

ABSTRACT
Embryonic formation and patterning of the vertebrate spinal column requires coordination of many molecular cues. After birth, the integrity of the spine is impacted by developmental abnormalities of the skeletal, muscular and nervous systems, which may result in deformities, such as kyphosis and scoliosis. We sought to identify novel genetic mouse models of severe spine deformity by implementing in vivo skeletal radiography as part of a high-throughput saturation mutagenesis screen. We report selected examples of genetic mouse models following radiographic screening of 54,497 mice from 1275 pedigrees. An estimated 30.44% of autosomal genes harbored predicted damaging alleles examined twice or more in the homozygous state. Of the 1275 pedigrees screened, 7.4% presented with severe spine deformity developing in multiple mice, and of these, meiotic mapping implicated N-ethyl-N-nitrosourea alleles in 21% of pedigrees. Our study provides proof of concept that saturation mutagenesis is capable of discovering novel mouse models of human disease, including conditions with skeletal, neural and neuromuscular pathologies. Furthermore, we report a mouse model of skeletal disease, including severe spine deformity, caused by recessive mutation in Scube3. By integrating results with a human clinical exome database, we identified a patient with undiagnosed skeletal disease who harbored recessive mutations in SCUBE3, and we demonstrated that disease-associated mutations are associated with reduced transactivation of Smad signaling in vitro. All radiographic results and mouse models are made publicly available through the Mutagenetix online database with the goal of advancing understanding of spine development and discovering novel mouse models of human disease.
KEY WORDS: ENU, N-ethyl-N-nitrosourea, Scoliosis, Kyphosis
Summary: We report selected mouse models of spine deformity following mutagenesis across 30% of autosomal genes, results of which are made publicly available to advance understanding of spine development and disease.
INTRODUCTION
Development of the spinal column is a highly regulated process that begins early in embryogenesis. Postnatal growth and maintenance of a structurally sound spine requires coordinated integration of the vertebrae (skeleton), intervertebral discs, nerves and muscles. Disruption of any of these components may lead to spinal malformations. In humans, scoliosis, defined as a lateral curvature of the spine, is the most common spine deformity and is characterized as idiopathic, congenital, neuromuscular or syndromic (Herring, 2013). Alternatively, kyphosis manifests as an anterior-posterior curvature of the spine, sometimes in combination with scoliosis (so-called kyphoscoliosis). Unlike idiopathic scoliosis, congenital scoliosis can be associated with skeletal malformations or segmentation defects of the vertebrae, and neuromuscular scoliosis is associated with neurological or muscular disease etiologies. Each of these disease types present with different scoliotic deformities and may require different treatment paradigms.
Population-based genome-wide association studies have identified several loci associated with idiopathic scoliosis, and next-generation sequencing studies have elucidated rare mutations underlying congenital scoliosis (Chen et al., 2020; Gao et al., 2007; Khanshour et al., 2018; Kou et al., 2013; Sharma et al., 2011, 2015; Takahashi et al., 2011; Wu et al., 2015). Despite the success of human genetic studies, modeling spine deformities for these loci in mice has proven to be challenging. For example, the correlation of human scoliosis with non-genetic factors, such as the adolescent growth spurt, hormonal influences during adolescence and the distribution of mechanical forces of a bipedal spine, may be difficult to recapitulate in mice or other model systems. Moreover, certain genes implicated in severe forms of scoliosis, such as TBX6, may be sub-viable in genetically engineered knockout mouse models (Chapman and Papaioannou, 1998; Wu et al., 2015). Alternative strategies, such as using Cre-inducible conditional knockout mouse models or engineering hypomorphic alleles, have been used successfully to investigate scoliosis-associated genes in vivo (Karner et al., 2015; Yang et al., 2019). However, these strategies are limited due to a lack of knowing a priori the relevant cell type for conditional models or which alleles may be hypomorphic and allow for viable mouse models of spine deformity in essential genes.
To facilitate discovery of genes required for proper spine development, we performed live-animal radiography to detect severe spine deformity as part of a saturation mutagenesis skeletal screen in mice (Rios et al., 2021; Wang et al., 2015). As proof of concept, we report selected results from 54,497 mice from 1275 pedigrees. As described previously (Wang et al., 2018), estimates of saturation based on the standard of detecting lethal effects in a curated collection of essential genes suggests that 30.44% of all protein-encoding autosomal genes were mutated to a state of detectable hypomorphism and examined in the homozygous state twice or more in this sample of mice. Among the mutations were 5746 putative null alleles in 4320 genes examined twice or more in the homozygous state. Mutations in genes regulating proper skeletal, muscular and nervous system development are all implicated in scoliotic manifestations discovered through our screen. Results from our study may inform human genetic studies, provide novel mouse models of human disease, and advance knowledge of spine development and deformity. As proof of principle, we cross-referenced results from our study with a database of undiagnosed subjects who underwent clinical exome sequencing, and we report a patient with a skeletal syndrome caused by recessive mutations in the SCUBE3 gene that resulted in reduced heterodimerization and reduced Smad signal transduction. All results and mouse models, including others not reported here, have been made publicly available through the online Mutagenetix database (https://mutagenetix.utsouthwestern.edu).
RESULTS
Radiographic screen for spine deformity
The breeding scheme for generating and screening mice harboring N-ethyl-N-nitrosourea (ENU)-induced alleles is shown in Fig. 1. C57BL/6J male mice are mutagenized with ENU and outcrossed to non-mutagenized C57BL/6J females, resulting in male pups (termed the G1 generation) heterozygous for germline ENU-induced alleles. G1 male mice are outcrossed to C57BL/6J female mice, and G2 pups are subsequently backcrossed to their G1 sire to produce G3 mice homozygous for the reference allele, heterozygous for the ENU allele, or homozygous for the ENU allele. All G3 mice undergo radiographic screening, including both dorsal and lateral radiography (Fig. 1). Phenotypic scoring is performed from radiographs as either presence or absence of any rigid spine deformity or malformation, such as kyphosis, scoliosis or kinked/curly tail.
Fig. 1.
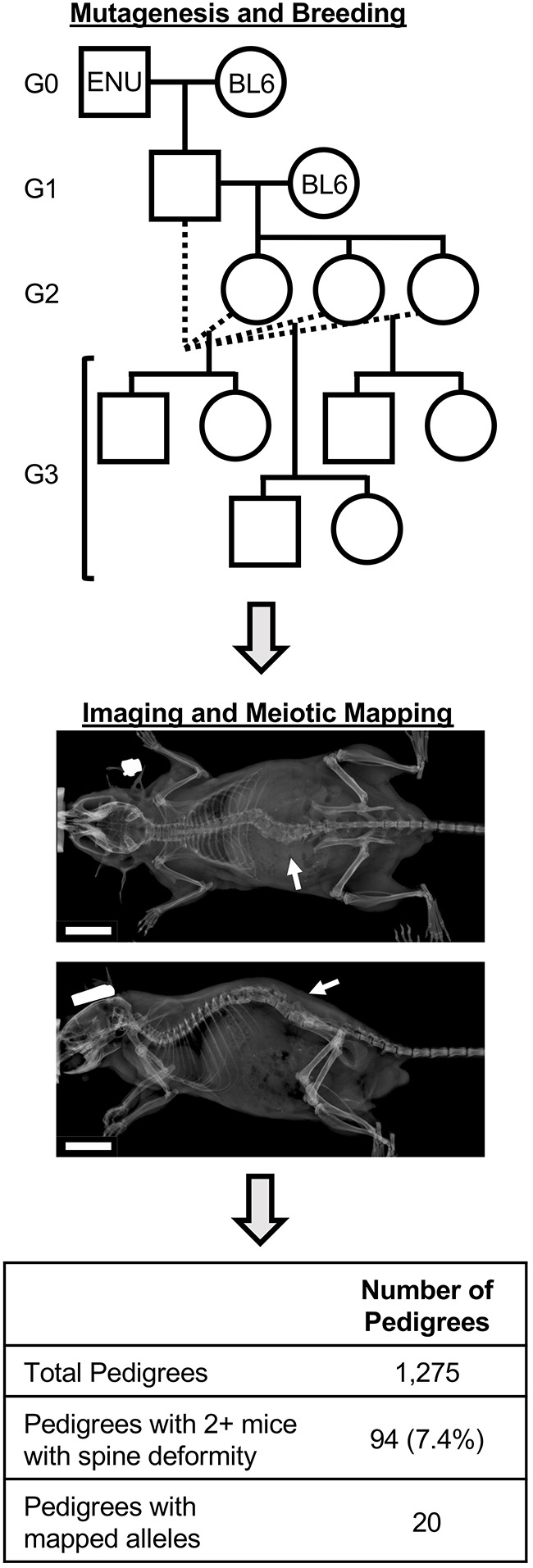
Schematic of the mutagenesis, breeding and screening strategy. Mutagenized G0 mice (ENU) are outcrossed to C57BL/6J (BL6) females. G1-generation pups are outcrossed, and subsequent G2-generation mice are backcrossed to their G1 sire to produce pedigrees of G3 mice for screening. Dorsal and lateral radiographs are obtained for all G3 mice, and mice are visually scored as affected or unaffected. Arrows indicate a spine deformity. Automated meiotic mapping tests whether ENU alleles are associated with the spine deformity phenotype after correction for the number of ENU alleles tested in the pedigree. Scale bars: 1 cm.
ENU-induced alleles are detected by exome sequencing G1 male mice, as described previously (Wang et al., 2015). Nonsynonymous variants identified relative to the C57BL/6J reference genome are genotyped by targeted capture and sequencing of all G2 and G3 mice. Automated meiotic mapping tests the null hypothesis that ENU-induced alleles segregating within the pedigree are not associated with a risk for spine deformity. Significantly associated loci are identified following Bonferroni correction for the number of ENU-induced alleles in the pedigree.
Of the 1275 pedigrees evaluated, 94 (7.3%) pedigrees were identified with at least two mice scored with a spine deformity phenotype. Of these, 20 (20.6%) were mapped to ENU-induced alleles (Fig. 1; Table S1), including 19 recessive and one dominant allele. We sought to evaluate variables limiting detection of significantly associated loci, such as the total number of mice in the pedigree and the total number of affected mice in the pedigree. Pedigrees with mapped alleles had significantly higher numbers of affected mice (Wilcoxon, P=1.59e−5) compared to unmapped pedigrees (Fig. S1A), and this remained significant among only recessive alleles (Wilcoxon, P=4.41e−5). Total numbers of mice were unchanged between pedigrees with mapped and unmapped alleles (Fig. S1B). Most (12/19; 63%) pedigrees with mapped recessive alleles had at least 80% penetrance of the spine deformity phenotype among homozygous G3 mice (Table S1).
Altered skeletal development leading to spine deformity in mice
Mouse model and case report of SCUBE3-associated skeletal disease
We identified the Scube3C301Y allele associated with recessive spine deformity, including severe kyphosis and kinked tail (Fig. 2A). Although the homozygous Scube3C301Y allele co-segregated with homozygous Rhot2V604A and Nrde2D607E alleles, we considered these to be less likely candidates based on the presence of skeletal phenotypes in mice homozygous for a previously reported Scube3N294K allele (Fuchs et al., 2016). Additionally, the Scube3C301Y allele was predicted to be more damaging by PolyPhen analysis than the Rhot2V604A and Nrde2D607E alleles.
Fig. 2.

Mutations in SCUBE3 are associated with skeletal deformity. (A) Representative radiographic imaging of spine deformity in 2-month-old female mice homozygous for the Scube3C301Y allele compared to a gender-matched littermate homozygous for the reference allele (Scube3WT). Enlarged radiographs of the kinked tail and spine deformities are shown (right). Arrows indicate location of the deformity. Scale bars: 1 cm. (B) Clinical radiographs of the hand (left) and feet (right) of a male patient with bi-allelic SCUBE3 mutations. Arrows indicate bone fusions that were evident in both the hands and feet. (C) Pedigree of a family with recessive skeletal disease. Affected individuals are shown with filled symbols. Two mutations were identified by exome sequencing, and genotype results for each mutation are shown for available subjects. Both mutations were confirmed to be compound heterozygous in the affected child by Sanger sequencing (Fig. S2). Circle, female; HET, heterozygous; square, male; WT, no mutation. (D) Schematic of the human SCUBE3 protein, including relative positions of annotated domains. Recently published variants identified in patients with skeletal disease (Lin et al., 2021) are shown in bold black. The human ortholog of the mouse ENU variant and the novel human variant reported here are shown in red. Location of the SCUBE3 p.(Thr231Ala) polymorphism is also shown. (E) Relative normalized quantification of SCUBE3 protein co-immunoprecipitated with SCUBE1. P=0.01. (F) The Smad signaling luciferase reporter MDA-scp28 cells were treated with conditioned medium from HEK293T cells expressing wild-type SCUBE3 (WT) and SCUBE3 containing the common polymorphism (T231A) or disease-associated mutations (as indicated in D). An untransfected NTC sample is shown as a negative control. P=2.695e−12. (G) Relative secretion of Myc-tagged SCUBE3 was evaluated in medium from HEK293 cells transiently expressing wild-type SCUBE3 (WT) and SCUBE3 containing the common polymorphism (T231A) or disease-associated mutations (as indicated in D). P=0.32. Data are mean±s.e.m. from three independent experiments. Statistically significant differences were evaluated by ANOVA and, in F, pairwise differences compared to NTC were determined by Dunnett's test (see Materials and Methods). A.U., arbitrary units.
The human SCUBE3 gene, encoding the signal peptide, CUB domain, and EGF-like domain containing 3 protein, is highly expressed in osteoblasts (Wu et al., 2004). The SCUBE3 protein was identified as a transforming growth factor-beta (TGF-β) receptor ligand and activator of Smad signaling (Wu et al., 2004, 2011). Proper regulation of the TGF-β signaling cascade is essential for normal skeletal development (Crane and Cao, 2014). Disruption of TGF-β signal transduction leads to defects in endochondral ossification via dysregulation of osteogenesis, which has been studied extensively using germline and conditional mouse models (Wu et al., 2016). Furthermore, disruption of TGF-β signaling affects the crosstalk between bone forming osteoblasts and bone resorbing osteoclasts during bone remodeling (Erlebacher and Derynck, 1996; Filvaroff et al., 1999). Mice homozygous for a Scube3N294K allele developed mild phenotypic abnormalities, including shorter hindlimbs, rib defects and auditory abnormalities, and Scube3−/− mice were recently shown to develop a postnatal dwarfism that was not apparent during embryonic development (Fuchs et al., 2016; Lin et al., 2021; Xavier et al., 2013).
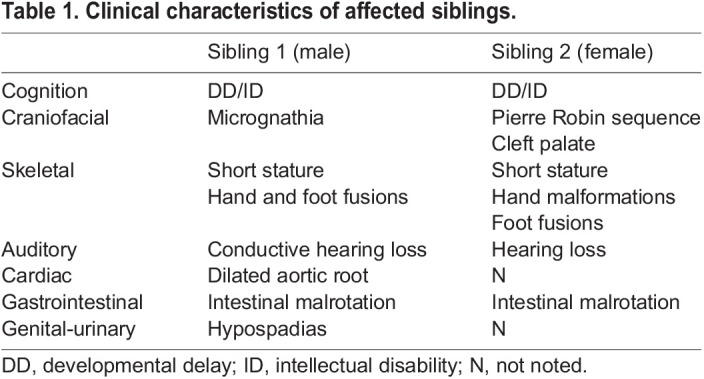
Although all three Scube genes (Scube1-3) are implicated in regulating proper skeletal development in mice (Fuchs et al., 2016; Lin et al., 2015; Tu et al., 2008), mutations in any one of three human Scube genes (SCUBE1-3) have yet to be associated with human disease. However, during the course of this study, multiple subjects with recessive SCUBE3 mutations were reported with short stature, oral-facial abnormalities, skeletal abnormalities and, although variable, spine deformity, suggesting a novel SCUBE3-associated skeletal disease (Lin et al., 2021). To identify additional patients with skeletal disease potentially attributable to mutations in SCUBE3, we queried the Baylor Genetics Laboratory database, which includes nonsynonymous variants identified from clinical exome sequencing and clinician-provided clinical synopses. We sought patients described with skeletal abnormalities, yet remaining without a genetic diagnosis following evaluation by clinical exome sequencing, among whom rare nonsynonymous variants in SCUBE3 were identified. We identified a now 16-year-old male patient presenting with GI malrotation, hearing loss, short stature, joint hyperextensibility, dilated aortic root with abnormal aortic valve, and oral-facial differences, including cupped ears, high-arched palate and micrognathia (Table 1). Skeletal abnormalities included asymmetric fusion of a pseudoepiphysis of the second metacarpal bilaterally (Fig. 2B). His feet were notable for an unusual fusion of the second metatarsal to the second cuneiform (Fig. 2B). The patient's parents were unaffected; however, an affected sibling was also noted to have similar clinical findings, consistent with a recessive disease presentation in this family (Table 1; Fig. 2C). The male patient previously underwent extensive genetic testing, including microarray analysis, testing for congenital disorders of glycosylation and targeted genetic testing, including COL1A1, COL2A1, COL3A1, COL11A1, COL11A2, CDH7, TGFBR1, and TGFBR2; however, all testing, including subsequent clinical exome sequencing, was negative. Through the database query, the patient was identified with two novel SCUBE3 variants, including a predicted loss-of-function frameshift mutation [NM_152753:c.1521_1522insGC, p.(Ile508AlafsTer74)] and a missense mutation [NM_152753:c.2785C>G, p.(Arg929Gly)]. Sanger sequencing performed in the patient and both parents confirmed compound heterozygous inheritance in the affected son; additional testing in the siblings was not available (Fig. 2C; Fig. S2A-C).
Table 1.
Clinical characteristics of affected siblings.
SCUBE3 was previously shown to heterodimerize with SCUBE1 (Wu et al., 2004), though it remains unclear whether this interaction is required for SCUBE3 function. We performed co-immunoprecipitation experiments to test potential deleterious effects of the novel SCUBE3 p.(Arg929Gly) variant on SCUBE1-SCUBE3 heterodimerization. Results were compared to an allelic series, including to recently reported SCUBE3 variants (Lin et al., 2021), to a common SCUBE3 p.(Thr231Ala) polymorphism (rs79753406) predicted to have normal function, and to the SCUBE3 p.(Cys301Tyr) variant orthologous to our mouse ENU allele associated with severe spine deformity (Fig. 2D). Compared to wild-type and SCUBE3 p.(Thr231Ala) proteins, SCUBE3 containing disease-associated variants showed reduced interaction with SCUBE1 (Fig. 2E; Fig. S2D). These results suggest disease-associated mutations alter the formation of the SCUBE1-SCUBE3 heterodimer.
SCUBE3 has also previously been shown to activate Smad signaling via direct interaction with the TGF-β type II receptor (TGFBR2) (Wu et al., 2011). Consistent with this, clinical features of the index patient reported here resembled Loeys–Dietz syndrome type 2 caused by mutations in TGFBR2 (Mizuguchi et al., 2004). We tested transactivation of Smad signaling by treating MDA-scp28 cells with conditioned medium from cells expressing recombinant SCUBE3 protein. Conditioned medium from cells expressing the wild-type or SCUBE3 p.(Thr231Ala) protein readily activated Smad signaling, whereas conditioned medium from cells expressing disease-associated SCUBE3 variants failed to activate Smad signaling (Fig. 2F). All samples showed similar levels of SCUBE3 secretion into the conditioned media (ANOVA, P=0.32), suggesting differences in transactivation were not due to differences in SCUBE3 secretion (Fig. 2G; Fig. S2D). These results demonstrate disease-associated SCUBE3 missense mutations are loss-of-function alleles causing SCUBE3-associated skeletal disease.
Mouse model of Hes7-associated spine deformity
During early embryonic development, the paraxial mesoderm gives rise to segmented bodies within the pre-somitic mesoderm and, ultimately, to the components of the vertebrate spinal column. The segmented bodies are formed through an intricate cyclical wave of gene regulation (i.e. segmentation clock), including the hairy and enhancer of split (HES)/HES-related (HER) family of transcription factors (Hubaud and Pourquie, 2014). Proper timing of the segmentation clock proceeds through feedback inhibition that controls Notch signaling periodicity. Disruption of the segmentation clock during early development causes various recognizable phenotypes, such as the spondylocostal dysostoses in humans. Spondylocostal dysostosis type 4, in which patients present with a variety of skeletal abnormalities, including spine and vertebral malformations, is caused by recessive loss-of-function mutations in the HES7 gene (Sparrow et al., 2008, 2010). Hes7 knockout mice died shortly after birth, whereas some heterozygous Hes7+/− mice displayed kinked tails but no other significant skeletal abnormalities (Bessho et al., 2001). We detected significant association of severe spine deformities with the heterozygous Hes7V20E allele, which co-segregated with a synonymous mutation in Slfn4 (Fig. 3A,B). A total of 15/21 (71%) heterozygous mice developed severe kyphoscoliosis with a sharp vertebral angulation resembling human patients with spine deformity associated with segmentation defects, and, of these, six also developed kinked tails. The Hes7V20E variant is located at the beginning of the basic-helix-loop-helix domain of the protein and may disrupt DNA binding, which is necessary for transcriptional feedback repression as part of the segmentation clock during mouse somitogenesis (Bessho et al., 2001). Interestingly, no additional skeletal abnormalities were observed in Hes7+/V20E mice. As the mice described here develop a more severe spine deformity compared to Hes7+/− mice (Bessho et al., 2001), it remains to be determined whether the Hes7V20E allele is loss of function or whether it has a dominant-negative effect on Hes7 transcription factor feedback signaling in vivo.
Fig. 3.
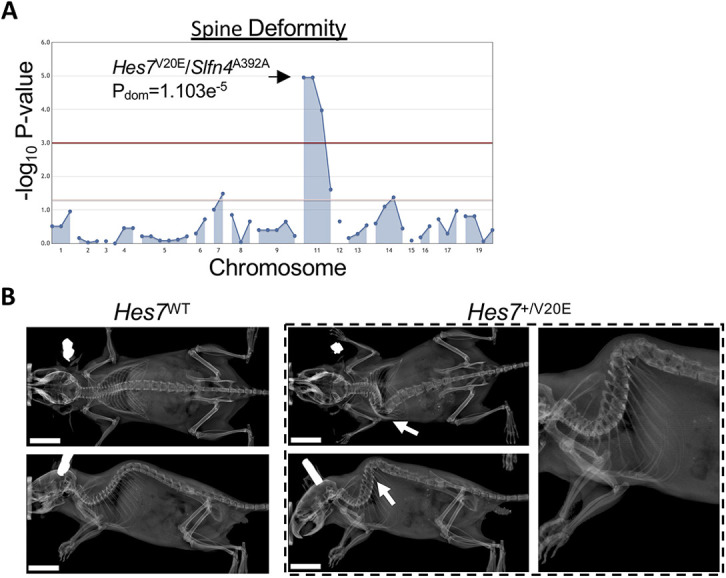
Mouse model of a dominant Hes7-associated spine deformity. (A) Manhattan plot showing statistical significance following automated meiotic mapping of ENU-induced alleles with a dominant spine deformity phenotype. The Hes7V20E allele was significantly associated with spine deformity in heterozygous mice. No homozygous mice were detected. (B) Representative radiographic images of the severe spine deformity in 3-month-old female mice heterozygous for the ENU allele (Hes7+/V20E) compared to a gender-matched littermate homozygous for the reference allele (Hes7WT). Arrows indicate the location of the deformity. Scale bars: 1 cm.
Neurogenic spine deformities in mice
Disruption of peripheral nervous system development
Some inborn errors of metabolism, such as lysosomal storage diseases, may manifest as neurodegenerative disease (Onyenwoke and Brenman, 2015). Krabbe disease is caused by recessive mutations in the GALC gene, which encodes galactosylceramidase, a lysosomal enzyme that hydrolyzes galactolipids prevalent in myelin of the central and peripheral nervous systems (Sakai et al., 1994; Wenger et al., 2000). Though the age of onset may vary, the majority of patients with Krabbe disease present during infancy with spasticity and developmental delay, leading to death by 2 years of age (Tappino et al., 2010). Among patients with infantile-onset Krabbe disease, most develop scoliosis by 2 years of age (Beltran-Quintero et al., 2019). Loss of Galc activity has been described in the ‘twitcher’ mouse line, which develops early-onset tremor, weakness, late-onset kyphosis and wasting with death by 3 months of age (Duchen et al., 1980; Kobayashi et al., 1980). We identified significant association between the GalcK207R allele and recessive spine deformity (Fig. 4A). All six homozygous mice displayed rigid cervicothoracic kyphosis (Fig. 4B) and wasting. The GalcK207R variant is located within an alpha-helix of the central triosephosphate isomerase (TIM) barrel domain located at the inner core of the Galc protein, near the GalcE198 catalytic residue involved in binding galactose (Deane et al., 2011). These results suggest that the GalcK207R variant results in loss of Galc protein function, possibly through altering substrate binding.
Fig. 4.

Neurogenic spine deformity. (A) Manhattan plot showing statistical significance of the GalcK207R allele following automated meiotic mapping of ENU-induced alleles with a recessive spine deformity phenotype. (B) Representative radiographic images of 2-month-old female mice heterozygous for the ENU allele (Galc+/K207R) or homozygous for the ENU allele (GalcK207R/K207R). Homozygous mice were smaller and developed a rigid thoracic kyphosis phenotype. Arrows indicate the location of the deformity. (C) Manhattan plot showing statistical significance of the Psma5V146G allele following automated meiotic mapping of ENU-induced alleles with a recessive spine deformity phenotype. (D) Representative radiographs of 5-month-old female mice heterozygous for the ENU allele (Psma5+/V146G) or homozygous for the ENU allele (Psma5V146G/V146G). Arrows indicate the location of the deformity. Scale bars: 1 cm.
Proteasome dysfunction
The 20S proteasome is a multimeric complex composed of seven alpha (PSMA1-7) and seven beta (PSMB1-7) subunits (Demartino and Gillette, 2007). Together with the PA700 regulatory component, the 20S protease degrades polyubiquitinated proteins. The genes encoding alpha and beta subunits of the 20S proteasome are highly conserved in humans, with high intolerance to loss-of-function mutations (Karczewski et al., 2020). Recently, a homozygous missense mutation in PSMB1 (PSMB1Y103H) was identified in a consanguineous family presenting with short stature, microcephaly, neurocognitive delay and other manifestations (Ansar et al., 2020). The PSMB1Y103 residue was predicted to interact with the PSMA5 subunit, and the PSMB1Y103H variant disrupted the formation and activity of the 20S proteasome in transient transfection assays. In mice, 20S proteasome subunits are largely unstudied; however, we detected significant association with spine deformity in mice homozygous for the Psma5V146G allele (Fig. 4C,D). These mice also variably developed ataxia and a wobbling gait (PREC=1.15e−6; Movie 1), suggestive of a neurologic deficit, and were significantly smaller (Fig. S3). It remains to be determined whether the Psma5V146G allele reported here is a complete loss of function or hypomorphic, as Psma5 is likely an essential gene, and knockout mice have not been characterized.
Mouse models of neuromuscular disease
Myotonic dystrophy
Neuromuscular conditions, such as muscular or myotonic dystrophies, present an increased risk for spine deformities. For example, Becker-type myotonic dystrophy is a skeletal muscle disorder caused by recessive mutations in the CLCN1 gene, which encodes the chloride voltage-gated channel 1 protein (Lorenz et al., 1994). In contrast, dominant-negative mutations in the same gene cause Thomsen-type myotonic dystrophy, though the recessive Becker type is more common and more severe. Both diagnoses present with a general inability to relax skeletal muscles throughout the body, and some patients with CLCN1 deficiency have been noted to develop spine deformity (Skalova et al., 2013). The CLCN1 homodimer functions as a chloride ion channel chiefly responsible for opposing excitatory signals in resting skeletal muscle. In mice, a spontaneous Clcn1 mutant line (Clcn1adr) was described with myotonia, muscle weakness, reduced growth and brittle bones (Watkins and Watts, 1984). We identified a significant association of the homozgygous Clcn1V292A allele with spine deformity (Fig. 5A,B). The Clcn1V292A allele co-segregated with a missense allele in the olfactory receptor 13 gene (Olfr13Y59H); however, the phenotypic resemblance of these mice with the Clcn1adr line and the lack of any published characterization of this olfactory receptor led us to implicate the Clcn1V292A allele in these mice. Similar to Clcn1adr mice, homozygous Clcn1V292A mice were noticeably smaller. It is notable that a human mutation (CLCN1E291K) in the residue adjacent to the orthologous human CLCN1V292 position was reported in three unrelated families with recessive myotonic dystrophy (Meyer-Kleine et al., 1995). To validate this mouse phenotype, we engineered Clcn1 knockout mice using CRISPR/Cas9. Knockout mice homozygous for a predicted frameshift mutation, p.(Phe337GlyfsTer4), developed significant spine deformity and reduced growth (Fig. 5C; Table S1), suggesting the Clcn1V292A allele is loss of function.
Fig. 5.

Neuromuscular spine deformity. (A) Manhattan plot showing statistical significance of the Clcn1V292A allele following automated meiotic mapping of ENU-induced alleles with a recessive spine deformity phenotype. (B) Representative radiographs of 2-month-old male mice heterozygous for the ENU allele (Clcn1+/V292A) or homozygous for the ENU allele (Clcn1V292A/V292A). Arrows indicate the location of the deformity. (C) Representative radiographs of 5-month-old male CRISPR-engineered mice heterozygous for the p.(Phe337GlyfsTer4) mutation (Clcn1+/ko) or homozygous for the mutation (Clcn1ko/ko). Arrows indicate the location of the deformity. (D) Manhattan plot showing statistical significance of the Large1Q359X allele following automated meiotic mapping of ENU-induced alleles with a recessive spine deformity phenotype. (E) Representative radiographs of 4-month-old male mice heterozygous for the ENU allele (Large1+/Q359X) or homozygous for the allele (Large1Q359X/Q359X). Arrows indicate the location of the deformity. Scale bars: 1 cm.
Muscular dystrophy
Dystroglycanopathies represent a subset of congenital muscular dystrophies, including Walker–Warburg syndrome and muscle-eye-brain disease. Both are among the most severe of the dystroglycanopathies and are caused by recessive mutations in LARGE1, encoding the LARGE xylosyl- and glucuronyltransferase 1 protein (Clement et al., 2008; van Reeuwijk et al., 2007). LARGE1 functions to glycosylate α-dystroglycan, an integral component of the skeletal muscle extracellular matrix (Inamori et al., 2012). α-dystroglycan binds to β-dystroglycan, a myofiber membrane protein that itself binds intracellular dystrophin (Martin, 2003). Disruption of different components of this complex results in several muscular dystrophies. Mice lacking Large1, as in the spontaneous myd mouse line, show reduced survival, reduced growth and kyphoscoliosis (Grewal et al., 2001; Lane et al., 1976). We identified a significant association of the Large1Q359X nonsense allele with recessive spine deformity (Fig. 5D,E). Interestingly, the Large1Q359X allele was not significantly associated with postnatal lethality. The variant is located in the middle of the first glycosyltransferase domain and is predicted to result in complete loss of function.
DISCUSSION
Discovering the genetic causes of human spine deformities has been elusive until recently, due, in part, to technological advances, such as genome-wide association studies and next-generation sequencing (Grauers et al., 2016; Wise et al., 2020). Understanding the genetic and mechanistic basis of human spinal disorders may be accelerated by forward genetic screens in model systems. To identify novel genetic mouse models of spine deformity, we performed a live-animal radiography screen in 54,497 mice collectively harboring predicted damaging or loss-of-function ENU-induced mutations in at least two homozygous mice across 30.44% of autosomal genes. ENU mutagenesis in mice provides unique advantages. First, the saturation mutagenesis approach employed here enables high-throughput generation and testing for associated phenovariance with predicted deleterious and loss-of-function alleles throughout the mouse genome (Wang et al., 2018; Wang et al., 2015). Second, ENU mutagenesis introduces single-base variants typically resulting in missense alterations that may discover hypomorphic effects (Arnold et al., 2012). Third, though less frequently, introduction of missense alleles may disrupt specific protein functions that result in detectable phenotypes not evident in knockout mouse models. Our results demonstrate the feasibility of detecting severe spine deformities by radiographic imaging within a high-throughput saturation mutagenesis screen in mice. Although our approach enables detection of spine deformities that may be missed with observation alone (i.e. kinked or curly tails), the relatively low percentage of pedigrees presenting with spine deformity suggest that challenges remain. For example, limitations from subjective radiographic evaluations may necessitate larger pedigrees to confidently map disease-causing ENU alleles or sexually dimorphic alleles. A recent saturation mutagenesis screen in zebrafish reported similarly low occurrence of pedigrees with spine deformity and with mapped alleles (Gray et al., 2020), suggesting such challenges may not be unique to mouse or other quadruped species. Although selected examples are presented here, results from the entire skeletal screen are deposited in the public online Mutagenetix database, which complements ongoing efforts of the IMPC (mousephenotype.org) to advance discovery of genes required for proper spine development in mice and, potentially, novel mouse models of human disease.
By integrating results with a clinical exome database of undiagnosed patients, we report a patient with bi-allelic SCUBE3 mutations presenting with skeletal abnormalities. The implication of SCUBE3 as a skeletal disease-causing gene was supported by our identification of mice homozygous for the Scube3C301Y allele developing severe skeletal defects. In the course of this study, multiple patients were reported with recessive SCUBE3 mutations presenting with skeletal and non-skeletal manifestations similar to the index patient reported here (Lin et al., 2021). Using two independent experimental assays, we show that missense variants identified in our mouse model and the undiagnosed patient reported here, as well as other missense variants recently reported (Lin et al., 2021), abrogated the interaction between SCUBE3 and SCUBE1 and reduced transactivation of Smad signaling. These results demonstrate that the characterization of novel mouse models may inform as-yet undiscovered rare human genetic diagnoses when integrated with patient-based exome sequencing studies.
Results from our study also suggest novel mouse models of human Mendelian skeletal diseases may be readily identified from the Mutagenetix database. As proof of concept, we present novel mouse models for multiple known human diseases representing different developmental etiologies, including segmentation defects (Hes7), leukodystrophy (Galc) and neuromuscular disease (Clcn1 and Large1). Though selected examples of severe spine deformity are presented here, the Mutagenetix database facilitates discovery of, and public access to, ENU-induced alleles and CRISPR-generated mouse lines associated with a spectrum of severe and mild (i.e. kinked tail) spine deformity phenotypes.
Other mutagenesis efforts have been undertaken to identify alleles associated with spine deformity in non-rodent model systems, such as zebrafish (Gray et al., 2020; Henke et al., 2017). Such large-scale mutagenesis efforts can rapidly resolve causal alleles with the integration of next-generation sequencing (Andrews et al., 2012; Wang et al., 2015). Furthermore, in vivo gene editing using CRISPR/Cas9 to simultaneously engineer knockout and allele-specific knock-in models provides for rapid confirmation of suspected disease-causing alleles. By integrating with ongoing large-scale human genome sequencing efforts, in vivo saturation mutagenesis screens have the potential to advance discovery of novel rare and ultra-rare human disease-causing genes.
MATERIALS AND METHODS
Skeletal mouse screen
The mutagenesis and breeding strategy have been described previously (Wang et al., 2018, 2015). Briefly, male mice (designated G0 generation) were mutagenized with ENU and outcrossed to C57BL/6J female mice. Transmitting ENU-induced alleles were detected in the resulting G1 male mice by exome sequencing. G1 male mice were outcrossed to C57BL/6J females, resulting in G2 mice heterozygous for ENU-induced alleles. Female G2 mice were backcrossed to their G1 sire, and all G2 and resulting G3 mice were genotyped by high-throughput targeted capture and next-generation sequencing.
All G3 mice were screened for spine deformity by live-animal radiography. Mice were anesthetized using inhaled isoflurane and imaged using a Faxitron UltraFocusDXA instrument. Spine deformities were scored as a binary variable by visualizing dorsal and lateral X-ray images. Following phenotype analysis, automated meiotic mapping identified ENU-induced alleles associated with the spine deformity phenotype using dominant and recessive inheritance models (Wang et al., 2015). All procedures were approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee.
Automated meiotic mapping of ENU alleles
ENU alleles were genotyped in G2 and G3 mice using a targeted capture and massively parallel sequencing approach. Genotypes of G2 and G3 mice were integrated with skeletal phenotypes, and the Linkage Analyzer tool tested the null hypothesis that each ENU allele within a pedigree has no association with the spine deformity phenotype using dominant and recessive models, as described previously (Wang et al., 2015). Statistical significance was assessed using logistic regression and P-values are reported using Wald tests. Results were visualized with Manhattan plots, and significantly associated alleles were those exceeding Bonferroni correction for the number of ENU alleles tested in the pedigree.
Generation of Clcn1 knockout mice
Clcn1 mutant mice were engineered using CRISPR/Cas9 as described previously (Ran et al., 2013). Female C57BL/6J mice were superovulated by injection of 6.5 U pregnant mare serum gonadotropin (Millipore), followed by injection of 6.5 U human chorionic gonadotropin (Sigma-Aldrich) 48 h later. The superovulated mice were subsequently mated overnight with C57BL/6J male mice. The following day, fertilized eggs were collected from the oviducts and in vitro-transcribed Cas9 mRNA (50 ng/μl) and Clcn1 small base-pairing guide RNA (50 ng/μl; 5′-GTTCAGAACGAATTTCCGAA-3′) were injected into the cytoplasm or pronucleus of the embryos. The injected embryos were cultured in M16 medium (Sigma-Aldrich) at 37°C in 5% CO2. For the production of mutant mice, two-cell-stage embryos were transferred into the ampulla of the oviduct (10-20 embryos per oviduct) of pseudopregnant Hsd:ICR (CD-1) female mice (Harlan Laboratories).
Founders were genotyped by Sanger sequencing. Mice carrying a predicted loss-of-function allele, p.(Phe337GlyfsTer4), were mated to C57BL/6J mice. Subsequent pups harboring heterozygous loss-of-function alleles were intercrossed, and resulting pups underwent skeletal screening. All mice were genotyped by Sanger sequencing by amplifying genomic DNA using the following primers: F, 5′-GTCTTGGTGGAGGTGGTATGAAA-3′, and R, 5′-ATGACTTGGCGATGCAGATAAAC-3′. PCR amplicons were sequenced using the following primers: F, 5′-AAATAGGGGTGATGGGAAGG-3′, and R, 5′-CGAAAACAGCTCCCAAGAAC-3′ (Fig. S4).
Exome sequencing and analysis
Clinical exome sequencing was performed as described previously (Yang et al., 2014). Briefly, sequencing libraries were generated following capture using the VCRome version 2.1 capture system. Paired-end sequencing was performed using the Illumina HiSeq system with 100 base pair reads, resulting in mean >100× coverage of targeted bases. Sequence reads were analyzed using software designed and implemented in the clinical laboratory. Variants in SCUBE3 were confirmed by Sanger sequencing (Fig. S2, Table S2). As per Institutional guidelines, informed consent was not obtained nor required for the case report included here.
Co-immunoprecipitation
The SCUBE1 coding sequence (NM_173050.3) was cloned into pcDNA3.1 introducing an N-terminal hemagglutinin (HA) tag, and the SCUBE3 coding sequence (NM_152753.2) was cloned into pcDNA3.1 introducing a C-terminal MYC tag. Site-directed mutagenesis (Takara, Mountain View, CA, USA) was used to introduce the following point mutations: SCUBE3 p.(Thr231Ala); SCUBE3 p.(Cys97Trp); SCUBE3 p.(Gly240Asp); SCUBE3 p.(Cys301Tyr); SCUBE3 p.(Ile815Thr); and SCUBE3 p.(Arg929Gly). HA-tagged SCUBE1 was co-transfected with MYC-tagged SCUBE3 using FuGene HD (Promega, Madison, WI, USA) into HEK-293 T cells cultured in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS) and antibiotic. After 48 h, cells were incubated with RIPA buffer (Santa Cruz Biotechnology, Dallas, TX, USA), scraped from the well, incubated on ice and then pelleted at 12,000 g. Cell lysate (10 µl) was used for western blotting (input).
Pure Proteome Protein A/G Mix Magnetic Beads (Sigma-Aldrich, St. Louis, MO, USA) were centrifuged at 1000 g for 30 s at 4°C, then incubated with 900 µl cell lysate and 10 µg HA-antibody (Sigma-Aldrich, St. Louis, MO, USA) at 4°C overnight with mixing. After incubation, the beads were pelleted by centrifugation at 1000 g for 30 s at 4°C. The pellet was washed three times with RIPA buffer. After, the pellet was resuspended in 20 µl Electrophoresis Sample Buffer (Santa Cruz Biotechnology, Dallas, TX, USA). The beads were boiled for 5 min, centrifuged at 1000 g for 30 s, and used for western blot analysis. Primary antibodies used for western blotting were anti-HA (1:1000; polyclonal, Sigma-Aldrich, St. Louis, MO, USA, H6908) and anti-Myc (1:1000; clone 9E10; Sigma-Aldrich, OP10). Secondary antibodies were IRDye goat anti-mouse IgG (1:5000; Li-Cor, 926-68070) and goat anti-rabbit IgG (1:5000; Li-Cor, 926-32211).
Western blots from three replicate experiments were quantified using ImageJ. For each experiment, the amount of SCUBE3 and SCUBE1 protein used for immunoprecipitation (by volume) was calculated based on the input sample, and the proportion of protein recovered following immunoprecipitation was calculated. The proportion of SCUBE3 protein recovered after immunoprecipitation was normalized to the proportion of SCUBE1 protein recovered after immunoprecipitation. These values were evaluated for statistically significant differences using ANOVA, and, to account for inter-experiment variation, the experimental replicate was included as a covariate. ANOVA P-values for each SCUBE3 variant are shown. For plotting, each variant SCUBE3 was then normalized to the SCUBE3WT control from the same experiment, and the mean and s.e.m. across the three replicate experiments were plotted.
Smad activation assays
The coding sequence of human SCUBE3 (NM_152753.2) was cloned into pcDNA3.1 introducing an N-terminal hemagglutinin (HA) tag. Site-directed mutagenesis was used to introduce the following point mutations: SCUBE3 p.(Thr231Ala); SCUBE3 p.(Cys97Trp); SCUBE3 p.(Gly240Asp); SCUBE3 p.(Cys301Tyr); SCUBE3 p.(Ile815Thr); and SCUBE3 p.(Arg929Gly). Each clone was transfected into HEK-293T cells, and 48 h later, conditioned medium was harvested and concentrated. The Smad reporter cell line MDA-scp28 was treated with conditioned medium to quantify transactivation of Smad signaling (Korpal et al., 2009). Briefly, 50,000 MDA-scp28 cells/well were plated in a 96-multiwell plate in high glucose DMEM supplemented with 10% FBS and 100 U/ml penicillin/streptomycin. Cells were not tested for contamination. MDA-scp28 cells were serum-starved for 24 h then treated with conditioned medium for 20 h. Luciferase activity was detected using a Dual Luciferase kit (Promega, Madison, WI, USA, E1910), following the manufacturer's instructions. Firefly luciferase activity was normalized by the Renilla luciferase activity (ratio F-Luc/R-Luc). Statistically significant differences were determined by one-way ANOVA and pairwise differences compared to no template control (NTC) were determined by Dunnett's test.
For testing SCUBE3 secretion, 5 ml of medium was concentrated to 200 µl by centrifugation in Amicon Ultra-4 filters (Sigma-Aldrich), then treated with an Aurum serum protein mini kit (Bio-Rad). For each sample, 60 µl was evaluated by western blotting using an anti-Myc antibody (1:1000; clone 9E10; Sigma-Aldrich, OP10). Western blots from three replicate experiments were quantified using ImageJ. These values were evaluated for statistically significant differences using ANOVA, and, to account for inter-experiment variation, the experimental replicate was included as a covariate. For plotting, each clone was normalized to the SCUBE3WT control from the same experiment, and the mean and s.e.m. across the three replicate experiments were plotted.
Supplementary Material
Acknowledgements
We thank members of the Rios–Wise and Beutler laboratories; Yibin Kang for providing the MDA-scp28 cell line (Korpal et al., 2009); and J. Kozlitina and J. Cohen for helpful discussion.
Footnotes
Competing interests
The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics Laboratories. P. L. is an employee of the Baylor College of Medicine and derives support through a professional services agreement with Baylor Genetics.
Author contributions
Conceptualization: J.J.R., B.B., C.A.W.; Methodology: J.J.R., B.B.; Validation: J.J.R.; Formal analysis: J.J.R., K.D.; Investigation: J.J.R., K.D., H.Y., K.M., S.G., J.R., S.L., J.A.R., P.L., J.M., D.J.S., C.A.W.; Resources: J.J.R., J.R., B.B.; Data curation: J.J.R., K.D.; Writing - original draft: J.J.R.; Writing - review & editing: J.J.R., K.D., K.M., J.A.R., B.B., C.A.W.; Supervision: J.J.R., B.B., C.A.W.; Project administration: J.J.R., B.B.; Funding acquisition: J.J.R., B.B.
Funding
This work was supported by Scottish Rite for Children (J.J.R.) and by the National Institutes of Health (R01AI125581 and U19AI100627 to B.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Data availability
Results and mouse models are available online via the Mutagenetix website (mutagenetix.utsouthwestern.edu) and the Mutant Mouse Resource and Research Center (mmrrc.org).
References
- Andrews, T. D., Whittle, B., Field, M. A., Balakishnan, B., Zhang, Y., Shao, Y., Cho, V., Kirk, M., Singh, M., Xia, Y.et al. (2012). Massively parallel sequencing of the mouse exome to accurately identify rare, induced mutations: an immediate source for thousands of new mouse models. Open Biol. 2, 120061. 10.1098/rsob.120061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansar, M., Ebstein, F., Ozkoc, H., Paracha, S. A., Iwaszkiewicz, J., Gesemann, M., Zoete, V., Ranza, E., Santoni, F. A., Sarwar, M. T.et al. (2020). Biallelic variants in PSMB1 encoding the proteasome subunit beta6 cause impairment of proteasome function, microcephaly, intellectual disability, developmental delay and short stature. Hum. Mol. Genet. 29, 1132-1143. 10.1093/hmg/ddaa032 [DOI] [PubMed] [Google Scholar]
- Arnold, C. N., Barnes, M. J., Berger, M., Blasius, A. L., Brandl, K., Croker, B., Crozat, K., Du, X., Eidenschenk, C., Georgel, P.et al. (2012). ENU-induced phenovariance in mice: inferences from 587 mutations. BMC Res. Notes 5, 577. 10.1186/1756-0500-5-577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran-Quintero, M. L., Bascou, N. A., Poe, M. D., Wenger, D. A., Saavedra-Matiz, C. A., Nichols, M. J. and Escolar, M. L. (2019). Early progression of Krabbe disease in patients with symptom onset between 0 and 5 months. Orphanet J. Rare Dis. 14, 46. 10.1186/s13023-019-1018-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessho, Y., Sakata, R., Komatsu, S., Shiota, K., Yamada, S. and Kageyama, R. (2001). Dynamic expression and essential functions of Hes7 in somite segmentation. Genes Dev. 15, 2642-2647. 10.1101/gad.930601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, D. L. and Papaioannou, V. E. (1998). Three neural tubes in mouse embryos with mutations in the T-box gene Tbx6. Nature 391, 695-697. 10.1038/35624 [DOI] [PubMed] [Google Scholar]
- Chen, W., Lin, J., Wang, L., Li, X., Zhao, S., Liu, J., Akdemir, Z. C., Zhao, Y., Du, R., Ye, Y.et al. (2020). TBX6 missense variants expand the mutational spectrum in a non-Mendelian inheritance disease. Hum. Mutat. 41, 182-195. 10.1002/humu.23907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement, E., Mercuri, E., Godfrey, C., Smith, J., Robb, S., Kinali, M., Straub, V., Bushby, K., Manzur, A., Talim, B.et al. (2008). Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann. Neurol. 64, 573-582. 10.1002/ana.21482 [DOI] [PubMed] [Google Scholar]
- Crane, J. L. and Cao, X. (2014). Bone marrow mesenchymal stem cells and TGF-beta signaling in bone remodeling. J. Clin. Invest. 124, 466-472. 10.1172/JCI70050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane, J. E., Graham, S. C., Kim, N. N., Stein, P. E., McNair, R., Cachon-Gonzalez, M. B., Cox, T. M. and Read, R. J. (2011). Insights into Krabbe disease from structures of galactocerebrosidase. Proc. Natl. Acad. Sci. USA 108, 15169-15173. 10.1073/pnas.1105639108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demartino, G. N. and Gillette, T. G. (2007). Proteasomes: machines for all reasons. Cell 129, 659-662. 10.1016/j.cell.2007.05.007 [DOI] [PubMed] [Google Scholar]
- Duchen, L. W., Eicher, E. M., Jacobs, J. M., Scaravilli, F. and Teixeira, F. (1980). Hereditary leucodystrophy in the mouse: the new mutant twitcher. Brain 103, 695-710. 10.1093/brain/103.3.695 [DOI] [PubMed] [Google Scholar]
- Erlebacher, A. and Derynck, R. (1996). Increased expression of TGF-beta 2 in osteoblasts results in an osteoporosis-like phenotype. J. Cell Biol. 132, 195-210. 10.1083/jcb.132.1.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filvaroff, E., Erlebacher, A., Ye, J., Gitelman, S. E., Lotz, J., Heillman, M. and Derynck, R. (1999). Inhibition of TGF-beta receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development 126, 4267-4279. 10.1242/dev.126.19.4267 [DOI] [PubMed] [Google Scholar]
- Fuchs, H., Sabrautzki, S., Przemeck, G. K., Leuchtenberger, S., Lorenz-Depiereux, B., Becker, L., Rathkolb, B., Horsch, M., Garrett, L., Ostereicher, M. A.et al. (2016). The First Scube3 Mutant Mouse Line with Pleiotropic Phenotypic Alterations. G3 (Bethesda) 6, 4035-4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, X., Gordon, D., Zhang, D., Browne, R., Helms, C., Gillum, J., Weber, S., Devroy, S., Swaney, S., Dobbs, M.et al. (2007). CHD7 gene polymorphisms are associated with susceptibility to idiopathic scoliosis. Am. J. Hum. Genet. 80, 957-965. 10.1086/513571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauers, A., Einarsdottir, E. and Gerdhem, P. (2016). Genetics and pathogenesis of idiopathic scoliosis. Scoliosis Spinal Disord 11, 45. 10.1186/s13013-016-0105-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray, R. S., Gonzalez, R., Ackerman, S. D., Minowa, R., Griest, J. F., Bayrak, M. N., Troutwine, B., Canter, S., Monk, K. R., Sepich, D. S.et al. (2020). Postembryonic screen for mutations affecting spine development in zebrafish. Dev. Biol. 471, 18-33. 10.1016/j.ydbio.2020.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal, P. K., Holzfeind, P. J., Bittner, R. E. and Hewitt, J. E. (2001). Mutant glycosyltransferase and altered glycosylation of alpha-dystroglycan in the myodystrophy mouse. Nat. Genet. 28, 151-154. 10.1038/88865 [DOI] [PubMed] [Google Scholar]
- Henke, K., Daane, J. M., Hawkins, M. B., Dooley, C. M., Busch-Nentwich, E. M., Stemple, D. L. and Harris, M. P. (2017). Genetic Screen for Postembryonic Development in the Zebrafish (Danio rerio): Dominant Mutations Affecting Adult Form. Genetics 207, 609-623. 10.1534/genetics.117.300187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring, J. A. (2013). Tachdjian's Pediatic Orthopaedics. Philadelphia: WB Saunders. [Google Scholar]
- Hubaud, A. and Pourquie, O. (2014). Signalling dynamics in vertebrate segmentation. Nat. Rev. Mol. Cell Biol. 15, 709-721. 10.1038/nrm3891 [DOI] [PubMed] [Google Scholar]
- Inamori, K., Yoshida-Moriguchi, T., Hara, Y., Anderson, M. E., Yu, L. and Campbell, K. P. (2012). Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 335, 93-96. 10.1126/science.1214115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alfoldi, J., Wang, Q., Collins, R. L., Laricchia, K. M., Ganna, A., Birnbaum, D. P.et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434-443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karner, C. M., Long, F., Solnica-Krezel, L., Monk, K. R. and Gray, R. S. (2015). Gpr126/Adgrg6 deletion in cartilage models idiopathic scoliosis and pectus excavatum in mice. Hum. Mol. Genet. 24, 4365-4373. 10.1093/hmg/ddv170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanshour, A. M., Kou, I., Fan, Y., Einarsdottir, E., Makki, N., Kidane, Y. H., Kere, J., Grauers, A., Johnson, T. A., Paria, N.et al. (2018). Genome-wide meta-analysis and replication studies in multiple ethnicities identify novel adolescent idiopathic scoliosis susceptibility loci. Hum. Mol. Genet. 27, 3986-3998. 10.1093/hmg/ddy306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, T., Yamanaka, T., Jacobs, J. M., Teixeira, F. and Suzuki, K. (1980). The Twitcher mouse: an enzymatically authentic model of human globoid cell leukodystrophy (Krabbe disease). Brain Res. 202, 479-483. 10.1016/0006-8993(80)90159-6 [DOI] [PubMed] [Google Scholar]
- Korpal, M., Yan, J., Lu, X., Xu, S., Lerit, D. A. and Kang, Y. (2009). Imaging transforming growth factor-beta signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat. Med. 15, 960-966. 10.1038/nm.1943 [DOI] [PubMed] [Google Scholar]
- Kou, I., Takahashi, Y., Johnson, T. A., Takahashi, A., Guo, L., Dai, J., Qiu, X., Sharma, S., Takimoto, A., Ogura, Y.et al. (2013). Genetic variants in GPR126 are associated with adolescent idiopathic scoliosis. Nat. Genet. 45, 676-679. 10.1038/ng.2639 [DOI] [PubMed] [Google Scholar]
- Lane, P. W., Beamer, T. C. and Myers, D. D. (1976). Myodystrophy, a new myopathy on chromosome 8 of the mouse. J. Hered. 67, 135-138. 10.1093/oxfordjournals.jhered.a108687 [DOI] [PubMed] [Google Scholar]
- Lin, Y. C., Roffler, S. R., Yan, Y. T. and Yang, R. B. (2015). Disruption of Scube2 Impairs Endochondral Bone Formation. J. Bone Miner. Res. 30, 1255-1267. 10.1002/jbmr.2451 [DOI] [PubMed] [Google Scholar]
- Lin, Y. C., Niceta, M., Muto, V., Vona, B., Pagnamenta, A. T., Maroofian, R., Beetz, C., van Duyvenvoorde, H., Dentici, M. L., Lauffer, P.et al. (2021). SCUBE3 loss-of-function causes a recognizable recessive developmental disorder due to defective bone morphogenetic protein signaling. Am. J. Hum. Genet. 108, 115-133. 10.1016/j.ajhg.2020.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz, C., Meyer-Kleine, C., Steinmeyer, K., Koch, M. C. and Jentsch, T. J. (1994). Genomic organization of the human muscle chloride channel CIC-1 and analysis of novel mutations leading to Becker-type myotonia. Hum. Mol. Genet. 3, 941-946. 10.1093/hmg/3.6.941 [DOI] [PubMed] [Google Scholar]
- Martin, P. T. (2003). Dystroglycan glycosylation and its role in matrix binding in skeletal muscle. Glycobiology 13, 55R-66R. 10.1093/glycob/cwg076 [DOI] [PubMed] [Google Scholar]
- Meyer-Kleine, C., Steinmeyer, K., Ricker, K., Jentsch, T. J. and Koch, M. C. (1995). Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am. J. Hum. Genet. 57, 1325-1334. [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi, T., Collod-Beroud, G., Akiyama, T., Abifadel, M., Harada, N., Morisaki, T., Allard, D., Varret, M., Claustres, M., Morisaki, H.et al. (2004). Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet. 36, 855-860. 10.1038/ng1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyenwoke, R. U. and Brenman, J. E. (2015). Lysosomal storage diseases-regulating neurodegeneration. J. Exp. Neurosci. 9, 81-91. 10.4137/JEN.S25475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A. and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281-2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios, J. J., Denton, K., Russell, J., Kozlitina, J., Ferreira, C. R., Lewanda, A. F., Mayfield, J. E., Moresco, E., Ludwig, S., Tang, M.et al. (2021). Germline saturation mutagenesis induces skeletal phenotypes in mice. J. Bone Miner. Res. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, N., Inui, K., Fujii, N., Fukushima, H., Nishimoto, J., Yanagihara, I., Isegawa, Y., Iwamatsu, A. and Okada, S. (1994). Krabbe disease: isolation and characterization of a full-length cDNA for human galactocerebrosidase. Biochem. Biophys. Res. Commun. 198, 485-491. 10.1006/bbrc.1994.1071 [DOI] [PubMed] [Google Scholar]
- Sharma, S., Gao, X., Londono, D., Devroy, S. E., Mauldin, K. N., Frankel, J. T., Brandon, J. M., Zhang, D., Li, Q. Z., Dobbs, M. B.et al. (2011). Genome-wide association studies of adolescent idiopathic scoliosis suggest candidate susceptibility genes. Hum. Mol. Genet. 20, 1456-1466. 10.1093/hmg/ddq571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, S., Londono, D., Eckalbar, W. L., Gao, X., Zhang, D., Mauldin, K., Kou, I., Takahashi, A., Matsumoto, M., Kamiya, N.et al. (2015). A PAX1 enhancer locus is associated with susceptibility to idiopathic scoliosis in females. Nat. Commun. 6, 6452. 10.1038/ncomms7452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalova, D., Zidkova, J., Vohanka, S., Mazanec, R., Musova, Z., Vondracek, P., Mrazova, L., Kraus, J., Reblova, K. and Fajkusova, L. (2013). CLCN1 mutations in Czech patients with myotonia congenita, in silico analysis of novel and known mutations in the human dimeric skeletal muscle chloride channel. PLoS ONE 8, e82549. 10.1371/journal.pone.0082549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow, D. B., Guillen-Navarro, E., Fatkin, D. and Dunwoodie, S. L. (2008). Mutation of Hairy-and-Enhancer-of-Split-7 in humans causes spondylocostal dysostosis. Hum. Mol. Genet. 17, 3761-3766. 10.1093/hmg/ddn272 [DOI] [PubMed] [Google Scholar]
- Sparrow, D. B., Sillence, D., Wouters, M. A., Turnpenny, P. D. and Dunwoodie, S. L. (2010). Two novel missense mutations in HAIRY-AND-ENHANCER-OF-SPLIT-7 in a family with spondylocostal dysostosis. Eur. J. Hum. Genet. 18, 674-679. 10.1038/ejhg.2009.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, Y., Kou, I., Takahashi, A., Johnson, T. A., Kono, K., Kawakami, N., Uno, K., Ito, M., Minami, S., Yanagida, H.et al. (2011). A genome-wide association study identifies common variants near LBX1 associated with adolescent idiopathic scoliosis. Nat. Genet. 43, 1237-1240. 10.1038/ng.974 [DOI] [PubMed] [Google Scholar]
- Tappino, B., Biancheri, R., Mort, M., Regis, S., Corsolini, F., Rossi, A., Stroppiano, M., Lualdi, S., Fiumara, A., Bembi, B.et al. (2010). Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease. Hum. Mutat. 31, E1894-E1914. 10.1002/humu.21367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, C. F., Yan, Y. T., Wu, S. Y., Djoko, B., Tsai, M. T., Cheng, C. J. and Yang, R. B. (2008). Domain and functional analysis of a novel platelet-endothelial cell surface protein, SCUBE1. J. Biol. Chem. 283, 12478-12488. 10.1074/jbc.M705872200 [DOI] [PubMed] [Google Scholar]
- van Reeuwijk, J., Grewal, P. K., Salih, M. A., Beltran-Valero de Bernabe, D., McLaughlan, J. M., Michielse, C. B., Herrmann, R., Hewitt, J. E., Steinbrecher, A., Seidahmed, M. Z.et al. (2007). Intragenic deletion in the LARGE gene causes Walker-Warburg syndrome. Hum. Genet. 121, 685-690. 10.1007/s00439-007-0362-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T., Zhan, X., Bu, C. H., Lyon, S., Pratt, D., Hildebrand, S., Choi, J. H., Zhang, Z., Zeng, M., Wang, K. W.et al. (2015). Real-time resolution of point mutations that cause phenovariance in mice. Proc. Natl. Acad. Sci. U.S.A. 112, E440-E449. 10.1073/pnas.1423216112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T., Bu, C. H., Hildebrand, S., Jia, G., Siggs, O. M., Lyon, S., Pratt, D., Scott, L., Russell, J., Ludwig, S.et al. (2018). Probability of phenotypically detectable protein damage by ENU-induced mutations in the Mutagenetix database. Nat. Commun. 9, 441. 10.1038/s41467-017-02806-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins, W. J. and Watts, D. C. (1984). Biological features of the new A2G--adr mouse mutant with abnormal muscle function. Lab. Anim. 18, 1-6. 10.1258/002367784780865036 [DOI] [PubMed] [Google Scholar]
- Wenger, D. A., Rafi, M. A., Luzi, P., Datto, J. and Costantino-Ceccarini, E. (2000). Krabbe disease: genetic aspects and progress toward therapy. Mol. Genet. Metab. 70, 1-9. 10.1006/mgme.2000.2990 [DOI] [PubMed] [Google Scholar]
- Wise, C. A., Sepich, D., Ushiki, A., Khanshour, A. M., Kidane, Y. H., Makki, N., Gurnett, C. A., Gray, R. S., Rios, J. J., Ahituv, N.et al. (2020). The cartilage matrisome in adolescent idiopathic scoliosis. Bone Res 8, 13. 10.1038/s41413-020-0089-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, B. T., Su, Y. H., Tsai, M. T., Wasserman, S. M., Topper, J. N. and Yang, R. B. (2004). A novel secreted, cell-surface glycoprotein containing multiple epidermal growth factor-like repeats and one CUB domain is highly expressed in primary osteoblasts and bones. J. Biol. Chem. 279, 37485-37490. 10.1074/jbc.M405912200 [DOI] [PubMed] [Google Scholar]
- Wu, Y. Y., Peck, K., Chang, Y. L., Pan, S. H., Cheng, Y. F., Lin, J. C., Yang, R. B., Hong, T. M. and Yang, P. C. (2011). SCUBE3 is an endogenous TGF-beta receptor ligand and regulates the epithelial-mesenchymal transition in lung cancer. Oncogene 30, 3682-3693. 10.1038/onc.2011.85 [DOI] [PubMed] [Google Scholar]
- Wu, N., Ming, X., Xiao, J., Wu, Z., Chen, X., Shinawi, M., Shen, Y., Yu, G., Liu, J., Xie, H.et al. (2015). TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N. Engl. J. Med. 372, 341-350. 10.1056/NEJMoa1406829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, M., Chen, G. and Li, Y. P. (2016). TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res 4, 16009. 10.1038/boneres.2016.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier, G. M., Panousopoulos, L. and Cobourne, M. T. (2013). Scube3 is expressed in multiple tissues during development but is dispensable for embryonic survival in the mouse. PLoS ONE 8, e55274. 10.1371/journal.pone.0055274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., Muzny, D. M., Xia, F., Niu, Z., Person, R., Ding, Y., Ward, P., Braxton, A., Wang, M., Buhay, C.et al. (2014). Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870-1879. 10.1001/jama.2014.14601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, N., Wu, N., Zhang, L., Zhao, Y., Liu, J., Liang, X., Ren, X., Li, W., Chen, W., Dong, S.et al. (2019). TBX6 compound inheritance leads to congenital vertebral malformations in humans and mice. Hum. Mol. Genet. 28, 539-547. 10.1093/hmg/ddy358 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.