

Abstract
Neocortex expansion during human evolution provides a basis for our enhanced cognitive abilities. Yet, which genes implicated in neocortex expansion are actually responsible for higher cognitive abilities is unknown. The expression of human‐specific ARHGAP11B in embryonic/foetal mouse, ferret and marmoset neocortex was previously found to promote basal progenitor proliferation, upper‐layer neuron generation and neocortex expansion during development, features commonly thought to contribute to increased cognitive abilities. However, a key question is whether this phenotype persists into adulthood and if so, whether cognitive abilities are indeed increased. Here, we generated a transgenic mouse line with physiological ARHGAP11B expression that exhibits increased neocortical size and upper‐layer neuron numbers persisting into adulthood. Adult ARHGAP11B‐transgenic mice showed altered neurobehaviour, notably increased memory flexibility and a reduced anxiety level. Our data are consistent with the notion that neocortex expansion by ARHGAP11B, a gene implicated in human evolution, underlies some of the altered neurobehavioural features observed in the transgenic mice, such as the increased memory flexibility, a neocortex‐associated trait, with implications for the increase in cognitive abilities during human evolution.
Keywords: basal progenitors, brain evolution, human‐specific gene, memory flexibility, neocortex expansion
Subject Categories: Neuroscience
Effects of human‐specific ARHGAP11B, linked to higher cognitive abilities, on basal progenitor proliferation and upper‐layer neuron generation in rodent embryos persist into adulthood, correlating with altered neurobehaviour including reduced anxiety levels.
Introduction
Neocortex expansion during human evolution likely enabled our enhanced cognitive abilities (Rakic, 2009; Lui et al, 2011; Geschwind & Rakic, 2013; Florio & Huttner, 2014; Dehay et al, 2015; Fernandez et al, 2016; Miller et al, 2019; Molnar et al, 2019). A number of genes have been implicated in neocortex expansion because they have been shown to increase basal progenitor (BP) proliferation and abundance, leading to greater production of cortical neurons (Stahl et al, 2013; Boyd et al, 2015; Florio et al, 2015; Liu et al, 2017; Cardenas et al, 2018; Fiddes et al, 2018; Florio et al, 2018; Suzuki et al, 2018; Kalebic et al, 2019; Kostic et al, 2019; Heide et al, 2020). One such gene is the human‐specific ARHGAP11B. In the developing human neocortex, ARHGAP11B is preferentially expressed in neural progenitor cells (NPCs) (Florio et al, 2015; Florio et al, 2018).
ARHGAP11B arose by partial duplication of the widespread gene ARHGAP11A ≈ 5 mya, that is, after segregation of the lineage leading to modern humans from that leading to the chimpanzee (Sudmant et al, 2010; Antonacci et al, 2014). Subsequently, ARHGAP11B underwent a single base substitution that endowed ARHGAP11B with the ability to increase BP proliferation and abundance. Specifically, this single base substitution generated a new splice‐donor site, the use of which results in a reading frame shift and the generation of a unique, human‐specific 47‐amino acid C‐terminal sequence (Florio et al, 2015; Florio et al, 2016). Due to this change in protein sequence, the ARHGAP11B protein, in contrast to the ARHGAP11A protein, is imported into the mitochondria of NPCs where it acts to increase glutaminolysis, a metabolic pathway that underlies the ARHGAP11B‐induced increase in BP proliferation and abundance (Namba et al, 2020). ARHGAP11B has emerged as a particularly promising candidate gene for the evolutionary expansion of the human neocortex because not only when overexpressed in embryonic mouse and ferret neocortex (Florio et al, 2015; Kalebic et al, 2018), but also when expressed to physiological levels in the foetal neocortex of a non‐human primate, the common marmoset, ARHGAP11B increases BP proliferation and abundance, raises the generation specifically of upper‐layer neurons and expands the neocortex during development (Heide et al, 2020).
However, two key questions regarding the potential role of ARHGAP11B in the evolutionary expansion of the human neocortex have remained unanswered. First, would the ARHGAP11B‐induced phenotype of increased cortical neuron numbers and an expanded neocortex during development persist into adulthood? Second, if so, would this phenotype be associated with higher cognitive abilities? To address these questions, we generated a stable transgenic mouse line that expresses an ARHGAP11B protein in the neocortical NPCs at a physiological level.
Results
Generation of an ARHGAP11B‐transgenic mouse line
To generate a stable transgenic mouse line that expresses an ARHGAP11B protein, we first determined the temporal and spatial expression patterns of human ARHGAP11A and human ARHGAP11B mRNAs by qPCR of foetal human neocortical tissue at various developmental stages (gestational weeks 12–21; Fig EV1A and B) and by analysing previously published RNA‐seq data sets of defined isolated NPC and neuron populations (Florio et al, 2015) (Fig EV1C and D), respectively. As the expression patterns of ARHGAP11A and ARHGAP11B mRNAs were found to be similar, we decided to generate the transgenic mouse line by converting one allele of the mouse Arhgap11a gene into a mutant mouse ARHGAP11B gene (mARHGAP11B), using the CRISPR/Cas9 genome editing technology (for details, see Materials and Methods). In mARHGAP11B, the 55 nucleotides of Arhgap11a that in humans would be deleted from the ARHGAP11B mRNA by splicing using the new splice‐donor site are replaced by the 141 nucleotides encoding the human‐specific 47‐amino acid sequence plus three nucleotides to generate a translational stop codon (Fig EV1E). Unless indicated otherwise, the ARHGAP11B‐transgenic mice obtained (referred to as 11B mice hereafter) were used as heterozygous animals, that is, with one mouse Arhgap11a allele being replaced by mARHGAP11B. The resulting ARHGAP11B protein will be expressed in developing mouse neocortex under the control of the native mouse Arhgap11a promotor.
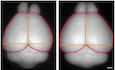
Figure EV1. Generation of 11B mice and expression of ARHGAP11A/Arhgap11a and ARHGAP11B in foetal human neocortex and embryonic wild‐type and 11B mouse neocortex.
-
A, BqPCR analysis of the expression of ARHGAP11A (A) and ARHGAP11B (B) in foetal human neocortex at the indicated developmental stages. The level of expression obtained at 12 wpc was set to 100, and the level of expression at the other stages is given relative to this. Data are the mean of 3 technical replicates from one foetus at each stage. Error bars indicate SD.
-
C, DFPKM values of ARHGAP11A (C) and ARHGAP11B (D) mRNAs in the indicated isolated cell populations of foetal (12–13 wpc) human neocortex; N, neuronal fraction, which includes bRG in G1. Data were taken from the previously reported transcriptome data set (Florio et al, 2015).
-
EDiagram showing the strategy of generating the ARHGAP11B‐transgenic mouse line. Numbers in the ARHGAP11B protein indicate the amino acid residues in the truncated GAP domain (red) and the human‐specific C‐terminal sequence (green).
-
F, GqPCR analysis on Arhgap11a mRNA expression in wild‐type (WT, solid line) and 11B (dashed line) mouse brain (F) and mARHGAP11B mRNA expression in the 11B mouse brain (G) at the indicated developmental and adult stages. The levels of expression obtained in 11B mouse embryos at E12.5 were set to 100, and the level of expression at the other stages is given relative to this. Data are the mean of 3 (WT) and 3 (11B) mice at each stage. Error bars indicate SD.
Accordingly, the expression patterns of the Arhgap11a mRNA in wild‐type mice (Fig EV1F), of the Arhgap11a mRNA in heterozygous ARHGAP11B‐transgenic mice (11B mice; Fig EV1F), and of the mARHGAP11B in 11B mice (Fig EV1G), were overall similar across the developmental stages examined, with highest mRNA levels at embryonic day (E) 12.5 and lowest mRNA levels at postnatal day (P) 56. Immunostaining of E12.5 and E14.5 11B mouse embryos revealed Arhgap11a and ARHGAP11B protein expression in the neocortex, in other parts of the central nervous system (CNS), and outside the CNS (Appendix Fig S1A–C). In the E14.5 neocortex, Arhgap11a and some of ARHGAP11B were found in the cell bodies of both VZ and SVZ NPCs (Appendix Fig S1C). The apparent differences in the pattern of immunofluorescence between Arhgap11a and ARHGAP11B most likely reflected primarily the different subcelluar localization of Arhgap11a, which is nuclear, and of ARHGAP11B, which is in mitochondria and hence in the cytoplasm extending into cell processes (Namba et al, 2020).
Furthermore, we noticed a reduction in Arhgap11a protein expression in 11B mouse embryos compared with wild type (Appendix Fig S1D). This reduction reflected the conversion of one allele of Arhgap11a to mARHGAP11B (Fig EV1E), resulting in a corresponding reduction in Arhgap11a mRNA levels (Fig EV1F), and was also evident upon Arhgap11a immunoblot analysis (Appendix Fig S1E and F). Given this reduction, we generated an Arhgap11a knockout mouse line, in order to be able to conclude that any phenotype we may observe in the 11B mice is due to the expression of ARHGAP11B rather than the reduction in Arhgap11a. This Arhgap11a knockout mouse line was generated by introducing a translational stop codon in exon 1, i.e., shortly 3′ to the translational start codon, again using the CRISPR/Cas9 genome editing technology (Appendix Fig S2A; for details, see Materials and Methods). As described below, heterozygous Arhgap11a knockout mice, which showed a similar level of Arhgap11a protein reduction as 11B mice (Appendix Figs S1C–F and S2B–D), were used as controls and compared with wild‐type mice to corroborate that the differences observed in the various parameters analysed in the 11B mice were due to ARHGAP11B protein expression and not the reduction in Arhgap11a protein expression.
Increased BP proliferation and abundance in the neocortex of 11B mouse embryos
We first examined the NPC levels in the developing neocortex of 11B mice. Analyses of NPCs at E14.5 for mitotic markers by phosphohistone H3 (PH3) and phosphovimentin (pVim) immunofluorescence (Fig 1A and B) revealed no change in mitotic apical progenitors (APs, Fig 1C), but a ≈ 50% increase in mitotic BPs (Fig 1D and E) in the neocortex of 11B mouse embryos compared with wild‐type littermate embryos. The latter increase comprised both mitotic basal (or outer) radial glia (bRG) (pVim+ & basal process+), a minor BP subpopulation in mouse (Shitamukai et al, 2011; Wang et al, 2011), and mitotic basal intermediate progenitors (bIPs) (pVim+ & basal process−), the major BP subpopulation in mouse (Florio & Huttner, 2014) (Fig 1E).
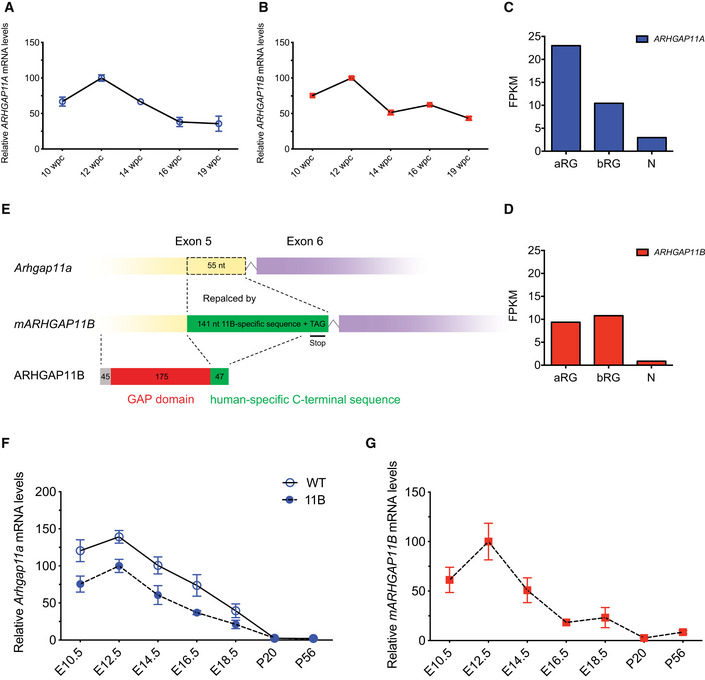
Figure 1. Increase in BP proliferation in 11B mouse neocortex at E14.5.
-
ARepresentative immunofluorescence for PH3 (green), combined with DAPI staining (white), of E14.5 wild‐type (WT) and 11B mouse dorsolateral neocortex rostrally.
-
BRepresentative immunofluorescence for pVim (yellow), combined with DAPI staining (white), of E14.5 WT and 11B mouse dorsolateral neocortex rostrally. Boxes (50 µm × 65 µm) in (B) indicate areas that are shown at higher magnification in (B′). Arrow, pVim+ BP with basal process (mitotic bRG); notched arrowheads, basal process; arrowhead, pVim+ BP without basal process (mitotic bIP).
-
C–EQuantification of PH3+ APs (C), PH3+ BPs (D) and pVim+ bRG (E, left) and bIPs (E, right) in a 200 µm‐wide field of E14.5 WT (white) and 11B (black) mouse dorsolateral neocortex rostrally. Note the difference in ordinate scales in (E).
-
FRepresentative immunofluorescence for Pax6 (cyan) and Tbr2 (magenta), combined with DAPI staining (white), of E14.5 WT and 11B mouse dorsolateral neocortex rostrally. Boxes (25 µm × 25 µm) in (F) indicate areas that are shown at higher magnification in (F′). Outlined by dashed white lines, Pax6+ & Tbr2+ BP; outlined by solid white lines, Pax6+ & Tbr2− BP.
-
G–KQuantification of Pax6+ & Tbr2− cells in VZ (G), Pax6+ & Tbr2+ cells in VZ (H), Pax6+ & Tbr2− cells in SVZ and IZ (I), Pax6+ & Tbr2+ cells in SVZ and IZ (J) and Pax6– & Tbr2+ cells in SVZ and IZ (K) in a 200 µm‐wide field of E14.5 WT (white) and 11B (black) mouse dorsolateral neocortex rostrally.
-
L–NSequential BrdU–EdU labelling. BrdU was injected into pregnant mice at E13.5, i.e., 24 h before sacrifice at E14.5, and EdU was injected into the same pregnant mice 0.5 h before sacrifice at E14.5. (L) Representative immunofluorescence for BrdU (magenta), combined with EdU staining (cyan) and DAPI staining (white), of E14.5 WT and 11B mouse dorsolateral neocortex rostrally. (M, N) Quantification of BrdU+ & EdU+ cells (M) and the percentage of BrdU+ cells that are BrdU+ & EdU+ (N) in VZ and SVZ in a 200 µm‐wide field of E14.5 WT (white) and 11B (black) mouse dorsolateral neocortex rostrally.
Data information: Images are single optical sections (A, B, B′, L) and Z projections of four optical sections (F, F′). Scale bars, 50 µm (A, B, F, L), 10 µm (B′, F′). Data are the mean of 6–19 (WT) and 6–26 (11B) littermate embryos, which were derived from 3–9 separate litters. Error bars indicate SD, two‐tailed unpaired Student's t‐test, *P < 0.05; **P < 0.01 (C, D, E, G‐K, M, N). P = 0.0269 (D); P = 0.0302 (E, left); P = 0.0139 (E, right); P = 0.0281 (I); P = 0.0088 (J); P = 0.0014 (K); P = 0.0049 (M, SVZ); P = 0.0323 (N, SVZ).
Upon immunofluorescence for Pax6 and Tbr2 (Fig 1F), two markers used to distinguish NPC subpopulations(Englund et al, 2005), the E14.5 11B mouse neocortex compared with wild‐type littermate embryos showed no change in Pax6+ & Tbr2− APs in the ventricular zone (VZ) (Fig 1G) nor in Pax6+ & Tbr2+ newborn BPs in the VZ (Fig 1H), but substantial increases in the abundance of Pax6+ & Tbr2− (Fig 1I) and Pax6+ & Tbr2+ (Fig 1J) BPs, two minor BP subpopulations likely to include bRG, and of Pax6– & Tbr2+ BPs (Fig 1K), i.e., bIPs, in the subventricular zone (SVZ) plus intermediate zone (IZ). Heterozygous Arhgap11a knockout mouse embryos showed no alteration in the level of the various BP subpopulations in interphase or at mitosis (Appendix Fig S2E–K). Hence, the increased BP levels in the neocortex of 11B mouse embryos are due to the expression of ARHGAP11B and not the reduction in Arhgap11a.
To determine whether the increase in BP levels in the neocortex of 11B mouse embryos involved increased cell cycle re‐entry, we carried out sequential labelling with two thymidine analogs, a single pulse with BrdU at E13.5 and a pulse with EdU 23.5 h later, half an hour prior to sacrifice at E14.5 (Fig 1L). In light of the known cell cycle parameters of APs and BPs in the neocortex of E14.5 mouse embryos(Arai et al, 2011), this approach allowed determining the abundance and proportion of the progeny of the BrdU‐labelled NPCs that were in S‐phase, i.e., cycling. We observed no difference in the abundance of BrdU+ & EdU+ progeny in the VZ, comprising APs and newborn AP‐derived BPs undergoing S‐phase in the VZ, but nearly a doubling in the abundance of BrdU+ & EdU+ progeny in the SVZ, i.e., BPs, in the neocortex of 11B mouse embryos compared with wild type (Fig 1M). These data are not consistent with a scenario in which the increased neocortical BP levels in the SVZ of 11B mouse embryos (Fig 1I–K) are due to an increased generation of BPs from APs in the VZ followed by the subsequent migration of the BPs to the SVZ where they undergo S‐phase, as one would then expect a decrease in the abundance of BrdU+ & EdU+ progeny in the VZ, which was not the case (Fig 1M). Rather, the data suggested that in contrast to the wild‐type situation, in which the vast majority of the BPs in the SVZ divide to generate two post‐mitotic neurons and only a minority generates cycling progeny (Haubensak et al, 2004; Miyata et al, 2004; Noctor et al, 2004), in the neocortex of 11B mouse embryos a significantly greater proportion of the BPs in the SVZ generated cycling progeny. We therefore conclude that BPs in the SVZ of 11B mouse embryos increasingly generate progeny that re‐enters the cell cycle, i.e., generate more BPs and hence self‐amplify. This conclusion was further supported when we expressed the BrdU+ & EdU+ progeny as a percentage of the originally BrdU‐labelled NPCs. This revealed no difference between E14.5 wild‐type and 11B embryos for the BrdU+ & EdU+ progeny in the VZ, indicative of unaltered AP self‐renewal, but a significant increase for the BrdU+ & EdU+ progeny in the SVZ of 11B embryos, indicative of increased BP self‐renewal (Fig 1N).
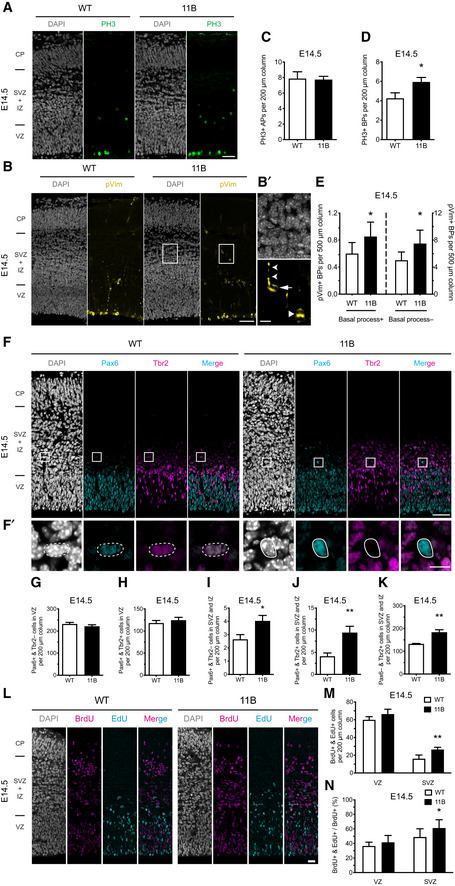
We sought to obtain independent evidence in support of our conclusion that the increased BP levels in the neocortical SVZ of 11B mouse embryos resulted from BPs in the SVZ undergoing self‐amplification, rather than from an increased generation of BPs from APs in the VZ followed by migration of the newborn BPs to the SVZ. To this end, we chose a labelling approach that, similar to cell lineage tracing, allowed us to follow the fate of the AP progeny. Specifically, we introduced a GFP‐expressing plasmid into neocortical APs in the VZ of E13.5 wild‐type and 11B mouse embryos by in utero electroporation (IUE) and then analysed the distribution of the GFP+ cells across the cortical wall at four different time points post‐IUE, that is, at 8, 18, 30 and 42 h (Fig EV2A–D). As expected, at 8 h post‐IUE, virtually all GFP+ cells were located in the VZ for both wild‐type and 11B embryos (Fig EV2E). Ten hours later, at 18 h post‐IUE, about half of the GFP+ cells had remained in the VZ while the other half was now found in the SVZ, equal for both wild‐type and 11B embryos (Fig EV2E), indicating that irrespective of the absence or presence of ARHGAP11B expression, a similar proportion of the progeny of the originally GFP‐expressing APs had become GFP+ BPs. In contrast, at 30 h post‐IUE, while still about half of the GFP+ cells were found in the VZ in both wild‐type and 11B embryos, the percentage of GFP+ progeny found in the SVZ was significantly increased in 11B embryos compared with wild type, at the expense of the percentage of GFP+ progeny appearing in the IZ (Fig EV2E). The same difference between wild‐type and 11B embryos was observed 42 h post‐IUE, with the GFP+ cells in the VZ now constituting only about 30% of total GFP+ progeny in both wild‐type and 11B embryos (Fig EV2E). Taken together, these data corroborate our conclusion that the increase in BP levels in the neocortex of 11B mouse embryos compared with wild type is not due to increased BP generation from APs, but to increased self‐renewal of BPs once they have reached the SVZ.
Figure EV2. Increased BP abundance results from BP self‐amplification.
-
A–DRepresentative immunofluorescence for GFP (green), combined with DAPI staining (white), of wild‐type (WT) and 11B mouse dorsolateral neocortex rostrally at 8 h (A), 18 h (B), 30 h (C) and 42 h (D) post‐IUE. Images are single optical sections. Scale bars, 20 µm.
-
EDistribution of GFP+ cells across the indicated zones of the neocortical wall, expressed as percentage of the total number of GFP+ cells in a 200 µm‐wide segment of the entire cortical wall, of WT and 11B mouse dorsolateral neocortex, using immunostained cryosections obtained as in (A–D). Data are the mean of 3–5 (WT) and 3–5 (11B) littermate embryos, which were derived from 3 to 4 separate litters at each time point analysed. Error bars indicate SD, two‐way ANOVA, followed by Bonferroni's multiple comparisons test, *P < 0.05, **P < 0.01. P = 0.0231 (30 h, SVZ); P = 0.0086 (30 h, IZ); P = 0.0466 (42 h, SVZ).
The immunofluorescence data with the mitotic marker PH3 and the NPC markers Pax6 and Tbr2 that had revealed an increased level of BPs, but not APs, in 11B mouse neocortex compared with wild type had all been obtained at E14.5 (Fig 1A–K). We performed similar immunofluorescence analyses on wild‐type and 11B mouse neocortex at E12.5, an earlier stage of BP generation. These analyses showed that the levels of apical mitoses (Appendix Fig S3A and B) and Tbr2+ cells in the VZ (Appendix Fig S3D and E) were not different between wild‐type and 11B embryos, but that the levels of basal mitoses (Appendix Fig S3A and C) and Tbr2+ cells in SVZ (Appendix Fig S3D and F) were increased in 11B embryos. These data not only indicated that also at this earlier developmental stage, the increase in BP levels in 11B embryos is not due to increased BP generation from APs but to increased BP self‐renewal in the SVZ, but also implied that the increase in BP levels in 11B embryos observed at E14.5 did not involve a greater BP generation earlier in neocortical development.
We also investigated whether reduced BP apoptosis contributed to the increased BP levels in 11B embryos. However, this was not the case as the level of Tbr2+ & Caspase‐3+ cells in the SVZ was not different between E14.5 wild‐type and 11B mouse embryos (Appendix Fig S3G and H).
Considering all these findings together, they indicate that 11B mouse embryos show an increased BP abundance that is the result of increased BP proliferation (rather than increased BP generation); this increased BP proliferation is due to the expression of ARHGAP11B at a physiological level (rather than the reduction of Arhgap11a).
Cortical expansion and increased neuron production in the developing 11B mouse neocortex
As a consequence of the increased BP proliferation and abundance in the neocortex of 11B mouse embryos, which are due to the expression of ARHGAP11B and not the reduction in Arhgap11a, 11B mouse embryos show an expanded neocortex and an increase in cortical neuron numbers. Measurement of the cortical perimeter, cortical thickness and ventricle length at E18.5 as shown in Fig 2A revealed increases in the lateral perimeter (Fig 2B) and radial thickness (Fig 2C), but not ventricle length (Fig 2D), of the neocortex of 11B mouse embryos compared with wild‐type littermate embryos. These increases were most pronounced in the rostral part of the brain (Fig 2E and F; compare to G). The increase in radial thickness was due to an increased thickness of the IZ and cortical plate (CP; Fig 2H and I). The latter increase in turn reflected increased numbers of Tbr1+, Ctip2+ and Satb2+ neurons in the CP of the E18.5 11B mouse neocortex (Fig 2H and J–L). These increased numbers of cortical neurons were not accompanied by alterations in cell density (Appendix Fig S4A and B) or in the abundance of cells expressing markers of gliogenesis (Appendix Fig S4C–E).
Figure 2. Neocortex expansion and increased cortical neuron production in embryonic 11B mouse neocortex at E18.5.
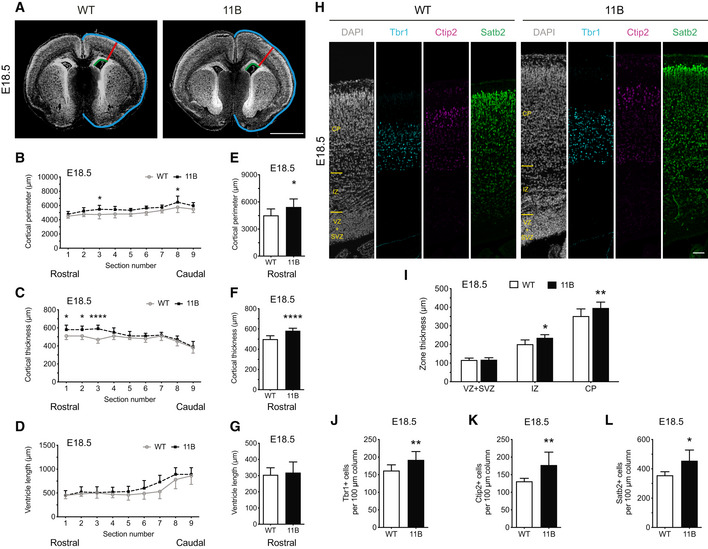
-
ARepresentative DAPI staining (white) of E18.5 wild‐type (WT) and 11B mouse neocortex illustrating the measurements of cortical perimeter (blue), cortical thickness (red) and ventricular length (green).
-
B–GQuantification across the rostral‐caudal axis (sections 1–9, B–D) and only the rostral part of the brain (sections 1–3, E–G) of cortical perimeter (B, E), cortical thickness (C, F) and ventricular length (D, G) of E18.5 WT (white) and 11B (black) mouse neocortex.
-
HRepresentative immunofluorescence for Tbr1 (cyan), Ctip2 (magenta) and Satb2 (green), combined with DAPI staining (white), of E18.5 WT and 11B mouse neocortex at the position where cortical thickness was measured (A, red lines).
-
IQuantification of zone thickness of E18.5 WT (white) and 11B (black) mouse neocortex at the position where cortical thickness was measured (A, red lines).
-
J–LQuantification of Tbr1+ (J), Ctip2+ (K) and Satb2+ (L) neurons in the CP in a 100 µm‐wide field of E18.5 WT (white) and 11B (black) mouse neocortex at the position where cortical thickness was measured (A, red lines).
Data information: Images are single optical sections (A, H). Scale bar, 1 mm (A), 20 µm (H). Data are the mean of 7 (WT) and 8–12 (11B) littermate embryos, which were derived from four separate litters. Error bars indicate SD. Two‐way ANOVA, followed by Bonferroni's multiple comparisons test (B–D); two‐tailed unpaired Student's t‐test (E–G, I–L), *P < 0.05, **P < 0.01, ****P < 0.0001. WT vs. 11B, P < 0.0001; section number 3, P = 0.0322; section number 8, P = 0.0411 (B). WT vs. 11B, P < 0.0001; section number 1, 2, P = 0.0233; section number 3, P < 0.0001 (C). P = 0.0476 (E); P < 0.0001 (F); P = 0.0287 (I, IZ); P = 0.0039 (I, CP); P = 0.0085 (J); P = 0.0093 (K); P = 0.0157 (L).
Cortical expansion and increased neuron numbers in the 11B mouse neocortex persist into adulthood
Key aspects of the neocortex expansion and increase in cortical neuron numbers observed in 11B mouse embryos were found to persist into adulthood. Thus, in contrast to the previous transient and locally confined overexpression of ARHGAP11B in embryonic mouse neocortex (Florio et al, 2015), the present ARHGAP11B‐transgenic mouse line that expresses ARHGAP11B physiologically in the neocortex allowed us to extend our studies to the adult stage. Specifically, we observed significant increases in neocortex width and neocortex area in adult 11B mice at P56 (Fig 3A–C). Moreover, the increases in cortical thickness and cortical perimeter observed in 11B mouse embryos persisted into adulthood (Fig 3D–F), notably the increase in CP thickness (Fig 3G–I). The latter increase was due to a specific thickening of the upper cortical layers, i.e., layers II + III (Fig 3J). Consistent with this, the adult 11B mouse neocortex at P56 showed increased numbers of Satb2+ and Brn2+ upper‐layer neurons (Fig 3G, H, K and L), but not of Ctip2+ deep‐layer neurons (Fig 3G, H and M). These increased numbers of upper‐layer neurons were not accompanied by alterations in cell density (Appendix Fig S4F and G). Importantly, as neither Arhgap11a nor ARHGAP11B is expressed in the adult wild‐type or 11B mouse brain, the differences between adult wild‐type and 11B mice with regard to neocortex size and upper‐layer neuron numbers can be assumed to be the consequence of the differences between wild‐type and 11B mice during embryonic brain development.
Figure 3. Persistence of neocortex expansion and increased cortical neuron numbers in adult 11B mouse brain at P56.
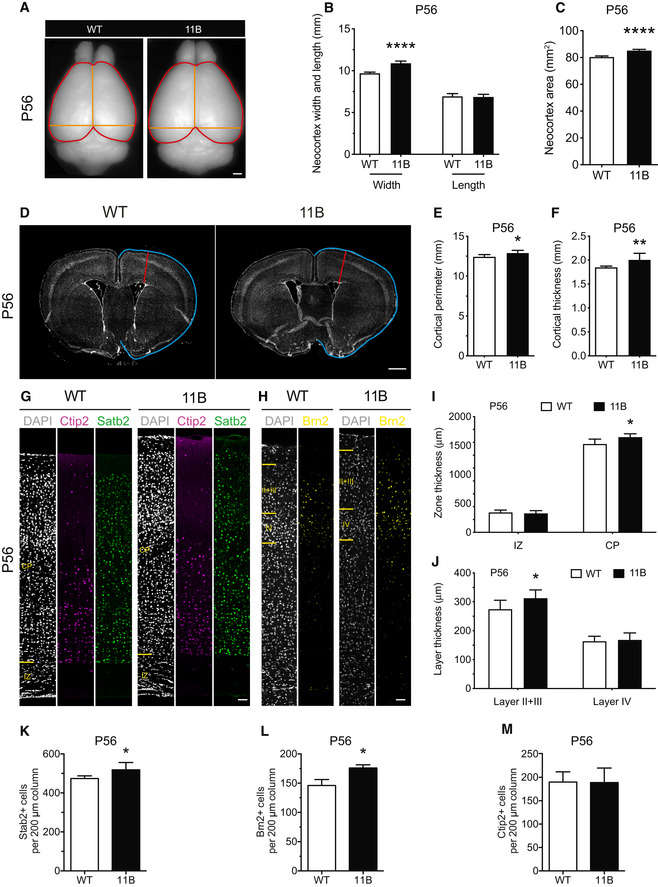
-
ADorsal view of adult wild‐type (WT) and 11B mouse brain at P56. Orange lines illustrate the measurements of neocortex width and length; red lines illustrate the measurements of neocortex area.
-
B, CQuantification of neocortex width, length and area.
-
DRepresentative DAPI staining (white) of P56 adult WT and 11B mouse neocortex illustrating the measurements of cortical perimeter (blue) and thickness (red).
-
E, FQuantification of cortical perimeter (E) and thickness (F) in the rostral part of P56 adult WT and 11B mouse brain.
-
G, HRepresentative immunofluorescence for Ctip2 (magenta), Satb2 (green) and Brn2 (yellow), combined with DAPI staining (white), of P56 adult WT and 11B mouse neocortex at the position where cortical thickness was measured (D, red lines).
-
I, JQuantification of zone thickness (I) and layer thickness (J) of P56 adult WT and 11B mouse neocortex at the position where cortical thickness was measured (D, red lines).
-
K–MQuantification of Satb2+ (K), Brn2+ (L) and Ctip2+ (M) neurons in a 200 µm‐wide field of P56 adult WT and 11B mouse neocortex.
Data information: Images are single optical sections (D, G). Scale bar, 1 mm (A, D), 20 µm (G, H). Data are the mean of 8 (WT) and 8 (11B) adult littermate mice. Error bars indicate SD, two‐tailed unpaired Student's t‐test (B, C, E, F, I–M), *P < 0.05, **P < 0.01, ****P < 0.0001. P < 0.0001 (B, neocortex width); P < 0.0001 (C); P = 0.0297 (E); P = 0.0092 (F); P = 0.0227 (I, CP); P = 0.0308 (J, layer II + III); P = 0.0278 (K); P = 0.0133 (L).
Importantly, key aspects of the morphology of layer II + III neurons in the somatosensory cortex of the rostral neocortex (Fig EV3A–C), that is, apical dendrite branching (Fig EV3D), basal dendrite branching (Fig EV3E), total dendrite branching (Fig EV3F), dendrite length (Fig EV3G), dendritic spine number (Fig EV3H) and spine morphology (Fig EV3I), were all unchanged in adult 11B mouse neocortex compared with adult wild‐type littermate mouse neocortex.
Figure EV3. Analysis of the dendritic and spine morphology of upper‐layer neurons in the adult WT and 11B mouse neocortex.
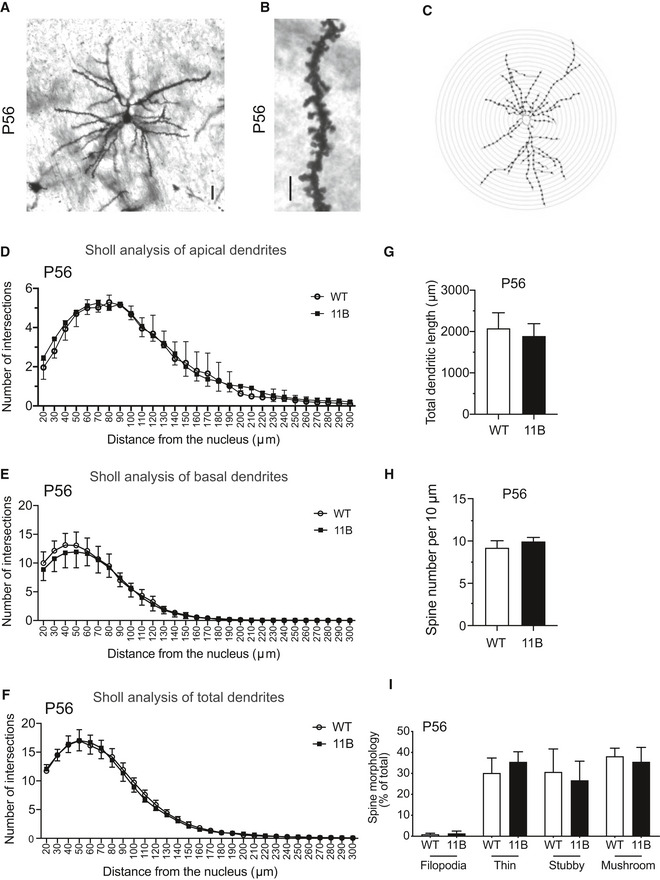
-
A, BExamples of a Golgi‐Cox stained upper‐layer pyramidal neuron (A) and dendritic spines of an upper‐layer neuron (B) in layers II + III of the somatosensory cortex of the rostral neocortex of P56 adult wild‐type (WT) mice. Images are a Z‐stack projection of five optical sections (A) and a single optical section (B). Scale bars, 10 µm (A); 5 µm (B).
-
CRepresentative Sholl analysis image showing the dendritic morphology of an adult mouse neocortical upper‐layer neuron. The cell mask was drawn using Fiji to represent the shape of the upper‐layer neuron. Concentric circles, with 10‐µm intervals, were placed on the upper‐layer neuron cell mask using the Sholl analysis plug‐in in Fiji. Intersections with the concentric circles are indicated by the dots.
-
D–GQuantification of the numbers of apical (D), basal (E) and total (F) dendritic intersections with the concentric circles and the total dendritic length (G) of adult WT (white) and 11B (black) mouse neocortical upper‐layer neurons, using Golgi‐Cox stained vibratome sections obtained as in (A). Data are the mean of 3 (WT) and 3 (11B) adult male mice. Error bars indicate SD. Two‐tailed unpaired Student's t‐test.
-
H, IQuantification of spine number in a 10 µm‐long dendrite (H) and of the percentage of each spine morphotype (I) of P56 adult WT (white) and 11B (black) mouse neocortical upper‐layer neurons, using Golgi‐Cox stained vibratome sections obtained as in (B). Data are the mean of 3 (WT) and 3 (11B) adult male mice. Error bars indicate SD, two‐tailed unpaired Student's t‐test.
In contrast to the previous overexpression of ARHGAP11B in embryonic mouse neocortex which resulted in cortical folding in about half of the cases (Florio et al, 2015), we did not observe cortical folding in the present 11B mice, neither at the embryonic (Fig 2A) stage nor at the adult (Fig 3A and D) stage. We explored whether this lack of cortical folding reflected a lesser increase in the level of upper‐layer neurons in the present 11B mice, which expressed ARHGAP11B at a physiological level, than in the mouse embryos that overexpressed ARHGAP11B upon IUE. To this end, we repeated the ARHGAP11B overexpression in the developing neocortex by IUE of mouse embryos at E13.5 with a plasmid driving ARHGAP11B expression from the strong constitutive CAG promoter, followed by analysis at E18.5, as done previously (Florio et al, 2015). This showed, again, that ARHGAP11B overexpression in embryonic mouse neocortex can induce folding (Appendix Fig S7A). This cortical folding was accompanied by an almost doubling (1.8‐fold increase) of upper‐layer neuron abundance among the progeny of the targeted APs (Appendix Fig S7B and C), which was a much greater increase in upper‐layer neuron abundance than that observed in the 11B embryos (1.2‐fold; Fig 2L). We conclude that the lack vs. the occurrence of cortical folding in 11B embryos vs. embryonic mouse neocortex overexpressing ARHGAP11B can be explained by the difference in the degree of increase in the level of upper‐layer neurons.
Adult 11B mice exhibit altered neurobehaviour
We exploited the fact that the present line of ARHGAP11B‐transgenic mice, which exhibited an expanded neocortex with increased upper‐layer neurons, allowed us to subject adult animals to a battery of behavioural tests to investigate their neurobehaviour. Thus, we conducted four separate behavioural tests targeting different aspects or types of learning and memory, including working memory (Y maze), spatial learning and memory (Barnes maze), associative memory (contextual fear conditioning) and memory flexibility (IntelliCage). The data obtained with the Barnes maze (Fig EV4A–F), Y maze (Fig EV4G–I) and contextual fear conditioning (Fig EV4J and K) revealed no statistically significant differences between adult wild‐type and 11B mice. Presumably, this reflected the fact that these tests focus mostly on the hippocampal‐dependent learning and memory. In line with this, we observed no significant changes in NPC abundance and newborn neuron numbers in the adult 11B mouse hippocampus compared with wild type (Appendix Fig S5A–C). Of note, neither 11A nor 11B protein expression was observed in adult wild‐type and 11B mouse hippocampus.
Figure EV4. Learning and memory analyses of 11B mice.
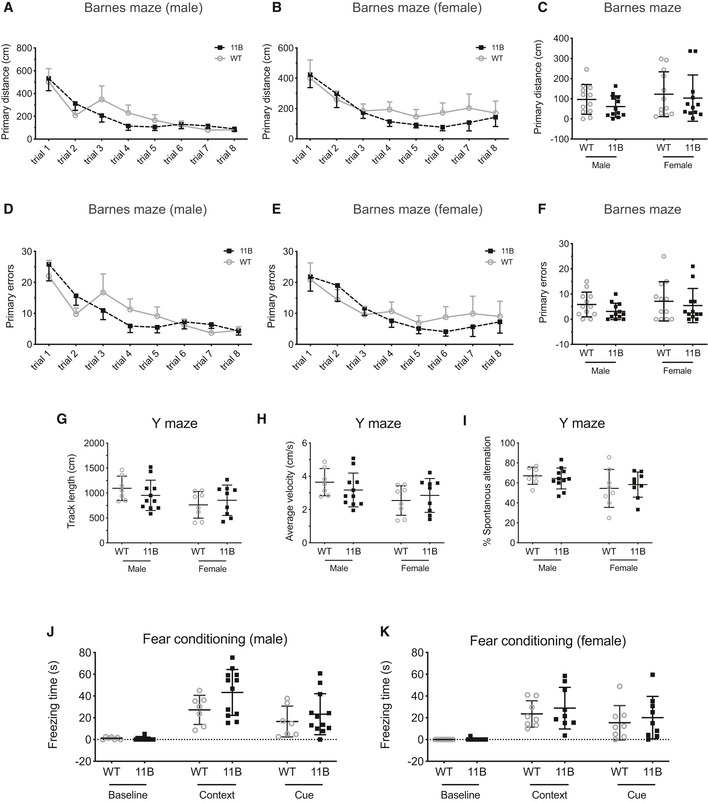
-
A–FQuantification of the primary distance (A–C) and primary error number (D–F) before reaching the target escape box during training sessions of adult wild‐type (WT) and 11B male (A, D) and female (B, E) mice in the Barnes Maze. Quantification of primary distance (C) and primary error number (F) before reaching the target hole during the probe test one day after the training sessions of adult WT and 11B male and female mice in the Barnes maze. Data are the mean of 12 male (WT), 12 male (11B), 12 female (WT) and 12 female (11B) mice. Error bars indicate SD. Two‐way ANOVA and two‐tailed unpaired Student's t‐test.
-
G–IQuantification of track length (G), average velocity (H) and % spontaneous alteration (I) of adult WT and 11B mice in the Y maze. Data are the mean of 7 male (WT), 11 male (11B), 8 female (WT) and 9 female (11B) mice. Error bars indicate SD, two‐tailed unpaired Student's t‐test.
-
J, KQuantification of freezing time of adult WT and 11B male (G) and female (H) mice in the fear conditioning test. Data are the mean of 7 male (WT), 12 male (11B), 8 female (WT) and 9 female (11B) mice. Error bars indicate SD, two‐way ANOVA.
IntelliCage, a fully automated system for the behavioural assessment of mice that live in social groups, was used to investigate the cognitive functions of 11B mice, especially the long‐term learning behaviour, which heavily relies on neocortex rather than hippocampus function. The discrimination error rate (visits to the incorrect corners per first 100 visits during the drinking phase) was taken as an indicator of the initial place learning and reversal place learning and was used to evaluate the memory flexibility. Four independent test sessions were carried out, two separate ones for male mice and two separate ones for female mice. We observed that both wild‐type and 11B mice learned the correct place to drink water over time during the acquisition stage, as it was evident that the discrimination error decreased markedly over the training days (Fig 4A and B). However, we did not observe any difference between wild‐type and 11B mice during the acquisition stage, which indicates that the initial place learning was not affected by ARHGAP11B. This finding is in agreement with our data from the Barnes maze on spatial learning and memory.
Figure 4. Enhanced memory flexibility in adult 11B mice.
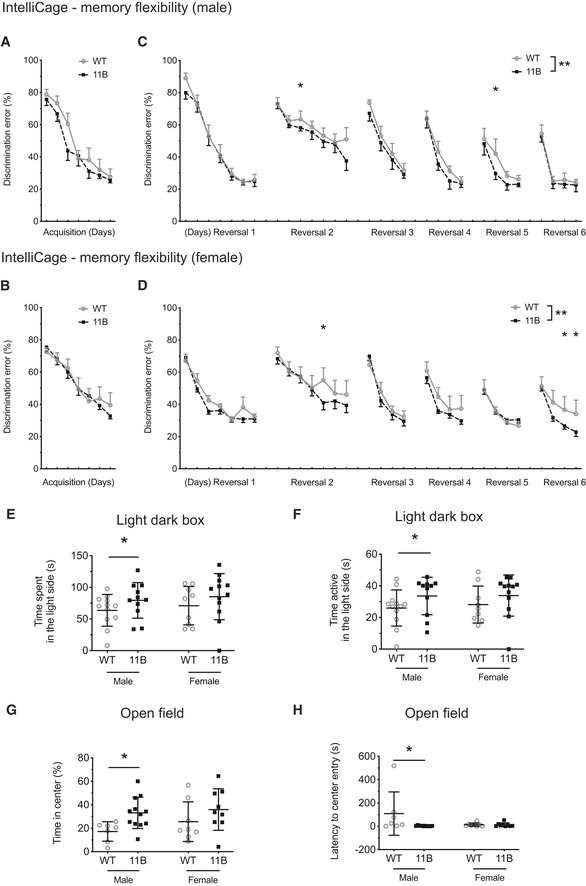
-
A–DIntelliCage was used to evaluate the memory flexibility of adult 11B mice. Four separate sessions were carried out: (i) five WT males together with five 11B males; (ii) five other WT males together with five other 11B males; (iii) five WT females together with seven 11B females; (iv) four other WT females together with eight other 11B females. Data are the mean of the total 10 males (WT), 10 males (11B), nine females (WT) and 15 females (11B) adult littermate mice. Data points were obtained at consecutive days; error bars indicate SD. (A, B) Quantification of the discrimination error rate in the first 100 visits during the drinking phase of the acquisition stage of adult wild‐type (WT) and 11B mice (male, A; female, B) in the IntelliCage. Overall place learning performance of WT and 11B mice was analysed using two‐way ANOVA followed by Fisher's LSD test; all data points during the acquisition phase were included. For males (A), WT vs. 11B, P > 0.05; data over time, ****P < 0.0001; interaction, P > 0.05. For females (B), WT vs. 11B, P > 0.05; data over time, ****P < 0.0001; interaction, P > 0.05. (C, D) Quantification of the discrimination error rate in the first 100 visits during the drinking phase of the six reversal sessions of adult wild‐type (WT) and 11B mice (male, C; female, D) in the IntelliCage. Overall reversal place learning performance and memory flexibility of WT and 11B mice were analysed using two‐way ANOVA. For males (C), WT vs. 11B, **P = 0.004358; data over time, ****P < 0.0001; interaction, P > 0.05. For females (D), WT vs. 11B, **P = 0.000578; data over time, ****P < 0.0001; interaction, P > 0.05. Differences between WT and 11B mice at each individual training day were compared by Fisher's LSD tests following two‐way ANOVA, *P < 0.05. P = 0.0037718 (C, day 10), P = 0.043855 (C, day 24); P = 0.018903 (D, day 12), P = 0.045954 (D, day 29); P = 0.041434 (D, day 330). (A–D) Note that (i) the two male sessions (pooled in panels A, C) and the two female sessions (pooled in panels B, D) yielded essentially the same results, and (ii) not shown in panels A–D, the two separate male sessions yielded essentially the same results, and the two separate female sessions yielded essentially the same results.
-
E, FQuantification of the time spent (E) and the time active (F) in the light side of the light–dark box. Data are the mean of 12 male (WT), 11 male (11B), nine female (WT) and 12 female (11B) adult littermate mice. Error bars indicate SD, two‐tailed unpaired Student's t‐test, *P < 0.05. P = 0. 0417 (E, male); P = 0. 0401 (F, male).
-
G, HQuantification of the percentage of the time spent in the centre area of the open‐field arena (G), and quantification of the latency to enter the centre area (H) of the open‐field arena. Data are the mean of 7 male (WT), 12 male (11B), 8 female (WT) and 9 female (11B) adult littermate mice. Error bars indicate SD, *P < 0.05. Two‐tailed unpaired Student's t‐test, P = 0. 0114 (G, male); Mann–Whitney test, P = 0. 0193 (H, male).
Data information: The lack of statistical significance for female WT vs. 11B mice in the light–dark box test (E, F) and the open‐field test (G, H) presumably reflects the greater variability among females as compared to males due to non‐synchronized oestrus cycles.
Next, we applied a serial reversal task to compare the memory flexibility of wild‐type and 11B mice. After each reversal, as expected, the mice committed a high number of errors, but the error rates on the first day of each reversal eventually declined as the mice learned the reversal task rule (Fig 4C and D). We found that the discrimination error rate during reversal place learning was overall significantly lower for both genders of 11B mice than wild‐type mice (Fig 4C and D). Not only did the two male sessions (pooled in Fig 4A and C) and the two female sessions (pooled in Fig 4B and D) yield essentially the same results, but (not shown in Fig 4A–D) the two male sessions yielded essentially the same results, and the two female sessions yielded essentially the same results. Using a modified IntelliCage protocol that scored the success rate during the drinking phase rather than the discrimination error rate, the place learning and reversal place learning experiments were repeated with another independent cohort of male wild‐type and 11B mice. This yielded essentially the same results. These data indicate that 11B mice exhibit increased memory flexibility and better adaptation to new rules. This increased memory flexibility was not due to any alteration in basic motor functions (Fig EV5A–C), pain response (Fig EV5D), social preference (Fig EV5E and F), compulsive and impulsive behaviours (Fig EV5G and H), or competitive dominant behaviours (Fig EV5I).
Figure EV5. Behavioural analyses of male and female adult 11B mice.
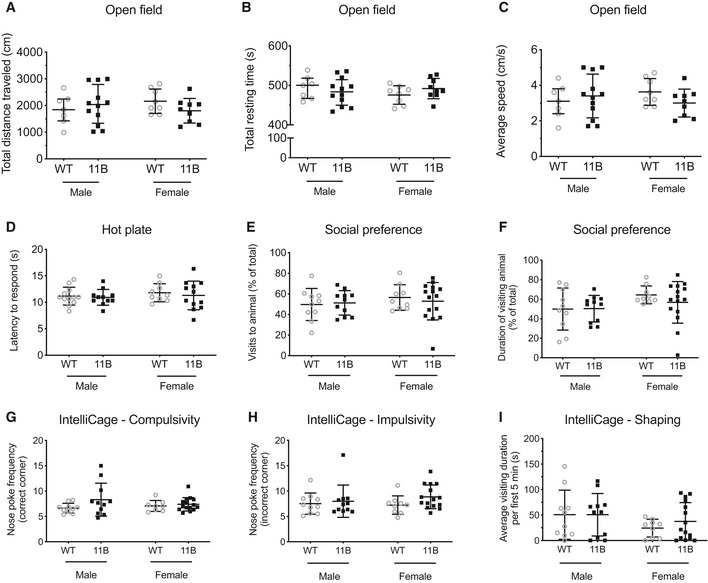
-
A–CQuantification of the total distance travelled (A), total resting time (B) and average speed (C) of adult wild‐type (WT) and 11B mice in open field. Data are the mean of 7 male (WT), 12 male (11B), 8 female (WT) and 9 female (11B) mice. Error bars indicate SD, two‐tailed unpaired Student's t‐test.
-
DQuantification of the latency of adult WT and 11B mice to respond in the hot plate test. Data are the mean of 12 male (WT), 12 male (11B), 9 female (WT) and 12 female (11B) mice. Error bars indicate SD, two‐tailed unpaired Student's t‐test.
-
E, FQuantification of the percentage of visits (E) and of the visiting duration (F) to the target animal of adult WT and 11B mice in the social preference test. Data are the mean of 10 male (WT), 11 male (11B), 9 female (WT) and 15 female (11B) mice. Error bars indicate SD, two‐tailed unpaired Student's t‐test.
-
G, HQuantification of the nose‐poke frequency in IntelliCage for compulsivity (G) and impulsivity (H) of adult WT and 11B mice. Data are the mean of 10 male (WT), 11 male (11B), 8 female (WT) and 15 female (11B) mice. Error bars indicate SD, two‐tailed unpaired Student's t‐test.
-
IQuantification of the average visiting duration within the 5 min after water is accessible in IntelliCage for competitive dominance behaviour of adult WT and 11B mice. Data are the mean of 10 male (WT), 11 male (11B), 9 female (WT) and 15 female (11B) mice. Error bars indicate SD, Mann–Whitney test.
A second set of behavioural phenotypes we observed pertained to the emotionality of adult mice. Both the light–dark box test (Fig 4E and F) and the open‐field test (Fig 4G and H) indicated that adult 11B mice exhibited a reduced level of anxiety compared with adult wild‐type mice.
Discussion
The present study not only corroborates previous findings showing that the human‐specific gene ARHGAP11B promotes BP proliferation and neocortex expansion during development (Florio et al, 2015; Kalebic et al, 2018; Heide et al, 2020), but also provides a crucial further insight—that adult mice exhibiting an ARHGAP11B‐induced increase in neocortex size and cortical neuron numbers show higher cognitive abilities. In this regard, two sets of findings of our study will be discussed in detail.
Increased neocortex size and cortical neuron numbers in adult 11B mice
The increased neocortex size and cortical neuron numbers that we observed in adult 11B mice had their basis in the mode of ARHGAP11B expression during mouse embryonic development. Thus, in contrast to previous studies in which ARHGAP11B was strongly overexpressed by electroporation of an exogenous vector, however only in a proportion of the NPCs present in the limited area of embryonic mouse neocortex that could be targeted (Florio et al, 2015), the present approach of expressing ARHGAP11B from an engineered endogenous genomic locus resulted in physiological levels of mRNA and protein throughout the embryonic mouse neocortex. This physiological ARHGAP11B expression caused a ≈ 50% increase in BP abundance, which pertained to both bIPs and bRG. This increased BP abundance in 11B mouse embryos in turn resulted in a corresponding increase in both deep‐layer and upper‐layer neuron generations by the end of embryonic cortical neurogenesis.
Importantly, the increase in upper‐layer neuron number induced by ARHGAP11B during embryonic corticogenesis, but not that in deep‐layer neuron number, persisted into adulthood. The lack of persistence of the ARHGAP11B‐induced increase in deep‐layer neurons reflected apoptosis during postnatal development (Appendix Fig S6A–E), presumably due to failure of these "extra‐neurons" to wire properly into the cortical circuits (Fricker et al, 2018). In contrast, the increased number, in the adult 11B mouse neocortex, of upper‐layer neurons, which exhibited a normal dendritic and spine morphology, provided a basis to explore whether the physiological ARHGAP11B expression would lead to enhanced cognitive abilities of these mice, as is discussed below. Upper‐layer neurons are known to connect cortical areas (Arlotta et al, 2005; Molyneaux et al, 2007). Hence, the fact that an increase in upper‐layer neurons has been implicated in enhanced cognitive performance (Fame et al, 2011; Fame et al, 2016) constituted a rationale for this exploration.
Concomitant with the persisting increase in upper‐layer neurons, the neocortex expansion in both the lateral and radial dimensions that we observed in the 11B mice by the end of embryonic corticogenesis also persisted into adulthood. The enlarged neocortex of adult 11B mice showed normal lamination, cell density and neuronal specification. However, the 11B mice showed no signs of cortical folding, neither at the embryonic stage nor at the adult stage. This apparently reflected the fact that in the 11B mice, ARHGAP11B was “only” expressed at a physiological level, which resulted in an increase in cortical neuron numbers that presumably did not surpass the “threshold” to induce cortical folding as seen previously (Florio et al, 2015). Consistent with this notion, the cortical folding induced by ARHGAP11B overexpression in mouse embryos (Appendix Fig S7A) was indeed accompanied by a greater increase in upper‐layer neuron abundance (1.8‐fold; Appendix Fig S7B and C) than that observed in embryonic and adult 11B mice (≤ 1.2‐fold; Figs 2L and, 3K and L).
It is interesting to compare these data obtained with 11B mice to the results of a previous study with another transgenic mouse line in which cortical expansion was achieved by the overexpression of cyclin‐dependent kinase 4 and cyclin D1 (4D) (Nonaka‐Kinoshita et al, 2013). In these 4D mice, too, the abundance of BPs and upper‐layer neurons was increased during development; however, no cortical folding was observed. Interestingly, and similar to the 11B mice, the increase in upper‐layer neurons in the 4D mice was also “only” ~ 1.2‐fold (Nonaka‐Kinoshita et al, 2013). These data, again, are consistent with our “threshold” hypothesis regarding the induction of cortical folding, that is, that a certain fold increase in upper‐layer neurons (> 1.2‐fold) is required for cortical folding to occur.
By showing neocortex expansion with increased upper‐layer neurons that persisted into adulthood, the 11B mice generated in the present study exhibited two key features assumed to be associated with the emergence of higher cognitive abilities in humans. However, this assumption is mainly based on comparative studies across various mammals. To obtain direct evidence showing that neocortex expansion and increased upper‐layer neurons indeed contribute to enhanced cognitive abilities has been challenging due to the lack of appropriate animal model systems. The behavioural analyses of the adult 11B mice described in the present study and discussed below therefore provide an opportunity to examine the potential link between neocortex expansion, increased upper‐layer neurons and higher cognitive abilities.
Increased memory flexibility and reduced anxiety in adult 11B mice
Higher cognitive functions, including spatial learning, reference memory and long‐term memory flexibility, involve (but are not limited to) brain areas such as the entorhinal cortex, the hippocampus, the amygdala and the frontal cortex (Clark, 2018; Yavas et al, 2019). Although the initial formation of memory is dependent on the hippocampus, the neocortex is believed to be the site of long‐term memory storage (Mercer et al, 2008; Vann & Albasser, 2011; Clark, 2018). Indeed, through the process of systems consolidation, the hippocampus guides the reorganization of the information that has been gradually stored in the neocortex such that it eventually becomes independent of the hippocampus (Mercer et al, 2008; Vann & Albasser, 2011; Richards & Frankland, 2017; Clark, 2018; Yavas et al, 2019). In our study, neither hippocampal neurogenesis nor hippocampus‐dependent learning and memory were different between wild‐type and 11B mice; however, the hippocampus‐independent memory flexibility was increased in the latter. Although further neural circuit analyses are required to assess the behavioural consequences of ARHGAP11B‐induced cortical expansion, the increased memory flexibility we observed in the adult 11B mice is fully in line with the expanded neocortex and increased abundance of upper‐layer neurons in these mice (Fig 3). However, other changes in neurobehaviour, notably the reduced anxiety, that were observed in the adult 11B mice may not necessarily reflect the effects of ARHGAP11B on embryonic development of the neocortex, but perhaps of other regions of the CNS that also showed ARHGAP11B expression in the embryo (Appendix Fig S1A and B).
Previous studies have reported that behavioural flexibility was associated with changes in the structure and function of a frontal cortical network in macaques (Sallet et al, 2020) and that changes in cortical pyramidal neuron abundance and morphology led to cognitive flexibility impairment in mice (Aung et al, 2016). Although enhanced memory flexibility does not necessarily mean higher cognitive abilities, it is conceivable that in a challenging environment, a greater reliance on learned information could indeed facilitate a better performance and hence be advantageous.
Other reports either have studied genes implicated in neocortex expansion transiently in mouse embryos (Stahl et al, 2013; Rani et al, 2016; Cardenas et al, 2018; Fiddes et al, 2018; Florio et al, 2018; Suzuki, 2019) or non‐human primate foetuses (Heide et al, 2020), or have established transgenic mouse lines carrying primate‐ or hominoid‐specific genes (Ju et al, 2016; Liu et al, 2017). However, in contrast to the present work, none of these studies has addressed the potential relationship between neocortex expansion and function, that is, cognitive abilities. In conclusion, the present study strongly suggests that the neocortex expansion induced by ARHGA11B, a gene implicated in the increase in brain size in the course of human evolution, contributes to altered neurobehaviour.
Materials and Methods
Mice
C57BL/6N mice were used throughout. All experimental procedures were designed and conducted according to the European directive 2010/63/EU and in agreement with the German Animal Welfare Legislation and the institutional guidelines of the Animal Care Committee of the Institute of Molecular Genetics. The licence number pertaining to the present experiments with mice is as follows: WH‐19‐ARGHAP11B Gehirnentwicklung (9/2019). Animals used for this study were kept pathogen‐free at the Biomedical Services Facility (BMS) of the Max Planck Institute of Molecular Cell Biology and Genetics (MPI‐CBG) and Czech Centre for Phenogenomics of the Institute of Molecular Genetics of the Czech Academy of Sciences under project licences (62/2016; 45/2017) (BIOCEV/IMG).
Human tissue
Foetal human brain tissue was obtained from the Human Development Biology Resource (HDBR), with the foetal human material being provided by the Joint MRC/Wellcome Trust (MR/R006237/1) Human Developmental Biology Resource (http://www.hdbr.org). The HDBR provided fresh tissue from foetuses aged 10–19 weeks postconception (wpc). Upon arrival, the tissue obtained from HDBR was flash‐frozen and stored at −80°C until further experimentation.
Generation of the ARHGAP11B‐transgenic and the Arhgap11a knockout mouse lines
The mouse Arhgap11a gene was engineered to generate mARHGAP11B which resembles the human ARHGAP11B gene as follows: we replaced the last 55 nucleotides of the mouse Arhgap11a exon 5, starting at the position where in human ARHGAP11B, the C–>G point mutation generates a new splice‐donor site, with the 141 nucleotides encoding the ARHGAP11B‐specific 47 amino acid sequence followed by three further nucleotides (TAG) to generate a translational stop codon (Fig EV1E). We achieved this by using CRISPR‐mediated homology directed repair (HDR). Specifically, a mixture containing (i) 111 ng/µl in vitro transcribed single guide RNA (5′‐AGAGAAGAAGCTACGTCTGC), (ii) 8 ng/µl synthetic double‐stranded HDR template DNA encoding the C‐terminal human‐specific 47 amino acid sequence in ARHGAP11B plus the stop codon TAG with 100 bp of homology arms (gBlock IDT), (iii) 334 ng/µl Cas9 nuclease (ToolGen) and (iv) 1 mM NU7026 (NHEJ‐inhibitor) was delivered into mouse zygotes ex vivo by pronuclear injection, and the zygotes were transferred to foster mothers as described previously (Fei et al, 2014). The resulting founder mouse, verified as described below, was found to have one allele of the Arhgap11a gene converted to mARHGAP11B. It was used to obtain, by sequential breeding steps, homozygous mice in which both alleles of the Arhgap11a gene were replaced by mARHGAP11B. The homozygous mARHGAP11B mice in turn were crossed with wild‐type mice to obtain heterozygous mARHGAP11B mice, which were routinely used in the experiments, unless indicated otherwise.
We used a similar strategy to generate an Arhgap11a knockout mouse line. The mouse Arhgap11a gene was engineered to contain a premature stop codon in exon 1 (75 nucleotide downstream of the start codon), by replacing nucleotides CCGCG with TTA (Appendix Fig S2A). Specifically, a mixture containing (i) 2.3 µM single guide RNA (5′‐TCCTGCGATCGCACTGACCGCGG), (ii) 2.3 µM synthetic double‐stranded HDR template DNA encoding TTA with 68‐nt 5′ and 36‐nt 3′ homology arms (gBlock IDT) and (iii) 2.1 µM Cas9 nuclease (MPI‐CBG) was used for pronuclear injection.
Positive founders were identified by PCR, and the engineering of Arhgap11a to generate mARHGAP11B and the Arhgap11a knockout was verified by DNA sequencing of the target region using the primers described in Appendix Table S1.
In utero electroporation of embryonic mouse neocortex
In utero electroporation was performed as described previously (Xing et al, 2020). Pregnant mice carrying E13.5 embryos were anaesthetized with 4% isoflurane (Baxter, HDG9623) in a narcosis box, followed by 2.5% isoflurane during the IUE procedure on a heated operation platform. Subsequently, the animals were injected subcutaneously with analgesic (0.1 ml, Metamizol, 200 mg/kg). The abdominal cavity was then surgically opened, and the uterus was exposed. Embryos were injected intraventricularly with a solution containing 0.1% Fast Green (Sigma) in PBS and 0.3–1.3 mg/ml plasmids (0.3 mg/ml of pCAGGS‐GFP alone; 0.3 mg/ml of pCAGGS‐GFP plus 1 mg/ml of either empty pCAGGS vector or pCAGGS‐ARHGAP11B (Namba et al, 2020)), using a borosilicate microcapillary (Sutter instruments, BF120‐69‐10). This was followed by six 50‐ms pulses of 27 V at 1‐s intervals (BTX Genetronics Inc., 45‐0052INT) delivered by 3‐mm diameter electrodes (BTX Genetronics Inc., 45‐0487), with anode and cathode positioned appropriately to promote DNA entry into the neocortex. After the IUE, the uterus was placed back into the abdominal cavity, and the peritoneum was sutured (VICRYL 5‐0, V493H). Abdominal skin was closed with clips. Animals received painkiller (metamizole 1.33 mg/ml) in drinking water.
For the cell linage tracing‐like analysis, pregnant mice carrying littermate wild‐type and 11B E13.5 embryos were used for IUE with GFP‐expressing plasmid and were then sacrificed by cervical dislocation at 8, 18, 30and 42 h post‐IUE. Embryonic brains were dissected and fixed in 4% paraformaldehyde in 120 mM phosphate buffer pH 7.4 (PFA‐PB), overnight at 4°C, for immunofluorescence.
For ARHGAP11B overexpression studies, pregnant mice carrying wild‐type E13.5 embryos were used for IUE with GFP‐expressing plus either control or ARHGAP11B‐expressing plasmids and were then sacrificed by cervical dislocation at E18.5. Embryonic brains were dissected and fixed in PFA‐PB, overnight at 4°C, for immunofluorescence.
Gene expression analysis by RT–qPCR
Total RNA was isolated from 10, 12, 14, 16 and 19 wpc human neocortical tissue and one hemisphere of E10.5, E12.5, E14.5, E16.5, E18.5, P20 and P56 mouse brains using the RNAeasy Mini Kit (Qiagen) according to the manufacturer's instructions. cDNA was synthesized using the Maxima first‐strand cDNA synthesis kit for RT–qPCR (Thermo Scientific). qPCR was performed using the FastStart essential DNA green master (Roche) and LightCycler® 96 Instrument (Roche). Primers used are indicated in Appendix Table S1.
Immunofluorescence
Immunofluorescence on cryosections was performed as previously described (Xing et al, 2020). Briefly, embryonic mouse brains were fixed in 4% paraformaldehyde in 120 mM phosphate buffer pH 7.4 (PFA‐PB) for 24 h at 4°C. Adult mouse brains were fixed by sequential intracardial perfusion with saline and then PFA‐PB at room temperature (RT). Next, brain tissue was infiltrated with 30% sucrose at 4°C before being embedded in Tissue‐Tek (Sakura Finetek) and stored at −20°C. Cryosections were cut at 20 µm and stored at −20°C until required. Cryosections were incubated in antigen retrieval solution (10 mM sodium citrate buffer pH 6.0) for 1 h at 70°C or 5 min at 95°C (Tbr2 immunostaining). After cooling down to RT, sections were permeabilized with 0.3% Triton X‐100 in phosphate‐buffered saline (PBS) for 30 min, PFA was quenched with 0.1 M glycine in PBS for 30 min and blocked with Tx buffer (0.2% gelatin, 300 mM NaCl, 0.3% Triton X‐100 in PBS) for 30 min at RT. Primary antibodies (active caspase‐3 [ab2302, Abcam, RRID:AB_302962]; Arhgap11a [ab113261, Abcam, 1:500, RRID:AB_10866587]; ARHGAP11B [MPI‐CBG, 1:200] (Namba et al, 2020); Brn2 [sc‐6029, Santa Cruz, 1:200, RRID:AB_2167385]; Ctip2 [ab18465, Abcam, 1:500, RRID:AB_2064130]; Dcx [ab18723, Abcam, 1:200, RRID:AB_732011]; GFP [ab13970, Abcam, RRID:AB_300798]; Ki67 [9129S, CST, 1:500, RRID:AB_2687446]; Pax6 [BLD‐901301, BioLegend, 1:300, RRID:AB_291612]; PH3 [ab10543, Abcam, 1:500, RRID:AB_2295065]; pVim [ab22651, Abcam, 1:500, RRID:AB_447222]; Satb2 [ab51502, Abcam, 1:100, RRID:AB_882455]; Tbr1 [ab31940, Abcam, 1:500, RRID:AB_220021]; Tbr2 [MPI‐CBG, 1:200]) were added to Tx buffer, and cryosections were incubated with primary antibodies overnight at 4°C. After washing the sections with Tx buffer for three times, 10 min each, the incubation with fluorophore‐conjugated secondary antibodies (A488‐, A594‐, A555‐, or A647‐labelled secondary antibodies (Alexa Fluor, Thermo Fisher Scientific), diluted 1:500) and DAPI (1:1,000) was carried out for 1 h at RT. After washing with PBS, sections were mounted in Mowiol (Merck Biosciences).
For cell cycle re‐entry analysis, 0.1 ml of BrdU (1 mg/ml in PBS) was injected intraperitoneally into pregnant mice at E13.5, i.e., 24 h before sacrifice at E14.5, and 0.1 ml of EdU (1 mg/ml in PBS) was injected intraperitoneally into the same pregnant mice 0.5 h before sacrifice at E14.5. BrdU staining was performed as previously described (Wong et al, 2015) with minor modifications. Briefly, permeabilized and quenched cryosections that had not yet been subjected to antigen retrieval were incubated with 2 M HCl three times in a water bath at 37°C, 15 min each time. Next, cryosections were incubated with 10% horse serum in PBS twice, 5 min each time, followed by Cy3‐conjugated BrdU antibody (MPI‐CBG, 1:500) incubation for 2 h at RT. After washing the cryosections with PBS, EdU staining was performed using the Click‐iT EdU Alexa Fluor 488 imaging kit (Invitrogen) according to the manufacturer's instructions. All cryosections subjected to BrdU and EdU staining were counterstained with DAPI (Sigma, 1:1,000) and mounted in Mowiol.
All stained sections were then imaged using a Zeiss LSM 880 Airy upright confocal microscope and Zeiss Plan‐Apochromat 20× 0.8 or Zeiss Plan‐Apochromat 40× 1.2 water objectives.
Immunoblotting
Immunoblotting was performed as previously described (Namba et al, 2020). Embryonic mouse neocortical tissue was homogenized in 100 μl of RIPA lysis buffer (Thermo Fisher Scientific) on ice using plastic pestles. The homogenates were sonicated at 20 kHz for 10 s and centrifuged for 12 min at 13,400 g in an Eppendorf benchtop centrifuge at 4°C. The supernatant was collected, and protein concentration was determined using the BCA assay (Thermo Fisher Scientific). Equal amounts (20 μg) of protein were subjected to SDS–PAGE (Thermo Fisher Scientific, NuPAGE 4–12% Bis–Tris Protein Gel 1.0 mm, 12‐well NP0322; NuPAGE MES SDS Running Buffer NP0002). After SDS–PAGE, proteins were transferred for 2 h at 222 mA in NuPAGE Transfer Buffer (Thermo Fisher Scientific, NP0006) containing 15% methanol to membranes (Merck Immobilon‐PVDF Membrane, IPVH00010, Millipore), which were then blocked with 5% BSA in Tris‐buffered saline pH 7.4 with Tween®20 (TBST, 20 mM Tris base, 150 mM NaCl, 0.5% Tween®20) and incubated overnight with primary antibodies (Arhgap11a [ab113261, Abcam, 1:1,000, RRID:AB_10866587]; β‐actin [4970, Biolabs, 1:1,000, RRID:AB_2223172]) in 5% BSA in TBST at 4°C with gentle shaking. Membranes were washed three times with TBST and incubated with appropriate HRP‐conjugated secondary antibodies (1:5,000; Jackson ImmunoResearch) in TBST for 1 h at RT with gentle shaking and then washed three times with TBST and developed with ECL Western Blotting Detection Reagents (GE Healthcare RPN 2106). Exposure time to X‐ray film was varied among individual experiments (30 s to 5 min) to obtain an appropriate intensity of the immunoreacted band of interest. Images were acquired using PERFECTION V750 PRO scanner (EPSON). Quantification of immunoblots was performed using Fiji.
Golgi‐Cox staining and neuronal dendrite and spine morphology analysis
Golgi‐Cox staining was performed on freshly dissected adult mouse brain using the FD Rapid GolgiStain™ Kit (FD NeuroTechonologies) according to manufacturer's instruction. Stained brain tissue was embedded in 3% low‐melt agarose, and vibratome sections were cut at 100 µm. Sections were mounted with Mowiol and imaged using a Zeiss Spinning Disc inverted microscope. Pyramidal neurons in layers II + III of the somatosensory cortex of the rostral neocortex were selected for further analysis. Dendritic tracings of upper‐layer neurons were quantified by Sholl analysis to estimate total dendritic length and dendritic arborization using the Sholl analysis Fiji plug‐in. The dendritic spine density was measured as previously reported (Parent et al, 2016). Briefly, secondary dendrites that were at least 10 μm long were traced, the exact length of the dendritic segment was calculated, and the number of spines along the dendrite was counted (to yield spines/10 µm). Also, dendritic spines were classified as previously reported (Parent et al, 2016; Laguesse et al, 2017) into filopodia, thin, stubby and mushroom.
Behavioural tests
Behavioural testing was performed during the light phase at BIOCEV/IMG. The wild‐type and heterozygous ARHGA11B‐transgenic male and female mice were tested at 9–11 weeks of age. Four separate cohorts were tested, with each cohort being used for 2–4 behavioural tasks to avoid carryover effects arising from the animal's experience gained in the previous tests (Bell, 2013). Male and female cohorts were tested separately on different days. Testing arenas, mazes, used objects, animal enclosures were thoroughly cleaned with 75% ethanol and then dried to remove olfactory traces before the first tested animal and between each tested animal.
Open field
Animal activity in a novel environment and the level of anxiety were evaluated in open‐field tests as previously described (Kulesskaya & Voikar, 2014). The area of the open field was a square of 42 × 42 cm uniformly illuminated with a light intensity of 200 lux in the centre of the field. The testing arena was divided into periphery and centre zones, where the centre zone constitutes 38% of the whole arena. Each mouse was placed in the corner of the arena for a 10‐min period. The time spent in each zone, the distance travelled and other indices were automatically computed based on a video tracking system (Viewer, Biobserve GmbH, Germany).
Light–dark box
Animal anxiety levels were also examined using another test based on an approach‐avoid conflict paradigm in a light–dark box (LDB) as previously described (Hascoet & Bourin, 1998). The LDB was divided into two compartments, the light part (light intensity of 430 lux, 67% of the area) and an enclosed dark part with a small entrance (7 cm × 7 cm) in between. Mice were placed individually into the dark compartment and allowed to freely explore the LDB for 5 min. The presence time, distance travelled, number of visits and latency to enter the light compartment were automatically computed based on a video tracking system (Viewer, Biobserve GmbH, Germany).
Hot plate
Nociceptive sensitivity was assessed by using an electronic Hot/Cold Plate apparatus (Ugo Basile, Gemonio, Italy), where the animal's reaction following the exposure to a heat stimulus (52°C warm plate) was recorded as previously described (Gunn et al, 2011). Each mouse was placed into the apparatus, from which it was removed immediately after noticing a reaction to the nociceptive stimulus (lifting, shaking or licking either hind paw). Three measurements of latency to the reaction to the heat stimulus were recorded with 15‐min intertrial intervals. The measurements were averaged for each animal.
Y maze
Working memory was assessed in a Y‐shaped maze with a light intensity of 50 lux in the centre (Moran et al, 1995). Animal behaviour was recorded by a digital video camera for a period of 5 min. The percentage of alternation was calculated automatically by the Biobserve software according to the equation %SA = (TA×100)/(TE‐2), where SA—spontaneous alternations, TA—total Alternations made by the animal, TE—total arm entries.
Barnes maze
The spatial learning and memory abilities were analysed according to the protocol published previously (Youn et al, 2012). Rather than using the classical Barnes maze, with holes arranged along the maze edge, the modified Barnes maze with 44 escape holes positioned symmetrically in each quadrant was used to prevent mice from using a serial strategy to find an escape box (O'Leary & Brown, 2012). The whole procedure consisted of four stages: habituation to the escape box, habituation to the maze, acquisition and probe trial. Except for the first habituation day, where a low‐light condition (70 lux) and no noise were used, the entire procedure was performed in high‐aversive conditions to motivate animals to search for the escape target box (illumination of 200 lux and moderate background noise).
In the first stage, mice were habituated to the escape box placed in their home cage for 7 days. On the 8th day, the escape box was removed and thoroughly cleaned to remove olfactory traces. During the second, two‐day habituation phase, mice were allowed to explore the maze for 7 min to find an escape box positioned in a different place each day. If an animal failed to find the escape box, it was gently guided in a glass container to the target and left there for 30 s.
The next stage was a 5‐day training session, during which 4 different escape box positions in the maze were used alternately as follows: the specific escape box position for each individual animal varied, but each individual animal was trained for the same escape box position during all 5 days of the training session. Each day of acquisition, mice were allowed to find the target hole during two 3‐min trials with an approximately 2‐h intertrial interval.
Animals were tested in probe trial, 24 h after the last day of acquisition. During the 1‐min probe trial, no escape box was present in the maze. Video recordings were automatically analysed by Viewer software (Biobserve GmbH, Germany). The primary distance, latency and errors to reach the target hole were used for further analysis. Additionally, for probe trial time and distance in the target quadrant were evaluated.
Contextual and delayed cued fear conditioning
The associative learning was assessed in the standard cage placed in a soundproof cabinet (Ugo Basile, Gemonio, Italy). The cage was equipped with a stainless‐steel rod for shock delivery. Experiments were performed based on the previously described protocol (Stiedl et al, 1999), with minor modifications. The acquisition trial started with a 4‐min adaptation period, after which mice were presented with two pairings of the conditioned stimulus (CS; 20 s of 4 kHz pure tone at 77 dB) that coterminated with the unconditioned stimulus (US; 1 s, 0.6 mA constant current to the cage floor). The interval between two pairings was 2 min. Mice remained in the chamber for 1 min after the last shock delivery. Animals were tested for associative memory 24 h after training. They were re‐introduced to the training context, and behaviour was recorded for 6 min with no CS or US presentation. Total freezing time was scored as a response to the context. Three hours later, the delayed cued memory was tested. The animal was placed in a novel context with a different cage wall pattern and a smooth floor texture with the presence of vanilla scent. The freezing response to the 2‐min CS was monitored. Freezing, defined as the absence of any movement other than that required for respiration, was detected automatically by ANY‐maze software and reviewed by a trained observer.
Social novelty preference test
The social novelty preference test was performed according to a previously described protocol, in which two wire cylindrical cages placed in an open field were used to test the social novelty preference in mice (Xuan & Hampson, 2014). Each cage (10.5 cm diameter, 15 cm high) was placed at an equal distance to the two diagonal corners of the open field (10 cm). All experimental procedures were carried out in a low‐illuminance condition (70 lux in the centre of the open field). The juvenile target animal (7‐week‐old C57Bl/6n mice) and the mouse to be tested (subject animal) were gender‐matched.
Habituation to the experimental setup: the target animals were habituated in the wire cages for 10 min without the presence of the conspecific mouse in the open field; the subject animals were habituated to the empty wire cages in the open field also for 10 min the day before testing.
On the day of testing, the target mouse was put into one of the wire cages and the object, Duplo block, to the second cage. The subject animal was introduced to the open field and allowed to explore both wire cages for 10 min. The latency, number of visits and time of interaction with the wire cage containing the target animal were automatically computed by Viewer software (Biobserve GmbH, Germany).
IntelliCage
Behavioural flexibility and competitive dominance behaviour were assessed using IntelliCage based on a previously described protocol (Benner et al, 2014). The IntelliCage (TSE Systems, GmbH, Germany) is designed for automatic, long‐term, high‐throughput investigation of cognitive abilities and the emotional state of group‐housed laboratory mice. The mice perform a variety of programmable behavioural tasks in the familiar environment of their home cage. Each IntelliCage contains four operant corners, and each corner has two door‐guided openings to drinking bottles. Each corner is also equipped with an radio‐frequency identification (RFID) antenna detecting an implanted transponder and allowing unequivocal identification whenever a mouse enters the corner. The system may reward the mouse by opening access to water, or it may punish the animals by denying access or even by delivering mildly aversive air‐puffs (www.newbehaviour.com).
Mice were implanted subcutaneously with transponders (ISO compliant transponders) and pooled together into experimental groups a week before transfer to IntelliCage. Males (10 animals per cage) and females (12 animals per cage) were tested in separate cages. Mice were introduced to the IntelliCage for a free‐adaptation regime for three consecutive days. Animals had free access to water. This protocol is the initial step of each IntelliCage design. Doors inside the conditioning corners open whenever an animal enters and close after it leaves the corner. Data collected during the free‐adaptation yield information about overall activity levels, exploratory behaviour and circadian rhythmicity. Augmented or reduced neophobic behaviour can also be detected during the early stages of that protocol (Vannoni et al, 2014).
The subsequent regime was nose‐poke adaptation and lasted 7 days. Animals were trained to nose poke to get access to water, 5 s long, once per visit. This regime follows free adaptation and precedes any other animal testing protocol or combination of protocols. This regime evaluates procedural memory/procedural conditioning and prepares animals for the following procedures.
The next procedure is shaping, which lasted 7 days. Animals had restricted access to water from four corners only between 10 pm and 1 am each day. Time with water availability was guided by a constant blue LED light in each corner. Doors opened for 7 s upon each visit. Restricted access to water within the time frame creates a competitive condition for the mice, which allowed the assessment of the competitive dominant behaviour (Benner et al, 2014). Visiting duration to each corner during the first 5 min of the water‐accessible session was measured for analysing competitive dominant behaviour.
The next procedure following shaping is corner alternation, which is a 7‐day learning phase (acquisition in Fig 4). Similar to shaping, water access was restricted to 3 h (between 10 pm and 1 am) per day in the corner alternation procedure. During the water‐accessible session, only one of the two diagonally positioned corners is “active” (door can be opened by nose poke to allow the animal to drink water, 4 s per visit), with the other corner being “inactive” (door is closed and cannot be opened by nose poke). After each drinking for each specific animal, the “active” corner becomes “inactive”, and the diagonally positioned “inactive” corner becomes “active”. Thus, the animal had to shuttle between the two diagonally positioned “active” and “inactive” corners to drink water during water‐accessible session each day. The two remaining corners were never rewarding (doors can never be opened to allow animals to drink water). During this corner alternation procedure, nose‐poke frequency per visit at the “active” corner during the drinking session was analysed for compulsive repetitive behaviour, since excessive nose poking was behaviourally useless for obtaining additional reward (water). Nose‐poke frequency per visit at the “inactive” and never rewarding corners was analysed for impulsive behaviour.
The next procedure following corner alternation is corner reversal, which consisted of 6 corner reversal phases (reversal 1–6 in Fig 4) and lasted 33 days in total. The first two corner reversal phases lasted 14, 7 days in each reversal phase, and the remaining four reversal phases lasted 16, 4 days in each reversal phase. The corner reversal procedure preserved all the conditions of corner alternation plus one more modification as follows. At the beginning of each reversal phase, the “active” and “inactive” corners in the previous phase became never rewarding, while previously never rewarding corners became rewarding (“active” and “inactive”‐alternating) corners.
Both corner alternation and corner reversal procedures were used to evaluate the memory flexibility of mice. Discrimination error rate was calculated as the number of visits to the incorrect (inactive and never rewarding) corners within the first 100 visits each day during the drinking session, or the percentage of the total visits that are incorrect when the total number of corner visits was less than 100.
A modified IntelliCage protocol was used to evaluate the memory flexibility of a different cohort of male mice. In this protocol, the corner alteration procedure lasted 4 days and the following corner reversal procedure lasted 4 days instead. The rest of the modified IntelliCage protocol remained the same as the original protocol described above. For the analysis of data derived from the modified IntelliCage protocol, the success rate was used to evaluate the memory flexibility. The success rate was calculated as the number of visits to the correct corners within the first 100 visits each day during the drinking session, or the percentage of the total visits that are correct when the total number of corner visits was less than 100.
Quantification and statistical analysis
Sample sizes (individual embryos or mice) used for statistical tests are reported in each figure legend. All cell counts were performed in standardized microscopic fields using the Fiji cell counter plug‐in. All quantifications were done blindly. All statistical analyses were conducted using GraphPad Prism. Data normality was tested by Shapiro–Wilk normality test or KS normality test, and variances between groups were tested using F‐test. Means between two groups were compared using two‐tailed unpaired Student's t‐test if data were normally distributed, or Mann–Whitney test if data were not normally distributed. Means from multiple groups were compared using one‐way analysis of variance (ANOVA) or two‐way ANOVA, followed by Bonferroni's or Fisher's LSD multiple comparison tests. Error bars in all figures indicate SD. Asterisks: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; no asterisk, not statistically significant.
Author contributions
LX, MF, WBH conceptualized the study; LX, MF, AK‐Z, MS and JP contributed to methodology; LX, AK‐Z, TN and AP performed formal analysis; LX, AK‐Z, TN and AP investigated the study; LX visualized the study; LX wrote the original draft, with input from AK‐Z, TN and AP; LX and WBH reviewed and edited the manuscript; RS and WBH acquired funding; WBH supervised the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We apologize to all researchers whose work could not be cited due to space limitations. We are grateful to the services and facilities of MPI‐CBG and BIOCEV/IMG for the outstanding support provided, notably Jana Kopkanová and her team of BIOCEV/IMG, as well as Sylke Winkler and her team of the DNA Sequencing and Genotyping Facility, Jussi Helppi and his team of the Biomedical Services, Jan Peychl and his team of the Light Microscopy Facility, Ronald Naumann and his team of the Transgenic Core Facility, Ina Nüßlein, Julia Jarrells and Christina Eugster Oegema of the Cell Technology Facility, Lena Hersemann of the Scientific Computing Facility and the Genome Engineering Facility of MPI‐CBG. RVO 68378050 from Czech Academy of Sciences, LM2018126, CZ.1.05/1.1.00/02.0109 and CZ.1.05/2.1.00/19.0395 for the Czech Center of Phenogenomics were provided to R.S. by MEYS. W.B.H. was supported by grants from the DFG (SFB 655 ‐ A2), the ERC (250197) and ERA‐NET NEURON (MicroKin). Open Access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2021) 40: e107093.
Data availability
This study includes no data deposited in external repositories.
References
- Antonacci F, Dennis MY, Huddleston J, Sudmant PH, Steinberg KM, Rosenfeld JA, Miroballo M, Graves TA, Vives L, Malig M et al (2014) Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nat Genet 46: 1293–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai Y, Pulvers JN, Haffner C, Schilling B, Nusslein I, Calegari F, Huttner WB (2011) Neural stem and progenitor cells shorten S‐phase on commitment to neuron production. Nat Commun 2: 154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD (2005) Neuronal subtype‐specific genes that control corticospinal motor neuron development in vivo . Neuron 45: 207–221 [DOI] [PubMed] [Google Scholar]
- Aung KH, Kyi‐Tha‐Thu C, Sano K, Nakamura K, Tanoue A, Nohara K, Kakeyama M, Tohyama C, Tsukahara S, Maekawa F (2016) Prenatal exposure to arsenic impairs behavioral flexibility and cortical structure in mice. Front Neurosci 10: 137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A (2013) Randomized or fixed order for studies of behavioral syndromes? Behav Ecol 24: 16–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner S, Endo T, Endo N, Kakeyama M, Tohyama C (2014) Early deprivation induces competitive subordinance in C57BL/6 male mice. Physiol Behav 137: 42–52 [DOI] [PubMed] [Google Scholar]
- Boyd JL, Skove SL, Rouanet JP, Pilaz LJ, Bepler T, Gordan R, Wray GA, Silver DL (2015) Human‐chimpanzee differences in a FZD8 enhancer alter cell‐cycle dynamics in the developing neocortex. Curr Biol 25: 772–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárdenas A, Villalba A, de Juan Romero C Picó E, Kyrousi C, Tzika AC, Tessier‐Lavigne M, Ma Le, Drukker M, Cappello S et al (2018) Evolution of cortical neurogenesis in amniotes controlled by Robo signaling levels. Cell 174: 590–606.e521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RE (2018) A history and overview of the behavioral neuroscience of learning and memory. Curr Top Behav Neurosci 37: 1–11 [DOI] [PubMed] [Google Scholar]
- Dehay C, Kennedy H, Kosik KS (2015) The outer subventricular zone and primate‐specific cortical complexification. Neuron 85: 683–694 [DOI] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF (2005) Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci 25: 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fame RM, MacDonald JL, Macklis JD (2011) Development, specification, and diversity of callosal projection neurons. Trends Neurosci 34: 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fame RM, MacDonald JL, Dunwoodie SL, Takahashi E, Macklis JD (2016) Cited2 regulates neocortical layer II/III generation and somatosensory callosal projection neuron development and connectivity. J Neurosci 36: 6403–6419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei JF, Haffner C, Huttner WB (2014) 3' UTR‐dependent, miR‐92‐mediated restriction of Tis21 expression maintains asymmetric neural stem cell division to ensure proper neocortex size. Cell Rep 7: 398–411 [DOI] [PubMed] [Google Scholar]
- Fernandez V, Llinares‐Benadero C, Borrell V (2016) Cerebral cortex expansion and folding: what have we learned? EMBO J 35: 1021–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiddes IT, Lodewijk GA, Mooring M, Bosworth CM, Ewing AD, Mantalas GL, Novak AM, van den Bout A Bishara A, Rosenkrantz JL et al (2018) Human‐specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell 173: 1356–1369.e1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, Huttner WB (2014) Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141: 2182–2194 [DOI] [PubMed] [Google Scholar]
- Florio M, Albert M, Taverna E, Namba T, Brandl H, Lewitus E, Haffner C, Sykes A, Wong Fk, Peters J et al (2015) Human‐specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 347: 1465–1470 [DOI] [PubMed] [Google Scholar]
- Florio M, Namba T, Pääbo S, Hiller M, Huttner WB (2016) A single splice site mutation in human‐specific ARHGAP11B causes basal progenitor amplification. Sci Adv 2: e1601941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, Heide M, Pinson A, Brandl H, Albert M, Winkler S, Wimberger P, Huttner WB, Hiller M (2018) Evolution and cell‐type specificity of human‐specific genes preferentially expressed in progenitors of fetal neocortex. eLife 7: e32332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC (2018) Neuronal cell death. Physiol Rev 98: 813–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Rakic P (2013) Cortical evolution: judge the brain by its cover. Neuron 80: 633–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn A, Bobeck EN, Weber C, Morgan MM (2011) The influence of non‐nociceptive factors on hot‐plate latency in rats. J Pain 12: 222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascoet M, Bourin M (1998) A new approach to the light/dark test procedure in mice. Pharmacol Biochem Behav 60: 645–653 [DOI] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W, Huttner WB (2004) Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci USA 101: 3196–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heide M, Haffner C, Murayama A, Kurotaki Y, Shinohara H, Okano H, Sasaki E, Huttner WB (2020) Human‐specific ARHGAP11B increases size and folding of primate neocortex in the fetal marmoset. Science 369: 546–550 [DOI] [PubMed] [Google Scholar]
- Ju XC, Hou QQ, Sheng AL, Wu KY, Zhou Y, Jin Y, Wen T, Yang Z, Wang X, Luo ZG (2016) The hominoid‐specific gene TBC1D3 promotes generation of basal neural progenitors and induces cortical folding in mice. Elife 5: e18197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalebic N, Gilardi C, Albert M, Namba T, Long KR, Kostic M, Langen B, Huttner WB (2018) Human‐specific ARHGAP11B induces hallmarks of neocortical expansion in developing ferret neocortex. eLife 7: e41241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalebic N, Gilardi C, Stepien B, Wilsch‐Bräuninger M, Long KR, Namba T, Florio M, Langen B, Lombardot B, Shevchenko A et al (2019) Neocortical expansion due to increased proliferation of basal progenitors is linked to changes in their morphology. Cell Stem Cell 24: 535–550.e539 [DOI] [PubMed] [Google Scholar]
- Kostic M, Paridaen J, Long KR, Kalebic N, Langen B, Grubling N, Wimberger P, Kawasaki H, Namba T, Huttner WB (2019) YAP activity is necessary and sufficient for basal progenitor abundance and proliferation in the developing neocortex. Cell Rep 27: 1103–1118.e1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulesskaya N, Voikar V (2014) Assessment of mouse anxiety‐like behavior in the light‐dark box and open‐field arena: role of equipment and procedure. Physiol Behav 133: 30–38 [DOI] [PubMed] [Google Scholar]
- Laguesse S, Morisot N, Shin JH, Liu F, Adrover MF, Sakhai SA, Lopez MF, Phamluong K, Griffin WC, Becker HC et al (2017) Prosapip1‐dependent synaptic adaptations in the nucleus accumbens drive alcohol intake, seeking, and reward. Neuron 96: 145–159.e148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JH, Hansen DV, Kriegstein AR (2011) Development and evolution of the human neocortex. Cell 146: 18–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Liu W, Yang Lu, Wu Q, Zhang H, Fang Ai, Li L, Xu X, Sun Le, Zhang J et al (2017) The primate‐specific gene TMEM14B marks outer radial glia cells and promotes cortical expansion and folding. Cell Stem Cell 21: 635–649.e638 [DOI] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Mariani J, Kosik KS, Mehler MF, Mattick JS (2008) Noncoding RNAs in long‐term memory formation. Neuroscientist 14: 434–445 [DOI] [PubMed] [Google Scholar]
- Miller DJ, Bhaduri A, Sestan N, Kriegstein A (2019) Shared and derived features of cellular diversity in the human cerebral cortex. Curr Opin Neurobiol 56: 117–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Saito K, Kawano M, Muto T, Ogawa M (2004) Asymmetric production of surface‐dividing and non‐surface‐dividing cortical progenitor cells. Development 131: 3133–3145 [DOI] [PubMed] [Google Scholar]
- Molnár Z, Clowry GJ, Šestan N, Alzu'bi A, Bakken T, Hevner RF, Hüppi PS, Kostović I, Rakic P, Anton Es et al (2019) New insights into the development of the human cerebral cortex. J Anat 235: 432–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD (2007) Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci 8: 427–437 [DOI] [PubMed] [Google Scholar]
- Moran PM, Higgins LS, Cordell B, Moser PC (1995) Age‐related learning deficits in transgenic mice expressing the 751‐amino acid isoform of human beta‐amyloid precursor protein. Proc Natl Acad Sci USA 92: 5341–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba T, Dóczi J, Pinson A, Xing L, Kalebic N, Wilsch‐Bräuninger M, Long KR, Vaid S, Lauer J, Bogdanova A et al (2020) Human‐specific ARHGAP11B acts in mitochondria to expand neocortical progenitors by glutaminolysis. Neuron 105: 867–881.e869 [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez‐Cerdeno V, Ivic L, Kriegstein AR (2004) Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7: 136–144 [DOI] [PubMed] [Google Scholar]
- Nonaka‐Kinoshita M, Reillo I, Artegiani B, Martinez‐Martinez MA, Nelson M, Borrell V, Calegari F (2013) Regulation of cerebral cortex size and folding by expansion of basal progenitors. EMBO J 32: 1817–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary TP, Brown RE (2012) The effects of apparatus design and test procedure on learning and memory performance of C57BL/6J mice on the Barnes maze. J Neurosci Methods 203: 315–324 [DOI] [PubMed] [Google Scholar]
- Parent AS, Pinson A, Woods N, Chatzi C, Vaaga CE, Bensen A, Gerard A, Thome JP, Bourguignon JP, Westbrook GL (2016) Early exposure to Aroclor 1254 in vivo disrupts the functional synaptic development of newborn hippocampal granule cells. Eur J Neurosci 44: 3001–3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P (2009) Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci 10: 724–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani N, Nowakowski TJ, Zhou H, Godshalk SE, Lisi V, Kriegstein AR, Kosik KS (2016) A primate lncRNA mediates Notch signaling during neuronal development by sequestering miRNA. Neuron 90: 1174–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards BA, Frankland PW (2017) The persistence and transience of memory. Neuron 94: 1071–1084 [DOI] [PubMed] [Google Scholar]
- Sallet J, Noonan MP, Thomas A, O’Reilly JX, Anderson J, Papageorgiou GK, Neubert FX, Ahmed B, Smith J, Bell AH et al (2020) Behavioral flexibility is associated with changes in structure and function distributed across a frontal cortical network in macaques. PLoS Biol 18: e3000605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitamukai A, Konno D, Matsuzaki F (2011) Oblique radial glial divisions in the developing mouse neocortex induce self‐renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J Neurosci 31: 3683–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl R, Walcher T, De Juan Romero C, Pilz G, Cappello S, Irmler M, Sanz‐Aquela J, Beckers J, Blum R, Borrell V et al (2013) Trnp1 regulates expansion and folding of the mammalian cerebral cortex by control of radial glial fate. Cell 153: 535–549 [DOI] [PubMed] [Google Scholar]
- Stiedl O, Radulovic J, Lohmann R, Birkenfeld K, Palve M, Kammermeier J, Sananbenesi F, Spiess J (1999) Strain and substrain differences in context‐ and tone‐dependent fear conditioning of inbred mice. Behav Brain Res 104: 1–12 [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Genomes P et al (2010) Diversity of human copy number variation and multicopy genes. Science 330: 641–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki IK, Gacquer D, Van Heurck R, Kumar D, Wojno M, Bilheu A, Herpoel A, Lambert N, Cheron J, Polleux F et al (2018) Human‐specific NOTCH2NL genes expand cortical neurogenesis through Delta/Notch regulation. Cell 173: 1370–1384.e1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki IK (2019) Molecular drivers of human cerebral cortical evolution. Neurosci Res 151: 1–14 [DOI] [PubMed] [Google Scholar]
- Vann SD, Albasser MM (2011) Hippocampus and neocortex: recognition and spatial memory. Curr Opin Neurobiol 21: 440–445 [DOI] [PubMed] [Google Scholar]
- Vannoni E, Voikar V, Colacicco G, Sanchez MA, Lipp HP, Wolfer DP (2014) Spontaneous behavior in the social homecage discriminates strains, lesions and mutations in mice. J Neurosci Methods 234: 26–37 [DOI] [PubMed] [Google Scholar]
- Wang X, Tsai JW, LaMonica B, Kriegstein AR (2011) A new subtype of progenitor cell in the mouse embryonic neocortex. Nat Neurosci 14: 555–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong FK, Fei JF, Mora‐Bermudez F, Taverna E, Haffner C, Fu J, Anastassiadis K, Stewart AF, Huttner WB (2015) Sustained Pax6 expression generates primate‐like basal radial glia in developing mouse neocortex. PLoS Biol 13: e1002217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L, Kalebic N, Namba T, Vaid S, Wimberger P, Huttner WB (2020) Serotonin receptor 2A activation promotes evolutionarily relevant basal progenitor proliferation in the developing neocortex. Neuron 108: 1113–1129.e1116 [DOI] [PubMed] [Google Scholar]
- Xuan IC, Hampson DR (2014) Gender‐dependent effects of maternal immune activation on the behavior of mouse offspring. PLoS One 9: e104433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavas E, Gonzalez S, Fanselow MS (2019) Interactions between the hippocampus, prefrontal cortex, and amygdala support complex learning and memory. F1000Research 8: 1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn J, Ellenbroek BA, van Eck I, Roubos S, Verhage M, Stiedl O (2012) Finding the right motivation: genotype‐dependent differences in effective reinforcements for spatial learning. Behav Brain Res 226: 397–403 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.