiment, such as varying sample conditions or varying time zero of the optical pump and the X-ray probe. The combination of XES from two different metals18,96–98 or XES with scattering/diffraction is well suited for this approach, as the incident X-ray energy can be freely selected (as long as it is above the absorption edge). We provide examples below of the simultaneous use of these techniques.
A study99 combined XES with X-ray diffuse scattering to monitor the ultrafast electron transfer in a Ru–Co dyad following excitation with a femtosecond pump laser (FIG. 3a). The dyad used is a representative model for many synthetic and natural photocatalysts, with a distance between the electron donor (Ru) and the acceptor (Co) of about 13 Å. While Co Kα XES was used to follow the changes in oxidation state of the Co centre and, hence, the kinetics of electron transfer, the X-ray diffuse scattering signal was used to follow structural changes in the dyad. The Kα XES, in addition to transient optical absorption spectra, revealed that the electron transfer occurs in a two-step mechanism with initial formation of a LS Co(II) species (500 fs) and subsequent conversion to a HS Co(II) within 2 ps. The simultaneously recorded X-ray scattering also revealed a fast change in the Co binding environment by 0.2 Å and showed that charge separation is faster (~2 ps) than the thermalization of the excitation energy with the surroundings, which was observed at around 12 ps (REF.99).
Another example of measurements of XES and XRD was done on Mn/Fe containing RNR18. In this case, XRD and XES of both Mn and Fe were measured, simultaneously, on crystals of RNR in the aerobic MnIIIFeIII state, confirming the oxidation state of the metal centres and showing the undamaged structure of the active site at room temperature (FIG. 3b).
The prime example for the simultaneous use of XES and XRD is the series of studies on PS II crystals57–59,100. PS II is a metalloenzyme that catalyses the light-driven oxidation of water to molecular oxygen in a four-step reaction cycle (FIG. 3c) and has been widely studied using XFELs (REFS57–59,100–105). The catalytic site contains a Mn4CaO5 cluster embedded in the large protein complex. Each step is driven by the absorption of one photon and the Mn4CaO5 cluster acts as a site of charge accumulation to finally catalyse the oxidation of two molecules of water to release molecular oxygen, four protons and four electrons. Samples such as PS II, which are very sensitive to radiation, are extremely difficult to study under functional conditions with X-rays106, but, as discussed above, the ultrashort XFEL pulses can outrun the damage they cause. Combined XRD and XES studies initially confirmed that the Mn4CaO5 cluster in PS II is in an intact, non-reduced state in the microcrystals used for the diffraction measurements57. Mn Kβ XES for the four stable intermediate states of the Mn cluster58 indicated proper in situ turnover of the PS II enzyme under the illumination conditions and allowed to estimate the population of each of the intermediate states in the sample. The XRD obtained in parallel showed that the Mn cluster undergoes a structural rearrangement in the S2 to S3 transition that consists of a widening of the cluster and insertion of an additional oxygen bridge between Mn and Ca to form a Mn4CaO6 cluster, different from earlier results at lower resolution103,104 and confirmed by a second independent study105. Subsequent time-resolved data collection of both XES and XRD59 revealed the time constant of this transition to be around 300 μs based on the XES data and showed that several amino acid residues in the vicinity of the Mn4CaO5 cluster first undergo structural changes, followed by an expansion of the metal cluster and subsequent insertion of the new oxygen bridge. Based on XES, it was possible to conclude that Mn oxidation and insertion of the new oxygen occur concomitantly59 (FIG. 3c).
Soft X-ray spectroscopy at XFELs
Soft X-ray spectroscopy of metalloenzymes and molecular catalysts at XFELs has the advantage of directly probing the local electronic structure of 3d transition metals via the dipole-allowed 2p → 3d transitions at the metal L-edge. The tunability and high intensity of the soft X-ray XFEL radiation are essential to address the dilute metal species in solution. The femtosecond duration of the XFEL pulses is necessary for avoiding X-ray damage and collecting time-resolved spectroscopy data, and this defines the two classes of applications we discuss (FIG. 4). The first is where high-valent metal sites in inorganic molecular complexes and metalloenzymes are studied with L-edge XAS (FIG. 4a–c). In this case, the high-valent metal species are probed with femtosecond pulses before X-ray-induced damage of the sample sets in. The second is time-resolved 2p3d RIXS, without X-ray-induced damage, where the femtosecond resolution in optical pump and X-ray probe experiments gives access to the frontier–orbital interactions in short-lived reaction intermediates (FIG. 4d–i). In this case, RIXS is used to probe ligand-field (LF) and charge-transfer (CT) transitions during molecular transformations and in excited-state dynamics. All examples are photon-in photon-out experiments on liquid samples, relying on the bulk sensitivity of the respective method. Liquid jets are used to prepare the solution samples in the vacuum environment necessary for soft X-rays with fast sample replenishment to avoid X-ray-induced sample damage from consecutive X-ray pulses. Many of the early soft X-ray investigations at XFELs aimed at proving that undistorted signals of intact samples could be measured (REF.4 and references therein). Although, in this Technical Review, we focus on homogeneous transition-metal catalysts, we note that there are other important applications of soft X-ray spectroscopy at XFELs in surface science and heterogeneous catalysis107–111, in photochemistry of small molecules and organic systems in solution112,113, and in the gas phase114–116
Fig. 4 |. Soft X-ray spectroscopy at transition-metal L-edges with X-ray free-electron lasers.

a | Mn L-edge absorption spectra (L3-edge partial fluorescence yield spectra) and structures of non-cubane reduced MnIIMnIII2CaO(OH) (red), non-cubane oxidized MnIII3CaO(OH) (dark blue), closed-cubane MnIV3CaO4 (green, measured in solution, Mn concentrations 6–15 mM) and of MnIII(acac)3 (light blue, measured in solution, Mn concentration 100 mM). b | Energy-level diagram for metal-specific L-edge partial fluorescence yield absorption spectroscopy. c | Top: calculated electron charge density in MnIII(acac)3 (acac = acetylacetonate) as a function of distance from the Mn centre. Bottom: accumulated electron charge and spin densities upon reduction of MnIII(acac)3 and haem a as a function of distance from the Mn and Fe centre, respectively. d | Resonant inelastic X-ray scattering (RIXS) intensities as a function of incident photon energy at the Fe L3-edge and as a function of energy transfer for Fe(CO)5 (left) and averaged for pump-probe delay times of 0–700 fs after optical excitation of Fe(CO)5 (right, measured in solution, Fe concentration 1 M). e | Integrated RIXS intensities as a function of the pump–probe delay time for regions 1 and 2 as marked in part d, with a kinetic model (solid lines) relating measured RIXS intensities and populations of fitted species. f | Energy-level diagram for RIXS at the Fe L3-edge of Fe(CO)5 with optical excitation (metal–ligand charge-transfer (MLCT) excitation initiating dissociation to Fe(CO)4) and frontier–orbital energies and populations in excited-state and triplet-state Fe(CO)4 and Fe(CO)4-ethanol (EtOH) solvent complexes (dπ and dσ denote Fe-centred 3d-derived orbitals of π and σ symmetry, respectively). g | RIXS intensities at the Fe L3-edge of ferric (FeIII) and ferrous (FeII) iron hexacyanide Fe(CN)6 (as measured in solution, Fe concentrations 0.5 and 0.33 M, respectively). h | RIXS intensity differences (pumped minus unpumped) averaged for pump–probe delay times of −70 to 110 fs after optical excitation of FeIII-cyanide, representative of the ligand–metal charge-transfer (LMCT) state, measured in solution with Fe concentration 0.3 M. Integration is for −70 to 110 fs because the temporal resolution in the experiment was 180 fs; saturated intensities are shown for better visualization. i | Energy-level diagram in octahedral (Oh) symmetry for FeIII-cyanide and FeII-cyanide and the LMCT state of ferric hexacyanide (*FeIII) with main RIXS transitions (*, 1 and 2 as indicated in parts g and h and calculated electron charge density difference between LMCT *FeIII-cyanide and FeIII-cyanide (LMCT minus ground state)). HOMO, highest occupied molecular orbital; LUMO, lowest occupied molecular orbital. Part a adapted with permission from REFS14,117. Part c adapted with permission from REFS117,171. Part d adapted with permission from REF.120. Part e adapted with permission from REF.120. Part g adapted with permission from REF.124. Parts h and i adapted with permission from REF.125.
Ligand-field (LF) and charge-transfer (CT) transitions.
The valence orbitals in metal complexes can often be classified as either being metal-centred (large amplitude on the metal) or ligand centred (large amplitude on one or several ligands). LF and CT transitions are electronic excitations in metal complexes. LF excitations, often also denoted d–d excitations, refer to one-electron transitions between metal-centred orbitals. CT excitations correspond to one-electron transitions between metal-centred and ligand-centred orbitals.
Partial fluorescence yield XAS.
Measuring fluorescence as a function of incident photon energy is often denoted partial fluorescence yield X-ray absorption spectroscopy (XAS), and it usually applies to soft X-rays.
X-ray absorption spectroscopy.
The Mn L-edge XAS study14 in FIG. 4a exemplifies the use of XFELs for probe-before-destroy spectroscopy at room temperature of solutions of high-valent Mn species in inorganic complexes and in PS II. The motivation is to study high-valent metal sites in inorganic molecular complexes and metalloenzymes at functional conditions (room-temperature solution) in order to relate their reactivity to the probed electronic structure. Successful implementation of such experiments requires specialized liquid-jet sample delivery, intense femtosecond X-ray pulses and an efficient soft X-ray spectrometer for partial fluorescence yield XAS. Low-consumption liquid jets are needed to prepare the often precious and dilute solutions with typical metal concentrations in the millimolar range16. The femtosecond pulse length is essential to avoid photoreduction and probe the sample before ions, radicals and electrons created by the ionizing soft X-ray radiation in the solvent, ligands or protein environment reduce the high-valent metal centres. As the level diagram in FIG. 4b shows, incident X-rays tuned to metal L-edges also ionize non-resonantly, light elements (C, N, O) in the sample (solvent, protein, ligands), followed by emission of their characteristic X-ray fluorescence. Efficient soft X-ray spectrometers are necessary to discriminate the metal fluorescence of interest from this light-element fluorescence background.
High throughput is essential to address the low (millimolar) concentrations of metals in inorganic catalysts and metalloenzymes. The successful implementation of the example in FIG. 4a at the LCLS XFEL shows that these requirements can all be met. The spectra of the high-valent MnIII and MnIV complexes in FIG. 4a do not contain significant amounts of the most reduced MnII state as present in the mixed-valent MnII–MnIII compound (peak around 639.5 eV). The spectra shift to higher energies with increasing Mn oxidation state, with a blue shift of 1.5–2 eV per unit increase of oxidation state. The investigation also included the Mn L-edge measurements of two states of the water oxidation catalyst Mn4CaO5 in PS II solution and the measured PS II spectra consistently shift, on illumination, to higher oxidation state14. This establishes probe-before-destroy soft X-ray spectroscopy at XFELs and opens the door to a new way of probing the electronic structure in inorganic catalysts and metalloenzymes at functional conditions in room-temperature solutions with soft X-ray L-edge spectroscopy.
Future prospects of X-ray L-edge spectroscopy include gaining more detailed insight into how the metal L-edge XAS spectrum probes charge and spin densities at high-valent metal sites. Quantum-chemical calculations indicate that the local charge at the metal site does not change considerably with changing oxidation state117,118, and the examples in FIG. 4c illustrate this. For the reduction of both the simple MnIII(acac)3, where acac stands for acetylacetonate, complex and the much larger iron haem a complex, the displayed calculations show that the additional charge density effectively spreads over the whole molecule, whereas the added spin density accumulates locally at the metal centre. Relating such calculations of charge and spin density distributions with measured L-edge XAS spectra as done in REF.117 for MnIII(acac)3 (see FIG. 4a for the spectrum of MnIII(acac)3) has led to a new understanding of metal L-edge XAS. With future developments in ab initio theoretical descriptions of the electronic structure and X-ray spectra119, in particular, the ability to describe multinuclear metal centres, a detailed understanding of charge and spin density changes in the redox reactions of inorganic compounds and metalloenzymes will be accessible with soft X-ray L-edge spectroscopy at XFELs.
Charge and spin densities.
Each electron as a particle carries one unit of charge and one unit of spin (alpha/spin up or beta/spin down). in the ensemble averages as described by quantum-chemical calculations, however, charge and spin density distributions in a metal complex or in the active centre of a metalloenzyme can differ, in that alpha (spin up) and beta (spin down) electrons are differently distributed in space.
Resonant inelastic X-ray scattering.
The essence of the optical pump and X-ray probe experiment is depicted in FIG. 4d–f: (REF.120): a MLCT excitation induces dissociation of a ligand from Fe(CO)5 in solution and creates the reactive intermediate Fe(CO)4 (FIG. 4f). As the Fe–CO bond breaks and new bonds are formed in the course of the reaction, covalent metal–ligand interactions change. This process was probed with time-resolved Fe 2p3d RIXS (for the ab initio calculations of X-ray spectra used to assign species; for more details, see REFS89,120–122). The RIXS intensities in FIG. 4d are plotted as a function of incident energy (hvin) versus energy transfer (ΔE = hvin − hvout). The encircled maxima are most informative and they are due to Fe 2p → LUMO(dσ⋆) transitions with inelastic scattering to final states, where ΔE corresponds to HOMO-LUMO (dπ → dσ⋆) excitations (FIG. 4f). ΔE decreases from Fe(CO)5 to Fe(CO)4 as covalent metal–ligand interactions and the HOMO-LUMO energy difference decrease (marked 1 in FIG. 4d, Fe(CO)4 is the most abundant species at pump–probe delays of 0–700 fs). Consistently, as the LUMO energy decreases, the Fe 2p → LUMO(dσ⋆) resonance red shifts. 2p3d RIXS, therefore, probes bonding at the Fe centre via the HOMO-LUMO (dπ → dσ⋆) LF transitions. They are selectively probed and resonantly enhanced by tuning the incident photon energy to the Fe 2p → LUMO (dσ⋆) resonance. A parallel pathway was also observed in which an ethanol solvent molecule attaches to the Fe centre in Fe(CO)4 to form an Fe(CO)4–EtOH solvent complex. This is seen in the RIXS measurement with intensities marked 2 in FIG. 4d: HOMO-LUMO excitation energies are increased compared with 1 because forming the bond in Fe(CO)4-EtOH partially restores the covalent Fe–ligand interactions and pushes apart the HOMO and the LUMO (FIG. 4f, the 2p → LUMO resonance energy is increased consistently as the LUMO energy is increased). In the discussion here, the different spin states of Fe(CO)4 (singlet excited state and triplet, FIG. 4f) are not distinguished in that they both contribute to the circled region 1. However, they can be distinguished by when they arise in the reaction, and the integrated RIXS intensities plotted versus time delay (FIG. 4e) are a measure of how the populations of singlet excited state and triplet Fe(CO)4 (1) and the Fe(CO)4–EtOH solvent complex (2) change over time. The combination of femtosecond 2p3d RIXS with ab initio calculations revealed that the reactivity of the transient intermediate Fe(CO)4 in terms of binding molecules is determined by the population of the LUMO (dσ⋆) orbital120. When it is populated, as in singlet excited state and triplet Fe(CO)4, ligand-to-Fe σ donation is impeded and these species are unreactive (do not bind any molecule) on the probed timescales. Fe(CO)4–EtOH, in turn, is formed by binding ethanol to singlet ground-state Fe(CO)4, which has an empty LUMO orbital and is formed by electronic relaxation. It will be interesting to see in future applications of 2p3d RIXS at XFELs how this approach can be used to understand, and, ultimately, control, the reactivity of new photocatalytic systems in solution.
HOMO-LUMO.
In molecular-orbital theory, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are frontier orbitals of molecules.
Ligand–metal charge-transfer (LMCT) excited state.
LMCT excitations are special cases of charge-transfer excitations in metal complexes. LMCT excited states result from one-electron transitions in which an electron is promoted from a ligand-centred to a metal-centred orbital.
10Dq.
The ligand-field splitting energy separating eg and t2g orbitals in coordination compounds with octahedral (Oh) symmetry.
Building on earlier L-edge XAS work at SR sources123, the studies on iron hexacyanide Fe(CN)6 in solution in FIG. 4g–i exemplify how time-resolved 2p3d RIXS at XFELs can be used to map transient charge distributions124,125. In contrast to the former example, the molecule stays intact, but valence-orbital occupations change. The 2p3d RIXS intensities for ferric (FeIII) and ferrous (FeII) hexacyanide (FIG. 4g) demonstrate how RIXS reflects valence-orbital occupations in the ground states of the two compounds (valence configurations of the Fe-centred orbitals are t2g5eg0 for FeIII-hexacyanide and t2g6eg0 for FeII-hexacyanide, both with octahedral, Oh, symmetry). The hole in the t2g orbital in ferric FeIII-hexacyanide (FIG. 4i) gives rise to a 2p → t2g resonance that is missing in ferrous FeII-hexacyanide, whereas both compounds exhibit 2p → eg and 2p → π⋆ resonances. 2p3d RIXS in this example probes the t2g → eg LF transitions (marked 1 in FIG. 4g–i) and the t2g → π⋆ CT transitions (marked 2 in FIG. 4g,i). By tuning the incident photon energy, the LF (1) or CT transitions (2) can be resonantly enhanced (evidently, ligand σ/π to t2g excitations only appear in FeIII-hexacyanide, marked ⋆ in FIG. 4g,i). FIGURE 4h shows how the Fe 2p3d RIXS intensities change when going from the ground state to the ligand–metal charge-transfer (LMCT) excited state of FeIII-cyanide, as generated by optical excitation. Femtosecond resolution was needed here to catch the transient intermediate LMCT state. As can be seen in the calculated charge densities in FIG. 4i, where charge decreases on two ligands and charge increases at the Fe centre, LMCT excitation of FeIII-hexacyanide promotes an electron from a ligand σ orbital to the Fe-centred t2g orbital, resulting in the LMCT excited-state configuration ⋆FeIII t2g6eg0. In the time-resolved RIXS measurement, the signatures of ground-state FeIII-hexacyanide decrease (blue in FIG. 4h, the 2p → t2g resonance disappears in particular, as the t2g shell is filled) and the new intensities (red) characterize the LMCT ⋆FeIII excited state. Since both FeII-hexacyanide and LMCT ⋆FeIII-hexacyanide have the same t2g6eg0 configuration, these intensities measure the transient effect of the ligand σ hole (FIG. 4i). In LMCT ⋆FeIII-hexacyanide, the 2p → eg resonance (1) is redshifted by 1 eV compared with FeII-hexacyanide, and this indicates that the 2p core-hole is better screened in the XAS final states of LMCT ⋆FeIII-hexacyanide. This stabilization of the 2p core-excited states indicates that the charge density locally at the Fe is higher in LMCT ⋆FeIII-hexacyanide compared with FeII-hexacyanide. Indeed, calculations confirm that the ligand σ hole in LMCT ⋆FeIII-hexacyanide induces an increase of charge density at Fe (REF.125). Calculations also suggest that this charge increase is due to an increase of ligand-to-Fe σ donation. This strengthens the Fe–ligand bond and increases the LF strength 10Dq (REFS125,126). 2p3d RIXS measures this by an increase of the t2g → eg transition energy of 0.3 eV in LMCT ⋆FeIII-hexacyanide compared with FeII-hexacyanide (1, FIG. 4g,h). Such detailed insight into transient charge distributions made accessible by time-resolved 2p3d RIXS could prove essential in the development of efficient photocatalysts by probing, for example, the effects of manipulating ligands in metal complexes to control excited-state dynamics127 or by resolving the CT dynamics between metal complexes and solid-state surfaces in dye-sensitized semiconductor devices.
The element-specific, site-specific and orbital-specific access to photochemical reaction mechanisms as demonstrated in these experiments enables correlating orbital symmetry and interactions, spin multiplicity and reactivity of metal compounds in solution. In future applications, this ability promises unique insight into the photochemical reaction dynamics of fundamentally important metal complexes, functional photocatalysts and dilute bioinorganic systems. High-repetition-rate XFELs, such as the European XFEL and LCLS-II, will enable systematic soft X-ray spectroscopy of functional systems in solution at the millimolar concentration level at metal L-edges with XAS and 2p3d RIXS.
New capabilities and methods at future XFELs
In the previous two sections, we focused on linear X-ray spectroscopy methods, in which the response of the signal of interest is independent of the X-ray intensity. Nonlinear X-ray spectroscopy accesses new aspects of matter, where the response depends on the X-ray intensity. These new forms of X-ray spectroscopy build upon the developments in nonlinear optics,and offer qualitative new scientific insights through element specificity, atomic spatial resolution and sub-femtosecond time resolution. In the following, we highlight a few emerging nonlinear X-ray spectroscopy methods that are particularly promising for a deeper understanding of molecular catalysts and metalloenzymes. Note that this section is not a comprehensive coverage of the emerging field of nonlinear X-ray science that has been opened by XFELs, which includes advances in nonlinear scattering128–130, two-photon absorption131, second-harmonic generation132 and transient-grating methods133–135, to name a few.
Amplified spontaneous emission.
The spontaneous emission in an ensemble of atoms or molecules, the majority of which are in an electronic excited state. In this scenario, initial spontaneous emission events trigger subsequent stimulated emission events along the propagation path, and the resulting amplified spontaneous emission signal grows exponentially.
Seeded stimulated emission.
A variation of the stimulated emission process, in which the incident photon is provided in the form of a coherent pulse. Photons with a specific wavelength stimulate the emission of photons with that same wavelength.
Spontaneous emission.
The process in which an atom or molecule in an electronic excited state relaxes to a lower-energy electronic state through the emission of a photon.
Core-hole lifetime broadening.
The (homogeneous) energy broadening of a core transition due to the finite lifetime of the core-hole. For a Lorentzian line shape, this can be expressed as ΔEFWHMΔT1/e = 0.6589 eV fs, where ΔEFWHM is the full width at half maximum of the linewidth and ΔT1/e is the exponential lifetime (also referred to as dephasing time) of the corresponding core-excited state.
Stimulated XES: extracting VtC, Kβ, soft XES and other weak emission lines.
Spontaneous XES is a powerful element-specific probe, but is subject to relatively weak emission cross sections, and photons are emitted in all directions. Stimulating the emission offers three potential advantages: signals are emitted along a well-defined X-ray beam direction, enabling efficient collection of the entire signal; strong signal enhancements that can out-compete other decay channels; spectral narrowing and selective stimulation may result in enhanced chemical sensitivity.
Strongly enhanced stimulated Kα emission and RIXS has been observed in Ne gas136,137 and in 3d transition-metal systems138,139. The two main approaches are amplified spontaneous emission (ASE)136,138,139 and seeded stimulated emission137,138 (FIG. 5a). In ASE, an intense X-ray pump creates a population inversion along the propagation path. Spontaneously emitted photons along the population inversion path can stimulate the emission of additional photons at the same wavelength and direction (FIG. 5a), resulting in an ASE signal that grows exponentially. Kα ASE can be used for spectroscopy, as it exhibits chemical shifts and strong spectral gain narrowing139. To stimulate the spectroscopically more sensitive Kβ and VtC emission lines, a second-colour seed pulse is tuned to the respective energy (FIG. 5a). The seeding approach is required because Kα amplification always dominates the weaker Kβ lines.
Fig. 5 |. Examples of future X-ray free-electron laser-based nonlinear spectroscopy methods.

a | Level diagram of various transition-metal K emission lines (top left) and schematics of amplified spontaneous emission (top right) and seeded stimulated emission (bottom right). The pump pulse is tuned above the K-edge to create 1s core-hole excited states in the sample (red dots). In amplified spontaneous emission, a randomly forward emitted photon stimulates emission when encountering an excited atom, leading to amplification along the path of atoms in the excited state, as created by the pump pulse. In seeded stimulated emission, the seed pulse energy is tuned to that of the emission line that one wants to enhance along the pump or seed pulse direction. Bottom left: single-shot seeded Kβ stimulated X-ray emission spectroscopy (S-XES) spectrum (red line) of NaMnO4 compared with conventional X-ray emission spectroscopy (XES) spectrum (blue line), showing the potential for spectral narrowing in S-XES. b | Level diagram and calculated valence-to-core (VtC) spectra in metal–ligand double-core-hole XES. Two photons from the X-ray free-electron laser pulse simultaneously create a double-core-hole (DCH) state, with one core-hole in the metal and one in the ligand. c | Calculated DCH spectra with N and Cl are compared with the conventional single-core-hole (SCH) VtC spectrum of MnII(terpy)Cl2 (terpy=2,2′:6′,2˝-terpyridine). d,e | Level diagram (part d) and representative calculated 2D X-ray core-hole correlation spectroscopy maps (part e) for the para and ortho isomers of aminophenol. The degree of orbital mixing of the N–2p and O–2p valence states gives rise to a nonlinear mixing of the polarizations associated with resonant excitation from the O–1s and N–1s core levels. The off-diagonal cross peaks (right map) indicate the enhanced mixing in the ortho conformation (effectively, mixing of the N–1s and O–1s X-ray absorption near edge structure (XANES) spectra). Such quantum effects are absent in the para isomer due to the separation of the O and N atoms. f | Generalized schematic of X-ray core-hole correlation spectroscopy using a four-wave mixing geometry with a three-pulse sequence (k1, k2, k3). The signal of interest is the nonlinear polarization, kS, shown here resolved in frequency (energy) using a spectrometer. The other energy axis (ω12) is determined by the Fourier transform of the signal with respect to the time delays of the first two phase-locked pulses. Part a adapted with permission from REF.140. Parts b and c adapted with permission from REF.159. Parts d and e adapted with permission from REF.162.
Seeding of Kβ emission from a NaMnO4 solution with an enhancement of more than 105 compared with spontaneous emission into the same solid angle was reported140, with strong spectral narrowing (FIG. 5a). Although these studies were not performed under optimal conditions, they show that seeded stimulated emission is, potentially, a powerful tool. Besides the practical advantages of larger signal and outcompeting other decay channels, we envision using seeded stimulated emission to enhance individual spectral features characteristic of the electronic structure of a species of interest, while potentially decreasing the core-hole lifetime broadening. Tuning to resonant features at the absorption edge can widen this approach to stimulated RIXS137. The enhanced spectral sensitivity and control will widen XFEL-based spectroscopy to reveal subtle electronic structure changes that are currently below the detection limit. Such studies will include complex systems and ultrafast intersystem crossings in 3d metal centres of light-harvesting and photocatalytic molecules.
Metal–ligand double-core-hole spectroscopy.
An important challenge for photocatalysis is understanding coupled valence states (where metal and ligand orbitals mix) that mediate charge separation and reactivity. A powerful element-specific approach relies on the simultaneous creation of two core-level transitions in adjacent atoms, for example, one from a metal and one from a bonded ligand atom or metal (FIG. 5). Theoretical studies of the creation of such double-core-hole (DCH) states date back to 1986, when it was predicted that the binding energies associated with DCHs at different atomic sites of small molecules could sensitively probe the chemical environment of the ionized atoms141. Since then, most theoretical studies142–146, synchrotron experiments147–152 (where the DCH states were created by one photon ejecting two core electrons) and XFEL experiments153–158 (where two photons create the DCH state) have focused on probing the chemical environment of atoms in small gas-phase molecules.
The application of DCH spectroscopy to 3d transition-metal compounds has been discussed in a theoretical study of VtC-XES in mononuclear Mn complexes and binuclear Co complexes with metal–metal direct bonds159. For the mononuclear Mn complexes, the VtC-XES signals of the metal-1s/ligand-1s DCH states, and, for the binuclear Co complexes, the VtC-XES signals for the two-site Fe and Co 1s DHC states were calculated and compared with the conventional single-core-hole (SCH) VtC-XES signals. Figure 5b,c show the schematics of DCH VtC-XES and a comparison of the DCH and the SCH VtC-XES signals of these systems.
Calculations show that, in the mononuclear Mn complexes, the XES signal of the metal-1s/ligand-1s DCH states reveals additional information about the chemical bonds between the metal centre and the coordinating atoms. Similarly, the two-site 1s DHC states in dinuclear compounds show spectral changes of features, including those related to ligand orbitals, as compared with the SCH states. The size of these shifts also reveals that, in the dinuclear Co complex, the metal–ligand interaction is stronger than that in the dinuclear Fe complex. These simulations show that, through the perturbation introduced by a second core-hole near the studied core-hole, DCH VtC-XES can go beyond the conventional SCH VtC-XES techniques and provide further information on the local electronic structure of the core-holes and, especially, the interaction between the two atoms with core-holes. DCH VtC-XES has the potential to become a new research tool in transition-metal-complex chemistry. In the future, it may be possible to extend the DCH approach to RIXS and combine it with stimulated emission discussed in the previous section.
Wave-mixing and multidimensional spectroscopy.
In addition to stimulated emission and DCH methods, new nonlinear and multidimensional X-ray spectroscopies will provide important new insight into functioning photocatalysts by mapping coherent charge flow and energy relaxation on fundamental timescales, with access to the full range of valence states (unrestricted by dipole selection rules). The element specificity of core-level X-ray resonances can reveal the charge and energy flow between constituent atoms in molecular complexes. This insight is not achievable with conventional optical ultrafast probes, and will provide critical insight into molecular catalysts with strong coupling between electronic and nuclear dynamics. High-repetition-rate XFEL sources will be essential to realize the potential of these new approaches by enabling experiments in the perturbative small-signal regime, with pulses of lower peak power, thereby, avoiding the complications from higher-order nonlinearities that arise in the strongly nonlinear regime.
One promising example of this class of experiments is X-ray core-hole correlation spectroscopy (XCCS) (FIG. 5d–f). This approach probes the degree of mixing of excited molecular orbitals (correlations between valence-excited states) associated with different atomic sites in a molecule160–162 and is analogous to 2D electronic spectroscopy161,163,164, but with the element specificity of X-rays.
FIGURE 5 presents an example in aminophenol where XCCS probes the orbital mixing (quantum coupling) between nitrogen-associated and oxygen-associated valence states in different isomers. Here, two pulses probe the aminophenol molecule in resonance with the N−1s (ħωN ~400 eV, where ωN is the N−1s transition frequency) and the O−1s (ħωO ~535 eV, where ωO is the O−1s transition frequency) core excitations, respectively. In a coherent four-wave mixing implementation, the initial excitation is created by a pulse pair, and a third pulse (in a phase-matched geometry) reads out the scattered Raman signal. Thus, XCCS measures a third-order, χ(3), four-wave mixing process, whereby a sequence of three incident pulses (three fields), En(kn,ωn) (n = 1, 2, 3) (of wavenumber kn and frequency ωn) generates a stimulated signal, Esig(−ω1 + ω2 + ω3), in the momentum-matched direction, ksig = −k1 + k2 + k3. A 2D spectral map of the valence electronic structure is created consisting of the emission signal as a function of the excitation frequency, with one axis, ωS (signal frequency), determined by the spectrum of the emitted signal (measured with a dispersive X-ray spectrometer) and the other axis, ω12 (excitation frequency = 1/t12), determined by the Fourier transform of the signal with respect to the time delays of the first two phase-locked pulses. (Note that sub-femtosecond phase-lock pulse pairs in the X-ray regime have been demonstrated at LCLS40.) The presence of off-diagonal features in this 2D map reflects the degree of orbital mixing between the two excited valence states associated with the N and O atoms; no signal should be seen in the Hartree–Fock limit of independent orbitals. Calculations show that the extent of this orbital mixing depends not only on molecular structure (meaning that it is stronger in ortho-aminophenol with adjacent orbitals, compared with the para conformation) but also on the nature of the molecular orbitals excited within the energy envelopes (~10 eV) of ωN and ωO (REFS160,162).
Quantum coupling.
The coupling of electronic wavefunctions. As used here, this refers to the mixing of molecular orbitals.
Four-wave mixing.
A nonlinear optical (X-ray) process involving four electromagnetic fields (for example, three input fields E1, E2 and E3, contribution to the creation of a new polarization, P). In a formulation that describes the susceptibility χnonlinear in terms of a perturbation expansion, four-wave mixing is a third-order process: P(3)=χ(3)e1E2E3.
As discussed before, in the soft X-ray range, an incident fluence of ~1017 photons cm−2 can lead to appreciable nonlinear effects. In the case of XCCS, the pulse duration must be comparable to (or less than) the order of the core-hole lifetime (~4 fs for low-Z elements68,69), since Auger decay suppresses the correlation signal of interest. This corresponds to ~109 photons per pulse in a 1-μm focus (~1015 W cm−2 at 500 eV). Estimates based on X-ray nonlinear susceptibilities lead to similar conclusions for the required X-ray peak power density162,165. Note that modern nonlinear optical spectroscopy experiments typically operate in the perturbative regime, ~0.1 photons per cross section, in order to avoid the distortion of the spectral signal of interest from higher-order nonlinear processes, saturation, nonlinear waveguiding and other undesirable effects. This is important in order to extract readily interpretable new scientific information about the complex under study, and is in contrast with the strongly nonlinear regime, where signals may be larger but much more complicated and difficult to interpret.
Outlook
The first decade of XFEL science has brought us many striking results and the emergence of exciting new research areas20,24–26,166,167. In this Technical Review, we have discussed new XFEL-based spectroscopy methods on metalloenzymes and molecular catalysts, and their future opportunities. There has been tremendous progress in every aspect of XFEL research over the last decade, and much work still lies ahead when it comes to making the best use of this powerful tool. Many research techniques are not yet fully optimized and, compared with most synchrotron experiments, most XFEL experiments are still complex undertakings. As the performance parameters of the current and the next generation of XFELs have been defined, we believe that addressing three critical areas will help in optimizing future XFEL science on metalloenzymes and molecular catalysts:
Improved instrumentation for XFEL experiments, including beam diagnostics, pulse control, X-ray optics, sample synthesis and delivery, and data acquisition and control systems. The large shot-by-shot variations of XFEL pulses require precise diagnostics for data normalization and arrival-time jitter, especially when measuring small spectral changes. XFEL facilities are actively working on improving such diagnostics and enhancing the pulse control to reduce the fluctuations. Making the best use of each XFEL pulse requires to increase further the photon-out signal, including more efficient optics and detectors, and better sample quality and delivery. We estimate that there is an order of magnitude in signal that can be gained once these parameters are optimized.
Development of a more complete theoretical description of the systems under study, the interaction of pump–probe pulses with the sample and the resulting spectral or diffraction features. Applications of ab initio theories to effectively describe the electronic structure and X-ray spectra of metal complexes and metalloenzymes are rapidly evolving (see REF.119 and references therein). For the purposes discussed in this Technical Review, they seem best suited to relate X-ray spectroscopic observables and mechanistic understanding of reactivity in catalysis. Enhanced computational power and more efficient descriptions will enable the improvement of such theories and simulations that match the improved experiments. Challenges include effective descriptions of femtosecond excited-state molecular dynamics of transition-metal systems, efficient multiconfigurational approaches to the valence and core electronic structure in transition-metal complexes and photocatalysts with numerous 3d electrons and open shells, and the efficient description of bonding, orbital interactions and X-ray spectra in large inorganic systems and metalloenzymes, including those with multinuclear metal centres119.
Global analysis procedures for both spectroscopic and diffraction data for multicomponent evolution in time are, at present, rudimentary and will need to be developed, probably making use of the emerging fields of machine learning and large-scale computing. This could allow to identify and correlate large data sets exhibiting small changes with subtle and/or complex time-dependent chemical and structural changes of the system under investigation. Advancements in machine learning algorithms have made a dramatic surge in recent years in many areas of science and technology. We need to learn how to develop similar approaches, to efficiently analyse and reduce our large and complex data sets that might contain 109 individual spectra from a single day of running an XFEL experiment. This is not simply doing the same analysis faster or on a larger scale but requires spawning entirely new approaches that will extract the mechanistic understanding of catalytic reactions from the large XFEL data sets168.
Understanding the function of metalloenzymes and molecular catalysts will greatly benefit from current and future XFELs and the X-ray methods described above. We hope that this Technical Review stimulates the readers to think about how these approaches can best benefit their research needs, and how they can be enhanced with the ultimate goal of bringing out the full potential of these powerful machines.
Key points.
Femtosecond pulses from X-ray free-electron lasers have unique characteristics that enable X-ray spectroscopy to follow catalytic reactions at the metal centres in chemical and biological systems under functional conditions and in real time.
Hard X-ray spectroscopy (>5 keV) is used to study transitions from and to the 1s shell (K-edge) and valence electron orbitals of transition metals involved in catalysis to uncover the geometric and electronic structure of the metal centres.
Soft X-ray spectroscopy (<1 keV), transitions from and to the 2p shell (L-edge) of transition metals to the valence orbitals, is used to probe the charge and spin distribution and the degree of covalency of bonds, all of which are critical properties for transition-metal-based catalysis.
Nonlinear spectroscopic methods, well established in optical and magnetic resonance energy domains, are now being developed in the X-ray domain.
Acknowledgements
J.K., V.K.Y. and J.Y. thank the support from the Director, Office of Science, Office of Basic Energy Sciences (OBES), Division of Chemical Sciences, Geosciences, and Biosciences (CSGB) of the Department of Energy (DOE) under contract DE-AC02–05CH11231. The National Institutes of Health (NIH) provides funding through grants GM126289 (J.K.) and GM110501 (J.Y.) for instrumentation development for XFEL experiments and metalloenzyme studies, and GM055302 (V.K.Y.) for biochemical aspects of PS II research. This work was supported by the US Department of Energy, Office of Science, Basic Energy Sciences under contract no. DEAC02– 76SF00515 (U.B., R.W.S.). P.W. is grateful to Raphael Jay for providing access to some of the data in Fig. 4 and for fruitful discussions.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Physics thanks Makina Yabashi, Majed Chergui and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Röntgen WC On a new kind of rays. Science 3, 227–231 (1896). [DOI] [PubMed] [Google Scholar]
- 2.Dam HJW in McClure’s Magazine Vol. 6 (McClure SS, 1896). [Google Scholar]
- 3.Watson JD & Crick FHC Molecular structure of nucleic acids - a structure for deoxyribose nucleic acid. Nature 171, 737–738 (1953). [DOI] [PubMed] [Google Scholar]
- 4.Rossbach J, Schneider JR & Wurth W 10 years of pioneering X-ray science at the free-electron laser FLASH at DESY. Phys. Rep 808, 1–74 (2019). [Google Scholar]
- 5.Garman EF Radiation damage in macromolecular crystallography: what is it and why should we care? Acta Cryst. D Biol. Crystallogr 66, 339–351 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.March AM et al. Development of high-repetition-rate laser pump/x-ray probe methodologies for synchrotron facilities. Rev. Sci. Instrum 82, 073110 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Borfecchia E, Garino C, Salassa L & Lamberti C Synchrotron ultrafast techniques for photoactive transition metal complexes. Philos. Trans. A Math. Phys. Eng. Sci 371, 20120132 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Smolentsev G et al. Pump-probe XAS investigation of the triplet state of an Ir photosensitizer with chromenopyridinone ligands. Photochem. Photobiol. Sci 17, 896–902 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Chaussavoine I et al. The microfluidic laboratory at Synchrotron SOLEIL. J. Synchrotron Radiat. 27, 230–237 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Monteiro DCF et al. 3D-MiXD: 3D-printed X-ray-compatible microfluidic devices for rapid, low-consumption serial synchrotron crystallography data collection in flow. IUCrJ 7, 207–219 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neutze R, Wouts R, van der Spoel D, Weckert E & Hajdu J Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 406, 752–757 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Chapman HN et al. Femtosecond X-ray protein nanocrystallography. Nature 470, 73–77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alonso-Mori R et al. Energy-dispersive X-ray emission spectroscopy using an X-ray free-electron laser in a shot-by-shot mode. Proc. Natl Acad. Sci. USA 109, 19103–19107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kubin M et al. Soft x-ray absorption spectroscopy of metalloproteins and high-valent metal-complexes at room temperature using free-electron lasers. Struct. Dyn 4, 054307 (2017).This Mn L-edge soft X-ray spectroscopic study of photosystem II at physiological (in operando) conditions based on measurements at the LCLS XFEL establishes protocols for probe-before-destroy spectroscopy of dilute high-valent metal complexes and proteins at XFELs.
- 15.Weierstall U Liquid sample delivery techniques for serial femtosecond crystallography. Philos. Trans. R. Soc. Lond. B Biol. Sci 369, 20130337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sierra RG et al. Concentric-flow electrokinetic injector enables serial crystallography of ribosome and photosystem II. Nat. Methods 13, 59–62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DePonte D in X-Ray Free Electron Lasers: Applications in Materials, Chemistry and Biology (Energy and Environment Series) Ch. 16 (eds Bergmann U, Yachandra VK & Yano J) 325–336 (Royal Society of Chemistry, 2017). [Google Scholar]
- 18.Fuller FD et al. Drop-on-demand sample delivery for studying biocatalysts in action at X-ray free-electron lasers. Nat. Methods 14, 443–449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martiel I, Müller-Werkmeister HM & Cohen AE Strategies for sample delivery for femtosecond crystallography. Acta Cryst. D Struct. Biol 75, 160–177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bostedt C et al. Linac coherent light source: the first five years. Rev. Mod. Phys 88, 015007 (2016). [Google Scholar]
- 21.Zhu D et al. A single-shot transmissive spectrometer for hard x-ray free electron lasers. Appl. Phys. Lett 101, 034103 (2012). [Google Scholar]
- 22.Harmand M et al. Achieving few-femtosecond time-sorting at hard X-ray free-electron lasers. Nat. Photonics 7, 215–218 (2013). [Google Scholar]
- 23.Tono K et al. Beamline, experimental stations and photon beam diagnostics for the hard x-ray free electron laser of SACLA. New J. Phys 15, 083025 (2013). [Google Scholar]
- 24.Elsaesser T Introduction: ultrafast processes in chemistry. Chem. Rev 117, 10621–10622 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Marangos JP The measurement of ultrafast electronic and structural dynamics with X-rays. Philos. Trans. A Math. Phys. Eng. Sci. 377, 20170481 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asakura K, Gaffney KJ, Milne C & Yabashi M XFELs: cutting edge X-ray light for chemical and material sciences. Phys. Chem. Chem. Phys 22, 2612–2614 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Milne CJ, Penfold TJ & Chergui M Recent experimental and theoretical developments in time-resolved X-ray spectroscopies. Coord. Chem. Rev 277–278, 44–68 (2014). [Google Scholar]
- 28.No authors listed. The next decade of XFELs. Nat. Rev. Phys 2, 329 (2020). [Google Scholar]
- 29.Kondratenko AM & Saldin EL Generation of coherent radiation by a relativistic electron beam in an undulator. Part. Accel 10, 207–216 (1980). [Google Scholar]
- 30.Bonifacio R, Pellegrini C & Narducci LM Collective instabilities and high-gain regime in a free-electron laser. Opt. Commun 50, 373–378 (1984). [Google Scholar]
- 31.Allaria E et al. The FERMI free-electron lasers. J. Synchrotron Radiat. 22, 485–491 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Ishikawa T et al. A compact X-ray free-electron laser emitting in the sub-angstrom region. Nat. Photon 6, 540–544 (2012). [Google Scholar]
- 33.Ko IS et al. Construction and commissioning of PAL-XFEL facility. Appl. Sci 7, 479 (2017). [Google Scholar]
- 34.Prat E et al. A compact and cost-effective hard X-ray free-electron laser driven by a high-brightness and low-energy electron beam. Nat. Photonics 14, 748–754 (2020). [Google Scholar]
- 35.Ayvazyan V et al. First operation of a free-electron laser generating GW power radiation at 32 nm wavelength. Eur. Phys. J. D 37, 297–303 (2006). [Google Scholar]
- 36.Tschentscher T et al. Photon beam transport and scientific instruments at the European XFEL. Appl. Sci 7, 592 (2017). [Google Scholar]
- 37.SLAC National Accelerator Laboratory. LCLS-II Conceptual Design Report, 519 pp (2014). [Google Scholar]
- 38.SLAC National Accelerator Laboratory. Linac Coherent Light Source II High Energy Upgrade (LCLS-II-HE) Conceptual Design Report (SLAC-R-1098) (2017). [Google Scholar]
- 39.Huang S et al. Generating single-spike hard X-ray pulses with nonlinear bunch compression in free-electron lasers. Phys. Rev. Lett 119, 154801 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Duris J et al. Tunable isolated attosecond X-ray pulses with gigawatt peak power from a free-electron laser. Nat. Photonics 14, 30–36 (2019). [Google Scholar]
- 41.Amann J et al. Demonstration of self-seeding in a hard-X-ray free-electron laser. Nat. Photonics 6, 693–698 (2012). [Google Scholar]
- 42.Allaria E et al. Highly coherent and stable pulses from the FERMI seeded free-electron laser in the extreme ultraviolet. Nat. Photonics 6, 699–704 (2012). [Google Scholar]
- 43.Allaria E et al. Two-colour pump–probe experiments with a twin-pulse-seed extreme ultraviolet free-electron laser. Nat. Comm 4, 2476 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ackermann S et al. Generation of coherent 19- and 38-nm radiation at a free-electron laser directly seeded at 38 nm. Phys. Rev. Lett 111, 114801 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Ratner D et al. Experimental demonstration of a soft x-ray self-seeded free-electron laser. Phys. Rev. Lett 114, 054801 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Gauthier D et al. Generation of phase-locked pulses from a seeded free-electron laser. Phys. Rev. Lett 116, 024801 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Finetti P et al. Pulse duration of seeded free-electron lasers. Phys. Rev. X 7, 021043 (2017). [Google Scholar]
- 48.Inoue I et al. Generation of narrow-band X-ray free-electron laser via reflection self-seeding. Nat. Photonics 13, 319–322 (2019). [Google Scholar]
- 49.Hara T et al. Two-colour hard X-ray free-electron laser with wide tunability. Nat. Commun 4, 2919 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Lutman AA et al. Experimental demonstration of femtosecond two-color x-ray free-electron lasers. Phys. Rev. Lett 110, 134801 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Marinelli A et al. Multicolor operation and spectral control in a gain-modulated X-ray free-electron laser. Phys. Rev. Lett 111, 134801 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Lutman AA et al. Demonstration of single-crystal self-seeded two-color X-ray free-electron lasers. Phys. Rev. Lett 113, 254801 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Marinelli A et al. High-intensity double-pulse X-ray free-electron laser. Nat. Commun 6, 6369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prince KC et al. Coherent control with a short-wavelength free-electron laser. Nat. Photonics 10, 176–179 (2016). [Google Scholar]
- 55.Hartmann N et al. Attosecond time–energy structure of X-ray free-electron laser pulses. Nat. Photonics 12, 215–220 (2018). [Google Scholar]
- 56.Kraus PM, Zurch M, Cushing SK, Neumark DM & Leone SR The ultrafast X-ray spectroscopic revolution in chemical dynamics. Nat. Rev. Chem 2, 82–94 (2018). [Google Scholar]
- 57.Kern J et al. Simultaneous femtosecond X-ray spectroscopy and diffraction of photosystem II at room temperature. Science 340, 491–495 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kern J et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 563, 421–425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ibrahim M et al. Untangling the sequence of events during the S2 → S3 transition in photosystem II and implications for the water oxidation mechanism. Proc. Natl Acad. Sci. USA 117, 12624–12635 (2020).The application of combined X-ray crystallography and X-ray emission spectroscopy at an XFEL to understand the sequence of events during the water oxidation reaction in photosystem II.
- 60.Mara MW et al. Metalloprotein entatic control of ligand-metal bonds quantified by ultrafast x-ray spectroscopy. Science 356, 1276–1280 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bacellar C et al. Spin cascade and doming in ferric hemes: Femtosecond X-ray absorption and X-ray emission studies. Proc. Natl Acad. Sci. USA 117, 21914–21920 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kinschel D et al. Femtosecond X-ray emission study of the spin cross-over dynamics in haem proteins. Nat. Commun 11, 4145 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schreck S et al. Reabsorption of soft x-ray emission at high x-ray free-electron laser fluences. Phys. Rev. Lett 113, 153002 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Szlachetko J et al. Establishing nonlinearity thresholds with ultraintense X-ray pulses. Sci. Rep 6, 33292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tamasaku K et al. X-ray two-photon absorption competing against single and sequential multiphoton processes. Nat. Photonics 8, 313–316 (2014). [Google Scholar]
- 66.Spence JCH Outrunning damage: Electrons vs X-rays—timescales and mechanisms. Struct. Dyn 4, 044027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alonso-Mori R et al. Femtosecond electronic structure response to high intensity XFEL pulses probed by iron X-ray emission spectroscopy. Sci. Rep 10, 16837 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krause MO & Oliver JH Natural widths of atomic K and L levels, Kα X-ray lines and several KLL Auger lines. J. Phys. Chem. Ref. Data 8, 329–338 (1979). [Google Scholar]
- 69.Neeb M, Rubensson JE, Biermann M & Eberhardt W Coherent excitation of vibrational wave functions observed in core hole decay spectra of O2, N2 and CO. J. Electron. Spectrosc. Relat. Phenom 67, 261–274 (1994). [Google Scholar]
- 70.Zhang W et al. Tracking excited-state charge and spin dynamics in iron coordination complexes. Nature 509, 345–348 (2014).Demonstrates that femtosecond time resolution Fe K-edge X-ray emission spectroscopy can be used to investigate ultrafast spin-crossover dynamics in transition-metal systems resulting from photoinduced metal–ligand charge-transfer excitation.
- 71.Haldrup K et al. Observing solvation dynamics with simultaneous femtosecond X-ray emission spectroscopy and X-ray scattering. J. Phys. Chem. B 120, 1158–1168 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Kubin M et al. Direct determination of absolute absorption cross sections at the L-edge of dilute Mn complexes in solution using a transmission flatjet. Inorg. Chem 57, 5449–5462 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fransson T et al. X-ray emission spectroscopy as an in situ diagnostic tool for X-ray crystallography of metalloproteins using an X-ray free-electron laser. Biochemistry 57, 4629–4637 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Srinivas V et al. High resolution XFEL structure of the soluble methane monooxygenase hydroxylase complex with its regulatory component at ambient temperature in two oxidation states. J. Am. Chem. Soc 142, 14249–14266 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang W et al. Manipulating charge transfer excited state relaxation and spin crossover in iron coordination complexes with ligand substitution. Chem. Sci 8, 515–523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kjaer KS et al. Ligand manipulation of charge transfer excited state relaxation and spin crossover in [Fe(2,2′-bipyridine)2(CN)2]. Struct. Dyn 4, 044030 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kjaer KS et al. Finding intersections between electronic excited state potential energy surfaces with simultaneous ultrafast X-ray scattering and spectroscopy. Chem. Sci 10, 5749–5760 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tatsuno H et al. Hot branching dynamics in a light-harvesting iron carbene complex revealed by ultrafast X-ray emission spectroscopy. Angew. Chem. Int. Ed. Engl 59, 364–372 (2020). [DOI] [PubMed] [Google Scholar]
- 79.Kunnus K et al. Vibrational wavepacket dynamics in Fe carbene photosensitizer determined with femtosecond X-ray emission and scattering. Nat. Commun 11, 634 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vacher M, Kunnus K, Delcey MG, Gaffney KJ & Lundberg M Origin of core-to-core x-ray emission spectroscopy sensitivity to structural dynamics. Struct. Dyn 7, 044102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Glatzel P & Bergmann U High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes—electronic and structural information. Coord. Chem. Rev 249, 65–95 (2005). [Google Scholar]
- 82.Pollock CJ & DeBeer S Insights into the geometric and electronic structure of transition metal centers from valence-to-core X-ray emission spectroscopy. Acc. Chem. Res 48, 2967–2975 (2015). [DOI] [PubMed] [Google Scholar]
- 83.Katayama T et al. A versatile experimental system for tracking ultrafast chemical reactions with X-ray free-electron lasers. Struct. Dyn 6, 054302 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ledbetter K et al. Excited state charge distribution and bond expansion of ferrous complexes observed with femtosecond valence-to-core x-ray emission spectroscopy. J. Chem. Phys 152, 074203 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Katayama T et al. Femtosecond x-ray absorption spectroscopy with hard x-ray free electron laser. Appl. Phys. Lett 103, 131105 (2013). [Google Scholar]
- 86.Lemke HT et al. Femtosecond X-ray absorption spectroscopy at a hard X-ray free electron laser: application to spin crossover dynamics. J. Phys. Chem. A 117, 735–740 (2013). [DOI] [PubMed] [Google Scholar]
- 87.Levantino M et al. Observing heme doming in myoglobin with femtosecond X-ray absorption spectroscopy. Struct. Dyn 2, 041713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barends TR et al. Direct observation of ultrafast collective motions in CO myoglobin upon ligand dissociation. Science 350, 445–450 (2015). [DOI] [PubMed] [Google Scholar]
- 89.Wernet P Chemical interactions and dynamics with femtosecond X-ray spectroscopy and the role of X-ray free-electron lasers. Philos. Trans. A Math. Phys. Eng. Sci 377, 20170464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Katayama T et al. Tracking multiple components of a nuclear wavepacket in photoexcited Cu(I)-phenanthroline complex using ultrafast X-ray spectroscopy. Nat. Commun 10, 3606 (2019).This study on the femtosecond excited-state dynamics of a Cu complex in solution based on time-resolved Cu K-edge hard X-ray absorption spectroscopy at the SACLA XFEL reports the observation of hitherto unreported nuclear wavepacket dynamics and a new mechanism for the ultrafast relaxation in the photochemical reaction of this complex.
- 91.Shelby ML et al. Ultrafast excited state relaxation of a metalloporphyrin revealed by femtosecond X-ray absorption spectroscopy. J. Am. Chem. Soc 138, 8752–8764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miller NA et al. Polarized XANES monitors femtosecond structural evolution of photoexcited vitamin B12. J. Am. Chem. Soc 139, 1894–1899 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Miller NA et al. Ultrafast XANES monitors femtosecond sequential structural evolution in photoexcited coenzyme B12. J. Phys. Chem. B 124, 199–209 (2020). [DOI] [PubMed] [Google Scholar]
- 94.Chatterjee R et al. XANES and EXAFS of dilute solutions of transition metals at XFELs. J. Synchrotron Radiat. 26, 1716–1724 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Britz A et al. Resolving structures of transition metal complex reaction intermediates with femtosecond EXAFS. Phys. Chem. Chem. Phys 22, 2660 (2020). [DOI] [PubMed] [Google Scholar]
- 96.Gul S et al. Simultaneous detection of electronic structure changes from two elements of a bifunctional catalyst using wavelength-dispersive X-ray emission spectroscopy and in situ electrochemistry. Phys. Chem. Chem. Phys 17, 8901–8912 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Alonso-Mori R et al. Towards characterization of photo-excited electron transfer and catalysis in natural and artificial systems using XFELs. Faraday Discuss. 194, 621–638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Martinie RJ et al. Two-color valence-to-core X-ray emission spectroscopy tracks cofactor protonation state in a class I ribonucleotide reductase. Angew. Chem. Int. Ed. Engl 57, 12754–12758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Canton SE et al. Visualizing the non-equilibrium dynamics of photoinduced intramolecular electron transfer with femtosecond X-ray pulses. Nat. Commun 6, 6359 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kern J et al. Taking snapshots of photosynthetic water oxidation using femtosecond X-ray diffraction and spectroscopy. Nat. Commun 5, 4371 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kupitz C et al. Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature 513, 261–265 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Suga M et al. Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 517, 99–103 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Young ID et al. Structure of photosystem II and substrate binding at room temperature. Nature 540, 453–457 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Suga M et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 543, 131–135 (2017). [DOI] [PubMed] [Google Scholar]
- 105.Suga M et al. An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an x-ray free-electron laser. Science 366, 334–338 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Yano J et al. X-ray damage to the Mn4Ca complex in photosystem II crystals: a case study for metallo-protein X-ray crystallography. Proc. Natl Acad. Sci. USA 102, 12047–12052 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dell’Angela M et al. Real-time observation of surface bond breaking with an x-ray laser. Science 339, 1302–1305 (2013). [DOI] [PubMed] [Google Scholar]
- 108.Östrom H et al. Surface chemistry. Probing the transition state region in catalytic CO oxidation on Ru. Science 347, 978–982 (2015). [DOI] [PubMed] [Google Scholar]
- 109.Nilsson A et al. Catalysis in real time using X-ray lasers. Chem. Phys. Lett 675, 145–173 (2017). [Google Scholar]
- 110.Ismail ASM et al. Direct observation of the electronic states of photoexcited hematite with ultrafast 2p3d X-ray absorption spectroscopy and resonant inelastic X-ray scattering. Phys. Chem. Chem. Phys 22, 2685–2692 (2020). [DOI] [PubMed] [Google Scholar]
- 111.Siefermann KR et al. Atomic-scale perspective of ultrafast charge transfer at a dye–semiconductor interface. J. Phys. Chem. Lett 5, 2753–2759 (2014). [DOI] [PubMed] [Google Scholar]
- 112.Eckert S et al. Ultrafast independent N–H and N–C bond deformation investigated with resonant inelastic X-ray scattering. Angew. Chem. Int. Ed. Engl 56, 6088–6092 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Loh ZH et al. Observation of the fastest chemical processes in the radiolysis of water. Science 367, 179–182 (2020). [DOI] [PubMed] [Google Scholar]
- 114.Wolf TJA et al. Probing ultrafast ππ*/nπ* internal conversion in organic chromophores via K-edge resonant absorption. Nat. Commun 8, 29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wolf TJA & Guhr M Photochemical pathways in nucleobases measured with an X-ray FEL. Philos. Trans. A Math. Phys. Eng. Sci 377, 20170473 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gessner O & Guhr M Monitoring ultrafast chemical dynamics by time-domain X-ray photo- and Auger-electron spectroscopy. Acc. Chem. Res 49, 138–145 (2016). [DOI] [PubMed] [Google Scholar]
- 117.Kubin M et al. Probing the oxidation state of transition metal complexes: a case study on how charge and spin densities determine Mn L-edge X-ray absorption energies. Chem. Sci 9, 6813–6829 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Blomberg MRA & Siegbahn PEM A comparative study of high-spin manganese and iron complexes. Theor. Chem. Acc 97, 72–80 (1997). [Google Scholar]
- 119.Lundberg M & Delcey MG in Transition Metals in Coordination Environments: Computational Chemistry and Catalysis Viewpoints (eds Broclawik E, Borowski T. & Radon M) 185–217 (Springer, 2019). [Google Scholar]
- 120.Wernet P et al. Orbital-specific mapping of the ligand exchange dynamics of Fe(CO)5 in solution. Nature 520, 78–81 (2015).This femtosecond time-resolved resonant inelastic X-ray scattering (RIXS) study of a metal complex in solution based on Fe L-edge soft X-ray measurements at the LCLS XFEL establishes probing photoactivated metal complexes in solution with XFELs and motivates using XFEL pulses for probing ultrafast dynamics in photocatalysts.
- 121.Kunnus K et al. Identification of the dominant photochemical pathways and mechanistic insights to the ultrafast ligand exchange of Fe(CO)5 to Fe(CO)4EtOH. Struct. Dyn 3, 043204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lundberg M & Wernet P in Synchrotron Light Sources and Free-Electron Lasers 2nd edn (eds Jaeschke EJ, Khan S, Schneider JR & Hastings JB) 1–52 (Springer, 2019). [Google Scholar]
- 123.Hocking RK et al. Fe L-edge XAS studies of K4[Fe(CN)6] and K3[Fe(CN)6]: A direct probe of back-bonding. J. Am. Chem. Soc 128, 10442–10451 (2006).Example of the use of transition-metal L-edge soft X-ray spectroscopy for element-site-selective and chemical-site-selective and orbital-specific probing of the electronic structure of metal complexes, and details how the covalence of metal–ligand bonds can be quantified.
- 124.Kunnus K et al. Viewing the valence electronic structure of ferric and ferrous hexacyanide in solution from the Fe and cyanide perspectives. J. Phys. Chem. B 120, 7182–7194 (2016). [DOI] [PubMed] [Google Scholar]
- 125.Jay RM et al. Disentangling transient charge density and metal–ligand covalency in photoexcited ferricyanide with femtosecond resonant inelastic soft X-ray scattering. J. Phys. Chem. Lett 9, 3538–3543 (2018). [DOI] [PubMed] [Google Scholar]
- 126.Norell J et al. Fingerprints of electronic, spin and structural dynamics from resonant inelastic soft X-ray scattering in transient photo-chemical species. Phys. Chem. Chem. Phys 20, 7243–7253 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kunnus K et al. Chemical control of competing electron transfer pathways in iron tetracyano-polypyridyl photosensitizers. Chem. Sci 11, 4360–4373 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Glover TE et al. X-ray and optical wave mixing. Nature 488, 603–608 (2012). [DOI] [PubMed] [Google Scholar]
- 129.Fuchs M et al. Anomalous nonlinear X-ray compton scattering. Nat. Phys 11, 964–970 (2015). [Google Scholar]
- 130.Stohr J Two-photon X-ray diffraction. Phys. Rev. Lett 118, 024801 (2017). [DOI] [PubMed] [Google Scholar]
- 131.Tamasaku K et al. Nonlinear spectroscopy with X-ray two-photon absorption in metallic copper. Phys. Rev. Lett 121, 083901 (2018). [DOI] [PubMed] [Google Scholar]
- 132.Shwartz S et al. X-ray second harmonic generation. Phys. Rev. Lett 112, 163901 (2014). [DOI] [PubMed] [Google Scholar]
- 133.Foglia L et al. First evidence of purely extreme-ultraviolet four-wave mixing. Phys. Rev. Lett 120, 263901 (2018). [DOI] [PubMed] [Google Scholar]
- 134.Bencivenga F et al. Nanoscale transient gratings excited and probed by extreme ultraviolet femtosecond pulses. Sci. Adv 5, eaaw5805 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Svetina C et al. Towards X-ray transient grating spectroscopy. Opt. Lett 44, 574–577 (2019). [DOI] [PubMed] [Google Scholar]
- 136.Rohringer N et al. Atomic inner-shell X-ray laser at 1.46 nanometres pumped by an X-ray free-electron laser. Nature 481, 488–491 (2012).Reports the observation of an atomic inner-shell X-ray laser, creating strong gain of X-ray emission in the forward direction.
- 137.Weninger C et al. Stimulated electronic X-ray Raman scattering. Phys. Rev. Lett 111, 233902 (2013). [DOI] [PubMed] [Google Scholar]
- 138.Yoneda H et al. Atomic inner-shell laser at 1.5-ångström wavelength pumped by an X-ray free-electron laser. Nature 524, 446–449 (2015). [DOI] [PubMed] [Google Scholar]
- 139.Kroll T et al. Stimulated X-ray emission spectroscopy in transition metal complexes. Phys. Rev. Lett 120, 133203 (2018).A study of using stimulated X-ray emission as a new spectroscopy tool to study transition-metal complexes.
- 140.Kroll T et al. Observation of seeded Mn Kβ stimulated X-ray emission using two-color X-ray free-electron laser pulses. Phys. Rev. Lett 125, 037404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cederbaum LS, Tarantelli F, Sgamellotti A & Schirmer J On double vacancies in the core. J. Chem. Phys 85, 6513–6523 (1986). [Google Scholar]
- 142.Ågren H & Jensen HJA Relaxation and correlation contributions to molecular double core ionization energies. Chem. Phys 172, 45–57 (1993). [Google Scholar]
- 143.Tashiro M, Ueda K & Ehara M Double core–hole correlation satellite spectra of N2 and CO molecules. Chem. Phys. Lett 521, 45–51 (2012). [Google Scholar]
- 144.Thomas TD Single and double core-hole ionization energies in molecules. J. Phys. Chem. A 116, 3856–3865 (2012). [DOI] [PubMed] [Google Scholar]
- 145.Takahashi O & Ueda K Molecular double core-hole electron spectroscopy for probing chemical bonds: C60 and chain molecules revisited. Chem. Phys 440, 64–68 (2014). [Google Scholar]
- 146.Koulentianos D et al. KL double core hole pre-edge states of HCl. Phys. Chem. Chem. Phys 20, 2724–2730 (2018). [DOI] [PubMed] [Google Scholar]
- 147.Linusson P, Takahashi O, Ueda K, Eland JHD & Feifel R Structure sensitivity of double inner-shell holes in sulfur-containing molecules. Phys. Rev. A 83, 022506 (2011). [Google Scholar]
- 148.Lablanquie P et al. Evidence of single-photon two-site core double ionization of C2H2 molecules. Phys. Rev. Lett 107, 193004 (2011). [DOI] [PubMed] [Google Scholar]
- 149.Tashiro M et al. Auger decay of molecular double core-hole and its satellite states: comparison of experiment and calculation. J. Chem. Phys 137, 224306 (2012). [DOI] [PubMed] [Google Scholar]
- 150.Nakano M et al. Single photon K−2 and K−1K−1 double core ionization in C2H2n (n = 1−3), CO, and N2 as a potential new tool for chemical analysis. Phys. Rev. Lett 110, 163001 (2013). [DOI] [PubMed] [Google Scholar]
- 151.Carniato S et al. Single photon simultaneous K-shell ionization and K-shell excitation. I. Theoretical model applied to the interpretation of experimental results on H2O. J. Chem. Phys 142, 014307 (2015). [DOI] [PubMed] [Google Scholar]
- 152.Penent F et al. Double core hole spectroscopy with synchrotron radiation. J. Electron. Spectrosc. Relat. Phenom 204, 303–312 (2015). [Google Scholar]
- 153.Young L et al. Femtosecond electronic response of atoms to ultra-intense X-rays. Nature 466, 56–56 (2010). [DOI] [PubMed] [Google Scholar]
- 154.Cryan JP et al. Auger electron angular distribution of double core-hole states in the molecular reference frame. Phys. Rev. Lett 105, 083004 (2010). [DOI] [PubMed] [Google Scholar]
- 155.Fang L et al. Double core-hole production in N2: beating the Auger clock. Phys. Rev. Lett 105, 083005 (2010). [DOI] [PubMed] [Google Scholar]
- 156.Salen P et al. Experimental verification of the chemical sensitivity of two-site double core-hole states formed by an x-ray free-electron laser. Phys. Rev. Lett 108, 153003 (2012). [DOI] [PubMed] [Google Scholar]
- 157.Piancastelli MN K-shell double core-hole spectroscopy in molecules. Eur. Phys. J. Spec. Top 222, 2035–2055 (2013). [Google Scholar]
- 158.Zhaunerchyk V et al. Disentangling formation of multiple-core holes in aminophenol molecules exposed to bright X-FEL radiation. J. Phys. B At. Mol. Opt. Phys 48, 244003 (2015). [Google Scholar]
- 159.Zhang Y, Bergmann U, Schoenlein R, Khalil M & Govind N Double core hole valence-to-core x-ray emission spectroscopy: A theoretical exploration using time-dependent density functional theory. J. Chem. Phys 151, 144114 (2019). [DOI] [PubMed] [Google Scholar]
- 160.Hua W, Bennett K, Zhang Y, Luo Y & Mukamel S Study of double core hole excitations in molecules by X-ray double-quantum-coherence signals: a multiconfiguration simulation. Chem. Sci 7, 5922–5933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mukamel S et al. Coherent multidimensional optical probes for electron correlations and exciton dynamics: from NMR to X-rays. Acc. Chem. Res 42, 553–562 (2009).A review of multidimensional spectroscopy techniques with an outlook to future XFEL applications.
- 162.Schweigert IV & Mukamel S Coherent ultrafast core-hole correlation spectroscopy: X-ray analogues of multidimensional NMR. Phys. Rev. Lett 99, 163001 (2007). [DOI] [PubMed] [Google Scholar]
- 163.Li X, Zhang T, Borca CN & Cundiff ST Many-body interactions in semiconductors probed by optical two-dimensional Fourier transform spectroscopy. Phys. Rev. Lett 96, 057406 (2006). [DOI] [PubMed] [Google Scholar]
- 164.Brixner T et al. Two-dimensional spectroscopy of electronic couplings in photosynthesis. Nature 434, 625–628 (2005). [DOI] [PubMed] [Google Scholar]
- 165.Tanaka S & Mukamel S Coherent X-ray Raman spectroscopy: a nonlinear local probe for electronic excitations. Phys. Rev. Lett 89, 043001 (2002). [DOI] [PubMed] [Google Scholar]
- 166.Bergmann U, Yachandra VK & Yano J X-Ray Free Electron Lasers: Applications in Materials, Chemistry and Biology (Energy and Environment Series) (Royal Society of Chemistry, 2017). [Google Scholar]
- 167.Jaeschke EJ, Khan S, Schneider JR & Hastings JB Synchrotron Light Sources and Free-Electron Lasers (Springer, 2016). [Google Scholar]
- 168.Ourmazd A Science in the age of machine learning. Nat. Rev. Phys 2, 342–343 (2020). [Google Scholar]
- 169.Schoenlein R et al. Recent advances in ultrafast X-ray sources. Philos. Trans. A Math. Phys. Eng. Sci 377, 20180384 (2019). [DOI] [PubMed] [Google Scholar]
- 170.Bergmann U & Glatzel P X-ray emission spectroscopy. Photosynth. Res 102, 255–266 (2009). [DOI] [PubMed] [Google Scholar]
- 171.Johansson MP, Blomberg MRA, Sundholm D & Wikstrom M Change in electron and spin density upon electron transfer to haem. Biochim. Biophys. Acta Bioenerg 1553, 183–187 (2002). [DOI] [PubMed] [Google Scholar]
- 172.Agarwal BK X-Ray Spectroscopy: An Introduction (Springer, 1991). [Google Scholar]
- 173.Henke BL, Gullikson EM & Davis JC X-ray interactions: photoabsorption, scattering, transmission, and reflection at E = 50−30,000 eV, Z = 1−92. At. Data Nucl. Data Tables 54, 181–342 (1993). [Google Scholar]
- 174.Siegbahn K ESCA Applied to Free Molecules (North-Holland, 1969). [Google Scholar]
- 175.Gelius U Binding energies and chemical shifts in ESCA. Phys. Scr 9, 133–147 (1974). [Google Scholar]
- 176.Stohr J NEXAFS Spectroscopy (Springer, 2013). [Google Scholar]
- 177.de Groot FMF Ligand and metal X-ray absorption in transition metal complexes. Inorg. Chim. Acta 361, 850–856 (2008). [Google Scholar]
- 178.Koningsberger DC & Prins R Chemical Analysis Vol. 92 (Wiley, 1988). [Google Scholar]
- 179.de Groot FMF Multiplet effects in X-ray spectroscopy. Coord. Chem. Rev 249, 31–63 (2005). [Google Scholar]
- 180.de Groot FMF X-ray absorption and dichroism of transition metals and their compounds. J. Electron. Spectrosc. Relat. Phenom 67, 529–622 (1994). [Google Scholar]
- 181.Stern EA Musings about the development of XAFS. J. Synchrotron Radiat 8, 49–54 (2001). [DOI] [PubMed] [Google Scholar]
- 182.Meisel A, Leonhardt G & Szargan R X-Ray Spectra and Chemical Binding (Springer, 1989). [Google Scholar]
- 183.Kotani A & Shin S Resonant inelastic x-ray scattering spectra for electrons in solids. Rev. Mod. Phys 73, 203–246 (2001). [Google Scholar]
- 184.de Groot F & Kotani A Core Level Spectroscopy of Solids (CRC, 2008). [Google Scholar]
- 185.Hahn AW et al. Probing the valence electronic structure of low-spin ferrous and ferric complexes using 2p3d resonant inelastic X-ray scattering (RIXS). Inorg. Chem 57, 9515–9530 (2018). [DOI] [PubMed] [Google Scholar]
- 186.Van Kuiken BE, Hahn AW, Maganas D & DeBeer S Measuring spin-allowed and spin-forbidden d–d excitations in vanadium complexes with 2p3d resonant inelastic X-ray scattering. Inorg. Chem 55, 11497–11501 (2016). [DOI] [PubMed] [Google Scholar]