

Abstract
Tuberculosis (TB) in wildlife challenges epidemiological surveillance and disease control. An outbreak of TB was detected in a free‐ranging wild boar population of a Natural Park in Catalonia (Spain) and the outbreak investigation was conducted in the area. During the study period (2015–2020), 278 wild boars were analysed by gross pathology, histopathology, mycobacterial culture and DVR‐spoligotyping. In addition, all cattle (49) and goat (47) herds of the area were tested with tuberculin skin test. TB compatible lesions were detected in 21 wild boars, and Mycobacterium caprae was isolated in 17 of them with two different spoligotypes: SB0415 (13) and SB1908 (4). Only two goat herds showed TB positive animals that were subsequently slaughtered. M. caprae with the spoligotypes SB0416 and SB0415 were isolated from these animals. To investigate the phylogenetic relationships and the transmission chain of the outbreak, nine strains isolated from six wild boars and three goats of the study area were analysed by whole genome sequencing (WGS) followed by single nucleotide polymorphism (SNP) analysis by maximum likelihood and median‐joining network inference methods. Results indicated that infected wild boars maintained M. caprae strains circulation in their own population and have likely transmitted the infection to goats, thus acting as TB reservoirs, compromising the success of livestock TB eradication campaigns and posing a risk for public health. The results also highlighted the usefulness of WGS followed by SNP analysis in providing relevant epidemiological information when detailed contact data are missing.
Keywords: epidemiology, goats, Mycobacterium caprae, tuberculosis, whole genome sequencing, wild boar
1. INTRODUCTION
Tuberculosis (TB) is a chronic infectious disease caused by Mycobacterium tuberculosis complex (MTBC) that affects a wide range of mammals, including humans, livestock and wildlife species. In particular, the control of zoonotic TB has been identified as a major threat to achieve the goal of human TB eradication (World Health Organization, 2018). The eradication of TB in cattle represents one of the leading expenditures on veterinary programmes co‐funded by the European Union, although its complete eradication has not been achieved yet in many member states (European Commission, 2020).
Despite many efforts have been focused on bovine TB eradication programmes, the epidemiological surveillance of MTBC circulation in a non‐bovine wildlife–livestock interface is rarely addressed in a comprehensive way. It is well‐known that both wild boars and goats are reservoirs of MTBC in certain multi‐host contexts, hindering the eradication of bovine TB (Guta et al., 2014; Napp et al., 2013; Naranjo, Gortazar, Vicente, & de la Fuente, 2008). Goats are particularly susceptible to TB infection (Crawshaw et al., 2008; Daniel et al., 2009; Pérez de Val et al., 2011) and caprine TB is rapidly spread from infected to susceptible animals within positive herds (Vidal et al., 2017), being a risk of infection for livestock (Cano‐Terriza et al., 2018; Napp et al., 2013; Vidal et al., 2018) and for humans (Rodríguez et al., 2009). Mycobacterium caprae, a member of MTBC (Aranaz, Cousins, Mateos, & Domínguez, 2003), is the main causative agent of goat TB in Spain (Rodríguez et al., 2011). On the other hand, infected wild boars cause an important fraction of new bovine TB breakdowns in Spain (Guta et al., 2014; Hermoso de Mendoza et al., 2006).
Molecular tools to discriminate MTBC strains, such as DVR‐Spoligotyping (Kamerbeek et al., 1997) and multilocus variable number of tandem repeat analysis (MIRU‐VNTR), have been used worldwide as ancillary tools for animal TB epidemiological surveillance and outbreak investigations in multi‐host scenarios (Boniotti et al., 2009; Hauer et al., 2015; Rodriguez‐Campos, Gonzalez, et al., 2012). However, these techniques have a rather restricted discriminatory capacity since they analyse <1% of MTBC genome (Hauer et al., 2019). Thus, they show limitations to detect genomic divergences of MTBC strains belonging to the same regional clonal complex, sometimes making it difficult to trace the origin of a given outbreak (Hauer et al., 2016; Rodriguez‐Campos, Schürch, et al., 2012). Alternatively, the use of whole genome sequencing (WGS) has been extended in the last decade due to its higher discriminatory power to detect local transmission events missed by classical epidemiological investigations (Biek et al., 2012).
The aim of this study was to use WGS and epidemiological information to investigate the relationships of cattle and goat herds with free‐ranging wild boars in a TB outbreak area and gain insight into potential transmission patterns.
2. MATERIALS AND METHODS
2.1. Context, study area and period
In December 2015, a wild boar with TB suspicious lesions in mediastinal, tracheobronchial and pre‐scapular lymph nodes (Ln) of the Montseny Natural Park (Catalonia, Spain) was detected during inspection in a game meat processing facility. Samples were submitted to the laboratory of IRTA‐CReSA through the Catalan slaughterhouse support network—SESC (www.cresa.cat/blogs/sesc) for diagnostic investigation (Vidal et al., 2016). Histopathology showed TB compatible lesions (i.e. chronic granulomatous lymphadenitis) and M. caprae was isolated. DVR‐spoligotyping was performed according to a previously described method (Kamerbeek et al., 1997) and the spoligotype profile SB0415 was identified (www.Mbovis.org) (Figure 1).
Figure 1.
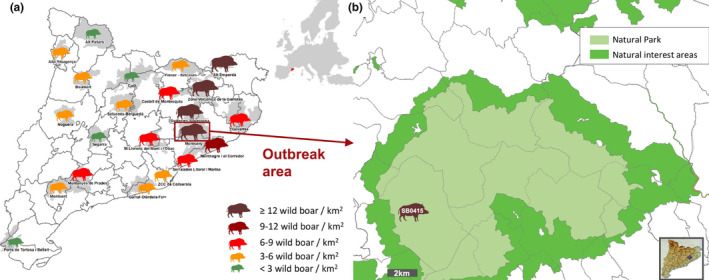
Study area. (a) Map of Catalonia (North‐eastern Spain) that shows the local densities of wild boars estimated for the hunting season 2018–19 (DARP 2019). The brown rectangle shows the outbreak area (14 wild boar/km2). (b) The studied outbreak area includes the Montseny Natural Park (light green) and surrounding areas. The different municipalities are delimited by dotted lines. The silhouette shows the approximate localization of the first detected wild boar with TB lesions and Mycobacterium caprae isolation (the spoligotype profile is indicated within the silhouette) [Colour figure can be viewed at wileyonlinelibrary.com]
After confirmation of this case, the TB outbreak investigation was performed in the affected area in both wild boar and livestock. The study area (SA) was defined as the whole Natural Park and the surrounding area (Figure 1). This is a woodland area with coastal Mediterranean climate with a high number of livestock farms, well‐preserved wildlife habitats and without artificial fencing. Free‐ranging wild boars are particularly abundant in this area, with an estimated density over 12 individuals/sq. km in a surface of 47,840 ha (Rosell, Pericas, Colomer, & Navàs, 2019).
2.2. Cattle and goat herds testing and sampling
Investigation of all cattle and goat herds of the SA (49 and 47, respectively) was carried out by the Official Veterinary Services through tuberculin skin testing (TST) between 2017 and 2018. TST was performed following the procedures and interpretations as prescribed guidelines of the Spanish Ministry of Agriculture, Fisheries and Food (Bezos, Romero, de Juan, Gonzalez, & Sáez, 2019). All positive animals were slaughtered and retropharyngeal, tracheobronchial and mediastinal Ln or other tissues with visible TB compatible lesions were collected for further pathological and bacteriological confirmation.
2.3. Wild boars inspection and sampling
Intensified sampling and subsequent analysis of hunted wild boars of the SA were conducted during five consecutive hunting seasons (i.e. from September to March, between 2015 and 2020). The main source of tissue samples was game meat processing facilities either through active sampling conducted by the specialized personnel (official veterinarians, game meat processing facility inspectors and forest guard officers) or through the submission of TB suspicious lesions using the SESC platform by meat inspectors (i.e. passive surveillance). Analyses were performed at IRTA‐CReSA laboratory. During the study period, 278 wild boars were sampled (mainly head and tracheobronchial Ln), but also other tissues when TB compatible lesions were observed (e.g. lungs or mesenteric Ln).
2.4. Pathological examination
All tissues were analysed by gross lesion evaluation performed by sectioning the sampled tissues 2–3 mm thick slices under appropriate light conditions in a biosafety cabinet. In case of a positive or non‐conclusive result, histopathology (haematoxylin–eosin staining) and Ziehl–Neelsen stain were carried out to confirm the diagnosis, and a fraction of the lesion was frozen for further mycobacterial culture. A lesion was considered compatible with TB when granulomatous inflammatory infiltrate with variable areas of necrosis and mineralization was observed. The presence of Langhan's cells and/or acid fast bacilli was assessed but not indispensable for a TB compatible diagnosis.
2.5. Mycobacterial isolation and identification
TB was confirmed by mycobacterial isolation in Löwenstein–Jensen with pyruvate and Coletsos solid media (BD Diagnostics) as previously described (Vidal et al., 2018), and BACTEC MGIT 320 system (BD Diagnostics). Positive growths were stored in brain heart infusion with 20% glycerol at −80°C. Isolates were then confirmed as MTBC by conventional multiplex PCR (Wilton & Cousins, 1992), and strains were subsequently typified by DVR‐Spoligotyping.
2.6. Whole genome sequencing
A representative subset of M. caprae isolates from wild boar (N = 6) and livestock (N = 3) of the SA, as well as other 43 M. caprae isolates from cattle (N = 18), goats (N = 22), sheep (N = 1) and wild boar (N = 2) in Catalonia (outside the SA) between 2008 and 2018, were sub‐cultured in 7H11 plates (BD diagnostics) for 28 days at 37 ± 1°C. Growth was suspended in 1ml PBS in O‐ring microtubes and was heat‐inactivated at 100°C for 30 min. Inactivated MTBC samples were sent to the National Veterinary Services Laboratories, United States Department of Agriculture (USA) to conduct the DNA purification and whole genome sequencing (WGS) as previously described (Salvador et al., 2019). Briefly, DNA from bacterial cultures was collected; libraries were prepared with Nextera XT DNA Library Preparation Kit according to manufacturer's instructions, and sequenced in an Illumina MiSeq device using paired‐end 250 bp segments. Sequences were mapped against the genome of the reference strain M. bovis AF2122/97 (RefSeq ID NC_002945.4, BioProject PRJNA57695, (Malone et al., 2017)) using the Burrows–Wheeler aligner algorithm (BWA, (Li & Durbin, 2010)) and genome analysis toolkit 2.5.2 (GATK, (Auwera et al., 2013)), applying filtering parameters and variant quality score recalibration according to GATK Best Practices recommendations.
2.7. SNP identification and phylogenetic analyses
Single nucleotide polymorphisms (SNPs) that had quality scores (QUAL) < 150 and Allele Counts (AC) = 1 were removed, and SNPs that fell into repeat regions (PPE, PE_PGRES gene families, transposable elements), or that were in loci with poor read density were removed similarly to previously described (Bryant et al., 2013; Kato‐Maeda et al., 2013). SNPs were visually validated using Integrated Genomics Viewer (IGV, Broad Institute and the Regents of the University of California) (Robinson et al., 2011), and those that fell in areas with low coverage, mapping issues or alignment problems were manually filtered out.
The evolutionary relationships among isolates were analysed by means of two different phylogenetic analyses; firstly, the concatenated SNP sequences for all isolates were aligned, and a maximum likelihood phylogeny was constructed using a GTR‐CAT model with RaxML version 8 (Stamatakis, 2014). The tree was rooted using the genome sequence of M. bovis AF2122/97 reference strain. The confidence of the internal branches of the tree was assessed by means of 1,000 Bootstrap replicates.
In second place, the whole genomic SNP sequences (i.e. DNA Alignment) were used to generate a median‐joining network (MJN) to further explore the genetic relatedness of isolates (Bandelt, Forster, & Rohl, 1999). The MJN was constructed with SplitsTree4 version 4.15.1 (Huson & Bryant, 2006) using the default parameters suggested by the program. The network was also rooted by specifying the genome sequence of M. bovis AF2122/97 as the out‐group in order to facilitate its interpretation as a genealogical history. Thereafter, we computed the maximum parsimony (MP) post‐processing in order to remove all superfluous median vectors and links that are not contained in the shortest trees of the network (Polzin & Daneshmand, 2003). The MP post‐processing calculation was available in Network Phylogenetic Software (version 4.6; available at www.fluxus‐engineering.com/sharenet.htm) which was also used to redraw the network.
3. RESULTS
3.1. Findings in livestock
Two out of the 47 goat herds showed positive animals to the TST, one meat aptitude herd with extensive management (detected in December 2017) and one dairy herd with intensive management (detected in January 2018). The meat herd showed 4/241 (1.7%) positive animals to TST, three of them were confirmed by culture and M. caprae SB0415 was identified. The dairy herd showed 37/255 (14.5%) positive animals to TST. All positive animals were slaughtered and 14 of them were sampled for post‐mortem examination. All examined animals showed TB compatible lesions and the isolated M. caprae strains all belonged to the unique spoligotype SB0416. None of the 49 tested cattle herds showed any positive animal to the TST.
3.2. Findings in wild boar population
Twenty‐one out of 278 (7.6%) wild boars showed TB compatible lesions confirmed by histopathology (Figure 2). Of those, 17 were confirmed by culture, identifying two distinct M. caprae spoligotype profiles: SB0415 (13) and SB1908 (4). No other mycobacteria or other pathological agents were isolated from the remaining four samples with histologically TB compatible lesions. The majority of cases with TB lesions were obtained during hunting seasons 2016–2017 and 2017–2018; the pathological and bacteriological annual results in wild boar are summarized in Table 1.
Figure 2.
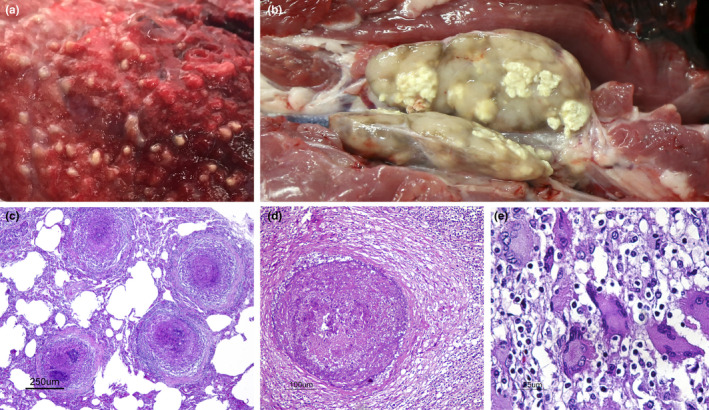
TB compatible lesions in wild boars. (a) Wild boar, Lung section. Miliary granulomatous pneumonia. This animal was likely excreting numerous mycobacteria. (b) Wild boar, submandibular lymph node section. Multifocal caseous granulomatous lymphadenitis. (c) Wild boar, lung (same as shown in a). Four neighbouring encapsulated granulomas in the lung parenchyma. H&E. Bar 250 μm. (d) Wild boar, submandibular lymph node. Chronic encapsulated granuloma in the lymph node parenchyma, mostly composed of necrosis, mineralization and minimal macrophage inflammatory infiltrate. H&E. Bar 100 μm. (e): Wild boar, submandibular lymph node. Cluster of multinucleated giant cells (Langhan's cells) diffusely infiltrating the lymph node parenchyma. H&E. Bar 25 μm [Colour figure can be viewed at wileyonlinelibrary.com]
Table 1.
Number of wild boar tissue samples analysed per hunting season in the study area
| Season | 2015–2016 | 2016–2017 | 2017–2018 | 2018–2019 | 2019–2020 |
|---|---|---|---|---|---|
| No. of wild boar tissue samples | 23 | 109 | 34 | 66 | 46 |
| No. samples with TB visible lesions (%) | 1 (4%) | 13 (12%) | 6 (18%) | 1 (2%) | 0 (0%) |
| Isolated spoligopatterns (No. of isolates) | Mycobacterium caprae SB0415 (1) | M. caprae SB0415 (10) | M. caprae SB0415 (2) | M. caprae SB1908 (1) | ‐ |
| No. of municipalities affected | 1 | 8 | 5 | 1 | 0 |
3.3. Phylogenetic relationships and transmission patterns
Six isolates from wild boars and three from goats (two dairy and one meat) of the SA were selected for the phylogenetic analyses. Overall, the DNA of 52 M. caprae isolates (nine inside and 43 outside the SA) were sequenced by WGS and the SNP distances between the isolates were analysed.
The phylogenetic tree showed the separation of the strains in two major groups (A and B) (Figure 3). All the strains isolated from the SA were included in group A, whereas group B was clearly differentiated from group A and was mostly composed by strains isolated from fattening cattle imported from Romania. Within group A, three different clusters (A.1, A.2 and A.3) were defined on the basis of number of SNPs separating each sequenced strain. This clustering discriminated the two strains isolated from the dairy goat herd (cluster A.2, blue arrows in Figure 3), from the rest of the SA strains that were included in cluster A.3. A clade composed by the two dairy goat strains was defined by 18 specific SNPs (clade SA‐I). The spoligotype SB0416 was identified in both of them. Within the cluster A.3., the meat goat and three wild boar strains of the SA formed another clade with genome sequences possessing 32 specific SNPs (clade SA‐II, red arrows in Figure 3). The spoligotype SB0415 was identified in the four strains. Another branch of the cluster A.3 contained the remaining three wild boar strains of the SA (green arrows in Figure 3). The spoligotype SB0415 was identified in two of them (WB‐2 and WB‐5), whereas SB1908 was identified in the third (WB‐3). Localizations of the SA strains with WGS and/or spoligotype information, as well as the localizations of the remaining TB cases detected in the SA, are shown in the map represented in Figure 4.
Figure 3.
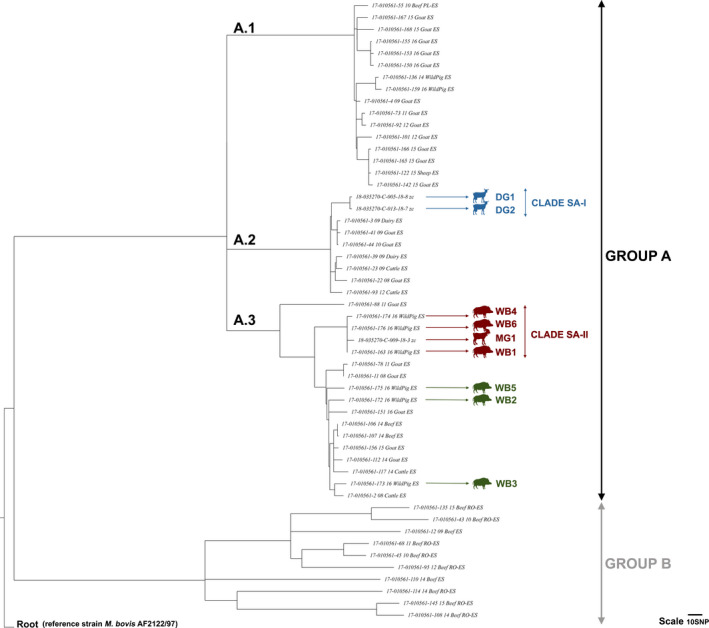
Phylogenetic relationships among Mycobacterium caprae strains genomes. Rooted phylogenetic tree based on maximum likelihood model (GTR‐CAT, RAxML) showing a SNP distance of the nine strains of the study area (SA) and 43 strains outside the SA isolated in Catalonia between 2008 and 2018. Root: Mycobacterium bovis AF 2122/97 reference strain sequence. Scale: 10 SNP. Two groups were identified: A, divided in three clusters (A.1, A.2 and A.3) and B. The nine strains of the SA are indicated by arrows as following: dairy goats forming the clade SA‐I within the cluster A.2 (blue), wild boars and meat goat forming the clade SA‐II within the cluster A.3 (red), and remaining wild boars of the cluster A.3 (green) [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 4.
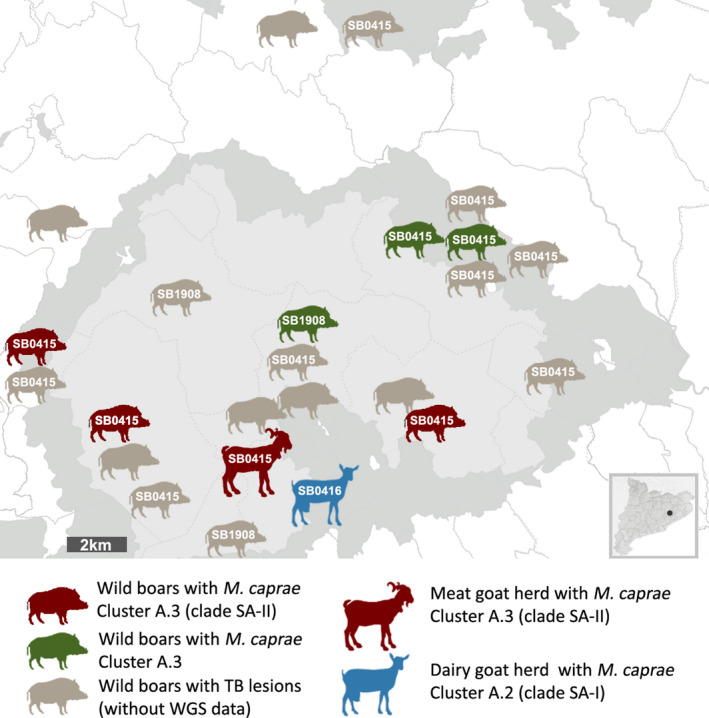
Localization of animal TB cases detected in the study area (SA). The map shows the approximate location of the animals from which the strains were isolated (precision level is the municipality, delimited by dotted lines). The number within the animal silhouettes indicates the spoligotype profile (when mycobacterial isolate was obtained) [Colour figure can be viewed at wileyonlinelibrary.com]
Putative local transmission patterns of M. caprae from the 52 analysed strains were inferred from the analysis of the phylogenetic relationships among taxa (Figure 5). The MJN results indicated that the six strains isolated from wild boars of the SA (from WB‐1 to WB‐6) were highly related to each other and with the strain from the meat goat (MG‐1). These strains showed genetic relatedness also with other goat and cattle strains isolated in Catalonia between 2008 and 2014 (mostly from meat‐producing animals) (Figure 5). The results also revealed the occurrence of transmission events inside the SA, between wild boars and from wild boar to the meat goat (at end‐point position along the network branch, forming the clade SA‐II marked in red in Figure 5). In particular, the wild boar strain WB‐6, isolated in 2015, showed a difference of five SNPs compared with the wild boar strain WB‐4 (isolated in 2016) and a difference of one SNP with the wild boar strain WB‐1 (isolated in 2016); the latter (WB‐1) was separated by seven SNPs from the meat goat strain (MG‐1), which was isolated in 2018 (Figure 5). Finally, the two dairy goat strains, sampled in the same herd, were genetically close to each other, differing only in 5 SNPs, but showed a distant genetic relationship to the rest of the strains isolated from the SA (Figure 5). Instead, they were genetically closer to other M. caprae strains (mostly from dairy animals of Spanish origin) sampled from outside the SA in previous years (2009–2012).
Figure 5.
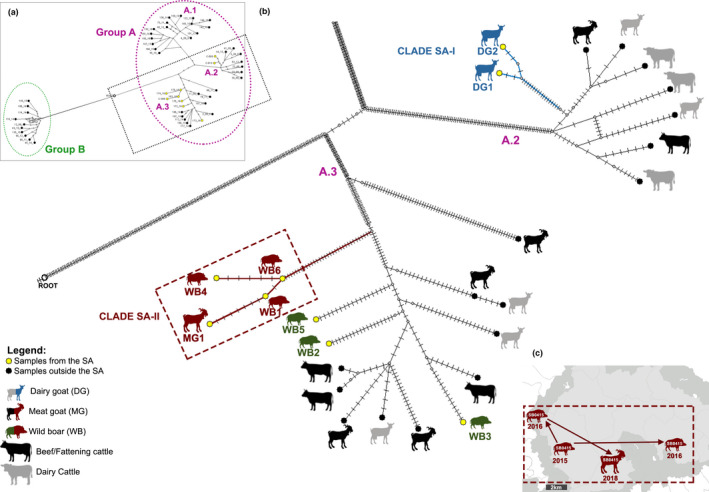
Evolutionary relationship of Mycobacterium caprae strains based on WGS. (a) Rooted median‐joining network analysis performed with the 52 M. caprae strains isolated in Catalonia between 2008 and 2018 (left‐upper insert). (b) In the zoomed main central image: Yellow circles show the nine strains of the study area (SA), black circles show the 43 strains isolated outside the SA. Root: Mycobacterium bovis AF2122/97 reference strain. Blue silhouettes indicate the dairy goat strains forming the clade SA‐I within the cluster A.2; Red silhouettes indicate the wild boars and meat goat strains forming the clade SA‐II within the cluster A.3; Green silhouettes indicate the remaining wild boars strains of the SA within the cluster A.3. The number of small perpendicular lines in each branch represents the SNPs separating sequences. Potential median vectors (hypothetical genotype) which connect the genome sequences are displayed as little circles in light grey. (c) The map (right‐bottom insert) represents the suggested evolutionary scenario within the clade SA‐II. Silhouettes represent the animal species, the year of M. caprae isolation is indicated below them and arrows indicate the transmission direction [Colour figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
The MJN results indicate that seven out of the nine sequences from the SA might have a common genealogical origin, and then the infection spread through different animal populations within the Natural Park. Even though additional data would be required to establish precise evolutionary relationships among strains from the SA, the results suggest a local transmission of M. caprae from wild boars to a goat herd of meat aptitude and extensive management. Indeed, the putative evolutionary history inferred from the rooted MJN was congruent with the isolation dates of the strains forming the clade SA‐II (M. caprae strains were first isolated in wild boars and later in the meat goat herd, see Figure 5). As far as we know, this is the first scientific study reporting infected wild boars as the most likely cause of a TB outbreak in goats. Local MTBC hotspots in free‐ranging wild boars were previously detected in Catalonia in relatively restricted natural areas, and were epidemiologically associated with cattle TB breakdowns (Mentaberre et al., 2014) or positive TST results (Pérez de Val et al., 2019). Now, our findings revealed a circulation of MTBC between non‐bovine species but still posing a risk of infection for cattle herds of the Natural Park that may interact with infected free‐ranging wild boars or share pastures with infected goats.
Data on TB status of the meat goat herd were unavailable prior to the first wild boar TB detection (in 2015), although the very low ratio of TST positive animals found in 2018 (1.7%) was strongly suggestive of a recent infection, occurring after that date. M. caprae is particularly adapted to domestic goats (Aranaz et al., 2003), showing fast animal‐to‐animal transmission within the herd, not only by direct contact through aerosols, but also by indirect oral contact by using shared feeders (Vidal et al., 2017). Therefore, the low number of positive goats detected by TST (four, only three with visible lesions and positive culture result) is consistent with a recent exogenous entry of the disease in the herd. This recent transmission chain could be also supported by the short genetic pairwise distance between strains (i.e. 7 SNPs between WB‐1 and MG‐1). Pairs differing less than 6 SNPs are considered indicative of recent person‐to‐person M. tuberculosis transmission (Nikolayevskyy, Kranzer, Niemann, & Drobniewski, 2016), but this threshold to delineate epidemiological links between strains could increase to 12 SNPs within 3 years of evolution in human M. tuberculosis outbreaks (Walker et al., 2013) and after 5–6 years in cattle M. bovis outbreaks (Price‐Carter et al., 2018).
The specific way of entry of the infection into the goat herd is still unknown, although the indirect contact in contaminated grazing areas, shared by both free‐ranging wild boars and goats in pasturage, seems the most likely source of the infection. In particular, aggregation points such as waterholes and baited places are considered hotspots for inter‐species transmission of MTBC (Barasona, Torres, Aznar, Gortázar, & Vicente, 2017; Payne, Philipon, Hars, Dufour, & Gilot‐Fromont, 2017). On the contrary, WGS results displayed a low genetic relatedness of the two strains of the dairy goat herd with the rest of the strains from the SA, indicating a separate origin of TB outbreak without any apparent interaction with wildlife reservoirs of the SA. The origin of this outbreak is probably related to other dairy goats from outside of the SA (i.e. animal movements). The low SNP variation between the two dairy goat strains (5 SNPs) is consistent with the intra‐herd spread of the infection, as previously described in M. caprae‐infected cattle (Broeckl et al., 2017), which might be facilitated by the intensive management of the sampled herd. The persistence of MTBC lineages in the livestock–wildlife interface within a local environment of a farm is common and can be explained by direct or indirect contact between farming animals and wildlife hosts (Biek et al., 2012). However, in this study, the M. caprae strains isolated from wild boars tended to involve the same genetic lineage (cluster A.3, Figure 5) across the whole SA, an extensive area with different microhabitats, rather than being differentiated by local farm environments (the SA involves almost 100 cattle and goats herds, together). This finding suggests a common origin of M. caprae strains at a specific time point in the past (probably from infected livestock) and subsequent maintenance of M. caprae circulation in the wild boar population of the SA, with mild genetic evolution within local sub‐populations (i.e. clade SA‐II formation).
The reduced genetic diversity observed among the 42 M. caprae strains included in the group A can be explained by the selective pressure carried out after years of bovine TB eradication campaigns based on test and slaughter of positive animals (Hauer et al., 2015). Yet, WGS analysis detected genetic variations of M. caprae strains of the SA and local transmission patterns that had been missed by the DVR‐spolgotyping analysis alone, which, in the last two decades, has been the most extended tool for molecular characterization of MTBC isolated from animals worldwide (Haddad, Masselot, & Durand, 2004; Milian‐Suazo et al., 2008; Rodríguez et al., 2010).
Interestingly, the WGS also enabled to genetically associate strains classified in apparently distant spoligotypes (i.e. SB1908 presents deletions in 9 oligonucleotide spacers compared to SB0415, www.Mbovis.org database), demonstrating that some of them were very much closely related in comparison with other strains, despite being classified within the same spoligotype (i.e. SB0415). These findings evidenced not only the lower discriminatory power of conventional molecular characterization tools compared to WGS, but also inaccurate epidemiological associations in outbreak investigations.
Specific non‐synonymous SNPs related with virulence were not investigated. Although previous studies showed that wild boars were even more susceptible to M. caprae than M. bovis strains (García‐Jiménez et al., 2013), in the present study, the M. carpae transmission and expansion could be mainly driven by the high wild boar population densities (i.e. increased contact rates) rather than host adaptation of M. caprae strains. Indeed, Catalonia, and particularly the Natural Park of the TB outbreak, shows one of the highest wild boar densities in Europe (Pittiglio, Khomenko, & Beltran‐Alcrudo, 2018; Rosell et al., 2019).
The increase in number of hunted animals in the SA after the outbreak detection did not reduce significantly the wild boar density (from 16 to 12 individuals/sq. km in seasons 2015–16 and 2018–19, respectively, (Rosell et al., 2019)). A previous study suggested a compensatory reproduction effect of wild boars as a response to an increased hunting pressure (Mentaberre et al., 2014). However, a higher hunting rate could contribute to the removal of animals in advanced stages of the disease from the environment, thus limiting the transmission rates of M. caprae within the wild boar population. This might explain the reduction of apparent prevalence based on post‐mortem examination of visible lesions in the two last hunting seasons included in the study. Nevertheless, a previous serological study in a high prevalent area of TB in wild boars showed that the increase of hunting pressure reduced the prevalence of TB lesions in adult wild boars but increased the reproduction rates, resulting in a significant number of seropositive piglets although they still had not developed TB visible lesions and were negative to culture (Pérez de Val et al., 2017). Thus, the evolution of wild boar TB in the SA should be carefully monitored in the upcoming seasons to confirm the apparent drop in TB prevalence or its reemergence as a potential risk for livestock herds in contact with infected wild boars.
In summary, our findings revealed that infected wild boars were able to maintain the mycobacteria in their own population and have likely transmitted the infection to sympatric goats. The study evidenced local M. caprae circulation between wild boars and goats without an apparent participation of cattle in the epidemiology, reinforcing the notion that animal TB should be approached as a multi‐host disease which requires holistic control measures. The present study evidences the need of a thorough laboratory diagnosis and epidemiological follow‐up in non‐bovine susceptible hosts since these can maintain the infection and compromise the long‐term success of bovine TB eradication programmes. The results also highlighted the usefulness of WGS analysis in providing relevant epidemiological information when detailed contact data are missing or difficult to obtain.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ETHICAL APPROVAL
The authors confirm that the ethical policies of the journal, as noted on the journal's author guidelines page, have been adhered to. No ethical approval was required for this study as all sampling and handling procedures were conducted by authorized personnel according to Spanish legislation (Royal Decree 2611/1996 and amendments) and European Union laws for protection of animals used for scientific purposes (2010/63/EU).
ACKNOWLEDGEMENTS
This work was supported by the Department of Agriculture, Livestock, Fisheries and Food (DARP) of the Government of Catalonia. IRTA is supported by Centres de Recerca de Catalunya (CERCA) Program/Generalitat de Catalunya (www.cerca.cat). DVR‐spoligotyping was performed at VISAVET Health Surveillance Centre, Universidad Complutense de Madrid, Spain. Whole genome sequencing was performed at the National Veterinary Services Laboratories, United States Department of Agriculture, USA. We thank the forest guard body of the Generalitat de Catalunya, Carniques Llorà S.L. and Senglar de Girona S.L., for their contribution in wild boar samples collection. We are also grateful to Zoraida Cervera, Abel Muñoz, Sierra Espinar and Mónica Pérez for their outstanding technical assistance.
Ciaravino G, Vidal E, Cortey M, et al. Phylogenetic relationships investigation of Mycobacterium caprae strains from sympatric wild boar and goats based on whole genome sequencing. Transbound Emerg Dis.2021;68:1476–1486. 10.1111/tbed.13816
Giovanna Ciaravino and Enric Vidal contributed equally to this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Aranaz, A. , Cousins, D. , Mateos, A. , Domínguez, L. Elevation of Mycobacterium tuberculosis subsp. caprae Aranaz et al 1999 to species rank as Mycobacterium caprae comb. nov., sp. nov. International Journal of Systematic and Evolutionary Microbiology, 53(Pt 6), 1785–1789. 10.1099/ijs.0.02532-0 [DOI] [PubMed] [Google Scholar]
- Auwera, G. A. , Carneiro, M. O. , Hartl, C. , Poplin, R. , del Angel, G. , Levy‐Moonshine, A. , … DePristo, M. A. (2013). From FastQ data to high‐confidence variant calls: The genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics, 43(1), 1–11. 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt, H. J. , Forster, P. , & Rohl, A. (1999). Median‐joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16(1), 37–48. 10.1093/oxfordjournals.molbev.a026036 [DOI] [PubMed] [Google Scholar]
- Barasona, J. A. , Torres, M. J. , Aznar, J. , Gortázar, C. , & Vicente, J. (2017). DNA detection reveals Mycobacterium tuberculosis complex shedding routes in its wildlife reservoir the Eurasian wild boar. Transboundary and Emerging Diseases, 64(3), 906–915. 10.1111/tbed.12458 [DOI] [PubMed] [Google Scholar]
- Bezos, J. , Romero, B. , de Juan, L. , Gonzalez, S. , & Sáez, J. (2019). Manual de Procedimiento Sanitario: Realización de las pruebas de intradermotuberculinización y gamma‐interferón. 2019–2020. Madrid, Spain: Ministerio de Agricultura, Pesca y Alimentación (MAPA) y Centro de Vigilancia Sanitaria Veterinaria (VISAVET). Universidad Complutense de Madrid. Retrieved from https://www.visavet.es/data/mapa/manual_procedimiento_IDTB_IFN_2019.pdf [Google Scholar]
- Biek, R. , O’Hare, A. , Wright, D. , Mallon, T. , McCormick, C. , Orton, R. J. , … Kao, R. R. (2012). Whole genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations. PLoS Pathogens, 8(11), e1003008. 10.1371/journal.ppat.1003008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boniotti, M. B. , Goria, M. , Loda, D. , Garrone, A. , Benedetto, A. , Mondo, A. , … Pacciarini, M. L. (2009). Molecular typing of Mycobacterium bovis strains isolated in Italy from 2000 to 2006 and evaluation of variable‐number tandem repeats for geographically optimized genotyping. Journal of Clinical Microbiology, 47(3), 636–644. 10.1128/JCM.01192-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broeckl, S. , Krebs, S. , Varadharajan, A. , Straubinger, R. K. , Blum, H. , & Buettner, M. (2017). Investigation of intra‐herd spread of Mycobacterium caprae in cattle by generation and use of a whole‐genome sequence. Veterinary Research Communications, 41(2), 113–128. 10.1007/s11259-017-9679-8 [DOI] [PubMed] [Google Scholar]
- Bryant, J. M. , Harris, S. R. , Parkhill, J. , Dawson, R. , Diacon, A. H. , van Helden, P. , … Bentley, S. D. (2013). Whole‐genome sequencing to establish relapse or re‐infection with Mycobacterium tuberculosis: A retrospective observational study. The Lancet Respiratory Medicine, 1(10), 786–792. 10.1016/S2213-2600(13)70231-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano‐Terriza, D. , Risalde, M. A. , Rodríguez‐Hernández, P. , Napp, S. , Fernández‐Morente, M. , Moreno, I. , … García‐Bocanegra, I. (2018). Epidemiological surveillance of Mycobacterium tuberculosis complex in extensively raised pigs in the south of Spain. Preventive Veterinary Medicine, 159, 87–91. 10.1016/J.PREVETMED.2018.08.015 [DOI] [PubMed] [Google Scholar]
- Crawshaw, T. , Daniel, R. , Clifton‐Hadley, R. , Clark, J. , Evans, H. , Rolfe, S. , & de la Rua‐Domenech, R. (2008). TB in goats caused by Mycobacterium bovis . The Veterinary Record, 163(4), 127. 10.1136/vr.163.4.127 [DOI] [PubMed] [Google Scholar]
- Daniel, R. , Evans, H. , Rolfe, S. , de la Rua‐Domenech, R. , Crawshaw, T. , Higgins, R. J. , Clifton‐Hadley, R. (2009). Outbreak of tuberculosis caused by Mycobacterium bovis in golden Guernsey goats in Great Britain. Veterinary Record, 165(12), 335–342. 10.1136/vr.165.12.335 [DOI] [PubMed] [Google Scholar]
- European Commission (2020). Outcome of the evaluation procedure of the eradication, control and surveillance programmes submitted by Member States for Union financial contribution for 2020: List of the programmes technically approved and amount allocated to each programme. Brussels, Belgium. Retrieved from https://ec.europa.eu/food/sites/food/files/safety/docs/cff_animal_vet‐progs_guidance_progs_erad_wd‐12728‐2019.pdf [Google Scholar]
- García‐Jiménez, W. L. , Benítez‐Medina, J. M. , Fernández‐Llario, P. , Abecia, J. A. , García‐Sánchez, A. , Martínez, R. , … Hermoso de Mendoza, J. (2013). Comparative pathology of the natural infections by Mycobacterium bovis and by Mycobacterium caprae in Wild Boar (Sus scrofa). Transboundary and Emerging Diseases, 60(2), 102–109. 10.1111/j.1865-1682.2012.01321.x [DOI] [PubMed] [Google Scholar]
- Guta, S. , Casal, J. , Napp, S. , Saez, J. L. , Garcia‐Saenz, A. , Perez De Val, B. , … Allepuz, A. (2014). Epidemiological investigation of bovine tuberculosis herd breakdowns in Spain 2009/2011. PLoS One, 9(8), e104383. 10.1371/journal.pone.0104383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad, N. , Masselot, M. , & Durand, B. (2004). Molecular differentiation of Mycobacterium bovis isolates. Review of main techniques and applications. Research in Veterinary Science, 76(1), 1–18. 10.1016/S0034-5288(03)00078-X [DOI] [PubMed] [Google Scholar]
- Hauer, A. , De Cruz, K. , Cochard, T. , Godreuil, S. , Karoui, C. , Henault, S. , … Boschiroli, M. L. (2015). Genetic evolution of Mycobacterium bovis causing tuberculosis in livestock and wildlife in France since 1978. PLoS One, 10(2), e0117103. 10.1371/journal.pone.0117103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer, A. , Michelet, L. , Cochard, T. , Branger, M. , Nunez, J. , Boschiroli, M.‐L. , & Biet, F. (2019). Accurate phylogenetic relationships among Mycobacterium bovis strains circulating in France based on whole genome sequencing and single nucleotide polymorphism analysis. Frontiers in Microbiology, 10, 955. 10.3389/fmicb.2019.00955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer, A. , Michelet, L. , De Cruz, K. , Cochard, T. , Branger, M. , Karoui, C. , … Boschiroli, M. L. (2016). MIRU‐VNTR allelic variability depends on Mycobacterium bovis clonal group identity. Infection, Genetics and Evolution: Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases, 45, 165–169. 10.1016/j.meegid.2016.08.038 [DOI] [PubMed] [Google Scholar]
- Hermoso de Mendoza, J. , Parra, A. , Tato, A. , Alonso, J. , Rey, J. , Peña, J. , Hermoso de Mendoza, M. (2006). Bovine tuberculosis in wild boar (Sus scrofa), red deer (Cervus elaphus) and cattle (Bos taurus) in a Mediterranean ecosystem (1992–2004). Preventive Veterinary Medicine, 74(2‐3), 239–247. 10.1016/j.prevetmed.2005.10.005 [DOI] [PubMed] [Google Scholar]
- Huson, D. H. , & Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution, 23(2), 254–267. 10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- Kamerbeek, J. , Schouls, L. , Kolk, A. , van Agterveld, M. , van Soolingen, D. , Kuijper, S. , … van Embden, J. (1997). Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. Journal of Clinical Microbiology, 35(4), 907–914. Retrieved from https://jcm‐asm‐org.are.uab.cat/content/35/4/907.long 10.1128/JCM.35.4.907-914.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato‐Maeda, M. , Ho, C. , Passarelli, B. , Banaei, N. , Grinsdale, J. , Flores, L. , … Hopewell, P. C. (2013). Use of whole genome sequencing to determine the microevolution of Mycobacterium tuberculosis during an outbreak. PLoS One, 8(3), e58235. 10.1371/journal.pone.0058235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2010). Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics (Oxford, England), 26(5), 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone, K. M. , Farrell, D. , Stuber, T. P. , Schubert, O. T. , Aebersold, R. , Robbe‐Austerman, S. , & Gordon, S. V. (2017). Updated reference genome sequence and annotation of Mycobacterium bovis AF2122/97. Genome Announcements, 5(14), e00157‐17. 10.1128/genomeA.00157-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentaberre, G. , Romero, B. , de Juan, L. , Navarro‐González, N. , Velarde, R. , Mateos, A. , … Serrano, E. (2014). Long‐term assessment of wild boar harvesting and cattle removal for bovine tuberculosis control in free ranging populations. PLoS One, 9(2), e88824. 10.1371/journal.pone.0088824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milian‐Suazo, F. , Harris, B. , Díaz, C. A. , Romero Torres, C. , Stuber, T. , Ojeda, G. A. , … Payeur, J. B. (2008). Molecular epidemiology of Mycobacterium bovis: Usefulness in international trade. Preventive Veterinary Medicine, 87(3–4), 261–271. 10.1016/j.prevetmed.2008.04.004 [DOI] [PubMed] [Google Scholar]
- Napp, S. , Allepuz, A. , Mercader, I. , Nofrarías, M. , López‐Soria, S. , Domingo, M. , … Pérez de Val, B. (2013). Evidence of goats acting as domestic reservoirs of bovine tuberculosis. The Veterinary Record, 172(25), 663. 10.1136/vr.101347 [DOI] [PubMed] [Google Scholar]
- Naranjo, V. , Gortazar, C. , Vicente, J. , & de la Fuente, J. (2008). Evidence of the role of European wild boar as a reservoir of Mycobacterium tuberculosis complex. Veterinary Microbiology, 127(1–2), 1–9. 10.1016/j.vetmic.2007.10.002 [DOI] [PubMed] [Google Scholar]
- Nikolayevskyy, V. , Kranzer, K. , Niemann, S. , & Drobniewski, F. (2016). Whole genome sequencing of Mycobacterium tuberculosis for detection of recent transmission and tracing outbreaks: A systematic review. Tuberculosis, 98, 77–85. 10.1016/j.tube.2016.02.009 [DOI] [PubMed] [Google Scholar]
- Payne, A. , Philipon, S. , Hars, J. , Dufour, B. , & Gilot‐Fromont, E. (2017). Wildlife Interactions on baited places and waterholes in a French area infected by bovine tuberculosis. Frontiers in Veterinary Science, 3, 122. 10.3389/fvets.2016.00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez de Val, B. , López‐Soria, S. , Nofrarías, M. , Martín, M. , Vordermeier, H. M. , Villarreal‐Ramos, B. , … Domingo, M. (2011). Experimental model of tuberculosis in the domestic goat after endobronchial infection with Mycobacterium caprae . Clinical and Vaccine Immunology, 18(11), 10.1128/CVI.05323-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez de Val, B. , Napp, S. , Velarde, R. , Lavín, S. , Cervera, Z. , Singh, M. , … Mentaberre, G. (2017). Serological follow‐up of tuberculosis in a wild boar population in contact with infected cattle. Transboundary and Emerging Diseases, 64(1), 10.1111/tbed.12368 [DOI] [PubMed] [Google Scholar]
- Pérez de Val, B. , Sanz, A. , Soler, M. , Allepuz, A. , Michelet, L. , Boschiroli, M. L. , & Vidal, E. (2019). Mycobacterium microti Infection in free‐ranging Wild Boar, Spain, 2017–2019. Emerging Infectious Diseases, 25(11), 2152–2154. 10.3201/eid2511.190746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittiglio, C. , Khomenko, S. , & Beltran‐Alcrudo, D. (2018). Wild boar mapping using population‐density statistics: From polygons to high resolution raster maps. PLoS One, 13(5), e0193295. 10.1371/journal.pone.0193295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polzin, T. , & Daneshmand, S. V. (2003). On Steiner trees and minimum spanning trees in hypergraphs. Operations Research Letters, 31(1), 12–20. 10.1016/S0167-6377(02)00185-2 [DOI] [Google Scholar]
- Price‐Carter, M. , Brauning, R. , de Lisle, G. W. , Livingstone, P. , Neill, M. , Sinclair, J. , … Collins, D. M. (2018). Whole genome sequencing for determining the source of Mycobacterium bovis infections in livestock herds and wildlife in New Zealand. Frontiers in Veterinary Science, 5, 272. 10.3389/fvets.2018.00272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J. T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E. S. , Getz, G. , & Mesirov, J. P. (2011). Integrative genomics viewer. Nature Biotechnology, 29(1), 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez, E. , Sánchez, L. P. , Pérez, S. , Herrera, L. , Jiménez, M. S. , Samper, S. , & Iglesias, M. J. (2009). Human tuberculosis due to Mycobacterium bovis and M. caprae in Spain, 2004–2007. The International Journal of Tuberculosis and Lung Disease: The Official Journal of the International Union against Tuberculosis and Lung Disease, 13(12), 1536–1541. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19919773 [PubMed] [Google Scholar]
- Rodríguez, S. , Bezos, J. , Romero, B. , de Juan, L. , Álvarez, J. , Castellanos, E. , … Aranaz, A. (2011). Mycobacterium caprae infection in livestock and wildlife, Spain. Emerging Infectious Diseases, 17(3), 532–535. 10.3201/eid1703.100618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez, S. , Romero, B. , Bezos, J. , de Juan, L. , Álvarez, J. , Castellanos, E. , … Aranaz, A. (2010). High spoligotype diversity within a Mycobacterium bovis population: Clues to understanding the demography of the pathogen in Europe. Veterinary Microbiology, 141(1–2), 89–95. 10.1016/j.vetmic.2009.08.007 [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Campos, S. , Gonzalez, S. , de Juan, L. , Romero, B. , Bezos, J. , Casal, C. , … Surveillance, S. N. (2012). A database for animal tuberculosis (mycoDB.es) within the context of the Spanish national programme for eradication of bovine tuberculosis. Infection, Genetics and Evolution, 12(4), 877–882. 10.1016/j.meegid.2011.10.008 [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Campos, S. , Schürch, A. C. , Dale, J. , Lohan, A. J. , Cunha, M. V. , Botelho, A. , … Smith, N. H. (2012). European 2 – A clonal complex of Mycobacterium bovis dominant in the Iberian Peninsula. Infection, Genetics and Evolution, 12(4), 866–872. 10.1016/j.meegid.2011.09.004 [DOI] [PubMed] [Google Scholar]
- Rosell, C. , Pericas, B. , Colomer, J. , & Navàs, F. (2019). Programa de seguiment de les poblacions de senglar a Catalunya. Temporada 2018–2019. Barcelona, Spain: Departament d’Agricultura, Ramadria, Pesca i Alimentació (DARP) de la Generalitat de Catalunya. Department d’Agricultura, Ramaderia, Pesca i Alimentació. Retrieved from http://agricultura.gencat.cat/web/.content/06‐medi‐natural/caca/enllacos‐documents/informes‐tecnics/programa‐seguiment‐poblacions‐senglar‐sus‐scrofa/fitxers‐binaris/seguiment_senglar_cat_2019.pdf [Google Scholar]
- Salvador, L. C. M. , O’Brien, D. J. , Cosgrove, M. K. , Stuber, T. P. , Schooley, A. M. , Crispell, J. , … Kao, R. R. (2019). Disease management at the wildlife‐livestock interface: Using whole‐genome sequencing to study the role of elk in Mycobacterium bovis transmission in Michigan, USA. Molecular Ecology, 28(9), 2192–2205. 10.1111/mec.15061 [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics (Oxford, England), 30(9), 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal, E. , Arrieta‐Villegas, C. , Grasa, M. , Mercader, I. , Domingo, M. , & Pérez de Val, B. (2017). Field evaluation of the efficacy of Mycobacterium bovis BCG vaccine against tuberculosis in goats. BMC Veterinary Research, 13(1), 252. 10.1186/s12917-017-1182-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal, E. , Grasa, M. , Perálvarez, T. , Martín, M. , Mercader, I. , & Pérez de Val, B. (2018). Transmission of tuberculosis caused by Mycobacterium caprae between dairy sheep and goats. Small Ruminant Research, 158, 22–25. 10.1016/j.smallrumres.2017.11.010 [DOI] [Google Scholar]
- Vidal, E. , Tolosa, E. , Espinar, S. , de Val, B. P. , Nofrarías, M. , Alba, A. , … Domingo, M. (2016). Six‐year follow‐up of slaughterhouse surveillance (2008–2013): The Catalan slaughterhouse support network (SESC). Veterinary Pathology, 53(3), 532–544. 10.1177/0300985815593125 [DOI] [PubMed] [Google Scholar]
- Walker, T. M. , Ip, C. L. , Harrell, R. H. , Evans, J. T. , Kapatai, G. , Dedicoat, M. J. , … Peto, T. E. (2013). Whole‐genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. The Lancet Infectious Diseases, 13(2), 137–146. 10.1016/S1473-3099(12)70277-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton, S. , & Cousins, D. (1992). Detection and identification of multiple mycobacterial pathogens by DNA amplification in a single tube. Genome Research, 1(4), 269–273. 10.1101/gr.1.4.269 [DOI] [PubMed] [Google Scholar]
- World Health Organization (2018). GLOBAL TUBERCULOSIS REPORT. Geneva, Switzerland. Retrieved from https://apps.who.int/iris/bitstream/handle/10665/274453/9789241565646‐eng.pdf?ua=1&ua=1 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.