

Abstract
Background:
Whether statin type influences hepatocellular carcinoma (HCC) incidence or mortality in chronic hepatitis B or C virus infection is unknown.
Objective:
To assess the relationship between lipophilic or hydrophilic statin use and HCC incidence and mortality in a nationwide population with viral hepatitis.
Design:
Prospective propensity score (PS)–matched cohort.
Setting:
Swedish registers, 2005 to 2013.
Participants:
A PS-matched cohort of 16 668 adults (8334 who initiated statin use [6554 lipophilic and 1780 hydrophilic] and 8334 nonusers) among 63 279 eligible adults.
Measurements:
Time to incident HCC, ascertained from validated registers. Statin use was defined from filled prescriptions as 30 or more cumulative defined daily doses (cDDDs).
Results:
Compared with matched nonusers, 10-year HCC risk was significantly lower among lipophilic statin users (8.1% vs. 3.3%; absolute risk difference [RD], –4.8 percentage points [95% CI, –6.2 to –3.3 percentage points]; adjusted subdistribution hazard ratio [aHR], 0.56 [CI, 0.41 to 0.79]) but not hydrophilic statin users (8.0% vs. 6.8%; RD, –1.2 percentage points [CI, –2.6 to 0.4 percentage points]; aHR, 0.95 [CI, 0.86 to 1.08]). The inverse association between lipophilic statins and HCC risk seemed to be dose-dependent. Compared with nonusers, 10-year HCC risk was lowest with 600 or more lipophilic statin cDDDs (8.4% vs. 2.5%; RD, –5.9 percentage points [CI, –7.6 to –4.2 percentage points]; aHR, 0.41 [CI, 0.32 to 0.61]), and 10-year mortality was significantly lower among both lipophilic (15.2% vs. 7.3%; RD, –7.9 percentage points [CI, –9.6 to –6.2 percentage points]) and hydrophilic (16.0% vs. 11.5%; RD, –4.5 percentage points [CI, –6.0 to –3.0 percentage points]) statin users.
Limitation:
Lack of lipid, fibrosis, or HCC surveillance data.
Conclusion:
In a nationwide viral hepatitis cohort, lipophilic statins were associated with significantly reduced HCC incidence and mortality. An association between hydrophilic statins and reduced risk for HCC was not found. Further research is needed to determine whether lipophilic statin therapy is feasible for prevention of HCC.
Primary Funding Source:
None.
Approximately 500 000 cases of hepatocellular carcinoma (HCC) are diagnosed worldwide each year (1), related primarily to chronic infection with hepatitis B virus (HBV) or hepatitis C virus (HCV) (2). In the United States and Europe, the incidence of HCC has tripled since the 1970s, and mortality is increasing more rapidly for HCC than for any other type of cancer (1, 3). The disease carries a grim prognosis, with limited treatment options and median survival of less than 1 year (2, 3). Although HCC risk is reduced with HBV suppression or HCV eradication, it may persist in high-risk patients or in those with advanced fibrosis (2, 4). Consequently, there is an urgent need to identify effective primary prevention strategies to reduce HCC-related morbidity and mortality.
Accumulating data suggest that in chronic liver disease, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) may improve clinical outcomes and reduce HCC risk (5, 6). However, the influence of different statin classes on HCC risk and survival is unknown. Recent experimental models (7) and observational studies (8–10) suggest that lipophilic statins (atorvastatin, simvastatin, fluvastatin, and lovastatin) may prevent hepatocarcinogenesis more effectively than hydrophilic statins (pravastatin or rosuvastatin). However, clinical evidence for the effectiveness of lipophilic or hydrophilic statins in chronic liver disease is scarce (8 – 15), and published studies have been limited by retrospective designs and a lack of detailed, updated data on statin class and dosage over time. As a result, the optimal statin type for effective HCC prevention remains unknown.
We examined the influence of lipophilic and hydrophilic statins on risk for incident HCC and death in a nationwide cohort of patients with confirmed chronic HBV or HCV infection in Sweden.
Methods
Data Sources
Hepatitis B and C are notifiable diseases under Swedish law, and the Register for Surveillance of Communicable Diseases maintains well-validated registers of all cases of acute and chronic HBV infection (since 1967) and HCV infection (since 1990) (16), including information on acute versus chronic infection (for HBV) and route of transmission. Cases are confirmed in duplicate by clinician report and serologic testing (hepatitis B surface antigen and HBV DNA, or HCV antibodies and HCV RNA) (16). We linked this database to the validated Patient Register, the Cause of Death Register, the Cancer Register, and the Prescribed Drug Register at the National Board of Health and Welfare, using the unique personal identity number assigned to Swedish residents. The Patient Register contains prospectively updated and detailed data on all hospitalizations (including surgeries and liver transplants), discharge diagnoses (since 1987), and specialty outpatient care (since 2001). All clinical diagnoses are coded using the International Classification of Diseases (ICD), with positive predictive values that exceed 85% in validation studies (17). This study was approved by the Ethics Review Board in Stockholm, Sweden (2017/707–31/2) on 15 June 2017.
Study Population
The population included patients aged 18 years or older with confirmed chronic HBV or HCV mono-infection who filled a first prescription for lipophilic or hydrophilic statins between 1 July 2005 and 31 December 2013. To ensure a new-user design (18), we required each statin user to fulfill a baseline period of at least 180 days between hepatitis B or C notification date and the date of the first filled statin prescription (index date). The index date for nonusers was randomly selected from recorded medical visits in the same calendar month and year as the matched statin user index date to ensure similar opportunities for care and treatment (19). We also required that nonusers fulfill the same entry criteria as statin users (19). We excluded patients prescribed a statin before diagnosis of HBV or HCV infection (n = 42 and 179, respectively), those with HCC diagnosed before or within the 180-day baseline period (n = 336), those with HIV infection (n = 4515) (8), and those who received lipophilic and hydrophilic statins (together or sequentially) during follow-up (n = 27), leaving 63 279 eligible patients for analysis (Supplement Figures 1A and 1B, available at Annals.org).
Exposures
Statins available in Sweden between 2005 and 2013 included simvastatin, atorvastatin, pravastatin, rosuvastatin, fluvastatin, and lovastatin. However, fluvastatin and lovastatin were rarely prescribed (<1%), a pattern that has been observed in other European countries (20–23). Our initial eligible population included no lovastatin users and 6 fluvastatin users, all of whom had previously used hydrophilic statins and therefore were excluded. Thus, statin exposures included simvastatin, atorvastatin, pravastatin, and rosuvastatin.
The Prescribed Drug Register has prospectively recorded all dispensed prescriptions from Swedish pharmacies since 1 July 2005 and is at least 99.7% complete (24). Statins are classified by Anatomical Therapeutic Chemical classification system code C10AA (Supplement Table 1, available at Annals.org). Each record includes prescription dates; dosage; number of pills; and defined daily dose (DDD), a standardized statistical measure of average daily drug consumption used by the World Health Organization. A single statin DDD is equivalent to a daily 30-mg dose of simvastatin, 20-mg dose of atorvastatin, 30-mg dose of pravastatin, or 10-mg dose of rosuvastatin. The sum of the total DDD over monthly intervals is the cumulative DDD (cDDD), a widely used measure of cumulative dose and duration of use (8, 11). Statin use was defined as 30 or more cDDDs, consistent with prior studies (8, 11). For analyses of dose and duration, we also created predefined, a priori categories: 0 to fewer than 30 (reference), 30 to 299, 300 to 599, and 600 or more cDDDs.
To construct the propensity score (PS), baseline demographic, clinical, and medication covariates were identified up to and including the index date (Supplement Methods and Supplement Table 1, available at Annals.org). For additional sensitivity analyses, we selected 10 prognostic covariates a priori on the basis of known or putative associations with study outcomes, including age (years); sex; duration of HBV or HCV infection (years); presence or absence of cirrhosis or diabetes; obesity; alcohol abuse; and prior use of antiviral therapy, aspirin, or metformin (Supplement Methods).
Outcomes
Study outcomes were ascertained from the validated Cancer Register and Cause of Death Register (25). The Cancer Register contains prospectively collected data from more than 96% of incident cancer cases, and HCC cases are confirmed by trained specialists using established pathologic or radiographic criteria (25). Secondary outcomes included all-cause and liver-specific mortality, ascertained from the Cause of Death Register, which is more than 99% complete. Liver-specific mortality was defined as a primary ICD code for chronic viral hepatitis, chronic liver disease, liver failure, variceal bleeding, spontaneous bacterial peritonitis, hepatorenal syndrome, or HCC.
Statistical Analysis
We used a PS-matched, prospective cohort design to balance baseline characteristics and minimize potential confounding (26). Exposure PSs were derived from the predicted probability of hydrophilic or lipophilic statin use, estimated in a logistic regression model, and conditioned on baseline covariates selected a priori (Supplement Methods). We used a published greedy nearest-neighbor within-caliper matching algorithm without replacement to select nonuser matches for each lipophilic or hydrophilic statin user in a 1:1 ratio (27) according to cohort (HBV or HCV) and index date (in the same month and year). Adequacy of matching was assessed by calculating standardized mean differences (28) and comparing PS distribution and balance (Supplement Figure 2 and Supplement Table 2, available at Annals.org).
Follow-up time accrued from the index date to the first recorded date of HCC diagnosis, all-cause death, liver transplant, emigration, or 31 December 2015. No heterogeneity was found between statin type and HCC risk by HBV or HCV cohort (P for heterogeneity = 0.37), and data were therefore pooled. Proportional hazards regression models that accounted for competing risks (29) were used to estimate adjusted cumulative incidences (percentages), adjusted subdistribution hazard ratios (aHRs), and corresponding 95% CIs for each outcome according to statin type and cDDD. We defined death, liver transplant, and emigration as the competing events for HCC, and we defined liver transplant and emigration as the competing events for death (Supplement Table 3A, available at Annals.org). We conducted the primary analysis of statin type in the PS-matched cohort, in which lipophilic or hydrophilic statin use was defined over study follow-up. In addition, we conducted analyses of statin dose and duration in the unmatched population using time-varying cDDD exposures and covariates to minimize potential immortal time bias. The main model was stratified by cohort (HBV or HCV) and PS quintile, from which we estimated adjusted absolute risk differences (RDs), with 95% CIs calculated via bootstrapping (30). In the unmatched population, we constructed an additional model that further accounted for age, sex, region, continuous PS, and a priori time-varying covariates, updated over each month of follow-up. In stratified models, we assessed the relationship between statin exposure and outcomes according to prespecified HCC risk factors, and we tested the significance of interactions using the log-likelihood ratio test. Schoenfeld residual tests identified no violations of the proportional hazards assumption.
We conducted several sensitivity analyses to test the robustness of our results. First, we evaluated statin use and type in the unmatched population using time-varying statin exposures with and without adjustment for PS, region, and time-updated covariates. Second, we repeated the cDDD analysis in the unmatched population after limiting the cohort to statin users. Third, within the PS-matched cohort, we compared our results using the following alternative analytic approaches: excluding HCC cases diagnosed during the first 4 years, extending the baseline period to 1 year, adjusting for baseline covariates and/or continuous PSs, using cDDD exposures defined in the PS-matched cohort over follow-up, using PS matching with data sorted in ascending (rather than descending) order of the PS, removing PS stratification, and restricting the cohort to lipophilic statin users (to compare current vs. former use) (31). In addition, we constructed separate models to assess atorvastatin, simvastatin, and nonstatin lipid-lowering agents. Finally, we performed an exploratory analysis of liver-specific mortality (Supplement Table 3B, available at Annals.org).
All P values were 2-tailed, and values less than 0.05 were considered statistically significant. All statistical analyses were performed using SAS, version 9.4 (SAS Institute). Propensity scores were estimated using the LOGISTIC procedure (Supplement Methods), with matching performed with the GREEDMTCH macro (27). The PHREG procedure was used to fit proportional hazards regression models.
Role of the Funding Source
Grants were received from the American College of Gastroenterology, the American Association for the Study of Liver Diseases, the Boston Nutrition Obesity Research Center, the National Institutes of Health, Nyckelfonden (Örebro, Sweden), Region Örebro County, and Karolinska Institutet. The funders had no role in the design, conduct, or reporting of this study.
Results
The unmatched population included 63 279 adults (15 104 with HBV infection and 48 175 with HCV infection), comprising 7827 lipophilic statin users and 2115 hydrophilic statin users (Supplement Tables 4A and 4B, available at Annals.org). Propensity score matching yielded a cohort of 16 668 patients (3906 with HBV infection and 12 762 with HCV infection), including 6554 lipophilic statin users and 1780 hydrophilic statin users, each matched in a 1:1 ratio with 8334 nonusers. After PS matching, all standardized mean differences were less than 0.1 between groups, indicating good covariate balance (Table 1) (28). Median follow-up was 8.0 years (range, 2 to 10 years) in nonusers and 7.9 years (range, 2 to 10 years) in lipophilic and hydrophilic statin users. Overall, we recorded 616 incident HCC cases and 1803 deaths.
Table 1.
Baseline Characteristics of Lipophilic and Hydrophilic Statin Users and Nonusers After Propensity Score Matching (n = 16 668)*
| Characteristic† | Statin Use |
Standardized Mean Difference‡ |
|||
|---|---|---|---|---|---|
| None (n = 8334) | Lipophilic (n = 6554)§ | Hydrophilic (n = 1780)|| | Lipophilic vs. None | Hydrophilic vs. None | |
| HBV infection, n (%) | 1953 (23.4) | 1540 (23.5) | 413 (23.2) | 0.000 | 0.000 |
| HCV infection, n (%) | 6381 (76.6) | 5014 (76.5) | 1367 (76.8) | 0.000 | 0.000 |
| Mean age at diagnosis (SD), y | |||||
| HBV infection | 47.3 (11.0) | 47.5 (11.5) | 47.5 (10.2) | 0.018 | 0.018 |
| HCV infection | 47.5 (13.7) | 47.6 (11.9) | 47.0 (12.5) | 0.008 | −0.04 |
| Female, % | 34.4 | 34.8 | 32.8 | 0.013 | −0.006 |
| Cirrhosis, % | 10.7 | 10.7 | 10.8 | 0.001 | 0.001 |
| Diabetes, % | 30.5 | 30.6 | 30.0 | 0.005 | −0.004 |
| Hypertension, % | 22.2 | 22.1 | 22.9 | −0.008 | 0.013 |
| Obesity, % | 6.2 | 6.2 | 6.0 | 0.001 | −0.002 |
| Coronary artery disease, % | 8.6 | 8.6 | 8.6 | −0.001 | −0.011 |
| Vascular disease, % | 4.1 | 4.2 | 3.7 | 0.002 | −0.004 |
| Congestive heart failure, % | 6.4 | 6.0 | 6.2 | −0.008 | −0.001 |
| Alcohol abuse history, % | 14.1 | 14.0 | 14.3 | −0.023 | 0.019 |
| Metformin use, % | 20.0 | 20.0 | 20.1 | 0.016 | 0.002 |
| Insulin use, % | 4.9 | 4.9 | 4.8 | 0.013 | −0.005 |
| Aspirin use, % | 35.5 | 35.4 | 36.0 | −0.007 | 0.002 |
| Current or prior antiviral therapy, n/N (%)¶ | |||||
| Anti-HBV | 450/1953 (23.0) | 356/1540 (23.1) | 94/413 (22.8) | 0.017 | −0.008 |
| Anti-HCV | 1286/6381 (20.2) | 1001/5014 (20.0) | 285/1367 (20.8) | −0.002 | 0.001 |
| Education, n (%) | |||||
| ≤9 y | 3052 (36.6) | 2403 (36.7) | 649 (36.4) | 0.011 | −0.016 |
| >9–12 y | 3281 (39.4) | 2596 (39.6) | 685 (38.5) | 0.024 | −0.007 |
| >12 y | 1764 (21.2) | 1378 (21.0) | 386 (21.7) | −0.028 | 0.035 |
| Unknown | 237 (2.8) | 177 (2.7) | 60 (3.4) | −0.006 | 0.020 |
HBV = hepatitis B virus; HCV = hepatitis C virus.
Baseline (index date) was defined as the first recorded date of a filled statin prescription after a 180-d statin-naive period.
Defined according to validated diagnosis codes in the International Classification of Diseases (Supplement Table 1, available at Annals.org).
Values <0.1 indicate good covariate balance (28).
Atorvastatin and/or simvastatin.
Rosuvastatin and/or pravastatin.
Use of antiviral therapy was assessed individually in the HBV and HCV cohorts and modeled up to the index date. Anti-HBV therapy was defined as ≥30 cumulative defined daily doses of interferon or a nucleos(t)ide reverse transcriptase inhibitor, and anti-HCV therapy was defined as ≥30 cumulative defined daily doses of interferon or direct-acting antiviral therapy.
Statin Use and Type
Figure 1 and Table 2 show cumulative incidence, adjusted RDs, and aHRs for incident HCC according to statin use and type in the PS-matched cohort. Compared with nonusers, lipophilic statin users had significantly lower 10-year cumulative incidence of HCC (8.1% vs. 3.3%; RD, –4.8 percentage points; aHR, 0.56 [95% CI, 0.41 to 0.79]). In contrast, compared with nonusers, the 10-year cumulative incidence of HCC was not significantly reduced among hydrophilic statin users (8.0% vs. 6.8%; RD, –1.2 percentage points; aHR, 0.95 [CI, 0.86 to 1.08]). These associations were similar in the individual HBV and HCV cohorts.
Figure 1.
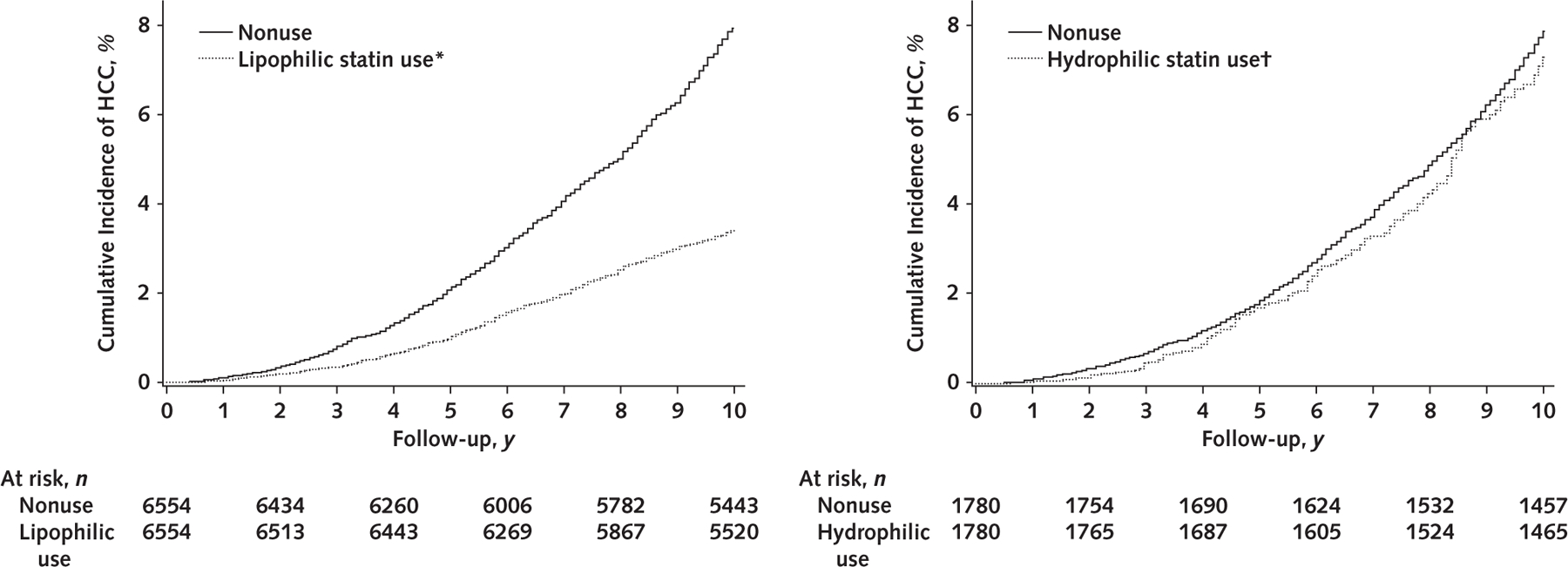
Cumulative incidence of HCC in the pooled propensity score–matched cohort (n = 16 668), by lipophilic statin use, hydrophilic statin use, or nonuse.
Statin use was defined as ≥30 cDDDs of filled statin prescriptions; nonuse was defined as <30 cDDDs or never-use. P < 0.001 for lipophilic statin users versus nonusers; P = 0.38 for hydrophilic statin users versus nonusers. P values were calculated using the Gray test for equality of the cumulative incidence functions between each group, with accounting for competing risks for death, emigration, and liver transplant. cDDD = cumulative defined daily dose; HCC = hepatocellular carcinoma.
* Atorvastatin and/or simvastatin.
† Rosuvastatin and/or pravastatin.
Table 2.
Ten-Year Cumulative Incidence, Absolute Risk Differences, and HRs for Incident HCC in the Propensity Score–Matched Cohort, by Statin Use and Type (n = 16 668)
| Variable* | Statin Use† |
|||
|---|---|---|---|---|
| No Lipophilic Statin Use | Lipophilic Statin Use‡ | No Hydrophilic Statin Use | Hydrophilic Statin Use§ | |
| Pooled cohort | ||||
| Events/person-years | 260/68 608 | 185/94 806 | 91/24 055 | 80/22 062 |
| Cumulative incidence, % | 8.1 | 3.3 | 8.0 | 6.8 |
| Absolute risk difference (95% CI), percentage points|| | 0 (reference) | −4.8 (−6.2 to −3.3) | 0 (reference) | −1.2 (−2.6 to 0.4) |
| HR (95% CI) | 1 (reference) | 0.56 (0.41 to 0.79) | 1 (reference) | 0.95 (0.86 to 1.08) |
| HCV cohort | ||||
| Events/person-years | 194/51 530 | 142/71 991 | 75/19 888 | 69/19 030 |
| Cumulative incidence, % | 8.1 | 3.1 | 7.9 | 6.7 |
| Absolute risk difference (95% CI), percentage points|| | 0 (reference) | −5.0 (−6.5 to −3.4) | 0 (reference) | −1.2 (−2.9 to 0.7) |
| HR (95% CI) | 1 (reference) | 0.54 (0.45 to 0.83) | 1 (reference) | 0.96 (0.87 to 1.10) |
| HBV cohort | ||||
| Events/person-years | 66/17 078 | 43/22 815 | 16/4167 | 11/3032 |
| Cumulative incidence, % | 8.2 | 3.4 | 8.0 | 6.8 |
| Absolute risk difference (95% CI), percentage points|| | 0 (reference) | −4.7 (−6.5 to −2.9) | 0 (reference) | −1.2 (−3.0 to 0.7) |
| HR (95% CI) | 1 (reference) | 0.58 (0.48 to 0.78) | 1 (reference) | 0.94 (0.84 to 1.14) |
HBV = hepatitis B virus; HCC = hepatocellular carcinoma; HCV = hepatitis C virus; HR = subdistribution hazard ratio.
Cumulative incidence, risk differences, and HRs were estimated using a proportional hazards regression model that was fitted to the propensity score–matched cohort, accounted for competing risks, and was stratified by cohort (HBV vs. HCV) and quintile of propensity score.
Defined as ≥30 cumulative defined daily doses.
Atorvastatin and/or simvastatin.
Rosuvastatin and/or pravastatin.
95% CI was calculated via bootstrapping.
In stratified models, the relationships between statin type and HCC incidence were similar across all prespecified subgroups, including sex, cirrhosis status, and antiviral therapy use (P for interaction > 0.05 for all) (Supplement Table 5, available at Annals.org). Moreover, the findings were consistent after statin type was compared according to cirrhosis status (P for heterogeneity = 0.40 and 0.31 for lipophilic and hydrophilic statins, respectively).
Next, we evaluated cumulative incidence, adjusted RDs, and aHRs for all-cause mortality according to statin use and type within the PS-matched cohort (Figure 2 and Table 3). Compared with nonusers, 10-year mortality was significantly lower among both lipophilic statin users (15.2% vs. 7.3%; RD, –7.9 percentage points; aHR, 0.62 [CI, 0.45 to 0.91]) and hydrophilic statin users (16.0% vs. 11.5%; RD, –4.5 percentage points; aHR, 0.88 [CI, 0.80 to 0.97]). These associations were similar in the individual HBV and HCV cohorts.
Figure 2.
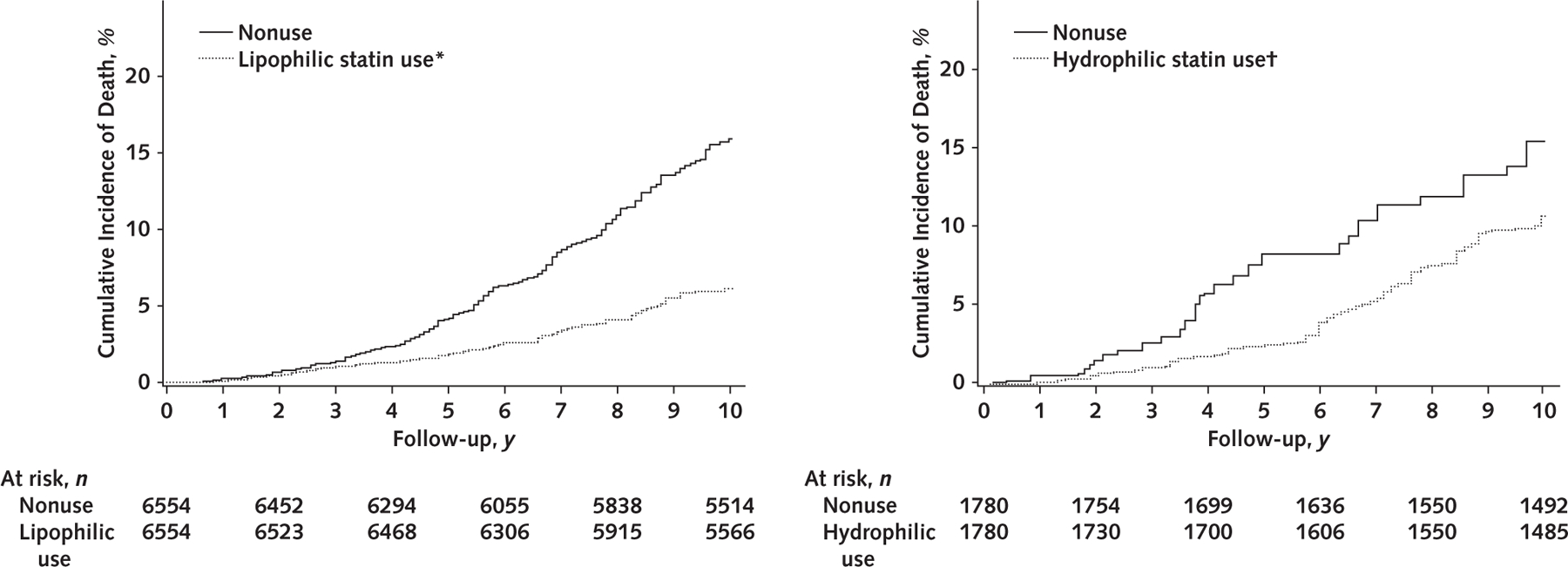
Cumulative incidence of all-cause mortality in the pooled propensity score–matched cohort (n = 16 668), by lipophilic statin use, hydrophilic statin use, or nonuse.
Statin use was defined as ≥30 cDDDs of filled statin prescriptions; nonuse was defined as <30 cDDDs or never-use. P < 0.001 for lipophilic statin users versus nonusers; P = 0.023 for hydrophilic statin users versus nonusers. P values were calculated using the Gray test for equality of the cumulative incidence functions between each group, with accounting for competing risks for death, emigration, and liver transplant. cDDD = cumulative defined daily dose.
* Atorvastatin and/or simvastatin.
† Rosuvastatin and/or pravastatin.
Table 3.
Ten-Year Cumulative Incidence, Absolute Risk Differences, and HRs for All-Cause Mortality in the Propensity Score–Matched Cohort, by Statin Use and Type (n = 16 668)
| Variable* | Statin Use† |
|||
|---|---|---|---|---|
| No Lipophilic Statin Use | Lipophilic Statin Use‡ | No Hydrophilic Statin Use | Hydrophilic Statin Use§ | |
| Pooled cohort | ||||
| Deaths/person-years | 798/87 653 | 644/100 162 | 199/22 056 | 162/20 114 |
| Cumulative incidence, % | 15.2 | 7.3 | 16.0 | 11.5 |
| Absolute risk difference (95% CI), percentage points|| | 0 (reference) | −7.9 (−9.6 to −6.2) | 0 (reference) | −4.5 (−6.0 to −3.0) |
| HR (95% CI) | 1 (reference) | 0.62 (0.45 to 0.91) | 1 (reference) | 0.88 (0.80 to 0.97) |
| HCV cohort | ||||
| Deaths/person-years | 684/75 082 | 568/87 417 | 175/19 366 | 146/18 112 |
| Cumulative incidence, % | 15.7 | 7.5 | 16.2 | 11.4 |
| Absolute risk difference (95% CI), percentage points|| | 1 (reference) | −8.2 (−9.9 to −6.4) | 1 (reference) | −4.8 (−6.3 to −3.2) |
| HR (95% CI) | 1 (reference) | 0.65 (0.41 to 0.85) | 1 (reference) | 0.87 (0.78 to 0.96) |
| HBV cohort | ||||
| Deaths/person-years | 114/12 201 | 76/12 745 | 24/2690 | 16/2002 |
| Cumulative incidence, % | 14.4 | 7.0 | 14.8 | 11.2 |
| Absolute risk difference (95% CI), percentage points|| | 1 (reference) | −7.4 (−9.0 to −5.8) | 1 (reference) | −3.6 (−5.4 to −1.8) |
| HR (95% CI) | 1 (reference) | 0.57 (0.33 to 0.92) | 1 (reference) | 0.91 (0.82 to 0.99) |
HBV = hepatitis B virus; HCV = hepatitis C virus; HR = subdistribution hazard ratio.
Cumulative incidence, risk differences, and HRs were estimated using a proportional hazards regression model that was fitted to the propensity score–matched cohort, accounted for competing risks, and was stratified by cohort (HBV vs. HCV) and quintile of propensity score.
Defined as ≥30 cumulative defined daily doses.
Atorvastatin and/or simvastatin.
Rosuvastatin and/or pravastatin.
95% CI was calculated via bootstrapping.
Statin Dose and Duration of Use
To characterize dose–response and duration–response relationships, we examined lipophilic and hydrophilic statin cDDDs in relation to study outcomes in the unmatched population (Table 4). We found a significant, dose-dependent inverse association between increased lipophilic statin cDDD and reduced HCC risk. Compared with nonusers, the adjusted 10-year RDs and corresponding aHRs were –3.4 percentage points (CI, –4.6 to –2.2 percentage points) and 0.75 (CI, 0.62 to 0.93) for 30 to 299 cDDDs, –4.6 percentage points (CI, –6.3 to –2.9 percentage points) and 0.52 (CI, 0.40 to 0.67) for 300 to 599 cDDDs, and –5.9 percentage points (CI, –7.6 to –4.2 percentage points) and 0.41 (CI, 0.32 to 0.61) for 600 or more cDDDs (P for trend < 0.001). In contrast, no significant reduction in HCC risk was found with increasing hydrophilic statin cDDD. Compared with nonusers, the adjusted 10-year RDs and aHRs were –1.1 percentage points (CI, –2.7 to 0.5 percentage points) and 0.97 (CI, 0.89 to 1.06) for 30 to 299 cDDDs, –1.7 percentage points (CI, –3.6 to 0.3 percentage points) and 0.92 (CI, 0.81 to 1.05) for 300 to 599 cDDDs, and –1.6 percentage points (CI, –3.4 to 0.2 percentage points) and 0.94 (CI, 0.82 to 1.03) for 600 or more cDDDs (P for trend = 0.60).
Table 4.
Ten-Year Cumulative Incidence, Absolute Risk Differences, and HRs for Incident HCC and All-Cause Mortality in the Unmatched Study Population, by Statin Type and cDDDs (n = 63 279)
| Variable* | Statin Use† |
P Value for Trend‡ | |||
|---|---|---|---|---|---|
| None | 30–299 cDDDs | 300–599 cDDDs | ≥600 cDDDs | ||
| Lipophilic statin use§ | |||||
| HCC | |||||
| Cumulative incidence (95% CI), % | 8.4 (7.8 to 9.0) | 5.0 (4.4 to 5.6) | 3.8 (3.3 to 4.3) | 2.5 (1.6 to 3.4) | |
| Absolute risk difference (95% CI), percentage points|| | 0 (reference) | −3.4 (−4.6 to −2.2) | −4.6 (−6.3 to −2.9) | −5.9 (−7.6 to −4.2) | |
| HR (95% CI) | 1 (reference) | 0.75 (0.62 to 0.93) | 0.52 (0.40 to 0.67) | 0.41 (0.32 to 0.61) | <0.001 |
| Death | |||||
| Cumulative incidence (95% CI), % | 15.1 (14.5 to 15.6) | 8.0 (7.3 to 8.8) | 7.9 (7.1 to 8.8) | 5.5 (5.0 to 6.0) | |
| Absolute risk difference (95% CI), percentage points|| | 0 (reference) | −7.1 (−8.3 to −5.9) | −7.2 (−8.5 to −6.0) | −9.6 (−11.2 to −8.1) | |
| HR (95% CI) | 1 (reference) | 0.79 (0.64 to 0.95) | 0.79 (0.64 to 0.94) | 0.58 (0.48 to 0.72) | 0.001 |
| Hydrophilic statin use¶ | |||||
| HCC | |||||
| Cumulative incidence (95% CI), % | 8.1 (7.9 to 8.8) | 7.0 (6.4 to 7.9) | 6.4 (5.7 to 7.1) | 6.5 (5.8 to 7.5) | |
| Absolute risk difference (95% CI), percentage points|| | 0 (reference) | −1.1 (−2.7 to 0.5) | −1.7 (−3.6 to 0.3) | −1.6 (−3.4 to 0.2) | |
| HR (95% CI) | 1 (reference) | 0.97 (0.89 to 1.06) | 0.92 (0.81 to 1.05) | 0.94 (0.82 to 1.03) | 0.60 |
| Death | |||||
| Cumulative incidence (95% CI), % | 15.9 (15.6 to 16.2) | 13.5 (13.1 to 13.9) | 10.6 (10.3 to 11.0) | 11.8 (11.1 to 12.5) | |
| Absolute risk difference (95% CI), percentage points|| | 0 (reference) | −2.4 (−3.5 to −1.3) | −5.3 (−6.4 to −4.2) | −4.1 (−5.5 to −2.7) | |
| HR (95% CI) | 1 (reference) | 0.92 (0.84 to 0.98) | 0.85 (0.79 to 0.91) | 0.88 (0.82 to 0.95) | 0.38 |
cDDD = cumulative defined daily dose; HBV = hepatitis B virus; HCC = hepatocellular carcinoma; HCV = hepatitis C virus; HR = subdistribution hazard ratio.
Cumulative incidence, risk differences, and HRs were estimated using a proportional hazards regression model that was fitted to the unmatched population, accounted for competing risks, and was stratified by cohort (HBV vs. HCV) and quintile of propensity score.
Defined as ≥30 cDDDs of filled statin prescriptions and modeled as a time-varying exposure updated at each month of follow-up.
P value for linear trend in the subdistribution hazards models was calculated using continuous cDDDs.
Atorvastatin and/or simvastatin.
95% CI was calculated via bootstrapping.
Rosuvastatin and/or pravastatin.
Increasing lipophilic statin cDDD was also significantly associated with dose-dependent reductions in all-cause mortality (Table 4). Compared with nonuse, the adjusted 10-year RDs and aHRs were –7.1 percentage points (CI, –8.3 to –5.9 percentage points) and 0.79 (CI, 0.64 to 0.95) for 30 to 299 cDDDs, –7.2 percentage points (CI, –8.5 to –6.0 percentage points) and 0.79 (CI, 0.64 to 0.94) for 300 to 599 cDDDs, and –9.6 percentage points (CI, –11.2 to –8.1 percentage points) and 0.58 (CI, 0.48 to 0.72) for 600 or more cDDDs (P for trend = 0.001). In contrast, dose-dependent inverse associations were not found with increasing hydrophilic statin cDDD. Compared with nonuse, the adjusted 10-year RDs and aHRs were –2.4 percentage points (CI, –3.5 to –1.3 percentage points) and 0.92 (CI, 0.84 to 0.98) for 30 to 299 cDDDs, –5.3 percentage points (CI, –6.4 to –4.2 percentage points) and 0.85 (CI, 0.79 to 0.91) for 300 to 599 cDDDs, and –4.1 percentage points (CI, –5.5 to –2.7 percentage points) and 0.88 (CI, 0.82 to 0.95) for 600 or more cDDDs (P for trend = 0.38).
Sensitivity Analyses
Our findings were similar when we assessed statin type in the unmatched population with and without adjustment for continuous PS, region, and potential confounders (Supplement Table 6A, available at Annals.org) and when we repeated the primary cDDD analyses after limiting the unmatched population to statin users (Supplement Table 6B, available at Annals.org). Furthermore, our findings were robust to several sensitivity analyses in the PS-matched cohort, including the following: after exclusion of patients with HCC diagnosed within 4 years (n = 176 excluded) (aHR, 0.62 [CI, 0.44 to 0.97]); after the baseline period was extended to 1 year (n = 372 excluded) (aHR, 0.68 [CI, 0.50 to 0.88]); after adjustment for baseline covariates and/or continuous PS (Supplement Table 7A, available at Annals.org); after alternative cDDD exposures that were defined over follow-up were applied (Supplement Tables 7B and 7C and Supplement Figures 3 and 4, available at Annals.org); when we examined atorvastatin (aHR, 0.40 [CI, 0.11 to 0.84]) and simvastatin (aHR, 0.60 [CI, 0.31 to 0.80]) separately; and when we used the matched cohort to directly compare current versus former lipophilic statin users (31) (aHR, 0.74 [CI, 0.55 to 0.94]). In an exploratory analysis of use of nonstatin lipid-lowering agents, we found no significant associations between HCC and mortality; however, this should be interpreted with caution because of the small numbers of cases and wide CIs (Supplement Table 8, available at Annals.org). An array-approach sensitivity analysis demonstrated that an unmeasured confounder would have to be very strongly associated with HCC risk and highly imbalanced between groups to fully attenuate the association between lipophilic statins and incident HCC risk (Supplement Tables 9A and 9B, available at Annals.org). Finally, we examined statin type and liver-specific mortality. Despite relatively few liver-related deaths (n = 462), we observed that compared with nonuse, liver-specific mortality was significantly lower with lipophilic statins (aHR, 0.76 [CI, 0.50 to 0.92]) but not hydrophilic statins (aHR, 0.98 [CI, 0.72 to 1.46]).
Discussion
In this nationwide, population-based cohort with viral hepatitis, lipophilic statin use was associated with substantially lower risk for incident HCC, all-cause mortality, and liver-related mortality compared with nonuse. The apparent benefits of lipophilic statins were dose- and duration-dependent, with the greatest reduction in HCC risk occurring after at least 600 cDDDs (the equivalent of taking a moderate-dose statin for approximately 2 years). Of note, our findings were robust across several sensitivity analyses and were similar in all predefined subgroups, including among men and women and persons with and without cirrhosis or antiviral therapy use. In contrast, we did not observe a significant association between hydrophilic statin use and HCC risk, suggesting that lipophilic statins may exert unique, class-specific hepatoprotective effects.
Our findings confirm prior data linking statins with improved survival (6, 15) and reduced HCC risk (5) in chronic liver disease. However, little is known about the relative impact of lipophilic or hydrophilic statins on HCC chemoprevention and survival. Two cohort studies showed equivalent HCC risk reduction with lipophilic and hydrophilic statins (11, 32), but others have not found similar associations (12, 13) or reported benefit exclusively with lipophilic statins (9, 10, 14). In Western populations, epidemiologic data focusing on statin dose and type are derived from just 2 published studies of predominantly male U.S. veterans (8, 32). However, both were limited by retrospective designs and included highly selected populations (male veterans with cirrhosis), and neither simultaneously evaluated dose–response relationships by statin type. In contrast, our study benefits from an unselected, population-based cohort of men and women with prospectively collected data over long-term follow-up, thus providing more compelling evidence of an inverse, dose-dependent relationship between lipophilic statin use and incident HCC.
Several lines of evidence support a role for lipophilic statins in HCC prevention. First, preclinical studies suggest that lipophilic statins prevent viral replication, potentiate antiviral therapy, and stimulate antitumor immunity more effectively than hydrophilic statins (33–37). Second, in the setting of inflammation and hepatocarcinogenesis, reduced hepatocyte surface expression of organic anion transporter proteins may prevent hydrophilic statins from entering hepatocytes (38, 39). In contrast, lipophilic statins passively diffuse across cell membranes and thus may provide more potent, cholesterol-dependent anti-HCC effects (34, 40). Third, unlike hydrophilic statins, lipophilic statins exert consistent and potent antitumor effects, including induction of G0/G1 cell cycle arrest (41), inhibition of Ras/Raf/Mek/ERK signaling (38), and triggering apoptosis (7). Finally, applying simvastatin to HepG2 cells induces the proapoptotic BAX gene and downregulates the antiapoptotic BCL-2 gene, suggesting that pretranslational events may further augment the hepatic response to lipophilic statins (42).
To our knowledge, this is the first population-based cohort study to comprehensively evaluate statin classes in relation to HCC risk and mortality. This work benefits from a nationwide population with confirmed hepatitis and from the high specificity and near-complete follow-up of the Swedish registers, which minimize selection bias. We applied several techniques to address potential bias from residual confounding, survival, and indication, including PS matching, a new-user design (18), and sensitivity analyses directly comparing current and former statin users (31). In addition, conducting time-dependent analyses of statin exposures in the unmatched cohort minimized potential exposure misclassification and immortal time bias (a follow-up period where study outcomes cannot occur) (43).
We acknowledge several limitations. First, we cannot exclude the possibility of residual confounding. Swedish registers lack information on smoking, HBV DNA, HCV eradication, fibrosis stage, and HCC screening and are subject to underreporting of conditions relevant to HCC risk, such as obesity. They also lack laboratory data, so we could not address the influence of changing cholesterol levels on statin use or outcomes. Further, because direct-acting antivirals were recently introduced in Sweden, numbers and follow-up for patients treated with those medications were limited. Collectively, these factors highlight the need for future research in highly phenotyped populations. Nevertheless, our findings were robust across multiple sensitivity analyses, including an array-based analysis that showed it would be highly unlikely for an unmeasured confounder to fully attenuate the association between lipophilic statins and HCC risk. Second, we acknowledge that temporal trends, regional variation, or provider concerns with regard to liver fibrosis could influence statin prescribing patterns and outcomes. However, patients were matched by index date, and our findings were similar after we accounted for cirrhosis and region. Third, we could not evaluate remote statin use before 1 July 2005 because Swedish prescription data were not available before that date. However, our findings were similar in time-varying analyses and in analyses limited to statin users. Fourth, we did not directly compare lipophilic and hydrophilic statins, and we also used cDDDs, which simultaneously account for cumulative dose and duration. Accordingly, short durations of use at high dosages can yield cDDDs that are equivalent to long durations at low dosages. Finally, the Swedish HCV cohort is primarily white, and some of our subgroups had small numbers of cases, underscoring the need for future research in larger, more diverse populations. Nevertheless, our estimated HCC incidence approximates that in published epidemiologic cohorts (8), and the influence of statins on all-cause mortality accords with prior cardiovascular studies (44), which supports the generalizability of our findings.
In conclusion, in a prospective, nationwide population with chronic viral hepatitis, lipophilic statin use was associated with significantly reduced risk for both incident HCC and death. A similar association with reduced HCC risk was not found with hydrophilic statin use. These findings support further research to characterize the potential hepatoprotective benefits of lipophilic statins.
Supplementary Material
Grant Support:
Dr. Simon was supported by a North American Training Grant from the American College of Gastroenterology. Dr. Duberg was supported by a research ALF grant from Region Örebro County (OLL-683801). Dr. Hagstrom was supported by a Stockholm County Council clinical postdoctoral appointment. Dr. Khalili was supported by grant K23 DK099681 from the National Institutes of Health. Dr. Chung was supported by grant K24 DK078772 from the National Institutes of Health.
Footnotes
Dr. Ludvigsson had access to all data in the study and was responsible for the decision to submit the manuscript for publication.
Disclosures: Dr. Duberg reports personal fees from AbbVie, Bristol-Myers Squibb, Gilead, Janssen, and MSD outside the submitted work. Dr. Aleman reports honoraria for lectures and expert group discussions from AbbVie, Bristol-Myers Squibb, Gilead, and MSD and grants from AbbVie and Gilead outside the submitted work. Dr. Chung reports grants from Gilead Sciences, MassBiologics, Merck, AbbVie, Bristol-Myers Squibb, Boehringer Ingelheim, Janssen, Synlogic, Roche, and Kaleido and personal fees from Alnylam outside the submitted work. Dr. Ludvigsson reports other support from Janssen outside the submitted work. Authors not named here have disclosed no conflicts of interest. Disclosures can also be viewedat www.acponline.org/authors/icmje/ConflictOfInterestForms.do?msNum=M18-2753.
Reproducible Research Statement: Study protocol: Available from Dr. Ludvigsson (jonasludvigsson@yahoo.com). Statistical code and data set: Not available.
Contributor Information
Tracey G. Simon, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts.
Ann-Sofi Duberg, Örebro University Hospital, Örebro, Sweden.
Soo Aleman, Karolinska University Hospital, Stockholm, Sweden.
Hannes Hagstrom, Karolinska University Hospital and Karolinska Institutet, Stockholm, Sweden.
Long H. Nguyen, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts.
Hamed Khalili, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, and Karolinska Institutet, Stockholm, Sweden.
Raymond T. Chung, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts.
Jonas F. Ludvigsson, Karolinska Institutet and Karolinska University Hospital, Stockholm, Sweden, Örebro University Hospital, Örebro, Sweden, and Columbia University College of Physicians and Surgeons, New York, New York.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210 [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–1273.e1. doi: 10.1053/j.gastro.2011.12.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryerson AB, Eheman CR, Altekruse SF, et al. Annual Report to the Nation on the Status of Cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer. 2016;122:1312–37. doi: 10.1002/cncr.29936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Terrault NA, Bzowej NH, Chang KM, et al. ; American Association for the Study of Liver Diseases. AASLD guidelines for treatment of chronic hepatitis B. Hepatology. 2016;63:261–83. doi: 10.1002/hep.28156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh S, Singh PP, Singh AG, et al. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. 2013;144:323–32. doi: 10.1053/j.gastro.2012.10.005 [DOI] [PubMed] [Google Scholar]
- 6.Kim RG, Loomba R, Prokop LJ, et al. Statin use and risk of cirrhosis and related complications in patients with chronic liver diseases: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 2017;15:1521–1530.e8. doi: 10.1016/j.cgh.2017.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menter DG, Ramsauer VP, Harirforoosh S, et al. Differential effects of pravastatin and simvastatin on the growth of tumor cells from different organ sites. PLoS One. 2011;6:e28813. doi: 10.1371/journal.pone.0028813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simon TG, Bonilla H, Yan P, et al. Atorvastatin and fluvastatin are associated with dose-dependent reductions in cirrhosis and hepatocellular carcinoma, among patients with hepatitis C virus: results from ERCHIVES. Hepatology. 2016;64:47–57. doi: 10.1002/hep.28506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai SW, Liao KF, Lai HC, et al. Statin use and risk of hepatocellular carcinoma. Eur J Epidemiol 2013;28:485–92. doi: 10.1007/s10654-013-9806-y [DOI] [PubMed] [Google Scholar]
- 10.McGlynn KA, Hagberg K, Chen J, et al. Statin use and risk of primary liver cancer in the Clinical Practice Research Datalink. J Natl Cancer Inst 2015;107. doi: 10.1093/jnci/djv009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsan YT, Lee CH, Wang JD, et al. Statins and the risk of hepatocellular carcinoma in patients with hepatitis B virus infection. J Clin Oncol 2012;30:623–30. doi: 10.1200/JCO.2011.36.0917 [DOI] [PubMed] [Google Scholar]
- 12.Chiu HF, Ho SC, Chen CC, et al. Statin use and the risk of liver cancer: a population-based case–control study. Am J Gastroenterol 2011;106:894–8. doi: 10.1038/ajg.2010.475 [DOI] [PubMed] [Google Scholar]
- 13.McGlynn KA, Divine GW, Sahasrabuddhe VV, et al. Statin use and risk of hepatocellular carcinoma in a U.S. population. Cancer Epidemiol 2014;38:523–7. doi: 10.1016/j.canep.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsushita Y, Sugihara M, Kaburagi J, et al. Pravastatin use and cancer risk: a meta-analysis of individual patient data from long-term prospective controlled trials in Japan. Pharmacoepidemiol Drug Saf 2010;19:196–202. doi: 10.1002/pds.1870 [DOI] [PubMed] [Google Scholar]
- 15.Stokkeland K, Lageborn CT, Ekbom A, et al. Statins and angiotensin-converting enzyme inhibitors are associated with reduced mortality and morbidity in chronic liver disease. Basic Clin Pharmacol Toxicol 2018;122:104–10. doi: 10.1111/bcpt.12844 [DOI] [PubMed] [Google Scholar]
- 16.Duberg AS, Törner A, Davidsdóttir L, et al. Cause of death in individuals with chronic HBV and/or HCV infection, a nationwide community-based register study. J Viral Hepat 2008;15:538–50. doi: 10.1111/j.1365-2893.2008.00982.x [DOI] [PubMed] [Google Scholar]
- 17.Ludvigsson JF, Andersson E, Ekbom A, et al. External review and validation of the Swedish National Inpatient Register. BMC Public Health. 2011;11:450. doi: 10.1186/1471-2458-11-450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ray WA. Evaluating medication effects outside of clinical trials: new-user designs. Am J Epidemiol 2003;158:915–20. [DOI] [PubMed] [Google Scholar]
- 19.Mohanty A, Tate JP, Garcia-Tsao G. Statins are associated with a decreased risk of decompensation and death in veterans with hepatitis C-related compensated cirrhosis. Gastroenterology. 2016;150: 430–40.e1. doi: 10.1053/j.gastro.2015.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Svensson E, Nielsen RB, Hasvold P, et al. Statin prescription patterns, adherence, and attainment of cholesterol treatment goals in routine clinical care: a Danish population-based study. Clin Epidemiol 2015;7:213–23. doi: 10.2147/CLEP.S78145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberfroid D, Dubois C, Vrijens F, et al. Les statines en Belgique: évolutions de l′utilisation et impact des politiques de remboursement. Health Services Research (HSR). KCE Reports 141B. D/2010/10.273/70. Brussels: Centre fédéral d′expertise des soins de santé; 2010. Accessed at https://kce.fgov.be/sites/default/files/atoms/files/kce_141b_statines_en_belgique.pdf on 19 June 2019.
- 22.Chapman SR, Fitzpatrick RW, Aladul MI. Has cost inhibited the uptake of more potent statins in England? Pharmacoepidemiol Drug Saf 2017;26:984–91. doi: 10.1002/pds.4231 [DOI] [PubMed] [Google Scholar]
- 23.Redlich C, Berk M, Williams LJ, et al. Statin use and risk of depression: a Swedish national cohort study. BMC Psychiatry 2014;14: 348. doi: 10.1186/s12888-014-0348-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wettermark B, Hammar N, Fored CM, et al. The new Swedish Prescribed Drug Register–opportunities for pharmacoepidemiological research and experience from the first six months. Pharmacoepidemiol Drug Saf 2007;16:726–35. [DOI] [PubMed] [Google Scholar]
- 25.Barlow L, Westergren K, Holmberg L, et al. The completeness of the Swedish Cancer Register: a sample survey for year 1998. Acta Oncol 2009;48:27–33. doi: 10.1080/02841860802247664 [DOI] [PubMed] [Google Scholar]
- 26.Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res 2011;46:399–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parsons LS. Reducing bias in a propensity score matched-pair sample using greedy matching techniques [Abstract]. In: Proceedings of the Twenty-Sixth Annual SAS Users Group International Conference, Long Beach, California, 22–25 April 2001. Cary, NC: SAS Institute; 2001:214. Abstract no. 214–26. Accessed at www2.sas.com/proceedings/sugi26/p214-26.pdf on 2 February 2018. [Google Scholar]
- 28.D′Agostino RB Jr. Propensity score methods for bias reduction in the comparison of a treatment to a non-randomized control group. Stat Med 1998;17:2265–81. [DOI] [PubMed] [Google Scholar]
- 29.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc 1999;94:496–509. doi: 10.2307/2670170 [DOI] [Google Scholar]
- 30.Austin PC. Absolute risk reductions and numbers needed to treat can be obtained from adjusted survival models for time-to-event outcomes. J Clin Epidemiol 2010;63:46–55. doi: 10.1016/j.jclinepi.2009.03.012 [DOI] [PubMed] [Google Scholar]
- 31.Mamtani R, Lewis JD, Scott FI, et al. Disentangling the association between statins, cholesterol, and colorectal cancer: a nested case-control study. PLoS Med 2016;13:e1002007. doi: 10.1371/journal.pmed.1002007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaplan DE, Serper MA, Mehta R, et al. ; VOCAL Study Group. Effects of hypercholesterolemia and statin exposure on survival in a large national cohort of patients with cirrhosis. Gastroenterology. 2019;156:1693–1706.e12. doi: 10.1053/j.gastro.2019.01.026 [DOI] [PubMed] [Google Scholar]
- 33.Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol Metab 2010;21:33–40. doi: 10.1016/j.tem.2009.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kapadia SB, Chisari FV. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc Natl Acad SciUS A 2005;102:2561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dorobantu C, Macovei A, Lazar C, et al. Cholesterol depletion of hepatoma cells impairs hepatitis B virus envelopment by altering the topology of the large envelope protein. J Virol 2011;85:13373–83. doi: 10.1128/JVI.05423-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xia Y, Xie Y, Yu Z, et al. The mevalonate pathway is a druggable target for vaccine adjuvant discovery. Cell 2018;175:1059–1073.e21. doi: 10.1016/j.cell.2018.08.070 [DOI] [PubMed] [Google Scholar]
- 37.Bader T, Korba B. Simvastatin potentiates the anti-hepatitis B virus activity of FDA-approved nucleoside analogue inhibitors in vitro. Antiviral Res 2010;86:241–5. doi: 10.1016/j.antiviral.2010.02.325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kato S, Smalley S, Sadarangani A, et al. Lipophilic but not hydrophilic statins selectively induce cell death in gynaecological cancers expressing high levels of HMGCoA reductase. J Cell Mol Med 2010;14:1180–93. doi: 10.1111/j.1582-4934.2009.00771.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thakkar N, Slizgi JR, Brouwer KLR. Effect of liver disease on hepatic transporter expression and function. J Pharm Sci 2017;106: 2282–94. doi: 10.1016/j.xphs.2017.04.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karlic H, Thaler R, Gerner C, et al. Inhibition of the mevalonate pathway affects epigenetic regulation in cancer cells. Cancer Genet 2015;208:241–52. doi: 10.1016/j.cancergen.2015.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sutter AP, Maaser K, Höpfner M, et al. Cell cycle arrest and apoptosis induction in hepatocellular carcinoma cells by HMG-CoA reductase inhibitors. Synergistic antiproliferative action with ligands of the peripheral benzodiazepine receptor. J Hepatol 2005;43:808–16. [DOI] [PubMed] [Google Scholar]
- 42.Spampanato C, De Maria S, Sarnataro M, et al. Simvastatin inhibits cancer cell growth by inducing apoptosis correlated to activation of Bax and down-regulation of BCL-2 gene expression. Int J Oncol 2012;40:935–41. doi: 10.3892/ijo.2011.1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Walraven C, Davis D, Forster AJ, et al. Time-dependent bias was common in survival analyses published in leading clinical journals. J Clin Epidemiol 2004;57:672–82. [DOI] [PubMed] [Google Scholar]
- 44.Ford I, Murray H, McCowan C, et al. Long-term safety and efficacy of lowering low-density lipoprotein cholesterol with statin therapy: 20-year follow-up of West of Scotland Coronary Prevention Study. Circulation 2016;133:1073–80. doi: 10.1161/CIRCULATIONAHA.115.019014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.