

Abstract
Cancerous tumours contain a rare subset of cells with stem‐like properties that are termed cancer stem cells (CSCs). CSCs are defined by their ability to divide both symmetrically and asymmetrically, to initiate new tumour growth and to tolerate the foreign niches required for metastatic dissemination. Accumulating evidence suggests that tumours arise from cells with stem‐like properties, the generation of CSCs is therefore likely to be an initiatory event in carcinogenesis. Furthermore, CSCs in established tumours exist in a dynamic and plastic state, with nonstem tumour cells thought to be capable of de‐differentiation to CSCs. The regulation of the CSC state both during tumour initiation and within established tumours is a desirable therapeutic target and is mediated by epigenetic factors. In this review, we will explore the epigenetic parallels between induced pluripotency and the generation of CSCs, and discuss how the epigenetic regulation of CSCs opens up novel opportunities for therapeutic intervention.
Keywords: cancer stem cells, cancer therapy, early detection of cancer, epigenetics, induced pluripotent stem cells, tumorigenesis
Abbreviations
- AML
acute myeloid leukaemia
- ATP
adenosine triphosphate
- BAF
BRG1‐ or BRM‐associated factors
- BER
base‐excision repair
- BET
bromodomain and extra terminal
- BRD
bromodomain
- CIMP
CpG island methylator phenotype
- CSC
cancer stem cell
- DNA
deoxyribose nucleic acid
- DNMT
DNA methyltransferase
- EMT
epithelial‐mesenchymal transition
- ESC
embryonic stem cell
- FACs
fluorescence‐activated cell sorting
- GBM
glioblastoma multiforme
- HDAC
histone deacetylase
- IL6/17
interleukin 6/17
- iPSC
induced pluripotent stem cell
- lncRNA
long noncoding RNA
- LSD1
lysine‐specific demethylase 1
- MET
mesenchymal‐epithelial transition
- miRNA/miR
microRNA
- ncRNA
noncoding RNA
- PDAC
pancreatic ductal adenocarcinoma
- PRC
polycomb repressive complex
- RISC
RNA‐induced silencing complex
- RNA
ribose nucleic acid
- SAM
S‐adenosyl methionine
- SWI/SNF
SWItch/sucrose nonfermentable
- TAM
tumour‐associated macrophage
- TET
ten‐eleven transcription factor
- TGF
transforming growth factor
- TME
tumour microenvironment
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
As with adult tissues, cancerous tumours also contain a rare subset of cells with stem‐like properties that can function to regenerate the heterogeneous cell populations observed therein. These cancer stem cells (CSCs) are defined by their ability to divide both symmetrically and asymmetrically, to initiate new tumour growth and to tolerate the foreign niches required for metastatic dissemination. As the tumour‐initiating population, CSCs underpin the very nature of malignancy and studying their regulation is essential for understanding tumour formation, metastasis and relapse after therapy.
As it is not possible to isolate CSCs based on functional properties, CSC identification can be achieved by FACs sorting based on surrogate cell surface marker profiles and subsequent transplantation into immune‐compromised mice to demonstrate enhanced tumourigenic potential. Using this strategy, CSCs have been identified in most cancers, first in acute myeloid leukaemia (AML) followed by breast cancer and other solid malignancies such as brain, colon and pancreatic cancer, and are purported to account for only a few per cent of the total cell population. 1 , 2 , 3 , 4 , 5 , 6 The existence of a rare population of CSCs supports the notion that a heterogeneous cancer arises from a single cell atop a cellular hierarchy. This also suggests that cancer arises from a cell with stem‐like properties, as the cell‐of‐origin would require the asymmetric division to initiate and maintain tumour growth. Furthermore, the existing properties and long lifespan of a stem cell make it more likely than a differentiated cell to acquire a tumourigenic phenotype. 7 Recent evidence supports the premise that cancer arises from the deregulation of existing stem cell populations. In an organ‐wide study, Zhu et al induced oncogenic mutations specifically in CD133+ cells in the mouse. Tumours only arose in those organs where CD133 was proven to have generative capacity, that is, was an effective marker of a normal stem cell population (the liver, small intestine and stomach but not brain, kidney or pancreas). Furthermore, liver injury increased CD133+ cells and tumourigenic potential after transformation, thus indicating environmental factors can converge with genetic mutations to increase cancer incidence. 8 Another study showed that deregulation of existing cell populations preceded tumour formation in an inflammatory model of bowel cancer. Chronic inflammation disrupted homeostasis in the large intestine so that the paneth cells de‐differentiated to a stem‐like population from which tumours could arise. 9 These two studies are both examples of cancer arising from stem cells, be that an existing stem cell pool or one generated by extrinsic factors. CSC plasticity also persists during tumour growth as both stem and nonstem‐like populations are capable of inter or intraconversion in response to extrinsic signals. 10 , 11 , 12 This de novo generation of the CSC phenotype has obvious implications for therapeutic strategies, however, the molecular mechanisms involved are poorly understood. Creation of CSCs by definition requires a reversible but heritable process (asymmetric division), which strongly suggests a role for epigenetic regulation and there is mounting evidence in support of this, not least the importance of epigenetics in induced pluripotency.
Epigenetics refers to a number of mechanisms that control the reversible regulation of gene expression by changing the chromosome without altering the DNA sequence: DNA can be altered epigenetically by methylation and demethylation of CpG nucleotides. Epigenetic changes in the overall structure of chromatin occur through at least three interrelated mechanisms: posttranslational modifications of histones, ATP‐dependent chromatin remodelling and the incorporation (or replacement) of specialised histone variants into chromatin. Finally, noncoding RNA can interact with transcriptional processes to alter gene expression. In addition to 2D processes, epigenetic regulation can also involve higher‐order chromatin organisation including promoter‐enhancer interactions, regulatory DNA loops and 3D chromatin localisation in the cell nucleus (Figure 1).
FIGURE 1.

Epigenetic regulation of cancer stem cells. A variety of epigenetic mechanisms have been implicated in the generation of CSCs including changes in (A) nucleosome remodelling and associated complexes (eg, mutations in SWI/SNF). B, Histone variant deposition including higher H2A.Z and lower macroH2A. C, Histone modifications, in particular, bivalent histone marks D. Hypomethylation mediated by TET proteins E Altered expression of noncoding RNAs for example, high Lin28, low let‐7 [Color figure can be viewed at wileyonlinelibrary.com]
As these epigenetic mechanisms are important mediators of cellular identity, we will explore how the restructuring of such epigenetic barriers reinforces the stem‐like state in both normal cells and cancer, and their relevance to tumour initiation (Figure 2). Furthermore, we will discuss how the epigenetic regulation of CSCs opens up novel opportunities for cancer detection and therapeutic intervention.
FIGURE 2.
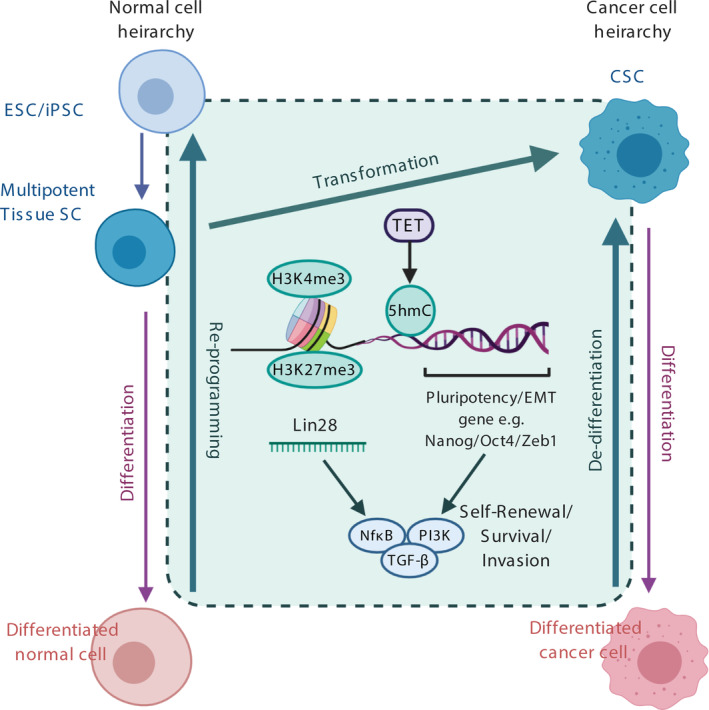
Epigenetic parallels between reprogramming, transformation and de‐differentiation. Transition to stem‐like states requires enhanced expression of pluripotency and EMT genes, which can be mediated at relevant loci by a combination of epigenetic processes including nucleosome loss at transcription start sites, histone variant deposition, histone modifications, in particular, bivalent histone marks to allow for rapid gene activation, demethylation by TET proteins and miRNA expression such as Lin‐28. All converge on downstream signalling pathways such as TGFβ and NfkB to promote stem‐like characteristics such as self‐renewal, survival and invasion [Color figure can be viewed at wileyonlinelibrary.com]
2. DNA METHYLATION AND DEMETHYLATION IN CSC PLASTICITY
DNA methylation is a form of gene silencing that occurs mainly to CpG dinucleotides which cluster in CpG islands; areas of high CpG density usually found at promoters. To methylate cytosines, DNA methyltransferases (DNMTs) catalyse the transfer of a methyl group from cofactor S‐adenosylmethionine to the carbon of the cytosine ring to generate 5‐methylcytosine (5mC). This functions to inhibit gene expression either by recruitment of methyl‐CpG‐binding domain proteins which in turn recruit histone‐modifying and chromatin‐remodelling complexes, or by preventing the recruitment of DNA‐binding proteins, that is, transcription factors. To reactivate expression after silencing, 5‐mCs can be oxidised to 5‐hydroxymethylcytosine (5hmC) and back to the unmodified state by TET proteins and base excision repair (BER), to ultimately restore the unmethylated cytosine. TET protein‐mediated demethylation can occur via either passive or active processes. Passive demethylation results from the failure to maintain 5mC marks across cell divisions (5hmC is not a substrate for DNMTs). Active demethylation is enzymatic, whereby TET proteins further oxidise 5hmC marks to 5‐formylcytosine (5fC) and 5‐carboxylcytosine (5caC) then back to the unmodified state. 5fC bases are recognised by thymine‐DNA‐glycosylases (TDG) that excise mismatched pyrimidines that would then be replaced by an unmodified cytosine by the BER pathway. Intermediate marks (5fC or 5caC) are much less abundant than 5hmC but may also have independent functions 13 (Figure 3A).
FIGURE 3.
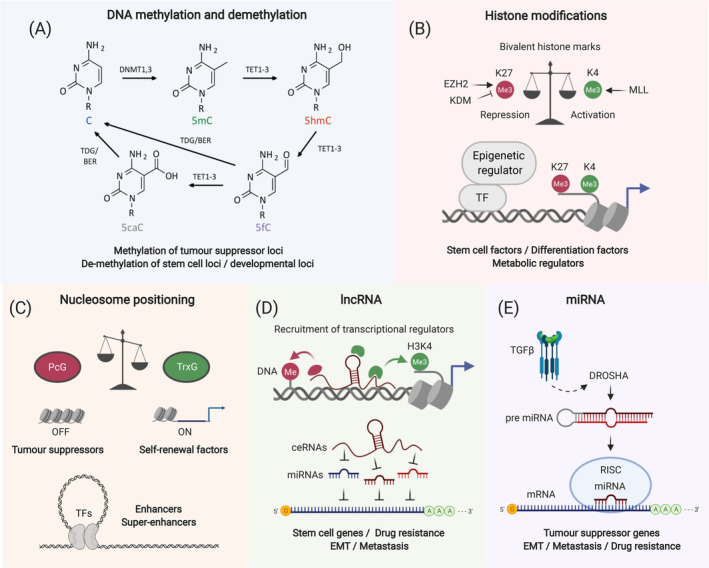
Epigenetic mechanisms in detail. A, Cycle of methylation and demethylation by DMNT and TET proteins, respectively. TET‐mediated demethylation generates intermediate 5hmC, 5fC and 5caC marks that are converted to unmodified cytosine by the BER pathway. B, Bivalent histone marks; the presence of both activating H3K4me3, and repressing H3K27me3 marks at certain loci allow for rapid gene activation and are characteristic of stem cells. C, Nucleosome remodelling complexes can affect gene expression at relevant loci: TrxG complexes allow expression of self‐renewal factors whereas PcG complexes prevent transcription of tumour suppressors. D, Long noncoding RNAs modify gene expression by affecting the recruitment of transcriptional regulators. E, MicroRNAs modify gene expression by directly affecting transcription [Color figure can be viewed at wileyonlinelibrary.com]
Methylation patterns are altered in cancer; localised hypermethylation occurs in CpG islands of the promoters of tumour suppressor genes (the so‐called “CpG island methylator phenotype”—CIMP), silencing their expression. 14 However, global hypomethylation in intergenic regions causes oncogene activation and ultimately results in genomic instability. 15 , 16 What happens in the CSC subset and what methylation changes are required to generate CSCs and initiate tumour growth?
In general, CpG methylation is required for differentiation whereas demethylation is essential for induction of the pluripotent state. 17 , 18 , 19 However non‐CpG methylation, although rarer, is associated with pluripotency and is lost upon development in all tissues except the brain. 20 There is evidence to suggest that loss of methylation is also required for the generation of CSCs and tumour initiation: data produced in mouse models demonstrate that global loss‐of‐imprinting (LOI) events alone promote the onset of cancer, including loss of growth inhibition by TGFβ. 21 CSC formation from cancer cells was found to depend on loss of methylation of the Nanog promoter via DNMT1 inhibition. 22 Loss‐of‐function mutations in DMNT3A also led to the expansion of preleukaemic SCs, indicating a role for dysregulated DNA methylation in inducing tumour‐initiating cells. 23
Some hypomethylation also continues to be associated with CSCs once the tumour has been established. For example, the CpG region of the CD133 gene promoter, a key gene involved in CSCs, was shown to be hypomethylated in several cancer types including breast, ovarian, colorectal and glioblastoma. 24 , 25 , 26 Also, some important demethylated regions occur exclusively in CSCs compared to nonstem tumour cells. For example, in pancreatic ductal adenocarcinoma (PDAC), genes involved in CSC pathways, including GATA6, SOX9 and BMP4, were demethylated in the invasive (more stem‐like) population. 27 Demethylation is highly important in pluripotency, both in ESCs and induced pluripotent stem cells (iPSCs). Reprogramming of somatic cells to a stem‐like state can be achieved by expression of the Yamanaka factors (Oct3/4, Sox2, Klf4 and c‐Myc), and is accompanied by epigenetic changes including demethylation. Reprogramming is dependent on TET1, which affects Nanog levels and can even act as a substitute for Oct4. However, knockout of TET1 in ESCs did not perturb the pluripotent state. 18 , 28 , 29 , 30 These studies show that TET1 is important in the establishment of pluripotency as opposed to the maintenance of this state, and suggests that TET proteins may also be involved in the generation of CSCs.
However, opposing roles for DMNT1 have been demonstrated as it can both promote or inhibit CSC formation. For example, DNMT1 was required for the initiation of colon cancer. 31 DNMT was also important for CSC function in established leukaemia, breast, lung and PDAC tumours 32 , 33 , 34 , 35 whereas inhibition of DNMT1 promoted CSCs and EMT in prostate cancer. 36 These discrepancies could be due to tumour‐specific effects or local hypermethylation of tumour suppressor genes which may promote the generation of CSCs in some cases. 37
The importance of both hypomethylation and DNMT1 in CSCs suggests that specific localization of 5mC and 5hmC marks is likely to be a more important determinant of cellular identity than the overall global patterning. In particular, hypomethylation at pluripotency loci and hypermethylation at tumour suppressor genes or those involved in differentiation is important in the generation of CSCs.
3. HISTONE MODIFICATIONS IN CSC PLASTICITY
The amino acid residues located on the N‐ and C‐terminal tails of histones can be modified to influence gene expression, including by acetylation, methylation and ubiquitylation. When occurring on promoters or enhancer regions, these modifications confer chromatin states that affect gene expression by altering the ability of protein complexes to bind. Chromatin formation that permits protein binding and gene expression is known as euchromatin, and repressive is known as heterochromatin. These states are mediated by the Trithorax group (TrxG) proteins and Polycomb group (PcG) proteins, respectively. Modifications are named by the histone type (eg, H3) followed by the amino acid and modification, for example, K4 me3 (trimethylation on lysine 4). The most studied modifications are those that occur to histone 3 found primarily at active enhancers (H3K9ac, H3K27ac), promoters (H3K4me3) and within the bodies of actively transcribed genes (H3K36me3). 38 Repressive marks include H3K27me3 and H3K9me3, also found on enhancers (Figure 3B).
Alterations of the histone modification landscape are widely associated with SCs with approximately one‐third of the genome differing in chromatin structure between differentiated cells and ESCs. 39 In general, the genome of pluripotent SCs is enriched for more transcriptionally permissive euchromatin and less heterochromatin relative to somatic cells. This means that PSCs contain more acetylated chromatin and smaller regions of the repressive marks H3K9me3 and H3K27me3 relative to differentiated cells. 39 Accordingly, certain protein modifying enzymes (eg, acetylases, demethylases) are important in PSCs and establishment of cell identity. 13 In particular, the repressive complexes PRC1 and PRC2 are both required for induction of pluripotency. 40
Although there are many parallels between PSCs and CSCs, histone‐modifying enzymes can have tumour‐specific effects. The subunit of the Polycomb group complex 2 (PRC2) that catalyses H3K27me3 is the histone methyltransferase EZH2 (enhancer of zeste homologue 2). EZH2 inhibition has been shown to be sufficient for tumourigenesis and is associated with the generation of CSCs in some cancers. 41 , 42 However, EZH2 has been found to be hyperactivated in breast cancer and PDAC. 43 , 44 , 45 Loss‐of‐function mutations of the H3K27 demethylases UTX3 and JMJD3 were also found to have completely opposite effects on tumour initiation in T‐ALL whereby loss of UTX3 acted as a tumour suppressor while the loss of JMJD3 promoted tumour initiation. 46
As well as methylation, other modifications have shown importance in CSCs. The BMI1 subunit of the polycomb repressive complex 1 (PRC1), which monoubiquitinates histone H2A on lysine 119 (H2AK119Ub1) was able to promote CSCs in leukaemia and GBM. 47 , 48 This was also the case for some HDACs: HDAC7 promoted tumourigenesis in the lung. 49 In addition to histone modifiers, dysregulation of some proteins that regulate chromatin structure, such as cohesins, are involved in promotion of stem cells and the generation of CSCs in leukaemias. 50 Furthermore, mutations that disrupt the function of chromatin‐remodelling complexes, and are found at high frequency in cancers, can cause aberrant activation of stem cell‐related pathways. 51 , 52
Nearly all the genes involved in ESC identity, including KLF4, Sox2, Oct4 and Nanog, are regulated by super‐enhancers; large genomic regions with very high levels of transcription factors that are highly important in controlling pluripotency and differentiation. 53 , 54 In turn, Sox2, Nanog and Oct4 co‐bind to various enhancers and super‐enhancers, thereby forming a self‐maintaining gene circuitry that maintains pluripotency. Aberrant activity of histone modifiers can impair the function of enhancer regions and effect cell identity, with certain modifications promoting the acquisition of stem‐like cells. 55 , 56 One study highlights the importance of enhancer reprogramming in the generation of stem cell identity by an oncogenic event. Overexpression of the oncogenic MYC in luminal human mammary epithelial cells (HMECs) led to downregulation of transcription factors and consequent loss of activity at enhancer regions important for maintaining luminal identity. At the same time, new enhancer regions were activated inducing transcriptional reprogramming to the stem‐like state. This enabled the HMECs to acquire stem‐like features including self‐renewal and multipotency. 57
Chromatin at promoters and enhancers can be classed as active, repressed or poised. Poised regions are bivalent in terms of histone modifications as they contain both H3K27me (repressive) and H3K4me (activating) histone marks. In ESCs, the majority of bivalency occurs in the promoters of transcription factors and half of bivalent domains have binding sites for pluripotency transcription factors. 58 In ESCs, bivalency tends to be lost upon differentiation with some enhancers becoming active (loss of H3K27me3, gain of H3K27ac) and some become repressed (H3K27me3 enrichment), this occurring in a cell type‐specific manner. 59 As the fluid interconversion between cellular states (normal and stem) depends on the capacity of a cell to switch on and off cell‐specific transcriptional programs, it is not surprising that poised chromatin is also important for determining cell identity and plasticity in CSCs. Indeed CSCs possess a more plastic and dynamic chromatin formation than differentiated cells. 60 In AML CSCs, genes involved in stem cell identity were bivalently marked with both H3K4me3 and H3K27me3, with H3K4me3 marks lost during differentiation. 61 Bivalent marks at pluripotency loci have also been identified in solid cancers. 62 , 63 In theory, poised chromatin can allow cellular identity to switch in any direction, that is, not only differentiation but also de‐differentiation, and may therefore be important in the generation of CSCs. Bivalency also allows for the highly dynamic state so characteristic of pluripotency, and may be the single most important epigenetic process for conferring CSC plasticity. A direct link between CSC generation and bivalency was demonstrated in breast cancer where CSCs marked by high CD44 expression were generated from CD44‐low cells by TGFβ stimulation. This process was dependent on poised, bivalent chromatin at the promoter of the ZEB1 gene, and conversion of bivalent to repressive marks rendered CD44‐low cells insensitive to TGFβ. 64
4. NUCLEOSOME POSITIONING AND HISTONE VARIANTS IN CSC PLASTICITY
The basic unit of chromatin is the nucleosome, which is formed of 147 bp of DNA wrapped around eight histones, two each of histones H2A, H2B, H3 and H4. These appear like beads on a string of chromatin. Gene expression is regulated at the level of chromatin structure in an ATP‐dependant process by chromatin modifiers which act to remove or slide assembled nucleosomes along with the DNA, and can also exchange histone H2A‐H2B dimers with dimers of histone variants. The presence of nucleosomes naturally represses gene expression by preventing the access of transcription factors. The absence or loss of nucleosomes at a transcription start site (nucleosome‐free region [NFR]) allows for assembly of the transcription machinery and rapid activation of gene expression. 65 , 66 The position of nucleosomes must therefore be precisely regulated at promoters, enhancers and repressors. This is achieved by four known families of ATP‐dependent chromatin remodelling complexes; switch/sucrose nonfermenting (SWI/ SNF), imitation switch (ISWI), inositol requiring 80 (INO80) and those with a NuRD/Mi‐2/CHD helicase binding domain. 52 , 67 , 68 , 69 Chromatin remodelling complexes use ATP hydrolysis to catalyse the assembly, sliding and ejecting of nucleosomes. 65 DNA‐sequence specificity is achieved by interaction with transcription factors. How nucleosome reorganisation is achieved is not fully understood but it is thought that remodelling complexes cause DNA to loop or twist to disrupt connection with histones and thus forcing translocation across a nucleosome. Chromatin modifiers can also interact with methylated DNA and covalent histone modifications to affect global gene expression patterns and chromatin architecture 70 , 71 (Figure 3C).
The chromatin architecture of SCs differs greatly from that of differentiated cells. While small‐scale chromatin changes at DNA regulatory elements occur during transcriptional regulation in most cells, extensive remodelling of chromatin structure is required for cellular differentiation during embryonic development. 72 , 73 , 74 In particular, the SWI/SNF remodelling complex is known to be important in ESCs and development: SWI/SNF complexes containing the ARID1A protein can remodel chromatin to inhibit expression of the pluripotency genes Sox2 and Oct4, and promote differentiation to mesoderm, but not ectoderm. 75 Nucleosome positioning is also important in the reprogramming of somatic cells to pluripotency. Nucleosomal landscapes of induced PSCs become extensively altered during reprogramming, in particular, nucleosome occupancy is reduced at enhancers which colocalise with binding sites of key pluripotency transcription factors, including Klf4, Oct4, Sox2 and c‐Myc. 76
As nucleosome repositioning is required for the generation of the pluripotent state it may be important for the formation of CSCs. This may occur via aberrant functions of ATP‐dependent chromatin remodelers, which has also been linked to cancer, especially in terms of the SWI/SNF complex, which is frequently found mutated. 77 , 78 , 79 In leukaemia, the SWI/SNF complex appears to function as it does in PSCs where the ATPase subunit BRG1 is required for nucleosome repositioning at the Myc enhancer to promote Myc expression, leading to enhanced self‐renewal. 80 , 81 However, in lung cancer, loss of either BRG1 or BRM ATPase subunits can promote cancer development. 82 The SNF5 (BAF47) subunit of the SWI/SNF complex is considered a tumour suppressor and its inhibition is sufficient to drive malignant transformation by inactivating p16 and p21. In the case of the rare paediatric rhabdoid tumour, SNF5 mutation is an inherited lesion that is considered the sole cause of this highly aggressive cancer. 83 Rhabdoid tumours are poorly differentiated and highly metastatic suggesting a high proportion of CSC features. This strongly links aberrant SWI/SNF‐dependent nucleosome remodelling with CSC generation. This process may also play a role in CSC generation in other cancers as mutations of SNF5 have been identified in epithelioid sarcoma and renal medullary carcinoma. 84 , 85
ATP‐dependent chromatin remodelling complexes can also regulate transcription via incorporation of histone variants into nucleosomes. 86 There are eight histone variants of H2A, and six of H3, which are deposited in specific locations along the genome. Histone variants can influence gene expression by directly altering the structure and stability of nucleosomes, or by recruiting readers of histone modifications to induce local chromatin changes. For example, nucleosomes that contain the histone variant H2A.Bbd, bind less DNA and are not as stable, resulting in less compact chromatin. Less stable nucleosomes including those that contain H2A.Bbd, H2A.Z or H3.3 are localised at active promoters, enhancers and insulators, and may serve to prevent the formation of stable nucleosomes around these regulatory regions and facilitate transcription. 87 In contrast, nucleosomes containing the histone variant macroH2A are relatively more stable and inhibit transcription. 88
High mobility and exchange of histone variants is a key feature of SCs and this dynamism is thought to contribute to SC plasticity. 89 , 90 The expression of histone variants is tightly regulated in ES cells and during development, and variants have specialised roles in cell fate decisions and lineage‐specification. 73 , 91 These variants, in turn, have a corresponding influence on reprogramming: those expressed in early development (the less stable H3.3, TH2A and TH2B) facilitate transcription and promote reprogramming whereas those expressed in somatic cells (the more stable macroH2A) inhibit transcription and prevent the induction of pluripotency. 91 As the histone variant macroH2A is considered an epigenetic barrier to reprogramming, a tumour suppressive function would be expected in cancer. 92 Accordingly, overexpression of macroH2A reduced metastatic potential of melanoma and inhibition of macroH2A1 generated CSCs in hepatocellular carcinoma. 93 , 94 Importantly, inhibition of macroH2A1 in bladder cancer increased stem‐like cells. 95 In contrast, the less stable H2A.Z and H3.3 variants promote transcription and reprogramming, and both have been found to be overexpressed in cancer. 96 H2A.Z correlates with poor prognosis in melanoma, breast cancer and hepatocellular carcinoma where it has been linked with EMT. 97 , 98 , 99 However, the role of H3.3 appears to differ in adult GBM where it is repressed in SCs and its overexpression impairs self‐renewal. Furthermore, repression of H3.3 in nonstem cancer cells is sufficient to induce cell plasticity and generate a CSC state. 100
These findings suggest that the overexpression of labile histone variants such H2A.Z (and in some cases H3.3), coupled with the reduction of stable histone variants such as macroH2A could contribute to cancer cell plasticity and the generation of CSCs.
4.1. Noncoding RNA and CSC plasticity
Noncoding RNAs (ncRNA) are transcribed from regions of the genome that do not encode for proteins. The resulting RNA transcripts function to regulate the expression of protein‐coding genes and are therefore essential for control of cellular function and identity. ncRNAs can be divided into two major groups based on their size: small ncRNAs being 200 nucleotides or less, and long ncRNAs (lncRNAs) 200 nucleotides or more. Small ncRNAs can be further subcategorized based on length, function and subcellular localization and include microRNAs (miRNAs) and short interfering RNAs (siRNAs) amongst others. 101 Long and short ncRNAs regulate gene expression by different modes of action.
4.2. Long ncRNAs
LncRNAs can physically associate with DNA or proteins to either promote or repress gene expression. To promote transcription, lncRNAs can function as guides or scaffolds for the assembly of protein complexes at specific loci. To repress transcription lncRNAs can function as decoys, binding and preventing functions of RNA or protein targets 102 (Figure 3D). Many lncRNAs are known to be involved in the regulation of pluripotency and cell fate transitions and are important in many types of adult stem cells. In particular, long intergenic ncRNAs (lincRNAs) which reside in gene deserts have tissue‐specific expression patterns that make them key players in the establishment of cellular identity. Knockdown of those lincRNAs associated with pluripotency results in differentiation of PSCs. 103 Furthermore, the expression of many lncRNAs is altered during the early stages of reprogramming to iPSCs. 104 Some lncRNAs associated with pluripotency have also been found to be upregulated in CSCs, for example, H19, which is involved in both mesenchymal SCs and glioblastoma CSCs. 105 , 106 Importantly, many lncRNAs have been implicated in the generation of CSCs via roles in cellular transformation and EMT. In particular, the lncRNA LINK‐RoR (regulator of reprogramming) is elevated in iPSCs and modulates iPS‐mediated reprogramming. 107 LINK‐RoR is also more highly expressed in CSCs as compared to more differentiated cancer cells and is important in a wide range of cancer types. 108 , 109 , 110 , 111 , 112 , 113 Furthermore, LINK‐RoR also induces EMT and may therefore be involved in the conversion of cancer cells to CSCs during tumourigenesis. 109 , 114 Many other lncRNAs play an important role in EMT. In particular, the lncRNA HOTAIR is involved in the recruitment of PRC2 to Hox genes during development and is also upregulated in TGF‐beta1‐responsive cancer cells during EMT. 115 , 116
4.3. Micro RNAs
MiRNAs function by recognising small sequences (6‐8 nucleotides) at the 3′‐untranslated region of multiple target mRNAs. 117 To do this, miRNAs are assembled in the RNA‐induced silencing complex (RISC), where complementarity between the miRNA and mRNA target sequence results in mRNA cleavage. If partial complementarity occurs, deadenylase complexes are recruited which remove or shorten the mRNA poly‐A tail to impair translation 101 (Figure 3E).
The importance of miRNAs in embryonic stem cells (ESCs) has been established by deletion of Dicer1 and DGCR8 (both critical for miRNA biogenesis) in mouse models. Dicer1 deletion is embryonic lethal whereas DGCR8‐deficient mice have impaired differentiation due to failure to silence the stem markers, Oct4, Sox2 and Nanog. 118 , 119 This phenotype can be rescued by expression of the let‐7 family miRNAs, which are not expressed in ESCs and appear during differentiation. 120 , 121 Let7 miRNA expression is controlled by a negative feedback loop with Lin‐28, which is a marker of pluripotency and is also important in reprogramming. 122 In PSCs and cancer cells, Lin‐28 binds to let‐7 precursors to block their maturation, whereas the let‐7 family miRNAs target Lin‐28 for degradation in differentiated cells. 121
Let7 is also known as a negative regulator of CSCs especially in breast cancer where, as expected, let‐7 was linked with differentiation. Inhibition of let‐7 promoted CSC properties, mediated by increased RAS and HMGA2 expression. 123 In turn, Lin‐28 has been shown to promote CSCs in breast and prostate cancer, 124 , 125 , 126 , 127 and even induce transformation in multiple cancers. 128 , 129 , 130 It was found that transformation of a normal breast cell line by the Src oncoprotein required an epigenetic switch whereby the resulting upregulation of NFkB by src directly activated transcription of Lin‐28, which in turn reduced let‐7 expression. Low let‐7 expression allowed for elevated levels of IL6, which in turn reactivated NFkB. This epigenetic positive feedback loop was found to be essential for maintaining the transformed state of breast cancer cells. 128 These findings strongly suggest that the Lin‐28/ let‐7 miRNA axis may control the generation of CSCs in cancer as it does with pluripotency of noncancerous cells.
As well as directly affecting the transcription of pluripotency‐associated genes, miRNAs can also affect CSC biology via cross‐talk with other CSC pathways and epigenetic mechanisms. For example, the maturation of miR‐21 via a DROSHA and RNA helicase p68 complex is induced by TGF‐β signalling. 131 The up‐regulation of miR‐21 promotes cancer invasion and metastasis by negatively regulating the expression of the tumour suppressor gene PDCD4. 132 Furthermore, miR‐22 promotes SCs in breast cancer through direct inhibition of TET activity, thus preventing demethylation of the mir‐200 promoter and silencing mir‐200 expression. 133 miRNA expression in SCs is also regulated by bivalent promoters. In particular, lineage‐specific miRNAs appear to be controlled in this manner, suggesting that an important mechanism underlying cellular plasticity is mediated by miRNAs. 134
5. THE ROLE OF EMT‐MET PLASTICITY IN THE EPIGENETIC REGULATION OF CSCS
Many studies have associated EMT with the generation of CSCs. 135 , 136 However two distinct but interconverting populations of CSCs have been identified in some cancers including breast and pancreatic; one being more epithelial‐like and one more mesenchymal‐like. 6 , 12 The functional relevance of two CSC populations is thought to lie in the fact that metastasis requires phenotypic plasticity: mesenchymal‐like properties are required for cellular dissemination whereas epithelial properties are required for attachment and proliferation within the foreign niche. 137 EMT‐MET plasticity can be conferred and maintained by epigenetic mechanisms. For example, a TGF‐β‐induced EMT resulted in a downregulation of p53 and the miRNAs 200c and 183, and concomitant elevation of the stem cell‐related genes Bmi1 and Klf4, which could be rescued by overexpression of miR‐ 200c or p53. 138 Bivalent histone markings could also mediate EMT‐MET plasticity as poised chromatin allows for flexible determination of gene expression.
CSC generation by EMT may also be influenced by the localised tissue environment via epigenetic modification. Cells present in the tumour CSC niche such as stromal and immune cells produce cytokines and growth factors which promote CSC characteristics. For example, tumour‐associated macrophages (TAMs) secrete VEGF and IL6, and T cells secrete IL17 and TGF‐β, all of which are able to promote CSCs via EMT. 139 , 140 In addition, myeloid‐derived stem cells in the TME secrete IL6 and nitric oxide that promote CSC characteristics via a Stat‐3 and mir101‐mediated EMT. 141 , 142 Environmental cross‐talk occurs both ways; epigenetic mechanisms in CSCs also alter their interaction with cells in the TME. For example, hypermethylation at the Tap promoter enhances the ability of CSCs to evade destruction by immune cells. 143
6. TRANSLATIONAL RELEVANCE OF CSC EPIGENETICS
Many epigenetic aberrations identified as being involved in the generation of CSCs are likely early markers of cancer and therefore potentially useful as early detection methods. Early cancer detection could be achieved by analysis of patient blood samples, which is considered a relatively noninvasive procedure compared to a biopsy. Patient blood can contain cancer cells and cell‐free DNA, which could be identified by characteristic genetic and epigenetic signatures as biomarkers of the cancerous state. Should such a process be found to be accurate, it may facilitate the detection of tumour signatures even when the tumour is not large enough to be picked up on a scan. This technique was originally designed for the identification of large genomic alterations, however, advances in the understanding of epigenetic aberrations in cancer have led to the pursuit of an epigenetic signature that may define the cancerous state. Although these may differ between cancers, epigenetic signatures associated with the generation of CSCs are likely candidates across cancer types due to the requirement for CSC generation in tumour initiation (Figure 4).
FIGURE 4.
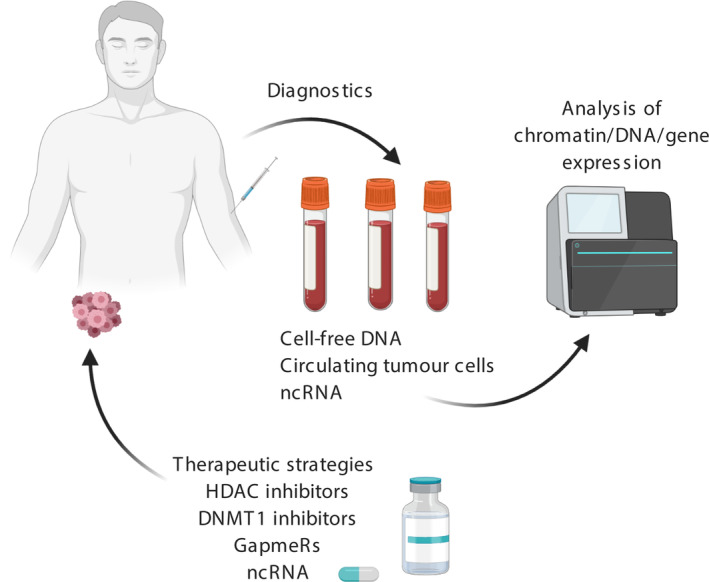
Translational relevance of epigenetic processes. Epigenetic mechanisms involved in CSC generation reveal factors with potential both as biomarkers for early diagnosis and as therapeutic targets. Early diagnosis could be achieved by analysis of cell‐free DNA, circulating tumour cells or noncoding RNAs for the presence of characteristic epigenetic marks. Epigenetic marks could also be used as targets for therapeutic agents, such as DMNT1 or HDAC inhibitors. Alternatively, ncRNAs or gapmeRs could be used as therapeutics to modify gene expression [Color figure can be viewed at wileyonlinelibrary.com]
Most approaches have focussed on the methylation status of particular gene promoters but it is likely that the methylation status of several promoters would be required to serve as an accurate and reliable signature. For example, hypermethylation of the combination of four genes: BCAT1, CDO1, TRIM58 and ZNF177, has been shown to predict lung cancer. 144 Recently, methylation patterns were used to detect and localise over 50 different cancer types. 22 Hypomethylation is a characteristic signature of cancer and CSCs, and considered an early event in tumourigenesis. Therefore, a global hypomethylation profile, defined by the presence of 5‐hmC marks, may be a useful sign of tumour presence. In particular, hypomethylation of SC‐associated genes such as Oct4, Sox2 and Nanog, together with or methylation of certain differentiation markers or tumour suppressor genes such as p53 may indicate the presence of CSCs and therefore early cancerous lesions.
An increase in histones and their associated covalent modifications have also been identified in the blood of cancer patients. 145 Bivalent histone signatures may be a promising avenue of exploration due to their specific association with CSCs. Unexpectedly, nucleosome positioning inferred from whole‐genome sequencing of plasma DNA correlates with RNA levels and furthermore with RNA in tumour tissue. This suggests that nucleosome positioning could also accurately reflect gene expression and be used to predict tumour incidence. 146
A useful form of epigenetic cancer detection is ncRNA. Noncoding RNAs are readily secreted by the tumour into the microenvironment and subsequently reach the bloodstream and even distant tissue sites. LncRNAs and miRNAs have been detected in blood, urine and saliva, all of which facilitate diagnostic testing and many have been identified as potential biomarkers. 147 , 148 Biomarkers have been identified through comparison of the pre and postoperative state, that is, a potentially accurate cancer biomarker should decrease upon removal of the tumour. Such candidates include mir‐221 and mir‐375 in breast and pancreatic cancer 149 , 150 and mir‐20, mir‐21, mir‐145 and mir‐223 in early‐stage lung cancer. 151 Some miRNAs may even be able to distinguish certain subtypes of cancer, for example, lower levels of miR‐16‐5p, miR‐21‐5p and miR‐199a‐5p were found to be associated specifically with the presence of triple‐negative breast cancer. 152 Many of those miRNAs found upregulated in cancer are also associated with CSCs. For example, mir‐221 was found to promote the generation of CSCs in breast cancer. Interestingly, this occurred via its role in the downregulation of DMNT3b, hence providing a link between a miRNA and reduced methylation, also associated with CSCs. 153 Mi‐21 is also a promoter of CSCs in many cancers especially colorectal. 154 , 155 These associations suggest that a role in CSCs may define what makes a useful and specific epigenetic biomarker for cancer.
Epigenetic targeting is an exciting area of translational cancer research offering many novel opportunities for therapeutic intervention. Many studies have explored the use of existing and novel compounds to target epigenetic marks to impair cancer cell growth and elicit an improved clinical response (Figure 4).
HDAC inhibitors are already widely considered to have anticancer effects and some are in clinical use. For example, SAHA (vorinostat) is approved for T‐cell lymphoma and many others are in clinical trials for various malignancies either alone or in combination with other therapeutics. 156 Despite promising anticancer effects, results have been mixed and in some cases accompanied by substantial toxicity. 157 , 158 However, preclinical studies have shown that a range of HDAC inhibitors can preferentially target CSCs in cancer cell lines. In breast cancer, these effects were attributed specifically to inhibition of the HDACs 1 and 7. 159 However, the HDAC inhibitor valproic acid, was actually able to increase the number of CSCs in breast cancer via inducing de‐differentiation to the stem‐like state (ie, CSC generation). 160 These effects were mediated by the promotion of wnt‐signalling. These conflicting results may reflect the specificities of different HDAC inhibitors, or the nonspecific effects of HDAC inhibitors in general, and may explain conflicting clinical data and toxicities. While there may be potential in pursuing HDAC inhibition as a CSC‐targeting approach, much further work is required to elucidate underlying mechanisms of action and avoid adverse effects. An alternative approach to targeting histone acetylation has been sought through bromodomain inhibitors. Bromodomains (BRDs) are readers of histone acetylation that target chromatin‐modifying enzymes to specific genomic sites to regulate transcription. The bromodomain and extra terminal (BET) family is made up of four different proteins that are important in cancer and also function as cell cycle regulators. The first developed inhibitor of the BET bromodomain family was JQ1. 161 , 162 A combination of JQ1 and the HDAC inhibitor SAHA promoted apoptosis in a number of cancers including PDAC. JQ1 also promoted apoptosis and cell‐cycle arrest of CSCs in glioma. 163 , 164
Targeting of histone modifications may also be achieved through LSD1 inhibitors. Lysine‐specific demethylase 1A (LSD1) removes methyl groups from the histone 3 proteins (H3K4me2/1 and H3K9me2/1) resulting in transcriptional repression or activation, respectively. 165 LSD1 is overexpressed in cancer, where it functions to inhibit differentiation and enhance proliferation, invasiveness and cell motility. 166 Effective LSD1 inhibitors have been developed by mimicking the lysine groups of the LSD phenyl ring at two positions (NCL‐1) or (NCL‐2). 167 NCL‐1 analogues have exhibited good anticancer activities across solid tumours including against CSCs. 168
Histone modification could also be targeted by inhibition of PRC components BMI‐1 or EZH2. BMI‐1 is a key regulatory component of (PRC1) and its function is strongly associated with the maintenance of CSCs. 169 , 170 Two small molecule inhibitors of BMI‐1, PTC‐209 and QW24, both impaired the self‐renewal of colorectal CSCs. 171 EZH2 is an enzymatic catalytic subunit of polycomb repressive complex 2 (PRC2), which is also associated with CSC function. 172 Several inhibitors of EZH2 methyltransferase activity have been developed with a 2‐pyridone core designed to occupy the binding pocket for cosubstrate S‐adenosyl‐methionine (SAM) in EZH2. GSK343 is one such SAM‐competitive inhibitor, which has been shown to inhibit stemness in patient‐derived glioma lines. 173 However, targeting of EZH2 may be unpredictable as it has been shown to have opposing effects across cancer types. 41 , 42 , 43 , 44 , 45 , 46
Although an increasing number of ncRNAs are known to be involved in CSCs and cancer in general, and are therefore attractive drug targets, it is considered difficult to specifically target aberrant mRNAs or RNA‐protein interactions with small molecules. Nevertheless, due to the high promise of such targets, a number of studies have attempted therapeutic modulation. The miRNA lin28 which is very important in CSC generation has been found to be targeted by the compound “N‐methyl‐N‐[3‐(3‐methyl[1,2,4]triazolo[4,3‐b]pyridazin‐6‐yl)phenyl]acetamide”, which acts by blocking the interaction between lin‐28 and let‐7. This compound impaired Lin‐28, rescued let‐7 function and resulted in differentiation in ESCs, and reduced tumorsphere formation in cancer cell lines. It is likely that the reduction in tumoursphere formation was caused by differentiation of CSCs. 174 An alternative approach is the administration of synthetic miRNAs to restore functions lost in cancer, for example, the introduction of tumour suppressor miRNAs. 175
Another way of targeting DNA and RNA moieties is by using peptide nucleic acids (PNA). These are synthetic analogues of DNA that are highly stable and have a hybridization affinity toward natural DNA and RNA, and are therefore a potential approach for the modification of gene expression and epigenetic states. 176 This approach has been applied to the targeting of the lncRNA HOTAIR to block its interaction with EZH2, resulting in reduced invasiveness and increased sensitivity to chemotherapy. 177 These effects strongly suggest inhibition of the stem‐cell compartment.
Despite promising results, therapeutic targeting of epigenetic marks should proceed with caution. A therapeutic strategy designed to target epigenetic readers and writers is by definition not a specific approach. Such an intervention would elicit gene expression changes on a global scale and is likely to incur off‐target and adverse effects. Therefore much further investigation is required to identify a clinically viable approach. Epigenetic targeting that focuses on a specific gene or pathway changes (eg, miRNA and lncRNA) may be more easily targeted to CSC inhibition and therefore have much greater potential for efficacy.
7. CONCLUSIONS AND FUTURE DIRECTIONS
The epigenetic processes involved in CSC generation have substantial implications for tumour initiation, metastasis and relapse. Many studies have focussed on the elucidation of mechanistic links between epigenetic processes and CSCs, and have identified attractive targets for cancer prevention, diagnosis and therapy. However, as always, these studies have highlighted many areas in which further investigation is required. In particular:
To obtain a greater understanding of the epigenetic mechanisms which drive CSC formation from normal cells.
To determine whether the mechanisms of CSC generation differ when deriving from normal or cancerous cells and their associated stem and nonstem subpopulations. Epigenetic mechanisms involved in the emergence of CSCs will likely differ depending on from which population they are derived, that is, from a normal cell, normal stem cell, pre‐CSC or bulk tumour cell, as each of these identities has distinct epigenetic profiles.
To ascertain the relative contribution of genetic and epigenetic effects to tumour initiation; can epigenetic events alone initiate the transformed state?
To determine which are the most important environmental signals involved in CSC generation and how these can be modified or targeted for prevention and treatment.
CONFLICT OF INTEREST
The authors note no conflict of interest.
French R, Pauklin S. Epigenetic regulation of cancer stem cell formation and maintenance. Int. J. Cancer. 2021;148:2884–2897. 10.1002/ijc.33398
Funding information Cancer Research UK, Grant/Award Number: C59392/A25064; Pancreatic Cancer UK, Grant/Award Number: 2018RIF_03
REFERENCES
- 1. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730‐737. [DOI] [PubMed] [Google Scholar]
- 2. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983‐3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396‐401. [DOI] [PubMed] [Google Scholar]
- 4. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106‐110. [DOI] [PubMed] [Google Scholar]
- 5. Ricci‐Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon‐cancer‐initiating cells. Nature. 2007;445:111‐115. [DOI] [PubMed] [Google Scholar]
- 6. Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313‐323. [DOI] [PubMed] [Google Scholar]
- 7. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105‐111. [DOI] [PubMed] [Google Scholar]
- 8. Zhu L, Finkelstein D, Gao C, et al. Multi‐organ mapping of Cancer risk. Cell. 2016;166:1132‐46.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmitt M, Schewe M, Sacchetti A, et al. Paneth cells respond to inflammation and contribute to tissue regeneration by acquiring stem‐like features through SCF/c‐kit signaling. Cell Rep. 2018;24:2312‐28.e7. [DOI] [PubMed] [Google Scholar]
- 10. Chaffer CL, Brueckmann I, Scheel C, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem‐like state. Proc Natl Acad Sci USA. 2011;108:7950‐7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gupta PB, Fillmore CM, Jiang G, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633‐644. [DOI] [PubMed] [Google Scholar]
- 12. Liu S, Cong Y, Wang D, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014;2:78‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boland MJ, Nazor KL, Loring JF. Epigenetic regulation of pluripotency and differentiation. Circ Res. 2014;115:311‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baylin S, Bestor TH. Altered methylation patterns in cancer cell genomes: cause or consequence? Cancer Cell. 2002;1:299‐305. [DOI] [PubMed] [Google Scholar]
- 15. Gaudet F, Hodgson JG, Eden A, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489‐492. [DOI] [PubMed] [Google Scholar]
- 16. Baylin SB, Jones PA. Epigenetic determinants of Cancer. Cold Spring Harb Perspect Biol. 2016;8(9):a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leitch HG, McEwen KR, Turp A, et al. Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol. 2013;20:311‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jackson SA, Sridharan R. The nexus of Tet1 and the pluripotency network. Cell Stem Cell. 2013;12:387‐388. [DOI] [PubMed] [Google Scholar]
- 19. Suelves M, Carrio E, Nunez‐Alvarez Y, Peinado MA. DNA methylation dynamics in cellular commitment and differentiation. Brief Funct Genomics. 2016;15:443‐453. [DOI] [PubMed] [Google Scholar]
- 20. Lister R, Mukamel EA, Nery JR, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holm TM, Jackson‐Grusby L, Brambrink T, Yamada Y, Rideout WM 3rd, Jaenisch R. Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell. 2005;8:275‐285. [DOI] [PubMed] [Google Scholar]
- 22. Liu S, Cheng K, Zhang H, et al. Methylation status of the Nanog promoter determines the switch between Cancer cells and Cancer stem cells. Adv Sci. 2020;7:1903035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mayle A, Yang L, Rodriguez B, et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2015;125:629‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yi JM, Tsai HC, Glockner SC, et al. Abnormal DNA methylation of CD133 in colorectal and glioblastoma tumors. Cancer Res. 2008;68:8094‐8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baba T, Convery PA, Matsumura N, et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene. 2009;28:209‐218. [DOI] [PubMed] [Google Scholar]
- 26. Kagara N, Huynh KT, Kuo C, et al. Epigenetic regulation of cancer stem cell genes in triple‐negative breast cancer. Am J Pathol. 2012;181:257‐267. [DOI] [PubMed] [Google Scholar]
- 27. Sun L, Mathews LA, Cabarcas SM, et al. Epigenetic regulation of SOX9 by the NF‐kappaB signaling pathway in pancreatic cancer stem cells. Stem Cells. 2013;31:1454‐1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Costa Y, Ding J, Theunissen TW, et al. NANOG‐dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013;495:370‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao Y, Chen J, Li K, et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell. 2013;12:453‐469. [DOI] [PubMed] [Google Scholar]
- 30. Doege CA, Inoue K, Yamashita T, et al. Early‐stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature. 2012;488:652‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morita R, Hirohashi Y, Suzuki H, et al. DNA methyltransferase 1 is essential for initiation of the colon cancers. Exp Mol Pathol. 2013;94:322‐329. [DOI] [PubMed] [Google Scholar]
- 32. Trowbridge JJ, Sinha AU, Zhu N, Li M, Armstrong SA, Orkin SH. Haploinsufficiency of Dnmt1 impairs leukemia stem cell function through derepression of bivalent chromatin domains. Genes Dev. 2012;26:344‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pathania R, Ramachandran S, Elangovan S, et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat Commun. 2015;6:6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu CC, Lin JH, Hsu TW, et al. IL‐6 enriched lung cancer stem‐like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int J Cancer. 2015;136:547‐559. [DOI] [PubMed] [Google Scholar]
- 35. Zagorac S, Alcala S, Fernandez Bayon G, et al. DNMT1 inhibition reprograms pancreatic Cancer stem cells via Upregulation of the miR‐17‐92 cluster. Cancer Res. 2016;76:4546‐4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee E, Wang J, Yumoto K, et al. DNMT1 regulates epithelial‐Mesenchymal transition and Cancer stem cells, which promotes prostate Cancer metastasis. Neoplasia. 2016;18:553‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wajed SA, Laird PW, DeMeester TR. DNA methylation: an alternative pathway to cancer. Ann Surg. 2001;234:10‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ernst J, Kheradpour P, Mikkelsen TS, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hawkins RD, Hon GC, Lee LK, et al. Distinct epigenomic landscapes of pluripotent and lineage‐committed human cells. Cell Stem Cell. 2010;6:479‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pereira CF, Piccolo FM, Tsubouchi T, et al. ESCs require PRC2 to direct the successful reprogramming of differentiated cells toward pluripotency. Cell Stem Cell. 2010;6:547‐556. [DOI] [PubMed] [Google Scholar]
- 41. Beguelin W, Popovic R, Teater M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Venkatesan N, Wong JF, Tan KP, et al. EZH2 promotes neoplastic transformation through VAV interaction‐dependent extranuclear mechanisms. Oncogene. 2018;37:461‐477. [DOI] [PubMed] [Google Scholar]
- 43. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB‐E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323‐5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kleer CG, Cao Q, Varambally S, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606‐11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. van Vlerken LE, Kiefer CM, Morehouse C, et al. Hurt EM. EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl Med. 2013;2:43‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arcipowski KM, Martinez CA, Ntziachristos P. Histone demethylases in physiology and cancer: a tale of two enzymes, JMJD3 and UTX. Curr Opin Genet Dev. 2016;36:59‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yuan J, Takeuchi M, Negishi M, Oguro H, Ichikawa H, Iwama A. Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia. 2011;25:1335‐1343. [DOI] [PubMed] [Google Scholar]
- 48. Abdouh M, Facchino S, Chatoo W, Balasingam V, Ferreira J, Bernier G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci. 2009;29:8884‐8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lei Y, Liu L, Zhang S, et al. Hdac7 promotes lung tumorigenesis by inhibiting Stat3 activation. Mol Cancer. 2017;16:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mazumdar C, Shen Y, Xavy S, et al. Leukemia‐associated Cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell. 2015;17:675‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jagani Z, Mora‐Blanco EL, Sansam CG, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the hedgehog‐Gli pathway. Nat Med. 2010;16:1429‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481‐492. [DOI] [PubMed] [Google Scholar]
- 53. Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super‐enhancers at key cell identity genes. Cell. 2013;153:307‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nord AS, Blow MJ, Attanasio C, et al. Rapid and pervasive changes in genome‐wide enhancer usage during mammalian development. Cell. 2013;155:1521‐1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Herz HM. Enhancer deregulation in cancer and other diseases. Bioessays. 2016;38:1003‐1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer. 2016;16:483‐493. [DOI] [PubMed] [Google Scholar]
- 57. Poli V, Fagnocchi L, Fasciani A, et al. MYC‐driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell‐like state. Nat Commun. 2018;9:1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315‐326. [DOI] [PubMed] [Google Scholar]
- 59. Bertero A, Madrigal P, Galli A, et al. Activin/nodal signaling and NANOG orchestrate human embryonic stem cell fate decisions by controlling the H3K4me3 chromatin mark. Genes Dev. 2015;29:702‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567‐1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yamazaki J, Estecio MR, Lu Y, et al. The epigenome of AML stem and progenitor cells. Epigenetics. 2013;8:92‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zaidi SK, Frietze SE, Gordon JA, et al. Bivalent epigenetic control of Oncofetal gene expression in Cancer. Mol Cell Biol. 2017;37(23):e00352‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dunican DS, Mjoseng HK, Duthie L, Flyamer IM, Bickmore WA, Meehan RR. Bivalent promoter hypermethylation in cancer is linked to the H327me3/H3K4me3 ratio in embryonic stem cells. BMC Biol. 2020;18:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chaffer CL, Marjanovic ND, Lee T, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013;154:61‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273‐304. [DOI] [PubMed] [Google Scholar]
- 66. Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen L, Cai Y, Jin J, et al. Subunit organization of the human INO80 chromatin remodeling complex: an evolutionarily conserved core complex catalyzes ATP‐dependent nucleosome remodeling. J Biol Chem. 2011;286:11283‐11289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vignali M, Hassan AH, Neely KE, Workman JL. ATP‐dependent chromatin‐remodeling complexes. Mol Cell Biol. 2000;20:1899‐1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011;11:588‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Harikrishnan KN, Chow MZ, Baker EK, et al. Brahma links the SWI/SNF chromatin‐remodeling complex with MeCP2‐dependent transcriptional silencing. Nat Genet. 2005;37:254‐264. [DOI] [PubMed] [Google Scholar]
- 71. Wysocka J, Swigut T, Xiao H, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86‐90. [DOI] [PubMed] [Google Scholar]
- 72. Gifford CA, Ziller MJ, Gu H, et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149‐1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. 2014;15:93‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang W, Li Y, Kulik M, et al. Nucleosome positioning changes during human embryonic stem cell differentiation. Epigenetics. 2016;11:426‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gao X, Tate P, Hu P, Tjian R, Skarnes WC, Wang Z. ES cell pluripotency and germ‐layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci USA. 2008;105:6656‐6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. West JA, Cook A, Alver BH, et al. Nucleosomal occupancy changes locally over key regulatory regions during cell differentiation and reprogramming. Nat Commun. 2014;5:4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Neely KE, Workman JL. The complexity of chromatin remodeling and its links to cancer. Biochim Biophys Acta. 2002;1603:19‐29. [DOI] [PubMed] [Google Scholar]
- 78. Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One. 2013;8:e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Oike T, Ogiwara H, Nakano T, Yokota J, Kohno T. Inactivating mutations in SWI/SNF chromatin remodeling genes in human cancer. Jpn J Clin Oncol. 2013;43:849‐855. [DOI] [PubMed] [Google Scholar]
- 80. Buscarlet M, Krasteva V, Ho L, et al. Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood. 2014;123:1720‐1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shi J, Whyte WA, Zepeda‐Mendoza CJ, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer‐mediated Myc regulation. Genes Dev. 2013;27:2648‐2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Marquez‐Vilendrer SB, Rai SK, Gramling SJ, Lu L, Reisman DN. Loss of the SWI/SNF ATPase subunits BRM and BRG1 drives lung cancer development. Onco Targets Ther. 2016;3:322‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chai J, Charboneau AL, Betz BL, Weissman BE. Loss of the hSNF5 gene concomitantly inactivates p21CIP/WAF1 and p16INK4a activity associated with replicative senescence in A204 rhabdoid tumor cells. Cancer Res. 2005;65:10192‐10198. [DOI] [PubMed] [Google Scholar]
- 84. McKenna ES, Roberts CW. Epigenetics and cancer without genomic instability. Cell Cycle. 2009;8:23‐26. [DOI] [PubMed] [Google Scholar]
- 85. McKenna ES, Sansam CG, Cho YJ, et al. Loss of the epigenetic tumor suppressor SNF5 leads to cancer without genomic instability. Mol Cell Biol. 2008;28:6223‐6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sarma K, Reinberg D. Histone variants meet their match. Nat Rev Mol Cell Biol. 2005;6:139‐149. [DOI] [PubMed] [Google Scholar]
- 87. Jin C, Zang C, Wei G, et al. H3.3/H2A.Z double variant‐containing nucleosomes mark ‘nucleosome‐free regions’ of active promoters and other regulatory regions. Nat Genet. 2009;41:941‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chakravarthy S, Luger K. The histone variant macro‐H2A preferentially forms "hybrid nucleosomes". J Biol Chem. 2006;281:25522‐25531. [DOI] [PubMed] [Google Scholar]
- 89. Boskovic A, Eid A, Pontabry J, et al. Higher chromatin mobility supports totipotency and precedes pluripotency in vivo. Genes Dev. 2014;28:1042‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Santenard A, Torres‐Padilla ME. Epigenetic reprogramming in mammalian reproduction: contribution from histone variants. Epigenetics. 2009;4:80‐84. [DOI] [PubMed] [Google Scholar]
- 91. Turinetto V, Giachino C. Histone variants as emerging regulators of embryonic stem cell identity. Epigenetics. 2015;10:563‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Creppe C, Janich P, Cantarino N, et al. MacroH2A1 regulates the balance between self‐renewal and differentiation commitment in embryonic and adult stem cells. Mol Cell Biol. 2012;32:1442‐1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kapoor A, Goldberg MS, Cumberland LK, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468:1105‐1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lo Re O, Fusilli C, Rappa F, et al. Induction of cancer cell stemness by depletion of macrohistone H2A1 in hepatocellular carcinoma. Hepatology. 2018;67:636‐650. [DOI] [PubMed] [Google Scholar]
- 95. Park SJ, Shim JW, Park HS, et al. MacroH2A1 downregulation enhances the stem‐like properties of bladder cancer cells by transactivation of Lin28B. Oncogene. 2016;35:1292‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zink LM, Hake SB. Histone variants: nuclear function and disease. Curr Opin Genet Dev. 2016;37:82‐89. [DOI] [PubMed] [Google Scholar]
- 97. Hua S, Kallen CB, Dhar R, et al. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol Syst Biol. 2008;4:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Vardabasso C, Gaspar‐Maia A, Hasson D, et al. Histone variant H2A.Z.2 mediates proliferation and drug sensitivity of malignant melanoma. Mol Cell. 2015;59:75‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yang HD, Kim PJ, Eun JW, et al. Oncogenic potential of histone‐variant H2A.Z.1 and its regulatory role in cell cycle and epithelial‐mesenchymal transition in liver cancer. Oncotarget. 2016;7:11412‐11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gallo M, Coutinho FJ, Vanner RJ, et al. MLL5 orchestrates a Cancer self‐renewal state by repressing the histone variant H3.3 and globally reorganizing chromatin. Cancer Cell. 2015;28:715‐729. [DOI] [PubMed] [Google Scholar]
- 101. Esteller M. Non‐coding RNAs in human disease. Nat Rev Genet. 2011;12:861‐874. [DOI] [PubMed] [Google Scholar]
- 102. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904‐914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Guttman M, Donaghey J, Carey BW, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kim DH, Marinov GK, Pepke S, et al. Single‐cell transcriptome analysis reveals dynamic changes in lncRNA expression during reprogramming. Cell Stem Cell. 2015;16:88‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Huang Y, Zheng Y, Jin C, Li X, Jia L, Li W. Long non‐coding RNA H19 inhibits adipocyte differentiation of bone marrow Mesenchymal stem cells through epigenetic modulation of histone Deacetylases. Sci Rep. 2016;6:28897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Jiang X, Yan Y, Hu M, et al. Increased level of H19 long noncoding RNA promotes invasion, angiogenesis, and stemness of glioblastoma cells. J Neurosurg. 2016;2016:129‐136. [DOI] [PubMed] [Google Scholar]
- 107. Loewer S, Cabili MN, Guttman M, et al. Large intergenic non‐coding RNA‐RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet. 2010;42:1113‐1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gao S, Wang P, Hua Y, et al. ROR functions as a ceRNA to regulate Nanog expression by sponging miR‐145 and predicts poor prognosis in pancreatic cancer. Oncotarget. 2016;7:1608‐1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lou Y, Jiang H, Cui Z, Wang L, Wang X, Tian T. Linc‐ROR induces epithelial‐to‐mesenchymal transition in ovarian cancer by increasing Wnt/beta‐catenin signaling. Oncotarget. 2017;8:69983‐69994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Peng WX, Huang JG, Yang L, Gong AH, Mo YY. Linc‐RoR promotes MAPK/ERK signaling and confers estrogen‐independent growth of breast cancer. Mol Cancer. 2017;16:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rezaei M, Emadi‐Baygi M, Hoffmann MJ, Schulz WA, Nikpour P. Altered expression of LINC‐ROR in cancer cell lines and tissues. Tumour Biol. 2016;37:1763‐1769. [DOI] [PubMed] [Google Scholar]
- 112. Wang L, Yu X, Zhang Z, et al. Linc‐ROR promotes esophageal squamous cell carcinoma progression through the derepression of SOX9. J Exp Clin Cancer Res. 2017;36:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zhang R, Hardin H, Huang W, Buehler D, Lloyd RV. Long non‐coding RNA Linc‐ROR is Upregulated in papillary thyroid carcinoma. Endocr Pathol. 2018;29:1‐8. [DOI] [PubMed] [Google Scholar]
- 114. Zhan HX, Wang Y, Li C, et al. LincRNA‐ROR promotes invasion, metastasis and tumor growth in pancreatic cancer through activating ZEB1 pathway. Cancer Lett. 2016;374:261‐271. [DOI] [PubMed] [Google Scholar]
- 115. Gupta RA, Shah N, Wang KC, et al. Long non‐coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Padua Alves C, Fonseca AS, Muys BR, et al. Brief report: the lincRNA Hotair is required for epithelial‐to‐mesenchymal transition and stemness maintenance of cancer cell lines. Stem Cells. 2013;31:2827‐2832. [DOI] [PubMed] [Google Scholar]
- 117. Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4:143‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bernstein E, Kim SY, Carmell MA, et al. Dicer is essential for mouse development. Nat Genet. 2003;35:215‐217. [DOI] [PubMed] [Google Scholar]
- 119. Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self‐renewal. Nat Genet. 2007;39:380‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Melton C, Judson RL, Blelloch R. Opposing microRNA families regulate self‐renewal in mouse embryonic stem cells. Nature. 2010;463:621‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wang L, Su Y, Huang C, et al. NANOG and LIN28 dramatically improve human cell reprogramming by modulating LIN41 and canonical WNT activities. Biol Open. 2019;8:bio047225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yu F, Yao H, Zhu P, et al. Let‐7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109‐1123. [DOI] [PubMed] [Google Scholar]
- 124. Albino D, Civenni G, Dallavalle C, et al. Activation of the Lin28/let‐7 Axis by loss of ESE3/EHF promotes a tumorigenic and stem‐like phenotype in prostate Cancer. Cancer Res. 2016;76:3629‐3643. [DOI] [PubMed] [Google Scholar]
- 125. Guo L, Cheng X, Chen H, et al. Induction of breast cancer stem cells by M1 macrophages through Lin‐28B‐let‐7‐HMGA2 axis. Cancer Lett. 2019;452:213‐225. [DOI] [PubMed] [Google Scholar]
- 126. Ottaviani S, Castellano L. microRNAs: novel regulators of the TGF‐beta pathway in pancreatic ductal adenocarcinoma. Mol Cell Oncol. 2018;5:e1499066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yang X, Lin X, Zhong X, et al. Double‐negative feedback loop between reprogramming factor LIN28 and microRNA let‐7 regulates aldehyde dehydrogenase 1‐positive cancer stem cells. Cancer Res. 2010;70:9463‐9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF‐kappaB, Lin28, Let‐7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Viswanathan SR, Powers JT, Einhorn W, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Wang YC, Chen YL, Yuan RH, et al. Lin‐28B expression promotes transformation and invasion in human hepatocellular carcinoma. Carcinogenesis. 2010;31:1516‐1522. [DOI] [PubMed] [Google Scholar]
- 131. Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA‐mediated microRNA maturation. Nature. 2008;454:56‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Asangani IA, Rasheed SA, Nikolova DA, et al. MicroRNA‐21 (miR‐21) post‐transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128‐2136. [DOI] [PubMed] [Google Scholar]
- 133. Song SJ, Poliseno L, Song MS, et al. MicroRNA‐antagonism regulates breast cancer stemness and metastasis via TET‐family‐dependent chromatin remodeling. Cell. 2013;154:311‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Iliou MS, Lujambio A, Portela A, et al. Bivalent histone modifications in stem cells poise miRNA loci for CpG Island hypermethylation in human cancer. Epigenetics. 2011;6:1344‐1353. [DOI] [PubMed] [Google Scholar]
- 135. Mani SA, Guo W, Liao MJ, et al. The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial‐mesenchymal transition. PLoS One. 2008;3:e2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ocana OH, Corcoles R, Fabra A, et al. Metastatic colonization requires the repression of the epithelial‐mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22:709‐724. [DOI] [PubMed] [Google Scholar]
- 138. Chang CJ, Chao CH, Xia W, et al. p53 regulates epithelial‐mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wan S, Zhao E, Kryczek I, et al. Tumor‐associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147:1393‐1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Nakano M, Kikushige Y, Miyawaki K, et al. Dedifferentiation process driven by TGF‐beta signaling enhances stem cell properties in human colorectal cancer. Oncogene. 2019;38:780‐793. [DOI] [PubMed] [Google Scholar]
- 141. Cui TX, Kryczek I, Zhao L, et al. Myeloid‐derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. 2013;39:611‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Peng D, Tanikawa T, Li W, et al. Myeloid‐derived suppressor cells endow stem‐like qualities to breast Cancer cells through IL6/STAT3 and NO/NOTCH cross‐talk signaling. Cancer Res. 2016;76:3156‐3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Sultan M, Vidovic D, Paine AS, et al. Epigenetic silencing of TAP1 in Aldefluor(+) breast Cancer stem cells contributes to their enhanced immune evasion. Stem Cells. 2018;36:641‐654. [DOI] [PubMed] [Google Scholar]
- 144. Diaz‐Lagares A, Mendez‐Gonzalez J, Hervas D, et al. A novel epigenetic signature for early diagnosis in lung cancer. Clin Cancer Res. 2016;22:3361‐3371. [DOI] [PubMed] [Google Scholar]
- 145. McAnena P, Brown JA, Kerin MJ. Circulating nucleosomes and nucleosome modifications as biomarkers in cancer. Cancers (Basel). 2017;9(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Murtaza M, Caldas C. Nucleosome mapping in plasma DNA predicts cancer gene expression. Nat Genet. 2016;48:1105‐1106. [DOI] [PubMed] [Google Scholar]
- 147. Bolha L, Ravnik‐Glavac M, Glavac D. Circular RNAs: biogenesis, function, and a role as possible Cancer biomarkers. Int J Genomics. 2017;2017:6218353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Filipow S, Laczmanski L. Blood circulating miRNAs as Cancer biomarkers for diagnosis and surgical treatment response. Front Genet. 2019;10:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Chen WX, Hu Q, Qiu MT, et al. miR‐221/222: promising biomarkers for breast cancer. Tumour Biol. 2013;34:1361‐1370. [DOI] [PubMed] [Google Scholar]
- 150. Kawaguchi T, Komatsu S, Ichikawa D, et al. Clinical impact of circulating miR‐221 in plasma of patients with pancreatic cancer. Br J Cancer. 2013;108:361‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zhang H, Mao F, Shen T, et al. Plasma miR‐145, miR‐20a, miR‐21 and miR‐223 as novel biomarkers for screening early‐stage non‐small cell lung cancer. Oncol Lett. 2017;13:669‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Shin VY, Siu JM, Cheuk I, Ng EK, Kwong A. Circulating cell‐free miRNAs as biomarker for triple‐negative breast cancer. Br J Cancer. 2015;112:1751‐1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Roscigno G, Quintavalle C, Donnarumma E, et al. MiR‐221 promotes stemness of breast cancer cells by targeting DNMT3b. Oncotarget. 2016;7:580‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Mamoori A, Gopalan V, Smith RA, Lam AK. Modulatory roles of microRNAs in the regulation of different signalling pathways in large bowel cancer stem cells. Biol Cell. 2016;108:51‐64. [DOI] [PubMed] [Google Scholar]
- 155. Sekar D, Krishnan R, Panagal M, Sivakumar P, Gopinath V, Basam V. Deciphering the role of microRNA 21 in cancer stem cells (CSCs). Genes Dis. 2016;3:277‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone Deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18:1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Goldstein LJ, Zhao F, Wang M, et al. A phase I/II study of suberoylanilide hydroxamic acid (SAHA) in combination with trastuzumab (Herceptin) in patients with advanced metastatic and/or local chest wall recurrent HER2‐amplified breast cancer: a trial of the ECOG‐ACRIN Cancer research group (E1104). Breast Cancer Res Treat. 2017;165:375‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Subramanian S, Bates SE, Wright JJ, Espinoza‐Delgado I, Piekarz RL. Clinical toxicities of histone Deacetylase inhibitors. Pharmaceuticals (Basel). 2010;3:2751‐2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Witt AE, Lee CW, Lee TI, et al. Identification of a cancer stem cell‐specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene. 2017;36:1707‐1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Debeb BG, Lacerda L, Xu W, et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/beta‐catenin signaling. Stem Cells. 2012;30:2366‐2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067‐1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Leal AS, Williams CR, Royce DB, Pioli PA, Sporn MB, Liby KT. Bromodomain inhibitors, JQ1 and I‐BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017;394:76‐87. [DOI] [PubMed] [Google Scholar]
- 163. Mazur PK, Herner A, Mello SS, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics‐based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21:1163‐1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Wen N, Guo B, Zheng H, et al. Bromodomain inhibitor jq1 induces cell cycle arrest and apoptosis of glioma stem cells through the VEGF/PI3K/AKT signaling pathway. Int J Oncol. 2019;55:879‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Wang J, Hevi S, Kurash JK, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125‐129. [DOI] [PubMed] [Google Scholar]
- 166. Pedersen MT, Helin K. Histone demethylases in development and disease. Trends Cell Biol. 2010;20:662‐671. [DOI] [PubMed] [Google Scholar]
- 167. Ueda R, Suzuki T, Mino K, et al. Identification of cell‐active lysine specific demethylase 1‐selective inhibitors. J Am Chem Soc. 2009;131:17536‐17537. [DOI] [PubMed] [Google Scholar]
- 168. Sareddy GR, Viswanadhapalli S, Surapaneni P, Suzuki T, Brenner A, Vadlamudi RK. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene. 2017;36:2423‐2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Jin M, Zhang T, Liu C, et al. miRNA‐128 suppresses prostate cancer by inhibiting BMI‐1 to inhibit tumor‐initiating cells. Cancer Res. 2014;74:4183‐4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Wang Y, Zhe H, Ding Z, Gao P, Zhang N, Li G. Cancer stem cell marker Bmi‐1 expression is associated with basal‐like phenotype and poor survival in breast cancer. World J Surg. 2012;36:1189‐1194. [DOI] [PubMed] [Google Scholar]
- 171. Kreso A, van Galen P, Pedley NM, et al. Self‐renewal as a therapeutic target in human colorectal cancer. Nat Med. 2014;20:29‐36. [DOI] [PubMed] [Google Scholar]
- 172. Gan L, Xu M, Hua R, et al. The polycomb group protein EZH2 induces epithelial‐mesenchymal transition and pluripotent phenotype of gastric cancer cells by binding to PTEN promoter. J Hematol Oncol. 2018;11:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Yu T, Wang Y, Hu Q, et al. The EZH2 inhibitor GSK343 suppresses cancer stem‐like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget. 2017;8:98348‐98359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Roos M, Pradere U, Ngondo RP, et al. A small‐molecule inhibitor of Lin28. ACS Chem Biol. 2016;11:2773‐2781. [DOI] [PubMed] [Google Scholar]
- 175. Ji W, Sun B, Su C. Targeting MicroRNAs in Cancer gene therapy. Genes (Basel). 2017;8(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Montazersaheb S, Hejazi MS, Nozad Charoudeh H. Potential of peptide nucleic acids in future therapeutic applications. Adv Pharm Bull. 2018;8:551‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Ozes AR, Wang Y, Zong X, Fang F, Pilrose J, Nephew KP. Therapeutic targeting using tumor specific peptides inhibits long non‐coding RNA HOTAIR activity in ovarian and breast cancer. Sci Rep. 2017;7:894. [DOI] [PMC free article] [PubMed] [Google Scholar]