

Abstract
Background
Pantothenate kinase–associated neurodegeneration (PKAN) currently has no approved treatments.
Objectives
The Fosmetpantotenate Replacement Therapy pivotal trial examined whether treatment with fosmetpantotenate improves PKAN symptoms and stabilizes disease progression.
Methods
This randomized, double‐blind, placebo‐controlled, multicenter study evaluated fosmetpantotenate, 300 mg oral dose three times daily, versus placebo over a 24‐week double‐blind period. Patients with pathogenic variants of PANK2, aged 6 to 65 years, with a score ≥6 on the PKAN‐Activities of Daily Living (PKAN‐ADL) scale were enrolled. Patients were randomized to active (fosmetpantotenate) or placebo treatment, stratified by weight and age. The primary efficacy endpoint was change from baseline at week 24 in PKAN‐ADL.
Results
Between July 23, 2017, and December 18, 2018, 84 patients were randomized (fosmetpantotenate: n = 41; placebo: n = 43); all 84 patients were included in the analyses. Six patients in the placebo group discontinued treatment; two had worsening dystonia, two had poor compliance, and two died of PKAN‐related complications (aspiration during feeding and disease progression with respiratory failure, respectively). Fosmetpantotenate and placebo group PKAN‐ADL mean (standard deviation) scores were 28.2 (11.4) and 27.4 (11.5) at baseline, respectively, and were 26.9 (12.5) and 24.5 (11.8) at week 24, respectively. The difference in least square mean (95% confidence interval) at week 24 between fosmetpantotenate and placebo was −0.09 (−1.69 to 1.51; P = 0.9115). The overall incidence of treatment‐emergent serious adverse events was similar in the fosmetpantotenate (8/41; 19.5%) and placebo (6/43; 14.0%) groups.
Conclusions
Treatment with fosmetpantotenate was safe but did not improve function assessed by the PKAN‐ADL in patients with PKAN. © 2020 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society.
Keywords: pantothenate kinase–associated neurodegeneration, fosmetpantotenate, treatment, randomized controlled trial
Pantothenate kinase–associated neurodegeneration (PKAN) is caused by pathogenic or likely pathogenic variants in PANK2, which encodes pantothenate kinase 2 (PanK2). 1 PKAN is the most common form of the neurodegeneration with brain iron accumulation (NBIA) disorders and is estimated to account for approximately half of all NBIA cases. 2 The prevalence of PKAN is not known, but it is estimated at 1 per million population (eg, US prevalence estimated as n = 318–6363). 2 The pathogenic or likely pathogenic variants in PANK2 lead to complete or partial dysfunction in PanK2 activity 4 and cause disruption in the cascade underlying the biosynthesis of coenzyme A (CoA), an important factor in numerous biochemical reactions. 5 The resulting dysfunction in the PanK2 enzyme disrupts the conversion of pantothenate (pantothenic acid, vitamin B5) to phosphopantothenate (4′‐phosphopantothenic acid), which is the first step in the mitochondrial CoA biosynthesis pathway. 6 The end result for patients with PKAN is postulated to be decreased concentrations of CoA in selectively vulnerable tissues, including the brain. 5 , 7 , 8
Currently, there are no approved treatments for PKAN, and available therapies remain symptom‐targeted, aimed at symptom management and palliation, such as deep brain stimulation for the treatment of dystonic features. 9 However, deferiprone, an oral iron chelator that crosses the blood–brain barrier, recently showed positive outcomes in a phase 3 randomized, double‐blind, controlled trial with a large number of patients with PKAN enrolled (88 patients). 10 Treatment with deferiprone versus placebo for up to 3 years significantly lowered iron in the globus pallidus of patients and at 18 months resulted in somewhat slower disease progression in patients with the atypical form of PKAN as assessed by the Barry–Albright Dystonia (BAD) scale.
Fosmetpantotenate was developed as a phosphopantothenate replacement therapy to target the underlying biochemical defect in PKAN. 11 Preliminary positive findings from uncontrolled, open‐label, fosmetpantotenate treatment of three adult patients with PKAN within international compassionate use case reports led to the design of the Fosmetpantotenate Replacement Therapy (FORT) pivotal trial. 12 , 13 The FORT pivotal trial was conducted to examine whether treatment with fosmetpantotenate could improve signs and symptoms of PKAN and slow or halt disease progression. The development of the FORT primary efficacy outcome measure, the patient/surrogate‐reported PKAN‐Activities of Daily Living (PKAN‐ADL) scale, has been described elsewhere. 14 , 15 In the double‐blind period, FORT examined the efficacy of fosmetpantotenate in improving disease‐related signs (ie, dystonia), symptoms, and functioning in patients with PKAN. The open‐label extension period was planned to examine safety and to determine whether any improvements in functioning were maintained within more real‐world therapeutic conditions with allowed adjustments in concomitant medications.
Patients and Methods
Study Design
The FORT study design has been described previously. 14 FORT was a pivotal, randomized, double‐blind, placebo‐controlled, international, multicenter, two‐arm study that evaluated treatment with fosmetpantotenate or placebo for a 24‐week double‐blind period. Patients who completed the double‐blind period could enroll in the 278‐week open‐label extension, in which all patients received fosmetpantotenate. The study was conducted in 20 hospitals and medical centers located in the United States, Canada, Czech Republic, France, Germany, Italy, Norway, Poland, and Spain. Ethics approval was obtained from the appropriate independent ethics committees/institutional review boards, applicable regulatory authorities, and host institutions.
Patients
Patients eligible for inclusion had a diagnosis of PKAN with confirmed pathogenic variants in PANK2 (ie, confirmed homozygous or compound heterozygous pathogenic variants in PANK2), had a score ≥6 on the PKAN‐ADL (indicating at least mild impairment as a result of PKAN), and were 6 to 65 years old (inclusive). Patients or parents/legal guardians (as appropriate) provided signed informed consent/assent before any screening procedures. PKAN maintenance treatments (eg, benzodiazepines, anticholinergics, baclofen, deep brain stimulation, botulinum toxin) were allowed without changes or modifications to any of the treatments from 30 days before randomization until completion of the double‐blind period, unless changes were medically necessary. Exclusion criteria are provided in Supporting Information Table S1.
Randomization and Masking
Patients were randomized via integrated web response system (IWRS) in a 1:1 ratio to receive either fosmetpantotenate or placebo for the 24‐week double‐blind period. An independent team generated the randomization sequence used to program the IWRS, and group allocation was concealed through the IWRS randomization and assignment process. Randomization was stratified by weight (≥40 kg, ≥20 kg but <40 kg, or <20 kg) and age group (pediatric: age 6 to <18 years or adult: age 18–65 years, inclusive) at screening. Study drug identity was concealed through identical packaging, labeling, schedule of administration, appearance, taste, and odor. Patients, parents/legal guardians, investigators, and study personnel were blinded to treatment assignment in the double‐blind treatment period.
Procedures
During the double‐blind period, in adults, dose escalation of orally administered fosmetpantotenate or placebo occurred on days 1 through 3, followed by the full dose of 900 mg, divided equally as 300 mg three times daily with food (approximately 8‐hour intervals), starting on day 4. No dose escalation was required for the open‐label period, regardless of randomization in the double‐blind period. During the double‐blind period, the daily dose for patients 6 to <18 years old was based on weight at screening. Pediatric patients weighing ≥40 kg received the same dosing regimen as adults. Patients weighing ≥20 kg but <40 kg received 450 mg of study drug daily, divided equally as 150 mg three times daily, and patients weighing <20 kg received 225 mg of study drug daily, divided equally as 75 mg three times daily. Assessment visits occurred at baseline (day −1), days 1 through 4, and weeks 3, 6, 12, 18, and 24. Patients were admitted to an inpatient facility for safety monitoring from baseline through day 4 or were closely followed in an outpatient setting. The primary and secondary efficacy endpoint measures were assessed at baseline and at weeks 3, 6, 12, 18, and 24. 14
Outcomes
The primary efficacy endpoint was the change in the patient‐ or surrogate‐reported PKAN‐ADL total score from baseline to the end of the 24‐week double‐blind period. The PKAN‐ADL is a validated PKAN‐specific clinical outcomes assessment adapted from the Unified Parkinson's Disease Rating Scale Part II (UPDRS II) that assesses the ability of patients to complete ADLs that are impacted by the diffuse motor manifestations of PKAN. 14 , 15 The PKAN‐ADL was validated for use in patients aged 6 years and older. The secondary efficacy endpoint was the change in the UPDRS III total score from baseline to the end of the 24‐week double‐blind period. 16 The BAD scale was included as an exploratory endpoint to examine changes in dystonia from baseline to week 24. 17 Centralized training was provided to all investigators who were raters for the PKAN‐ADL, UPDRS III, and BAD scale, and videotaped assessments were reviewed to determine any need for retraining. Other exploratory measures included the Clinician Global Impression of Improvement, the Quality of Life in Neurological Disorders adult and pediatric versions, the EuroQoL 5‐dimension 3 level and youth versions, the Functional Independence Measure/Functional Independence Measure for Children, the 25‐foot walk test, and diadochokinetic speech assessments.
Study drug safety and tolerability were evaluated by examining treatment‐emergent adverse events (TEAEs), treatment‐emergent serious adverse events, serial vital signs, weight, physical examination findings, clinical laboratory parameters (chemistry, hematology, coagulation, and urinalysis), the Columbia Suicide Severity Rating Scale score (in assessable patients), and 12‐lead electrocardiograms. Pharmacokinetic assessments of fosmetpantotenate in whole blood were evaluated using a sparse sampling design.
Statistical Analyses
The study power analysis estimated that a total of 74 patients would provide approximately 80% power to detect a three‐point difference between treatment groups in the average change from baseline scores of the PKAN‐ADL, based on Student t test with α = 0.05 (two‐sided). The calculation assumed a standard deviation of 4.5 points for the change from baseline scores. Analysis of primary and secondary efficacy endpoints used the double‐blind full analysis set (all randomized patients with a baseline assessment and at least one postbaseline assessment of the primary efficacy endpoint). A mixed model for repeated‐measures analysis assessed data from all visits simultaneously. The model included fixed effects for treatment, age group, visit, and treatment‐by‐visit interaction, with visit as the repeating factor, patient as a random effect, and baseline total score as a covariate. Hypothesis testing was performed at the two‐sided 5% significance level using SAS Version 9.2. Safety and tolerability were examined using the double‐blind safety population (all patients who received at least one dose of double‐blind study medication). An independent Data Monitoring Committee reviewed safety data as the trial began, to determine whether the study could continue as planned, and continued to periodically review safety data throughout the study. 14 The trial was registered with ClinicalTrials.gov (NCT03041116) and with EudraCT (2016–001955‐29).
Results
The first patient was enrolled on July 23, 2017, and enrollment was completed on December 18, 2018. A total of 84 patients were enrolled: 41 patients were randomized to receive fosmetpantotenate and 43 were randomized to placebo (Fig. 1). All patients receiving fosmetpantotenate completed the double‐blind period, whereas six patients receiving placebo discontinued before the week 24 evaluation. Among these six patients, two patients died of PKAN‐related complications (aspiration during feeding and disease progression with respiratory failure, respectively), and four patients withdrew patient/guardian assent/consent (two discontinued because of the adverse event of worsened dystonia and two discontinued because of poor compliance related to the taste of the study drug).
FIG. 1.
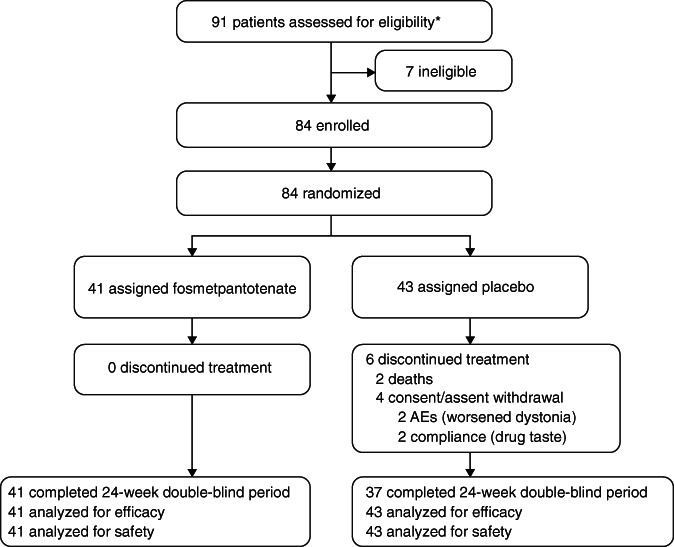
Patient disposition. *Including the two patients who were screened twice, the total number of screens was 93. Abbreviation: AE, adverse event.
Patient demographics (Table 1) and PKAN‐specific medical history at baseline (Table 2) were similar in the fosmetpantotenate and placebo groups. The groups were well balanced in the stratification factors of age and weight at baseline. Patients randomized to fosmetpantotenate versus placebo had comparable PKAN‐related clinical characteristics at baseline, including similar age at symptom onset and duration of disease. Problems in walking and speech were the most common presenting signs of PKAN. Dystonia, especially involving the mouth and foot, was also a common presenting sign of PKAN. Both groups reported a high burden of illness during the last year as documented by the numbers of medical and therapy visits (Table 2). Treatment compliance was confirmed in all patients receiving fosmetpantotenate and in 90.7% (39/43) of patients receiving placebo.
TABLE 1.
Patient demographics
| Fosmetpantotenate (n = 41) | Placebo (n = 43) | |
|---|---|---|
| Age, years, mean (SD) | ||
|
minimum maximum |
22.6 (10.6) 6, 58 |
23.1 (13.6) 6, 58 |
| Age group, n (%) | ||
| Pediatric | 14 (34.1) | 16 (37.2) |
| Adult | 27 (65.9) | 27 (62.8) |
| Sex, n (%) | ||
| Male | 21 (51.2) | 24 (55.8) |
| Female | 20 (48.8) | 19 (44.2) |
| Ethnicity a | ||
| Hispanic or Latino | 8 (19.5) | 2 (4.7) |
| Not Hispanic or Latino | 28 (68.3) | 36 (83.7) |
| Not reported | 5 (12.2) | 5 (11.6) |
| Racea | ||
| American Indian or Alaska Native | 0 | 0 |
| Asian | 1 (2.4) | 2 (4.7) |
| Black or African American | 0 | 4 (9.3) |
| Native Hawaiian or Other Pacific Islander | 0 | 0 |
| White | 25 (61.0) | 28 (65.1) |
| Multiple | 2 (4.9) | 0 |
| Other | 4 (9.8) | 3 (7.0) |
| Unknown | 6 (14.6) | 6 (14.0) |
| Missing | 3 (7.3) | 0 |
| Weight, kg, mean (SD) | 49.2 (18.9) | 49.0 (20.6) |
| Pediatric (n = 14 and 16) | 31.6 (13.0) | 30.8 (11.9) |
| Adult (n = 27 and 27) | 58.3 (14.5) | 59.7 (16.7) |
| BMI, kg/m2, mean (SD) b | 19.1 (4.0) | 19.6 (4.6) |
Race and ethnicity data were not available for up to 14.6% of patients, including country‐specific limitation on collection of these data within clinical trials.
Data are not available for one patient receiving fosmetpantotenate and two patients receiving placebo.
Abbreviations: SD, standard deviation; BMI, body mass index.
TABLE 2.
Patient clinical characteristics
| Fosmetpantotenate (n = 41) | Placebo (n = 43) | |
|---|---|---|
| Age at PKAN onset, years, mean (SD) | 7.8 (7.1) | 8.5 (6.6) |
| Duration of PKAN, years, mean (SD) | 14.8 (8.2) | 14.6 (10.5) |
| First PKAN‐related problem, n (%) | ||
| Walking | 28 (68.3) | 32 (74.4) |
| Speech | 29 (70.7) | 32 (74.4) |
| Swallowing | 11 (26.8) | 10 (23.3) |
| Writing | 18 (43.9) | 20 (46.5) |
| Emotional/behavioral problems | 11 (26.8) | 13 (30.2) |
| Vision | 9 (22.0) | 9 (20.9) |
| Dressing self | 10 (24.4) | 12 (27.9) |
| Coordination | 23 (56.1) | 24 (55.8) |
| Dystonia | 25 (61.0) | 33 (76.7) |
| Mouth/tongue | 16 (39.0) | 21 (48.8) |
| Neck | 13 (31.7) | 12 (27.9) |
| Hand | 15 (36.6) | 27 (62.8) |
| Foot | 19 (46.3) | 24 (55.8) |
| Back/trunk | 15 (36.6) | 15 (34.9) |
| Other dystonia | 2 (4.9) | 7 (16.3) |
| Other problem | 10 (24.4) | 12 (27.9) |
| Time from first symptom to diagnosis, years, mean (SD) | 5.8 (6.9) | 6.3 (5.4) |
| Age at first MRI, years, mean (SD) | 11.4 (6.7) | 14.0 (8.4) |
| Number of doctors seen before diagnosis, mean (SD) a | 3.9 (3.3) | 5.1 (4.4) |
| Number of medical visits in last year, mean (SD) b | 7.8 (6.5) | 9.3 (11.4) |
| Number of therapy visits in last year, mean (SD) c | 44.4 (83.8) | 57.5 (55.4) |
| Baseline PKAN‐ADL total score, mean (SD) | 28.2 (11.4) | 27.4 (11.5) |
| Baseline UPDRS Part III total score, mean (SD) | 45.3 (21.2) | 43.5 (21.1) |
| Baseline BAD total score, mean (SD) | 19.5 (7.8) | 17.2 (9.1) |
Data are not available for five patients receiving fosmetpantotenate and five patients receiving placebo.
Therapy visits (eg, physical, speech, and occupational therapies) data were not available for two patients receiving fosmetpantotenate and two patients receiving placebo.
Data are not available for four patients receiving fosmetpantotenate and five patients receiving placebo.
Abbreviations: PKAN‐ADL, Pantothenate Kinase–Associated Neurodegeneration‐Activities of Daily Living; SD, standard deviation; UPDRS, Unified Parkinson's Disease Rating Scale; BAD, Barry–Albright Dystonia.
Mean PKAN‐ADL total scores were similar between treatment groups at baseline, indicating a comparable degree of impaired daily functioning caused by PKAN. Mean PKAN‐ADL scores per visit stayed within the range of 23 to 28 in both treatment groups from baseline to week 24. After 24 weeks of treatment, fosmetpantotenate did not demonstrate statistically significant or clinically different effects on the primary efficacy endpoint PKAN‐ADL total score in the total patient group (Fig. 2A), the subgroup of pediatric patients (Fig. 2B), or the subgroup of adult patients (Fig. 2C). Thus, the primary efficacy endpoint was not met. The mean change from baseline at week 24 for both groups was a decrease (improvement) of −1.4 points, with a mean percent change from baseline of −6.9% in the fosmetpantotenate group and −6.6% in the placebo group (see Supporting Information Table S2 for mean baseline and week 24 scores for the PKAN‐ADL, UPDRS III, and BAD). Among pediatric patients, the mean change from baseline at week 24 was a decrease (improvement) of −0.6 point (mean percent change from baseline of −4.8%) in the fosmetpantotenate group and an increase (worsening) of 0.3 point (mean percent change from baseline of 4.1%) in the placebo group. In adults, the mean change from baseline at week 24 was a decrease (improvement) of −1.8 points (mean percent change from baseline of −8.0%) in the fosmetpantotenate group and a decrease of −2.5 points (mean percent change from baseline of −13.8%) in the placebo group. Similarly, there were no statistically significant or clinically meaningful differences between groups when examining subgroup analyses by time of diagnosis, age at onset of symptoms, severity of symptoms at baseline, and patient age at baseline.
FIG. 2.
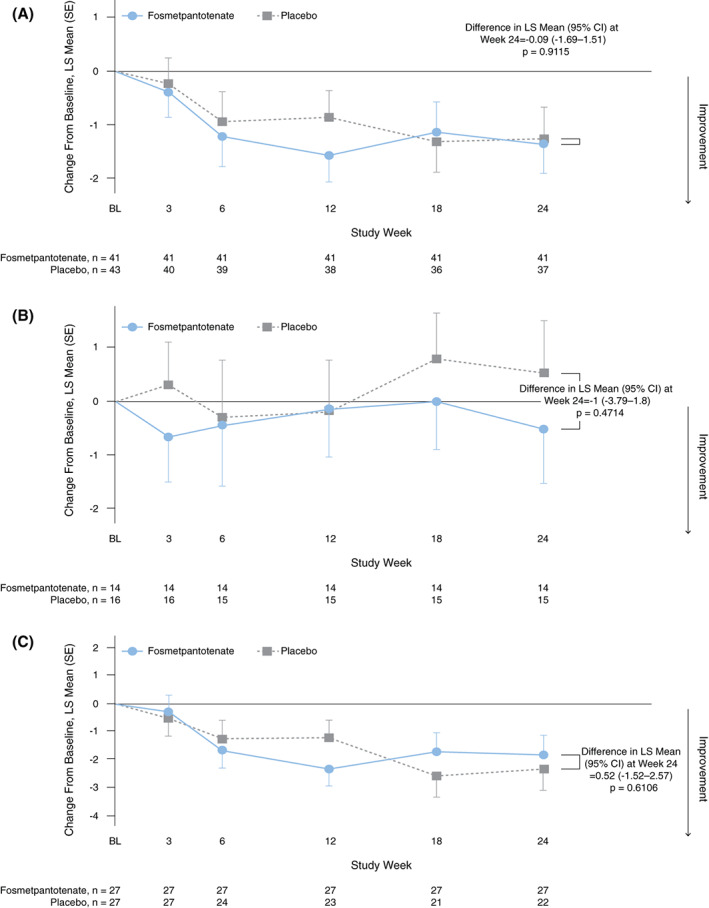
Pantothenate Kinase–Associated Neurodegeneration‐Activities of Daily Living (PKAN‐ADL) total score change from baseline at week 24 in all patients (A), pediatric patients (B), and adult patients (C). Primary endpoint analysis (double‐blind period full analysis set). BL, baseline; CI, confidence interval; LS, least square; SE, standard error. [Color figure can be viewed at wileyonlinelibrary.com]
The secondary efficacy endpoint analysis, comparing UPDRS III total score change from baseline to week 24 (Fig. 3A), and the exploratory analysis of the BAD scale total score change from baseline to week 24 (Fig. 3B) also found no significant difference or clinically different effect between the fosmetpantotenate and placebo groups. The mean change from baseline at week 24 in the UPDRS III total score for the fosmetpantotenate group was an increase (worsening) of 0.7 point (mean percent change from baseline of 7.0%) and for the placebo group was a decrease (improvement) of −1.0 point (mean percent change from baseline of −4.8%). In pediatric patients, the mean change from baseline at week 24 was an increase (worsening) of 0.7 point (mean percent change from baseline of 6.9%) in the fosmetpantotenate group and an increase of 1.6 points (mean percent change from baseline of 0.9%) in the placebo group. In adults, the mean change from baseline at week 24 was an increase (worsening) of 0.7 point (mean percent change from baseline of 7.0%) in the fosmetpantotenate group and a decrease (improvement) of −2.7 points (mean percent change from baseline of −8.7%) in the placebo group.
FIG. 3.
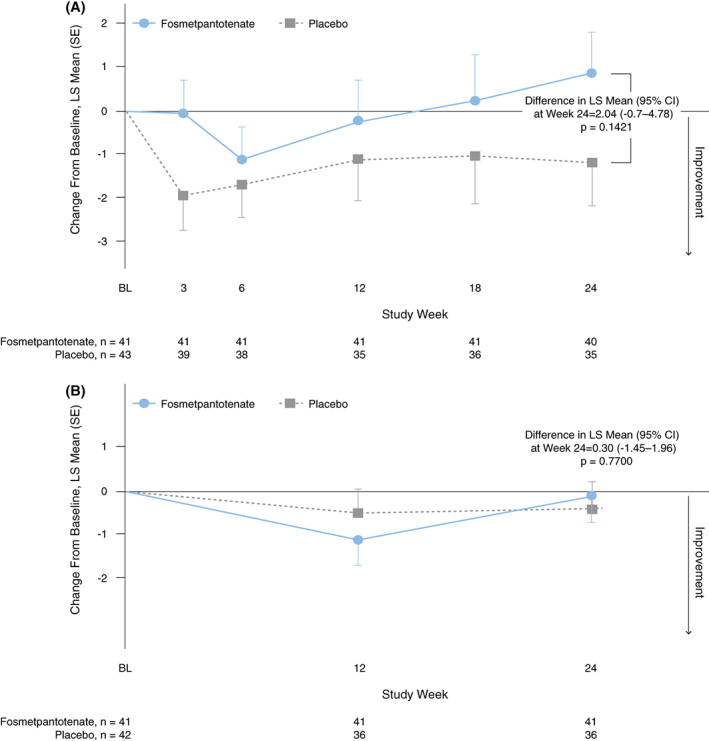
Unified Parkinson's Disease Rating Scale (UPDRS) Part III total score (A) and BAD total score (B) change from baseline at week 24. Secondary endpoint analysis (A) and exploratory analysis (B) (double‐blind period full analysis set, respectively). BL, baseline; CI, confidence interval; LS, least square; SE, standard error. [Color figure can be viewed at wileyonlinelibrary.com]
The mean change from baseline at week 24 in the BAD scale total score for both groups was a decrease (improvement) of −0.2 point (fosmetpantotenate group mean percent change from baseline of −1.4%; placebo group mean percent change from baseline of 10.8%). In pediatric patients, the mean change from baseline at week 24 was −0.0 points (mean percent change from baseline of 0.9%) in the fosmetpantotenate group and an increase (worsening) of 0.9 point (mean percent change from baseline of 2.7%) in the placebo group. In adults, the mean change from baseline at week 24 was a decrease (improvement) of −0.3 point (mean percent change from baseline of −2.6%) in the fosmetpantotenate group and a decrease of −1.0 point (mean percent change from baseline of 16.6%) in the placebo group.
Preplanned exploratory endpoint analysis of PKAN‐ADL domains found no significant differences between fosmetpantotenate and placebo groups in any domain (Supporting Information Fig. S1A). Similarly, there were no significant differences in the individual items of the UPDRS III (Supporting Information Fig. S1B).
There were no significant differences or clinically meaningful effects between the fosmetpantotenate and placebo groups in any of the exploratory measures (see Supporting Information Tables S3–S8).
Fosmetpantotenate appeared to be generally safe and well tolerated. No patients discontinued fosmetpantotenate permanently as a result of an adverse event or for any other reason. The overall incidence rates of treatment‐emergent serious adverse events (Supporting Information Table S9) were similar in the fosmetpantotenate (8 of 41 patients, 19.5%) and placebo (6 of 43 patients, 14.0%) groups, and the most common were dystonia (3.6%, 3 of 84 patients) and aspiration (2.4%, 2 of 84 patients) (also with a similar incidence in both groups). The most common TEAEs (≥10%) were dystonia (17.9%, 15 of 84 patients), vomiting (11.9%, 10 of 84 patients), diarrhea (10.7%, 9 of 84 patients), and pyrexia (10.7%, 9 of 84 patients), with a similar incidence in fosmetpantotenate and placebo groups. Dystonia TEAEs occurred in 9 (22.0%) patients receiving fosmetpantotenate and 10 (23.3%) patients receiving placebo. The 17 dystonia TEAEs experienced by patients receiving fosmetpantotenate had a median duration of 16.0 days, whereas the 16 dystonia TEAEs experienced by patients receiving placebo had a median duration of 32.5 days.
There was no evidence of fosmetpantotenate‐related safety concerns, including gastrointestinal, neurological, or liver enzyme elevation. Potentially clinically significant laboratory values for hematology, coagulation, and chemistry tests occurred in some patients within both the fosmetpantotenate and placebo groups (Supporting Information Table S10). Mean (standard deviation) body weight gain from baseline to week 24 was numerically greater in the fosmetpantotenate (1.0 [2.7] kg) versus the placebo (0.5 [2.3] kg) group, both in the pediatric patients (fosmetpantotenate, 1.4 [2.8] kg; placebo, 0.7 [1.7] kg) and in the adult patients (fosmetpantotenate, 0.9 [2.6] kg; placebo, 0.4 [2.7] kg).
Discussion
Fosmetpantotenate was designed as a prodrug that delivers phosphopantothenate to cells, with the aim to provide a substrate for CoA biosynthesis to bypass the underlying biochemical defect in PKAN. 11 Preclinical studies supported the ability of fosmetpantotenate to partially restore CoA; its ability to cross the blood–brain barrier was supported by fosmetpantotenate reaching striatal dialysate in monkeys after oral administration. 11 The FORT trial in patients with PKAN is among the largest clinical trials to date for this ultra‐rare neurodegenerative disease. The FORT trial did not demonstrate a difference between fosmetpantotenate and placebo for the primary or secondary efficacy endpoints. After 24 weeks of treatment with fosmetpantotenate, no difference was observed versus placebo in change from baseline on the patient‐ or surrogate‐reported PKAN‐ADL measure of patient function in daily activities. Similarly, no difference versus placebo was observed in change from baseline on the clinician‐reported UPDRS III measure of patient motor function. Fosmetpantotenate appeared to be safe and well tolerated. All patients receiving fosmetpantotenate continued treatment through the week 24 evaluation, and there were not more specific adverse events in patients receiving fosmetpantotenate versus placebo. Six patients in the placebo group did not complete the double‐blind period, including two patients who died as a result of PKAN‐related complications (aspiration while feeding and respiratory failure, respectively).
To date there has been one other randomized controlled trial with large enrolment of patients with PKAN that has examined a potentially disease‐modifying treatment, the trial that compared treatment with deferiprone, an iron chelating agent, versus placebo for up to 3 years. 10 A significant reduction in globus pallidus iron, assessed using MRI‐R2* imaging, occurred in patients receiving deferiprone, whereas patients receiving placebo showed no change. In general, patients receiving deferiprone showed less worsening on outcome measures over 18 months versus patients receiving placebo. Although statistical significance was not achieved for many group comparisons, the pattern of response across the secondary endpoints suggested a small improvement with deferiprone across wide‐ranging functions. Notably, the predefined subgroup analysis examining patients with atypical PKAN found a significantly slower progression of clinical symptoms (assessed using the BAD) in patients receiving deferiprone versus placebo. Challenges noted in the deferiprone trial included a slower rate of worsening in patients treated with placebo than had been anticipated based on the limited availability of natural history studies. 10
In the FORT trial, there was no difference between patients receiving fosmetpantotenate and placebo over 24 weeks in the BAD, an exploratory outcome. On average, there was minimal change in the BAD. Limitations of the BAD as an outcome measure include its low sensitivity to change, with a relatively large smallest detectable difference of 17.72%, which may limit its ability to detect smaller clinical improvements in treatment trials, as well as poor reliability in assessment of eyes, mouth, and neck regions. 18
Therapies that target the underlying biochemical defect in PKAN and cross both cell membranes and the blood–brain barrier to affect the central nervous system are needed to slow disease progression and to stabilize, or possibly improve, patient function. One compound in early development, PZ‐2891, has demonstrated these characteristics in a mouse model of PKAN. 19 , 20 PZ‐2891, a pantazine molecule, is an allosteric activator of PanK1 and PanK3 that targets augmentation of CoA levels through activation of these alternative isoforms of the Pank enzyme family. 19 Oral administration in the PKAN mouse model resulted in increased brain CoA and improvement in locomotor, growth, and lifespan outcomes. 19 , 20 Another compound in early development, 4′‐phosphopantetheine, corrected CoA‐associated defects and secondary abnormalities when orally administered in a mouse model of PKAN, with CoA, iron, and dopamine metabolic defects. 21 Treatment with 4′‐phosphopantetheine also rescued human primary PKAN fibroblasts. Compared with fosmetpantotenate, 4′‐phosphopantetheine targets CoA biosynthesis further downstream along the cascade, at the point where 4′‐phosphopantetheine is endogenously produced. 21
Heterogeneity of PKAN severity of clinical signs and symptoms, as well as varying rates of disease progression, follows from the wide range of PANK2 pathological genetic variants that continue to be identified. 22 Certain types of genetic variants in the PANK2 gene are proposed to lead to reductions in CoA biosynthesis. 23 , 24 Complete loss of function of the PANK2 gene protein is presumed to lead to classic PKAN, and partial loss is presumed to lead to atypical PKAN. 2 , 25 , 26 , 27 Such heterogeneity among patients within this ultra‐rare disease presents challenges for the conduct of clinical trials examining treatment outcomes. Greater understanding of genotype‐to‐phenotype associations and the natural history of disease progression is needed. The wide‐ranging heterogeneity of the PKAN phenotype and the relatively short duration of treatment within a clinical trial are possible reasons underlying the failed FORT trial examining fosmetpantotenate for treatment of PKAN; however, subanalyses and exploratory outcomes did not show any trend or evidence in support of fosmetpantotenate efficacy. Exploratory subgroup analyses by time of diagnosis, age at onset of symptoms, severity of symptoms at baseline, and patient age at baseline were used to approximate groups of patients with possible differences in their underlying PANK2 pathological genetic variants and showed no support for fosmetpantotenate efficacy. The absence of any support of efficacy while using higher fosmetpantotenate dosing versus that received by the three international compassionate use patients (from 120 to 210 mg/day) who showed clinical improvements in motor behaviors within 8 weeks of treatment start 12 , 13 indicate that a higher dose or longer treatment period would not result in clinical benefit.
Another potential explanation is the lack of adequate brain target engagement because of the inability of fosmetpantotenate to sufficiently cross the blood–brain barrier. No appropriate measure of brain target engagement was available during this study. Thus, within the context of understanding this negative trial outcome, it is not possible to know whether any brain target engagement occurred. Identification of a biomarker for PKAN‐related target engagement in the brain to directly assess blood–brain barrier penetration efficiency remains an important unmet need.
Consideration of several elements of the FORT study design may help inform future clinical trials. A 6‐month double‐blind study duration may not be long enough to show significant changes in currently available dystonia and motor function measures, such as the BAD and UPDRS. The deferiprone trial did not find a significant difference in BAD scores for all patients at 18 months, but did find significant improvement at 18 months in patients with the atypical PKAN phenotype. 10 Using the Burke–Fahn and Marsden Dystonia Rating Scale, another deferiprone study found no difference in dystonia outcome at 6 months of treatment. 28 These studies underscore the need for a better understanding of the natural history of PKAN progression and suggest motor function may worsen more slowly than projected. 10 In part this could be related to study exclusion criteria. In the FORT trial, patients who were unable to remain on their prestudy dose of concomitant PKAN medications or prestudy deep brain stimulation settings during the double‐blind period were excluded. Thus, patients with a rapidly evolving disease, as with the classic phenotype of PKAN, could not participate in the study. The patients who were included in the study may have had a less aggressive phenotype with the need of a longer study duration to see significant changes in disease progression. In the FORT trial, eligible patients were at least 6 years old, and it is likely that some patients with a severe classic phenotype at baseline already had severe neurodegenerative lesions that could prevent possible improvement with a metabolic therapeutic approach. For patients with a more severe classic phenotype, intervention at the time of symptom onset in earlier stages of the disease may be needed for meaningful treatment benefit. In addition, more sensitive assessment measures may need to be developed to capture the PKAN‐specific changes in movement and dystonia that indicate improved function for patients, especially over a shorter study duration. Although the PKAN‐ADL was developed as a disease‐specific measure with PKAN‐relevant items, whether the PKAN‐ADL is sensitive to small changes over time has not yet been shown. In addition, because of the focus on deficits in motor function related to daily living, the behaviors that are measured in the PKAN‐ADL may be similar to motor functions that are still developing in young children, and the ability to communicate such differences is limited in very young children. For these reasons, children younger than 6 years were not included in the PKAN‐ADL validation study, and this may limit the use of the PKAN‐ADL with interventions aimed at earlier stages of classic disease onset.
In summary, the FORT trial did not find treatment benefit in daily activities or motor function with fosmetpantotenate treatment compared with placebo in patients with PKAN. Therapies that prevent the progression of PKAN remain urgently needed.
Author Roles
Research Project: Conception, Thomas Klopstock and Feriandas Greblikas; Organization, Thomas Klopstock and Feriandas Greblikas; Execution: Thomas Klopstock, Aleksandar Videnovic, Almut Turid Bischoff, Cecilia Bonnet, Laura Cif, Cynthia Comella, Marta Correa‐Vela, Maria L. Escolar, Jamie L. Fraser, Victoria Gonzalez, Neal Hermanowicz, Robert Jech, Hyder A. Jinnah, Tomasz Kmiec, Anthony Lang, Maria J. Martí, Saadet Mercimek‐Andrews, Migvis Monduy, Graeme A. M. Nimmo, Belen Perez‐Dueñas, Helle Cecilie Viekilde Pfeiffer, Lluis Planellas, Emmanuel Roze, Nivedita Thakur, Laura Tochen, Nora Vanegas‐Arroyave, Giovanna Zorzi, Colleen Burns, Feriandas Greblikas
Statistical Analysis: Design: Thomas Klopstock, Feriandas Greblikas, Colleen Burns. Execution: Colleen Burns. Review and Critique: all authors.
Manuscript: Writing of the First Draft: all authors. Review and Critique: all authors.
Thomas Klopstock and Feriandas Greblikas conceived and designed the work that led to the submission. With the exceptions of Colleen Burns and Feriandas Greblikas, all authors executed the study and acquired data. Colleen Burns performed statistical analyses. All authors had access to and interpreted the data. All authors reviewed and revised drafts of the manuscript and approved the final version for submission.
Financial Disclosures (for Preceding 12 Months)
Thomas Klopstock serves as coordinating investigator of the FORT trial; receives research funding from Retrophin, Inc.; serves as coordinating investigator of the deferiprone in PKAN randomized and extension trial; receives research funding from ApoPharma Inc.; receives support from the European Commission 7th Framework Programme (FP7/2007–2013, HEALTH‐F2‐2011; grant agreement no. 277984, TIRCON) and from the European Reference Network for Rare Neurological Diseases (ERN‐RND), co‐funded by the European Commission (ERN‐RND: 3HP 767,231); provides consulting services to CoA Therapeutics, TM3 Therapeutics, and Retrophin, Inc.; receives travel support from ApoPharma Inc.
Aleksandar Videnovic provides consulting services to Retrophin, Inc., and receives active grant support from the National Institutes of Health (NIH); provides consulting services to Acadia, Jazz, and Roche; and has served on DSMB for Wilson Therapeutics and Acorda.
Almut Turid Bischoff has no conflict of interest.
Cecilia Bonnet has no conflict of interest.
Laura Cif has received honoraria for lectures from Boston Scientific, International Parkinson's Disease and Movement Disorders Society, and Medtronic, Inc.
Cynthia Comella serves on the editorial board of Clinical Neuropharmacology and Sleep Medicine; received compensation/honoraria for services as a consultant or an advisory committee member from Acorda Therapeutics, Allergan, Inc.; Lundbeck Ltd.; Merz Pharmaceuticals; Acadia Pharmaceuticals; Ipsen Pharmaceuticals, Jazz Pharmaceuticals, Neurocrine Biosciences Inc., Revance Therapeutic, Sunovion, and AEON Biopharma; and receives royalties from Cambridge, Wolters Kluwer.
Marta Correa‐Vela has no conflict of interest.
Maria L. Escolar provides consulting services to Retrophin, Inc.
Jamie L. Fraser received research support for the study site supervision and administration of FORT trial from Retrophin, Inc.
Victoria Gonzalez provided consulting services in the context of an Advisory Board organized by Retrophin, Inc., and received travel grant from Allergan Inc.
Neal Hermanowicz has received honoraria for lectures, advisory boards, and consulting from Abbott Laboratories, Acadia Pharmaceuticals, Acorda Therapeutics, Adamas Pharmaceuticals, Inc., Allergan Inc., Amneal Pharmaceuticals, and Kyowa Kirin.
Robert Jech received honoraria for lectures or consultancy support from Medtronic Czechia, Abbvie, Ipsen Pharma, Desitin, and Cardion.
HyderA. Jinnah provides consulting services to Retrophin, Inc.; has received active or recent grant support from the US government (NIH), academically oriented institutions (the Dystonia Study Group), and industry (Cavion Therapeutics, Ipsen Pharmaceuticals, Retrophin, Inc.); serves on advisory boards or as a consultant for Allergan Inc., CoA Therapeutics, Psyadon Pharmaceuticals, and Medtronic, Inc.; has received honoraria or stipends for lectures or administrative work from the American Academy of Neurology, Dystonia Medical Research Foundation, International Neurotoxin Society, International Parkinson's Disease and Movement Disorders Society, Parkinson's Disease Foundation; serves on scientific advisory boards for Cure Dystonia Now, Dystonia Medical Research Foundation, Tourette Association of America, and Tyler's Hope for a Cure; and serves as principal investigator for the Dystonia Coalition, which receives the majority of its support through NIH grants from the NINDS and the Office of Rare Diseases Research at the National Center for Advancing Translational Sciences. The Dystonia Coalition has received additional material or administrative support from industry sponsors (Allergan, Inc. and Merz Pharmaceuticals) and from private foundations (American Dystonia Society, Beat Dystonia, Benign Essential Blepharospasm Foundation, Cure Dystonia Now, Dystonia Inc., Dystonia Ireland, Dystonia Medical Research Foundation, European Dystonia Federation, Foundation for Dystonia Research, National Spasmodic Dysphonia Association, and National Spasmodic Torticollis Association).
Tomasz Kmiec has no conflict of interest.
Anthony Lang reports consultancy support from Abbvie, AFFiRis, Biogen, Janssen, Lilly, Lundbeck, Merck, Paladin, Roche, Sun Pharma, Theravance, and Corticobasal Degeneration Solutions; advisory board support form Jazz Pharma, PhotoPharmics, Sunovion; other honoraria from Sun Pharma, AbbVie, Sunovion, American Academy of Neurology, and the International Parkinson and Movement Disorder Society; grants from Brain Canada, Canadian Institutes of Health Research, Corticobasal Degeneration Solutions, Edmond J Safra Philanthropic Foundation, Michael J. Fox Foundation, the Ontario Brain Institute, Parkinson Foundation, Parkinson Canada, and W. Garfield Weston Foundation, and royalties from Elsevier, Saunders, Wiley‐Blackwell, Johns Hopkins Press, and Cambridge University Press.
Maria J. Martí has received honoraria for lectures from Allergan Inc. and Merz Pharmaceutical, and grants from Fundacio la Marato TV3, Fondo de Investigación Sanitaria‐ISCIII, and Michael J. Fox Foundation.
Saadet Mercimek‐Andrews received research support for the study site supervision and administration of the FORT trial from Retrophin, Inc.; received honoraria for lectures and advisory board for epilepsy genetics from Biomarin Pharmaceutical; received investigator‐initiated funding from Genzyme; and received honoraria from Recordati Rare Diseases for organizing a workshop and being an advisory board member.
Migvis Monduy provides consulting services to Retrophin, Inc., and serves on advisory boards or receives honoraria for lectures from Ipsen, PTC Therapeutics, Sarepta Pharmaceuticals, Avexis, and Biogen.
Graeme A. M. Nimmo provides consulting services to Shire/Takeda and has received honoraria for lectures from Shire/Takeda and Pfizer.
Belen Perez‐Dueñas receives research grant support from Retrophin, Inc., to Hospital Universitari Vall d'Hebron, Barcelona, Spain.
Helle Cecilie Viekilde Pfeiffer reports advisory board support from Octapharma AG and has received grants from the Danish Parkinson's Disease Association and the Norwegian Research Council, as well as the South‐Eastern Norway Regional Health Authority Research Trust.
Lluis Planellas has no conflict of interest.
Emmanuel Roze served on scientific advisory boards for Orkyn, Aguettant, and Merz‐Pharma; received honoraria for speeches from Orkyn, Aguettant, Merz‐Pharma, Everpharma, International Parkinson and Movement Disorder Society; received research support from Merz‐Pharma, Orkyn, Aguettant, Elivie, Ipsen, Everpharma, Fondation Desmarest, AMADYS, Fonds de Dotation Brou de Laurière, and Agence Nationale de la Recherche; and received travel grants from Vitalair, PEPS development, Aguettant, Merz‐Pharma, Ipsen, Merck, Orkyn, Elivie, Adelia Medical, Dystonia Medical Research Foundation, International Parkinson and Movement Disorder Society, European Academy of Neurology, and International Association of Parkinsonism and Related Disorders.
Nivedita Thakur provides consulting services to Retrophin, Inc. and receives grant support from Retrophin, Inc.
Laura Tochen has no conflict of interest.
Nora Vanegas‐Arroyave received consulting honoraria from Neurocrine and research support from the NIH and the Parkinson's Disease Foundation.
Giovanna Zorzi receives grant support from Retrophin, Inc., and provides consulting services to Medtronic, Inc.
Colleen Burns is an employee of Retrophin, Inc., and may have an equity or other financial interest in Retrophin, Inc.
Feriandas Greblikas is an employee of Retrophin, Inc., and may have an equity or other financial interest in Retrophin, Inc.
Financial Disclosures
Thomas Klopstock serves as coordinating investigator of the FORT trial and receives research funding from Retrophin, Inc.
Aleksandar Videnovic provides consulting services to Retrophin, Inc.
Almut Turid Bischoff has no conflict of interest concerning the research related to the manuscript.
Cecilia Bonnet has no conflict of interest concerning the research related to the manuscript.
Laura Cif has no conflict of interest concerning the research related to the manuscript.
Cynthia Comella has no conflict of interest concerning the research related to the manuscript.
Marta Correa‐Vela has no conflict of interest concerning the research related to the manuscript.
Maria L. Escolar provides consulting services to Retrophin, Inc.
Jamie L. Fraser received research support for the study site supervision and administration of FORT trial from Retrophin, Inc.
Victoria Gonzalez provided consulting services in the context of an Advisory Board organized by Retrophin, Inc.
Neal Hermanowicz has no conflict of interest concerning the research related to the manuscript.
Robert Jech has no conflict of interest concerning the research related to the manuscript.
H. A. Jinnah provides consulting services to Retrophin, Inc.
Tomasz Kmiec has no conflict of interest concerning the research related to the manuscript.
Anthony Lang has no conflict of interest concerning the research related to the manuscript.
Maria J. Martí has no conflict of interest concerning the research related to the manuscript.
Saadet Mercimek‐Andrews received research support for the study site supervision and administration of the FORT trial from Retrophin, Inc.
Migvis Monduy provides consulting services to Retrophin, Inc.
Graeme A. M. Nimmo has no conflict of interest concerning the research related to the manuscript.
Belen Perez‐Dueñas receives research grant support from Retrophin, Inc.
Helle Cecilie Viekilde Pfeiffer has no conflict of interest concerning the research related to the manuscript.
Lluis Planellas has no conflict of interest concerning the research related to the manuscript.
Emmanuel Roze has no conflict of interest concerning the research related to the manuscript.
Nivedita Thakur provides consulting services to Retrophin, Inc., and receives grant support from Retrophin, Inc.
Laura Tochen has no conflict of interest concerning the research related to the manuscript.
Nora Vanegas‐Arroyave has no conflict of interest concerning the research related to the manuscript.
Giovanna Zorzi receives grant support from Retrophin, Inc.
Colleen Burns is an employee of Retrophin, Inc., and may have an equity or other financial interest in Retrophin, Inc.
Feriandas Greblikas is an employee of Retrophin, Inc., and may have an equity or other financial interest in Retrophin, Inc.
Supporting information
Appendix S1: Supporting Information
Acknowledgments
We thank all the patients and their families for their participation in the FORT pivotal trial. At Friedrich Baur Institute, Department of Neurology, University Hospital, LMU Munich, Germany, we thank Dr. Boriana Büchner for administrative support, Dr. Ivan Karin and Dr. Florentine Radelfahr for clinical support, and Ira Brandstetter for technical support. The FORT trial was supported by Retrophin, Inc. Writing and editorial support was provided by Lynanne McGuire, PhD, CMPP, of MedVal Scientific Information Services, LLC (Princeton, NJ, USA), and was funded by Retrophin, Inc.
Relevant conflicts of interest/financial disclosures: T. Klopstock serves as coordinating investigator of the FORT trial and receives research funding from Retrophin, Inc. A.V. provides consulting services to Retrophin, Inc. M.L.E. provides consulting services to Retrophin, Inc. J.L.F. received research support for the study site supervision and administration of FORT trial from Retrophin, Inc. V.G. provided consulting services in the context of an Advisory Board organized by Retrophin, Inc. H.A.J. provides consulting services to Retrophin, Inc. S.M.‐A. received research support for the study site supervision and administration of the FORT trial from Retrophin, Inc. M.M. provides consulting services to Retrophin, Inc. B.P.‐D. receives research grant support from Retrophin, Inc. N.T. provides consulting services to Retrophin, Inc., and receives grant support from Retrophin, Inc. G.Z. receives grant support from Retrophin, Inc. C. Burns is an employee of Retrophin, Inc., and may have an equity or other financial interest in Retrophin, Inc. F.G. is an employee of Retrophin, Inc., and may have an equity or other financial interest in Retrophin, Inc. A.T.B., C. Bonnet, L.C., C.C., M.C.‐V., N.H., R.J., T. Kmiec, A.L., M.J.M., G.A.M.N., H.C.V.P., L.P., E.R., L.T., and N.V.‐A. have no conflict of interest concerning the research related to the manuscript.
Funding agencies: The FORT trial was supported by Retrophin, Inc.
Full financial disclosures and author roles may be found in the online version of this article.
References
- 1. Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden‐Spatz syndrome. Nat Genet 2001;28(4):345–349. [DOI] [PubMed] [Google Scholar]
- 2. Hayflick SJ, Kurian MA, Hogarth P. Neurodegeneration with brain iron accumulation. In: Geschwind DH, Paulson HL, Klein C, eds. Handb Clin Neurol. Amsterdam, The Netherlands: Elsevier BV; 2018:293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marshall RD, Collins A, Escolar ML, et al. Diagnostic and clinical experience of patients with pantothenate kinase‐associated neurodegeneration. Orphanet J Rare Dis 2019;14(1):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hartig MB, Hortnagel K, Garavaglia B, et al. Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation. Ann Neurol 2006;59(2):248–256. [DOI] [PubMed] [Google Scholar]
- 5. Leonardi R, Zhang YM, Rock CO, Jackowski S. Coenzyme a: back in action. Prog Lipid Res 2005;44(2–3):125–153. [DOI] [PubMed] [Google Scholar]
- 6. Di Meo I, Carecchio M, Tiranti V. Inborn errors of coenzyme a metabolism and neurodegeneration. J Inherit Metab Dis 2019;42(1):49–56. [DOI] [PubMed] [Google Scholar]
- 7. Hortnagel K, Prokisch H, Meitinger T. An isoform of hPANK2, deficient in pantothenate kinase‐associated neurodegeneration, localizes to mitochondria. Hum Mol Genet 2003;12(3):321–327. [DOI] [PubMed] [Google Scholar]
- 8. Kotzbauer PT, Truax AC, Trojanowski JQ, Lee VM. Altered neuronal mitochondrial coenzyme a synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2. J Neurosci 2005;25(3):689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hogarth P, Kurian MA, Gregory A, et al. Consensus clinical management guideline for pantothenate kinase‐associated neurodegeneration (PKAN). Mol Genet Metab 2017;120(3):278–287. [DOI] [PubMed] [Google Scholar]
- 10. Klopstock T, Tricta F, Neumayr L, et al. Safety and efficacy of deferiprone for pantothenate kinase‐associated neurodegeneration: a randomised, double‐blind, controlled trial and an open‐label extension study. Lancet Neurol 2019;18(7):631–642. [DOI] [PubMed] [Google Scholar]
- 11. Elbaum D, Beconi MG, Monteagudo E, et al. Fosmetpantotenate (RE‐024), a phosphopantothenate replacement therapy for pantothenate kinase‐associated neurodegeneration: mechanism of action and efficacy in nonclinical models. PLoS One 2018;13(3):e0192028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christou YP, Tanteles GA, Kkolou E, et al. Open‐label fosmetpantotenate, a phosphopantothenate replacement therapy in a single patient with atypical PKAN. Case Rep Neurol Med 2017;2017:3247034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roa P, Stoeter P, Perez‐Then E, Santana M, Marshall RD. A pilot study of a potential phosphopantothenate replacement therapy in 2 patients with pantothenate kinase‐associated neurodegeneration. Int J Rare Dis Orphan Drugs 2017;2(2):1006. [Google Scholar]
- 14. Klopstock T, Escolar ML, Marshall RD, et al. The FOsmetpantotenate replacement therapy (FORT) randomized, double‐blind, placebo‐controlled pivotal trial: study design and development methodology of a novel primary efficacy outcome in patients with pantothenate kinase‐associated neurodegeneration. Clin Trials 2019;16(4):410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marshall RD, Collins A, Escolar ML, et al. A scale to assess activities of daily living in pantothenate kinase‐associated neurodegeneration. Mov Disord Clin Pract 2019;6(2):139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goetz CG, Tilley BC, Shaftman SR, et al. Movement Disorder Society‐sponsored revision of the unified Parkinson's disease rating scale (MDS‐UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008;23(15):2129–2170. [DOI] [PubMed] [Google Scholar]
- 17. Barry MJ, VanSwearingen JM, Albright AL. Reliability and responsiveness of the Barry‐Albright dystonia scale. Dev Med Child Neurol 1999;41(6):404–411. [DOI] [PubMed] [Google Scholar]
- 18. Monbaliu E, Ortibus E, Roelens F, et al. Rating scales for dystonia in cerebral palsy: reliability and validity. Dev Med Child Neurol 2010;52(6):570–575. [DOI] [PubMed] [Google Scholar]
- 19. Sharma LK, Subramanian C, Yun M‐K, et al. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat Commun 2018;9(1):4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jackowski S. Proposed therapies for pantothenate‐kinase‐associated neurodegeneration. J Exp Neurosci 2019;13:1179069519851118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jeong SY, Hogarth P, Placzek A, et al. 4'‐Phosphopantetheine corrects CoA, iron, and dopamine metabolic defects in mammalian models of PKAN. EMBO Mol Med 2019;11(12):e10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hartig MB, Prokisch H, Meitinger T, Klopstock T. Pantothenate kinase‐associated neurodegeneration. Curr Drug Targets EMBO Mol Med 2019;11(12):e10489. [DOI] [PubMed] [Google Scholar]
- 23. Meyer E, Kurian MA, Hayflick SJ. Neurodegeneration with brain iron accumulation: genetic diversity and pathophysiological mechanisms. Annu Rev Genomics Hum Genet 2015;16:257–279. [DOI] [PubMed] [Google Scholar]
- 24. Venco P, Dusi S, Valletta L, Tiranti V. Alteration of the coenzyme a biosynthetic pathway in neurodegeneration with brain iron accumulation syndromes. Biochem Soc Trans 2014;42(4):1069–1074. [DOI] [PubMed] [Google Scholar]
- 25. Hayflick SJ. Defective pantothenate metabolism and neurodegeneration. Biochem Soc Trans 2014;42(4):1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hayflick SJ, Westaway SK, Levinson B, et al. Genetic, clinical, and radiographic delineation of Hallervorden‐Spatz syndrome. N Engl J Med 2003;348(1):33–40. [DOI] [PubMed] [Google Scholar]
- 27. Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet 2009;46(2):73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zorzi G, Zibordi F, Chiapparini L, et al. Iron‐related MRI images in patients with pantothenate kinase‐associated neurodegeneration (PKAN) treated with deferiprone: results of a phase II pilot trial. Mov Disord 2011;26(9):1756–1759. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information