

Abstract
Powdery mildew (PM) caused by Podosphaera aphanis is a major fungal disease of cultivated strawberry. Mildew Resistance Locus O (MLO) is a gene family described for having conserved seven-transmembrane domains. Induced loss-of-function in specific MLO genes can confer durable and broad resistance against PM pathogens. However, the genomic structure and potential role of MLO genes for PM resistance have not been characterized yet in the octoploid cultivated strawberry. In the present study, MLO gene families were characterized in four diploid progenitor species (Fragaria vesca, F. iinumae, F. viridis, and F. nipponica) and octoploid cultivated (Fragaria ×ananassa) strawberry, and potential sources of MLO-mediated susceptibility were identified. Twenty MLO sequences were identified in F. vesca and 68 identified in F. ×ananassa. Phylogenetic analysis divided diploid and octoploid strawberry MLO genes into eight different clades, in which three FveMLO (MLO10, MLO17, and MLO20) and their twelve orthologs of FaMLO were grouped together with functionally characterized MLO genes conferring PM susceptibility. Copy number variations revealed differences in MLO composition among homoeologous chromosomes, supporting the distinct origin of each subgenome during the evolution of octoploid strawberry. Dissecting genomic sequence and structural variations in candidate FaMLO genes revealed their potential role associated with genetic controls and functionality in strawberry against PM pathogen. Furthermore, the gene expression profiling and RNAi silencing of putative FaMLO genes in response to the pathogen indicate the function in PM resistance. These results are a critical first step in understanding the function of strawberry MLO genes and will facilitate further genetic studies of PM resistance in cultivated strawberry.
Subject terms: Plant breeding, Genomics
Introduction
The Mildew Resistance Locus O (MLO) gene family is present in several crop species and was described for having conserved seven-transmembrane (TM) and C-terminal calmodulin-binding (CaMB) domains that are functionally important for powdery mildew resistance1,2. The genetic diversity among MLO gene members has been explored in a wide variety of plant species3–5, but still little information is known about its origin and biochemical function associated with plant development and response to biotic and abiotic stresses. A recent study revisited the biological origin of MLO gene family that traced it back from the ancestral species of unicellular photosynthetic eukaryote and, that in the course of evolution diversified to the present MLO proteins6. In modern plants, the divergence of MLO proteins was demonstrated through phylogenetic analysis and classified them to different clades ranging from four to eight5,7–9.
The MLO gene family has been the focus of attention in many crop species because transgenic downregulation or elimination of specific endogenous MLO genes has led to powdery mildew (PM) resistance10. However, alteration of MLO gene sequences can trigger negative phenotypic effects including premature leaf chlorosis, altered root growth and pollen tube germination11. The first MLO-based resistance trait was first characterized in barley (HvMLO), where a loss-of-function mutation in an MLO gene conferred broad resistance against PM pathogens12. This discovery led to subsequent comparative genomic studies of MLO gene families in several plant species to find suitable candidate genes. In the model plant Arabidopsis thaliana, three MLO genes (AtMLO2, AtMLO6, and AtMLO12) were functionally characterized as conferring PM susceptibility13. Expression analysis of these homologs upon pathogen challenge suggested functional redundancy. The Atmlo2 single mutant has only partial resistance while triple mutants (Atmlo2, Atmlo6, and Atmlo12) have full resistance against PM pathogens7. Since identification of functional MLO genes is enabled by DNA sequence information, the advent of reference genomes for several crop species provides an opportunity to identify MLO orthologs as targets for functional studies. MLO genes have been genetically characterized across many crops including apple (MdMLO11 and MdMLO19)5, pepper (CaMLO2)14, rose (RhMLO1)15, grapevine (VvMLO6 and VvMLO7)16, melon (CmMLO2)17, cucumber (CsMLO1)18, pea (PsMLO1)19, tobacco (NtMLO1)20, tomato (SlMLO1)21, rice (OsMLO2)22, corn (ZmMLO1)23, and wheat (TaMLO1)22,24. Recently, targeted-genome mutation of SlMLO1 resulted in the development of a PM-resistant Slmlo1 tomato variety25. The potential functional role of these genes as susceptibility factors has not been highlighted yet in the octoploid cultivated strawberry. Thus, in the present study, the orthologs of AtMLO2, AtMLO6 and AtMLO12 were identified from the octoploid strawberry genome and explored their possible function for PM resistance.
Strawberry PM caused by an obligate parasite Podosphaera aphanis (Wallr.) (former Sphaerotheca macularis f. sp. fragariae)26 is a major fungal plant disease in strawberry. PM primarily affects the leaf and, depending on severity, can also affect other organs27. Initially, a white powdery mycelium develops on the underside of the leaves, followed by upward curling of the leaf edges, while severe leaf infection can cause burning at the leaf margin28. Infected flowers and fruits may result in fruit deformation and delayed ripening29. PM is widespread in many strawberry growing regions worldwide, such that both open-field and high-tunnel growing systems may experience severe yield losses when infected fields are left untreated30. Most farmers rely on multiple pesticide applications to manage PM. Hence, developing cultivars with improved resistance is highly advantageous.
The modern cultivated strawberry (Fragaria ×ananassa) is an allo-octoploid (2n = 8x = 56) resulting from hybridization between a Chilean strawberry (F. chiloensis) and a North American native strawberry (F. virginiana)31. Further domestication of F. ×ananassa produced large and flavorful berries that have become the world’s most widely grown fruit crop. In 2010, the genome of the diploid progenitor species F. vesca was sequenced32 and has been used as a diploid reference genome towards molecular marker development33 and for gene-trait association studies in F. ×ananassa34,35. Recently, three other diploid reference sequences, F. iinumae36, F. nipponica37, and F. viridis38, have been published (available at https://www.rosaceae.org/). However, the octoploid F. ×ananassa genome is far more complicated than its diploid progenitor. In 2019, the chromosome-scale F. ×ananassa ‘Camarosa’ reference genome was developed, and F. vesca was shown to be the dominant diploid progenitor in terms of gene content, expression abundance and genetic control for metabolic and disease-resistance traits39. This new reference sequence will serve as a powerful genetic resource to unravel the complexity of octoploid strawberry genome for gene-trait association studies, including causal MLO genes in strawberry breeding programs.
The goals of this study were to characterize the MLO gene family in four diploid progenitors (F. vesca, F. iinumae, F. viridis, and F. nipponica) and octoploid (F. ×ananassa) strawberry using currently available high-quality genome sequences. The genomic structures of MLO genes were compared between diploid and octoploid strawberry, and patterns of transcript levels were explored throughout different strawberry plant tissues. Furthermore, we determined potential MLO genes associated with PM resistance in cultivated strawberry. These data and insights generated here are a critical first step in understanding the function of MLO genes in strawberry and will facilitate further genetic studies of PM resistance in cultivated strawberry.
Results
Genome-wide identification of the MLO gene family in diploid and octoploid strawberry
Using the Arabidopsis AtMLO1 (AT4G02600) protein sequence as a BLAST query, 20 putative MLO genes were identified using the latest diploid genome annotation F. vesca v4.0.a140. These 20 MLO genes were renamed FveMLO1 through FveMLO20 based on their ordered chromosomal positions (Fig. 1A, Table S1-A). Predicted proteins ranged between 200 and 903 aa with an average of 524 aa. Three truncated MLO proteins were identified as putative pseudogenes (Table S2-B). The number of predicted MLO genes in diploid strawberry agrees with the previous characterization of strawberry MLO genes using the first F. vesca draft genome41.
Fig. 1. Chromosomal localization and distribution of FveMLO.

A and FaMLO B genes in F. vesca and F. ×ananassa genomes, respectively. The seven chromosomes of F. vesca were named as Fvb1–Fvb7 while F. ×ananassa was named as Fvb1–1 to Fvb1–4, Fvb2–1 to Fvb2–4, Fvb3–1 to Fvb3–4, Fvb4–1 to Fvb4–4, Fvb5–1 to Fvb5–4, Fvb6–1 to Fvb6–4 and Fvb7–1 to Fvb7–4, respectively, to indicate four subgenomes within each chromosome. Different color of F. ×ananassa subgenomes indicates their respective diploid progenitors, namely F. viridis (yellow), F. iinumae (green), F. nipponica (gray), and F. vesca (orange)39 Putative strawberry MLO gene chromosome locations were visualized using MapChart 2.3 software26. The relative chromosome size was indicated by the unit Mbp.
The FveMLO genes identified in F. vesca were then used to identify gene orthologs in the octoploid strawberry-annotated genome F. ×ananassa v1.0.a1(hardmasked)39. Analysis revealed 68 predicted MLO genes across 28 chromosomes (Fig. 1B, Table S1-B). Of 20 FveMLO genes, 17 were matched with high amino acid sequence identity to a putative ortholog; however, conserved sequences could not be found for FveMLO6, FveMLO7, or FveMLO8. Based on their putative orthology to F. vesca, the octoploid MLO genes were named FaMLO1 through FaMLO20, respectively. To distinguish homeologous MLO genes, FaMLO1–1 to FaMLO1–4 were used for each FveMLO1 homolog. Predicted MLO proteins have amino acid sequence lengths ranging between 144 and 2,365 with an average of 560 amino acids (Table S2).
Twenty FveMLO genes were distributed randomly across six chromosomes of the diploid F. vesca genome: three in Fvb1, one in Fvb2, seven in Fvb3, four in Fvb5, three in Fvb6, and two in Fvb7 (Fig. 1, Table S1-A). The coding DNA sequence composition ranged from 1 to 16 exons (Fig. S1-A). In the octoploid genome, 68 FaMLO genes were distributed across every chromosome, except Fvb4–1 and Fvb4–3 (Fig. 1B, Table S1-B). The intron–exon structures of FaMLO genes have more variation as compared with the diploid progenitor F. vesca, with coding DNA sequence (CDS) composition ranging from 1 to 23 exons (Fig. S1-B). Despite this structural complexity, many FaMLO genes demonstrated high DNA sequence identity with the diploid progenitors, F. vesca, F. iinumae, and F. nipponica (Fig. 2 and Fig. S2). Two additional FaMLO sequences in chromosome 4–2 (FaMLO19-i) and 4–4 (FaMLO12-iii) were identified in F. ×ananassa that was not present in F. vesca (Fig. 1A, B).
Fig. 2. Phylogenetic analysis of MLO gene family in strawberry and other related crop species.
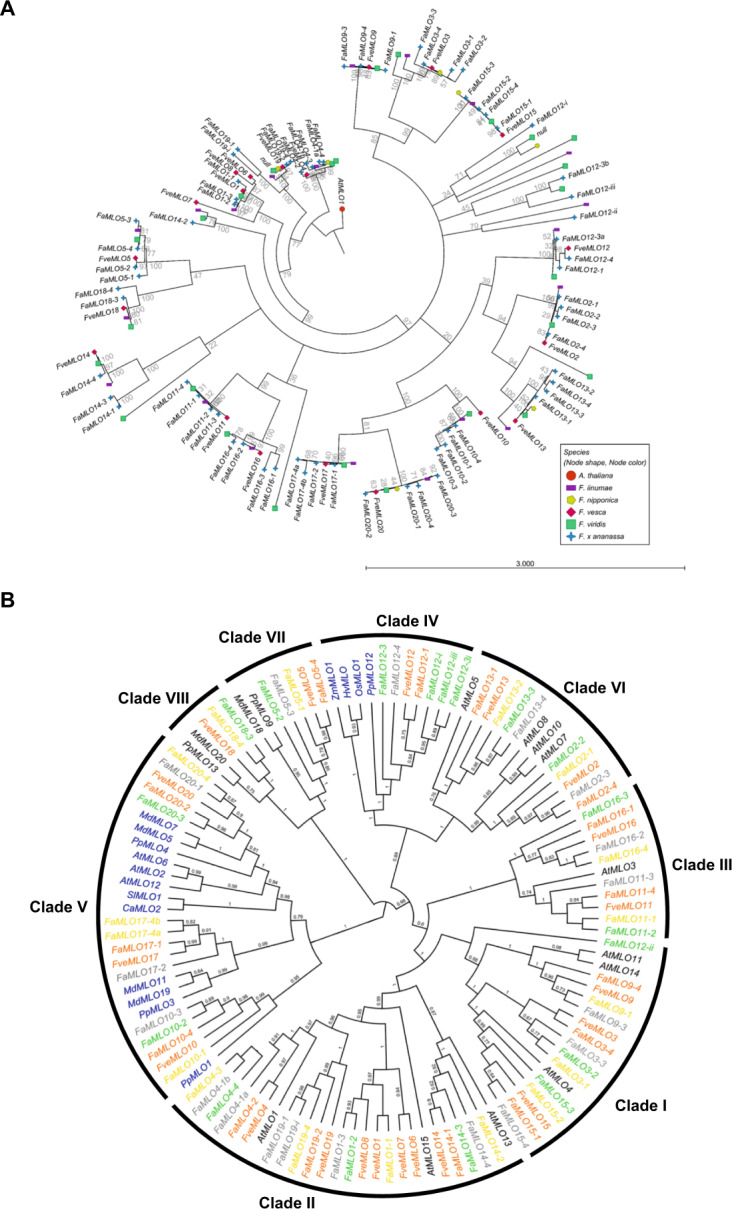
A The maximum likelihood tree of MLO coding sequences from F. ×ananassa and four putative diploid progenitors, F. vesca, F. iinumae, F. nipponica, and F. viridis. Arabidopsis MLO1 was used as an outgroup. Fragaria spp. are indicated by node shapes and node color. B Phylogenetic tree of MLO protein sequences of strawberry species F. vesca and F. ×ananassa, and other plant species such as Arabidopsis, tomato, pepper, rice, barley, maize, apple, and peach. Strawberry MLO genes together with other MLOs were grouped into eight different clades. Unrooted phylogenetic tree of 15 AtMLOs, 20 FveMLOs, 59 FaMLOs, five PpMLOs, six MdMLOs, CaMLO2, SlMLO1, HvMLO, OsMLO1, and ZmMLO1 was constructed by using FastTree via Geneious software. FaMLO proteins were highlighted in different colors according to their subgenomic location, the respective diploid progenitors such as the F. viridis-like (yellow), F. vesca-like (orange), F. nipponica-like (gray) and F. iinumae-like (green) subgenomes. MLO proteins highlighted in blue were previously characterized MLO-susceptible genes.
Structural gene variation, especially between homeologous genes, could imply that some homeologs evolved differently in regulation of gene expressions and could result in novel function42,43. Putative strawberry MLO genes were selected for further phylogenetic analysis of functional divergence.
Phylogenetic relationship analysis of MLO genes in diploid and octoploid strawberry
To study the evolutionary relationships of MLO genes in F. ×ananassa with its diploid progenitors, we created the maximum likelihood tree of MLO coding sequences from octoploid F. ×ananassa and diploids F. vesca, F. iinumae, F. nipponica, and F. viridis (Fig. 2A). This phylogeny demonstrates that a majority FaMLO genes, as well as their homeologs, are most similar to a Fragaria vesca ortholog. The overrepresentation of F. vesca-like FaMLO genes supports the recent finding of F. vesca subgenome dominance in octoploid strawberry, which occurred via homeologous exchange between subgenomes of octoploid strawberry and resulted in an octoploid genome that is broadly more F.vesca-like39. Strong homology with F. iinumae can be observed within a few octoploid MLO homeologous groups, including FaMLO3, MLO16, and MLO20, and possibly signify instances of conversion by the F. iinumae subgenome. Homology with the F. nipponica MLO sequences is rarer, likely due to the noted conversion away from this subgenome in the modern octoploid39 and due to the incompleteness of the F. nipponica genome (Fig. 2A).
To estimate the direction and magnitude of evolutionary pressures on FaMLO proteins, we applied a pairwise comparison of MLO coding sequences from octoploid and diploid strawberry spp., and calculated the rate of nonsynonymous (dN) and synonymous (dS) mutations among putative orthologs (Table S3). Most FaMLO genes demonstrate evidence of negative selection, with their low dN/dS ratios indicating a functional protein constraint. A few FaMLO homeologs, including FaMLO16-1 and FaMLO20-3, appear to be under neutral selection (Table S3). No FaMLO genes showed evidence of positive selection (dN/ dS » 1).
To study the evolutionary relationships between strawberry MLO genes and functional MLO genes from other plant species, we aligned the deduced amino acid sequence of 20 FveMLOs and 68 from FaMLOs with previously characterized MLOs from Arabidopsis (AtMLOs), corn (ZmMLO1), rice (OsMLO1), barley (HvMLO), tomato (SlMLO1), pepper (CaMLO2) and other rosaceous crops such as apple (MdMLOs) and peach (PpMLOs). Phylogenetic analysis using MUSCLE and FastTree divided MLO genes into eight different clades (Fig. 2B). Functionally characterized MLO genes that are associated with PM susceptibility from selected monocots and dicots were clustered in clades IV and V, respectively7. Among strawberry MLO genes, FveMLO12 and FaMLO12 together with HvMLO, OsMLO1, and ZmMLO1 were grouped in clade IV, while FveMLO10, FveMLO17, FveMLO20, FaMLO10, FaMLO17, and FaMLO20 together with SlMLO1, CaMLO1, AtMLO2, AtMLO6, and AtMLO12 were grouped in clade V. The remaining putative MLO sequences were distributed to six other groups. Thus, three FveMLO (FveMLO10, FveMLO17, and FveMLO20) and 12 FaMLO (FaMLO10-1, FaMLO10-2, FaMLO10-3, FaMLO10-4, FaMLO17-1, FaMLO17-2, FaMLO17-4a, FaMLO17-4b, FaMLO20-1, FaMLO20-2, FaMLO20-3, and FaMLO20-4) proteins have a close evolutionary relationship with the MLO proteins known to confer PM susceptibility in diploid plants (Fig. 2B).
Synteny analysis of octoploid strawberry MLO genes
The four subgenomes of the modern cultivated strawberry are a result of allopolyploidization with four specific diploid progenitor genomes, which have subsequently undergone substantial subgenome conversion39. To elaborate on this recent discovery, we display synteny networks for putative MLO genes between diploid (F. vesca) and octoploid (F. ×ananassa) strawberry (Fig. 3). Most putative FaMLO orthologs co-localized to the same chromosome of the F. vesca genome. Additional FaMLO orthologs of FveMLO12 and FveMLO19 were identified in subgenomes Fvb4–4 and Fvb4–2, respectively. Because of the octoploidy of the cultivated strawberry, four homeologous genes are expected for each FveMLO gene. This was filled for 11 out of the 20 FveMLO genes: FaMLO2, FaMLO3, FaMLO5, FaMLO10, FaMLO11, FaMLO13, FaMLO14, FaMLO15, FaMLO16, FaMLO17, and FaMLO20. For the other FaMLO gene, variants in copy number of homeologous genes for each FaMLO were identified with two homeologs each for FaMLO18, three for FaMLO1, FaMLO9, and FaMLO19, five for FaMLO4, and six for FaMLO12 (Fig. 3, Table S1), which suggest rearrangement in the octoploid genome. Variation in MLO gene distributions illustrates the wide diversity in the genome composition of octoploid strawberry. Furthermore, there were at least two FaMLO homeologs with 80% or more sequence similarity with FveMLO orthologs (Fig. S2).
Fig. 3. Synteny analysis of MLO genes between F. vesca and F. ×ananassa. Syntenic regions present in each chromosome of F. vesca were filled with red, light blue, green, dark blue, yellow, and purple sequentially.

A total of 68 connecting lines between two genomes denote syntenic chromosomal regions. The F. vesca chromosomes are highlighted in orange while F. ×ananassa subgenomes were highlighted according to their diploid progenitors namely F. viridis (yellow), F. vesca (orange), F. nipponica (gray) and F. iinumae (green). Chromosomes Fvb4 from F. vesca, and Fvb4–1 and Fvb4–3 from F. ×ananassa, were not included since no putative MLO genes were identified in those regions. The relative chromosome size was indicated by the unit, Mbp. Circular visualization of syntenic regions between F. vesca and F. ×ananassa was constructed using an R-package “Circlize”.
Domain organization and structure characterization of octoploid strawberry MLO genes
The first MLO gene was identified in barley and was characterized as membrane protein with seven transmembrane (TM) domains and a uniquely-identified “MLO-functional domain”1. To examine the conserved protein domains of strawberry MLO genes, the deduced amino acid sequence of the predicted MLO proteins found in diploid and octoploid strawberries was subjected to theoretical domain prediction using online software InterProScan (https://www.ebi.ac.uk) and NCBI’s conserved domain database (https://www.ncbi.nlm.nih.gov /Structure/cdd). Most MLO proteins from either diploid or octoploid strawberry contain the conserved domain of MLO and TM, which covers a large portion of the protein (Fig. 4). To predict TM domains and subcellular localization of strawberry MLO proteins, CCTOP44 and WoLF PSORT45 software were used to find differences in the number of TM domains and subcellular localization among FveMLO and FaMLO proteins (Fig. 4, Table S2). All FveMLO proteins were predicted to localize within the plasma membrane, except for FveMLO7, which was predicted to localize in the extracellular matrix. Out of 68 FaMLO genes, 58 were predicted to localize within the plasma membrane, while nine were predicted to localize in other organelles: four in the chloroplast, two in ER, two in the nucleus, and one in Golgi bodies (Table S2). Thirteen out of 20 FveMLO proteins have seven TM domains, while seven have three to six TM domains. The FaMLO gene family has a high degree of variation in TM domain composition (Table S2). Only 35 FaMLO proteins have seven TM domains, while the remaining FaMLO proteins have TM domains ranging between zero and eight (Fig. 4, Table S2).
Fig. 4. Domain organization of deduced MLO protein sequences of F. vesca and F. ×ananassa.

Visualization of protein domains was constructed using IBS 1.0.3 (Illustrator for Biological Sequences) program29. The positions of conserved transmembrane (blue) and MLO (yellow) domains were predicted using online CCTOP prediction server (http://cctop.enzim.ttk.mta.hu/) and CDD: NCBI’s conserved domain database (https://www.ncbi.nlm.nih.gov /Structure/cdd).
The domain organization of some FaMLO proteins showed high levels of conservation with the diploid ancestor species, F. vesca. For example, there were at least two homeologs of FaMLO2, FaMLO4, FaMLO5, FaMLO9, FaMLO11, FaMLO12, FaMLO15, FaMLO16, FaMLO17, and FaMLO20 that showed F. vesca-like domain structures (Fig. 4). However, one to three homeologs of some FaMLO proteins, including FaMLO3, FaMLO4, FaMLO5, FaMLO10, FaMLO12, FaMLO13, FaMLO14, FaMLO16, FaMLO17, FaMLO18, FaMLO19, and FaMLO20, showed more diverse protein structures indicating a distinct origin from different diploid progenitors (Fig. 4).
To further investigate the candidate susceptibility-conferring FaMLO genes, namely FaMLO10-1, FaMLO10-2, FaMLO10-3, FaMLO10-4, FaMLO17-1, FaMLO17-2, FaMLO174a, FaMLO17-4b, FaMLO20-1, FaMLO20-2, FaMLO20-3, and FaMLO20-4, their deduced amino acid sequences were aligned with their orthologs from F. vesca and AtMLO2, AtMLO6, and AtMLO12 from Arabidopsis (Fig. S3-A). We identified conserved domains, including seven TM, CaMB and two C-terminal protein (I and II) domains of MLO genes2,46. FaMLO17-2, FaMLO17-4a, FaMLO20-1, FaMLO20-2, FaMLO20-3, and FaMLO20-4 proteins possessed seven TM domains and conserved CaMB and C-terminal I and II domains; however, FaMLO10-1, FaMLO10-2, FaMLO10-3, FaMLO10-4, FaMLO17-1, and FaMLO-17a- and FaMLO17-4b showed variable truncated protein sequences at the C-terminal end, resulting in the loss of these domains (Fig. S3-A). Among candidate MLO proteins, FaMLO10-1, FaMLO17-1, FaMLO17-2, FaMLO17-4a, FaMLO20-1, FaMLO20-2, FaMLO20-3, and FaMLO20-4 protein sequences are most identical to known susceptibility-conferring Arabidopsis MLO proteins, and therefore could have potential association with PM resistance in octoploid strawberry. MLO susceptibility factors were functionally conserved between monocot and dicots47. Among FaMLO12 proteins that were clustered in clade IV, FaMLO12-1, FaMLO12-3a and FaMLO12-4 showed high protein sequence identity and with functional MLO genes in distant relative monocot species (Fig. S3-B).
Expression profiling of FaMLO genes in different tissues of cultivated strawberry
Expression patterns of previously characterized MLO genes suggested diverse biological functions for the MLO gene family31. To characterize MLO transcript accumulation patterns in octoploid strawberry, raw data from the strawberry gene expression atlas study32 were reassembled using the recently published octoploid strawberry genome39. The steady state accumulation of several MLO transcripts is specific to different tissues, with FaMLO genes such as FaMLO1 and FaMLO17 showing root-specific gene expression (Fig. 5, Table S4). Variation in gene expression between homeologs is also observed. For example, FaMLO1–1 is highly expressed specifically in root, but the other homeologs, FaMLO1–2 and FaMLO1–3, are not expressed. For candidate FaMLOs that are potentially associated with PM disease, the expression pattern and levels of transcript abundance also vary. The four homeologs of FaMLO10 are expressed in roots at different levels while only FaMLO10-1 and FaMLO10-4 are preferably expressed in leaf and other developing fruit tissues. In root, FaMLO17-2 also shows higher gene expression as compared with its other three homeologs, FaMLO17-1 and FaMLO174a. The expression of FaMLO17-4b was not detected. The homeologs FaMLO20-1, FaMLO20-2, and FaMLO20-3 show stable expression in root, leaf, and developing fruit tissues; however, FaMLO20-4 is not expressed in any of these tissues (Fig. 5, Table S4).
Fig. 5. Expression profile of putative strawberry MLO genes in different plant tissues, e.g., leaf, roots, green (GR), turning (TR), white (WR), and red (RR) receptacles, and green (GA), turning (TA), white (WA), and red (RA) achenes extracted from RNA-seq data published by Sánchez-Sevilla, et al. (2017).

The mean gene expression level was normalized using transcripts per million (TPM). Gene expression profile was visualized with heatmap using R-package “ggplot2”.
Structural variation analysis of putative FaMLO genes using long-read sequencing
Genetic mutation that causes allelic variation is a major driver of genetic diversity and change in gene function. To investigate gene diversity among putative FaMLO gene members of clade V, we determined genomic sequences of FaMLO10, FaMLO17, and FaMLO20, including their respective homeologs using long-read sequencing from one resistant University of Florida (UF) strawberry advance breeding selection, ‘13.55-195’ and compared them to the genomic reference sequences of the susceptible cultivar, ‘Camarosa’. A total of 455,111 PacBio long reads (6,735,339,589 bp) were generated representing about 15× coverage of the F. ×ananassa genome (805 Mb) and the sequence-length distribution was summarized in Figure S4 with an average length of 17,642 bp.
The full length of 10 FaMLO-like sequences out of 12 candidate FaMLO genes was successfully extracted from the assembly of long read sequencing data from the resistant accession ‘13.55-195’ (Fig. S5). These include FaMLO10-1, FaMLO10-3, FaMLO10-4, FaMLO17-1, FaMLO17-2, FaMLO17-4a, and FaMLO20-1 to 4. The FaMLO-like genes were then used for pairwise comparison of genomic sequences with the reference genome ‘Camarosa’ and presented major sequence variations (SV) in seven FaMLO-like genes of resistant (‘13.55195’) accession (Fig. 6A). In FaMLO10, a wide SV with up to 269 bp was detected at the noncoding regions including a 24 bp deletion of TC repeats in 5′ UTR region of homeolog FaMLO10-1. Fewer SVs were detected in FaMLO17 genes with indels ranging from 3 bp to 9 bp, while FaMLO20 had a wider SV that reached up to 283 bp and homeolog FaMLO20-1 having the most SVs. Furthermore, premature termination codon was detected from SV in FaMLO17-4a and FaMLO20-1 (Table S5). Overall, these data provided evidence of high genetic differentiation of putative functional FaMLO genes that might be associated with adaptive response to PM.
Fig. 6. FaMLO associated with powdery mildew susceptibility in strawberry.

A Structural variation of FaMLO10, FaMLO17 and FaMLO20 genes between ‘Camarosa’ and UF accession, ‘13.55-195’. The gene structure of FaMLO genes is shown with exons in blue boxes, UTR in gray boxes, and introns with a black line. The nucleotide insertions and deletions are presented by vertical lines with the corresponding number of indels. B Light micrographs of UF accessions, ‘13.55-195’, ‘Florida127’, ‘12.55-220’, and ‘14.34-33’ visualizing fungal infection at 0 (control) and 96 hpi (top). Leaf tissues were cleared in destaining solution and subsequently, the leaf segments were mounted on the slides and a drop of lactophenol cotton blue was added. The fungal structure can be visualized with cotton blue that specifically stains chitin in the fungal cell walls. Phenotype of four UF accessions in field conditions showing variable susceptibility/resistance to PM (bottom). C Relative gene expression of FaMLO10 (top), FaMLO17 (middle) and FaMLO20 (bottom) in five UF accessions at 0, 12, 24, and 48 hpi by real-time qPCR. FaGAPDH2 was used as internal control and plants at 0 hpi were used as sample control. Error bars indicate standard error.
Expression analysis of FaMLO genes after Podosphaera aphanis inoculation
To evaluate functional putative FaMLO gene members of clade V, the level of transcript accumulation was determined using quantitative real-time PCR in leaf tissues of four strawberry accessions, including two susceptible, Sweet Sensation® ‘Florida127’ and ‘12.55-220’, and two resistant accessions, ‘13.55-195’ and ‘14.33-34’, after PM infection. Three biological replicates were used for each following a randomized complete block design. Plants were inoculated with PM spores from naturally infected plants using a fine artist brush (Figure S6-B). Disease progression was observed among genotypes after PM infection and susceptible genotypes, ‘12.55-220’ and Sweet Sensation® ‘Florida127’ showed rapid growth of conidia as compared with resistant genotypes (Fig. 6B, Fig. S6-C, D). Leaf tissues collected at 0, 12, 24, and 48 h post inoculation (hpi) were used for gene expression analysis.
The data result illustrated a variable transcript accumulation at different time points post inoculation among FaMLO10, FaMLO17, and FaMLO20 genes (Fig. 6). FaMLO10 was induced at 24 hpi in both susceptible genotypes, while the transcript level in resistant genotypes remained low (Fig. 6C). Surprisingly, FaMLO17 that indicates a root-specific endogenous gene expression from RNA-seq analysis was highly upregulated in one susceptible genotype from 24 hpi to 48 hpi, while the resistant genotypes showed a low level of transcripts across multiple time points (Fig. 6C). A slight upregulation of the transcript was observed in one susceptible genotype, ‘12.55-220’ at 24 hpi but the rest did not show any obvious differences in FaMLO20 gene expression observed between susceptible and resistant genotypes after PM infection (Fig. 6C). The primer pairs used in this experiment were not homeologous-specific, and use of homeologous-specific primer pairs could facilitate identification of specific homeologs of functional FaMLO genes (Table S6). Overall, the gene expression analysis in this study provided insights of potential FaMLO genes associated with susceptibility response to PM infection.
Transient gene silencing of FaMLO10 and FaMLO20 genes in strawberry
The constitutive expression of FaMLO10 and FaMLO20 in the leaf and the upregulation of these genes after pathogen inoculation showed potential effects in PM response. To further validate the functional role of these genes in PM susceptibility, we designed three RNAi constructs targeting FaMLO10, FaMLO20, and both FaMLO10 and FaMLO20 genes and transiently expressed each gene into a susceptible UF strawberry accession, ‘Sensation®Florida127’. The RNAi vector construction is summarized in Fig. 7A. A total of 11–12 seedlings were transformed for each construct, including the empty vector that was used as the nonsilenced control. The mRNA level reduced by more than 50% in silenced individuals compared with the control (Fig. 7B). However, the individual RNAi construct was sufficient to silence the other MLO gene, indicating a relatively high coding DNA sequence similarity between FaMLO10 and FaMLO20. The transformed individuals were then inoculated with powdery mildew that was previously discussed and each transformant was evaluated for PM symptoms. The onset of PM symptoms of the nonsilenced control showed as early as five days after inoculation (DAI) and the symptoms rapidly progressed up to 90% at 16 DAI. On the other hand, the silenced individuals substantially delayed the progression of PM symptoms with only 50% disease incidence at 16 DAI (Fig. 7C). The nonsilenced control also showed higher DSI of more than 50% as compared with the silenced genotypes with DSI between 10% and 15%. (Fig. 7D, E). This finding suggests a major role of FaMLO10 and FaMLO20 genes in strawberry susceptibility against PM pathogen.
Fig. 7. Transient gene expression silencing of FaMLO10 and FaMLO20 genes.

A Construction of RNAi-expression vectors for transcriptional gene silencing in F. ×ananassa using UF accession ‘Sensation®Florida127’. B mRNA levels for FaMLO10 and FaMLO20 in silenced and nonsilenced strawberry. FaGAPDH2 was used as internal gene control for −∆∆Ct normalization and gene expression level was calculated using 2. Bars represent mean ± standard error (SE) of three biological replicates with three technical replicates. C Progression of PM from 1 to 16 days after inoculation (DAI). D Evaluation of disease severity of leaf samples based on mycelial leaf coverage, and E leaf phenotypes of silenced and nonsilenced strawberry. Photos were taken at 16 DAI.
Discussion
The MLO gene family is an important target in many agricultural crops for the improvement of resistance against PM pathogens. Previous characterizations identified that some MLO genes are involved in PM susceptibility, and that loss-of-function of those genes can confer durable and broad-spectrum resistance. In this study, it was found that variants of F. vesca MLO genes and F. ×ananassa MLO genes were distributed across the genome. However, we observed copy number variation of FaMLO homeologs showing differences in composition among subgenomes, supporting the distinct origin of each subgenome during the evolution of octoploid strawberry29. The unique FaMLO sequences in chromosome four (Fig. 1B and Fig. 3) that are not present in F. vesca might suggest an acquisition of chromosome segments from other diploid progenitors. Also, we did not find any significant homologs for FveMLO6, FveMLO7, and FveMLO8 using the F. ×ananassa ‘Camarosa’ reference genome (Fig. 1B). An over representation of some F. vesca-like MLO gene homeologs in octoploid strawberry was detected and this coincides with the previous findings showing an evidence of the dominance of F. vesca subgenome during homeologous exchange39. The loss and gain of genes could be evolutionarily significant in the development of polyploid species48 or other possible functional divergence during domestication. Additional octoploid reference genomes would explain such phenomena, as the chromosomal localization of FaMLO genes and possible misorientation in genome sequence for some subgenomes can be identified.
Phylogenetic analysis grouped strawberry MLO genes into eight different clades (Fig. 2B), which is consistent with previously classified MLO gene families in diploid strawberry41 and other Rosaceous crops5. The phylogenetic analysis of MLO genes between octoploid and diploid strawberry spp. illustrated the genetic diversity among FaMLO genes, and often indicated an F. vesca-like sequence ancestry for most FaMLO genes due to subgenome conversion (Fig. 2A). FveMLO genes were closely related to FaMLO genes localized in F. vesca-like subgenomes (Fig. 2B), while FaMLO gene localized in either F. iinumae- and F. nipponica-like and F. viridis-like subgenomes that appeared to have greater diversity of MLO genes in terms of potential gene duplication (Figs. 2B and 3). Unique homeologs of FaMLO4, FaMLO12, FaMLO17, and FaMLO19 were identified in these subgenomes supporting a greater genomic diversity of the genomes derived from these diploid progenitors (F. iinumae and F. viridis)49 that might explain the additional MLO copies in these subgenomes. Since genomes of homoploid hybrid lineages of F. ×ananassa were colinear and diploidized49, the genetic variation identified between FaMLO homeologs could be mainly attributed to their ancestral diploid progenitors and not by allelic diversity during chromosomal exchange between subgenomes.
Functional MLO genes associated with PM resistance are exclusively grouped in clades IV and V for monocots and dicots, respectively7. Interestingly, we found FveMLO12, six homeologs of FaMLO12 grouped together in clade IV with previously characterized HvMLO, ZmMLO1, and PpMLO125. In the present study, we found at least three clade V FveMLO and FaMLO genes, which are potential candidate genes involved in PM resistance. MLO-based resistance to the PM pathogen P. aphanis in a strawberry was successfully tested through RNAi-induced gene silencing of FaMLO genes using antisense MLO from peach (PpMLO1)50. In Arabidopsis, mutation of AtMLO2 showed only partial resistance, while complete resistance was obtained after knocking out both AtMLO6 and AtMLO1213.
The average MLO protein length (~500 amino acids) was similar in both F. vesca and F. ×ananassa, although few FaMLO proteins had more structural variations (SV) as compared with other homologous genes (Fig. 4). SV is defined as a change in genome sequence such as deletion, insertion, and inversion caused by mutations which in turn can affect gene function51. Furthermore, SV between homeologs of FaMLO10 and FaMLO12 causes changes in their gene expression profile (Fig. 5) and subcellular localization (Table S2). SV between homeologs might result in different gene regulation of gene expression and consequently lead to novel functions43. Diverse gene expression patterns among FaMLOs across different tissues (Fig. 5) suggest that MLO proteins might be specialized for different biological responses. This may be analogous to the unique expression patterns observed for Arabidopsis MLO genes, where transcription of each MLO gene is distinct and regulated differently by various biotic and abiotic stimuli52. In the present study, we found that the number of the TM domains of FveMLOs and FaMLOs varied between zero and eight. This variation was also observed in previous genome-wide MLO studies in other Rosaceous crops such as apple and peach5. Redundancy of gene function among homologs could arise from gene duplication for adaptation to varying selection pressures and changing environments53,54. High gene copy number is commonly observed in polyploid crop species due to the presence of homeologous genes, posing a considerable challenge for determining the biological contribution of specific genes. The genes FaMLO10-1, FaMLO17-1, FaMLO17-2, FaMLO17-4a, FaMLO17-4b, and FaMLO20-1 to 4 showed the highest sequence identity with known Arabidopsis MLO genes. Like functional AtMLO genes, we detected a transcriptional upregulation of FaMLO genes upon challenge of strawberry powdery mildew (Fig. 6C). Silenced MLO genes in strawberry also showed substantial delay in disease progression with reduced PM symptoms (Fig. 7C–E), indicating potential role in PM susceptibility16,55. Furthermore, genomic sequence variations in these FaMLO genes between resistant and susceptible genotypes were detected, including a truncation in TC repeats in the promoter region of the gene (Fig. 6B) that is known be evolutionarily conserved among functional MLO genes56. Like in the previous studies, silenced MLO genes in strawberry showed substantial delay in disease progression with reduced PM severity (Fig. 7C–E)16,55. Meanwhile, three homeologs of FaMLO12 genes that were closely related and shared conserved protein domains with the known MLO genes to confer to PM susceptibility in monocot were also identified as potential functional MLO-susceptible genes. Since, the susceptibly factors in MLO genes were functionally conserved between monocot and dicot47, this suggests that they might also contribute to strawberry MLO susceptibility. Overall, we identified a total of nine and three FaMLO genes with high homology to functional disease-susceptible MLO in dicot and monocot, respectively, representing candidate genes for breeding new cultivars with improved PM resistance. These putative FaMLOs should be further validated for their functional roles in PM resistance with more breeding accessions and cultivars.
Conclusions
Here, we identified a total of 20 MLO homologs in F. vesca and 68 in F.×ananassa. Three FveMLO genes and ten FaMLO genes were clustered with previously characterized MLO genes known to be in PM resistance/susceptibility in other plant species. The deduced amino acid sequences of putative strawberry MLO genes showed conserved protein characteristics, including transmembrane and calmodulin-binding domains that have been previously described. The considerable amino acid-level variation between MLO homoeologous copies was observed, suggesting possible non-redundant functions of MLOs in different subgenomes. FaMLO10, FaMLO17 and FaMLO20 are the most identical to functionally characterized MLO genes associated with PM susceptibility. The expression of FaMLO10 and FaMLO17 was substantially induced in response to the infection of P. aphanis in susceptible strawberry varieties and RNAimediated silencing of FaMLO10 and FaMLO20 greatly delayed disease progression and reduced PM severity suggesting a potential functional role in the PM resistance. Moreover, sequence variations in these MLO genes were detected between resistant and susceptible cultivars, and can be a potential target for functional characterization via CRISPR gene editing in future studies. Taken together, these data are a critical first step in understanding the allele function of the strawberry MLO gene family and should be useful for future functional studies to better understand their role in powdery mildew resistance in strawberry.
Materials and methods
Identification of the strawberry MLO genes in diploid and octoploid strawberry
To identify MLO gene orthologs in diploid strawberry, F. vesca, Arabidopsis MLO genes were searched to the latest F. vesca v4.0.a140 genome. Consequently, each FveMLO gene identified was used to search for predicted MLO genes in octoploid strawberry using the masked version of the F. ×ananassa v1.0.a139 reference genome. The deduced diploid and octoploid strawberry protein sequences were validated by reciprocal BLAST searches obtained from NCBI data sets of Arabidopsis reference genome. The chromosomal localization of each predicted MLO genes in F. vesca and F. ×ananassa was identified using available information in GDR database and visualized using MapChart 2.3 software57. Gene structure featuring introns, exons and UTR of predicted MLO genes was constructed using Gene Structure Display Server 2.0 (GSDS) (http://gsds.cbi.pku.edu.cn/)58.
For phylogenetic analysis of MLO genes in Fragaria species, full-length MLO–like coding sequences were extracted from the F. nipponica37, F. iinumae36, F. vesca40, F.viridis38, and F. ×ananassa39 genomes. The CIPRES Science Gateway59 was utilized for full-length coding sequence alignment using MUSCLE v3.760 and Maximum likelihood analysis using RAxML v8.2.1061. Tree construction was performed with 100 bootstrap replicates and rooted with Arabidopsis MLO1 (NM_116494), and this process was replicated five times using different random number seeds. Trees were visualized with a 50% bootstrap threshold using CLC Genomic Workbench 11.
For phylogenetic analysis of strawberry MLO and functional MLO genes, the protein sequences of MLO genes from F. vesca and F. ×ananassa were aligned to other available sequences of plant species, and phylogenetic relationship was constructed using FastTree consensus tree protein alignment via Geneious® 11.0.5 software62. Phylogenetic tree was constructed by adding 15 MLO genes from Arabidopsis, six MLO from other Rosaceous crops, apple (MdMLOs) and peach (PpMLOs), barley (HvMLO), corn (ZmMLO1), rice (OsMLO1), tomato (SlMLO1) and pepper (CaMLO2).
dN/dS analysis of MLO genes in Fragaria species
Putative orthologous MLO genes from the F. ×ananassa39, F. vesca v440, F. iinumae36, and F. nipponica37 genomes were identified from the Maximum Likelihood phylogenetic tree analysis. Alignment of each orthologous protein pair was performed using MUSCLE v3.759 followed by PAL2NAL (v14)63 supplied with the corresponding DNA sequences to convert the peptide alignment to a codon alignment. The dN and dS values were computed using the codeml program from PAML64 implemented on the PAL2NAL web server (http://www.bork.embl.de/pal2nal).
Synteny analysis of strawberry MLO genes
Pairwise comparisons of coding DNA sequence (CDS) between predicted FveMLO and FaMLO genes were obtained using ClustalW2 multiple-sequence alignment via Geneious software62 followed by heat map visualization to determine closely related MLO genes using Rpackage “Lattice”65. Synteny analysis of MLO genes between F. vesca and F. ×ananassa was summarized using R-package “Circlize”66.
Protein characterization and domain prediction
The deduced amino acid sequences of putative FveMLO and FaMLO genes were analyzed by different prediction software to identify functional domains and determine protein topologies and sub-cellular localizations. Functional MLO domains of protein sequences were predicted using CDD: NCBI’s conserved domain database (https://www.ncbi.nlm.nih.gov /Structure/cdd)67. Protein topology and number of transmembrane domains were predicted using online software CCTOP Prediction Server44 while protein sub-cellular localization was analyzed using WoLF PSORT program45. Default setting was used to run for all prediction software. Visualization of protein domains was constructed using IBS 1.0.3 (Illustrator for Biological Sequences) program68. To analyze conserved amino acids of MLO genes associated with PM resistance, protein sequences of FveMLO10, FveMLO17, and FveMLO20 from F. vesca and FaMLO10, FaMLO17, and FaMLO20 from F. ×ananassa were aligned against functionally characterized AtMLO2, AtMLO6 and AtMLO12 from Arabidopsis using MultAlin software (http://multalin.toulouse.inra.fr/multalin/)69.
Expression profile of putative strawberry MLO genes
To examine transcript accumulation patterns of putative strawberry MLO genes, RNAseq libraries from various ‘Camarosa’ tissues70 with the study reference PRJEB12420 were downloaded from the European Nucleotide Archive (https://www.ebi.ac.uk/ena). The complete 54 libraries RNA-seq experiment consisted of six independent green receptacle libraries, six white receptacle libraries, six turning receptacle libraries, six red receptacle libraries, three root libraries, three leaf libraries, and six achene libraries each for all the corresponding fruit stages. For both libraries, raw RNA-seq reads were assembled to the ‘Camarosa’ reference genome using CLC Genomic Workbench 11 (mismatch cost of 2, insertion cost of 3, deletion cost of 3, length fraction of 0.8, similarity fraction of 0.8, and 1 maximum hit per read). Reads that mapped equally well to more than one locus were discarded from the analysis. RNA-seq counts were quantified in transcripts per million (TPM).
Genomic DNA extraction and PacBio sequencing
Leaves of UF strawberry-breeding selection, ‘13.55-195’, were kept in the dark for a week and etiolated leaf tissues were collected for DNA extraction. The DNA was extracted using a modified CTAB method71 and sheared to about 20 Kb. The construction of SMRTbell libraries and sequencing a total of 14 SMRT cells using a PacBio RSII sequencer (Pacific Biosciences) were conducted at UC Davis Genome Center. After sequencing, RSII raw bam files were converted into subreads in FASTA format using the standard PacBio SMRT software package and produced a total of 11.97 Gb (~15x coverage of F. ×ananassa genome) with average read length of 19,957 bp.
Genome assembly of long-read PacBio sequences
The 11.97-Gb subreads generated from PacBio sequencing were assembled using Canu assembler72 (corrected error rate = 0.105, cor out coverage = 200). A total of 12,174 contigs were generated with an N50 of 17.64 Kb (Fig. S4).
Podosphaera aphanis inoculation using naturally infected strawberry leaves
In this study, a total of five UF strawberry accessions, including one cultivar (‘Sensation®Florida127’) and four advance breeding selections, ‘12.55-220’, ‘13.55-195’, ‘13.42-5’, and ‘14.33-34’, were used for FaMLO gene expression analysis. Of these, two genotypes, ‘Sensation®Florida127’ and ‘12.55-220’ were susceptible while the rest were resistant to PM. All accessions were transplanted in small pots in the greenhouse and were maintained pathogen-free for few weeks before fungal inoculation.
The inoculation using naturally infected strawberry leaves was performed following a modified method described by Calis, et al.73. The PM isolates were propagated and maintained on a susceptible strawberry genotype in a growth chamber (Fig. S6-A, B). Inoculation with PM was done using a fine artist’s paintbrush to carefully brush conidia from heavily infected leaves onto the leaves. Brushing was carried out at a height of ∼5–10 cm above the canopy to achieve an even distribution of conidia. The plots were arranged in an RCB design with three independent biological replicates and strawberry plants were well-watered and were maintained in a desirable condition with a temperature and humidity of ~70°F and 60–70%, respectively, throughout the experiments. The deposition of conidia was estimated by counting the number of spores in a 1-mm2 area under a microscope (Figure S6-C). Leaf tissues (S1 leaf) were collected at 0, 12, 24, 48, 72, and 96 h post inoculation (hpi) and immediately kept in a −80 °C freezer before processing.
RNA extraction and expression analysis using Real-Time Quantitative PCR (qPCR)
The leaf tissues were ground using liquid nitrogen and the total RNA was extracted using Spectrum™ Plant Total RNA Kit (Sigma–Aldrich, MO, USA) as recommended by the manufacturer. To remove any DNA contaminants, the isolated RNA was treated with DNase I (Invitrogen, MA, USA). The RNA samples from 0, 12, 24 and 96 hpi were selected for expression analysis using Real-Time qPCR. A total of 1 µg of total RNA was used for firststrand cDNA using LunaScript® RT SuperMix Kit (New England Biolabs, MA, USA). The target genes were FaMLO10, FaMLO17, and FaMLO20, while FaGAPDH2 was selected as endogenous control. Primers were designed using IDT PrimerQuest tool (https://www.idtdna.com/PrimerQuest/Home/Index) and all sequences were presented in Table S5. The qRT-PCR experiment was performed using LightCycler® 480 system (Roche, Swizerland) using Forget-Me-Not™ EvaGreen® qPCR Master Mix (Biotium, CA, USA). The Real-Time qPCR reaction was performed in triplicates of 100 ng of cDNA, 0.4 µl of each primer (400 nmol), and 3 µl of EvaGreen master mix in a final volume of 5 µl. The primer sequences were presented in Table S5. The reaction conditions were 95 °C for 5 min, followed by 40 cycles of 95 °C for 20 s, 60 °C for 20 s and 72 °C for 20 s followed by melting curve analysis to validate the single-amplicon product. The relative gene expression was calculated by using the Livak method (2−∆ΔCT)74.
RNAi vector construction and bacterial transformation
Using pssRNAit75, two ~200-bp RNAi fragments of genes FaMLO10 and FaMLO20 and one combined ~400-bp fragment were synthesized (Eurofins, Des Moines, IA, USA) and cloned in pDONR221™ vector (Thermo Fisher Scientific, Waltham, MA, USA) using standard procedures (Table S7). The insert identity was confirmed by sequencing (GENEWIZ, South Plainfield, NJ, USA) of three clones for each target gene. After checking, the fragments were inserted into silencing RNAi Gateway® vector pK7GWIWG2(II)76 and the transformants were confirmed by sequencing both strands. The constructs were then inserted into Agrobacterium tumefaciens strain EHA105 and the transformed cells were tested by PCR using specific primer pairs for the presence of RNAi constructs (Table S7).
Agrobacterium-mediated leaf transformation
The method for transient leaf transformation was performed as described by Cui et al.77 with slight modifications. One-month-old cultivated octoploid strawberry seedlings, ‘Sensation®Florida127’ from the University of Florida were used for leaf transformation. The seedlings were submerged in a 400-ml glass beaker with 350-ml bacterial suspension and a vacuum was applied. The vacuum was kept at 1.3 × 10−8 MPa for 3 min and was slowly released to facilitate Agrobacterium infiltration in the leaf. The excess bacterial suspension on the leaf surfaces was removed using filter paper. The seedlings were placed in the growth chamber with ambient and normal conditions (22 °C with RH of ~70%, 14 h of light, and 10 h of dark daily cycle). The seedlings were kept for five days in the growth room, and then leaf samples were collected for RNA isolation.
Transcript and phenotypic analysis of silenced FaMLO genes
As described previously, mRNA levels of genes FaMLO10 and FaMLO20 were quantified using qRT-PCR in silenced and nonsilenced strawberry. The remaining seedlings were then infected by PM pathogen, as previously described and were assessed for disease progression and PM severity symptoms up to 16 DAI. The disease severity was rated as described by Kennedy et al.78 using a modified Horsfall–Barratt scale of 0–6 based on mycelial leaf coverage with zero and six as the least (0%) and most severe (100%), respectively. The disease incidence (DI) and disease severity index (DSI)79 were calculated using the following formula:
Supplementary information
Acknowledgements
This work was supported by the Florida Strawberry Research and Education Foundation (FSREF), and the “Next-generation Disease Resistance Breeding and Management Solutions for Strawberry” under award no. 2017-51181-26833. We thank all the members of the strawberry genetics and breeding program, and the strawberry molecular genetics and genomics program for their technical assistance.
Author contributions
R. T., C. R. B. and S. L conceived and designed experiments for the genomic characterization of MLO genes in strawberry. S. C. assisted R. T. in synteny analysis. R. T. conducted gene expression profiling and transient gene expression silencing experiments of MLO genes against the PM pathogen. R. T., C. R. B., K. M. F., V. M. W., and S. L. wrote the paper, and all the remaining authors contributed to the final version of the paper.
Conflict of interest
The authors declare no competing interests.
Supplementary information
The online version contains supplementary material available at 10.1038/s41438-021-00587-y.
References
- 1.Devoto A, et al. Topology, subcellular, localization, and sequence diversity of the Mlo family in plants. J. Biol. Chem. 1999;274:13. doi: 10.1074/jbc.274.49.34993. [DOI] [PubMed] [Google Scholar]
- 2.Kim MC, et al. Mlo, a modulator of plant defense and cell death, is a novel calmodulinbinding protein. Isolation and characterization of a rice Mlo homologue. J. Biol. Chem. 2002;277:19304–19314. doi: 10.1074/jbc.M108478200. [DOI] [PubMed] [Google Scholar]
- 3.Ablazov A, Tombuloglu H. Genome-wide identification of the mildew resistance locus O (MLO) gene family in novel cereal model species Brachypodium distachyon. Eur. J. Plant Pathol. 2015;145:239–253. doi: 10.1007/s10658-015-0833-2. [DOI] [Google Scholar]
- 4.Liu Q, Zhu H. Molecular evolution of the MLO gene family in Oryza sativa and their functional divergence. Gene. 2008;409:1–10. doi: 10.1016/j.gene.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 5.Pessina S, et al. Characterization of the MLO gene family in Rosaceae and gene expression analysis in Malus domestica. BMC Genomics. 2014;15:618. doi: 10.1186/1471-2164-15-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kusch S, Pesch L, Panstruga R. Comprehensive phylogenetic analysis sheds light on the diversity and origin of the MLO family of integral membrane proteins. Genome Biol. Evol. 2016;8:878–895. doi: 10.1093/gbe/evw036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Acevedo-Garcia J, Kusch S, Panstruga R. Magical mystery tour: MLO proteins in plant immunity and beyond. New Phytol. 2014;204:273–281. doi: 10.1111/nph.12889. [DOI] [PubMed] [Google Scholar]
- 8.Feechan A, Jermakow A, Torregrosa L, Panstruga R, Dry I. Identification of grapevine MLO gene candidates involved in susceptibility to powdery mildew. Funct. Plant Biol. 2008;35:1255–1266. doi: 10.1071/FP08173. [DOI] [PubMed] [Google Scholar]
- 9.Deshmukh R, Singh VK, Singh BD. Comparative phylogenetic analysis of genome-wide Mlo gene family members from Glycine max and Arabidopsis thaliana. Mol. Genet Genomics. 2014;289:345–359. doi: 10.1007/s00438-014-0811-y. [DOI] [PubMed] [Google Scholar]
- 10.Kusch S, Panstruga R. mlo-Based resistance: an apparently universal “Weapon” to defeat powdery mildew disease. Mol. Plant Microbe Interact. 2017;30:179–189. doi: 10.1094/MPMI-12-16-0255-CR. [DOI] [PubMed] [Google Scholar]
- 11.Pastruga R. Serpentine plant MLO proteins as entry portals for powdery mildew fungi. Biochem. Soc. Trans. 2005;33:389–392. doi: 10.1042/BST0330389. [DOI] [PubMed] [Google Scholar]
- 12.Jørgensen IH. Discovery, characterization and exploitation of Mlo powdery mildew resistance in barley. Euphytica. 1992;63:141–152. doi: 10.1007/BF00023919. [DOI] [Google Scholar]
- 13.Consonni C, et al. Conserved requirement for a plant host cell protein in powdery mildew pathogenesis. Nat. Genet. 2006;38:716–720. doi: 10.1038/ng1806. [DOI] [PubMed] [Google Scholar]
- 14.Zheng Z, et al. Loss of function in Mlo orthologs reduces susceptibility of pepper and tomato to powdery mildew disease caused by Leveillula taurica. PLoS ONE. 2013;8:e70723. doi: 10.1371/journal.pone.0070723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu X, et al. Antisense RhMLO1 genetransformation enhances resistance to the powdery mildew pathogen in Rosa multiflora. Plant Mol. Biol. Report. 2015;33:1659–1665. doi: 10.1007/s11105-015-0862-1. [DOI] [Google Scholar]
- 16.Pessina S, et al. Knockdown of MLO genes reduces susceptibility to powdery mildew in grapevine. Hortic. Res. 2016;3:16016. doi: 10.1038/hortres.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong C, Weiping K, Junfen L, Jiping L. Analysis of powdery mildew resistance in wild melon MLO mutants. Horticultural Plant J. 2015;1:165–171. [Google Scholar]
- 18.Nie J, et al. Loss-of-function mutations in CsMLO1 confer durable powdery mildew resistance in cucumber (Cucumis sativus L.) Front. Plant Sci. 2015;6:1155–1155. doi: 10.3389/fpls.2015.01155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pavan S, et al. Pea powdery mildew er1 resistance is associated to loss-of-function mutations at a MLO homologous locus. Theor. Appl. Genet. 2011;123:1425–1431. doi: 10.1007/s00122-011-1677-6. [DOI] [PubMed] [Google Scholar]
- 20.Appiano M, et al. Identification of candidate MLO powdery mildew susceptibility genes in cultivated Solanaceae and functional characterization of tobacco NtMLO1. Transgenic Res. 2015;24:847–858. doi: 10.1007/s11248-015-9878-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai Y, et al. Naturally occurring broad-spectrum powdery mildew resistance in a Central American tomato accession is caused by loss of mlo function. Mol. Plant Microbe Interact. 2008;21:30–39. doi: 10.1094/MPMI-21-1-0030. [DOI] [PubMed] [Google Scholar]
- 22.Elliott C, Zhou F, Spielmeyer W, Panstruga R, Schulze-Lefert P. Functional conservation of wheat and rice Mlo orthologs in defense modulation to the powdery mildew fungus. Mol. Plant Microbe Interact. 2002;15:1069–1077. doi: 10.1094/MPMI.2002.15.10.1069. [DOI] [PubMed] [Google Scholar]
- 23.Devoto A, et al. Molecular phylogeny and evolution of the plant-specific seventransmembrane MLO family. J. Mol. Evol. 2003;56:77–88. doi: 10.1007/s00239-002-2382-5. [DOI] [PubMed] [Google Scholar]
- 24.Acevedo-Garcia J, et al. mlo-based powdery mildew resistance in hexaploid bread wheat generated by a non-transgenic TILLING approach. Plant Biotechnol. J. 2017;15:367–378. doi: 10.1111/pbi.12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nekrasov V, et al. Rapid generation of a transgene-free powdery mildew resistant tomato by genome deletion. Sci. Rep. 2017;7:482. doi: 10.1038/s41598-017-00578-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peries OS. Studies on strawberry mildew, caused by Sphaerotheca macularis (Wallr. ex Fries) Jaczewski*. Ann. Appl. Biol. 1962;50:211–224. doi: 10.1111/j.1744-7348.1962.tb06004.x. [DOI] [Google Scholar]
- 27.Xiao CL, et al. Comparison of epidemics of botrytis fruit rot and powdery mildew of strawberry in large plastic tunnel and field production systems. Plant Dis. 2001;85:901–909. doi: 10.1094/PDIS.2001.85.8.901. [DOI] [PubMed] [Google Scholar]
- 28.Paulus A. Fungal disease of strawberry. HortScience. 1990;25:885–889. doi: 10.21273/HORTSCI.25.8.885. [DOI] [Google Scholar]
- 29.Peres, N. A. & Mertely. J. C. Powdery Mildew of Strawberries. Electronic Data Information Source UF/IFAS Extension (2018).
- 30.Nelson MD, Gubler WD, Shaw DV. Relative resistance of 47 strawberry cultivars to powdery mildew in california greenhouse and field environments. Plant Dis. 1996;80:326–328. doi: 10.1094/PD-80-0326. [DOI] [Google Scholar]
- 31.Njuguna W, Liston A, Cronn R, Ashman T-L, Bassil N. Insights into phylogeny, sex function and age of Fragaria based on whole chloroplast genome sequencing. Mol. Phylogenet. Evol. 2013;66:17–29. doi: 10.1016/j.ympev.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 32.Shulaev V, et al. The genome of woodland strawberry (Fragaria vesca) Nat. Genet. 2011;43:109–116. doi: 10.1038/ng.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anciro A, et al. FaRCg1: a quantitative trait locus conferring resistance to Colletotrichum crown rot caused by Colletotrichum gloeosporioides in octoploid strawberry. Theor. Appl Genet. 2018;131:2167–2177. doi: 10.1007/s00122-018-3145-z. [DOI] [PubMed] [Google Scholar]
- 34.Salinas N, Verma S, Peres N, Whitaker VM. FaRCa1: a major subgenome-specific locus conferring resistance to Colletotrichum acutatum in strawberry. Theor. Appl. Genet. 2019;132:1109–1120. doi: 10.1007/s00122-018-3263-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roach JA, et al. FaRXf1: a locus conferring resistance to angular leaf spot caused by Xanthomonas fragariae in octoploid strawberry. Theor. Appl. Genet. 2016;129:1191–1201. doi: 10.1007/s00122-016-2695-1. [DOI] [PubMed] [Google Scholar]
- 36.Hirakawa H, et al. Dissection of the octoploid strawberry genome by deep sequencing of the genomes of fragaria species. DNA Res. 2013;21:169–181. doi: 10.1093/dnares/dst049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edger PP, et al. Reply to: revisiting the origin of octoploid strawberry. Nat. Genet. 2020;52:5–7. doi: 10.1038/s41588-019-0544-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng C, et al. Tracing the diploid ancestry of the cultivated octoploid strawberry. Mol. Biol. Evol. 2020;38:478–485. doi: 10.1093/molbev/msaa238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edger PP, et al. Origin and evolution of the octoploid strawberry genome. Nat. Genet. 2019;51:541–547. doi: 10.1038/s41588-019-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edger PP, et al. Single-molecule sequencing and optical mapping yields an improved genome of woodland strawberry (Fragaria vesca) with chromosome-scale contiguity. Gigascience. 2018;7:1–7. doi: 10.1093/gigascience/gix124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miao LX, et al. Genomic identification, phylogeny, and expression analysis of MLO genes involved in susceptibility to powdery mildew in Fragaria vesca. Genet. Mol. Res. 2016;15:gmr.15038400. doi: 10.4238/gmr.15038400. [DOI] [PubMed] [Google Scholar]
- 42.Shitsukawa N, et al. Genetic and epigenetic alteration among three homoeologous genes of a class E MADS box gene in hexaploid wheat. Plant Cell. 2007;19:1723. doi: 10.1105/tpc.107.051813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mutti JS, Bhullar RK, Gill KS. Evolution of gene expression balance among homeologs of natural polyploids. G3 Genes\Genomes\Genet. 2017;7:1225–1237. doi: 10.1534/g3.116.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobson L, Reményi I, Tusnády GE. CCTOP: a consensus constrained topology prediction web server. Nucleic acids Res. 2015;43:W408–W412. doi: 10.1093/nar/gkv451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horton P, et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Panstruga R. Discovery of novel conserved peptide domains by ortholog comparison within plant multi-protein families. Plant Mol. Biol. 2005;59:485–500. doi: 10.1007/s11103-005-0353-0. [DOI] [PubMed] [Google Scholar]
- 47.Appiano M, et al. Monocot and dicot MLO powdery mildew susceptibility factors are functionally conserved in spite of the evolution of class-specific molecular features. BMC Plant Biol. 2015;15:257. doi: 10.1186/s12870-015-0639-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiao Y, Paterson AH. Polyploidy-associated genome modifications during land plant evolution. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014;369:20130355. doi: 10.1098/rstb.2013.0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hardigan MA, et al. Genome Synteny has been conserved among the octoploid progenitors of cultivated strawberry over millions of years of evolution. Front. Plant Sci. 2020;10:1789. doi: 10.3389/fpls.2019.01789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiwan D, Roalson EH, Main D, Dhingra A. Antisense expression of peach mildew resistance locus O (PpMlo1) gene confers cross-species resistance to powdery mildew in Fragaria xananassa. Transgenic Res. 2013;22:1119–1131. doi: 10.1007/s11248-013-9715-6. [DOI] [PubMed] [Google Scholar]
- 51.Tao Y, Zhao X, Mace E, Henry R, Jordan D. Exploring and exploiting pangenomics for crop improvement. Mol. Plant. 2019;12:156–169. doi: 10.1016/j.molp.2018.12.016. [DOI] [PubMed] [Google Scholar]
- 52.Chen Z, et al. Expression analysis of the AtMLO gene family encoding plant-specific seven-transmembrane domain proteins. Plant Mol. Biol. 2006;60:583–597. doi: 10.1007/s11103-005-5082-x. [DOI] [PubMed] [Google Scholar]
- 53.Leister D. Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance gene. Trends Genet. 2004;20:116–122. doi: 10.1016/j.tig.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 54.Panchy N, Lehti-Shiu M, Shiu SH. Evolution of gene duplication in plants. Plant Physiol. 2016;171:2294–2316. doi: 10.1104/pp.16.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng Z, et al. Genome-wide study of the tomato SlMLO Gene family and its functional characterization in response to the powdery mildew fungus Oidium neolycopersici. Front Plant Sci. 2016;7:380. doi: 10.3389/fpls.2016.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andolfo G, et al. Evolutionary conservation of MLO gene promoter signatures. BMC Plant Biol. 2019;19:150. doi: 10.1186/s12870-019-1749-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Voorrips RE. MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002;93:77–78. doi: 10.1093/jhered/93.1.77. [DOI] [PubMed] [Google Scholar]
- 58.Guo A-Y, et al. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2014;31:1296–1297. doi: 10.1093/bioinformatics/btu817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. in 2010 Gateway Computing Environments Workshop (GCE) 1–8 (2010).
- 60.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kearse M, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 65.Sarkar, D. Lattice: Multivariate Data Visualization with R. (Springer, 2008).
- 66.Brors B, Gu L, Schlesner M, Eils R, Gu Z. circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 67.Marchler-Bauer A, et al. CDD: a Conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu W, et al. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015;31:3359–3361. doi: 10.1093/bioinformatics/btv362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mitchell C. MultAlin–multiple sequence alignment. Bioinformatics. 1993;9:614–614. [Google Scholar]
- 70.Sánchez-Sevilla JF, et al. Gene expression atlas of fruit ripening and transcriptome assembly from RNA-seq data in octoploid strawberry (Fragaria ×ananassa) Sci. Rep. 2017;7:13737. doi: 10.1038/s41598-017-14239-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Porebski S, Bailey LG, Baum BR. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Report. 1997;15:8–15. doi: 10.1007/BF02772108. [DOI] [Google Scholar]
- 72.Koren S, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Calis O, Cekic C, Soylu S, Tör M. Identification of new resistance sources from diploid wild strawberry against powdery mildew pathogen. Pak. J. Agric. Sci. 2015;52:677–683. [Google Scholar]
- 74.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quatitative PCR and the 2−ddCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 75.Ahmed F, et al. A web server for designing effective and specific plant siRNAs with genome-wide off-target assessment. Plant Physiol. 2020;184:65. doi: 10.1104/pp.20.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karimi M, Inzé D, Depicker A. GATEWAY™ vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci. 2002;7:193–195. doi: 10.1016/S1360-1385(02)02251-3. [DOI] [PubMed] [Google Scholar]
- 77.Wei W, et al. Ectopic expression of FvWRKY42, a WRKY transcription factor from the diploid woodland strawberry (Fragaria vesca), enhances resistance to powdery mildew, improves osmotic stress resistance, and increases abscisic acid sensitivity in Arabidopsis. Plant Sci. 2018;275:60–74. doi: 10.1016/j.plantsci.2018.07.010. [DOI] [PubMed] [Google Scholar]
- 78.Kennedy C, et al. Evaluation of strawberry species and cultivars for powdery mildew resistance in open-field and high tunnel production systems. HortScience. 2013;48:5. doi: 10.21273/HORTSCI.48.9.1125. [DOI] [Google Scholar]
- 79.Chaube, H. S. & Singh, U. S. Plant Disease Management: Principles and Practices (CRC Press, Boca Raton, 1991).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.