

Abstract
Human cytomegalovirus (HCMV) spreads and establishes a persistent infection within a host by stimulating the survival of carrier myeloid cells via the upregulation of Mcl-1, an antiapoptotic member of the Bcl-2 family of proteins. However, the lack of potent Mcl-1-specific inhibitors and a targetable delivery system has limited to ability to exploit Mcl-1 as a therapeutic strategy to eliminate HCMV-infected monocytes. In this study, we found a lead compound from a novel class of Mcl-1 small-molecule inhibitors rapidly induced death of HCMV-infected monocytes. Moreover, encapsulation of Mcl-1 antagonists into myeloid cell-targeting nanoparticles was able to selectively increase the delivery of inhibitors into HCMV-activated monocytes, thereby amplifying their potency. Our study demonstrates the potential use of nanotechnology to target Mcl-1 small-molecule inhibitors to HCMV-infected monocytes.
Keywords: Cytomegalovirus, Monocytes, Myeloid Cell Leukemia 1, Nanoparticles, Small-Molecule Inhibitor
Summary
Human cytomegalovirus (HCMV) is clinically the most important opportunistic infectious cause of transplant failure. Symptomatic HCMV infections can lead to overt inflammation-mediated end-organ dysfunction (Rollag et al., 2012; Sagedal et al., 2000). Without preventive anti-HCMV therapy, 30-75% of transplant recipients will exhibit an active HCMV infection within the first 100 days after transplantation (Sagedal et al., 2004). The use of prophylactic antiviral agents, such as ganciclovir, to limit virus replication has significantly reduced the incidence of early infection to ≤10% (Fishman et al., 2007; Sagedal et al., 2004). However, drug toxicities associated with long-term antiviral therapy limit treatment duration (Humar et al., 2010). Consequently, prophylactic treatment appears to have simply shifted the kinetics of HCMV infection to later after transplantation (≥100 days) with HCMV disease occurring in 20-30% of transplant patients at risk for infection (Karuthu and Blumberg, 2012). The multi-organ inflammatory diseases associated with symptomatic HCMV infections are a direct consequence of the viral persistence strategy (Bissinger et al., 2002; Gnann et al., 1988; Smith et al., 2010).
HCMV utilizes the myeloid compartment of the immune system to facilitate both hematogenous dissemination and the establishment of viral persistence within infected hosts (Goodrum et al., 2012; Sinclair and Sissons, 2006; Smith et al., 2004). During a primary/acute infection by either contact with HCMV-containing bodily fluids from an individual actively shedding virus or a transplanted organ from a seropositive donor, peripheral blood monocytes are responsible for the systemic dissemination of the virus (Gnann et al., 1988; Smith et al., 2010). HCMV enters a “quiescent” (non-replicative) state during the initial infection of monocytes (Sinclair and Sissons, 1996; Smith et al., 2004; Soderberg-Naucler et al., 2001). However, HCMV stimulates the survival and differentiation of short-lived monocytes into long-lived macrophages, which are permissive for virus replication (Fish et al., 1995; Ibanez et al., 1991; Sinclair and Sissons, 1996; Smith et al., 2004). Following extravasation of infected cells into tissue and completion of the differentiation process, a chronic HCMV infection within organs can be established resulting in long-term viral secretion and widespread organ inflammation seen in symptomatic patients (Noyola et al., 2000). Lastly, the migration of infected monocytes into the bone marrow leads to viral latency within CD34+ myeloid progenitor cells (Goodrum et al., 2012; Sinclair and Sissons, 2006). During reactivation of HCMV, infected progenitor cells are released into the circulation and undergo myeloid differentiation allowing for reseeding of the virus in organ tissue and inflammation (Kondo et al., 1994; Maciejewski et al., 1992; Sinclair and Sissons, 1996). Since the survival and differentiation of myeloid cells are essential for HCMV dissemination and underlie the pathogenesis of HCMV diseases, eliminating these cells are essential to new anti-HCMV strategies.
Current anti-HCMV drugs block specific steps along the virus replication cycle (Biron, 2006). However, the lack of HCMV replication within monocytes and CD34+ cells limits the effectiveness of these drugs at long-term clearance of the virus due to the inability to eliminate infiltrating infected inflammatory myeloid cells, which are the principle cell types found in the infected organs of transplant patients (Gnann et al., 1988). Moreover, the emergence of antiviral drug resistant HCMV stains has put an impetus on finding new anti-HCMV therapies (Gohring et al., 2015). We and others have shown HCMV to specifically upregulate Mcl-1, a member of the antiapoptotic B-cell lymphoma (Bcl)-2 family, in order to drive the survival of carrier myeloid cells in absence of viral antiapoptotic protein expression (Chan et al., 2010; Chan et al., 2012; Reeves et al., 2012). The use of Mcl-1 appears to play a particularly critical role during HCMV-induced monocyte survival as normal myeloid growth factors utilize different Bcl-2 family members to promote survival (Peppenelli et al., 2016). Indeed, we previously found the unique signaling network generated during the viral entry process mediates the selective upregulation of Mcl-1 within infected monocytes (Peppenelli et al., 2016). These data indicate Mcl-1 as a potential therapeutic target to prevent/treat HCMV-associated diseases in high-risk transplant patients.
A new class of Mcl-1 inhibitor selectively induces apoptosis of HCMV-infected monocytes.
The development of Mcl-1 inhibitors has proven to be a difficult task due to the minor structural differences within the BH3 binding grooves and/or the BH3 helix relative to other related antiapoptoic Bcl-2 proteins (Czabotar et al., 2014). Currently, MIM1 is the only commercially available Mcl-1 small-molecule inhibitor but exhibits relatively weak binding affinity (Ki = 4.8 μM) and does not induce cell death of all Mcl-1-dependent tumor cells (Cohen et al., 2012; Varadarajan et al., 2013). We have identified a novel chemical class of Mcl-1 inhibitors based off a hydroxyl-naphthalenylsulfonamide scaffold that exhibited nanomolar-binding affinity to Mcl-1 (Abulwerdi et al., 2014). Using C10 as a representative of this chemical library of Mcl-1 inhibitors, we found C10 stimulated ~3 fold more death of HCMV-infected monocytes when compared to MIM1 (Fig. 1A), which is consistent with our previous study (Cojohari et al., 2015). Furthermore, C10 failed to induce death of Bcl-xL-dependent EBV-infected B cells (Akata) while MIM1 stimulated moderate levels of cell death, demonstrating the increased specificity and efficacy of this new class of Mcl-1 antagonists at eliminating HCMV-infected monocytes. HCMV infection of monocytes blocks the two proteolytic cleavage steps necessary for the activation of caspase 3 and the subsequent triggering of apoptosis (Chan et al., 2012). We have shown HCMV-upregulated Mcl-1 functions to specifically block the first cleavage step of pro-caspase 3 (Chan et al., 2012). Accordingly, we found HCMV infection resulted in the accumulation of pro-caspase 3 (Fig. 1B). Consistent with the on-target effects of C10 at inhibiting Mcl-1, increasing concentrations of C10 treatment led to increased cleavage and subsequent loss of pro-caspase 3 (Fig. 1B). Because monocytes rapidly undergo apoptosis within 24 hours of isolation (Cojohari et al., 2016), which is consistent with their naturally short lifespan (Whitelaw, 1972), we were unable evaluate the effects of C10 on uninfected monocytes. Despite the promising potential use of Mcl-1 inhibitors to prevent and treat HCMV infections, clinical studies of small-molecule inhibitors against the Bcl-2 family as a cancer therapy have shown challenging pharmacokinetic profiles and side effects due to bystander killing (Scarfo and Ghia, 2013; Vogler, 2014). Thus, concomitant with the development of Mcl-1 antagonists, delivery strategies to target infected myeloid cells must also be developed to improve pharmacokinetic profiles and to minimize the non-specific toxic effects of these small-molecule inhibitors.
Fig. 1.
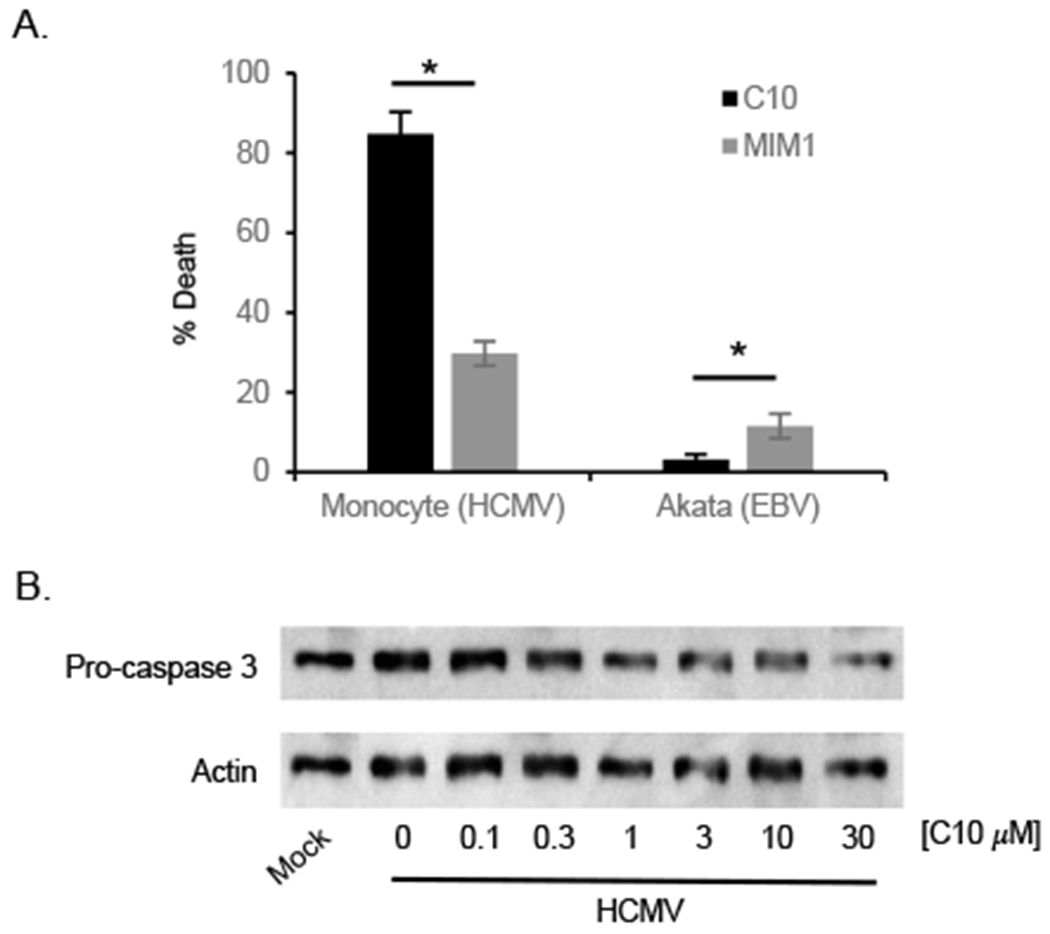
A novel class of Mcl-1 inhibitor induces death of HCMV-infected monocytes. HCMV-infected monocytes or EBV-infected Akata cells were treated with 30 μM of C10 or MIM1. After 24 hours of treatment with inhibitor, cell viability was measured by a Trypan blue exclusion assay. Shown is the mean of 3-4 independent experiments from different blood donors. Differences between experimental groups were evaluated by one-way analysis of variance (ANOVA) with pair-wise multiple comparison procedures. Asterisk (*) represents a P value of <0.05. (B) Monocytes were infected with HCMV at an MOI of 5. Following a 24 h incubation period, infected monocytes were treated with increasing concentrations of C10 for an additional 24 h. Pro-caspase 3 was then detected by immunoblotting and membranes reprobed for β-actin as a loading control. Results are representative of 3 independent experiments from different blood donors.
HCMV-infected monocytes efficiently take up nanoparticles.
Nanotechnology has now taken center stage for delivery of therapeutics for inflammatory-based diseases by selective ablation of infiltrating monocytes/macrophages or inhibition of their inflammatory activity leading to resolution of inflammation within the diseased organ (Kelly et al., 2011; Patel and Janjic, 2015; Weissleder et al., 2014). Pan-macrophage targeting nanoparticles have also been shown to increase the efficacy of anti-HIV agents by increasing drug delivery into macrophages serving as reservoirs for HIV (Oussoren et al., 1999). Large charged lipidoid nanoparticles are taken up by mononuclear phagocyte system (MPS) and can be utilized to target aberrantly activated myeloid cells involved in the pathogenesis of inflammatory diseases. However, poor stability and overly large particle sizes lead to non-specific cytoxicity of nanoparticles resulting in limitations to in vivo applications (Cabral et al., 2011; Lee et al., 2010).
We have recently engineered two novel architectures of telodendrimers, G3 and G2 telodendrimers, for rational nanocarrier design (Cai et al., 2015; Huang et al., 2015; Shi et al., 2015; Shi et al., 2014; Xu et al., 2015) (Fig. 2A). Various drug-binding moieties (DBM) identified by computational virtual screening can be introduced on the periphery as core-forming segments to optimize drug-loading properties. The unique facial amphiphilic structure of cholic acid (CA) stabilize nanoparticles as co-core forming building blocks, which allows the free engineering of DBMs into G3 and G2 telodendrimers to optimize drug loading properties without sacrificing nanoparticle stability. Using G3 telodendrimers with cholesterol (CHO) as a DBM (Shi et al., 2015) and cationic lipidoids, we generated hybrid G3-CHO telodendrimer/lipidoid nanoparticles (noted as TL-NP) at appropriate particle sizes (~100 nm) while concurrently sheltering positive charges to reduce non-specific toxicity (Wu et al., 2014). The specific lipidoid used in this study was synthesized via an epoxy-amino chemistry using 1,2-epoxyohexadecane and N,N’-dimethyl-1,3-propanediamine as starting materials (Wang et al., 2014). We found TL-NPs had larger particle sizes (hydrodynamic diameter [Dh]: ~64 nm) as measured by dynamic light scattering (DLS) (Fig. 2B) and a positive surface charge (zeta potential: 4.5 mV). Next, the rate of nanoparticle uptake into HCMV-infected monocytes was assessed using FITC-labeled TL-NPs (Fig. 2C). We found HCMV-infected monocytes were able to rapidly engulf TL-NP by 1 hour and that maximum uptake was achieved by 2 hours. More importantly, consistent with our previous finding that HCMV-infected monocytes and macrophages exhibit increased phagocytic activity (Smith et al., 2004), absorption of TL-NP infected monocytes was 6-7 fold higher when compared to uninfected monocytes. However, the larger size and positive charge also resulted in significant non-specific uptake of TL-NP in mock-infected monocytes. Nonetheless, these data demonstrate the potential use of hybrid nanoparticles to rapidly deliver drugs to HCMV-activated monocytes in the circulation.
Fig. 2.
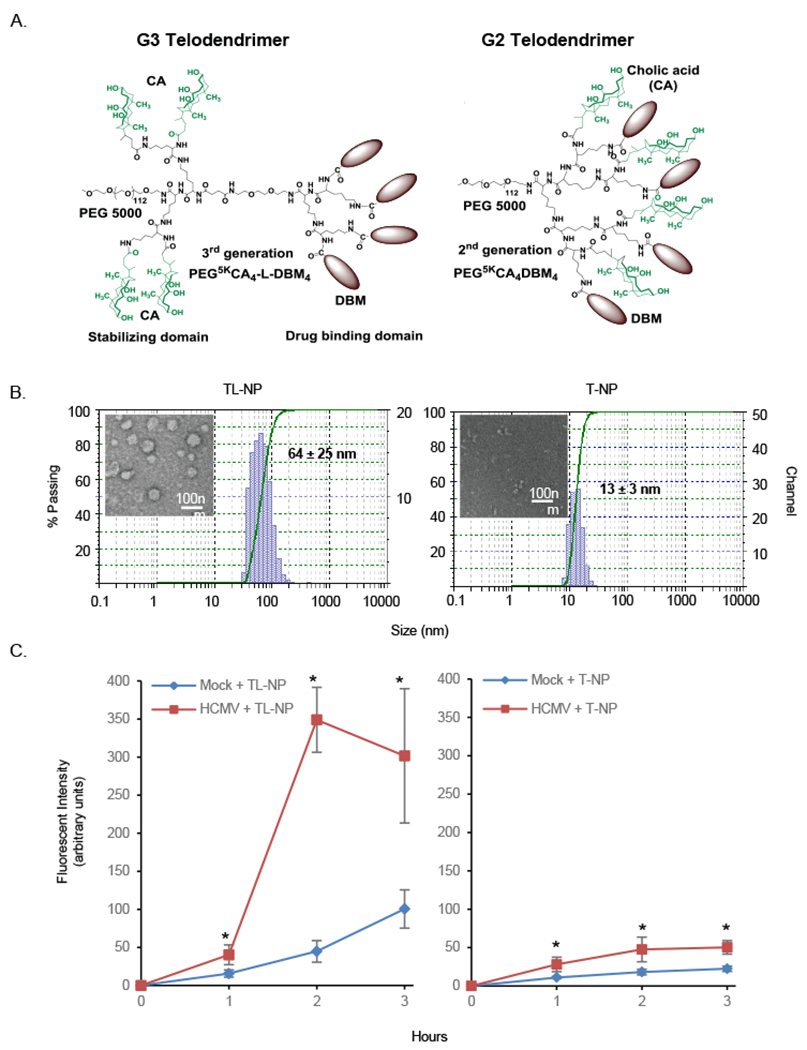
Nanoparticles are rapidly engulfed into HCMV-infected monocytes. (A) Structures of the functional-segregated G3 (left) and G2 (right) telodendrimers. (B) Hydrodynamic size distributions and TEM images of blank TL-NP and T-NP nanoparticles. (C) Peripheral blood monocytes were mock infected or HCMV infected for 24 hours (h). Following infection, FITC-labeled TL-NP or T-NP was added to infected cells for 1, 2, and 3 h then washed with PBS to remove the nanocarriers and fluorescent intensities immediately determined with a Nikon Eclipse Ti-U inverted fluorescent microscope. Results are from 3 independent experiments from different blood donors. Differences between experimental groups were evaluated by one-way analysis of variance (ANOVA) with pair-wise multiple comparison procedures. Asterisk (*) represents a P value of <0.05.
Although lipid-based nanoparticles readily target peripheral blood monocytes, the larger particle sizes along with the positive surface charge restrict extravasation and tissue diffusion (Luo et al., 2010; Xiao et al., 2011), thereby limiting drug delivery to monocytes that have penetrated deep into inflamed tissue. Alternatively, we generated nanoparticles formed from only G2 telodendrimers containing aromatic rhein (Rh) as a DBM (noted as T-NP) (Shi et al., 2015), which form small particle sizes (Dh: ~13 nm) (Fig. 2B) with a neutral surface charge (zeta potential: 0.1 mV) that readily penetrate leaky inflamed blood vessels and diffuse through extracellular matrix as demonstrated in the intratumoral drug delivery (Cabral et al., 2011; Lee et al., 2010). Next, we tested the capacity of HCMV-infected monocytes to phagocytose T-NP. FITC-labeled T-NP were engulfed into HCMV-infected monocytes by 1 hour and reached maximum uptake at 2 hours. Moreover, T-NP was phagocytosed 2-fold more efficiently into HCMV-infected monocytes than uninfected cells (Fig. 2C). Although T-NP was taken up less efficiently than TL-NP into infected monocytes, its smaller size and neutral charge limited non-specific uptake into uninfected monocytes (Fig. 2C). Taken together, these data highlight the possibility of selectively delivering Mcl-1 small-molecule inhibitors into infected myeloid cells found both in the circulation and within diseased tissues using G2 telodendrimer- and hybrid G3 telodendrimer/lipidoid-based nanoformulations.
Encapsulation of Mcl-1 small-molecule inhibitors enhances cytotoxicity.
Next, we tested C10 with nanocarriers TL-NP and T-NP for aromatic drug delivery. Nanoprecipitation and thin-film hydration methods were used to load C10 into TL-NP and T-NP, respectively (Shi et al., 2015; Whitehead et al., 2014). At a 1:10 (w:w) ratio of C10 to nanoparticle macromonomers, 100% of C10 was found to be complexed into TL-NP or T-NP nanoparticles as determined by mass spectrometry. The hydrodynamic diameters of the compound-loaded nanoparticles in aqueous solutions were measured by DLS. Encapsulation of C10 in TL-NP nanocarriers formed particle sizes of ~109 nm (Fig. 3A) with the positive surface charge (zeta potential: 2.0 mV). Transmission electron microscopy (TEM) images indicated that TL-NP had spherical shape before and after loading of C10 (Fig. 3A), although encapsulation of C10 into TL-NP led to a ~40% increase in particle size. To test if TL-NP encapsulated C10 exhibited increased cytotoxicity, HCMV-infected monocytes were treated with C10, empty TL-NP, or C10/TL-NP for 2 hours, a time at which the maximum difference between TL-NP uptake between mock- and HCMV-infected monocytes was observed (Fig. 2C). The molar concentration of free C10 was calculated based on the weight of C10. An equivalent amount of C10 loaded into TL-NP was then used to treat infected monocytes. The weight concentration of the TL-NP component of the loaded nanoparticles was determined from the feed amount of drug and based on the 1:10 (w/w) ratio of C10 to nanoparticle macromonomers. The concentration of empty TL-NP used to treat infected monocytes was equivalent to the polymer weight concentration used to load C10. Following the 2-hour incubation, cells were washed to remove inhibitors and incubated for an additional 24 hours prior to cell viability staining. We found C10/TL-NP induced greater cell death compared to free C10 and empty TL-NP (Fig. 3B). Further, at 3 μM of C10 (the concentration at which the greatest difference in killing activity between C10/TL-NP and TL-NP was observed) C10/TL-NP, but not empty TL-NP, led to caspase 3 activation, thus confirming the delivery of C10 into HCMV-infected monocytes (Fig. 3D). However, TL-NP still exhibited significant cytotoxicity indicating that further modifications to the composition of TL-NP to reduce the size and/or cationic surface charge of the nanoparticle are needed to reduce the non-specific killing by the nanoparticle itself. Nonetheless, because free C10 alone did not stimulate death of infected monocytes at any concentration following a 2-h absorption period, TL-NPs represents a mode of drug delivery to significantly increase the rate and amount of Mcl-1 inhibitors delivered into infected peripheral blood monocytes.
Fig. 3.
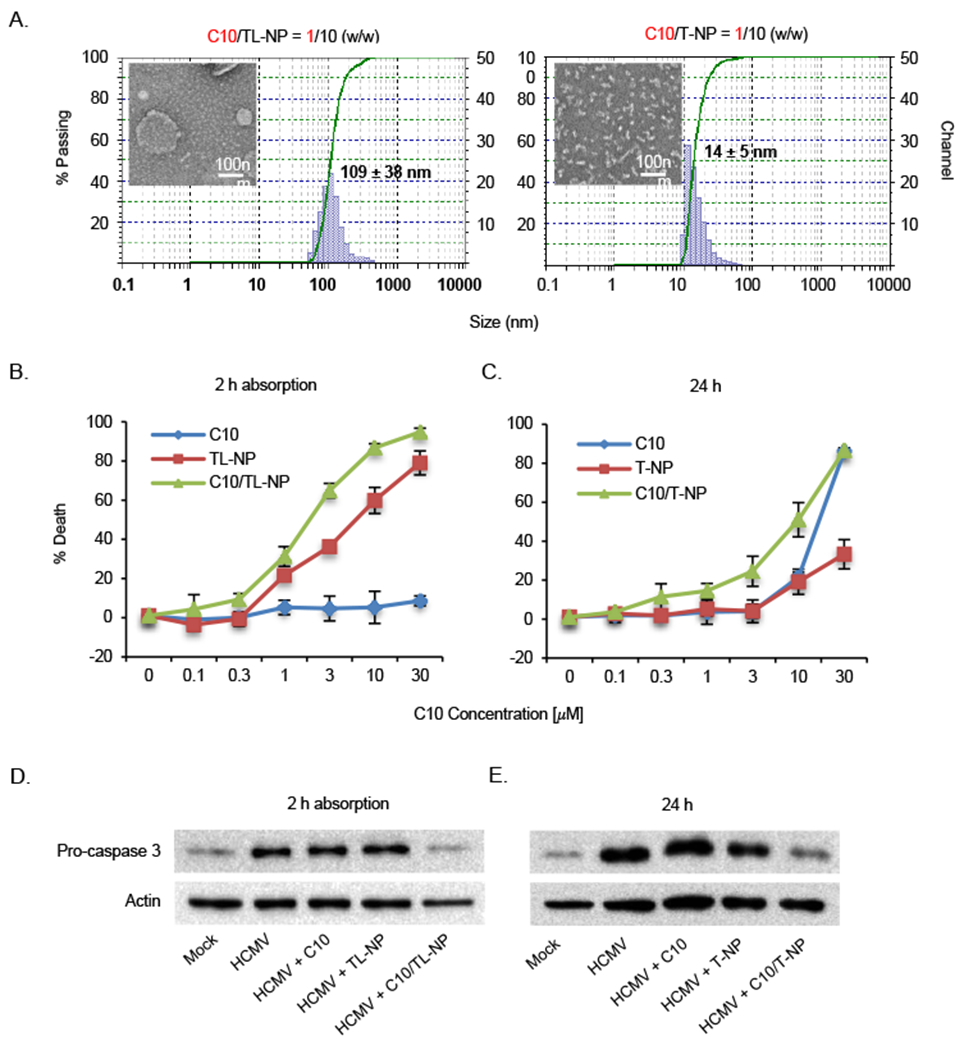
Encapsulation enhances the efficacy of Mcl-1 small-molecule inhibitor to induce death of HCMV-infected monocytes. (A) Hydrodynamic size distributions and TEM images of C10-loaded TL-NP and T-NP nanoparticles. (B) HCMV-infected monocytes were treated with increasing concentrations of C10, TL-NP, or C10/TL-NP for 2 hours. Following a wash to remove inhibitors, cells were further incubated for 24 h. (C) HCMV-infected monocytes were treated with increasing concentrations of C10, T-NP, or C10/T-NP for 24 hours. (B, C) Cell viability was determined by a trypan blue exclusion assay. (D) Infected monocytes were treated with 3 μM of C10, TL-NP, and C10/TL-NP for 2 hours, washed with PBS, and then incubated a further 24 h. (E) Infected monocytes were treated with 10 μM of C10, T-NP, and C10/T-NP for 24 hours. (D, E) Pro-caspase 3 was detected by immunoblotting and membranes reprobed for β-actin as a loading control. Results are representative of 3 independent experiments from different blood donors.
In contrast, loading of Mcl-1 inhibitor C10 into neutrally charged T-NP formed particle sizes of 14 nm with a zeta potential of −1.3 mV) (Fig. 3A), which are ideal nanoparticle characteristics that allow for prolonged circulation time (Shi et al., 2015). T-NP before and after loading of C10 exhibited a worm-like shape due to the molecular stacking of Rh (Shi et al., 2015) (Fig. 3A). Because T-NP exhibits significantly lower non-specific cytotoxicity than TL-NP (Fig. 3B), cells were treated for 24 h with T-NP versus the 2-h absorption period used for TL-NP. The concentrations of C10-loaded and empty T-NP were determined as described for TL-NP. We found C10/T-NP induced higher cell death of infected monocytes relative to free C10 and empty T-NP, albeit C10 and C10/T-NP exhibited similar toxicity at 30 μM (Fig. 3C). Moreover, C10/T-NP treatment triggered the cleavage of pro-caspase 3 at 10 μM demonstrating C10’s Mcl-1 inhibitory activity within HCMV-infected monocytes (Fig 3E). Overall, these data indicate both nanoformulations are able to lower the LD50 of C10 and provide a proof-of-concept for the further development of nanotechnology to target HCMV-infected cell types and deliver Mcl-1 small-molecule inhibitors.
Conclusion:
Nanoparticles with large particle sizes and surface charges efficiently target monocytes in the blood; however, these same properties limit extravasation and tissue diffusion (Lee et al., 2010; Xiao et al., 2011). Alternatively, nanocarriers with small particle sizes and neutral surface charges are able to penetrate deep into inflamed, but not normal, tissues through leaky blood vessels, thus able to distinguish between diseased and normal organs (Cabral et al., 2011; Weissleder et al., 2014). Given the heterogeneity of HCMV inflammatory diseases, a combination of two types of nanoparticles will likely be needed to effectively target HCMV-infected myeloid cells in the circulation and tissue (Fig. 4). In this study, we tested two series of nanocarriers for delivery a Mcl-1 inhibitor into HCMV-infected monocytes: (a) positively-charged TL-NP with large particle sizes, and (b) T-NP with a neutral surface and small particle sizes. We found both nanoformaulations were preferentially taken up by HCMV-activated monocytes versus unactivated monocytes and consequently able to increase the killing efficiency of C10. Thus, optimization of these two pan-macrophage targeting nanoparticles may hold promising therapeutic potential to treat acute HCMV infections. Indeed, monocyte/macrophage subset-specific nanoparticles have been recently developed by introducing subset-specific moieties onto the surface of the nanocarrier capable of differentiating between inflammatory (M1) and anti-inflammatory (M2) monocytes/macrophages (Weissleder et al., 2014). Our previous finding that HCMV induces unique M1/M2 changes to the surface of infected monocytes suggest adding moieties onto nanoparticles that target these virus-specific changes may enhance the specificity of nanocarriers to delivery Mcl-1 inhibitors to infected cells while minimizing bystander killing (Chan et al., 2008; Smith et al., 2007). Furthermore, bone marrow-homing nanoparticles may also allow for delivery of Mcl-1 small-molecule inhibitors into latently infected CD34+ cells (Sou et al., 2011), which are also dependent on Mcl-1 for viability (Reeves et al., 2012). Because survival of quiescently/latently infected myeloid cells are largely dependent on the exploiting antiapoptotic Bcl-2 family members (Chan et al., 2010; Reeves et al., 2012), the use of nanoparticle technology may allow for the selective delivery of small-molecule antagonist into infected myeloid cells in order to eliminate these cells while reducing the typical side effects of traditional chemotherapeutic agents.
Fig. 4.
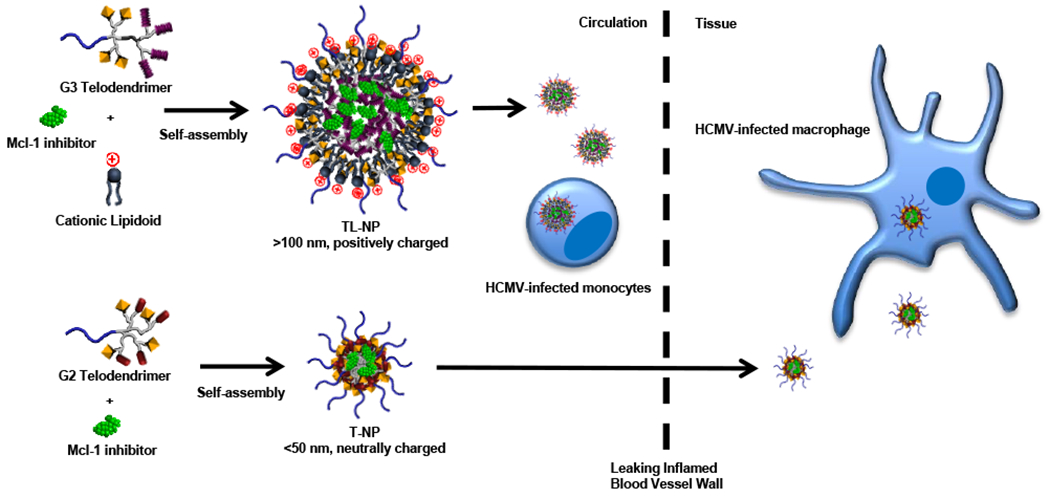
Model of nanoformulations for two-stage drug delivery. G3 telodendrimers and cationic lipidoids self-assemble into positively-charged nanoparticles (TL-NP) to encapsulate Mcl-1 inhibitors with large particle sizes (>100 nm), which readily target circulating HCMV-infected blood monocytes to delivery Mcl-1 small-molecule inhibitors. G2 telodendrimers self-assemble into nanoparticles (T-NP) to encapsulate Mcl-1 inhibitors with small particle sizes (<50 nm) and a neutral surface. T-NP is able to penetrate through leaky blood vessels and diffuse deep into the inflamed tissue to deliver Mcl-1 inhibitors into HCMV-infected monocytes/macrophages within diseased organs.
Acknowledgments
This work was supported by grants from the Sinsheimer Scholar Award to G.C. Chan, American Heart Association to G.C. Chan, National Institute of Allergy and Infectious Disease (R56AI110803-01) to G.C. Chan, National Cancer Institute (R01CA149442) to Z. Nikolovska-Coleska, National Cancer Institute (R01CA140449) to J. Luo, and National Institute of Biomedical Imaging and Bioengineering (1R21EB019607) to J. Luo.
Footnotes
We have no conflicting financial interests.
References
- Abulwerdi FA, Liao C, Mady AS, Gavin J, Shen C, Cierpicki T, Stuckey JA, Showalter HD, Nikolovska-Coleska Z, 2014. 3-Substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamides as a novel class of selective Mcl-1 inhibitors: structure-based design, synthesis, SAR, and biological evaluation. J Med Chem 57, 4111–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron KK, 2006. Antiviral drugs for cytomegalovirus diseases. Antiviral Res 71, 154–163. [DOI] [PubMed] [Google Scholar]
- Bissinger AL, Sinzger C, Kaiserling E, Jahn G, 2002. Human cytomegalovirus as a direct pathogen: correlation of multiorgan involvement and cell distribution with clinical and pathological findings in a case of congenital inclusion disease. J Med Virol 67, 200–206. [DOI] [PubMed] [Google Scholar]
- Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K, 2011. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol 6, 815–823. [DOI] [PubMed] [Google Scholar]
- Cai L, Xu G, Shi C, Guo D, Wang X, Luo J, 2015. Telodendrimer nanocarrier for co-delivery of paclitaxel and cisplatin: A synergistic combination nanotherapy for ovarian cancer treatment. Biomaterials 37, 456–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G, Bivins-Smith ER, Smith MS, Smith PM, Yurochko AD, 2008. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation toward an M1 macrophage. J Immunol 181, 698–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G, Nogalski MT, Bentz GL, Smith MS, Parmater A, Yurochko AD, 2010. PI(3)K-dependent upregulation of Mcl-1 by human cytomegalovirus is mediated by epidermal growth factor receptor and inhibits apoptosis in short-lived monocytes. J Immunol 184, 3213–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G, Nogalski MT, Yurochko AD, 2012. Human cytomegalovirus stimulates monocyte-to-macrophage differentiation via the temporal regulation of caspase 3. J Virol 86, 10714–10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SR, Koss B, Opferman JT, Walensky LD, 2012. A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chemistry & biology 19, 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cojohari O, Burrer CM, Peppenelli MA, Abulwerdi FA, Nikolovska-Coleska Z, Chan GC, 2015. BH3 profiling reveals selectivity by herpesviruses for specific Bcl-2 proteins to mediate survival of latently infected cells. J Virol 89, 5739–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cojohari O, Peppenelli MA, Chan GC, 2016. Human Cytomegalovirus Induces an Atypical Activation of Akt To Stimulate the Survival of Short-Lived Monocytes. J Virol 90, 6443–6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM, 2014. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15, 49–63. [DOI] [PubMed] [Google Scholar]
- Fish KN, Depto AS, Moses AV, Britt W, Nelson JA, 1995. Growth kinetics of human cytomegalovirus are altered in monocyte-derived macrophages. J Virol 69, 3737–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman JA, Emery V, Freeman R, Pascual M, Rostaing L, Schlitt HJ, Sgarabotto D, Torre-Cisneros J, Uknis ME, 2007. Cytomegalovirus in transplantation - challenging the status quo. Clin Transplant 21, 149–158. [DOI] [PubMed] [Google Scholar]
- Gnann JW Jr., Ahlmen J, Svalander C, Olding L, Oldstone MB, Nelson JA, 1988. Inflammatory cells in transplanted kidneys are infected by human cytomegalovirus. Am J Pathol 132, 239–248. [PMC free article] [PubMed] [Google Scholar]
- Gohring K, Hamprecht K, Jahn G, 2015. Antiviral Drug- and Multidrug Resistance in Cytomegalovirus Infected SCT Patients. Comput Struct Biotechnol J 13, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrum F, Caviness K, Zagallo P, 2012. Human cytomegalovirus persistence. Cell Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Wang X, Shi C, Guo D, Xu G, Wang L, Bodman A, Luo J, 2015. Fine-tuning vitamin E-containing telodendrimers for efficient delivery of gambogic acid in colon cancer treatment. Molecular pharmaceutics 12, 1216–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humar A, Lebranchu Y, Vincenti F, Blumberg EA, Punch JD, Limaye AP, Abramowicz D, Jardine AG, Voulgari AT, Ives J, Hauser IA, Peeters P, 2010. The efficacy and safety of 200 days valganciclovir cytomegalovirus prophylaxis in high-risk kidney transplant recipients. Am J Transplant 10, 1228–1237. [DOI] [PubMed] [Google Scholar]
- Ibanez CE, Schrier R, Ghazal P, Wiley C, Nelson JA, 1991. Human cytomegalovirus productively infects primary differentiated macrophages. J Virol 65, 6581–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuthu S, Blumberg EA, 2012. Common infections in kidney transplant recipients. Clinical journal of the American Society of Nephrology : CJASN 7, 2058–2070. [DOI] [PubMed] [Google Scholar]
- Kelly C, Jefferies C, Cryan SA, 2011. Targeted liposomal drug delivery to monocytes and macrophages. J Drug Deliv 2011, 727241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Kaneshima H, Mocarski ES, 1994. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc Natl Acad Sci U S A 91, 11879–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Fonge H, Hoang B, Reilly RM, Allen C, 2010. The effects of particle size and molecular targeting on the intratumoral and subcellular distribution of polymeric nanoparticles. Molecular pharmaceutics 7, 1195–1208. [DOI] [PubMed] [Google Scholar]
- Luo J, Xiao K, Li Y, Lee JS, Shi L, Tan YH, Xing L, Holland Cheng R, Liu GY, Lam KS, 2010. Well-defined, size-tunable, multifunctional micelles for efficient paclitaxel delivery for cancer treatment. Bioconjugate chemistry 21, 1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejewski JP, Bruening EE, Donahue RE, Mocarski ES, Young NS, St Jeor SC, 1992. Infection of hematopoietic progenitor cells by human cytomegalovirus. Blood 80, 170–178. [PubMed] [Google Scholar]
- Noyola DE, Demmler GJ, Williamson WD, Griesser C, Sellers S, Llorente A, Littman T, Williams S, Jarrett L, Yow MD, 2000. Cytomegalovirus urinary excretion and long term outcome in children with congenital cytomegalovirus infection. Congenital CMV Longitudinal Study Group. Pediatr Infect Dis J 19, 505–510. [DOI] [PubMed] [Google Scholar]
- Oussoren C, Magnani M, Fraternale A, Casabianca A, Chiarantini L, Ingebrigsten R, Underberg WJ, Storm G, 1999. Liposomes as carriers of the antiretroviral agent dideoxycytidine-5′-triphosphate. Int J Pharm 180, 261–270. [DOI] [PubMed] [Google Scholar]
- Patel SK, Janjic JM, 2015. Macrophage targeted theranostics as personalized nanomedicine strategies for inflammatory diseases. Theranostics 5, 150–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppenelli MA, Arend KC, Cojohari O, Moorman NJ, Chan GC, 2016. Human cytomegalovirus stimulates the synthesis of select Akt-dependent antiapoptotic proteins during viral entry to promote survival of infected monocytes. J Virol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves MB, Breidenstein A, Compton T, 2012. Human cytomegalovirus activation of ERK and myeloid cell leukemia-1 protein correlates with survival of latently infected cells. Proc Natl Acad Sci U S A 109, 588–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollag H, Asberg A, Ueland T, Hartmann A, Jardine AG, Humar A, Pescovitz MD, Bignamini AA, Aukrust P, 2012. Treatment of cytomegalovirus disease in solid organ transplant recipients: markers of inflammation as predictors of outcome. Transplantation 94, 1060–1065. [DOI] [PubMed] [Google Scholar]
- Sagedal S, Hartmann A, Nordal KP, Osnes K, Leivestad T, Foss A, Degre M, Fauchald P, Rollag H, 2004. Impact of early cytomegalovirus infection and disease on long-term recipient and kidney graft survival. Kidney Int 66, 329–337. [DOI] [PubMed] [Google Scholar]
- Sagedal S, Nordal KP, Hartmann A, Degre M, Holter E, Foss A, Osnes K, Leivestad T, Fauchald P, Rollag H, 2000. A prospective study of the natural course of cytomegalovirus infection and disease in renal allograft recipients. Transplantation 70, 1166–1174. [DOI] [PubMed] [Google Scholar]
- Scarfo L, Ghia P, 2013. Reprogramming cell death: BCL2 family inhibition in hematological malignancies. Immunol Lett 155, 36–39. [DOI] [PubMed] [Google Scholar]
- Shi C, Guo D, Xiao K, Wang X, Wang L, Luo J, 2015. A drug-specific nanocarrier design for efficient anticancer therapy. Nature communications 6, 7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Yuan D, Nangia S, Xu G, Lam KS, Luo J, 2014. A structure-property relationship study of the well-defined telodendrimers to improve hemocompatibility of nanocarriers for anticancer drug delivery. Langmuir : the ACS journal of surfaces and colloids 30, 6878–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair J, Sissons P, 1996. Latent and persistent infections of monocytes and macrophages. Intervirology 39, 293–301. [DOI] [PubMed] [Google Scholar]
- Sinclair J, Sissons P, 2006. Latency and reactivation of human cytomegalovirus. J Gen Virol 87, 1763–1779. [DOI] [PubMed] [Google Scholar]
- Smith MS, Bentz GL, Alexander JS, Yurochko AD, 2004. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J Virol 78, 4444–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MS, Bivins-Smith ER, Tilley AM, Bentz GL, Chan G, Minard J, Yurochko AD, 2007. Roles of phosphatidylinositol 3-kinase and NF-kB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: strategy for viral persistence. J Virol 81, 7683–7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MS, Goldman DC, Bailey AS, Pfaffle DL, Kreklywich CN, Spencer DB, Othieno FA, Streblow DN, Garcia JV, Fleming WH, Nelson JA, 2010. Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell host & microbe 8, 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg-Naucler C, Streblow DN, Fish KN, Allan-Yorke J, Smith PP, Nelson JA, 2001. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J Virol 75, 7543–7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sou K, Goins B, Oyajobi BO, Travi BL, Phillips WT, 2011. Bone marrow-targeted liposomal carriers. Expert Opin Drug Deliv 8, 317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan S, Vogler M, Butterworth M, Dinsdale D, Walensky LD, Cohen GM, 2013. Evaluation and critical assessment of putative MCL-1 inhibitors. Cell Death Differ 20, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler M, 2014. Targeting BCL2-Proteins for the Treatment of Solid Tumours. Adv Med 2014, 943648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Alberti K, Sun S, Arellano CL, Xu Q, 2014. Combinatorially designed lipid-like nanoparticles for intracellular delivery of cytotoxic protein for cancer therapy. Angew Chem Int Ed Engl 53, 2893–2898. [DOI] [PubMed] [Google Scholar]
- Weissleder R, Nahrendorf M, Pittet MJ, 2014. Imaging macrophages with nanoparticles. Nature materials 13, 125–138. [DOI] [PubMed] [Google Scholar]
- Whitehead KA, Dorkin JR, Vegas AJ, Chang PH, Veiseh O, Matthews J, Fenton OS, Zhang Y, Olejnik KT, Yesilyurt V, Chen D, Barros S, Klebanov B, Novobrantseva T, Langer R, Anderson DG, 2014. Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nature communications 5, 4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitelaw DM, 1972. Observations on human monocyte kinetics after pulse labeling. Cell Tissue Kinet 5, 311–317. [DOI] [PubMed] [Google Scholar]
- Wu J, Kamaly N, Shi J, Zhao L, Xiao Z, Hollett G, John R, Ray S, Xu X, Zhang X, Kantoff PW, Farokhzad OC, 2014. Development of multinuclear polymeric nanoparticles as robust protein nanocarriers. Angew Chem Int Ed Engl 53, 8975–8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS, 2011. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials 32, 3435–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Shi C, Guo D, Wang L, Ling Y, Han X, Luo J, 2015. Functional-segregated coumarin-containing telodendrimer nanocarriers for efficient delivery of SN-38 for colon cancer treatment. Acta biomaterialia 21, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]