

Summary
Primary human bone marrow adipocytes (BM-Ads) display a specific metabolism that is not recapitulated by in vitro differentiated bone marrow mesenchymal stromal cells. These findings highlight the need for using primary BM-Ads in studies of the metabolic impact of BM-Ads on surrounding cells. Here, we present a protocol for isolating human BM-Ads from bone marrow aspirates and verifying adipocyte suspension purity. These isolated and purified BM-Ads can be used for functional assays or frozen for molecular analyses.
For complete details on the use and execution of this protocol, please refer to Attane et al. (2020).
Subject areas: Cell Biology, Cell isolation, Cancer, Metabolism, Microscopy
Graphical abstract
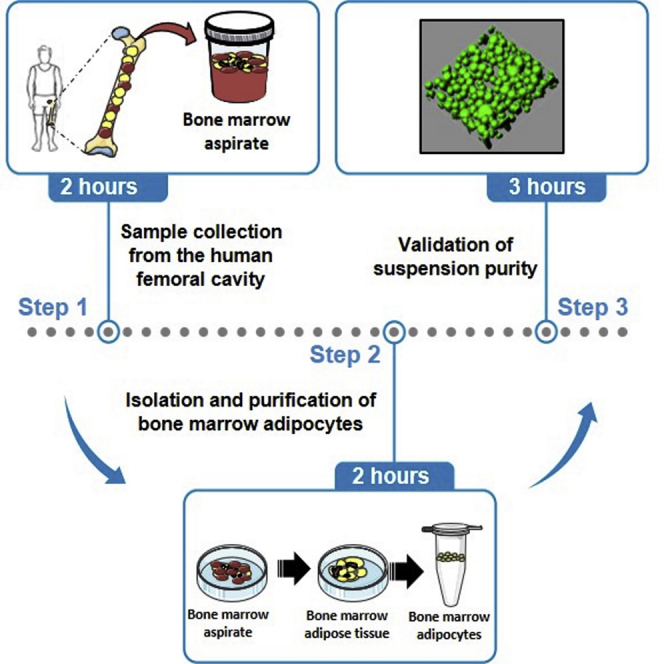
Highlights
-
•
Isolation of human bone marrow adipocytes (BM-Ads) from femoral cavity
-
•
Purification of BM-Ads to eliminate contaminant cells
-
•
Validation of BM-Ad quality and suspension purity with immunofluorescence imaging
Primary human bone marrow adipocytes (BM-Ads) display a specific metabolism that is not recapitulated by in vitro differentiated bone marrow mesenchymal stromal cells. These findings highlight the need for using primary BM-Ads in studies of the metabolic impact of BM-Ads on surrounding cells. Here, we present a protocol for isolating human BM-Ads from bone marrow aspirates and verifying adipocyte suspension purity. These isolated and purified BM-Ads can be used for functional assays or frozen for molecular analyses.
Before you begin
-
1.
All experiments using human tissue must be performed with the approval of national ethic committee and all patients must give informed consent. For this article and our previous work (Attane et al., 2020), all patients gave informed consent and the samples were obtained according to national ethics committee rules (authorization DC-2017-2914).
-
2.
Set up a collaboration with orthopedic surgeons to organize sample collection from patients undergoing hip surgery.
-
3.
As temperature changes should be avoided to limit the lysis of adipocytes, it is better to keep sample at 37°C for the transport. Prepare transport bag at least 3 h before going to the surgical theater: warm 3 hot packs at 37°C (to maintain the temperature in the bag during the transport of samples) and 50 mL tube containing 20 mL KRBHA for tissue collection (Figure 1).
-
4.
If you plan to use BM-Ads for cell culture, sterilize tweezers and scissors.
-
5.
Make sure you have enough stock solutions (described in material and equipment) for all procedures.
-
6.
Before going to the surgical theater, warm 100 mL KRBHA to 37°C.
Figure 1.

Transport system
(A) Transport bag must mention legal annotation according local legislation.
(B) Warmpack, 50 mL collection tube containing 20 mL KRBHA, tube holder and transport bag required for BM-aspirate transport. Warm KRBHA and warmpack to 37°C before leaving the lab.
(C) Organization of the bag for the transport
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Bone marrow aspirate | Orthopedic Surgery Unit | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Krebs-Ringer Bicarbonate Buffer powder | Sigma-Aldrich | K4002 |
| HEPES | Sigma-Aldrich | H3375 |
| NaHCO3 | Sigma-Aldrich | S5761 |
| BSA, fatty acid free | Sigma-Aldrich | A7030 |
| PBS, no calcium, no magnesium | Thermo Fisher Scientific | 14190-144 |
| Collagenase from Clostridium histolyticum (lot: 107M4042V) | Sigma-Aldrich | C6885-500MG |
| Cell strainer 100 μm | Greiner Bio-One | 542000 |
| NaCl | Sigma-Aldrich | S7653 |
| CaCl2·2H2O | Sigma-Aldrich | 1023820250 |
| Fibrinogen from bovine plasma | Sigma-Aldrich | F8630 |
| Thrombin from bovine plasma | Sigma-Aldrich | T6634 |
| Paraformaldehyde 32% aqueous solution | Electron Microscopy Sciences | 15714 |
| Rhodamine-coupled phalloidin | Thermo Fisher Scientific | R415 |
| TO-PRO3 iodide 642/661 | Thermo Fisher Scientific | T3605 |
| BODIPY 493/503 | Thermo Fisher Scientific | D3922 |
| Software and algorithms | ||
| ImageJ software | https://imagej.nih.gov/ij | N/A |
| Imaris software (v9.2) | Bitplane (https://imaris.oxinst.com/) | N/A |
| Other | ||
| Stainless steel dissecting scissors | N/A | N/A |
| Stainless steel dissecting tweezers | N/A | N/A |
| 20 mL Syringes | N/A | N/A |
| 21G Needles | N/A | N/A |
| 150 μm Coverslips | N/A | N/A |
| Orbital shaker | N/A | N/A |
| Transport bag for biological samples | N/A | N/A |
| Micro centrifuge for 1.5–2.0 mL tubes | N/A | N/A |
| Zeiss LSM 710 confocal microscope | Zeiss | N/A |
Materials and equipment
Krebs Ringer Buffer HEPES Albumin (KRBHA)
| Reagent | Final concentration | Amount |
|---|---|---|
| KRBH | n/a | One vial resuspended with 900 mL deionized H2O (dH2O) |
| NaHCO3 | 150 mM | 1.26 g |
| HEPES | 10 mM | 2.38 g |
| BSA | 0.5% | 5 g |
| dH2O | n/a | Qsp 1L |
Stir to dissolve.
Adjust the pH to 7.4
Filter the solution through a 0.22 μm filter under a laminar flow hood.
Stock solution should be sterile and can be stored for 2 months at 4°C.
PBS 2% BSA
| PBS 2% BSA | 20 g BSA, fill up to 1L with PBS. |
Filter the solution through a 0.22 μm filter under a laminar flow hood.
Stock solution can be stored for 2 months at 4°C.
Collagenase 5× (1250 CDU/mL)
| Collagenase 5× | Calculate the volume of PBS 2% BSA to obtain a solution at 1250 CDU/mL. Each collagenase lot possesses a specific activity and the dilution volume is therefore specific for each lot |
Filter the solution through a 0.22 μm filter under a laminar flow hood.
Prepare aliquots and store at −20°C.
Stock solution should be sterile and can be stored for one year at −20°C.
CRITICAL: To be reproducible, it is important to use the same lot of collagenase for the different experiments within a study. Thus, the necessary number of aliquots of 5× collagenase should be prepared and stored at −20°C. Before preparing large stocks, it is important to validate collagenase efficiency and determine the optimal digestion time since differences exist among collagenase types and providers. To evaluate collagenase efficiency, it is recommended to work with adipocytes that possess lipolytic activity (i.e. subcutaneous adipocytes from lean individuals), since lipolysis assays are easy to perform and well calibrated. First, it is important to determine the optimal digestion duration to obtain the expected amount of adipocytes after digestion (we typically obtain around 600 μL of subcutaneous adipocytes per gram of tissue). Digestion must be stopped when there are no more detectable pieces of intact tissue and before the occurrence of oil (see step 6, critical point). Usually, optimal digestion time should not exceed 30 minutes for subcutaneous adipocytes. This digestion duration can be decreased for BM-Ads which are much more fragile than subcutaneous adipocytes. Quality of digestion can be assessed by microscopy approaches as described below (steps 9–12). Then, it is mandatory to verify that collagenase digestion does not affect SC-Ads function. In vitro lipolysis assays can be performed as described in our previous manuscript (Attane et al., 2020). Furthermore, if researchers are interested in the characterization of the bone marrow adipose tissue microenvironment, stromal-vascular cells should be recovered as well. Stromal-vascular cell identity and viability can be assessed by flow cytometry (Decaunes et al., 2011).
Alternatives: Collagenase from other suppliers or other enzymes such as Liberase can be used for the digestion step. Their efficiency as well as the optimal concentration to use and digestion time must be determined in a pilot experiment as described above. Moreover, addition of collagenase and liberase could also be considered to optimize digestion, limit the need for extensive washing and therefore limit the lysis of the adipocytes. The efficiency of this procedure must also be tested and optimized in a pilot experiment.
Fibrinogen (18 g/L)
| Fibrinogen (18 g/L) | Dissolve 180 mg of fibrinogen in 10 mL NaCl 0.9%. Spread fibrinogen powder on the NaCl solution and mix gently with a magnetic stirrer at 20°C–25°C. |
Filter the solution through a 0.22 μm filter under a laminar flow hood. Avoid vacuum filtration system but use syringe with a 0.22 μm filter.
Prepare aliquots and store at −20°C.
Stock solution should be sterile and can be stored for 6 months at −20°C.
Thrombin (100 U/mL)
| Thrombin (100 U/mL) | Dissolve one vial of 250 U in 2.5 mL sterile NaCl 0.9% under a laminar flow hood. |
Prepare aliquots and store at −20°C.
Stock solution should be sterile and can be stored for 6 months at −20°C.
Fibrinogen and thrombin are sensitive to freeze-thawing. Aliquot volume must be determined according to the experiments planned. Thawed fibrinogen aliquots are stable at 4°C for 5 days.
PBS 2 mM CaCl2
| PBS 2 mM CaCl2 | Dissolve 29.4 mg CaCl2 in 100 mL PBS under a laminar flow hood. |
Filter the solution through a 0.22 μm filter under a laminar flow hood.
Store at 4°C for up to 2 weeks.
Paraformaldehyde (PFA) 3.7%
| Paraformaldehyde 3.7% | Mix 10 mL PFA 32% with 76 mL PBS. |
Aliquot and store at −20°C for up to 6 months. Thawed PFA is stable for at 4°C for up to 1 week.
Step-by-step method details
Sample collection and transport
Timing: 2 h
Surgeons perform sample collection. Bone marrow (BM) containing bone marrow adipose tissue (BM-AT) is harvested from the femoral cavity of patients undergoing hip surgery. Then, the collected sample is rapidly transferred to the lab for processing.
-
1.Aspiration of BM from the femoral proximal metaphysis and diaphysis
-
a.During the surgical procedure, exposure is made as routinely. During femur preparation, after the femoral neck is cut, the surgeon has access to the intramedullary canal. If required, the first broach can open the canal, especially in young patients with dense trabecular bone
-
b.Aspirate BM cautiously with 60 mL syringe and a soft cannula, e.g., a 4.0 mm tracheal tube, in the femoral proximal metaphysis and diaphysis.
-
a.
-
2.
Immediately place the sample (10–20 mL bone marrow aspirate) in the 37°C pre-warmed tube containing 20 mL KRBHA.
-
3.
Place the tube in the transport bag (Figure 1) and bring the sample back to the laboratory within 1 h for processing.
Note: This procedure describes how to collect BM from femoral cavity. It can be adapted to collect BM from other long bones such as tibia during knee surgery.
BM-Ad isolation and purification
This step consists of isolating bone marrow adipocytes (BM-Ads) from BM-AT and removing hematopoietic cells to obtain a pure BM-Ad suspension.
-
4.Separately, wash and collect BM samples (Figures 2A–2E). As BM aspirate often contain a significant amount of blood, it is necessary to rinse the aspirate with KRBHA to facilitate dissection.
-
a.Place a 100 μm cell strainer on top of a 50 mL tube, rinse the strainer with KRBHA, and pour the tissue sample on top.
-
b.Pour 20 mL KRBHA on the sample to remove blood.
-
c.Collect BM sample present on the strainer with tweezers and transfer it to a petri dish.
-
a.
-
5.Dissect and weigh the BM-AT. BM samples contain “red marrow”, an area rich in hematopoietic cells, as well as “yellow marrow”, an area rich in BM-Ads present in the BM-AT. To avoid contamination with hematopoietic cells and increase digestion efficiency, it is recommended to keep only the BM-AT (Figures 2F and 2G).
-
a.Using tweezers and scissors isolate the areas of BM-AT that are recognized by their yellow color and transfer this material to a new petri dish.
-
b.Add 10 mL KRBHA to the petri dish to wash the BM-AT.
-
c.Transfer BM-AT to a pre-weighed 14 mL tube.
-
d.Weigh the tube containing BM-AT and calculate the tissue weight.
-
a.
-
6.Digest BM-AT (Figures 3A–3D).
-
a.Use 2.5 mL collagenase 1× solution per gram of tissue. Calculate the volume of collagenase 1× solution necessary for the experiment and prepare it by diluting the 5× stock solution in PBS 2% BSA.
-
b.Add the collagenase 1× solution to the tube containing the BM-AT, close the tube and seal it with parafilm.
-
c.Place the tube in a horizontal position on an orbital shaker at 37°C, 150 rpm.
-
d.Incubate for roughly 20 min.CRITICAL: Depending on the size of BM-AT pieces, digestion can be more or less rapid. At the end of the digestion period, the digested adipose tissue should have a “soup-like” consistency. If the digestion is not finished, pieces of intact tissue will remain, whereas if the digestion takes too long, adipocytes will burst and release oil. We suggest checking the digestion regularly from 15 minutes onwards. As mentioned above, digestion efficiency depends on the collagenase lot and type.
-
e.Stop the digestion (Figure 3E).
-
i.Place a 100 μm cell strainer on top of a 50 mL tube, rinse the strainer with KRBHA, and filter the suspension through it to remove cellular debris, undigested fragments, and bone trabeculae.
-
ii.Pour 20 mL KRBHA on the strainer to recover a maximum of BM-Ads.
-
i.
-
f.Wait 1–3 min to allow the lipid-laden adipocytes to rise up and float at the surface (Figure 3F)
-
g.Remove 15–18 mL KRBHA from under the floating BM-Ads with a syringe and a 21G needle to facilitate the collection of the adipocytes and their transfer to a new tube (Figure 3G)
-
h.Collect gently floating BM-Ads with a 1 mL pipet with standard P1000 pipette tip and transfer them to a 2 mL tubes.CRITICAL: Isolated BM-Ads can remain stuck to the cell strainer or pipette tips. To limit this effect and avoid losing BM-Ads, rinse the cell strainer and the pipette tips with KRBHA before pouring or pipetting adipocytes. Moreover, as isolated adipocytes are fragile, the suspension must be poured on the filter and not pipetted through the filter with a culture pipet.
-
a.
-
7.Wash and centrifuge BM-Ads. Even after cell dissociation with collagenase and separation of adipocytes by flotation, some contaminant cells remain attached to adipocytes. To purify adipocytes, it is important to wash and centrifuge the adipocyte suspension to pellet contaminant cells (Figure 3H)
-
a.Wash adipocytes by repeating the following steps 3 times.
-
i.Add 1 mL KRBHA and mix the suspension gently by tilting.
-
ii.Wait until adipocyte rise again to form an upper layer, KRBHA being in the lower layer.
-
iii.Using a 2 mL syringe with a 21G needle, aspirate the KRBHA and discard it.
-
i.
-
b.Add 1 mL KRBHA and wait until adipocyte rise again to form an upper layer.
-
c.Centrifuge for 5 min at 200 g, 22°C. This step allows to pellet contaminant cells.
-
d.Transfer floating adipocytes to a new tube. At this step, BM-Ads can be used for cell culture and functional experiments or can be frozen (step 8).
-
e.Keep 30 μL of adipocytes in a separate 2 mL tube to validate the purity of the cell population (see steps 9–12).
-
a.
Note: Step 7 is essential to obtain pure adipocyte cell suspension but needs to be done as quickly as possible, as BM-Ads are fragile cells and can burst rapidly. Pipet gently the adipocytes to limit the lysis.
-
8.
Preparation of adipocytes for freezing. Freezing leads to adipocyte lysis. Thus, for functional assays, culture or imaging, adipocytes must be used directly after isolation and washing steps (step 7). For molecular analyses (RNA, protein, lipid content), we recommend freezing a known volume of adipocytes (without KRBHA) in liquid nitrogen and storing these samples at −80°C until use. Make several aliquots when possible.
Note: For Western blotting, BSA must be removed to allow a correct protein quantification of the samples. To do this, wash cells 2 times with PBS before freezing (step 7a).
Figure 2.

Dissection and washing of the bone marrow adipose tissue
Bone marrow aspirate is very bloody (A). Rinse the cell strainer (B), pour the tissue on it (C), and wash the tissue with 20 mL warm KRBHA (D). Transfer the BM sample to a petri dish (E) and dissect the yellow BM-AT with tweezers and scissors (F). Transfer the dissected BM-AT to a new 15 mL tube and weight the tissue prior BM-Ad isolation (G).
Figure 3.

Bone marrow adipocyte isolation
Incubate BM-AT with collagenase (A) for 20 min shaking horizontally on an orbital shaker (B). Digested BM-AT should have a “soup-like” consistency (C). Adipocytes rapidly float to the surface of the tube (D). Filter the suspension through a 100 μm cell strainer (E) and rinse the strainer with warm KRBHA. Wait a few minutes to allow adipocytes to float at the surface (F) and remove a maximum of KRBHA to facilitate adipocyte collection (G). Transfer BM-Ads with a 1 mL pipet to a new 2 mL tube and centrifuge for 5 min at 200 g at 22°C to pellet contaminant cells (H).
Validation of adipocyte suspension purity
This step allows for verifying the quality and purity of the BM-Ad suspension. For each preparation, we recommend preparing fibrinogen gel containing BM-Ads to check the shape of BM-Ads (lipid staining) and the absence of contaminant cells attached to adipocytes (actin and nucleus staining) by confocal microscopy.
-
9.Embed BM-Ads in fibrinogen matrix (Figures 4A–4C)
-
a.Thaw fibrinogen and thrombin at 20°C–25°C.
-
b.Dilute thrombin ¼ with PBS 2 mM CaCl2.
-
c.Gently mix the 30 μL of adipocytes kept from step 7e with 30 μL fibrinogen, then add 30 μL diluted thrombin and mix with up and down motions until the gel polymerizes (within seconds). Avoid forming bubbles.
-
a.
-
10.Fixation.
-
a.Keep the gel for 2 min at 20°C–25°C.
-
b.Under a chemical hood, add 400 μL 3.7% PFA. Incubate 20 min on an orbital shaker at 100 rpm.
-
c.Remove PFA and wash 2 times with PBS.
-
a.
Pause point: At this step, samples can be stored in PBS at 4°C for up to 1 week until staining.
-
11.Staining.
-
a.Remove PBS.
-
b.Add staining solution containing 10 ng/mL BODIPY 493/503, 2 U/mL rhodamine-coupled phalloidin, and 2 μM of TO-PRO3 iodide 642/661 diluted in PBS.
-
c.Incubate for 1 h on an orbital shaker at 100 rpm. Protect from light.
-
d.Remove staining solution and wash 2 times with PBS.
-
a.
Note: BODIPY 493/503 stock solution must be dissolved in DMSO or ethanol at a concentration of 1 μg/mL as recommended by supplier. Aliquots in dark tubes can be stored at −20°C for several months. TO-PRO3 iodide 642/661 is ready to use but as it is unstable, it is mandatory to avoid freeze-thawing. It is highly recommended to make small aliquots of up to 10 μL in dark tubes.
Alternatives: Fluorescent immunostaining can be performed. Saturation of non-specific antigen should be done using PBS containing 3% BSA and 0.1% Triton ×100 for 1–2 hours at 20°C–25°C. Staining with primary antibodies should be done in PBS containing 3% BSA and 0.1% Triton ×100 for 16 hours at 4°C (antibody dilution should be determined by user for each antibody used). Wash gel 5 times with PBS containing 0.05% Tween20 for 10 minutes each. Incubate the gel with secondary antibodies in PBS containing 3% BSA and 0.1% Triton ×100 for 2 hours at 20°C–25°C. Wash gel 5 times with PBS containing 0.05% Tween20 for 10 min each. All incubations should be performed on an orbital shaking at 100 rpm or rotative shaker at 20 rpm. Alternative dyes can be used to stain nuclei including classical DNA intercalant such as DAPI or Hoechst33342 if UV or blue laser light are available on the used imaging system (see below).
-
12.Imaging.
-
a.With a curved clamp, handle the gel carefully and place it on a 150 μm coverslip. Do not pinch the gel firmly so as not to break it (Figures 4D–4F).
-
b.Put the coverslip on an inverted confocal microscope.
-
c.Set up confocal light path with correct excitation and emission wavelengths according to the dyes used for staining. If lipids are stained with BODIPY 493/503, excitation should be performed with a 488 nm blue laser and emission signals should be collected at 510 ± 15 nm. If the actin cytoskeleton is stained with rhodamine-coupled phalloidin, excitation should be performed with a 532 nm green laser and emission signals should be collected at 550 ± 15 nm. If nuclei are stained with TO-PRO3 iodide 642/661, excitation should be performed with a 635 nm red laser and emission signals should be collected around 660 ± 20 nm.
-
a.
Note: The cytoskeleton of adipocytes may have dim fluorescence due to the specific organization of adipocyte cytoplasm. The use of an objective lens with higher resolution and/or with a high numerical aperture can improve the quality of the signal collected for phalloidin staining.
Figure 4.

Embed BM-Ads in fibrinogen matrix
Mix BM-Ads with fibrinogen (A) and then add thrombin (B) to obtain a fibrinogen matrix containing BM-Ads (C). After fixation and staining, place the gel on a coverslip for imaging (D–F). It is mandatory to handle the gel containing adipocytes carefully (D) to avoid any damage (F) that could affect sample quality.
Expected outcomes
A successful isolation process should yield 200–500 μL BM-Ads per gram of tissue. The average diameter of BM-Ads is 95 ± 6 μm, as we have previously reported (Attane et al., 2020). BM-Ad yield is dependent on the digestion efficiency but also on the different washing steps. BM-Ads are fragile (more fragile than subcutaneous adipocytes for which we are used to obtain routinely an average of 600 μL of adipocytes per gram of tissue) and can burst during digestion. It is therefore important to determine the optimal digestion duration (see step 6). Fat cells can also burst during the following purification steps that include several washing steps and centrifugation. These steps are nevertheless necessary to obtain a pure adipocyte suspension. We therefore advise to perform these steps as quickly as possible by following our recommendations (step 7).
BM-Ads should contain a unique large lipid droplet surrounded by actin cytoskeleton. The BM-Ad nucleus is located at the cell periphery. Thus, the staining of neutral lipids, actin, and nucleus allow to assess whether the isolation and purification steps have preserved BM-Ad morphology. In addition, actin and nucleus staining enable the detection of contaminant cells that may remain attached to BM-Ads. An example of the expected images of pure BM-Ad suspension is shown Figure 5A. Conversely, Figure 5B shows contaminating cells attached to BM-Ads due to insufficient digestion and/or insufficient washing.
Figure 5.

Validation of adipocyte suspension purity
Representative maximal intensity projection of Z-stack images taken on a confocal microscope with a 40× objective. Images represent individual channels for lipid or actin stainings and merged channels for lipid, actin, and nuclei stainings.
Upper set of images (A) represents a pure sample of isolated BM-Ad and lower set of images (B) represents isolated BM-Ad contaminated with stromal-vascular cells (arrowhead). Scale bar, 50 µm.
Limitations
The presence of BM-AT is variable within BM samples (Figure 6) due to inter-patient heterogeneity but also on the manner in which the BM is collected.
Figure 6.

Variability in bone marrow samples
Representative images showing sample heterogeneity from 4 patients with different amount of BM-AT.
The protocol described here can be adapted to BM collected from other depots (e.g., from the femoral head, tibia, or other long bones).
For the isolation of adipocytes from the tibia or other long bones, the removal of the marrow should be performed with a cannula as done for femur and the purification of adipocytes will be similar to that described here. However, for BM-Ad isolation from the femoral head, bone should be cut into several pieces to facilitate digestion (Mattiucci et al., 2018). As the digested sample contains bone fragments, the digestion time and the shaking speed should be adapted to avoid BM-Ad lysis. At the end of the digestion, bone fragments and trabeculae must be removed.
Troubleshooting
Problem 1
Low adipocyte yield after digestion and purification.
Potential solution
This problem could be due to a low amount of starting material but also to the loss of BM-Ads during the digestion and purification steps. To ensure proper digestion and obtaining at least 300 μL of BM-Ads, we recommend digesting at least 1 gram of tissue (step 5). This is not possible for some samples which have a low amount of BM-AT (Figure 6). To decrease cell loss, always rinse the cell strainer and the pipette tips with KRBHA before pouring or pipetting adipocytes (steps 6 and 7).
Problem 2
Lysis of adipocyte
Potential solution
To limit the lysis of BM-Ads during the digestion, reduce digestion time and shaker speed which can damage the cells (step 6). For all steps in this protocol, work only with plastic and not glass tubes which promote the lysis of adipocytes, use KRBHA warmed to 37°C (even in step 2 for the sample transport), pipet isolated BM-Ads gently, and work as quickly as possible.
Problem 3
Contamination with non-adipocyte cells.
Potential solution
Use 100 μm cell strainers and not larger cell strainer meshes. Pipet the suspension gently on the cell strainer to keep debris from seeping through (steps 6 and 7).
Change the type or lot of collagenase and increase the digestion time (step 6).
Add wash and centrifugation steps (see step 7).
Problem 4
Weak nuclei or cytoskeleton staining.
Potential solution
The thickness and structure of the fibrin gel may slow down the penetration rate of the fluorescent probes resulting in poor labeling of the cytoskeleton or nuclei. Adding 0.1% triton ×100 to the labeling solution (i.e., 10 ng/mL BODIPY 493/503, 2 U/mL rhodamine-coupled phalloidin, and 2 μM of TO-PRO3 iodide 642/661) will increase probe penetration and staining intensity (step 11). Alternatively, increase labeling incubation time up to 2 h.
Problem 5
Strong background or non-specific staining.
Potential solution
Depending on the amount of background noise, several strategies can be implemented to reduce it (step 11d):
Increase the number of washes from 2 to 5.
Add 0.05% Tween 20 to the wash solution.
Increase the time between washes. Leave the samples in the wash solution for 10 min under agitation before renewing it.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Catherine Muller (catherine.muller@ipbs.fr).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate any unique datasets or code.
Acknowledgments
In addition to the “Ligue Nationale contre le Cancer” (Equipe Labellisée) and INCa PLBIO20-028, this work was supported by the program “Initiative D'EXcellence” Toulouse (IDEX) from the University of Toulouse and the “Fondation de France” for running costs and post-doctoral fellowship for C.A. D.E. received a post-doctoral fellowship from the Fondation pour La Recherche Médicale (SPF201809007124).
Author contributions
N.R. set up the conditions for harvesting BM-AT and SC-AT in close collaboration with C.A. and D.E. and supervised the sample collection. C.A., D.E., and M.M. handled the AT samples and isolated adipocytes. D.E. performed the immunofluorescence experiments. C.A., D.E., and C.M. designed the protocol and wrote the manuscript. C.M. supervised the study.
Declaration of interests
The authors declare no competing interests.
References
- Attane C., Esteve D., Chaoui K., Iacovoni J.S., Corre J., Moutahir M., Valet P., Schiltz O., Reina N., Muller C. Human bone marrow is comprised of adipocytes with specific lipid metabolism. Cell Rep. 2020;30:949–958.e6. doi: 10.1016/j.celrep.2019.12.089. [DOI] [PubMed] [Google Scholar]
- Decaunes P., Esteve D., Zakaroff-Girard A., Sengenes C., Galitzky J., Bouloumie A. Adipose-derived stromal cells: cytokine expression and immune cell contaminants. Methods Mol. Biol. 2011;702:151–161. doi: 10.1007/978-1-61737-960-4_12. [DOI] [PubMed] [Google Scholar]
- Mattiucci D., Maurizi G., Izzi V., Cenci L., Ciarlantini M., Mancini S., Mensa E., Pascarella R., Vivarelli M., Olivieri A. Bone marrow adipocytes support hematopoietic stem cell survival. J. Cell. Physiol. 2018;233:1500–1511. doi: 10.1002/jcp.26037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique datasets or code.