

Abstract
CD8+ T lymphocytes are pivotal cells in the host response to antitumor immunity. Tumor-driven microenvironments provide the conditions necessary for regulating infiltrating CD8+ T cells in favor of tumor survival, including weakening CD8+ T cell activation, driving tumor cells to impair immune attack, and recruiting other cells to reprogram the immune milieu. Also in tumor microenvironment, stromal cells exert immunosuppressive skills to avoid CD8+ T cell cytotoxicity. In this review, we explore the universal function and fate decision of infiltrated CD8+ T cells and highlight their antitumor response within various stromal architectures in the process of confronting neoantigen-specific tumor cells. Thus, this review provides a foundation for the development of antitumor therapy based on CD8+ T lymphocyte manipulation.
KEY WORDS: CD8+ T lymphocyte, Stromal cell, Tumor microenvironment, Immunosuppression, Immunotherapy, Antitumor
Graphical abstract
CD8+ T cells are key killing immune cells for antitumor immunity. At the same time, tumor-driven microenvironments including CAFs, TAMs, and tumor vessels can also participate to modulate infiltrating CD8+ T cells.
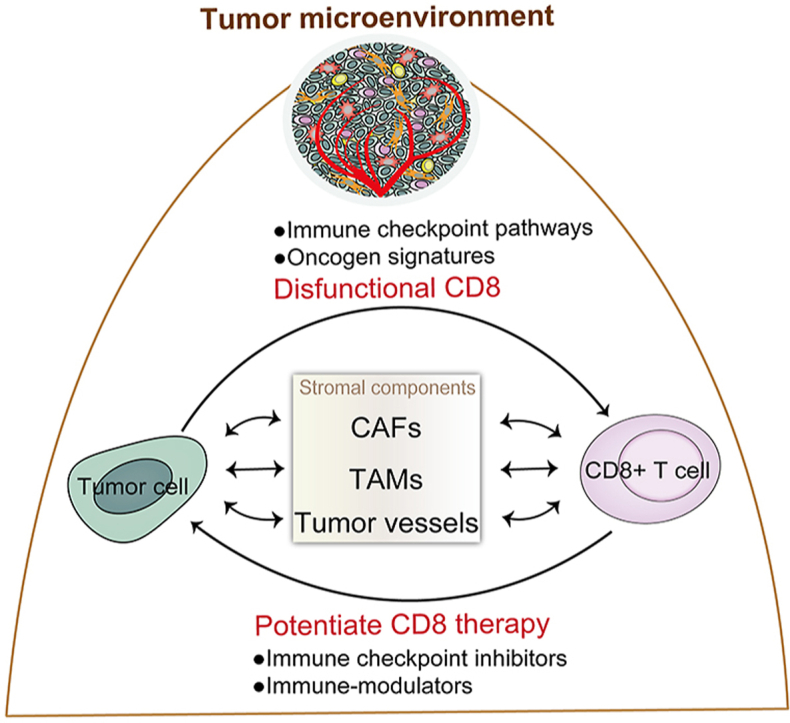
1. Introduction
Tumors are highly intricate, heterogeneous, and malignant tissue, characterized by abnormal invading and growing cells with a variety of biological and genetic aberrations. Many early studies have focused on tumor cells themselves to explain the drug resistance and poor prognosis of certain tumors, including gene mutations and activated signal pathways, rather than the integral status of the microenvironment of these types of cells. However, research is increasingly proving that the homeostasis of the cellular microenvironment is an important condition to foster normal cell proliferation, differentiation, and maturation, which ensures cell metabolism and functional activities. The concept of tumor microenvironment (TME) has been proposed particularly for solid tumors, which are specifically composed of tumor cells, growth factors, stromal cells, immunoinflammatory cells, and electrolytes, together remodeling the dynamic extracellular matrix progression1. The complex evolution of malignant tumors, from their occurrence to metastasis, is the outcome of the relationship between mutually reinforcing and antagonistic mechanisms between tumor cells and the TME components. Via these mechanisms, either the tumor cells adapt to the TME or the TME becomes capable of fostering tumor cells, depending on a sequence of adhesion molecules, receptors, and signals, including matrix metalloproteinases, cytoskeletal proteins, and receptor tyrosine kinase pathways.
With the development of tumor immunology, tumor cells have been found to be capable of developing resistance to the host's immunoreactivity. Immune status is governed by tumor-imbalanced microenvironment and may thus function plastically. The properties of the TME are prone to tumor-promoting immunosuppression due to an imbalance of the immune system impaired by counter-regulatory mechanisms2. These malignant tumors are able to domesticate immune cells in a state of low immunogenic activity, resulting in tumor resistance, a vital obstacle for therapeutic efficacy. The T lymphocyte cells in the TME are potential candidates for the generation of inflammatory factors that help tumor cells to escape suppressive environments. On the other hand, tumor cells are good at disguising themselves by reprogramming immune-related characteristics to avoid surveillance. Foremost, the activation of checkpoint inhibitory pathways by tumors regulates the ratio of renewing T lymphocytes, such as cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed cell death-1 (PD-1), as these two negative immune checkpoint regulators could be attracted in excess by the corresponding CD80 and PD-L1 molecule expression, respectively3. Moreover, other immune populations which harbor immunosuppressive abilities can also antagonize antitumor immune attacks, such as macrophages, dendritic cells (DCs), and regulatory T cells (Tregs)4.
Malignant cells are captured and eliminated by the tumor-infiltrated lymphocytes (TILs) cytotoxic responses. CD8+ T cells are key lymphocyte cells, responsible for inhibiting tumor proliferation and disrupting metastasis by directly recognizing and killing tumor cells via intracellular antigens. Previous studies have found that CD8+ T cells are positively relevant with cancer prognosis, controlling infections and cancer. However, the immunization mechanisms of tumor-penetrated T cells and tumor elimination are not yet fully understood. The amount of CD8+ T lymphocytes, as an antigen-specific effector, is considered an indicator of cancer regression5. Nevertheless, tumor cells are restricted with fewer CD8+ immunogenic signatures due to intrinsic heterogeneity6. The combination of immunosuppressive stromal components, immune cells, and factors in the TME results in a poor CD8+ T cell activation7. Additionally, evidence has made clear that CD8+ T lymphocyte activity may be inhibited by TME-stromal components. The majority of CD8+ T cells are dynamically adjusted into differentiation of complete CD8+ cytotoxic T lymphocytes. However, this process can be limited by a dysfunctional CD8+ T cell pool, which renegades towards tumor reactivity, benefiting cell proliferation8. Therefore, it is indispensable to determine the detailed regulatory mechanisms of the interaction between CD8+ T cells and TME components, as well as the role of stromal cells in local tumor inflammation, to characterize immune resistance disorder and develop immunotherapeutic strategies.
2. Immunological features of CD8+ T cells: Priming, differentiation, and cytotoxicity
Naïve T lymphocytes are initially inactive, and are programmed into mature T cells within thymus. The T cell receptors (TCRs) are the most important characteristic molecules on T cells and have crucial impact on realizing molecular binding and antigen recognition9. T cells only express a mutually exclusive subtype of CD4 or CD8 proteins after their differentiation, development, and maturation, which depends on a precise lineage from the double-positive stage of CD3+ CD4+ CD8+ T cells to a single-positive stage of CD3+CD4‒CD8+ or CD3+CD4+CD8‒ subtype. CD4+ T lymphocytes, also named helper T cells, play vital roles in immune regulation and antitumor effect. The CD4 molecules participate in the activation of TCR antigen-recognization and increasing the sensitive interaction of antigen-presenting cells (APC) and T cells via binding to β2 domain of self-major histocompatibility complex class Ⅱ molecules (MHC-Ⅱ). The naïve CD4+ T cells can be differentiated into multiple subsets under specific cytokines: Tregs, T helper cells (Th1, Th2, Th9, Th17, and Th22), and follicular helper T cell. CD4+ T lymphocytes exert antitumor by directly eliminating tumor cells in cytolytic-dependent manners or indirectly modulating the TME. Different CD4+ T subsets produce various cytokines for inflammation modulation10. For example, Th1 can release interferon-γ (IFN-γ), interleukin 2 (IL-2), and tumor necrosis factor (TNF), which promote defensive immunity mediated by macrophages, and mainly kill intracellular pathogens. Th2 can secrete IL-4, IL-5, IL-6, and IL-10, which promote specific B cells activation and antibody generation. Th17 can release cytokine IL-17 to withstand bacteria and fungi. While, Treg releases immunosuppressive cytokine IL-10 and TGF-β, which negatively regulate immunity by repressing CD4+ and CD8+ T cells activation and proliferation. In addition, CD4+ T lymphocytes can also elevate the antitumor response of CD8+ T cells by initiating their gene program to enhance cytotoxic T lymphocytes’ (CTL) function11,12. CD8+ T lymphocytes, as the main killing immune cell, secrete cytokines and recognize specific endogenous antigens of self-major histocompatibility complex class Ⅰ molecules (MHC-Ⅰ), and differ from CD4+ T cells, which engaged with recognizing exogenous antigens by restriction of self-MHC-Ⅱ molecules. During CD8+ T cell signal activation, TCRs bind to antigenic peptides in MHC-Ⅰ molecules to initiate signal activation. This process is transmitted to cells via CD3 molecules and supported by the binding of the CD8 co-receptor with the α3 domain of MHC-Ⅰ molecules to intensify signal transduction. Apart from CD3-TCR and CD8 molecule recognition, a series of costimulatory molecules also participate in T cell activation in the form of second interaction signals to match the APCs, which mediate antigen presentation via the formation of immunological synapses13. Once any of these costimulatory molecules successfully binds to their associated ligands on APCs, the activity of CD8+ T cells are influenced. This highlights that the connection with CD8+ T lymphocytes and APC is dynamic and can lead to diverse phenotypes, cytolytic activity, and immune functionality.
As a subtype of T lymphocytes, CD8+ T cells are activated immediately upon T cells initiation (Fig. 1). Once being activated, they are programmed to proliferate and differentiate into effector cells, known as CD8+ CTLs. CTLs play critical roles in targeting infected cells and eliminating tumor cells by generating an associated-antigen immune response. Thus, these cells take part in wide processes of physiological and pathological. The transmembrane glycoprotein CD8 is the main molecule expressed on the surface of CTLs, and can also be found in natural killer cells (NKs), cortical thymocytes, and DCs. Cytokine secretion and costimulatory molecule expression, alone with TCRs, are the key factors for CTL amplification, as well as differentiated into effector or memory CD8+ T lymphocytes. After antigen-specific recognition, unique cytolytic mediators are generated by active CTLs to exert an effective response against targeted tumors. To achieve more killing power, CD8+ CTLs have mastered two independent mechanisms of the perforin/granzyme pathway and FAS/FAS ligand (FASL) pathway, directly killing targeted cells without causing auto-injury14. Both pathways trigger caspase apoptosis in the cytosol of targeted cells. For the exocytosis perforin/granzyme pathway, perforin factors are released as pore-forming protein monomers in the granules of CD8+ CTLs cells, which can be inserted into the targeted cell membrane and polymerized to form an aqueous channel in a Ca2+-dependent manner, further destroying the membrane structure for targeted cells dying in osmotic swelling. In addition, granzymes enter targeted cells to activate caspase pathways via perforin-aqueous channels, inducing targeted cell apoptosis. In the FAS/FASL pathway, CD8+ CTLs express FAS ligands that identify FAS proteins expressed on targeted cells. This results in the activation of caspase-8 and caspase-3, subsequently inducing downstream apoptosis pathways. Conventionally, CD8+ CTLs prefer to utilize the perforin/granzyme pathway. Once the targeted cells are eradicated, CD8+ CTLs are immediately broken from the targeted cells synapses, allowing for an increased killing efficacy. TNF-α and IFN-γ are the major cytokines that eliminate malignant cells produced by activated CD8+ CTLs15. Once an infection or tumor is fully eradicated, most CD8+ CTLs are exhausted, and only a few will be conserved as memory CD8+ CTLs in the long-term, which allows for the self-renewal of a secondary response to antigen presentation16. As a result, CD8+ CTLs are comprised of powerful sensors exhibiting antitumor and anti-infection effects in the immune system.
Figure 1.

Infiltration of CD8+ T cells into tumors: differentiation, cytotoxicity, and dysfunction. (a) CD8+ T cells are programmed into CTLs after their activation and proliferation within the TME. The tumor cells are eventually eliminated, depending on IFN-γ and TNF-α secretion and apoptosis pathways. (b) Left: Various immune checkpoints in the divided region involved in CD8+ T cell dysfunction. Once the related ligand–receptor bindings are activated, CD8+ T cells are disabled and lose killing efficacy. Right: Oncologic signatures for suppressing tumor-infiltrating CD8+ T cells. Alternatively, WNT/β-catenin inhibits the differentiation and recruitment of CD8+ T cells against tumors. Other pathways include PI3K, STAT3, NF-κB, MYC, TP53, PTEN, LKB1, TOX, TCF-1, IDO, and CD39.
3. CD8+ T cells are exhausted by immune checkpoint pathways
During the course of CD8+ T cell activation, immune checkpoints are widely acknowledged as the main “brake signaling axis” responsible for tumor-peculiar CD8+ T-cell exhaustion, restricting their cytotoxic effect3,17. Generally, CD8+ T cells infiltrated in TME can be programmed into a dysfunction status either via an accumulated deficiency of cytolytic factors or with enhanced levels of immune checkpoint pathways, including PD-1, B- and T-lymphocyte attenuator (BTLA), CTLA-4, T cell Ig and ITIM domain (TIGIT), T-cell immunoglobulin and mucin-domain containing 3 (TIM-3), and lymphocyte-activation gene 3 (LAG-3), among multiple tumor types (Fig. 1)18. These inhibitory signaling pathways belong to intrinsic receptors that regulate both extracellular and intracellular signals in order to physiologically inhibit CD8+ T cell function. Understanding their mechanism will provide insights into how tumors escape immune surveillance and eradication, allowing for the utilization of immune checkpoint blockers to develop strategies for immunotherapy and augment CD8+ T-cell efficacy in tumors.
3.1. Programmed cell death-1
PD-1 play as a gatekeeper and master inhibitory modulator of the impact on stimulated CD8+ T cells within tumor tissues19. Ligand PD-L1/2 exists abundantly in both tumors and tumor-infiltrated T cells, B cells, macrophages, and DCs20. The PD-1/PD-L1 signaling within the TME plays an important role in abrogating CD8+ T cell activation, contributing to the exhaustion of proliferation and cytokine production, as well as promoting tumor escape in multiple tumors, such as ovarian cancer, melanoma, and lung cancer, among others21. The PD-1/PD-L1 axis mediates increased IL-10 production, inhibiting the immune response of antigen-specific CD8+ T cells22. High populations of PD-1 frequently exist in head and neck tumor, which can modulate CD8+ T cells into the “exhausted” phenotype more easily than that of low PD-1 populations23. Indeed, the threshold level of PD-1 in CD8+ TILs act as a potent marker, reflecting its conformity with clinical antitumor immunotherapy based on PD-1 interventions among spectrums of tumor types3.
The blocking of PD-1/PD-L1 interactions is generally taken advantages to restore the persisting killing-responses for CD8+ T cells during tumor-driven infiltration in the TME24. Moreover, the pool of PD-1+CD8+ T lymphocytes cooperates with certain epigenetic milieu to determine the efficacy of anti-PD-1 treatment. The genetic lineage of exhausted CD8+ T cells is distinct from that of effector and memory CD8+ T cells, and are transcriptionally re-engaged under treatment with anti-PD-125. Epigenetic analysis has suggested that “exhausted” CD8+ T cells preferentially express different chromatin profiles derived from active memory CD8+ T lymphocytes. The enhancer of RAR, T-bet, and SOX3 motifs is selective for PD-1 up-regulation in CD8+ T lymphocytes26. Strong evidence has emphasized that genomic landscapes, including mutations of molecular signature, DNA repair pathway, tumor-associated antigen burden, as well as immune heterogeneity and immunophenotype, are crucial factors to manipulate the sensitivity of anti-PD-1 to neoantigen-specific CD8+ T-cell recognition and surveillance for cancer patients27.
3.2. Cytotoxic T lymphocyte antigen 4
CTLA-4 is also an essential immune checkpoint in CD8+ T lymphocytes, responsible for dysregulating CD8+ T cell activation and expansion28. Functionally, the cytoplasmic domain of CTLA-4 is tasked with regulating the precise location of the second receptor CD28 in a trans-endocytosis manner28, competing with the costimulatory molecule CD28 for two ligands of B7 molecules, CD80 and CD86, on APC with higher appetencies29. In contrast, CTLA-4 is mainly existed on CD4+ T-cell surface rather than CD8+ T lymphocytes. As such, the antitumor effect of CTLA-4 monoclonal antibody may indirectly accelerate CD8+ T cell function, but enhancing CD4+ T lymphocytes30. CTLA-4 could curtail T-cell differentiation into an inactivated status. The mechanisms underlying CTLA-4 manipulation in CD8+ T cells include the control of cytokine excretion and cell cycle arrest, rather than directly induced apoptosis31. Novel mechanisms have revealed that the roles of CTLA-4 in dysregulating CD8+ T-cell expansion and proliferation represent dynamic outcomes of delaying the cell cycle upon the resting T-cell activation via IL-2 accumulation suppression32. Notably, previous studies have confirmed that CTLA-4 is distinct from PD-1 in terms of their cellular phenotypes, including the differential regulation of PI3K activity, which provides opportunities for their combination33. Similar with PD-1, cumulative CTLA-4 in the TME is closely related to CD8+ T cells eliminating obstacles by elevating the T cell activation threshold and impairing their expansion, ultimately worsening the outcome of tumor patients34. The effect of anti-CTLA-4 antibodies owes to the consequences of the directly-stimulated CD4+ and CD8+ effector T cells, rather than Treg damage35.
3.3. T-cell immunoglobulin and mucin-domain containing 3
TIM-3 is expressed on activated human T cells to exacerbate the “exhausted” condition in CD8+ T cells, benefiting cancer progression by dampening T cell liveness within TME36. It is emerging as an important immune target, similar to other T cell checkpoint receptors that mediate immune tolerance, and selectively up-regulated in infiltrating CD4+ and cytotoxic CD8+ T cells within TME than in CD11b+ CD45mid cells compared with peripheral blood mononuclear cells in glioblastoma and lung cancer37. It has been suggested that PD-1 and TIM-3 may limit CD8+ T-cell magnitude and activation depending on the secreted cytokine IL-10, in addition to blocking TNF-α, IL-2, and IFN-γ production, significantly enhancing the potential of PD-1 and TIM-3 as antitumor treatments by preventing CD8+ T cell consumption38. In colon cancer, the galectin-9/TIM-3 signaling axis enforces the apoptosis ratio of tumor-infiltrated CD8+ T cells39. Moreover, carcinoembryonic antigen cell adhesion molecule 1 (Ceacam1) is a heterodimer serving as TIM-3′ ligand40. This coexisting mechanism is required for CD8+ T-cell depletion in tumors. As a result, the dual repression of Ceacam1 and TIM-3 is feasible for the activated-CD8+ T cell’ enforcement with imposed IFN-γ in TME. In addition, TIM3+FOXP3+ Tregs have been found to shape tumor immunosuppression via sustainable dysfunctional CD8+ T cells41.
3.4. T cell Ig and ITIM domain
TIGIT signaling was discovered in 2009 as a key coinhibitory receptor analogous to the PD-1 signaling42. It is highly expressed in both human tumor-infiltrating T cells and chronic infection, responsible for restricting T cell activity. An abundance of TIGIT on CD8+ TILs is preferable to promote a less cytotoxic group of CD8+ T cells, and is co-expressed with higher PD-1 and TIM-3, plus low IL-2 and TNF-α43. TIGIT is expressed with PD-1 molecules, and is responsible for tumor-infiltrating lymphocytes to exhaust immune responses, possessing the ability to mark the dysfunctional phenomenon of effector CD8+ T cells, reducing the expansion and cytotoxicity subsets when confronted with viruses and tumors. This implies that TIGIT inhibition alone or its co-blockade with PD-1 are promising strategies to restore responses to antigen-specific CD8+ T cells in solid tumors, including in advanced melanoma44 and multiple myeloma45. Tregs with high levels of TIGIT have been found to elicit high levels of TIM-3 accumulation in local tumors, disrupting CD8+ T cells against tumors. In addition, TIGIT in CD8+ T cells could be restrained in a fibroblast activation protein 2-dependent manner within the TME46.
3.5. Lymphocyte activation gene 3
Similar with other inhibitory receptors, LAG-3 is an immune checkpoint molecule that is capable of negatively regulating signal transduction. LAG-3 is induced in effector T cells, NKs, and Tregs with conventional ligands of MHC-Ⅱ, as well as alternative ligands of fibrinogen-like protein 147. Studies have demonstrated that LAG-3 in TILs are independent markers, high levels of which indicate a poor diagnosis and tumor metastasis in cancer patients48. LAG-3 molecules assist in tumor escape thanks to being endowed with a powerful immunosuppressive microenvironment via the dysregulation of the homeostasis of tumor-infiltrated CD8+ T cells. MHC-Ⅱ aberrance is reason to impair CD8+ T-cell antitumor activity49. Preclinical evidence had indicated that the dual expression of protein PD-1 and LAG-3 exerts a synergistic effect on amplifying the exhausted subset of antigen-specific CD8+ T cells, promoting tumor malignancy, and making co-blockade a promising antitumor strategy50. The levels of LAG-3 combined with CTLA-4 content in CD8+ TILs represent prognostic indicators for the stratification of patients into distinct subgroups51. In addition, the co-existing of TIGIT and LAG-3 in TILs is also on behalf of an adverse prognostic factor, and has been accordant to PD-1 in melanoma52.
3.6. B and T lymphocyte attenuator
The overexpression of BTLA is another novel immunoregulatory receptor predominantly present in mature lymphocytes, such as T cells, B cells, DCs, macrophages and Tregs53. Functionally, BTLA molecules account for T cell exhaustion, characterized by depressed populations of CD8+ T cells, indicating a poor prognosis in cancer patients54. Herpesvirus entry mediator (HVEM) was initially identified as a specific ligand for BTLA. Since the BTLA/HVEM signaling axis is a vital pathway for sustaining the T cell immune response, its strong co-stimulation is capable of directly curtaining CD8+ T cell proliferation and activation53. Meanwhile, for melanoma tumors, the roles of BTLA pathway activation in manipulating CD8+ T cells are contradictory with not only defending CD8+ T cell from apoptosis, but also curtailing CD8+ T cell differentiation55. Further analysis revealed that low levels of BTLA together with low levels of PD-L1 resulted in a better rate of relapse-free survival for lung cancer patients56. Notably, the cytotoxic activity of CD8+ T cells in TME has been found to be elevated by the joint blockade of BTLA, TIM-3, and PD-1 in preclinical solid tumor models57.
3.7. V-domain Ig suppressor of T cell activation
VISTA was first confirmed to be a ligand in IGSF molecules (B7 family)58. During the adaptive immune response, VISTA is enriched in the TME and suppresses T cell activation, leading to tumor-induced immune suppression59. As a result, VSIG-3/IGSF11 has been described as a novel ligand that specifically interacts with VISTA60. Once the VSIG-3/VISTA signaling pathway is activated, the magnitude of T cells is subsequently suppressed, accompanied by remarkably reduced T cell-driven cytokine factors IL-2, IL-17, and IFN-γ, and decreased chemokines CCL3, CCL5, and CXCL11. It has been previously highlighted that VISTA expressed on APCs is able to directly depress CD8+ T cell enrichment61. Furthermore, anti-VISTA treatment has been found to reinvigorate effector CD8+ T cells and secretion of granzyme B and IFN-γ within TME62.
4. Dysfunctional tumor-infiltrated CD8+ T cells by oncologic signatures
The extent and location of CD8+ T cells in tumor site is positively correlated with the clinical diagnosis of cancer and patient prognosis in multi-type tumors, involving hepatocellular carcinoma, melanoma, colorectal cancer, breast cancer, and lung cancer, among others63. During the spread of metastatic tumors, tumor-penetrating CD8+ T cells are mainly determined by their trafficking and recruitment into local neoplasia tissues, which is directly in line with their immune response and killing efficacy. Nevertheless, when targeting tumors, most CD8+ T cells are vulnerable to efficient antitumor responses due to an impaired potency. Although our understanding of the underlying mechanisms is limited, their immunosuppressive status has been widely elucidated via a variety of dynamic adaptive changes within the TME8. Several researches have shown that CD8+ T-cell maintainability, especially for CTLs, is highly sensitive to inflammatory co-stimulators and co-inhibitors induced by the TME64. Specific cytokine secretion and signal transduction could also manipulate CD8+ CTL exhaustion, ultimately resulting in tumor-induced immune tolerance65. Tumors are motivated to interfere with antitumor immune responses. The molecules and genetic properties of activated oncogenic signaling and genotypes are intrinsically tumor aberrations, and are frequently reported to influence CD8+ T cell attraction and homeostasis across tumor types66. In what follows, several well-studied signaling pathways will be introduced (Fig. 1).
The WNT/β-catenin signaling axis is a valuable target for tumor immunotherapy because of its suppressive property for tumor-infiltrated T cells. This signaling has great significance in predicting immunotherapeutic implications and poor prognosis in patients by reducing CD8+ T cell recruitment into metastatic BRAF-mutated primary melanoma67. Meanwhile, WNT/β-catenin can prevent effector CD8+ T cells from differentiating into killing cells, resulting in immune exclusion with depleted CD8+ T cell trafficking into tumors68. Studies have also shown that the gain functions of the PI3K pathway show a significant disadvantage to immune surveillance, in part by depleting CD8+ T cells69. PI3Kγ accompanied with its downstream AKT and mTOR pathways acts as a switch to modulate immunosuppressive inflammation during tumor progression, mainly depending on the transcriptional procedure to restrain the activation of nuclear factor kappa B (NF-κB), but facilitates C/EBP-β activation70. mTOR signaling activation is related to memory CD8+ T cell differentiation71. Furthermore, in ER-positive breast cancer, PIK3CA-mutated tumor cells have shown increased PI3K downstream phosphorylation, which is adversely affiliated with tumoral-infiltrated CD8+ T cells, contributing to poor clinical results72. PI3K activation in CD8+ T cells in turn could rescue their exhaustion upon the blockade of PD-1/PD-L1 in gastrointestinal stromal tumors73. STAT3 signaling is regularly activated in both tumors and tumor-educated immune cells within the TME. These observations have shown that CD8+ T cell responses are markedly attenuated after inhibiting STAT3 activity due to abnormal DC differentiation74. The STAT3 pathway has been identified as being critical for the repression of the antitumoral functionality of CD8+ T cells by inhibiting IFN-γ/CXCR3/CXCL10 axis75. In addition, CD8+ CTLs are regulated by JAK–STAT3 signaling during glycolysis ablation in a fatty acid oxidation-dependent manner, further promoting fat-driven breast tumor progression76. In the evolution of hepatocellular carcinoma, high levels of STAT3 phosphorylation in CD8+ T cells are positively associated with the cumulative IL-6, IL-10, and IL-4 secretion, and negatively associated with IFN-γ77. STAT3 in tumor-infiltrated CD8+ T cells is induced by B cell-driven factor IL-35, which enables the ablation of CD8+ T-cell efficacy by limiting the generation of CXCR3, IFN-γ, and CCR5 in pancreatic ductal adenocarcinoma78. Furthermore, NF-κB activity in the TME has been reported to contribute to specific TNF-α and cyclooxygenase-2 (COX2) induction, which limits the cytotoxic type-1 response of CD8+ CTLs79. Genome-wide CRISPR screening for CD8 T cells has revealed that NF-κB signaling could be regulated by RNA helicase Dhx37 and thus modulate effector CD8+ T cells against triple-negative breast cancer80. On the other hand, RIPK1, driven by dying cells, activates NF-κB and initiates the immune responses of CD8+ T cells to associated antigens81.
In addition, a variety of intrinsic gene alterations are thought to be involved in modifying the tumor immune landscape2, such as MYC, TP53, PTEN, LKB1, TOX, TCF-1, IDO, and CD39. They have been reported to influence the accessibility of CD8+ TILs to local tumors and the activity of CTLs82. In myeloma mouse models, upon successful MYC activation, infiltrated effector CD8+ T cells were found to be substantially accelerated83. At the same time, MYC protein, a transcription factor, is able to activate CD8+ T cells by modulating metabolic reprogramming84. Studies have also shown that the greater the loss function of PTEN, TP53, and LKB1, the more CD8+ T cell exclusion is displayed via diverse downstream pathways. For instance, when PTEN is absent in melanoma cells, CD8+ T cell infiltration is sharply delayed in contrast with PTEN expression groups85. Otherwise, inactivating TP53, one of the most frequently mutated loss function genes for tumorigenesis, is able to harness CD8+ T cell capacity and free tumors of immunosurveillance86. The local activation of P53 in the TME has been shown to benefit tumor regression depending on the degree of reinforced tumoral infiltration of CD8+ T cells87. The LKB1 protein is a serine/threonine kinase mainly associated with cellular metabolism for cell viability. In a lung KRAS/LKB1 mouse model, profiling showed that STK11/LKB1 deficiency suppresses the infiltration ability of CD4 and CD8 T cells by recruiting neutrophil cells and secreting proinflammatory cytokines IL-6 and CXC-chemokine ligands88. The nuclear factor TOXs in CD8+ T cells act on the behalf of crucial transcriptional regulators that program the differentiation state toward the exhaustion state89. Additionally, TOXs are capable of dampening the functionality of CD8+ T cells by attenuating PD-1 degradation and releasing their translocation into the surface of tumoral-infiltrated CD8+ T cells90. TOX2, together with the orphan nuclear receptor 4 A family, has the potential to dysregulate effector CD8+ T cells, which are characterized by an exhausted phenotype of CD8+CAR+ PD-1 high TIM-3 high91. TCF-1 is another vital transcription factor that is committed to repressing CD8+ CTLs and is needed for the early stage fate of exhausted CD8+ T cells by upregulating a series of essential genes92, or in a WNT/β-catenin dependent manner93. Moreover, TCF-1 has been found to be a novel biomarker for responses to immune checkpoint blockades. The stem cell population of exhausted CD8+ T cells combined with evidence from single-cell RNA sequencing have revealed that PD-1 collaborates with TFC-1 to maintain CD8+ T cell precursor pools94. In addition, indoleamine 2,3-dioxygenase (IDO) and IDO2, key enzymes that break tryptophan into kynurenine in the metabolic pathway95, have also been found to be referred in the dysregulation of T cells. IDO is likely to be recognized by CD8+ T lymphocytes, and constitutes dominant anti-inflammatory responses against the immune system. Overexpressed IDO in various tumors has been found to be closely related to the abolishment of effector CD8+ T cells and invalid immunity96. Since the IDO gene is coordinately stimulated by IFN-γ, CTLA-4 may induce the generation of IDO in an IFN-γ dependent manner by binding to the CD80 and CD86 ligands on DCs97. Cholesterol has been reported as another positively linker of TME-metabolism factor, attenuating tumor-infiltrated CD8+ T cells via the ER–stress–XBP1 pathway98. After further mining the antigen-specific signatures of CD8+ T cell exhaustion, the surface molecule CD39 was abundantly and highly-expressed, leading to CD8+ T cells with less and less TNF-α and IL-2 secretion, similar to that observed in dysfunctional CD8+ T cells99.
5. Interconnectivity of CD8+ T cells with TME stromal cells
Generally, CD8+ T cells serve as the guarders which are naturally protective to host normal tissues and reject to external encroachment. But in tumors, it is more complicated. A dynamic interaction between tumor cells and the TME. For example, CD8+ T cells shapes the tumor progress, the antitumor immunity and the responsiveness to immunotherapy at the same time. Biological evidences have showed that the architecture of the tumor microenvironment displays suppressive immune characteristics, with tumors progressing via local invasion and distant metastasis within a complex and diverse environment comprised of tumor cells, immune cells, and stromal cells. Therein, tumor-relevant stromal cells belong to the family of local infiltrating cells comprised of tumor-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), and tumor angiogenesis. Their signatures are closely correlated with the negative accumulation of effector CD8+ T cells (Fig. 2). The innate and adaptive immunity within TME is responsible for monitoring tumorigenesis, progression, and invasion. Nevertheless, the TME may exacerbate tumor-inclined inflammation responses by reshaping the functionality of both stroma and immune cells in favor of malignant tumor ontogeny. In addition, a variety of inflammatory cytokine networks induced by the TME are also critical determinants of CD8+ T cell functionality, responsible for anti-infection and anti-tumor cells100. In this section, recent findings regarding the manipulation of the differentiation fate of CD8+ T cells are summarized, and the killing effect between different stromal participators and detail influencing factors.
Figure 2.

Suppressive immunization regulation of CD8+ T cells with stromal cells in the TME. The suppressive immunization regulation of CD8+ T cells for pro-tumoral microenvironment with TME-related stromal components is depicted in three parts: CAFs, TAMs, and tumor vessels. (1) For CAFs, the immune checkpoint molecules CTLA-4, TIM-3, PD-1, LAG-3, and CD73 are induced to attenuate CD8+ T cells. NF-κB prevents CD8+ T cells by upregulating CXCL12. IL-6 is secreted to downregulate CD8+ T cell infiltration and IL-6/STAT3 can master the PD-1/PD-L1 pathway to impair T cells by upregulating CXCR7. Tumor-specific CD8+ T cells are inhibited by TGF-β, along with two auto-stimulatory signaling of TGF-β and SDF-1. The CXCL12/CXCR4 axis induces FAP to diminish CD8+ T cells. FAS/FASL on T cells leads to CD8+ T cell apoptosis. CD8+ T cells are excluded with HDAC6 to activate STAT3 by targeting COX2. CAF-driven ROS, Chi3L1, βig-h3, and arginase II are capable of impairing CD8+ T cell activity. (2) For the TME, the immune checkpoint pathways PD-1/PD-L1/2 and CTLA-4/CD80/86 are observed on TAMs to confine CD8+ T cell initiation. NF-κB P65 is validated to impair CD8+ CTLs by inducing B7–H4/B7S1 and anti-apoptosis gene, as well as activating PD-1. Arginase and NO activity are modulators responsible for CD8+ T cell apoptosis via IFN-γ and TNF-α. IL-10 limits cytotoxic CD8+ T cells by suppressing DC-driven IL-12 or selectively reducing B7 upregulation. In addition, STAT1, CSF1, HLA-G, HLA-E, arginase I, and SHH from macrophages are crucial regulators to deplete the CD8+ T cell response. (3) During neovascularization, VEGF inhibits CD8+ T cell homing and induces apoptosis. HIF-1α may modulate vascularization via VEGF-A. FASL is selectively expressed in tumor-driven vasculatures to hinder CD8+ T cells with soluble VEGF-A, IL-10, and PGE2. NF-κB is capable of activating FASL and downregulating cFLIP for apoptosis. Intratumoral CD8+ T cells are rejected by RGS5, resulting in the formation of abnormal blood vessels and hypoxia. HIF-1α, ETBR, B7–H3 β-AR, PDPN, and TNF-α are also pivotal vascular molecules, impeding CD8+ T cell penetration into tumor sites.
5.1. Cancer-associated fibroblasts
As the most abundant stromal cell type, CAFs serve as a key immune inhibitory intermediator in the TME and have the capacity to orchestrate tumorigenesis proportions for tumor escape, as well as being highly associated with poor patient prognosis101. CAFs secrete large amounts of continuously activated paracrine and autocrine factors, chemokines, and signals via multiple mechanisms to facilitate a myriad of pro-tumorigenic stages and antitumor resistance, thereby influencing tumor-associated immune responses102. Studies have shown that CAF enrichment is inversely correlated with CD8+ T-cell content, counteracting CTLs’ efficacy against cancer. In immunotherapy, CD8+ T cells play key roles in local antitumor inflammation. However, their infiltration, cytokine production, and cytotoxic effects are indivisibly hindered by CAF activity. In return, CD8+ T cells may negatively modulate stromal fibroblast function. For instance, CD8+ T cells can overcome platinum-induced chemoresistance by regulating glutathione and cystine metabolism in fibroblasts in patients with ovarian cancer103. After CD8+ T-cells activation in TME, cytotoxic extracellular vesicles are preferentially produced to suppress CAFs in apoptosis104.
Human pancreatic CAFs express high levels of TIM-3, PD-1, CTLA-4, and LAG-3 in CD8+ T cells under proliferation, relying on activated prostaglandin E2 (PGE2)105. Furthermore, NF-κB driven by CAFs acts as a crucial tumor-promoting inflammatory factor and prevent CD8+ T cells from being recruited into tumors by upregulating CXCL12106. CAF-derived chitinase 3-like 1 (Chi3L1) is another signaling axis that is abundantly upregulated in cancer. It plays important roles in crippling tumor-infiltrated CD8+ T cells, which affects the recruitment and morphology of macrophages, CD4+ T cells, and TH1 cells, thereby shaping an immune milieu favorable for tumor survival and metastases107. When tumor cells and fibroblast cells are co-cultured, high expressions of IL-6 are secreted to downregulate CD8+ T cells infiltration and upregulate Treg population, further promoting an immunosuppressive TME108. The IL-6/STAT3 pathway masters CAF to induce the activation of PD-1/PD-L1 signaling in neutrophils, thus impairing T cells’ ability to inhibit hepatocellular carcinoma109. In esophageal squamous cell carcinoma, IL-6 secreted by CAFs promotes chemoresistance by upregulating CXCR7 expression110. In addition, CAFs may strongly impair CD8+ T-cell proliferation, as well as IFN-γ production by reprogramming monocytes to an immuno-inhibitory MDSC phenotype in the presence of reactive oxygen species (ROS)111. It has been reported that arginase II expression in CAFs is closely correlated with the diminished CD8+ T-cell recruitment, and could be used to predict poor prognosis in pancreatic cancer patients.
TGF-β is another typical regulatory factor in the TME. As a negative regulator, TGF-β can reduce the expansion and cytotoxicity of CD8+ T cells in vivo112. Low levels of TGF-β pathway activity indicate a strong cytotoxic efficacy of CD8+ T cells in tumor site after treatment with cyclophosphamide113. TGF-β and stromal cell-derived factor-1 (SDF-1) are two self-activated signaling molecules that form crosstalk loops, which are necessary for stromal fibroblasts to differentiate into myofibroblasts phenotype, thus stimulating tumor progression114. TGF-β signaling in stromal cells was previously found to attenuate effector CD8+ T cell penetration and inhibit their response to PD-1 inhibition115. In pancreatic ductal adenocarcinoma model, the CXCL12/CXCR4 axis was found to induce CAF surface marker FAP expression. FAP is able to aggregate the immunosuppressive atmosphere, further diminishing the accumulation of CD8+ T cells in tumors116. During non-small cell lung cancer (NSCLC) development, high levels of podoplanin-positive CAFs (PDPN+ CAFs) are consistent with the low ratio of CD8/FOXP3 T cells, indicating that CD8+ T-cell activation is blocked at that stage117. Observations in breast tumor models also found that FAP+PDPN+ CAFs cooperated with tumor cells, directly abrogating the proliferation of intratumoral CD8+ T cells based on the nitric oxide-dependent manner118. Additionally, CAFs play an immunosuppressive role by upregulating FAS/FASL in T cells and PD-1/PD-L2 signaling in CAFs in an antigen-dependent manner, resulting in CD8+ T cell dysfunction119. CAF-driven CD73 is a novel immune checkpoint that reduces the killing effect of CD8+ T cells with decreased IFN-γ production120. The presence of stromal protein βig-h3 from CAFs directly impairs the content and quality of intratumoral CD8+ T cells in pancreatic cancer121. High levels of histone deacetylase 6 (HDAC6) in CAFs are closely associated with poor survival in cancer patients. Mechanistically, HDAC6 activates CAF-driven STAT3 and targets the COX2 pathway, thus forming the immunosuppressive TME and excluding CD8+ T cell activity122.
5.2. Tumor-associated macrophages
Macrophages are a type of non-malignant stromal cell and on behalf of the first-line defense against tumors and infections123. Macrophages possess dynamic plasticity, encompassing two major morphologies, M1 and M2, whose polarization occurs under complex activation signaling. M1-like macrophages are activated macrophages that contribute to inflammatory responses, and indicate prolonged overall survival in cancer patients124. M2-like macrophages, on the contrary, damage the immune response. Notably, it is now well accepted that TAMs in the TME are inclined to M2 macrophage polarization. TAMs are classified into a variety of pro-tumor macrophages, promoting the development of malignant tumors and indicating a poor prognosis125.
Extensive studies have suggested that TAMs are able to directly modulate the tumor-immunosuppressive microenvironment in various aspects, making TAMs an important target in antitumor immunotherapy126. Significantly, tumor-associated macrophages possess diverse support networks, including that of the surrounding immune milieu, with inflammatory cytokines, regulatory factors, and pathways, to sustain tumor initiation and survival, as well as limit the cytotoxicity of T cells126. Since TAMs act as a crucial dominant immune supervisor in CD8+ T cells at tumor sites, the mechanism of the interaction between these two components was explored to elucidate how TAMs manipulate CD8+ T cell recruitment and cytotoxicity.
TAMs are programmed to express immune checkpoint ligands of PD-L1 and PD-L2, which represent major inhibitory functions that inhibit PD-1 molecules on introtumoral CD8+ T cells127. Similarly, immune checkpoint molecules CD80 and CD86 have also been observed to confine CD8+ T cell initiation in TAMs upon binding with CTLA-4128. Based on these findings, it has become clear that TAM intervention is feasible for immune checkpoint inhibition. Furthermore, TAMs are important resources, enriched in the hypoxic tumor microenvironments by generating hypoxia-inducible factor-1α (HIF-1α), which hinders T-cell activation and enhances tumor progression129. In addition, under hypoxic conditions, HIF-1α has been shown to upregulate PD-L1 accumulation in macrophages in the TME130. Thus, NF-κB appears to play key roles in the immune inflammatory. Evidence has also shown that intrinsic NF-κB P65 is capable of impairing CD8+ CTLs activity for promoting tumor progression, on one hand, by directly inducing the expression of T-cell inhibitory molecule B7x (B7-H4/B7S1) in TAMs. On the other hand, TAMs evade apoptosis by upregulating the anti-apoptosis gene resistant to CD8+ CTLs131. Additionally, NF-κB is a necessary modulator of PD-1 gene expression and activation in macrophages by binding to a specific upstream location in conserved region-C132. Studies have shown that increased arginase and NO activity generated by tumor-associated F4/80+ macrophages are the two main factors responsible for CD8+ T cell apoptosis via IFN-γ and TNF-α. This article also demonstrated that STAT1 signaling is a crucial regulator of TAM-inclined immunosuppressive activity, thereby depleting CD8+ T cell proliferation133. Colony-stimulating factor 1 (CSF1) is a key modulator of the differentiation and accumulation of M2 type-TAM polarization, thus representing a good target for anti-CSF-1R therapy134. In melanoma, the enrichment of CD8+ CTLs likely triggers enhanced CSF1 secretion, leading to TAM expansion. This recruiting mechanism for TAMs is in line with CD8+ T cell suppression, ultimately resulting in to resistance to immune checkpoint blockades135. As a result, the interruption of the CSF1/CSF1R axis could alter macrophage polarization, resulting in the enhancement of infiltrating CD8+ CTLs, which underlies chemotherapy and checkpoint immunotherapy in antitumor immunity136.
Infiltrating macrophages in the TME are an important source of IL-10 production, and aid in establishing a tolerant microenvironment. Studies have shown that IL-10 straightforwardly limits CD8+ cytotoxic T-cell responses by suppressing intratumoral DC-driven IL-12 or selectively reducing B7 upregulation137. Furthermore, HLA-G is an impressive molecule expressed on the surface of TAMs, allowing tumors to evade immunosurveillance138. LILRs promote T-cell dysfunction and proliferation by enhancing inhibitory cytokines IL-10 and TGF-β while reducing IFN-γ and IL-2. For instance, the cell surface receptors LIT2 and LIT4 have been found to compete with CD8 for MHC-Ⅰ binding139. Other studies have shown that inhibitory macrophages are modulated to express HLA-G antigens, binding to ILR2 on T cells, and function to diminish CD8+ CTL activity140. Moreover, most activated CD8+ T cells have been found to be able to rapidly obtain HLA-G1 from HLA-G+ APCs via trogocytosis with direct interaction, transferring from the effector state into immunosuppressive regulatory cells141. In addition, ILT4 expressed on infiltrating macrophages is recognized by HLA-G1, thus influencing subsequent immune activity139. Moreover, the enrichment of HLA-E on inflammatory macrophages is responsible for their interaction with the inhibitory receptor CD94/NKG2A in CD8+ TILs. In addition to suppressing the cytotoxic functions of NKs, this mechanism is further exploited by tumors to dampen CD8+ CTL antitumor responses142. On the other hand, arginase I production by macrophages contributes to a vital pro-tumorigenic mechanism143. The interaction of IL-4 and IL-13 with TAMs allows for the secretion of arginase I by l-arginine deprivation, which subsequently restricts the CD8+ T cell killing effect by inhibiting TCR CD3 ζ chain accumulation in activated T cells144. In addition, tumor-derived sonic hedgehog (SHH) has been demonstrated to drive pro-tumoral phenotype TAMs in a Krüppel-like factor 4 (KLF4)-dependent manner. This signaling further depletes CD8+ T cell recruitment into tumors by suppressing TAM-mediated CXCL9 and CXCL10 secretion145.
5.3. Tumor-associated vasculatures
TME-supported vasculatures are abnormal-stromal drivers that are crucial for the creation of an immune-suppressive microenvironment and are necessary for inducing tumor occurrence and recurrence. When the volume of tumors exceeds 2 mm3, they provide nutrition and oxygen by forming the necessary new microvascular coverage to maintain the barrier system for the survival, proliferation, and metastasis of tumors. At the same time, abnormal tumor vascularization can eliminate the localization, adhesion, and extravasation of T lymphocytes to endothelial cells and tumor tissue146. Even in cases where numerous CD8+ T cells are recruited to the tumor site, endowed with appropriate cytokine and chemokine attraction, abnormal blood vessels are capable of excluding CD8+ TILs, mainly owing to the orchestration effects of serious extracellular factors coordinated by immune checkpoint signaling activation. In this scenario, the interaction between CD8+ T cells and tumor cells will be abrogated by markedly inhibiting CD8+ T cells entering the solid tumor site from the circulatory system147. In addition to malignant tumors, the TME is also comprised of tight networks of surrounding cells, factors, and angiogenesis. This communication suggests that multi-step cellular establishment is involved in immune resistance. For instance, macrophages are able to harness T cells by dysfunctional perivascular regions to limit their extravasation into tumor sites. Therefore, a better understanding of its mechanism will help to guide clinical anti-tumor immunotherapy, relying on appropriate anti-angiogenesis agents to reshape inflammatory vascular normalization and enhance T lymphocyte infiltration. Mechanistically, a series of endothelium-relevant molecules have been found to be related to the deletion of CD8+ T cells. Foremost, the overexpression of circulating vascular endothelial growth factor (VEGF) is negatively correlated with the homing of host CD8+ T cells into tumors, inducing apoptosis in CD8+ T cells148. Therefore, the blockade of VEGF alone or in combination has attracted attention, since it creates an opportunity for the development of cytotoxic T lymphocyte immunotherapy. At the same time, HIF-1α may modulate vascularization by targeting VEGF-A, which is essential for cytotoxic CD8+ T cell deletion within the TME149. Moreover, the death mediator FASL is selectively expressed in human tumor-driven vasculatures in large numbers of solid cancer samples compared to normal organs150. In this context, the upregulated FASL is acquired to prevent the migration of effector CD8+ T cells into tumors while Tregs accumulate, which are cooperatively modulated by TME-inclined immunosuppressive factors, such as soluble VEGF-A, IL-10, and PGE2. In addition, NF-κB activates vascular FASL by recruiting HAT P300 and acetylated histones H3 and H4 to their promoter151. NF-κB also inversely downregulates cFLIP expression via the chromatin remodeling of increased HDAC1, decreased P300 histone acetylation, and reduced recruitment of transcription factor NFAT, ultimately resulting in apoptosis152. By contrast, the overexpression of VEGF-C results in an increased intratumoral CD8 T cell priming and migration ability in brain tumors, and shows synergistic effects with immune checkpoint inhibition therapy153. Furthermore, the regulator of G-protein signaling 5 (RGS5) has been identified as a pivotal vascular molecule for rejecting intratumoral CD8+ T cells in tumors via the formation of abnormal blood vessels and hypoxia154. During neovascularization, endothelin B receptor (ETBR) is another meaningful endothelial cell, whose upregulation hinders the inactivation of CD8+ TILs155. In addition, the overexpression of cell-surface protein B7-H3 is widely observed in tumor vasculatures, and has been found to inhibit tumor CD8+ T cell infiltration156. B7-H3-mediated drugs are thought to target the vasculature system to restore T cell function and eradicate tumors156. Researchers have found that β-adrenergic receptor (β-AR) signaling also strengthens tumor-related angiogenesis, accelerating pro-tumor features via AMPK pathway activation in a mouse ovarian carcinoma model. Notably, the blockade of the β-AR pathway has been found to enhance the initiation of CD8+ T cells157. Lastly, TNF-α in tumor tissues is a crucial adverse factor for the modulation of neovascularization. It resists the infiltration of CD8+ T lymphocytes and the uptake for anticancer drugs, whereby targeting TNF-α at low doses may allow for the switch to be made from stabilizing tumor-resultant vessels to active immunotherapy158.
6. Antitumor immunotherapy for targeting CD8+ T cells
Many basic research and clinical studies have investigated the regulation and function of CD8+ T cells in TME, providing the possibility of modulating CD8+ T cells to cure cancer. And some efficient approaches have been succeeded in clinical, such as immune checkpoint inhibitors and some immune-modulators targeting TME.
Tumor infiltrated CD8+ T cells are the main immune cells that respond to the immune checkpoint signaling pathways due to their high levels of immune checkpoints. They make CD8+ T cells easier to be “cheated” by tumor cells and exhausted with less IFN-γ secretion. Based on this, immune checkpoint inhibition is a valuable antitumor immunotherapy manner for improving the killing efficacy of T lymphocytes. The density of CD8+ T cells existing in TME is essential for immune checkpoint blockade to impair tumor growth, and may predicate the response to the checkpoint blockade therapy. Therefore, targeting immune checkpoints are promising strategies for CD8+ T cell-targeted immunotherapy. At present, the most successful immune checkpoint targets for drug development are PD-1, PD-L1, and CTLA-4. CTLA-4 restricts the priming of naive T cells in the lymphoid tissues, while PD-1/PD-L1 could cause the exhaustion of the effector T cells located in the TME159. Three PD-1 monoclonal antibodies nivolumab, pembrolizumab, and cemiplimab have been approved by FDA160. They can induce the augmentation and efficacy of exhausted-CD8+ T lymphocytes. And atezolizumab, avelumab, and durvalumab are three FDA-approved monoclonal antibodies against PD-L1. PD-L1 signaling blockade in TME can release inhibitory immunity and make CD8+ T cells normalized-activation for killing tumor cells. And CTLA-4 blockade with monoclonal antibodies ipilimumab or tremelimumab can augment the density of CTLs within the tumors, potentiate the activity of CD4+ T cells for further enhancement of CD8+ T cell-mediated immune responses159, as well as reduce Treg proportion and induce peripheral TCR reshaping in tumor tissues161. In addition, the mTOR signaling has also been reported to negatively regulate CD8+ T cells activation159. And some epigenetic targeting drugs such as DNA methyltransferase inhibitors have been demonstrated the potential to reverse immune suppression in various cancer models162. For example, EZH2 plays an important role in maintaining the survival of effector T cells, and EZH2 inhibitors and DNA methyltransferase inhibitor azacitidine can affect intratumoral CD8+ T cells infiltration159. Moreover, recent studies have also explored that some small molecule inhibitors affect CD8+ T cells by targeting the components of TME. For example, CSF-1R inhibitors can reprogram TAMs, and enhance T-cell mediated tumor eradication163.
7. Concluding remarks and perspectives
Tumor cells acquire adaptive immune resistance by manipulating the immune response, most likely bypassing immune cell surveillance and effectiveness by immuno-editing. In this review, research conducted on the characteristics and regulation of infiltrating CD8+ T cells during tumor progression and metastasis in recent decades was discussed. In addition, the intricate relationship between CD8+ T cells and TME-driven stromal components was also addressed. On the one hand, tumor-related stromal cells and infiltrated immune leukocytes cooperate to create an inflammatory environment to promote tumor cells survival. On the other hand, they are also able to reprogram the TME to impair CD8+ T-cell activity.
In host, CD8+ T cells are programed to differentiate into CD8+ CTLs and traffic into the tumor site to wipe out tumor cells. Herein the CAFs, TAMs, and endothelial cells act as three central TME-related stromal contributors to weaken CD8+ T cells cytotoxicity. Various factors, cytokines, and chemokines are secreted by TME-related stromal components, such as inflammatory factor (ILs, CCLs, CXCLs, and TGF-β, etc.), immune checkpoint molecules (PD-1, CTLA-4, and TIM-3, etc.), stromal molecules (SDF-1, βig-h3, and ETBR, etc.), as well as some signaling pathways (STAT3, AMPK, and HIF-1α, etc.). It is undisputable that these factors may be reciprocal to reshape the TME and work together to impair CD8+ T cells recruitment, activation, and cytotoxicity. And we believe that CD8+ T cell-targeted strategies provide hope for the success of TME immunotherapy. In conclusion, different TME components may be independent or co-existent to impede the killing efficacy of CD8+ T cells. Therefore, targeting CD8+ T cells, or CAFs, TAMs, and other TME components, to change the immunosuppressive state of TME could have important implications and a high clinical value in the development of cancer treatments. However, the interaction between CD8+ T cells and the tumor microenvironment will need to be explored further to provide breakthroughs for anticancer immunotherapy.
Acknowledgments
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Science (No. XDA12020101, China), and the National Natural Science Foundation of China (81773763). We would like to thank Editage (www.editage.cn) for English language editing.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Jian Ding, Email: jding@simm.ac.cn.
Yi Chen, Email: ychen@simm.ac.cn.
Author contributions
Qin Xie and Yi Chen drafted the manuscript. Jian Ding and Yi Chen revised the manuscript. Jian Ding and Yi Chen obtained funding. All authors approved the final version of the paper.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Quail D.F., Joyce J.A. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wellenstein M.D., Visser K.E. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity. 2018;48:399–416. doi: 10.1016/j.immuni.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Kim J.M., Chen D.S. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure) Ann Oncol. 2016;27:1492–1504. doi: 10.1093/annonc/mdw217. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y., Zeng G. Cancer and innate immune system interactions: translational potentials for cancer immunotherapy. J Immunother. 2012;35:299–308. doi: 10.1097/CJI.0b013e3182518e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ascierto P.A., Lewis K.D., Giacomo A.M., Demidov L., Mandala M., Bondarenko I. Prognostic impact of baseline tumour immune infiltrate on disease-free survival in patients with completely resected, BRAF(v600) mutation-positive melanoma receiving adjuvant vemurafenib. Ann Oncol. 2020;31:153–159. doi: 10.1016/j.annonc.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Evans R.A., Diamond M.S., Rech A.J., Chao T., Richardson M.W., Lin J.H. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight. 2016;1:88328–88344. doi: 10.1172/jci.insight.88328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galon J., Bruni D. Tumor immunology and tumor evolution: intertwined histories. Immunity. 2020;52:55–81. doi: 10.1016/j.immuni.2019.12.018. [DOI] [PubMed] [Google Scholar]
- 8.Li H., Leun A.M., Yofe I., Lubling Y., Solodkin D.G., Akkooi A.C. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019;176:775–789. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coulie P.G., Eynde B.J., Bruggen P., Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14:135–146. doi: 10.1038/nrc3670. [DOI] [PubMed] [Google Scholar]
- 10.Golubovskaya V., Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers. 2016;8:36–48. doi: 10.3390/cancers8030036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castellino F., Germain R.N. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu Rev Immunol. 2006;24:519–540. doi: 10.1146/annurev.immunol.23.021704.115825. [DOI] [PubMed] [Google Scholar]
- 12.Borst J., Ahrends T., Babala N., Melief C.M., Kastenmuller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18:635–647. doi: 10.1038/s41577-018-0044-0. [DOI] [PubMed] [Google Scholar]
- 13.VanGool S.W., Vandenberghe P., DeBoer M., Ceuppens J.L. CD80, CD86 and CD40 provide accessory signals in a multiple-step T-cell activation model. Immunol Rev. 1996;153:47–83. doi: 10.1111/j.1600-065x.1996.tb00920.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhang N., Bevan M.J. CD8+ T cells: foot soldiers of the immune system. Immunity. 2011;35:161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slaney C.Y., Kershaw M.H., Darcy P.K. Trafficking of T cells into tumors. Cancer Res. 2014;74:7168–7174. doi: 10.1158/0008-5472.CAN-14-2458. [DOI] [PubMed] [Google Scholar]
- 16.Mami C.F., Blanc C., Corgnac S., Hans S., Malenica I., Granier C. Resident memory T cells, critical components in tumor immunology. J Immunother Cancer. 2018;6:87–97. doi: 10.1186/s40425-018-0399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sade F.M., Yizhak K., Bjorgaard S.L., Ray J.P., Boer C.G., Jenkins R.W. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2019;176:404–407. doi: 10.1016/j.cell.2018.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson A.C., Joller N., Kuchroo V.K. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saito H., Kuroda H., Matsunaga T., Osaki T., Ikeguchi M. Increased PD-1 expression on CD4+ and CD8+ T cells is involved in immune evasion in gastric cancer. J Surg Oncol. 2013;107:517–522. doi: 10.1002/jso.23281. [DOI] [PubMed] [Google Scholar]
- 20.Keir M.E., Butte M.J., Freeman G.J., Sharpe A.H. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thommen D.S., Koelzer V.H., Herzig P., Roller A., Trefny M., Dimeloe S. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med. 2018;24:994–1004. doi: 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong H.D., Zhu G.F., Tamada K., Chen L.P. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 23.Kansy B.A., Concha B.F., Srivastava R.M., Jie H.B., Shayan G., Lei Y. PD-1 status in CD8+ T cells associates with survival and anti-PD-1 therapeutic outcomes in head and neck cancer. Cancer Res. 2017;77:6353–6364. doi: 10.1158/0008-5472.CAN-16-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J., Robert L. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pauken K.E., Sammons M.A., Odorizzi P.M., Manne S., Godec J., Khan O. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354:1160–1165. doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sen D. The epigenetic landscape of T cell exhaustion. Science. 2016;354:1165–1169. doi: 10.1126/science.aae0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype pelationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 28.Walker L.S., Sansom D.M. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. 2015;36:63–70. doi: 10.1016/j.it.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zang X., Allison J.P. The B7 family and cancer therapy: costimulation and coinhibition. Clin Cancer Res. 2007;13:5271–5279. doi: 10.1158/1078-0432.CCR-07-1030. [DOI] [PubMed] [Google Scholar]
- 30.Gattinoni L., Ranganathan A., Surman D.R., Palmer D.C., Antony P.A., Theoret M.R. CTLA-4 dysregulation of self/tumor-reactive CD8+ T-cell function is CD4+ T-cell dependent. Blood. 2006;108:3818–3823. doi: 10.1182/blood-2006-07-034066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chambers C.A., Kuhns M.S., Egen J.G., Allison J.P. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001;19:565–594. doi: 10.1146/annurev.immunol.19.1.565. [DOI] [PubMed] [Google Scholar]
- 32.Krummel M.F., Allison J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parry R.V., Chemnitz J.M., Frauwirth K.A., Lanfranco A.R., Braunstein I., Kobayashi S.V. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu F., Huang J., Liu X., Cheng Q., Luo C., Liu Z. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int. 2020;20:7–17. doi: 10.1186/s12935-019-1085-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Egen J.G., Kuhns M.S., Allison J.P. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol. 2002;3:611–618. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- 36.Liu F., Liu Y., Chen Z. Tim-3 expression and its role in hepatocellular carcinoma. J Hematol Oncol. 2018;11:126–138. doi: 10.1186/s13045-018-0667-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim H.S., Chang C.Y., Yoon H.J., Kim K.S., Koh H.S., Kim S.S. Glial TIM-3 modulates immune responses in the brain tumor microenvironment. Cancer Res. 2020;80:1833–1845. doi: 10.1158/0008-5472.CAN-19-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakuishi K., Apetoh L., Sullivan J.M., Blazar B.R., Kuchroo V.K., Anderson A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang C.W., Dutta A., Chang L.Y., Mahalingam J., Lin Y.C., Chiang J.M. Apoptosis of tumor infiltrating effector TIM-3+CD8+ T cells in colon cancer. Sci Rep. 2015;5:15659–15671. doi: 10.1038/srep15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Y.H., Zhu C., Kondo Y., Anderson A.C., Gandhi A., Russell A. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2015;517:386–390. doi: 10.1038/nature13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakuishi K., Ngiow S.F., Sullivan J.M., Teng M.W., Kuchroo V.K., Smyth M.J. TIM3+ FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. OncoImmunology. 2013;2:23849–23858. doi: 10.4161/onci.23849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu X., Harden K., Gonzalez L.C., Francesco M., Chiang E., Irving B. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- 43.Dougall W.C., Kurtulus S., Smyth M.J., Anderson A.C. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. 2017;276:112–120. doi: 10.1111/imr.12518. [DOI] [PubMed] [Google Scholar]
- 44.Chauvin J.M., Pagliano O., Fourcade J., Sun Z., Wang H., Sander C. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest. 2015;125:2046–2058. doi: 10.1172/JCI80445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guillerey C., Harjunpaeae H., Carrie N., Kassem S., Teo T., Miles K. TIGIT immune checkpoint blockade restores CD8+ T-cell immunity against multiple myeloma. Blood. 2018;132:1689–1694. doi: 10.1182/blood-2018-01-825265. [DOI] [PubMed] [Google Scholar]
- 46.Gur C., Ibrahim Y., Isaacson B., Yamin R., Abed J., Gamliel M. Binding of the Fap2 protein of fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42:344–355. doi: 10.1016/j.immuni.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J., Sanmamed M.F., Datar I., Su T.T., Ji L., Sun J. Fibrinogen-like protein 1 Is a major immune inhibitory ligand of LAG-3. Cell. 2019;176:334–347. doi: 10.1016/j.cell.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hald S.M., Rakaee M., Martinez I., Richardsen E., Saad S.A., Paulsen E.E. LAG-3 in non-small-cell lung cancer: expression in primary tumors and metastatic lymph nodes Is associated with improved survival. Clin Lung Cancer. 2018;19:249–259. doi: 10.1016/j.cllc.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 49.Donia M., Andersen R., Kjeldsen J.W., Fagone P., Munir S., Nicoletti F. Aberrant expression of MHC classⅡ in melanoma attracts inflammatory tumor-specific CD4+ T-cells, which dampen CD8+ T-cell antitumor reactivity. Cancer Res. 2015;75:3747–3759. doi: 10.1158/0008-5472.CAN-14-2956. [DOI] [PubMed] [Google Scholar]
- 50.Woo S.R., Turnis M.E., Goldberg M.V., Bankoti J., Selby M., Nirschl C.J. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–927. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang W., Chen D., Zhao Y., Zhao T., Wen J., Mao Y. Characterization of LAG-3, CTLA-4, and CD8+ TIL density and their joint influence on the prognosis of patients with esophageal squamous cell carcinoma. Ann Transl Med. 2019;7:776–788. doi: 10.21037/atm.2019.11.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee W.J., Lee Y.J., Choi M.E., Yun K.A., Won C.H., Lee M.W. Expression of lymphocyte-activating gene 3 and T-cell immunoreceptor with immunoglobulin and ITIM domains in cutaneous melanoma and their correlation with programmed cell death 1 expression in tumor-infiltrating lymphocytes. J Am Acad Dermatol. 2019;81:219–227. doi: 10.1016/j.jaad.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 53.Yu X., Zheng Y., Mao R., Su Z., Zhang J. BTLA/HVEM signaling: milestones in research and role in chronic hepatitis B virus infection. Front Immunol. 2019;10:617–625. doi: 10.3389/fimmu.2019.00617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qin S., Xu L., Yi M., Yu S., Wu K., Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer. 2019;18:155–169. doi: 10.1186/s12943-019-1091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haymaker C.L., Wu R.C., Ritthipichai K., Bernatchez C., Forget M.A., Chen J.Q. BTLA marks a less-differentiated tumor-infiltrating lymphocyte subset in melanoma with enhanced survival properties. OncoImmunology. 2015;4:1014246–1014261. doi: 10.1080/2162402X.2015.1014246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li X., Xu Z., Cui G., Yu L., Zhang X. BTLA expression in stage I–III non-small-cell lung cancer and its correlation with PD-1/PD-L1 and clinical outcomes. OncoTargets Ther. 2018;13:215–224. doi: 10.2147/OTT.S232234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fourcade J., Sun Z., Pagliano O., Guillaume P., Luescher I.F., Sander C. CD8+ T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012;72:887–896. doi: 10.1158/0008-5472.CAN-11-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ceeraz S., Nowak E.C., Noelle R.J. B7 family checkpoint regulators in immune regulation and disease. Trends Immunol. 2013;34:556–563. doi: 10.1016/j.it.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lines J.L., Sempere L.F., Broughton T., Wang L., Noelle R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. 2014;2:510–517. doi: 10.1158/2326-6066.CIR-14-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J., Wu G., Manick B., Hernandez V., Renelt M., Erickson C. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology. 2019;156:74–85. doi: 10.1111/imm.13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang L., Rubinstein R., Lines J.L., Wasiuk A., Ahonen C., Guo Y. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208:577–592. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nowak E.C., Lines J.L., Varn F.S., Deng J., Sarde A., Mabaera R. Immunoregulatory functions of VISTA. Immunol Rev. 2017;276:66–79. doi: 10.1111/imr.12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 64.He Q.F., Xu Y., Li J., Huang Z.M., Li X.H., Wang X. CD8+ T-cell exhaustion in cancer: mechanisms and new area for cancer immunotherapy. Brief Funct Genomics. 2019;18:99–106. doi: 10.1093/bfgp/ely006. [DOI] [PubMed] [Google Scholar]
- 65.Kallies A., Zehn D., Utzschneider D.T. Precursor exhausted T cells: key to successful immunotherapy?. Nat Rev Immunol. 2020;20:128–136. doi: 10.1038/s41577-019-0223-7. [DOI] [PubMed] [Google Scholar]
- 66.Rooney M.S., Shukla S.A., Wu C.J., Getz G., Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Massi D., Romano E., Rulli E., Merelli B., Nassini R., De Logu F. Baseline beta-catenin, programmed death-ligand 1 expression and tumour-infiltrating lymphocytes predict response and poor prognosis in BRAF inhibitor-treated melanoma patients. Eur J Cancer. 2017;78:70–81. doi: 10.1016/j.ejca.2017.03.012. [DOI] [PubMed] [Google Scholar]
- 68.Li X., Xiang Y., Li F., Yin C., Li B., Ke X. WNT/beta-catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front Immunol. 2019;10:2293–2305. doi: 10.3389/fimmu.2019.02293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sai J., Owens P., Novitskiy S.V., Hawkins O.E., Vilgelm A.E., Yang J. PI3K inhibition reduces mammary tumor growth and facilitates antitumor immunity and anti-PD1 responses. Clin Cancer Res. 2017;23:3371–3384. doi: 10.1158/1078-0432.CCR-16-2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaneda M.M., Messer K.S., Ralainirina N., Li H., Leem C.J., Gorjestani S. PI3K gamma is a molecular switch that controls immune suppression. Nature. 2016;539:437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Araki K., Turner A.P., Shaffer V.O., Gangappa S., Keller S.A., Bachmann M.F. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leite M.S., Salomon I., Opdam M., Kruger D.T., Beelen K.J., Noort V. Cancer-immune interactions in ER-positive breast cancers: PI3K pathway alterations and tumor-infiltrating lymphocytes. Breast Cancer Res. 2019;21:90–102. doi: 10.1186/s13058-019-1176-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao R., Song Y., Wang Y., Huang Y., Li Z., Cui Y. PD-1/PD-L1 blockade rescue exhausted CD8+ T cells in gastrointestinal stromal tumours via the PI3K/Akt/mTOR signalling pathway. Cell Prolif. 2019;52:12571–12581. doi: 10.1111/cpr.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu H., Kortylewski M., Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 75.Yue C., Shen S., Deng J., Priceman S.J., Li W., Huang A. STAT3 in CD8+ T cells inhibits their tumor accumulation by downregulating CXCR3/CXCL10 axis. Cancer Immunol Res. 2015;3:864–870. doi: 10.1158/2326-6066.CIR-15-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang C., Yue C., Herrmann A., Song J., Egelston C., Wang T. STAT3 activation-induced fatty acid oxidation in CD8+ T effector cells is critical for obesity-promoted breast tumor growth. Cell metab. 2020;31:148–161. doi: 10.1016/j.cmet.2019.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang X., Xin W., Zhang H., Zhang F., Gao M., Yuan L. Aberrant expression of p-STAT3 in peripheral blood CD4+ and CD8+ T cells related to hepatocellular carcinoma development. Mol Med Rep. 2014;10:2649–2656. doi: 10.3892/mmr.2014.2510. [DOI] [PubMed] [Google Scholar]
- 78.Mirlekar B., Michaud D., Lee S.J., Kren N.P., Harris C., Greene K. B cell-derived IL35 drives STAT3-dependent CD8+ T-cell exclusion in pancreatic cancer. Cancer Immunol Res. 2020;8:292–308. doi: 10.1158/2326-6066.CIR-19-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Theodoraki M.N., Yerneni S., Sarkar S.N., Orr B., Muthuswamy R., Voyten J. Helicase-driven activation of NF kappa B-COX2 pathway mediates the immunosuppressive component of dsRNA-driven inflammation in the human tumor microenvironment. Cancer Res. 2018;78:4292–4302. doi: 10.1158/0008-5472.CAN-17-3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dong M.B., Wang G., Chow R.D., Ye L., Zhu L., Dai X. Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell. 2019;178:1189–1204. doi: 10.1016/j.cell.2019.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yatim N., Saklani H.J., Orozco S., Schulz O., Silva R.B., Sousa C.R. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8+ T cells. Science. 2015;350:328–334. doi: 10.1126/science.aad0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Spranger S., Gajewski T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer. 2018;18:139–147. doi: 10.1038/nrc.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anderson D.A., Murphy T.L., Eisenman R.N., Murphy K.M. The MYCL and MXD1 transcription factors regulate the fitness of murine dendritic cells. Proc Natl Acad Sci U S A. 2020;117:4885–4893. doi: 10.1073/pnas.1915060117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Saravia J., Zeng H., Dhungana Y., Blanco D.B., Nguyen T.M., Chapman N.M. Homeostasis and transitional activation of regulatory T cells require c-Myc. Sci Adv. 2020;6:6443–6455. doi: 10.1126/sciadv.aaw6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Peng W., Chen J.Q., Liu C., Malu S., Creasy C., Tetzlaff M.T. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blagih J., Zani F., Chakravarty P., Hennequart M., Pilley S., Hobor S. Cancer-specific loss of p53 leads to a modulation of myeloid and T cell responses. Cell Rep. 2020;30:481–496. doi: 10.1016/j.celrep.2019.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guo G., Yu M., Xiao W., Celis E., Cui Y. Local activation of p53 in the tumor microenvironment overcomes immune suppression and enhances antitumor immunity. Cancer Res. 2017;77:2292–2305. doi: 10.1158/0008-5472.CAN-16-2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koyama S., Akbay E.A., Li Y.Y., Aref A.R., Skoulidis F., Herter G.S. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76:999–1008. doi: 10.1158/0008-5472.CAN-15-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Scott A.C., Dundar F., Zumbo P., Chandran S.S., Klebanoff C.A., Shakiba M. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571:270–274. doi: 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang X., He Q., Shen H., Xia A., Tian W., Yu W. TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J Hepatol. 2019;71:731–741. doi: 10.1016/j.jhep.2019.05.015. [DOI] [PubMed] [Google Scholar]
- 91.Seo H., Chen J., Avalos E.G., Castruita D.S., Das A., Wang Y.H. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc Natl Acad Sci U S A. 2019;116:12410–12415. doi: 10.1073/pnas.1905675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weber B.N., Chi A.W., Chavez A., Ohtani Y.Y., Yang Q., Shestova O. A critical role for TCF-1 in T-lineage specification and differentiation. Nature. 2011;476:63–68. doi: 10.1038/nature10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tiemessen M.M., Baert M.R., Kok L., Eggermond M.C., Elsen P.J., Arens R. T Cell factor 1 represses CD8+ effector T cell formation and function. J Immunol. 2014;193:5480–5487. doi: 10.4049/jimmunol.1303417. [DOI] [PubMed] [Google Scholar]
- 94.Chen Z., Ji Z., Ngiow S.F., Manne S., Cai Z., Huang A.C. TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity. 2019;51:840–855. doi: 10.1016/j.immuni.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Prendergast G.C. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene. 2008;27:3889–3900. doi: 10.1038/onc.2008.35. [DOI] [PubMed] [Google Scholar]
- 96.Prendergast G.C., Smith C., Thomas S., Nayak L.M., Kleintop L.L., Metz R. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. 2014;63:721–735. doi: 10.1007/s00262-014-1549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Grohmann U., Orabona C., Fallarino F., Vacca C., Calcinaro F., Falorni A. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3:1097–1101. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- 98.Ma X., Bi E., Lu Y., Su P., Huang C., Liu L. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metabol. 2019;30:143–156. doi: 10.1016/j.cmet.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Canale F.P., Ramello M.C., Nunez N., Furlan C.A., Bossio S.N., Serran M.G. CD39 expression defines cell exhaustion in tumor-infiltrating CD8+ T cells. Cancer Res. 2018;78:115–128. doi: 10.1158/0008-5472.CAN-16-2684. [DOI] [PubMed] [Google Scholar]
- 100.Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Yang T.H. The Immune landscape of cancer. Immunity. 2018;48:812–830. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Monteran L., Erez N. The dark side of fibroblasts: cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol. 2019;10:1835–1850. doi: 10.3389/fimmu.2019.01835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huelsken J., Hanahan D. A subset of cancer-associated fibroblasts determines therapy resistance. Cell. 2018;172:643–644. doi: 10.1016/j.cell.2018.01.028. [DOI] [PubMed] [Google Scholar]
- 103.Wang W., Kryczek I., Dostal L., Lin H., Tan L., Zhao L. Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell. 2016;165:1092–1105. doi: 10.1016/j.cell.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Seo N., Shirakura Y., Tahara Y., Momose F., Harada N., Ikeda H. Activated CD8+ T cell extracellular vesicles prevent tumour progression by targeting of lesional mesenchymal cells. Nat Commun. 2018;9:435–446. doi: 10.1038/s41467-018-02865-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gorchs L., Fernandez M.C., Bankhead P., Kern K.P., Sadeak I., Meng Q. Human pancreatic carcinoma-associated fibroblasts promote expression of co-inhibitory markers on CD4+ and CD8+ T-cells. Front Immunol. 2019;10:847–865. doi: 10.3389/fimmu.2019.00847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Garg B., Giri B., Modi S., Sethi V., Castro I., Umland O. NF kappa B in pancreatic stellate cells reduces infiltration of tumors by cytotoxic T cells and killing of cancer cells, via up-regulation of CXCL12. Gastroenterology. 2018;155:880–891. doi: 10.1053/j.gastro.2018.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cohen N., Shani O., Raz Y., Sharon Y., Hoffman D., Abramovitz L. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of chitinase 3-like 1. Oncogene. 2017;36:4457–4468. doi: 10.1038/onc.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kato T., Noma K., Ohara T., Kashima H., Katsura Y., Sato H. Cancer-associated fibroblasts affect intratumoral CD8+ and FoxP3+ T cells via IL6 in the tumor microenvironment. Clin Cancer Res. 2018;24:4820–4833. doi: 10.1158/1078-0432.CCR-18-0205. [DOI] [PubMed] [Google Scholar]
- 109.Cheng Y., Li H., Deng Y., Tai Y., Zeng K., Zhang Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018;9:422–433. doi: 10.1038/s41419-018-0458-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Qiao Y., Zhang C., Li A., Wang D., Luo Z., Ping Y. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene. 2018;37:873–883. doi: 10.1038/onc.2017.387. [DOI] [PubMed] [Google Scholar]
- 111.Xiang H., Ramil C.P., Hai J., Zhang C., Wang H., Watkins A.A. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol Res. 2020;8:436–450. doi: 10.1158/2326-6066.CIR-19-0507. [DOI] [PubMed] [Google Scholar]
- 112.Yang L., Pang Y., Moses H.L. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–227. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhong H., Lai Y., Zhang R., Daoud A., Feng Q., Zhou J. Low dose cyclophosphamide modulates tumor microenvironment by TGF-beta signaling pathway. Int J Mol Sci. 2020;21:957–974. doi: 10.3390/ijms21030957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kojima Y., Acar A., Eaton E.N., Mellody K.T., Scheel C., Ben P.I. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107:20009–20014. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mariathasan S., Turley S.J., Nickles D., Castiglioni A., Yuen K., Wang Y. TGF beta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fearon D.T. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res. 2014;2:187–193. doi: 10.1158/2326-6066.CIR-14-0002. [DOI] [PubMed] [Google Scholar]
- 117.Sakai T., Aokage K., Neri S., Nakamura H., Nomura S., Tane K. Link between tumor-promoting fibrous microenvironment and an immunosuppressive microenvironment in stage I lung adenocarcinoma. Lung Cancer. 2018;126:64–71. doi: 10.1016/j.lungcan.2018.10.021. [DOI] [PubMed] [Google Scholar]
- 118.Cremasco V., Astarita J.L., Grauel A.L., Keerthivasan S., MacIsaac K., Woodruff M.C. FAP delineates heterogeneous and functionally divergent stromal cells in immune-excluded breast tumors. Cancer Immunol Res. 2018;6:1472–1485. doi: 10.1158/2326-6066.CIR-18-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lakins M.A., Ghorani E., Munir H., Martins C.P., Shields J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8+ T Cells to protect tumour cells. Nat Commun. 2018;9:948–957. doi: 10.1038/s41467-018-03347-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yu M., Guo G., Huang L., Deng L., Chang C.S., Achyut B.R. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat Commun. 2020;11:515–532. doi: 10.1038/s41467-019-14060-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Goehrig D., Nigri J., Samain R., Wu Z., Cappello P., Gabiane G. Stromal protein beta ig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut. 2019;68:693–707. doi: 10.1136/gutjnl-2018-317570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li A., Chen P., Leng Y., Kang J. Histone deacetylase 6 regulates the immunosuppressive properties of cancer-associated fibroblasts in breast cancer through the STAT3-COX2-dependent pathway. Oncogene. 2018;37:5952–5966. doi: 10.1038/s41388-018-0379-9. [DOI] [PubMed] [Google Scholar]
- 123.Wynn T.A., Chawla A., Pollard J.W. Macrophage biology in development, homestasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Biswas S.K., Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 125.Noy R., Pollard J.W. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Quaranta V., Schmid M.C. Macrophage-mediated subversion of anti-tumour immunity. Cells. 2019;8:747–764. doi: 10.3390/cells8070747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu Y., Zugazagoitia J., Ahmed F.S., Henick B.S., Gettinger S.N., Herbst R.S. Immune cell PD-L1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin Cancer Res. 2020;26:970–977. doi: 10.1158/1078-0432.CCR-19-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mantovani A., Marchesi F., Malesci A., Laghi L., Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Doedens A.L., Stockmann C., Rubinstein M.P., Liao D., Zhang N., DeNardo D.G. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70:7465–7475. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Noman M.Z., Desantis G., Janji B., Hasmim M., Karray S., Dessen P. PD-L1 is a novel direct target of HIF-1 alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li L., Han L., Sun F., Zhou J., Ohaegbulam K.C., Tang X. NF-kappa B RelA renders tumor-associated macrophages resistant to and capable of directly suppressing CD8+ T cells for tumor promotion. OncoImmunology. 2018;7:250–263. doi: 10.1080/2162402X.2018.1435250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bally A.P., Lu P., Tang Y., Austin J.W., Scharer C.D., Ahmed R. NF-κB regulates PD-1 expression in macrophages. J Immunol. 2015;194:4545–4554. doi: 10.4049/jimmunol.1402550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kusmartsev S., Gabrilovich D.I. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174:4880–4891. doi: 10.4049/jimmunol.174.8.4880. [DOI] [PubMed] [Google Scholar]
- 134.Ries C.H., Cannarile M.A., Hoves S., Benz J., Wartha K., Runza V. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 135.Neubert N.J., Schmittnaegel M., Bordry N., Nassiri S., Wald N., Martignier C. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci Transl Med. 2018;10:436–466. doi: 10.1126/scitranslmed.aan3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cannarile M.A., Weisser M., Jacob W., Jegg A.M., Ries C.H., Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5:53–66. doi: 10.1186/s40425-017-0257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ruffell B., Chang S.D., Chan V., Rosenbusch A., Ho C.M., Pryer N. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26:623–637. doi: 10.1016/j.ccell.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Paul P., Rouas F.N., Khalil D.I., Moreau P., Riteau B., Gal F.A. HLA-G expression in melanoma: a way for tumor cells to escape from immunosurveillance. Proc Natl Acad Sci U S A. 1998;95:4510–4515. doi: 10.1073/pnas.95.8.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shiroishi M., Tsumoto K., Amano K., Shirakihara Y., Colonna M., Braud V.M. Human inhibitory receptors Ig-like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC classⅠbinding and bind preferentially to HLA-G. Proc Natl Acad Sci U S A. 2003;100:8856–8861. doi: 10.1073/pnas.1431057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Onno M., Pangault C., Friec G.L., Guilloux V., Andre P., Fauchet R. Modulation of HLA-G antigens expression by human cytomegalovirus: specific induction in activated macrophages harboring human cytomegalovirus infection. J Immunol. 2000;164:6426–6434. doi: 10.4049/jimmunol.164.12.6426. [DOI] [PubMed] [Google Scholar]
- 141.LeMaoult J., Caumartin J., Daouya M., Favier B., Rond S.L., Gonzalez A. Immune regulation by pretenders: cell-to-cell transfers of HLA-G make effector T cells act as regulatory cells. Blood. 2007;109:2040–2048. doi: 10.1182/blood-2006-05-024547. [DOI] [PubMed] [Google Scholar]
- 142.Hamid M.A., Wang R.Z., Yao X., Fan P., Li X., Chang X.M. Enriched HLA-E and CD94/NKG2A interaction limits antitumor CD8+ tumor-infiltrating T lymphocyte responses. Cancer Immunol Res. 2019;7:1293–1306. doi: 10.1158/2326-6066.CIR-18-0885. [DOI] [PubMed] [Google Scholar]
- 143.Pekarova M., Lojek A. The crucial role of l-arginine in macrophage activation: what you need to know about it. Life Sci. 2015;137:44–48. doi: 10.1016/j.lfs.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 144.Rodriguez P.C., Quiceno D.G., Zabaleta J., Ortiz B., Zea A.H., Piazuelo M.B. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 145.Petty A.J., Li A., Wang X., Dai R., Heyman B., Hsu D. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J Clin Invest. 2019;129:5151–5162. doi: 10.1172/JCI128644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Woods A.N., Wilson A.L., Srivinisan N., Zeng J., Dutta A.B., Peske J.D. Differential expression of homing receptor ligands on tumor-associated vasculature that control CD8 effector T-cell entry. Cancer Immunol Res. 2017;5:1062–1073. doi: 10.1158/2326-6066.CIR-17-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Albini A., Bruno A., Noonan D.M., Mortara L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: implications for immunotherapy. Front Immunol. 2018;9:527–546. [Google Scholar]
- 148.Shrimali R.K., Yu Z., Theoret M.R., Chinnasamy D., Restifo N.P., Rosenberg S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70:6171–6180. doi: 10.1158/0008-5472.CAN-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Palazon A., Tyrakis P.A., Macias D., Velica P., Rundqvist H., Fitzpatrick S. An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell. 2017;32:669–683. doi: 10.1016/j.ccell.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Motz G.T., Santoro S.P., Wang L.P., Garrabrant T., Lastra R.R., Hagemann I.S. Tumor endothelium FASL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20:607–615. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Aurora A.B., Biyashev D., Mirochnik Y., Zaichuk T.A., Sanchez M.C., Renault M.A. NF-kappa B balances vascular regression and angiogenesis via chromatin remodeling and NFAT displacement. Blood. 2010;116:475–484. doi: 10.1182/blood-2009-07-232132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sung B., Pandey M.K., Ahn K.S., Yi T., Chaturvedi M.M., Liu M. Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-κB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood. 2008;111:4880–4891. doi: 10.1182/blood-2007-10-117994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Song E., Mao T., Dong H., Boisserand L.S.B., Antila S., Bosenberg M. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature. 2020;577:689–694. doi: 10.1038/s41586-019-1912-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hamzah J., Jugold M., Kiessling F., Rigby P., Manzur M., Marti H.H. Vascular normalization in RGS5-deficient tumours promotes immune destruction. Nature. 2008;453:410–414. doi: 10.1038/nature06868. [DOI] [PubMed] [Google Scholar]
- 155.Buckanovich R.J., Facciabene A., Kim S., Benencia F., Sasaroli D., Balint K. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14:28–36. doi: 10.1038/nm1699. [DOI] [PubMed] [Google Scholar]
- 156.Seaman S., Zhu Z., Saha S., Zhang X.M., Yang M.Y., Hilton M.B. Eradication of tumors through simultaneous ablation of CD276/B7-H3-positive tumor cells and tumor vasculature. Cancer Cell. 2017;31:501–515. doi: 10.1016/j.ccell.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Daher C., Vimeux L., Stoeva R., Peranzoni E., Bismuth G., Wieduwild E. Blockade of beta-adrenergic receptors improves CD8+ T-cell priming and cancer vaccine efficacy. Cancer Immunol Res. 2019;7:1849–1863. doi: 10.1158/2326-6066.CIR-18-0833. [DOI] [PubMed] [Google Scholar]
- 158.Calcinotto A., Grioni M., Jachetti E., Curnis F., Mondino A., Parmiani G. Targeting TNF-alpha to neoangiogenic vessels enhances lymphocyte infiltration in tumors and increases the therapeutic potential of immunotherapy. J Immunol. 2012;188:2687–2694. doi: 10.4049/jimmunol.1101877. [DOI] [PubMed] [Google Scholar]
- 159.Farhood B., Najafi M., Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol. 2019;234:8509–8521. doi: 10.1002/jcp.27782. [DOI] [PubMed] [Google Scholar]
- 160.Mpakali A., Stratikos E. The role of antigen processing and presentation in cancer and the efficacy of immune checkpoint inhibitor immunotherapy. Cancers. 2021;13:134–164. doi: 10.3390/cancers13010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Robert L., Tsoi J., Wang X., Emerson R., Homet B., Chodon T. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin Cancer Res. 2014;20:2424–2432. doi: 10.1158/1078-0432.CCR-13-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhou L., Xu N., Shibata H., Saloura V., Uppaluri R. Epigenetic modulation of immunotherapy and implications in head and neck cancer. Cancer Metastasis Rev. 2021;55:20–32. doi: 10.1007/s10555-020-09944-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Osipov A., Saung M.T., Zheng L., Murphy A.G. Small molecule immunomodulation: the tumor microenvironment and overcoming immune escape. J Immunother Cancer. 2019;7:224–236. doi: 10.1186/s40425-019-0667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]