

Abstract
Background:
An increasing number of NFKB1 variants are being identified in patients with heterogeneous immunologic phenotypes.
Objective:
To characterize the clinical and cellular phenotype as well as the management of patients with heterozygous NFKB1 mutations.
Methods:
In a worldwide collaborative effort, we evaluated 231 individuals harboring 105 distinct heterozygous NFKB1 variants. To provide evidence for pathogenicity, each variant was assessed in silico; in addition, 32 variants were assessed by functional in vitro testing of nuclear factor of kappa light polypeptide gene enhancer in B cells (NF-κB) signaling.
Results:
We classified 56 of the 105 distinct NFKB1 variants in 157 individuals from 68 unrelated families as pathogenic. Incomplete clinical penetrance (70%) and age-dependent severity of NFKB1-related phenotypes were observed. The phenotype included hypogammaglobulinemia (88.9%), reduced switched memory B cells (60.3%), and respiratory (83%) and gastrointestinal (28.6%) infections, thus characterizing the disorder as primary immunodeficiency. However, the high frequency of autoimmunity (57.4%), lymphoproliferation (52.4%), noninfectious enteropathy (23.1%), opportunistic infections (15.7%), autoinflammation (29.6%), and malignancy (16.8%) identified NF-κB1–related disease as an inborn error of immunity with immune dysregulation, rather than a mere primary immunodeficiency. Current treatment includes immunoglobulin replacement and immunosuppressive agents.
Conclusions:
We present a comprehensive clinical overview of the NF-κB1–related phenotype, which includes immunodeficiency, autoimmunity, autoinflammation, and cancer. Because of its multisystem involvement, clinicians from each and every medical discipline need to be made aware of this autosomal-dominant disease. Hematopoietic stem cell transplantation and NF-κB1 pathway–targeted therapeutic strategies should be considered in the future.
Keywords: NFKB1 variant, NFKB1 mutation, common variable immunodeficiency, reduced penetrance, NF-κB1-related phenotype, autosomal dominant
Graphical Abstract
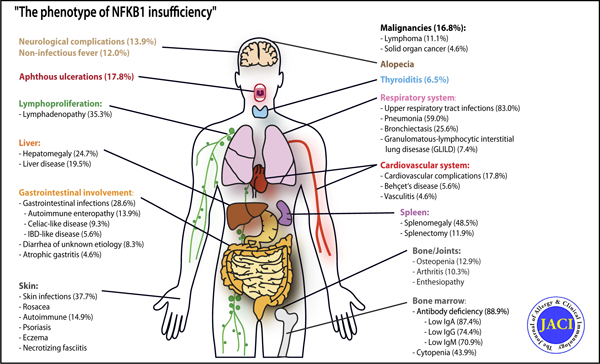
The nuclear factor of kappa light polypeptide gene enhancer in B cells (NF-κB) signaling pathway has been implicated in several biological processes, including cell survival and proliferation, inflammation, and the adaptive immune response.1 Its activation in lymphocytes is triggered by antigens, molecular patterns, and cytokines. NF-κB transcription factors can form various homo- or heterodimers containing the following 5 subunits: NF-κB1 (also known as p105, which is processed to p50), NF-κB2 (also known as p100, processed to p52), RelA, RelB, and c-Rel.
In unstimulated cells, p50 predominantly assembles with RelA, and remains inactive in the cytoplasm when complexed with the inhibitor NF-kappa-B alpha. On stimulation of the canonical (NF-κB1) pathway, the inhibitory inhibitor NF-kappa-B alpha protein is phosphorylated and degraded by the 26S proteasome, thereby releasing the active transcription factor heterodimer p50-RelA, which enters the nucleus and regulates the expression of its target genes. The noncanonical (NF-κB2) pathway is activated after the engagement of a small group of receptors such as the B-cell–activating factor receptor and CD40; this leads to proteasomal processing of p100 to generate p52, which preferentially pairs with RelB. The p52/RelB complex is mainly involved in B-cell survival and activation.2
The NFKB1 gene (MIM: 164011) encodes the precursor p105, which is cotranslationally processed into the transcriptionally active p50 subunit.3 Heterozygous NFKB1 mutations causing p50 haploinsufficiency have previously been associated with common variable immunodeficiency (CVID12 [MIM: 616576]),4 autoinflammatory, and rheumatologic features such as Behcet disease,5 EBV-driven lymphoproliferation,6,7 severe gastrointestinal manifestations,8 and susceptibility to opportunistic and viral infections.9,10 However, these reports covered only a few cases each, and an overview and understanding of the broader clinical spectrum of this NF-κB1–related condition is still lacking.
In a worldwide collaborative effort, we identified 231 individuals harboring 105 distinct heterozygous NFKB1 variants (see Fig E1 in this article’s Online Repository at www.jacionline.org). Sequence variants were classified into pathogenicity categories on the basis pof genetic and molecular criteria (see Table E1 in this article’s Online Repository at www.jacionline.org). Here, we describe the clinical and immunologic features of the largest to date cohort of patients (n = 157) with 56 distinct NFKB1 mutations (see Tables E2 and E3 in this article’s Online Repository at www.jacionline.org).
METHODS
We analyzed 105 heterozygous NFKB1 variants, identified in 231 individuals, from 129 unrelated families. For each variant, the following criteria were assessed: the predicted effect on the resulting protein (haploinsufficiency mutations, precursor-skipping mutations, missense variants affecting the p105 precursor and the mature p50, and missense variants probably affecting only the functions of the precursor), its localization in a functional domain of NF-κB1, the allele frequency in the Exome Aggregation Consortium data set, supportive functional studies, and the inheritance and segregation data (see Table E1). Detailed clinical and laboratory data can be found in Table E3. Multiple in silico tools (PolyPhen 2, Sorting Intolerant From Tolerant, Combined Annotation Dependent Depletion, Mutation Taster) have been used to predict the impact of missense changes. Functional assays evaluated the p105 and/or p50 levels in peripheral blood mononuclear leukocytes, neutrophils, or green fluorescent protein (GFP)-fused p105 and/or p50 in transfected human embryonic kidney 293T cells by Western blotting. In addition, we determined the nuclear localization and transcriptional activating function in human embryonic kidney 293T cells followingafter transfection of selected GFP-fused p105 and/or p50-like mutant proteins by fluorescence microscopy and by using an NF-κB–responsive fluorescence-based reporter assay or a dual luciferase reporter assay, respectively (see Table E4 in this article’s Online Repository at www.jacionline.org).
RESULTS
Genetic and functional assays
Of 105 variants, our combined in vitro and in silico assessment identified 56 distinct variants in 157 patients from 68 unrelated kindred as damaging. Of these 56 mutations, 28 have already been described,4,5,7–13 whereas 28 are novel. Thirty-four variants of the 56 mutations were located in the Rel homology domain, 17 in the central part of p105, whereas 3 affected the ankyrin repeat domain; in addition, 2 large deletions were identified (Fig 1).
FIG 1.
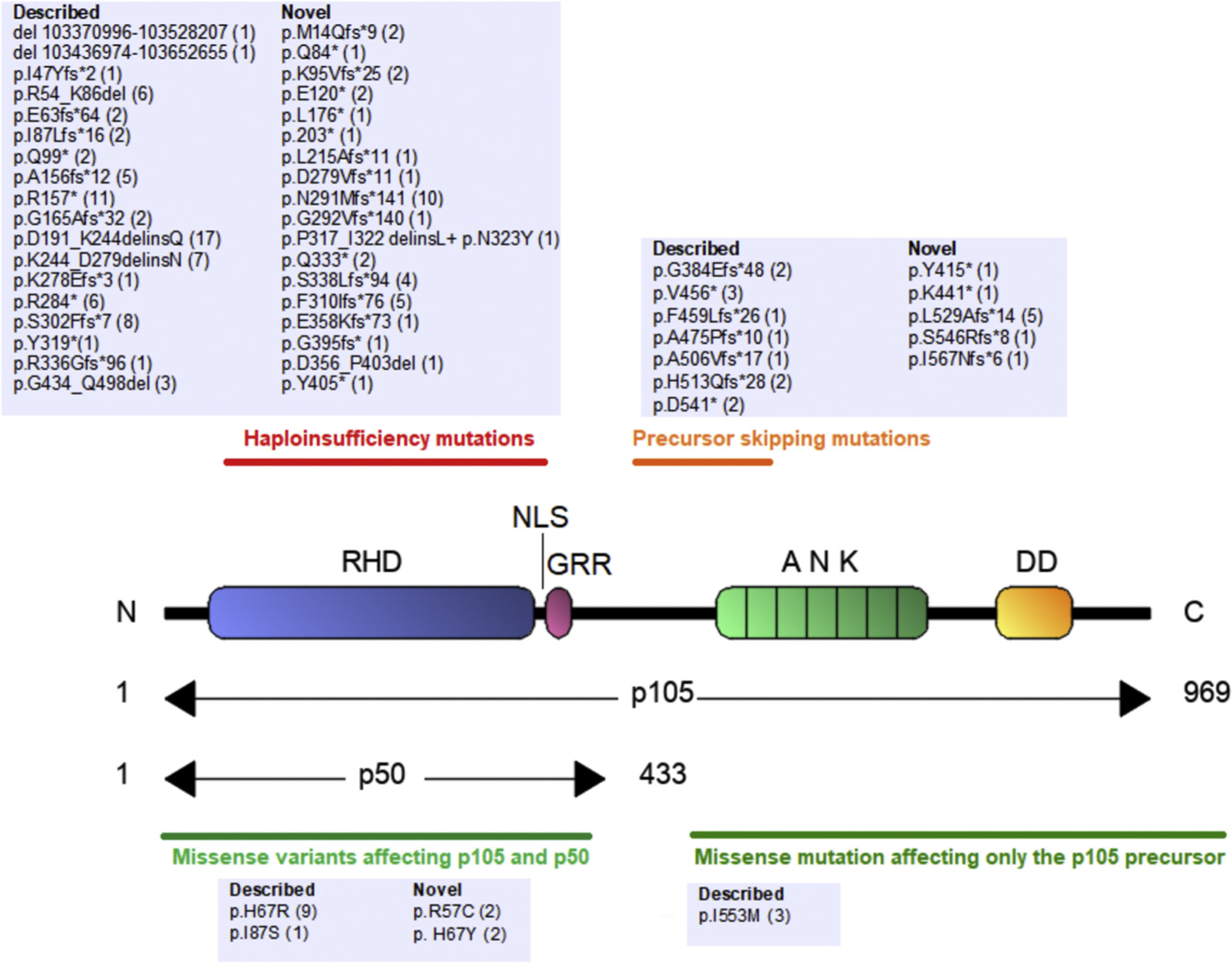
Localization of NFKB1 mutations. Numbers indicate amino acid positions. Horizontal black bars delineate the location of 4 different groups of damaging NFKB1 mutations. For each mutation, the number of carriers is indicated. GAPDH, Glyceraldehyde-3-phosphate dehydrogenase.
Generally, a haploinsufficiency mutation may cause either the lack of expression of the respective allele, or the expression of a severely truncated protein, which rapidly undergoes decay. As expected, the novel variant p.Ser338Leufs*94 revealed that the mutant p105 and p50 had reduced fluorescence intensity and aberrant localization, whereas wild-type (WT) p105 localized to the cytoplasm and WT p50 to the nucleus (Fig 2, A; see Table E4).
FIG 2.
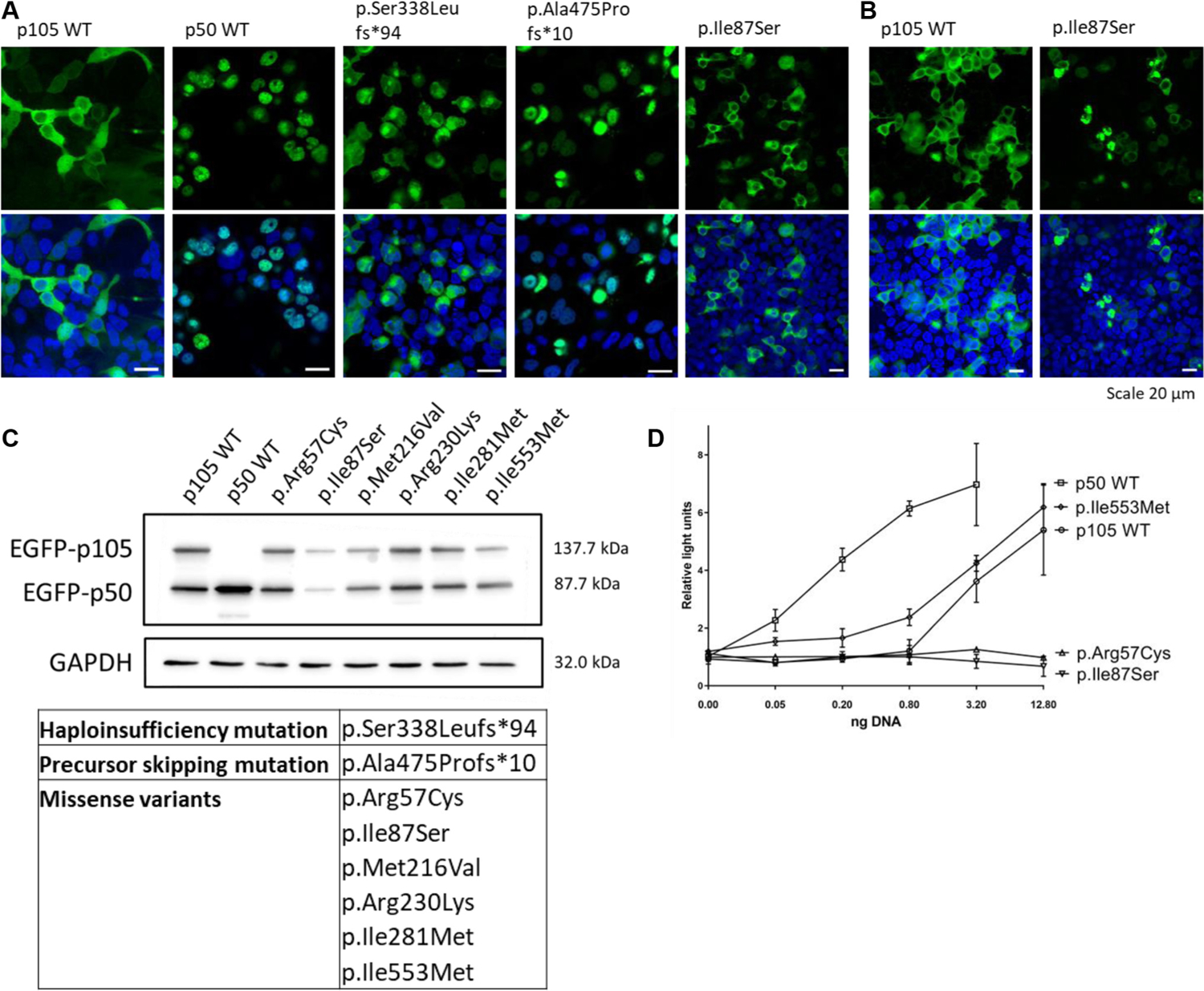
Subcellular localization, expression, and activity of distinct types of NFKB1 variants. HEK293T cells were transiently transfected with N-terminal GFP-fused constructs, as indicated. Nuclei were stained with Hoechst 33342 (blue). A, Haploinsufficiency mutations caused aberrant signals, whereas p50-like proteins (precursor-skipping variants) were localized to the nucleus. Missense variants (introduced into the full-length p105) produced signals that were indistinguishable from WT p105. B, PMA/ionomycin treatment caused clumping of the p.Ile87Ser mutant. C, Western Blot analysis (1) confirmed that transfected WT and transfected mutant p105 each underwent processing to p50, and (2) revealed the limited expression of the p.Ile87Ser variant. GAPDH was used as loading control. D, Loss of luciferase reporter activity with p.Arg57Cys and p.Ile87Ser mutants. Relative light units were normalized to cotransfected Renilla luciferase. Mock not shown. DNA amounts were compensated with nonrelated plasmid DNA. Depicted data represent the results from 2 to 4 experimental repeats, and additional data can be found in Fig E6. HEK293T, Human embryonic kidney 293T; PMS, phorbol myristate acetate.
Precursor-skipping NFKB1 mutations affect the central part of p105. These truncating mutations cause a lack of p105, but lead to the expression of a p50-like protein. On transfection of 4 mutant GFP-fused constructs, p50-like proteins localized to the nucleus and were indistinguishable from WT p50, potentially interfering with target gene transcription (Fig 2, A, and Table E4).
Transfection of 12 GFP-fused missense variants of p105 (Fig 2, A and B) revealed normal expression and cytoplasmic localization of the full-length p105, but 1 of the tested variants (p.Ile87Ser) showed a reduced fluorescence intensity in the cytoplasm and an abnormal accumulation of the signal in high-intensity spots. After stimulation with phorbol myristate acetate/ionomycin, a marginal increase in nuclear fluorescence was observed in cells transfected with WT p105, indicating increased processing to p50 (Fig 2, B). In contrast, the p.Ile87Ser mutant p105 was associated with cytoplasmic clumping on stimulation, indicating accelerated decay (Fig 2, B). Accordingly, Western blot analysis showed a reduced expression of the mutant p.Ile87Ser in transfected cells (Fig 2, C). In luciferase reporter assays, 2 of the missense mutations (p.Arg57Cys and p.Ile87Ser, both located in the N-terminal part of the Rel homology domain) showed reduced promoter activation (Fig 2, D). In agreement with the American College of Medical Genetics and Genomics (ACMG) classification, the remaining missense variants might only cause subtle rather than deleterious effects (see Table E4).
Patient characteristics
Among the 157 mutation carriers, 121 were classified as affected, whereas 36 were considered healthy (see Table E2). The median age of the whole cohort at the time of evaluation (June 2018) was 38 years (range, 6 months-79 years). The median age of healthy subjects (21.5 years) was lower than that of affected patients (39 years) (P <.001) (Fig 3, A). Because genetic screening could not be performed in all first-degree relatives of the affected patients, clinical penetrance was estimated to be 70% (see Fig E2 in this article’s Online Repository at www.jacionline.org). We found an increasing age-dependent penetrance (76.7% in individuals aged ≥10 years, 85.7% in individuals aged ≥30 years, and 100% in individuals aged ≥60 years), suggesting that the disease may manifest over time rather than having a bona fide reduced penetrance.
FIG 3.
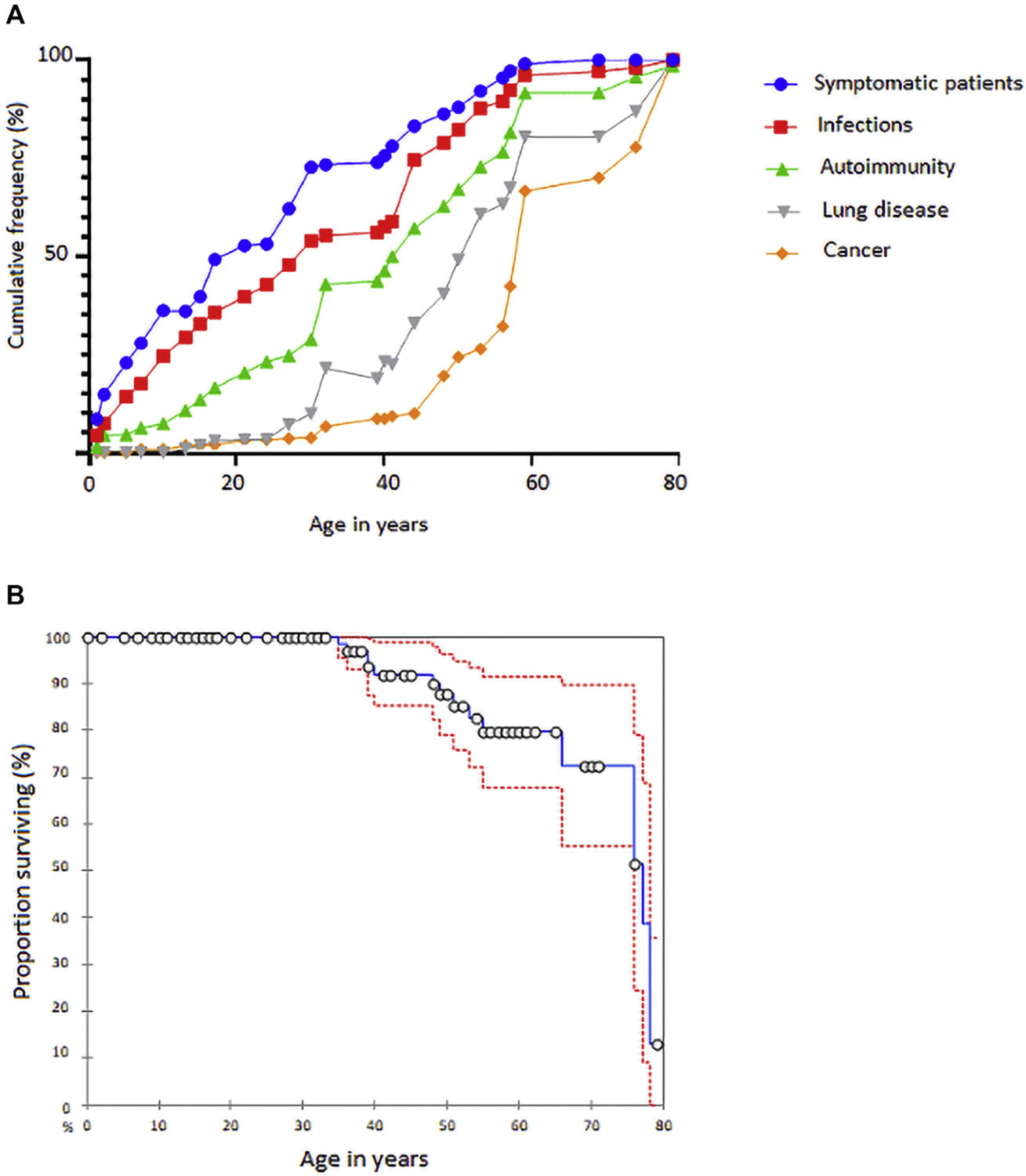
Clinical course and survival rate of NFKB1 cohort. A, Cumulative percentage of symptomatic patients who developed infections, autoimmunity, lung disease, and cancer. B, Kaplan-Meier survival curve with 95% CI (dotted lines).
The median age at which the first characteristic clinical manifestation occurred (mostly infections, autoimmune manifestations, and inflammatory symptoms) was 12 years (mean, 17.2 years; range, birth-69 years). The median age at NF-κB1–related disease diagnosis was 23 years (mean, 27.1 years; range, 1 month-73 years). Primary diagnoses at the time of disease onset were predominantly antibody deficiency (89.5%), diseases primarily characterized by autoimmunity (57.4%) and immune dysregulation (17.8%), and autoinflammatory disorders including Behçet disease (5.6%). The median follow-up time was 9 years (mean, 11.7 years; range, 0–50 years).
At the time of clinical data analysis (June 2018), 17.1% of patients of the affected carriers were deceased (Fig 3, B). Mortality rates were higher among males (21.5%) than among females (12.9%) (P = .22). Death occurred at a median age of 52 years (range, 35–78 years). The most frequent causes of death were infections on the background of a chronic illness (12 of 20) and complications from malignancies (5 of 20).
Respiratory involvement
Upper respiratory tract infections occurred in 83% of the 106 affected mutation carriers with definite mutations in NFkB1, on whom we had clinical data: 59.8% had sinusitis, 30.4% otitis, and 16.7% pharyngotonsillitis. Chronic sinusitis and nasal polyps led to sinus surgery in 6.9% of patients (Fig 4, A and C), whereas recurrent otitis was treated with tympanostomy tube placement in 5.9% of patients. Lower respiratory tract infections presented as pneumonia in 59% of patients and as bronchitis in 41.7% (Fig 5, A). Recurrent pneumonia (>3 episodes during the observation period) occurred in 24.2% of patients with any pneumonia; in 4.9% of patients, lung infection was complicated by pleural empyema. In 39.6% of patients, chronic lung disease was confirmed by pathological pulmonary function test and/or the detection of structural abnormalities by radiology or lung biopsy, and was associated with reduced survival (P = .003) (Fig 4, E–J and R). Patients with a history of pneumonia were at increased risk of developing lung disease (odds ratio [OR], 8.9; 95% CI, 3.1–25.9; P <.001). Bronchiectasis was detected by computed tomography in 25.6% of patients (Fig 4, G, and Fig 5, A). Interstitial lung disease had both granulomatous and interstitial histological patterns (granulomatous-lymphocytic interstitial lung disease) in 7.4% of patients (Fig 4, E, and Fig 5, A), whereas lung fibrosis and granuloma were detected in 6.4% and 3.2%, respectively. Five patients had pulmonary surgery, 3 patients with bronchiectasis underwent lobectomy, but 1 pneumonectomy was complicated by empyema, and the remaining 2 patients with empyema had lung decortication.
FIG 4.
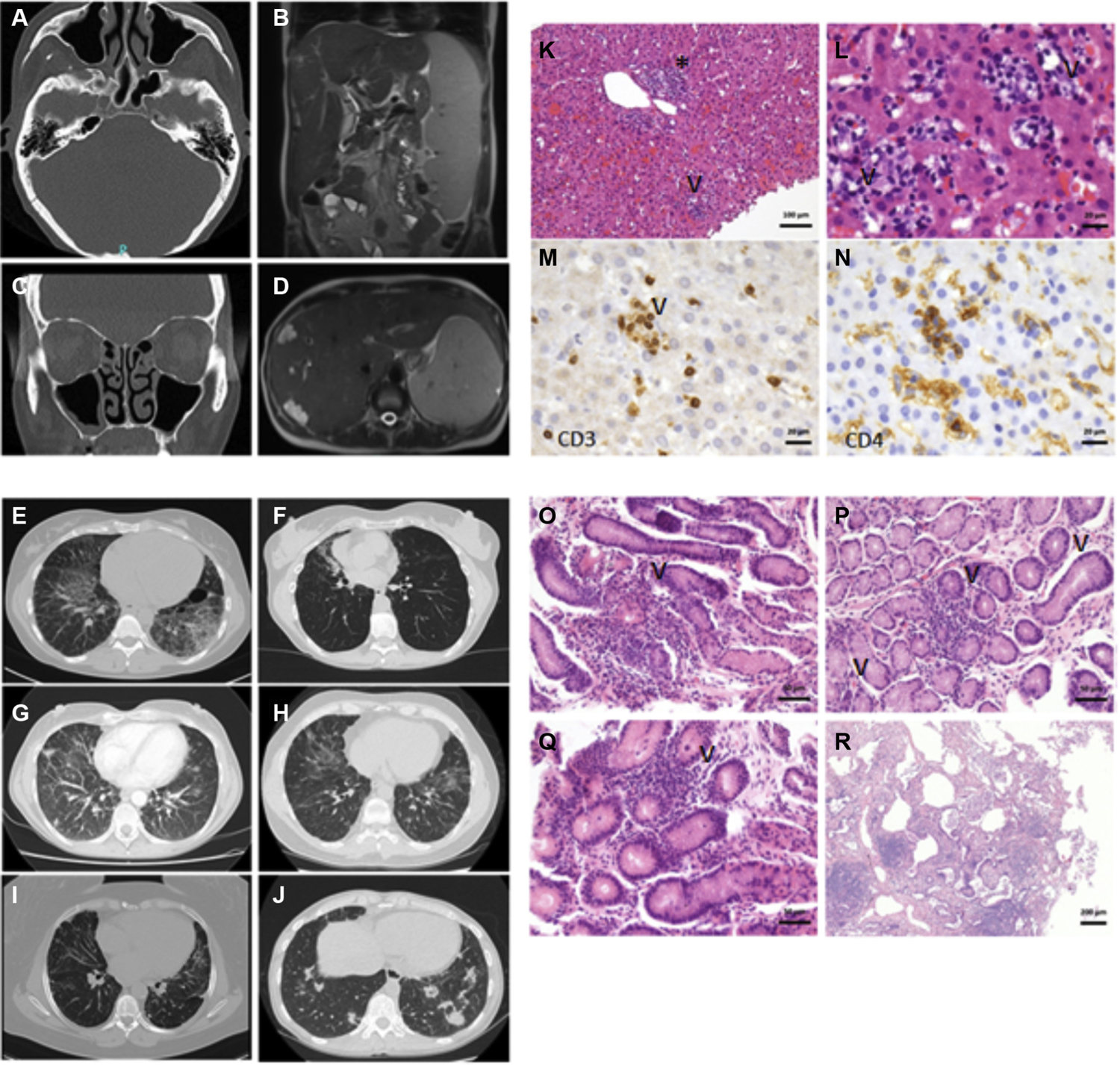
Exemplary CT and MRI findings and histopathology in patients with damaging heterozygous NFKB1 mutations. (A) and (C) from the same patient. Polypoid shifting of the ethmoidal cells as well as both sinus maxillares. Lower displacement of the frontal sinus and the sphenoid sinus. (B) and (D) from the same patient. Hepatosplenomegaly. Multiple liver hemangiomas and small liver cysts. Additional signs of focal nodular hyperplasia. Individual cystic lesions of the spleen. Widening of the portal vein due to possible portal venous hypertension. E-J, Several CT scans from different patients showing multiple pulmonary nodules, bronchiectasis with inflammatory changes, and interstitial lung disease. K and L, Hepatitis with T-cell–dominant lymphocytic inflammation. Fig 4, K, Portal (asterisk) and intralobular (arrowhead) inflammation. Fig 4, L, Higher magnification image showing intralobular lymphocytes and epithelioid cells, reminiscent of microgranulomas, with apoptosis of hepatocytes (nuclear remnants highlighted by arrowhead). M, CD3-positive T cells encircling an apoptotic hepatocyte, suggestive of T-cell–driven damage. N, Corresponding area to Fig 4, M, showing CD4-positive T cells, few monocytes, and intrasinusoidal macrophages (Kupffer cells). O-Q, Slightly chronic gastritis with patchy lymphocytic inflammation of the antrum (Fig 4, O, P highlighted by arrowhead) and corpus (Fig 4, Q highlighted by arrowhead). R, Chronic lymphocytic peribronchitis. Magnifications indicated by bars. CT, Computed tomography; MRI, magnetic resonance imaging.
FIG 5.
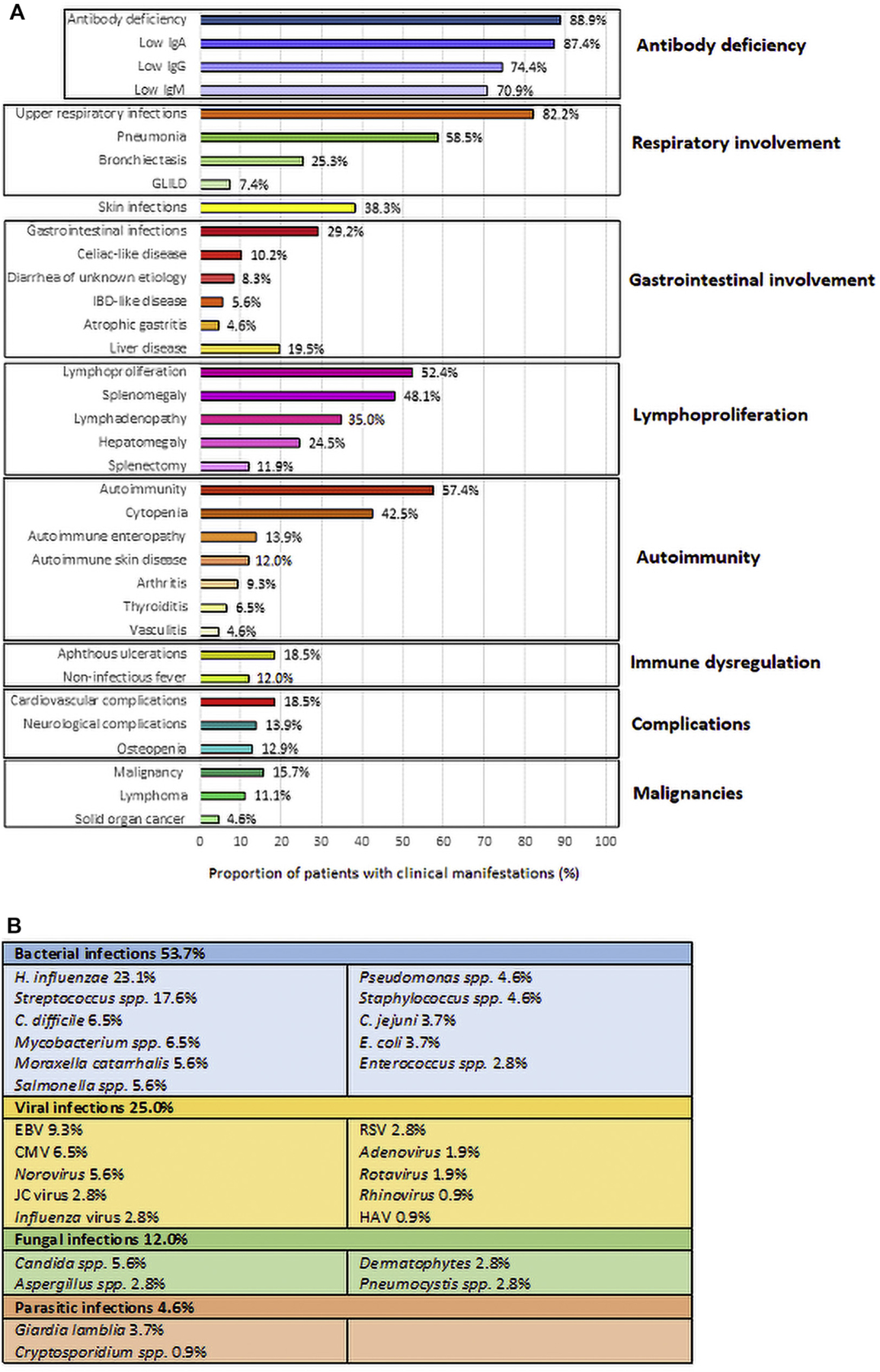
Main clinical findings in patients with damaging NFKB1 mutations. Percentage distribution of clinical manifestations (A), and infection types (B). CMV, Cytomegalovirus; GLILD, granulomatous-lymphocytic interstitial lung disease; HAV, hepatitis A virus; IBD, inflammatory bowel disease; JC virus, John Cunningham virus.
Gastrointestinal involvement
Diverse gastrointestinal involvement was observed in 54.2% of patients (Fig 5, A). Gastrointestinal infections occurred in 28.6% (n = 30) of patients. The histopathologic analysis of gastrointestinal biopsies, obtained in 28% of the 107 patients, revealed Herpes esophagitis (1.9%), eosinophilic esophagitis (0.9%), celiac-like disease (9.3%), chronic enteropathy mimicking inflammatory bowel disease (5.6%), lymphocytic or collagenous colitis (4.6%), and cytomegalovirus colitis (0.9%). In 8.3% of patients, no cause for chronic diarrhea was identified, despite extensive fecal examination and normal or nonspecific intestinal biopsies. The liver was involved in 24.1% of patients, with cirrhosis (3.7%), nodular regenerative hyperplasia (4.6%), hepatic hemangioma (3.7%), and hepatitis (7.5%) being the major pathologies. Hepatitis was classified as autoimmune in 3 patients (Fig 4, K–N), as drug-related in 3 patients, or virally induced in 2 patients (hepatitis A virus–related and hepatitis C virus–related in AD.I.1 and C.II.5, respectively).
Autoimmunity and immune dysregulation
Autoimmune conditions affected 57.4% of patients (Fig 5, A). The most common autoimmune conditions were cytopenia (43.9%), enteropathy (13.9%), skin disease (14.9%; see Appendix E2.II in this article’s Online Repository at www.jacionline.org), arthritis (10.3%), thyroiditis (6.5%), vasculitis (4.6%), hepatitis (2.8%), pernicious anemia (2.8%), type I diabetes (1.8%), and Addison disease (0.9%) (Fig 5, A). Autoantibodies against red blood cells or granulocytes were detected in 17.6% of patients. Subjects with autoimmune cytopenia were more likely to have lymphoproliferation (OR, 41.2; 95% CI, 12.3–137.6; P < .001), splenomegaly (OR, 36.4; 95% CI, 12–111; P < .001), or interstitial lung disease (OR, 8.3; 95% CI, 0.9–72.2; P = .05).
Lymphoproliferation and malignancies
Splenomegaly, lymphadenopathy, and hepatomegaly were detected by clinical assessment or ultrasonography in 48.5%, 35.3%, and 24.7% of patients, respectively (Fig 4, B and D, and Fig 5, A). Generalized expansion of the lymphoid compartment was associated with lung, liver, and gastrointestinal tract infiltration (OR, 9.3; 95% CI, 2.5–34.2; P <.001). Malignancies occurred in 18 of 107 (16.8%) patients (Fig 5, A); non-Hodgkin B-cell lymphomas were the most common, 8 patients (7.5%). Solid-organ cancer occurred in 5 patients (4.6% of all patients, including skin, lung, and cervical cancer). The median age at diagnosis of cancer was 46 years (range, 11–77 years). Death, primarily cancer-related or secondary to sepsis, occurred in 41.2% of patients with malignancies. Langerhans cell histiocytosis occurred in 1 child (patient AF.II.1).
Types of infections
Pathogenic bacteria, viruses, and fungi were identified in 53.7%, 25%, and 12% of patients, respectively (Fig 5, B). Bacteria were isolated from expectorated sputum samples in 31.5% of patients, with the most common being Haemophilus influenzae (23.1%), Streptococcus species (17.6%), Moraxella catarrhalis (5.6%), or Pseudomonas species (4.6%). Stool cultures were positive in 18$.5% of patients, with Clostridium difficile (6$.5%), Salmonella species (5.6%), and Campylobacter jejuni (3.7%). Ten patients with NFKB1 mutations developed sepsis (9.3%), 4 after surgical procedures, 3 secondary to pneumonia. Bacteria were isolated from blood samples in only 3 cases (Escherichia coli, Enterococcus faecalis, and Staphylococcus epidermidis). Five patients were diagnosed with Mycobacterium avium complex infection, affecting the lungs in 4 patients and the lymph nodes in 1 child. Disseminated bacillus Calmette-Guérin disease after vaccination and Mycobacterium genavense infection occurred in 1 patient each (W.I.1 and AF.II.1, respectively). In 6.5% of patients with respiratory symptoms, viral pathogens (influenza virus, respiratory syncytial virus, rhinovirus, and adenovirus) were isolated. In stool samples, 9.3% of patients had norovirus (5.6%), rotavirus (1.9%), adenovirus (0.9%), or hepatitis A virus (0.9%). EBVinfection presented as a low-grade/reactivating EBVinfection (viral load <500 copies/mL) and EBV-associated lymphoproliferative disease (viral load >1000 copies/mL) in 7 and 3 patients, respectively. Cytomegalovirus reactivation caused hepatitis, cytopenia, and retinitis in 1 patient each. A patient with colitis and diarrhea had a colon biopsy that was positive for the cytomegalovirus antigen (see Table E5 in this article’s Online Repository at www.jacionline.org). John Cunningham virus was detected in the cerebrospinal fluid of 3 patients with progressive multifocal leukoencephalopathy. They had normal levels of CD4 and CD8 T cells, but 2 of them had B-cell depletion therapy (see Table E5). Progressive multifocal leukoencephalopathy was the cause of death in 1 patient (AR.I.4). Candida species were isolated from skin swabs, expectorated sputum samples, and stool samples in 5.6% of patients. Dermatophytes accounted for noninvasive skin infections in 3 patients. Undetectable serum IgE (<2 IU/mL) was found in 54.2% of patients. Respiratory fungal opportunistic infections were caused by Aspergillus species and Pneumocystis jirovecii in 3 patients each; 2 of these were under immunosuppressive therapy (see Table E5).
Immunologic assessment
At the time of diagnosis, most (88.9%) symptomatic patients older than 4 years presented with serum IgG levels below 5 g/L (median, 3.6 g/L; range, 0–9.9 g/L), and a marked decrease in at least 1 of the IgA or IgM isotypes (<0.8 g/L and <0.4 g/L, respectively). In 10.7% of patients, all classes of immunoglobulins were found to be normal. A poor response to T-dependent (tetanus and diphtheria toxoid) and T-independent (pneumococcus) antigens was found in 65.2% of individuals. In 50% of patients, the levels of circulating B cells were still within the lower normal range of 6% to 19%, or 100 to 500 cells/µL, respectively (see Fig E3, A, in this article’s Online Repository at www.jacionline.org). In 60.3% of patients, the percentage of IgM‒IgD‒CD27+ switched memory B cells was less than or equal to 2% (normal range, 6.5%–29.2%) (Fig E3, A). An expansion of CD21lowCD38lowCD19hi B cells to above 10% was found in 56.1% of affected individuals (normal range, 1.1%–6.9%) (Fig E3, A). We observed a significant correlation between the expansion of CD21low B cells above 10% and both autoimmune cytopenia (OR, 5; 95% CI, 1.1–22.3; P = .03) and lymphoproliferation (OR, 5.7; 95% CI, 1.4–23.5; P = .01). Overall, opportunistic infections occurred in 15.7% of the patients and were associated with median CD4+ T-cell count not as low as expected (588/µL), a profound B-cell defect (median B-cell count, 46/µL), ongoing immunosuppressive treatments, and a poor outcome (see Table E5). Low numbers of circulating natural killer cells less than 100/µL, found in 33.3% of patients (Fig E3, A), were associated with an increased risk of viral infections (OR, 2.8; 95% CI, 1.1–6.9; P = .02).
Treatment
IgG replacement therapy alone was sufficient to treat 14.5% of the patients who needed medical intervention, whereas 85.5% of the patients required additional therapy. Antibiotic prophylaxis, antifungal agents, and antiviral drugs were added to treat 44.8%, 12.5%, and 12.4% of the patients, respectively (see Fig E4 in this article’s Online Repository at www.jacionline.org). In addition to IgG replacement, 60.1% of patients with autoimmune cytopenia were treated with systemic corticosteroids. For refractory or recurrent cytopenia, 17.4% of the patients had anti-CD20 mAb (rituximab), 15.2% splenectomy, and 8.7% mycophenolate mofetil. Granulomatous-lymphocytic interstitial lung disease was treated with oral corticosteroids alone in 5 patients, or in combination with immunosuppressive agents (cyclophosphamide, mycophenolate mofetil, rituximab, and cyclosporine), to which there was only a partial response. Noninfectious enteropathy was treated with systemic corticosteroids in 13 of 17 patients, whereas 3 patients received azathioprine (M.II.1, AH.I.1, and BB.I.1), eliciting a partial response. In addition to systemic corticosteroids, 3 patients with inflammatory bowel disease–like exacerbations received mesalazine. In patients (84.6%) with oral and genital ulcers, systemic corticosteroids induced a good response. Three individuals with lymphoproliferative disease were treated with anti-CD20 (rituximab), which led to complete remission. Abatacept, a cytotoxic T-lymphocyte antigen 4 fusion protein, was used to treat 1 patient with refractory autoimmunity and lymphoproliferation, eliciting a good response (Q.I.1). Hematopoietic stem cell transplantation is currently planned for 4 patients with EBV-lymphoproliferative disease, refractory cytopenia with lymphoproliferation, and mycobacterial disease (AB.II.1, S.I.1, AP.I.1, and BL.II.1).
DISCUSSION
Heterozygous NFKB1 mutations causing p50 haploinsufficiency have previously been reported to be associated with various phenotypes ranging from mere antibody deficiency to multiorgan autoinflammatory conditions.4–10 However, a comprehensive clinical description of the extended phenotype of the NF-κB1–related phenotype has been lacking. Here, we show that antibody deficiency was the main finding in patients with NFKB1 mutations (88.9%). However, this may well present as an ascertainment bias, because this survey was initiated by clinical immunology centers of the European Society for Immunodeficiencies. Only 76.9% of patients fulfilled the revised European Society for Immunodeficiencies registry criteria for CVID, indicating that patients with normal or only mildly affected humoral immunity may also have an impaired canonical NF-κB signaling. The median age was lower in healthy mutation carriers than in affected patients, and progressive development of humoral immunodeficiency was observed in some individuals, suggesting an age-dependent manifestation and expressivity of NFKB1-related phenotypes. Hypogammaglobulinemia is the reason for the high incidence of bacterial infections in our cohort, especially those affecting the upper (83%) and lower (59%) respiratory tract, the skin (37.7%), and the gastrointestinal tract (28.6%). Notably, 15.7% of patients developed opportunistic infections, a much higher percentage than expected in CVID.14,15 The observed difference can be explained by our definition of CVID,16 which did not exclude patients with a T-cell defect.17 However, a measurable CD4 T-cell defect was observed in some of our patients (15.1%), but was not necessarily associated with opportunistic infections (P = .57).18 Our observations suggest that in addition to the NFKB1 mutation, an immunosuppressive treatment may impair the T-cell response and, in combination with the lack of B cells, contribute to the pathogenesis of opportunistic infections. The clinical phenotype was also dominated by lymphoproliferation, particularly splenomegaly (48.5%) and lymphadenopathy (35.3%), and by autoimmunity (mainly cytopenia) (43.9%). Low serum IgA and IgM levels, which reflect the loss of switched memory B cells, were associated with an increased risk of developing autoimmunity and splenomegaly. Thus, defective isotype switching and somatic hypermutation may each account for the increased presence of autoreactive B cells.19 Autoimmune cytopenia and lymphoproliferation were also associated with an elevated proportion of CD21low B cells (>10%). CD21low B cells develop after chronic stimulation and have been found to be enriched in autoreactive clones.20 Impaired canonical NF-κB signaling has been observed not only in NFKB1-haploinsufficient patients but also in patients with CVID with the CVID 21low phenotype, thus potentially contributing to the accumulation of CD21low B cells.21 Conversely, a TH1-skewed profile in peripheral blood T cells, combined with the overexpression of proinflammatory cytokines such as IL-1β and TNF-α, may contribute to the autoinflammatory symptoms.6,8
Therapeutic strategies for individuals with antibody deficiency include immunoglobulin replacement therapy. Patients with autoimmunity and immune dysregulation shall be treated with steroids and rituximab. However, the beneficial effect has to be weighed against the infectious risk of immunosuppression. The cytotoxic T-lymphocyte antigen 4 fusion protein abatacept was used to treat 1 patient with good response. Additional therapeutic options that still require evaluation include hematopoietic stem cell transplantation, and targeted therapeutic strategies such as proteasome inhibitors. The potential therapeutic effect of anti-TNF, which inhibits TNF-mediated NF-κB activation, suggests that NF-κB1–related diseases result in dysregulated, rather than defective, NF-κB signaling.
However, immune dysregulation may result not only from a defective but also from an increased NF-κB activation.22,23 Indeed, p50 homodimers, stabilized by B-cell leukemia 3 protein, function as inhibitory factors for NF-κB1 transcriptional activity because they do not contain the transcriptional activation domain that is otherwise exclusively present in RelA (and RelB and c-Rel); however, they do compete with p50/RelA heterodimers for binding to DNA.24
While studying this cohort, it became clear that the development of drugs specifically interfering with the NF-κB signaling pathway will be an important step forward not only for the personalized treatment of patients with NF-κB–related disease but also for patients with more common autoimmune or inflammatory conditions.
Supplementary Material
Clinical implications:
The aim of this work was to aid diagnosis, management, and treatment of patients with NFKB1 mutations. Clinical features, complications, current treatment options, and future targeted therapeutic strategies are illustrated.
Acknowledgments
This study was supported by the German Research Foundation (DFG) (SFB1160 – IMPATH to B.G.), and under Germany’s Excellence Strategy (CIBSS - EXC-2189 - Project ID 390939984 and RESIST - EXC 2155 - Project ID 39087428), by the E-rare program of the European Union, managed by the DFG, grant code GR1617/14–1/iPAD; by the “Netzwerke Seltener Erkrankungen” of the German Ministry of Education and Research (BMBF) (grant code: GAIN_ 01GM1910A); and by research funding from the German Ministry of Education and Research (BMBF, grant nos. 01E01303 and 01ZX1306F).
We thank all patients and their families for participation in this study. We are grateful to Mary Buchta, Katrin Hübscher, Pavla Mrovecova, and Jessica Rojas-Restrepo for their excellent technical assistance. We are grateful to Francisco Bonilla, Isil B. Barlan, Katharine Halligan, Andrew MacGinnitie, and Anna Simon for their contribution to the patient recruitment process, and Katharina Thoma for helping with the graphical abstract. Some samples have been taken from the CCI-biobank, a partner of the Freeze Biobank Freiburg. NIHR BioResource authors: Zoe Adhya, Hana Alachkar, Ariharan Anantharachagan, Richard Antrobus, Gururaj Arumugakani, Sofie Ashford, William J. Astle, Anthony Attwood, Chiara Bacchelli, Joana Batista, Helen E. Baxendale, Claire Bethune, Shahnaz Bibi, Marta Bleda, Barbara Boardman, Claire Booth, John R. Bradley, Gerome Breen, Matthew Brown, Michael J. Browning, Mary Brownlie, Matthew S. Buckland, Siobhan O. Burns, Oliver S. Burren, Keren Carss, John Chambers, Anita Chandra, Naomi Clements Brod, Hayley Clifford, Nichola Cooper, Louise C. Daugherty, E. G. Davies, Sophie Davies, John Davis, Sarah Deacock, Sri V. V. Deevi, John Dempster, Lisa A. Devlin, Eleanor F. Dewhurst, Kate Downes, Elizabeth Drewe, Daniel Duarte, J. David M. Edgar, Karen Edwards, William Egner, Tariq El-Shanawany, Marie Erwood, Debra Fletcher, James Fox, Amy J. Frary, Mattia Frontini, Abigail Furnell, H. Bobby Gaspar, Rohit Ghurye, Kimberly C. Gilmour, Nicholas S. Gleadall, Sarah Goddard, Pavels Gordins, Stefan Gräf, Luigi Grassi, Daniel Greene, Sofia Grigoriadou, Scott Hackett, Rosie Hague, Matthias Haimel, Lorraine Harper, Grant Hayman, Archana Herwadkar, Fengyuan Hu, Stephen Hughes, Aarnoud P. Huissoon, Roger James, Stephen Jolles, Jennifer Jolley, Julie Jones, Yousuf Karim, Mary A. Kasanicki, Peter Kelleher, Carly Kempster, Sorena Kiani, Nathalie Kingston, Nigel Klein, Myrto Kostadima, Roman Kreuzhuber, Taco W. Kuijpers, Dinakantha Kumararatne, James Laffan, Hana Lango Allen, Sara E. Lear, Rachel Linger, Hilary Longhurst, Lorena E. Lorenzo, Paul A. Lyons, Jesmeen Maimaris, Ania Manson, Rutendo Mapeta, Jennifer Martin, Mark I. McCarthy, Elizabeth M. McDermott, Harriet McKinney, Stuart Meacham, Karyn Megy, Hazel Millar, Anoop Mistry, Valerie Morrisson, Sai H. K. Murng, Iman Nasir, Sergey Nejentsev, Sadia Noorani, Eric Oksenhendler, Willem H. Ouwehand, Sofia Papadia, Christopher J. Penkett, Romina Petersen, Mark J. Ponsford, Waseem Qasim, Ellen Quinn, Isabella Quinti, F. Lucy Raymond, Paula J. Rayner-Matthews, Alex Richter, Nilesh Samani, Crina Samarghitean, Alba Sanchis-Juan, Ravishankar B. Sargur, Sinisa Savic, Suranjith L. Seneviratne, W. A. Carrock Sewell, Denis Seyres, Fiona Shackley, Olga Shamardina, Ilenia Simeoni, Michael A. Simpson, Kenneth G. C. Smith, Simon Staines, Emily Staples, Hannah Stark, Hans Stauss, Cathal L. Steele, Jonathan Stephens, Kathleen E. Stirrups, James E. Thaventhiran, David Thomas, Moira J. Thomas, Patrick Thomas, Adrian J. Thrasher, Tobias Tilly, Catherine Titterton, Paul Treadaway, Salih Tuna, Ernest Turro, Rafal Urniaz, Julie von Ziegenweidt, Neil Walker, Christopher Watt, Steven B. Welch, Deborah Whitehorn, Lisa Willcocks, Nicholas Wood, Yvette Wood, Sarita Workman, Austen Worth, Katherine Yates, Nigel Yeatman, Patrick F. K. Yong, Timothy Young, Ping Yu, Eliska Zlamalova.
Abbreviations used
- CVID
Common variable immunodeficiency
- GFP
Green fluorescent protein
- NF-κB
Nuclear factor of kappa light polypeptide gene enhancer in B cells
- OR
Odds ratio
- WT
Wild-type
Footnotes
Disclosure of potential conflict of interest: All authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Beinke S, Ley SC. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem J 2004;382:393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayden MS, Ghosh S. NF-κB in immunobiology. Cell Res 2011;21:223–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pereira SG, Oakley F. Nuclear factor-κB1: regulation and function. Int J Biochem Cell Biol 2008;40:1425–30. [DOI] [PubMed] [Google Scholar]
- 4.Fliegauf M, Bryant VL, Frede N, Slade C, Woon ST, Lehnert K, et al. Haploinsufficiency of the NF-κB1 subunit p50 in common variable immunodeficiency. Am J Hum Genet 2015;97:3–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaustio M, Haapaniemi E, Göös H, Hautala T, Park G, Syrjänen J, et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J Allergy Clin Immunol 2017;140:782–96. [DOI] [PubMed] [Google Scholar]
- 6.Hoeger B, Serwas NK, Boztug K. Human NF-κB1 haploinsufficiency and Epstein-Barr virus-induced disease — molecular mechanisms and consequences. Front Immunol 2017;8:1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boztug H, Hirschmugl T, Holter W, Lakatos K, Kager L, Trapin D, et al. NF-κB1 haploinsufficiency causing immunodeficiency and EBV-driven lymphoproliferation. J Clin Immunol 2016;36:533–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dieli-Crimi R, Martínez-Gallo M, Franco-Jarava C, Lakatos K, Kager L, Trapin D, et al. Th1-skewed profile and excessive production of proinflammatory cytokines in a NFKB1-deficient patient with CVID and severe gastrointestinal manifestations. Clin Immunol 2018;195:49–58. [DOI] [PubMed] [Google Scholar]
- 9.Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol 2016;7:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lougaris V, Patrizi O, Baronio M, Tabellini G, Tampella G, Damiati E, et al. NFKB1 regulates human NK cell maturation and effector functions. Clin Immunol 2017;175:99–108. [DOI] [PubMed] [Google Scholar]
- 11.Schipp C, Nabhani S, Bienemann K, Simanovsky N, Kfir-Erenfeld S, Assayag-Asherie N, et al. Specific antibody deficiency and autoinflammatory disease extend the clinical and immunological spectrum of heterozygous NFKB1 loss-of-function mutations in humans. Haematologica 2016;101:e392–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rae W, Ward D, Mattocks CJ, Gao Y, Pengelly RJ, Patel SV, et al. Autoimmunity/inflammation in a monogenic primary immunodeficiency cohort. Clin Transl Immunol 2017;6:e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol 2018;142:1285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol 1999;92:34–48. [DOI] [PubMed] [Google Scholar]
- 15.Oksenhendler E, Gérard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis 2008;46:1547–54. [DOI] [PubMed] [Google Scholar]
- 16.European Society for Immunodeficiences. Clinical Working Party diagnostic criteria for PID. Available at: https://esid.org/Working-Parties/Clinical-Working-Party/Resources/Diagnostic-criteria-for-PID2#Q3. Accessed April 17, 2020.
- 17.Ameratunga R, Brewerton M, Slade C, Jordan A, Gillis D, Steele R, et al. Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front Immunol 2014;5:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertinchamp R, Gérard L, Boutboul D, Malphettes M, Fieschi C, Oksenhendler E. Exclusion of patients with a severe T-cell defect improves the definition of common variable immunodeficiency. J Allergy Clin Immunol Pract 2016;4:1147–57. [DOI] [PubMed] [Google Scholar]
- 19.Patuzzo G, Barbieri A, Tinazzi E, Veneri D, Argentino G, Moretta F, et al. Autoimmunity and infection in common variable immunodeficiency (CVID). Autoimmun Rev 2016;15:877–82. [DOI] [PubMed] [Google Scholar]
- 20.Isnardi I, Ng Y-S, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement receptor 2/CD21-human naive B cells contain mostly autoreactive unresponsive clones. Blood 2010;115:5026–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keller B, Cseresnyes Z, Stumpf I, Wehr C, Fliegauf M, Bulashevska A, et al. Disturbed canonical nuclear factor of κ light chain signaling in B cells of patients with common variable immunodeficiency. J Allergy Clin Immunol 2017;139:220–31.e8. [DOI] [PubMed] [Google Scholar]
- 22.O’Reilly LA, Putoczki TL, Mielke LA, Low JT, Lin A, Preaudet A, et al. Loss of NF-κB1 causes gastric cancer with aberrant inflammation and expression of immune checkpoint regulators in a STAT-1-dependent manner. Immunity 2018;48: 570–83.e8. [DOI] [PubMed] [Google Scholar]
- 23.Etzioni A, Ciechanover A, Pikarsky E. Immune defects caused by mutations in the ubiquitin system. J Allergy Clin Immunol 2017;139:743–53. [DOI] [PubMed] [Google Scholar]
- 24.Collins PE, Kiely PA, Carmody RJ. Inhibition of transcription by B cell leukemia 3 (Bcl-3) protein requires interaction with nuclear factor κB (NF-κB) p50. J Biol Chem 2014;2:7059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.