

Abstract
The actin cytoskeleton is a dynamic network that regulates cellular behavior from development to disease. By rearranging the actin cytoskeleton, cells are capable of migrating and invading during developmental processes; however, many of these cellular properties are hijacked by cancer cells to escape primary tumors and disseminate to distant organs in the body. In this review article, we highlight recent work describing how cancer cells regulate the actin cytoskeleton to achieve efficient invasion and metastatic colonization. We also review new imaging technologies that are capable of revealing the complex architecture and regulation of the actin cytoskeleton during motility and invasion of tumor cells.
1. Introduction
In metazoans, cell motility is required for key developmental processes, including gastrulation (Keller, 2005), neurulation (Theveneau and Mayor, 2012), the maintenance of tissue integrity and wound repair (Friedl and Gilmour, 2009), and immune cell trafficking (Friedl and Weigelin, 2008). Cell invasion is also conserved during embryonic development and homeostasis, as well as immune cell function (Medwig and Matus, 2017; Stuelten et al., 2018). The ability of a cell to move and invade is primarily dependent on the reorganization of the actin cytoskeleton, which requires spatiotemporal coordination of signaling pathways with actin regulatory and binding proteins (Blanchoin et al., 2014; Lauffenburger and Horwitz, 1996; Pollard and Cooper, 2009). Multiple human diseases and pathologies, including cancer cell invasion and metastasis, are associated with aberrant and deregulated cell motility and invasion (Hanahan and Weinberg, 2011).
The acquisition of motile and invasive phenotypes is a characteristic of aggressive tumors. Migration and invasion are needed for local invasion, intravasation into the vasculature, and extravasation at distant sites (Chaffer and Weinberg, 2011). The formation of actin-rich protrusions is a key feature of cancer cells that allows them to disseminate and colonize other organs (Bravo-Cordero et al., 2012). Thus, tumor cell motility and invasion are key rate-limiting steps during cancer progression and metastasis formation. Tumor cells are capable of activating different molecular mechanisms to remodel the actin cytoskeleton in order to leave primary tumors and travel to other organs (Bravo-Cordero et al., 2013a; Yamaguchi and Condeelis, 2007). From pseudopodia to filopodia and invadopodia protrusions, cancer cells display a repertoire of subcellular actin-rich structures that facilitate overcoming different barriers during tumor dissemination.
In this review, we address key modes of cancer cell motility and underlying signaling pathways, effects of the tumor microenvironment on cancer invasion, and recent technological advances that have been developed to visualize the invasion-metastasis cascade (Fig. 1).
Fig. 1.
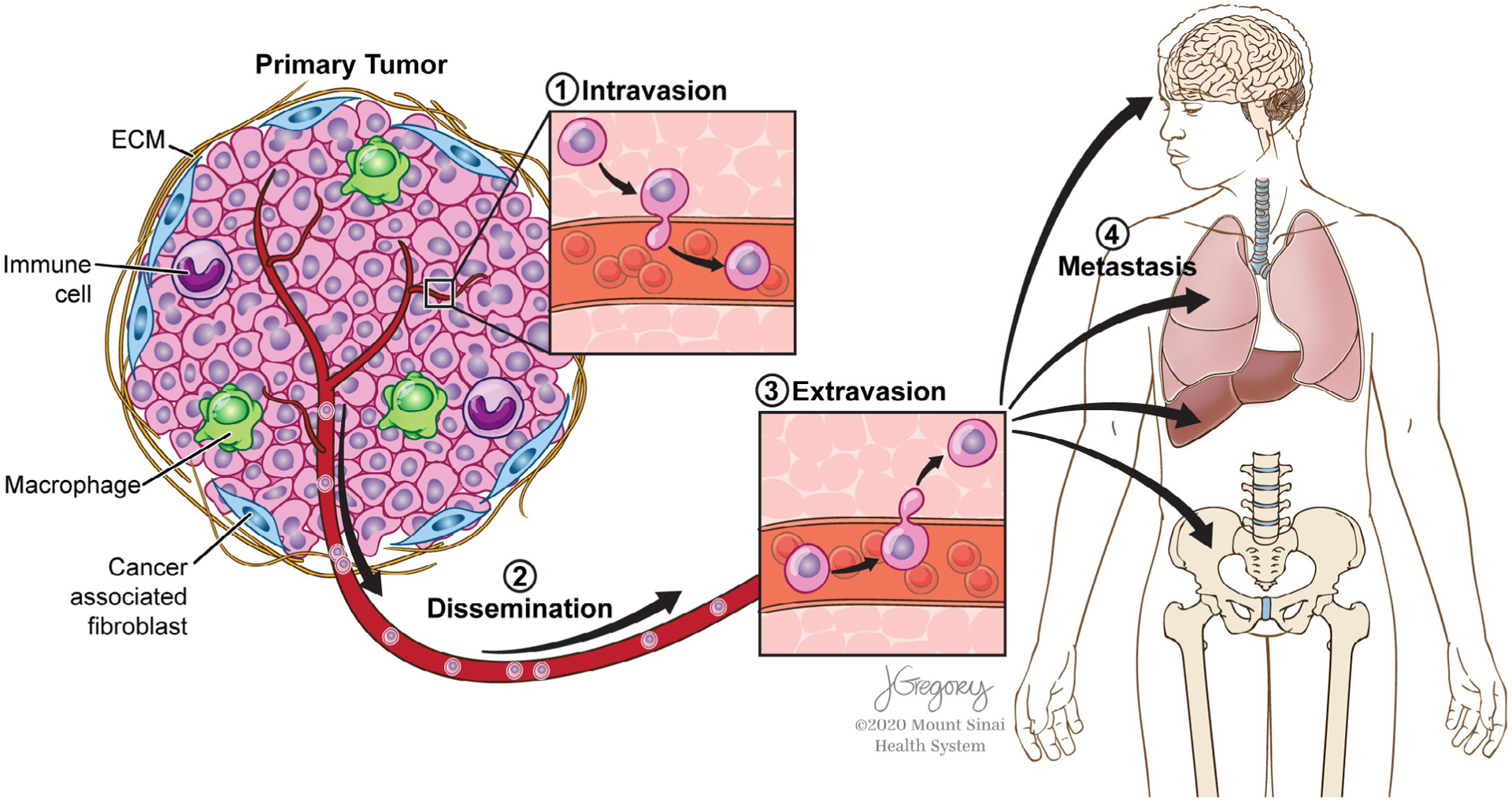
Invasion-metastasis cascade. Primary tumors have a complex tumor microenvironment (e.g., immune cells, cancer-associated fibroblasts, the extracellular matrix) that plays a dynamic role in affecting tumor cell dissemination and how tumor cells intravasate into the vasculature (1), disseminate through the circulatory system (2), extravasate out of the vasculature (3), and colonize distant organs (4).
2. Tumor dissemination and metastasis
In most epithelial cancers, tumor cells must acquire an invasive phenotype in order to escape the primary tumor and have the potential to invade locally, intravasate into the vasculature, survive circulation, extravasate into distant organs and colonize (Chaffer and Weinberg, 2011). The tumor microenvironment (TME) is a complex milieu of tumor cells, a dynamic extracellular matrix (ECM), and many stromal cells, including cancer-associated fibroblasts and immune cells (Fig. 1); in an orchestrated effort, components of the TME may act in concert to drive tumor cell invasion and metastasis formation (Clark and Vignjevic, 2015; Di Martino et al., 2019).
2.1. Epithelial-to-mesenchymal transition (EMT)
One mechanism by which epithelial tumor cells can become more invasive is by co-opting an epithelial-to-mesenchymal transition (EMT) developmental program (Dongre and Weinberg, 2019). An EMT induction results in the expression of core EMT transcription factors (e.g., Twist, Slug, Snail, Zeb1, Zeb2) which upregulate genes that promote a mesenchymal-like cell migratory phenotype. An EMT or partial-EMT can be induced through TGF-β, Notch, and Wnt signaling pathways, and is often dependent on the secretion of specific chemokines and cytokines by the TME (Dongre and Weinberg, 2019).
Signaling through TGF-β1, which can be produced by tumor cells, cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), myeloid-derived suppressor cells, and regulatory T cells (Tregs), as well as STAT3 signaling downstream of Il-6 and Il-23 secreted by leukocytes (Smith and Kang, 2013) has been shown to induce EMT. Tumor-associated macrophages, which are often found at the invasive front of tumors (Condeelis and Pollard, 2006) secrete pro-inflammatory cytokines such as TNF-α, which regulate the NF-κB pathway (Chen et al., 2018b). Multiple studies have demonstrated that EMT can be activated by TNF-α; TNF-α secreted by TAMs activated NF-κB signaling and stabilized Snail, which resulted in increased cancer cell invasion in vitro (Wu et al., 2009). TNF-α has also been demonstrated to upregulate Twist1 through NF-κB signaling in breast cancer cells (Li et al., 2012), induce EMT in renal cell carcinoma in a GSK3β-dependent manner (Ho et al., 2012), and stabilize Slug through NF-κB signaling in head and neck squamous cell carcinoma (HNSCC) (Liu et al., 2018a).
Induction of an EMT program can upregulate matrix metalloproteinases that are capable of degrading the basement membrane (Olmeda et al., 2007), which consists mainly of laminin and type IV collagen (Bosman et al., 1985). The Snail1 transcription factor is capable of inducing an invasion program dependent on MMPs; Snail1 induction increases both MT1-MMP and MT2-MMP expression in breast carcinoma cells (Ota et al., 2009). In addition, a subset of invasive cancer cells can form invadopodia (see Section 4.3) structures, which are capable of recruiting MT1-MMP, MMPs, and ADAMs, which allow them to degrade the extracellular matrix (Eddy et al., 2017). EMT has been demonstrated to drive invadopodia formation in a Twist1-dependent manner (Eckert et al., 2011).
Recently, the concept of an EMT has been challenged in that it may not be necessary for metastasis formation (Aiello and Kang, 2019). Using an EMT lineage tracing system in the PyMT model of spontaneous breast cancer, lung metastases were shown to form when EMT was inhibited (Fischer et al., 2015), and the tracing system was validated using single-cell RNAseq (Lourenco et al., 2020). In a study on invasive ductal carcinomas, expression of E-cadherin promoted tumor cell survival and the establishment of metastases (Padmanaban et al., 2019); loss of E-cadherin is one of the hallmarks of an epithelial-to-mesenchymal transition (Dongre and Weinberg, 2019). In the KPC model of pancreatic ductal adenocarcinoma, loss of Snail or Twist did not have a significant reduction in metastases formation (Zheng et al., 2015), yet Zeb1 depletion in the same background did (Krebs et al., 2017), suggesting the requirement for EMT may vary based on the context (Aiello and Kang, 2019).
Additional factors in the tumor microenvironment can directly impact tumor cell dissemination; an important aspect of tumor biology is the mechanical properties of the surrounding ECM (Mohammadi and Sahai, 2018). It has been shown that in vivo, tumor cells move along highly aligned collagen fibers (Condeelis and Segall, 2003) to facilitate local invasion (Provenzano et al., 2006). Also, increased matrix stiffness can induce EMT through Twist1 activation (Wei et al., 2015).
2.2. Intravasation
After cancer cells invade locally, they can intravasate into the vasculature and travel through the hematogenous system or more rarely, through the lymphatic vasculature (Chiang et al., 2016; Olmeda et al., 2017) (Fig. 1). Capturing intravasation events in vivo is rare and challenging (Wyckoff et al., 2007); work using intravital microscopy combined with a mammary imaging window and photoconvertible Dendra2 to mark and track breast tumor cells demonstrated that vascularized regions had a greater number of photoconverted tumor cells lining up around the blood vessels, presumably intravasating into the vasculature, as characterized by more lung metastases (Kedrin et al., 2008). A proxy for intravasation events in vivo is to quantify circulating tumor cells in the blood. This type of analysis in a breast cancer model demonstrated that ERBB2 has a greater effect on intravasation than ERBB1; both are highly altered and aberrantly expressed receptors in aggressive breast cancers (Kedrin et al., 2009). Recent work in a PyMT model of breast cancer showed that TIE2hi macrophages were able to promote intravasation of tumor cells through the secretion of VEGFA, which resulted in transient permeability of blood vessels (Harney et al., 2015). The intravasation events characterized in this study were restricted to tripartite structures known as TMEMs (tumor microenvironment of metastasis), where a tumor cell, macrophage, and an endothelial cell are in direct contact with each other (Harney et al., 2015).
Additional studies on intravasation have used in vitro assays and microfluidic devices to characterize signaling pathways promoting intravasation. One mechanism by which tumor cells have access to the vasculature is through endothelial barrier impairment during tumor progression, which was demonstrated through macrophage-secreted TNF-α, which increased vasculature permeability and the rate of tumor cell intravasation (Zervantonakis et al., 2012). Invadopodia formation has also been linked with intravasation events; macrophages in direct heterotypic contact with breast tumor cells are able to induce global activation of RhoA signaling in tumor cells, resulting in tumor cells forming an increased number of invadopodia which are necessary for transendothelial migration (Roh-Johnson et al., 2014). A Notch1/MenaINV signaling program has been demonstrated to regulate macrophage-induced invadopodium formation and transendothelial migration of breast cancer cells (Pignatelli et al., 2016).
2.3. Extravasation
Extravasation out of the vasculature has been visualized using ex ovo chicken embryos (Leong et al., 2014), zebrafish embryos (Berens et al., 2016), and tail vein injections in the mouse (Mohanty and Xu, 2010). One of the early studies in optically transparent zebrafish embryos showed that over-expression of Twist or VEGFA in highly invasive breast tumor cells increased the percentage of cells that were able to extravasate (Stoletov et al., 2010), and Twist expression induced a change in the mode of extravasation to β1-integrin independent (Stoletov et al., 2010). To examine the effects of inflamed neutrophils on tumor cell dissemination, LPS-stimulated neutrophils were co-injected with melanoma cells in zebrafish, resulting in increased extravasation (Chen et al., 2018a). Using an ex ovo chicken embryo model, human epidermoid cancer cells, as well as a series of other cancer cell lines were demonstrated to extravasate at endothelial junctions. Extravasation was dependent on invadopodia formation, as determined by localization of cortactin, Tks4, and Tks5, which are invadopodia components, using intravital imaging (Leong et al., 2014).
Tropism, or the homing of cancer cells to specific organs is often dependent on the tumor of origin; for example, the vast majority of patients with metastatic breast or prostate cancer have metastases in the bone (Weilbaecher et al., 2011). Cells in the bone secrete factors that attract cancer cells to the bone marrow, including RANKL, CXCL12, OPN, and BMPs (Jones et al., 2006; Obenauf and Massagué, 2015). Many breast cancer cells express EREG, MMP1, MMP2 and COX2 (Gupta et al., 2007), which allow them to selectively metastasize to the lung, as well as ANGPTL4 and SPARC (Padua et al., 2008; Tichet et al., 2015). Interestingly, COX2 and MMP1 signaling also allows breast cancer cells to overcome the blood brain barrier and form brain metastases (Wu et al., 2015), suggesting that some factors for tropism are organ-specific, and others are not as restrictive.
3. Cancer cell migration
3.1. Single-cell migration and the cell motility cycle
3.1.1. Mesenchymal motility
Within tumors, cancer cells can move as single, distinct entities or collectively as multicellular sheets or multicellular streams (Di Martino et al., 2019; Friedl and Wolf, 2010; Lintz et al., 2017; Roussos et al., 2011b). The tumor micro-environment is a key component that regulates the different modes of tumor cell migration; for example, the ECM stiffness, density, and orientation that a tumor cell encounters is one parameter that determines whether cells move in a mesenchymal or amoeboid manner (Friedl and Wolf, 2010; Talkenberger et al., 2017). When cancer cells are in contact with stiff substrata, they are capable of adopting an elongated, mesenchymal-based mode of motility. The extension of a leading edge protrusion (lamellipodium in 2D or pseudopodia in 3D) is the first step of the cell motility cycle (Fig. 2). In vitro, flat, veil-like lamellipodia form as a result of membrane deformation due to force generated by dynamic actin polymerization (Bravo-Cordero et al., 2013a; Yamaguchi and Condeelis, 2007). As the lamellipodia extends forward and the actin network moves backward through retrograde actin flow, the cell body is able to make new attachments to the substratum through coupling actin stress fibers to adhesion receptors (e.g., integrins). The cell body initially makes transient focal contacts with the substratum that can mature into focal adhesions, which are active signaling platforms that regulate mechanotransduction (Huttenlocher, 1995; Huttenlocher and Horwitz, 2011). As the cell is adhering in the front, it begins to disassemble focal adhesions in the rear and detach from the substratum. The cell body is able to translocate through the retraction force generated in the rear in a myosin II-dependent manner, which is regulated by Rho GTPase signaling (Friedl and Alexander, 2011).
Fig. 2.
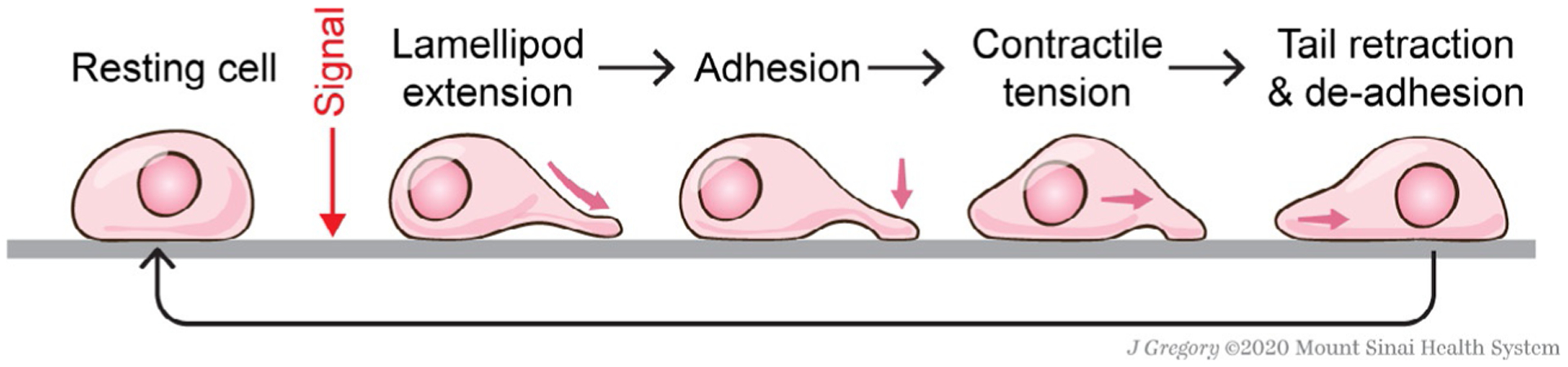
Cell motility cycle. Upon integrating signaling cues, a resting cell can form lamellipodia, or protrusive actin-rich structures, and adhere to the substrata with nascent adhesions. In order for the cell body to translocate, the cell experiences contractile tension and rear detachment through de-adhesion.
In vivo, cancer cells can polarize (resulting in cellular asymmetry) and extend pseudopodial protrusions (Fig. 3) (Bravo-Cordero et al., 2012). Cancer cell polarization can be achieved through sensing external cues or gradients, including chemotactic (Roussos et al., 2011b), haptotactic (King et al., 2016), durotactic (DuChez et al., 2019), and galvanotactic (Huang et al., 2016), as well as mechanical and topographical constraints (Northcott et al., 2018). In response to these stimuli, polarized cancer cells are capable of activating signaling pathways that primarily converge on the Rho GTPase family to initiate actin polymerization and generate protrusions that extend outward from the leading edge, or front of the cell to facilitate cell movement (Bravo-Cordero et al., 2013b; Haga and Ridley, 2016; Lawson and Ridley, 2018; Yamaguchi and Condeelis, 2007).
Fig. 3.
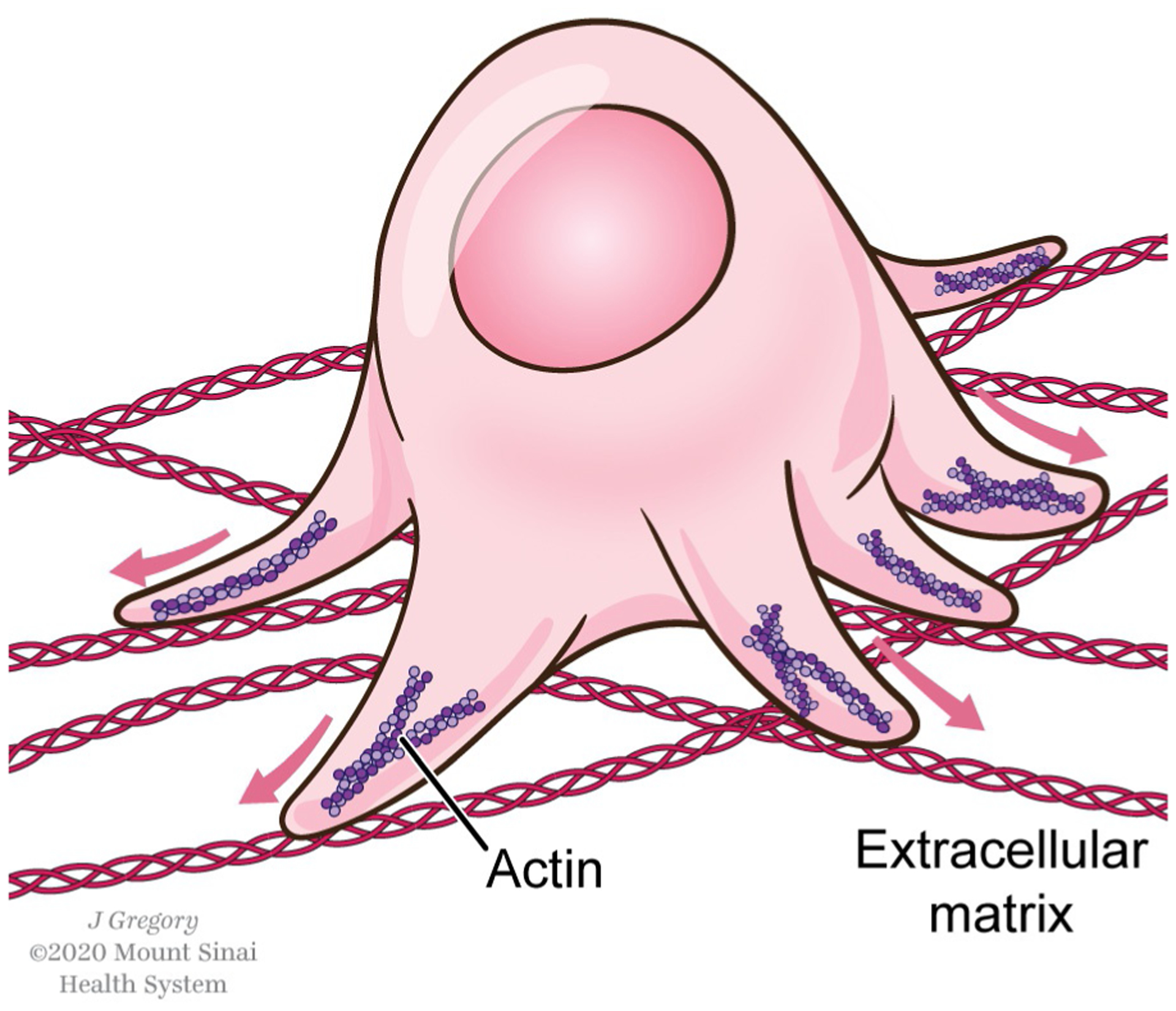
Tumor cell in a 3D context. In the tumor microenvironment, cancer cells encounter a complex extracellular matrix, and are capable of forming different types of protrusive structures, including pseudopodia.
In order for cancer cells to invade, they must remodel the extracellular matrix through expression of MMPs and other proteases (either cancer cell-intrinsic, or through stromal cells (Egeblad and Werb, 2002)) and in some cases, may recruit MT1-MMP, MMP2 and MMP9 through the formation of invadopodia structures, which are specialized F-actin-rich protrusions that degrade the extracellular matrix (Clark and Weaver, 2008; Clark et al., 2007; Eddy et al., 2017; Murphy and Courtneidge, 2011) (see Section 4.3). Recent work has shown that MMP activity can be regulated by cell density through an IL-6,8 paracrine loop (Jayatilaka et al., 2018), suggesting that MMP activity can be regulated locally through the homo-typic interactions between tumor cells.
3.1.2. Amoeboid motility
Another mode of single-cell movement is amoeboid motility, which is characterized by cells with a rounded morphology, and can present in multiple forms, including bleb-based (Petrie and Yamada, 2012). Bleb-based motility is movement that requires high levels of cell contractility, and resembles the motility of the single-cell organism Dictyostelium discoideum (Pinner and Sahai, 2008). Unlike mesenchymal-like migration, blebbing motility is defined as protease-independent, and relies on the cell’s capability to deform rapidly through dynamic alterations of the cortical actin cytoskeleton in the rear in a Rho-ROCK dependent manner (Sahai and Marshall, 2003). The high levels of contractility necessary for the cell to propel forward is dependent on the phosphorylation of myosin II-light chain (MLC2) via ROCK kinases, which are effectors of the Rho GTPases (Wilkinson et al., 2005). In addition, amoeboid motility usually occurs in areas with soft matrix, and amoeboid cells often have a decrease in integrin signaling and form weak adhesions (Brábek et al., 2010; Talkenberger et al., 2017). Interestingly, the use of protease inhibitors in HT-1080 fibrosarcoma cells and MDA-MB-231 breast carcinoma cells resulted in a transition from mesenchymal-to-amoeboid motility, rather than an abrogation of cellular invasion, demonstrating the plasticity of cancer cell movement (Wolf et al., 2003).
Cancer cells are able to interconvert between different motility patterns (Wolf et al., 2003). In triple-negative breast cancer cells, loss of the NEDD9 scaffolding protein resulted in more bleb-driven motility in vitro, including a loss of pFAK/pPaxillin mature focal adhesions, an increase in pMLC2, and a concurrent decrease in active Rac1 and increase in active RhoA (Jones et al., 2017). Similarly, in melanoma cells, bleb-based movement was driven through an active Rac GAP, ARHGAP22, which inactivated Rac signaling; conversely, mesenchymal-like motility was regulated by the NEDD9/DOCK3 complex which activated Rac signaling (Sanz-Moreno et al., 2008).
In mammary adenocarcinoma tumors in vivo, single-cell motility is characterized by the rapid movement of cancer cells that display an amoeboid morphology with the presence of F-actin-rich protrusions named pseudopodia at the leading front (Bravo-Cordero et al., 2012; Condeelis and Segall, 2003). These pseudopodia protrusions are characteristic of fast moving amoeboid cancer cells and are involved in chemotaxis toward blood vessels prior to intravasation (Condeelis and Segall, 2003).
3.2. Multicellular movement
In a 3D environment or in vivo, cancer cells encounter a complex microenvironment and are highly plastic in their motile behavior. Individual cancer cells are capable of moving in multicellular streams (Friedl and Alexander, 2011; Roussos et al., 2011a,b). In human orthotopic breast xenografts, cancer cells have been visualized to move in a multicellular stream (where a minimum of two cells follow each other in a directed manner) (Patsialou et al., 2013), or in a stream with host cells, such as tumor-associated macrophages (TAMs) (Patsialou et al., 2013). Co-migration of TAMs and breast cancer cells has also been demonstrated in a rat mammary adenocarcinoma model, and the transgenic PyMT spontaneous model of breast cancer. The interaction between these two cell types is dependent on an EGF/CSF-1 paracrine signaling loop (Wyckoff et al., 2004). MenaINV, a splice isoform of Mena, an actin regulatory protein, is spontaneously upregulated in invasive carcinoma cells; expression of MenaINV in a rat mammary adenocarcinoma model promotes multicellular streaming between tumor cells, as well as co-migration between tumor cells and TAMs in an EGF/CSF-1 dependent manner in vivo (Roussos et al., 2011a). Intravital imaging of B16 F2 tumors shows tumor cells following each other on the same tracks, in a multicellular stream. The streaming cells have increased SRF reporter activity (when compared to non-motile cells) (Manning et al., 2015); SRF is a master regulator of the actin cytoskeleton, and regulates the transcription of 200+ actin-related genes (Olson and Nordheim, 2010). Multicellular streaming has also been observed in a human orthotopic glioblastoma xenograft model, where tumor cells at the “invasive” margin between the tumor and brain parenchyma are capable of moving in succession, and overall, migrate with a lower velocity and increased persistence when compared to other motile tumor cells (Alieva et al., 2019). Intravital imaging of melanoma xenografts demonstrates that melanoma cells are capable of both single cell motility (Fig. 4A), as well as streaming, multicellular motility (Fig. 4B).
Fig. 4.
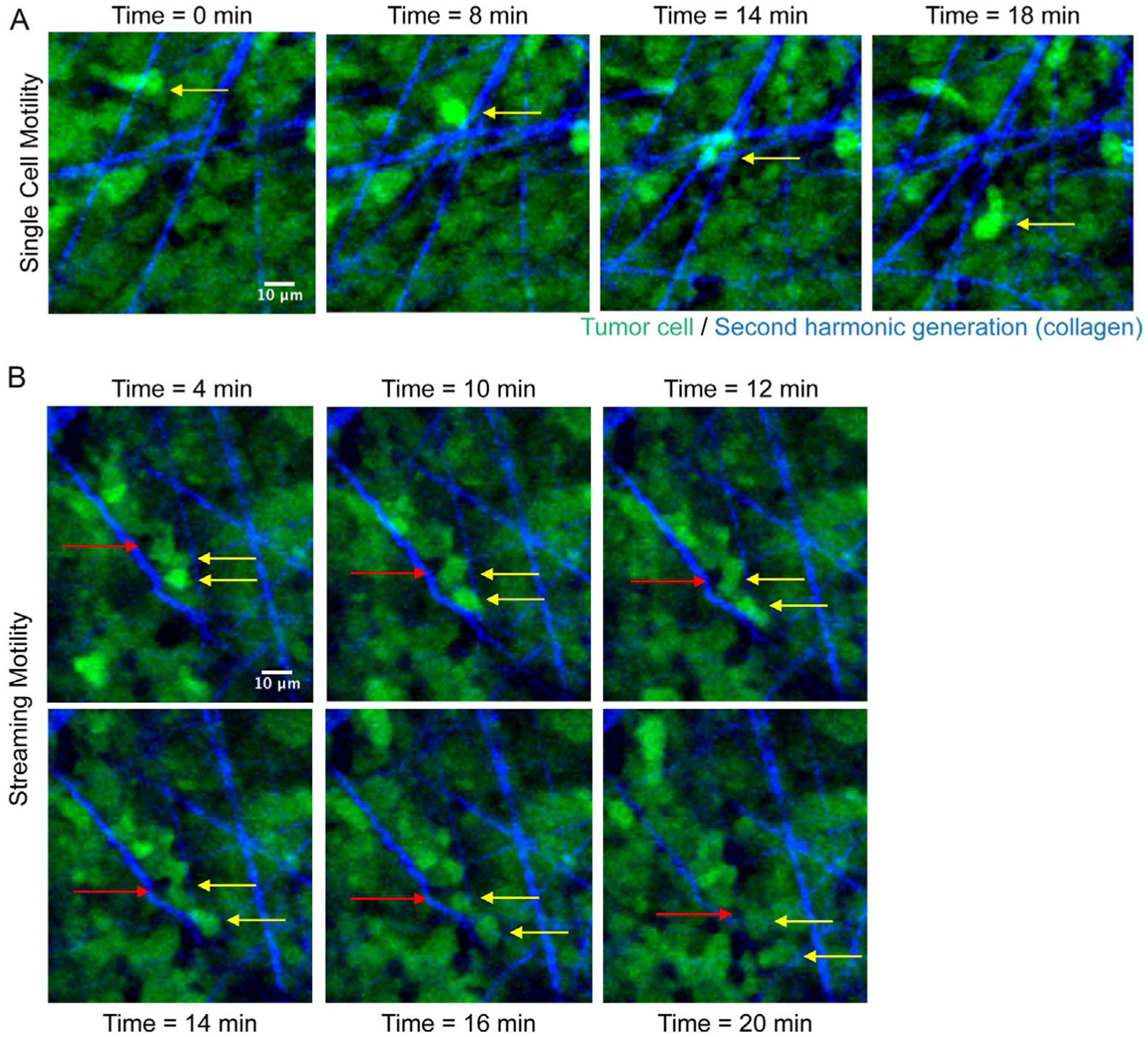
Intravital imaging of a melanoma xenograft. Two-photon imaging of tumor cells (in green) and second harmonic generation of fibrillar collagen (in blue). (A) Yellow arrow points to a single cell moving over time (indicated above each panel). (B) Example of streaming motility; yellow arrows point to cancer cells following each other, and red arrow points to another cell type in the tumor microenvironment moving within the multicellular stream. Scale Bar: 10μm. SK-Mel-147 GFP-labeled melanoma cells were injected subcutaneously in 6-week old female nude mice and tumors were allowed to grow up to 1cm3. Intravital imaging of the primary tumor was performed as in Patsialou et al. (2013) and collagen fibers were visualized by second harmonic generation. Images were acquired every 2min for 30min, with a step-size of 5μm.
3.2.1. Collective cancer cell migration
Collective cancer cell invasion is another mode of tumor cell movement found in many cancers, including breast (Cheung et al., 2013), squamous cell carcinoma (Hidalgo-Carcedo et al., 2011), liver cancer (Han et al., 2019), melanoma (Hegerfeldt et al., 2002), colorectal cancer (Chung et al., 2016), and lung cancer (Kuriyama et al., 2016). Collective cancer cell migration is sheet or strand-like multicellular movement that requires cancer cells to maintain cell:cell cohesion mechanisms. In some instances, cells at the front of the strand polarize and become “leader” cells, followed by a stream of “follower” cells (Friedl et al., 2012). Invasive fibrosarcoma and breast cancer cells have been demonstrated to move collectively upon large-scale MT1-MMP mediated proteolysis in a spheroid invasion model; tracks were initially created by “leader” cells, followed by larger tracks created by multicellular invasion strands (Wolf et al., 2007). Using 3D organoid and in vivo model systems of luminal breast cancer, multicellular invasion strands were characterized by “leader” cells that were K14+, and preferentially turned on basal epithelial markers (Cheung et al., 2013). Cancer cells are highly plastic; breast cancer cells in vivo have been demonstrated to switch from collective to single cell migration through increased local TGF-β signaling (Giampieri et al., 2009).
The mechanisms underlying collective cancer cell migration are not well understood; there is some evidence that the Wnt/PCP (planar cell polarity) non-canonical Wnt pathway can be co-opted from developmental processes to promote collective cell migration in gastric, ovarian and melanoma cancers (VanderVorst et al., 2019). The DDR1 receptor has been demonstrated to be required for collective cell migration of A431 squamous cell carcinoma through regulation of the actomyosin network and interaction with the Par3/Par6 cell polarity complexes (Hidalgo-Carcedo et al., 2011). The tumor microenvironment has also contributed to collective cell migration signaling. Cancer associated fibroblasts (CAFs) are able to transmit force to human A431 squamous carcinoma cells and mediate collective cancer cell invasion in 3D through the formation of heterophilic N-cadherin/E-cadherin adhesions (Labernadie et al., 2017).
4. Actin structures in cancer cell migration
4.1. Lamellipodia
Lamellipodia (as well as pseudopodia in a 3D context or in vivo) are protrusive structures formed at the leading edge of cells that can drive cancer cell migration. The generation of lamellipodia requires nascent branched actin polymerization in order to generate sufficient force to push the cell membrane forward; this occurs through multiple mechanisms, including de novo nucleation via activation of the Arp2/3 complex through nucleation-promoting factors (i.e., WASP and WAVE proteins) and the generation of free barbed ends (polymerization competent-ends of F-actin) through cofilin severing of pre-existing filaments (Bravo-Cordero et al., 2013a; Yamaguchi and Condeelis, 2007). Upstream regulation of the actin machinery that form lamellipodia include activation of migratory signaling pathways through extracellular stimuli, which converge on the Rho GTPase signaling node (Ridley, 2015). For example, Rac1 can promote membrane ruffling through interaction with WAVE complexes via IRSp53 (Miki et al., 2000), resulting in Arp2/3-mediated actin polymerization (Ridley, 2015).
Lamellipodium-driven migration is regulated directly by intricate coordination of the Rho GTPases RhoA, Rac1 and Cdc42, as shown by FRET biosensor imaging studies (Machacek et al., 2009) (see Section 6.2). In the context of cancer, the use of an optogenetic system with a photoactivatable Rac1 biosensor in prostate cancer cells demonstrated that Rac1-dependent lamellipodium extension functioned downstream of active PI3K signaling (Kato et al., 2014). RhoA has also been shown to play an important role during lamellipodium protrusion formation. By using a RhoA biosensor in breast cancer, studies have shown that the activity of this GTPase is highly confined to the first micron of the leading edge where it mediates lamellar extension (Bravo-Cordero et al., 2013b). Work with Rho GTPase FRET biosensors also revealed that another isoform from the Rho subfamily, RhoC, displays a particular spatial activation during protrusion formation. RhoC is activated in areas behind the leading edge where it regulates cofilin phosphorylation to confine cofilin activity (Bravo-Cordero et al., 2011). These studies showed that RhoA and RhoC GTPases have a unique spatiotemporal activation pattern that is necessary in order to achieve efficient lamellipodium extension.
GTPase signaling and activation is dependent on cycling between GDP-and GTP- bound states. This process is regulated by guanine nucleotide exchange factors (GEFs), GTPase activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs) (Haga and Ridley, 2016), many of which are mutated and aberrantly expressed in different cancer types (Porter et al., 2016). For example, P-rex1, a Rac1-specific GEF which can be regulated through PI3K-PI(3,4,5)P3 and GPCR signaling, has been demonstrated to promote invasion in melanoma in a Rac1-dependent manner (Lindsay et al., 2011), and is required for ErbB2-driven breast cancer cell migration (Sosa et al., 2010).
At the level of actin binding and regulatory proteins, there are changes in expression of many key actin regulators in multiple cancers, including the WASP/WAVE family (Iwaya et al., 2007; Kulkarni et al., 2012) and Mena proteins, amongst others (Gertler and Condeelis, 2011; Olson and Sahai, 2008; Yamaguchi and Condeelis, 2007). For example, in invasive breast cancer cells derived from rat mammary adenocarcinomas and the PyMT model of breast cancer, signaling through chemotactic factors, such as EGF, can directly affect lamellipodia formation by regulating the gene expression of actin nucleators, including several Arp2/3 subunits, as well as actin regulatory proteins that antagonize capping, including Mena (Wang et al., 2007). Mena, an Ena/VASP protein that binds the barbed ends of actin filaments and delays termination by capping proteins, has splice isoforms with distinct functions in breast cancer cells (Gertler and Condeelis, 2011); the Mena11a isoform dampens growth-factor elicited lamellipodial protrusions (Balsamo et al., 2016), whereas the MenaINV isoform promotes lamellipodial protrusions (Hughes et al., 2015; Philippar et al., 2008). In invasive breast cancer cells, Lamellipodin, a binding partner of Ena/VASP proteins, is required for EGF-dependent lamellipodial protrusion and can promote 3D cancer cell invasion through specific interactions with Scar/WAVE and Ena/VASP complexes (Carmona et al., 2016).
4.2. Filopodia
Filopodia are thin projections that require the elongation of bundled, parallel actin filaments; they arise primarily from de novo actin nucleation by formins, or through an Arp2/3-mediated convergent elongation model (Gupton and Gertler, 2007; Jacquemet et al., 2015). In migrating cells, filopodia have been found at the leading edge, where they are able to emerge out of the lamellipodium meshwork downstream of Rho GTPase signaling, which results in the regulation of proteins including Ena/VASP (which are enriched at filopodia tips) (Lebrand et al., 2004) and IRSp53 (an effector of Cdc42 that can induce membrane curvature) (Disanza et al., 2013). Filopodia are bundled by actin bundling proteins, such as fascin or alpha-actinin, and are capable of extracellular sensing and cargo transport (Jacquemet et al., 2015).
Filopodia-like protrusions (FLPs) have been observed in mouse mammary carcinoma cells that have extravasated into the lung parenchyma. FLPs are regulated by Rif and mDia2 and are decorated with β1 integrin. FLP contact with the ECM initiates adhesion-dependent signaling and results in increased tumor cell proliferation (Shibue et al., 2012). By using quantitative microscopy, recent work showed that filopodia density increases as breast cancer progresses (Jacquemet et al., 2017). In addition, filopodia stabilization through the L-type calcium channel is required for directed migration and invasion (Jacquemet et al., 2016). In a separate study, it was also identified that upregulation of Myosin-X in p53-driven cancers is needed for invasion through the formation of filopodia (Arjonen et al., 2014).
4.3. Invadopodia
Invadopodia are F-actin-rich protrusive structures that are formed by invasive cancer cells in contact with the extracellular matrix. Invadopodia have proteolytic function and can focalize the secretion and accumulation of metalloproteinases, such as MMP2, MMP9 and MT1-MMP (Eddy et al., 2017; Jacob and Prekeris, 2015). The ability of invadopodia to degrade the ECM promotes local invasion of tumor cells, intravasation, and extravasation events (Bravo-Cordero et al., 2012; Gligorijevic et al., 2012, 2014; Leong et al., 2014; Roh-Johnson et al., 2014).
Invadopodia are induced by a variety of stimuli. Growth factors such as EGF (DesMarais et al., 2009) and TGF-β1 (Mandal et al., 2008) stimulate invadopodia formation in breast tumor cells. GABA and EGFR, which are chemotaxis receptors, are involved in invadopodia dynamics in vivo and can guide cancer cell extravasation and promote brain tropism in breast cancer metastasis (Williams et al., 2019). Recently, IKKε has been described as a novel regulator of invadopodia formation and can promote metastasis in colorectal cancer (Liu et al., 2020). The tumor microenvironment can also regulate invadopodia; extracellular fibrillar collagen I was demonstrated as stimuli for cancer cells to form invadopodia in both 2D and 3D (Juin et al., 2012). Interestingly, collagen I induces invadopodia formation through the DDR1 collagen receptor in a kinase independent manner. The DDR1 receptor aligns along collagen I fibers, establishing linear invadosomes that recruit Cdc42 via the Tuba RhoGEF resulting in increased proteolytic activity (Juin et al., 2014). Other components of the extracellular matrix, including SERPINB5 and CSTB, can increase invadopodia formation and in vivo extravasation in pancreatic ductal adenocarcinoma (PDAC) (Tian et al., 2020). Adipocyte-derived lipid uptake by FATP proteins overexpressed in melanoma cells was able to induce invadopodia formation and drive melanoma progression (Zhang et al., 2018). Mechanosensing of the extracellular matrix can also form invadopodia, as invadopodia can contain integrin receptors (Mueller et al., 1999; Peláez et al., 2019) and CD44 (Petropoulos et al., 2018).
The formation of invadopodia occurs in steps, and is regulated temporally; briefly, invadopodium precursor structures assemble downstream of signaling cues (Beaty and Condeelis, 2014). The invadopodium precursor core is composed of cortactin, N-WASP, cofilin, and actin; invadopodium precursors are incapable of matrix degradation. Within seconds, Tks5 is recruited to the early invadopodium precursor where it stabilizes the structure. Subsequently, cortactin is phosphorylated and promotes the maturation of invadopodia, which endows them with the capability to polymerize new actin filaments and degrade the ECM (Eddy et al., 2017).
Rho GTPases, including Rac1, RhoA and Cdc42, have functional consequences during invadopodia formation (Beaty and Condeelis, 2014). Overexpression of the active form of Cdc42 and Rac1 induces invadopodia formation in cancer cells (Dutartre et al., 1996; Nakahara et al., 2003). RhoA drives invadopodium maturation (Bravo-Cordero et al., 2011; Sakurai-Yageta et al., 2008). Cdc42 is also a critical regulator of invadopodia dynamics, and affects invadopodium precursor assembly and maturation (DesMarais et al., 2009; Sakurai-Yageta et al., 2008; Yamaguchi et al., 2005; Eddy et al., 2017). A minimal signature to define invadopodia was proposed in 2014; actin structures that colocalize with Tks5 and the active form of Ccd42 are considered invadopodia (Di Martino et al., 2014).
Rho GTPases play an important role in regulating invadopodium dynamics. Work using Rho FRET biosensors showed that RhoC activation regulates cofilin activity at invadopodia (Bravo-Cordero et al., 2011). Use of FRET biosensor technology also demonstrated that Rac1 is required for invadopodium disassembly (Moshfegh et al., 2014) and Rac3 regulates integrin signaling at invadopodia and adhesion to the extracellular matrix (Donnelly et al., 2017). In relation to invadopodia formation, there is a small body of work describing GEF and GAP activity: one study has described how RhoC, which is important for metastasis formation (Clark et al., 2000), is spatially regulated at invadopodia by p190RhoGEF and p190RhoGAP (Bravo-Cordero et al., 2011). p190RhoGAP inactivates RhoC within the invadopodium core, and p190RhoGEF activates RhoC in areas surrounding the invadopodia (Bravo-Cordero et al., 2011). A few GEFs have been shown to be important for invadopodia function, including Vav1 (Razidlo et al., 2014), β-PIX (Donnelly et al., 2017; Md Hashim et al., 2013), Fgd1 (Ayala et al., 2009), Frabin (Nakahara et al., 2003), Trio (Moshfegh et al., 2014) and SGEF (Goicoechea et al., 2017), as well as some GAPs, including p190RhoGAP (Bravo-Cordero et al., 2011) and ArhGAP12 in melanoma and breast cancer cells (Diring et al., 2019).
4.4. Focal adhesions
Focal adhesions are dynamic signaling nodes that connect the actin cytoskeleton directly to the extracellular matrix (Huttenlocher, 1995). The main adhesion receptors that link the ECM to actin stress fibers are integrins, which are bidirectional signaling molecules that can be activated in an “outside-in” or “inside-out” manner (Hynes, 1992). Integrins can be activated by binding to their respective ligands (e.g., collagens, fibronectin), and recruit adaptor proteins, such as talin, kindlin (Sun et al., 2019) and paxillin (Turner, 2000), F-actin binding proteins (e.g., vinculin, alpha-actinin), receptor tyrosine kinases such as FAK (Hanks et al., 1992) and Src (Schaller et al., 1999), as well as the many proteins that make up the “adhesome” (Horton et al., 2016; Zaidel-Bar et al., 2007).
Cells are capable of forming multiple types of ECM-adhesions, including focal complexes, classic focal adhesions, and fibrillar adhesions in a 2D setting (Geiger and Yamada, 2011), as well as cell-matrix adhesions in 3D (Geiger et al., 2009). At the leading edge of a motile cell in 2D (within 1–2μm), nascent adhesions, or focal complexes (FCs) can form underneath the lamellipodium (Geiger et al., 2001; Nobes and Hall, 1995; Geiger and Yamada, 2011). Although not fully characterized, the molecular composition of focal complexes contains a few hundred proteins, including integrins, actin binding proteins (e.g., talin), and signaling molecules (i.e., FAK) (Zaidel-Bar et al., 2003; Geiger and Yamada, 2011). Focal complexes have short lifetimes, and can either disassemble rapidly or mature into focal adhesions. Focal adhesions, which are more elongated than focal complexes, remain primarily under the lamella (>2μm from lamellipodial tips) at the ends of stress fibers (Geiger et al., 2009; Geiger and Yamada, 2011). The maturation of focal complexes into focal adhesions requires tension and force, through actomyosin contractility (Choi et al., 2008; Giannone et al., 2007), tyrosine phosphorylation of certain proteins (i.e., paxillin), alterations in protein composition through recruitment of scaffolding proteins and increased adhesion-based signaling (Geiger et al., 2009; Geiger and Yamada, 2011). Rho GTPase proteins have a role in the formation of ECM-adhesions; Rac1 can control focal complex formation, and RhoA has a role in the maturation of focal adhesions (Parsons et al., 2010). Force generation can convert focal adhesions into fibrillar adhesions, which are mainly composed of α5β1 integrin and tensin, and regulate processes such as fibronectin fibrillogenesis (Danen et al., 2002; Zamir et al., 1999; Geiger and Yamada, 2011). Focal adhesion turnover, which can regulate cell migration, requires Src activity and the phosphorylation of FAK (Wozniak et al., 2004).
Cell-matrix adhesions have also been observed in 3D, which behave differently than adhesions in 2D. In cell-derived matrix, fibroblasts were able to make adhesions that were α5 integrin and paxillin positive (Cukierman et al., 2001). Breast epithelial cells in an attached 3D collagen gel are able to form small 3D adhesions with phosphorylated FAK Y397, whereas the same epithelial cells in a floating 3D matrix have adhesions that are absent of FAK phosphorylation (Wozniak et al., 2003); thus, 3D adhesions are mechanosensors that behave differently based on the ECM rigidity.
Cancer associated fibroblasts (CAFs) are key regulators of ECM deposition and remodeling during tumor progression, and are involved in paracrine signaling with tumor cells (Attieh and Vignjevic, 2016). In CAFs, Hic-5 was shown to promote the formation of fibrillar adhesions through interaction with tensin1, which are conserved in 3D cell-derived matrices (Goreczny et al., 2018). Interestingly, during breast cancer cell invasion, FAK, a key kinase at focal adhesions, differentially regulates tyrosine phosphorylation at focal adhesion and invadopodia components (Chan et al., 2009), suggesting there may be crosstalk between the signaling pathways that regulate the formation of actin-rich structures during cancer cell invasion.
5. The cell cycle and cancer cell invasion
In the hallmarks of cancer, a deregulated cell cycle state and cancer cell invasion have been regarded as distinct programs (Hanahan and Weinberg, 2011); however, recent evidence suggests that there is a closer link between regulators of proliferation and invasion during cancer progression than previously thought (Kohrman and Matus, 2017). In breast carcinoma cells, it has been elucidated that invadopodia preferentially form in the G1 phase of the cell cycle (Bayarmagnai et al., 2019). p27, a cell cycle inhibitor that binds to Cdk-cyclin complexes in the nucleus, is able to regulate tumor cell invasion when it mislocalizes to the cytoplasm (Chu et al., 2008). In addition, p27 has been demonstrated to localize to invadopodia (Bayarmagnai et al., 2019; Jeannot et al., 2017) and regulates its activity through a Rac1-PAK1-cortactin signaling axis (Jeannot et al., 2017).
In melanoma, tumor cells are able to switch between a high-proliferative/low invasive state to a low-proliferative/high invasive state, known as phenotype switching, as the disease progresses (Arozarena and Wellbrock, 2019). Tumor cells expressing high levels of the MITF transcription factor, low levels of the Axl receptor and the associated transcriptional program remain in the high-proliferative/low invasive state; a switch to an Axl high and MITF low state shifts cells into a high-invasive/low-proliferative program (Rambow et al., 2019). The reduction of transcription factor MITF has been shown to induce a G1 cell cycle arrest through p27, and concurrently leads to the downregulation of Dia1 and promotes ROCK-mediated invasion (Carreira et al., 2006). In the PyMT model of breast cancer, loss of p21CIP1 suppressed invasion and increased cell proliferation (Qian et al., 2013), suggesting that p21CIP1 may mediate switching between proliferation and invasion.
6. Imaging advances and future directions in studying tumor cell invasion
6.1. Single-molecule superresolution imaging
The advances in the study of the actin cytoskeleton have been driven by the implementation of different high-resolution imaging techniques. Actin-rich structures are complex subcellular entities that contain several actin regulatory molecules. Recent proteomics studies (Attanasio et al., 2011; Ezzoukhry et al., 2018) have elucidated the composition of invadopodia and invadosomes; however, these techniques are limited in that the spatial distribution of the components are unable to be characterized. Recently, the development of superresolution microscopy has revealed the organization of structures such as focal adhesions and podosomes (matrix-degrading protrusions similar to invadopodia formed in cells with a monocytic lineage; Linder and Wiesner, 2015).
A seminal study from the Waterman lab revealed the supramolecular organization of focal adhesions using superresolution microscopy, specifically PALM (Kanchanawong et al., 2010). This study showed that focal adhesions are multilaminar structures formed by three layers: an integrin layer, a force transduction layer, and an actin layer. PALM microscopy (Stubb et al., 2019) has been utilized to show the architecture of focal adhesions of stem cells, demonstrating that the organization of cornerstone adhesions and central adhesions are different at the nanoscale level; similar superresolution techniques have been used to image podosomes (Cox and Jones, 2013).
PALM, STORM, and other single molecule superresolution techniques can be applied to study the spatial organization of lamellipodium and invadopodium structures in cancer cells. MDA-MB-231 triple-negative breast cancer cells plated on a gelatin matrix were imaged with both widefield fluorescent microscopy and direct STORM (Fig. 5, left and middle panel); the use of dSTORM greatly increases the image resolution of subcellular structures and provides more detailed structural information. As shown in Fig. 5, the lamellipodium of cancer cells show a distinct localization of cortactin and F-actin at the leading edge. Interestingly, dSTORM imaging of invadopodia reveals complex spatial distribution of cortactin and F-actin, where F-actin is strongly enriched in the invadopodia core and cortactin molecules are scattered throughout the invadopodia structure (Fig. 5).
Fig. 5.
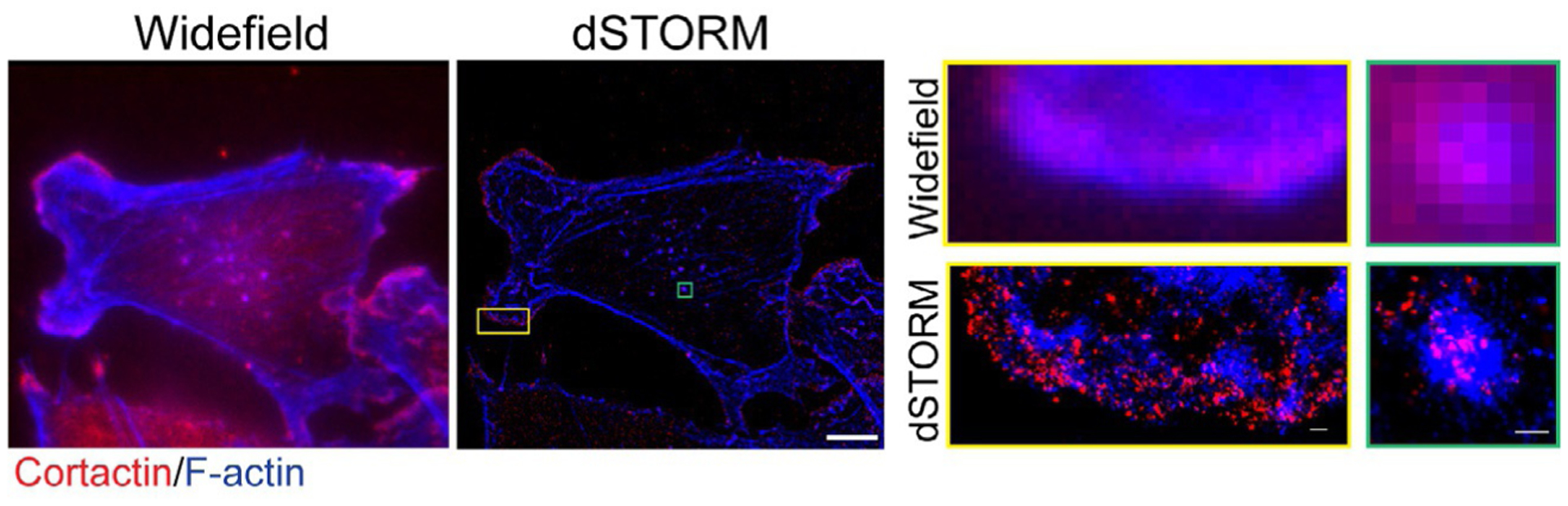
dSTORM image of a breast cancer cell. (Left panel): Widefield image of a cancer cell on gelatin matrix. F-actin in blue, cortactin in red. (Middle panel): dSTORM image of the same cancer cell. Top and bottom inset #1 (yellow box): Widefield and dSTORM of a lamellipodia structure. Top and bottom inset #2 (green box): Widefield and dSTORM showing spatial distribution of F-actin and cortactin in an invadopodia structure. Left and middle panel, scale bar: 10μm. Insets, scale bar: 1μm. Data acquisition for dSTORM was carried out on the Nanoimager S (Oxford NanoImaging, ONI, Oxford, UK). Signals from Alexa 647 and Alexa 488 were recorded sequentially for 10,000 frames each. Localization and image rendering were performed in the NimOS v1.4 software, and the final reconstruction displayed in a precision mode.
6.2. FRET-based imaging
To analyze signaling pathways of tumor cell motility, FRET-based imaging of Rho GTPases has been utilized both in vitro and in vivo and provides spatial information on Rho GTPase signaling and activation. Single-chain FRET biosensors, a more recent development in the field, consist of an N-terminal GTPase effector fragment, two fluorophores (an acceptor and donor FRET pair) separated by a linker, and a C-terminal GTPase (e.g., RhoA) (Donnelly et al., 2014; Mondal et al., 2020). When the GTPase is active (GTP-bound), it binds to the GTPase effector, resulting in the FRET pair coming into close proximity of each other and increasing the FRET signal (the donor fluorophore emission overlaps with the acceptor fluorophore excitation). Additional modifications of this technology include a near-IR FRET pair that allows for compatible imaging with other FRET-based biosensors (including CFP-YFP FRET pairs), as well as potential usage in vivo due to the optimal properties of near-IR fluorophores for deep imaging (Shcherbakova et al., 2018). In this particular study, the use of a near-IR Rac1 biosensor with a RhoA CFP-YFP FRET biosensor revealed that antagonistic RhoA and Rac1 activity in motile cells is dependent on ROCK signaling (Shcherbakova et al., 2018).
FRET-based imaging allows for spatial localization of Rho GTPase activity at a subcellular level and links it directly to tumor cell motility and invasion. For example, FRET biosensor technology has demonstrated that macrophage-tumor cell contact increases RhoA activity in tumor cells to promote invadopodia formation and intravasation (Roh-Johnson et al., 2014), whereas Rac1 activity can increase as invadopodia structures disassemble (Moshfegh et al., 2014). FRET biosensors are now being utilized in vivo to analyze RhoA activity in invasive breast and pancreatic cancer (Nobis et al., 2017); further exploration with FRET biosensors in vivo will be essential to elucidate signaling mechanisms activated during invasion and metastasis formation.
6.3. Intravital imaging and the tumor microenvironment
Techniques used to image in vivo, such as two photon intravital imaging and lattice light-sheet microscopy, are revealing the dynamics of tumor cells at the single-cell level during the metastatic cascade. Two-photon microscopy has visualized cancer cell motility in tumors (reviewed in Mondal et al., 2020), and more recently, behaviors of the surrounding microenvironment (reviewed in Di Martino et al., 2019). For example, the extracellular matrix has a key effect on modes of tumor cell migration; a combination of intravital two-photon imaging and computational modeling was used to delineate how tumor cells move based on what extracellular matrix structures they encounter (Tozluoğlu et al., 2013). Immune cells within the TME have also been characterized with two-photon imaging. Longitudinal studies using two-photon microscopy of tumor-associated macrophages in glioblastoma (GBM) has clearly defined two distinct types of TAMs, brain-resident microglia and bone marrow-derived macrophages, that are demonstrated to have distinct migratory behaviors (Chen et al., 2019).
Recently, the development of lattice light-sheet microscopy has allowed for imaging of tumor cell extravasation events. Lattice-light sheet imaging of zebrafish xenografts with labeled vasculature can capture the dynamics of cancer cells during extravasation with high temporal resolution in 3D (Liu et al., 2018b). Further studies using new tools of high-resolution imaging will help to illuminate the interplay between the TME and cancer cells, and how the TME affects tumor cell motility and invasion.
7. Conclusion
Understanding the dynamics of the actin cytoskeleton will help to develop targeted therapeutics that may prevent the dissemination of cancer cells and metastasis formation. The application of superresolution microscopy to investigate the macromolecular organization of invasive structures will provide valuable information about the spatial and temporal formation of invadopodia and pseudopodia and how the different components organize. We can envision that drugs that perturb the spatial organization of these molecules may interfere with the function of these actin-rich structures and may prevent the invasion and migration of cancer cells. As more work in the imaging field is developed, our understanding of actin dynamics at actin-rich structures will reveal possible candidates and additional signaling pathways to target during tumor cell dissemination.
Acknowledgments
We would like to thank Jill Gregory for her illustrations (Figs. 1–3), Linnea Olofsson for the acquisition and analysis of the widefield and STORM images (Fig. 5), and the Microscopy CoRE facility at Mount Sinai. This work was supported by a Susan G. Komen Career Catalyst Research Grant (CCR18547848), an NCI Career Transition Award (K22CA196750), an NCI R01 (CA244780), the Schneider-Lesser Foundation Award, a Stony Brook-Mount Sinai pilot award, an ACCRF award (to J.J.B.C); the Tisch Cancer Institute NIH Cancer Center grant (P30 CA196521). Chandrani Mondal received support from an NIH T32 CA078207 Training Program in Cancer Biology.
References
- Aiello NM, Kang Y, 2019. Context-dependent EMT programs in cancer metastasis. J. Exp. Med 216 (5), 1016–1026. 10.1084/jem.20181827. Rockefeller University Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alieva M, et al. , 2019. Intravital imaging of glioma border morphology reveals distinctive cellular dynamics and contribution to tumor cell invasion. Sci. Rep 9 (1), 2054. 10.1038/s41598-019-38625-4. Nature Publishing Group UK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjonen A, et al. , 2014. Mutant p53—associated myosin-X upregulation promotes breast cancer invasion and metastasis. J. Clin. Investig 124 (3), 1069–1082. 10.1172/JCI67280DS1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arozarena I, Wellbrock C, 2019. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 19 (7), 377–391. 10.1038/s41568-019-0154-4. [DOI] [PubMed] [Google Scholar]
- Attanasio F, et al. , 2011. Novel invadopodia components revealed by differential proteomic analysis. Eur. J. Cell Biol 90, 115–127. 10.1016/j.ejcb.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Attieh Y, Vignjevic DM, 2016. The hallmarks of CAFs in cancer invasion. Eur. J. Cell Biol 95 (11), 493–502. 10.1016/j.ejcb.2016.07.004. [DOI] [PubMed] [Google Scholar]
- Ayala I, et al. , 2009. Faciogenital dysplasia protein Fgd1 regulates invadopodia biogenesis and extracellular matrix degradation and is up-regulated in prostate and breast cancer. Cancer Res. 69 (3), 747–752. 10.1158/0008-5472.CAN-08-1980. United States. [DOI] [PubMed] [Google Scholar]
- Balsamo M, et al. , 2016. The alternatively-included 11a sequence modifies the effects of Mena on actin cytoskeletal organization and cell behavior. Sci. Rep 6, 35298. 10.1038/srep35298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayarmagnai B, et al. , 2019. Invadopodia-mediated ECM degradation is enhanced in the G1 phase of the cell cycle. J. Cell Sci 132, 20. 10.1242/jcs.227116. England. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty BT, Condeelis J, 2014. Digging a little deeper: the stages of invadopodium formation and maturation. Eur. J. Cell Biol 93 (10), 438–444. 10.1016/j.ejcb.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berens EB, Sharif GM, Wellstein A, Glasgow E, 2016. Testing the vascular invasive ability of cancer cells in zebrafish (Danio Rerio). J. Vis. Exp 117. 10.3791/55007, e55007.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin L, et al. , 2014. Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev 94 (1), 235–263. 10.1152/physrev.00018.2013. [DOI] [PubMed] [Google Scholar]
- Bosman FT, Havenith M, Cleutjens JP, 1985. Basement membranes in cancer. Ultrastruct. Pathol 8 (4), 291–304. 10.3109/01913128509141519. England. [DOI] [PubMed] [Google Scholar]
- Brábek J, et al. , 2010. The role of the tissue microenvironment in the regulation of cancer cell motility and invasion. Cell Commun. Signal 8, 22. 10.1186/1478-811X-8-22. BioMed Central. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, et al. , 2011. A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr. Biol 21 (8), 635–644. 10.1016/j.cub.2011.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, Hodgson L, Condeelis J, 2012. Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol 24 (2), 277–283. 10.1016/j.ceb.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, Magalhaes MA, Eddy RJ, Hodgson L, Condeelis J, 2013a. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol 14 (7), 405–415. 10.1038/nrm3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, et al. , 2013b. Spatial regulation of RhoC activity defines protrusion formation in migrating cells. J. Cell Sci 126 (15), 3356–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona G, et al. , 2016. Lamellipodin promotes invasive 3D cancer cell migration via regulated interactions with Ena/VASP and SCAR/WAVE. Oncogene 35 (39), 5155–5169. 10.1038/onc.2016.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S, et al. , 2006. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 20 (24), 3426–3439. 10.1101/gad.406406. Cold Spring Harbor Laboratory Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA, 2011. A perspective on cancer cell metastasis. Science (New York, N.Y.) 331 (6024), 1559–1564. 10.1126/science.1203543. United States. [DOI] [PubMed] [Google Scholar]
- Chan KT, Cortesio CL, Huttenlocher A, 2009. FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J. Cell Biol 185 (2), 357–370. 10.1083/jcb.200809110. The Rockefeller University Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MB, et al. , 2018a. Inflamed neutrophils sequestered at entrapped tumor cells via chemotactic confinement promote tumor cell extravasation. Proc. Natl. Acad. Sci 115 (27), 7022 LP–7027. 10.1073/pnas.1715932115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Tan W, Wang C, 2018b. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial-mesenchymal transition. OncoTargets Ther. 11, 3817–3826. 10.2147/OTT.S168317. Dove Medical Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Ross JL, Hambardzumyan D, 2019. Intravital 2-photon imaging reveals distinct morphology and infiltrative properties of glioblastoma-associated macrophages. Proc. Natl. Acad. Sci 116 (28), 14254 LP–14259. 10.1073/pnas.1902366116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, et al. , 2013. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 155 (7), 1639–1651. 10.1016/j.cell.2013.11.029. United States. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang SPH, Cabrera RM, Segall JE, 2016. Tumor cell intravasation. Am. J. Physiol. Cell Physiol 311 (1), C1–C14. 10.1152/ajpcell.00238.2015. American Physiological Society. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CK, et al. , 2008. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol 10 (9), 1039–1050. 10.1038/ncb1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu IM, Hengst L, Slingerland JM, 2008. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 8 (4), 253–267. 10.1038/nrc2347. England. [DOI] [PubMed] [Google Scholar]
- Chung Y-C, et al. , 2016. Rab11 collaborates E-cadherin to promote collective cell migration and indicates a poor prognosis in colorectal carcinoma. Eur. J. Clin. Investig 46 (12), 1002–1011. 10.1111/eci.12683. England. [DOI] [PubMed] [Google Scholar]
- Clark AG, Vignjevic DM, 2015. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol 36, 13–22. 10.1016/j.ceb.2015.06.004. Elsevier Ltd. [DOI] [PubMed] [Google Scholar]
- Clark ES, Weaver AM, 2008. A new role for cortactin in invadopodia: regulation of protease secretion. Eur. J. Cell Biol 87 (8–9), 581–590. 10.1016/j.ejcb.2008.01.008. Germany. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EA, et al. , 2000. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 406 (6795), 532–535. 10.1038/35020106. England. [DOI] [PubMed] [Google Scholar]
- Clark ES, et al. , 2007. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 67 (9), 4227–4235. 10.1158/0008-5472.CAN-06-3928. United States. [DOI] [PubMed] [Google Scholar]
- Condeelis J, Pollard JW, 2006. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124 (2), 263–266. 10.1016/j.cell.2006.01.007. United States. [DOI] [PubMed] [Google Scholar]
- Condeelis J, Segall JE, 2003. Intravital imaging of cell movement in tumours. Nat. Rev.Cancer 3 (12), 921–930. 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- Cox S, Jones GE, 2013. Imaging cells at the nanoscale. Int. J. Biochem. Cell Biol 10.1016/j.biocel.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Cukierman E, et al. , 2001. Taking cell-matrix adhesions to the third dimension. Science (New York, N.Y.) 294 (5547), 1708–1712. 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- Danen EHJ, et al. , 2002. The fibronectin-binding integrins alpha5beta1 and alphavbeta3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. J. Cell Biol 159 (6), 1071–1086. 10.1083/jcb.200205014. The Rockefeller University Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DesMarais V, et al. , 2009. N-WASP and cortactin are involved in invadopodium-dependent chemotaxis to EGF in breast tumor cells. Cell Motil. Cytoskeleton 66 (6), 303–316. 10.1002/cm.20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Martino J, et al. , 2014. Cdc42 and Tks5: a minimal and universal molecular signature for functional invadosomes. Cell Adhes. Migr 8 (3), 280–292. 10.4161/cam.28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Martino JS, Mondal C, Bravo-Cordero JJ, 2019. Textures of the tumour microenvironment. Essays Biochem. 63 (5), 619–629. 10.1042/EBC20190019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diring J, et al. , 2019. RPEL-family rhoGAPs link Rac/Cdc42 GTP loading to G-actin availability. Nat. Cell Biol 21 (7), 845–855. 10.1038/s41556-019-0337-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disanza A, et al. , 2013. CDC42 switches IRSp53 from inhibition of actin growth to elongation by clustering of VASP. EMBO J. 32 (20), 2735–2750. 10.1038/emboj.2013.208. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre A, Weinberg RA, 2019. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol 20 (2), 69–84. 10.1038/s41580-018-0080-4. England. [DOI] [PubMed] [Google Scholar]
- Donnelly SK, Bravo-Cordero JJ, Hodgson L, 2014. Rho GTPase isoforms in cell motility: don’t fret, we have FRET. Cell Adhes. Migr 8 (6), 526–534. 10.4161/cam.29712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly SK, Cabrera R, Mao SPH, et al. , 2017. Rac3 regulates breast cancer invasion and metastasis by controlling adhesion and matrix degradation. J. Cell Biol 216 (12), 4331–4349. 10.1083/jcb.201704048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuChez BJ, et al. , 2019. Durotaxis by human cancer cells. Biophys. J 116 (4), 670–683. 10.1016/j.bpj.2019.01.009. United States. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutartre H, et al. , 1996. Cytokinesis arrest and redistribution of actin-cytoskeleton regulatory components in cells expressing the Rho GTPase CDC42Hs. J. Cell Sci 109 (Pt. 2), 367–377. England. Available at: https://pubmed.ncbi.nlm.nih.gov/8838660. [DOI] [PubMed] [Google Scholar]
- Eckert MA, et al. , 2011. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 19 (3), 372–386. 10.1016/j.ccr.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy RJ, et al. , 2017. Tumor cell invadopodia: invasive protrusions that orchestrate metastasis. Trends Cell Biol. 27 (8), 595–607. 10.1016/j.tcb.2017.03.003. Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Werb Z, 2002. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2 (3), 161–174. London: Nature Publishing Group; 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Ezzoukhry Z, Henriet E, Cordelières FP, et al. , 2018. Combining laser capture micro-dissection and proteomics reveals an active translation machinery controlling invadosome formation. Nat. Commun 9, 2031. 10.1038/s41467-018-04461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KR, et al. , 2015. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527 (7579), 472–476. 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Alexander S, 2011. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147 (5), 992–1009. 10.1016/j.cell.2011.11.016. United States. [DOI] [PubMed] [Google Scholar]
- Friedl P, Gilmour D, 2009. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol 10 (7), 445–457. Nature Publishing Group. Available at: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- Friedl P, Weigelin B, 2008. Interstitial leukocyte migration and immune function. Nat. Immunol 9 (9), 960–969. 10.1038/ni.f.212. [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K, 2010. Plasticity of cell migration: a multiscale tuning model. J. Cell Biol 188 (1), 11–19. 10.1083/jcb.200909003. The Rockefeller University Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, et al. , 2012. Classifying collective cancer cell invasion. Nat. Cell Biol 14 (8), 777–783. London: Nature Publishing Group; 10.1038/ncb2548. [DOI] [PubMed] [Google Scholar]
- Geiger B, Yamada KM, 2011. Molecular architecture and function of matrix adhesions. Cold Spring Harb. Perspect. Biol 3 (5), a005033. 10.1101/cshperspect.a005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, et al. , 2001. Transmembrane crosstalk between the extracellular matrix—cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol 2 (11), 793–805. 10.1038/35099066. England. [DOI] [PubMed] [Google Scholar]
- Geiger B, Spatz JP, Bershadsky AD, 2009. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol 10 (1), 21–33. 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- Gertler F, Condeelis J, 2011. Metastasis: tumor cells becoming MENAcing. Trends Cell Biol. 21 (2), 81–90. Department of Biology and Koch Institute for Integrative Cancer Research at Massachusetts Institute of Technology (MIT), 77 Massachusetts Ave, Cambridge, MA 02138, USA. fgertler@MIT.EDU. Available at: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=21071226&retmode=ref&cmd=prlinks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampieri S, et al. , 2009. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol 11 (11), 1287–1296. 10.1038/ncb1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone G, et al. , 2007. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 128 (3), 561–575. 10.1016/j.cell.2006.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gligorijevic B, et al. , 2012. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J. Cell Sci 125 (Pt. 3), 724–734. 10.1242/jcs.092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gligorijevic B, Bergman A, Condeelis J, 2014. Multiparametric classification links tumor microenvironments with tumor cell phenotype. PLoS Biol. 12 (11), e1001995. Public Library of Science, Available at: 10.1371/journal.pbio.1001995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goicoechea SM, et al. , 2017. A RhoG-mediated signaling pathway that modulates invadopodia dynamics in breast cancer cells. J. Cell Sci 130 (6), 1064–1077. 10.1242/jcs.195552. England. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goreczny GJ, Forsythe IJ, Turner CE, 2018. Hic-5 regulates fibrillar adhesion formation to control tumor extracellular matrix remodeling through interaction with tensin1. Oncogene 37 (13), 1699–1713. 10.1038/s41388-017-0074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, et al. , 2007. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 446 (7137), 765–770. 10.1038/nature05760. England. [DOI] [PubMed] [Google Scholar]
- Gupton SL, Gertler FB, 2007. Filopodia: the fingers that do the walking. Sci. Signal 2007 (400), re5. Available at: http://stke.sciencemag.org/content/2007/400/re5.abstract. [DOI] [PubMed] [Google Scholar]
- Haga RB, Ridley AJ, 2016. Rho GTPases: regulation and roles in cancer cell biology. Small GTPases 7 (4), 207–221. 10.1080/21541248.2016.1232583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, et al. , 2019. Netrin-1 promotes the collective cell migration of liver cancer cells in a 3D cell culture model. J. Physiol. Biochem 75 (4), 489–498. 10.1007/s13105-019-00701-8. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA, 2011. Hallmarks of cancer: the next generation. Cell 144 (5), 646–674. Elsevier Inc. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21376230. [DOI] [PubMed] [Google Scholar]
- Hanks SK, et al. , 1992. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc. Natl. Acad. Sci. U. S. A 89 (18), 8487–8491. 10.1073/pnas.89.18.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harney AS, et al. , 2015. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage–derived VEGFA. Cancer Discov. 5 (9), 932–943. 10.1158/2159-8290.CD-15-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegerfeldt Y, et al. , 2002. Collective cell movement in primary melanoma explants. Cancer Res. 62 (7), 2125 LP–2130. Available at: http://cancerres.aacrjournals.org/content/62/7/2125.abstract. [PubMed] [Google Scholar]
- Hidalgo-Carcedo C, et al. , 2011. Collective cell migration requires suppression of actomyosin at cell–cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat. Cell Biol 13 (1), 49–59. 10.1038/ncb2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M-Y, et al. , 2012. TNF-α induces epithelial–mesenchymal transition of renal cell carcinoma cells via a GSK3β-dependent mechanism. Mol. Cancer Res 10 (8), 1109 LP–1119. 10.1158/1541-7786.MCR-12-0160. [DOI] [PubMed] [Google Scholar]
- Horton ER, et al. , 2016. The integrin adhesome network at a glance. J. Cell Sci 129 (22), 4159 LP–4163. 10.1242/jcs.192054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-J, et al. , 2016. Cellular microenvironment modulates the galvanotaxis of brain tumor initiating cells. Sci. Rep 6, 21583. 10.1038/srep21583. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SK, Oudin MJ, Tadros J, Neil J, Del Rosario A, Joughin BA, Ritsma L, Wyckoff J, Vasile E, Eddy R, Philippar U, Lussiez A, Condeelis JS, van Rheenen J, White F, Lauffenburger DA, Gertler FB, 2015. PTP1B-dependent regulation of receptor tyrosine kinase signaling by the actin-binding protein Mena. Mol. Biol. Cell 26 (21), 3867–3878. 10.1091/mbc.E15-06-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher A, 1995. Adhesion in cell migration. Curr. Opin. Cell Biol 7 (5), 697–706. 10.1016/0955-0674(95)80112-X. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A, Horwitz AR, 2011. Integrins in cell migration. Cold Spring Harb. Perspect. Biol 3 (9), a005074. 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO, 1992. Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69 (1), 11–25. 10.1016/0092-8674(92)90115-S. [DOI] [PubMed] [Google Scholar]
- Iwaya K, Norio K, Mukai K, 2007. Coexpression of Arp2 and WAVE2 predicts poor outcome in invasive breast carcinoma. Mod. Pathol 20 (3), 339–343. 10.1038/modpathol.3800741. [DOI] [PubMed] [Google Scholar]
- Jacob A, Prekeris R, 2015. The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Biol 3, 4. 10.3389/fcell.2015.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemet G, Hamidi H, Ivaska J, 2015. Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr. Opin. Cell Biol 36, 23–31. 10.1016/j.ceb.2015.06.007. [DOI] [PubMed] [Google Scholar]
- Jacquemet G, Baghirov H, Georgiadou M, et al. , 2016. L-type calcium channels regulate filopodia stability and cancer cell invasion downstream of integrin signalling. Nat. Commun 7, 13297. 10.1038/ncomms13297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemet G, Paatero I, Carisey AF, Padzik A, Orange JS, Hamidi H, Ivaska J, 2017. FiloQuant reveals increased filopodia density during breast cancer progression. J. Cell. Biol 216 (10), 3387–3403. 10.1083/jcb.201704045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayatilaka H, Umanzor FG, Shah V, et al. , 2018. Tumor cell density regulates matrix metalloproteinases for enhanced migration. Oncotarget 9 (66), 32556–32569. 10.18632/oncotarget.25863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeannot P, et al. , 2017. p27(Kip1) promotes invadopodia turnover and invasion through the regulation of the PAK1/Cortactin pathway. eLife 6, e22207. 10.7554/eLife.22207. eLife Sciences Publications, Ltd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DH, et al. , 2006. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 440 (7084), 692–696. 10.1038/nature04524. England. [DOI] [PubMed] [Google Scholar]
- Jones BC, et al. , 2017. Dual targeting of mesenchymal and amoeboid motility hinders metastatic behavior. Mol. Cancer Res 15 (6), 670–682. 10.1158/1541-7786.MCR-16-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juin A, et al. , 2012. Physiological type I collagen organization induces the formation of a novel class of linear invadosomes. Mol. Biol. Cell 23 (2), 297–309. 10.1091/mbc.E11-07-0594. The American Society for Cell Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juin A, et al. , 2014. Discoidin domain receptor 1 controls linear invadosome formation via a Cdc42-Tuba pathway. J. Cell Biol 207 (4), 517–533. 10.1083/jcb.201404079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera A, et al. , 2010. Nanoscale architecture of integrin-based cell adhesions. Nature 468, 580–584. 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, et al. , 2014. Rac1-dependent lamellipodial motility in prostate cancer PC-3 cells revealed by optogenetic control of Rac1 activity. PLoS One 9 (5), e97749. Public Library of Science. Available at: 10.1371/journal.pone.0097749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedrin D, et al. , 2008. Intravital imaging of metastatic behavior through a mammary imaging window. Nat. Meth 5 (12), 1019–1021. Nature Publishing Group. Available at: 10.1038/nmeth.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedrin D, et al. , 2009. ERBB1 and ERBB2 have distinct functions in tumor cell invasion and intravasation. Clin. Cancer Res 15 (11), 3733–3739. 10.1158/1078-0432.CCR-08-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R, 2005. Cell migration during gastrulation. Curr. Opin. Cell Biol 17 (5), 533–541. 10.1016/j.ceb.2005.08.006. England. [DOI] [PubMed] [Google Scholar]
- King SJ, et al. , 2016. Lamellipodia are crucial for haptotactic sensing and response. J. Cell Sci 129 (12), 2329–2342. 10.1242/jcs.184507. England. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohrman AQ, Matus DQ, 2017. Divide or conquer: cell cycle regulation of invasive behavior. Trends Cell Biol 27 (1), 12–25. 10.1016/j.tcb.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs AM, et al. , 2017. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol 19 (5), 518–529. 10.1038/ncb3513. England. [DOI] [PubMed] [Google Scholar]
- Kulkarni S, et al. , 2012. Increased expression levels of WAVE3 are associated with the progression and metastasis of triple negative breast cancer. PLoS One 7 (8), e42895. 10.1371/journal.pone.0042895. Public Library of Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama S, et al. , 2016. LPP inhibits collective cell migration during lung cancer dissemination. Oncogene 35 (8), 952–964. 10.1038/onc.2015.155. England. [DOI] [PubMed] [Google Scholar]
- Labernadie A, et al. , 2017. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol 19 (3), 224–237. 10.1038/ncb3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF, 1996. Cell migration: a physically integrated molecular process. Cell 84 (3), 359–369. 10.1016/s0092-8674(00)81280-5. United States. [DOI] [PubMed] [Google Scholar]
- Lawson CD, Ridley AJ, 2018. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol 217 (2), 447–457. 10.1083/jcb.201612069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrand C, et al. , 2004. Critical role of Ena/VASP proteins for filopodia formation in neurons and in function downstream of netrin-1. Neuron 42 (1), 37–49. 10.1016/s0896-6273(04)00108-4. United States. [DOI] [PubMed] [Google Scholar]
- Leong HS, Robertson AE, Stoletov K, Leith SJ, Chin CA, Chien AE, Hague MN, Ablack A, Carmine-Simmen K, McPherson VA, Postenka CO, Turley EA, Courtneidge SA, Chambers AF, Lewis JD, 2014. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 8 (5), 1558–1570. 10.1016/j.celrep.2014.07.050. [DOI] [PubMed] [Google Scholar]
- Li C-W, et al. , 2012. Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res. 72 (5), 1290–1300. 10.1158/0008-5472.CAN-11-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S, Wiesner C, 2015. Tools of the trade: podosomes as multipurpose organelles of monocytic cells. Cell. Mol. Life Sci 72 (1), 121–135. 10.1007/s00018-014-1731-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay CR, et al. , 2011. P-Rex1 is required for efficient melanoblast migration and melanoma metastasis. Nat. Commun 2 (1), 555. 10.1038/ncomms1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lintz M, Muñoz A, Reinhart-King CA, 2017. The mechanics of single cell and collective migration of tumor cells. J. Biomech. Eng 139 (2), 210051–210059. 10.1115/1.4035121. American Society of Mechanical Engineers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, et al. , 2018a. Stabilization of slug by NF-κB is essential for TNF-α-induced migration and epithelial-mesenchymal transition in head and neck squamous cell carcinoma cells. Cell. Physiol. Biochem 47 (2), 567–578. 10.1159/000489990. Germany. [DOI] [PubMed] [Google Scholar]
- Liu TL, Upadhyayula S, Milkie DE, Singh V, Wang K, Swinburne IA, Mosaliganti KR, Collins ZM, Hiscock TW, Shea J, Kohrman AQ, Medwig TN, Dambournet D, Forster R, Cunniff B, Ruan Y, Yashiro H, Scholpp S, Meyerowitz EM, Hockemeyer D, Drubin DG, Martin BL, Matus DQ, Koyama M, Megason SG, Kirchhausen T, Betzig E, 2018b. Observing the cell in its native state: imaging subcellular dynamics in multicellular organisms. Science 360 (6386). 10.1126/science.aaq1392. eaaq1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, et al. , 2020. IKKε phosphorylates kindlin-2 to induce invadopodia formation and promote colorectal cancer metastasis. Theranostics 10 (5), 2358–2373. 10.7150/thno.40397. Ivyspring International Publisher. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco AR, Ban Y, Crowley MJ, Lee SB, Ramchandani D, Du W, Elemento O, George JT, Jolly MK, Levine H, Sheng J, Wong ST, Altorki NK, Gao D, 2020. Differential contributions of pre- and post-EMT tumor cells in breast cancer metastasis. Cancer Res. 80 (2), 163–169. 10.1158/0008-5472.CAN-19-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G, 2009. Coordination of Rho GTPase activities during cell protrusion. Nature 461 (7260), 99–103. 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S, Johnson KR, Wheelock MJ, 2008. TGF-beta induces formation of F-actin cores and matrix degradation in human breast cancer cells via distinct signaling pathways. Exp. Cell Res 314 (19), 3478–3493. 10.1016/j.yexcr.2008.09.013. United States. [DOI] [PubMed] [Google Scholar]
- Manning CS, Hooper S, Sahai EA, 2015. Intravital imaging of SRF and Notch signalling identifies a key role for EZH2 in invasive melanoma cells. Oncogene 34 (33), 4320–4332. 10.1038/onc.2014.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Md Hashim NF, et al. , 2013. Hypoxia-induced invadopodia formation: a role for β-PIX. Open Biol. 3 (6), 120159. 10.1098/rsob.120159. The Royal Society. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medwig TN, Matus DQ, 2017. Breaking down barriers: the evolution of cell invasion. Curr. Opin. Genet. Dev 47, 33–40. 10.1016/j.gde.2017.08.003. Elsevier Ltd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H, et al. , 2000. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature 408 (6813), 732–735. 10.1038/35047107. England. [DOI] [PubMed] [Google Scholar]
- Mohammadi H, Sahai E, 2018. Mechanisms and impact of altered tumour mechanics. Nat. Cell Biol 20 (7), 766–774. 10.1038/s41556-018-0131-2 (Erratum in: Nat. Cell Biol. 20 (9), 2018, 1099). [DOI] [PubMed] [Google Scholar]
- Mohanty S, Xu L, 2010. Experimental metastasis assay. J. Vis. Exp (42), 1942. 10.3791/1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal C, Di Martino JS, Bravo-Cordero JJ, 2020. Imaging cell adhesion and migration. In: Barroso M, Intes X (Eds.), Imaging from Cells to Animals in Vivo. Chapman and Hall/CRC, New York. [Google Scholar]
- Moshfegh Y, et al. , 2014. A Trio-Rac1-Pak1 signalling axis drives invadopodia disassembly. Nat. Cell Biol 16 (6), 574–586. 10.1038/ncb2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SC, et al. , 1999. A novel protease-docking function of integrin at invadopodia. J. Biol. Chem 274 (35), 24947–24952. 10.1074/jbc.274.35.24947. United States. [DOI] [PubMed] [Google Scholar]
- Murphy DA, Courtneidge SA, 2011. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat. Rev. Mol. Cell Biol 12 (7), 413–426. 10.1038/nrm3141. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara H, et al. , 2003. Involvement of Cdc42 and Rac small G proteins in invadopodia formation of RPMI7951 cells. Genes Cells 8 (12), 1019–1027. England. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A, 1995. Rho, rac, and cdc42 GTPases regulate the assembly of multi-molecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81 (1), 53–62. 10.1016/0092-8674(95)90370-4. United States. [DOI] [PubMed] [Google Scholar]
- Nobis M, et al. , 2017. A RhoA-FRET biosensor mouse for intravital imaging in normal tissue homeostasis and disease contexts. Cell Rep. 21 (1), 274–288. 10.1016/j.celrep.2017.09.022. United States. [DOI] [PubMed] [Google Scholar]
- Northcott JM, Dean IS, Mouw JK, Weaver VM, 2018. Feeling stress: the mechanics of cancer progression and aggression. Front. Cell Dev. Biol 6, 17. 10.3389/fcell.2018.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauf AC, Massagué J, 2015. Surviving at a distance: organ-specific metastasis. Trends Cancer 1 (1), 76–91. 10.1016/j.trecan.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmeda D, et al. , 2007. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene 26 (13), 1862–1874. 10.1038/sj.onc.1209997. England. [DOI] [PubMed] [Google Scholar]
- Olmeda D, Cerezo-Wallis D, Riveiro-Falkenbach E, Pennacchi PC, Contreras-Alcalde M, Ibarz N, Cifdaloz M, Catena X, Calvo TG, Cañón E, Alonso-Curbelo D, Suarez J, Osterloh L, Graña O, Mulero F, Megías D, Cañamero M, Martínez-Torrecuadrada JL, Mondal C, Di Martino J, Lora D, Martinez-Corral I, Bravo-Cordero JJ, Muñoz J, Puig S, Ortiz-Romero P, Rodriguez-Peralto JL, Ortega S, Soengas MS, 2017. Whole-body imaging of lymphovascular niches identifies pre-metastatic roles of midkine. Nature 546 (7660), 676–680. 10.1038/nature22977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EN, Nordheim A, 2010. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol 11 (5), 353–365. 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MF, Sahai E, 2008. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 26 (4), 273. 10.1007/s10585-008-9174-2. [DOI] [PubMed] [Google Scholar]
- Ota I, et al. , 2009. Induction of a MT1-MMP and MT2-MMP-dependent basement membrane transmigration program in cancer cells by Snail1. Proc. Natl. Acad. Sci. U. S. A 106 (48), 20318–20323. 10.1073/pnas.0910962106. National Academy of Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanaban V, et al. , 2019. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573 (7774), 439–444. 10.1038/s41586-019-1526-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padua D, et al. , 2008. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133 (1), 66–77. 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons J, Horwitz A, Schwartz M, 2010. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell. Biol 11, 633–643. 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsialou A, et al. , 2013. Intravital multiphoton imaging reveals multicellular streaming as a crucial component of in vivo cell migration in human breast tumors. Intravital 2 (2), e25294. 10.4161/intv.25294. United States. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peláez R, Pariente A, Pérez-Sala Á, Larrayoz IM, 2019. Integrins: moonlighting proteins in invadosome formation. Cancers (Basel) 11 (5), 615. 10.3390/cancers11050615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Yamada KM, 2012. At the leading edge of three-dimensional cell migration. J. Cell Sci 125 (24), 5917 LP–5926. 10.1242/jcs.093732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulos C, Guichet PO, Masliantsev K, Wager M, Karayan-Tapon L, 2018. Functional invadopodia formed in glioblastoma stem cells are important regulators of tumor angiogenesis. Oncotarget 9 (29), 20640–20657. 10.18632/oncotarget.25045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippar U, et al. , 2008. A Mena invasion isoform potentiates EGF-induced carcinoma cell invasion and metastasis. Dev. Cell 15 (6), 813–828. Massachusetts Institute of Technology, Koch Institute, Cambridge, MA 02139, USA. Available at: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=19081071&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli J, et al. , 2016. Macrophage-dependent tumor cell transendothelial migration is mediated by Notch1/Mena(INV)-initiated invadopodium formation. Sci. Rep 6, 37874. 10.1038/srep37874. England. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinner S, Sahai E, 2008. Imaging amoeboid cancer cell motility in vivo. J. Microsc 231 (3), 441–445. 10.1111/j.1365-2818.2008.02056.x. England. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA, 2009. Actin, a central player in cell shape and movement. Science 326 (5957), 1208–1212. 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter AP, Papaioannou A, Malliri A, 2016. Deregulation of Rho GTPases in cancer. Small GTPases 7 (3), 123–138. 10.1080/21541248.2016.1173767. Taylor & Francis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ, 2006. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 4 (1), 38. 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, et al. , 2013. p21CIP1 mediates reciprocal switching between proliferation and invasion during metastasis. Oncogene 32 (18), 2292–2303.e7. 10.1038/onc.2012.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Marine J-C, Goding CR, 2019. Melanoma plasticity and phenotypic diversity: therapeutic barriers and opportunities. Genes Dev. 33 (19–20), 1295–1318. 10.1101/gad.329771.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razidlo GL, et al. , 2014. Vav1 as a central regulator of invadopodia assembly. Curr. Biol 24 (1), 86–93. 10.1016/j.cub.2013.11.013. England. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, 2015. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol 36, 103–112. 10.1016/j.ceb.2015.08.005. Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh-Johnson M, et al. , 2014. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene 33 (33), 4203–4212. 10.1038/onc.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos ET, et al. , 2011a. Mena invasive (MenaINV) promotes multicellular streaming motility and transendothelial migration in a mouse model of breast cancer. J. Cell Sci 124 (Pt. 13), 2120–2131. Department of Anatomy and Structural Biology, Albert Einstein College of Medicine, Bronx, NY 10461, USA. Available at: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=21670198&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos ET, Condeelis JS, Patsialou A, 2011b. Chemotaxis in cancer. Nat. Rev. Cancer 11 (8), 573–587. 10.1038/nrc3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ, 2003. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol 5 (8), 711–719. 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- Sakurai-Yageta M, et al. , 2008. The interaction of IQGAP1 with the exocyst complex is required for tumor cell invasion downstream of Cdc42 and RhoA. J. Cell Biol 181 (6), 985 LP–998. Available at: http://jcb.rupress.org/content/181/6/985.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Moreno V, et al. , 2008. Rac activation and inactivation control plasticity of tumor cell movement. Cell 135 (3), 510–523. 10.1016/j.cell.2008.09.043. United States. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Parsons JT, 1999. Complex formation with focal adhesion kinase: a mechanism to regulate activity and subcellular localization of Src kinases. Mol. Biol. Cell 10 (10), 3489–3505. 10.1091/mbc.10.10.3489. The American Society for Cell Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova DM, et al. , 2018. Direct multiplex imaging and optogenetics of Rho GTPases enabled by near-infrared FRET. Nat. Chem. Biol 14 (6), 591–600. 10.1038/s41589-018-0044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, et al. , 2012. The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov. 2 (8), 706 LP–721. 10.1158/2159-8290.CD-11-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HA, Kang Y, 2013. The metastasis-promoting roles of tumor-associated immune cells. J. Mol. Med. (Berl) 91 (4), 411–429. 10.1007/s00109-013-1021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MS, et al. , 2010. Article identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol. Cell 40 (6), 877–892. 10.1016/j.molcel.2010.11.029. Elsevier Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, Klemke R, 2010. Visualizing extravasation dynamics of metastatic tumor cells. J. Cell Sci 123 (Pt 13), 2332–2341. 10.1242/jcs.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubb A, Guzmán C, Närvä E, Aaron J, Chew TL, Saari M, Miihkinen M, Jacquemet G, Ivaska J, 2019. Superresolution architecture of cornerstone focal adhesions in human pluripotent stem cells. Nat. Commun 10 (1), 4756. 10.1038/s41467-019-12611-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuelten CH, Parent CA, Montell DJ, 2018. Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat. Rev. Cancer 18 (5), 296–312. 10.1038/nrc.2018.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Costell M, Fässler R, 2019. Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol 21 (1), 25–31. 10.1038/s41556-018-0234-9. [DOI] [PubMed] [Google Scholar]
- Talkenberger K, Cavalcanti-Adam EA, Voss-Böhme A, et al. , 2017. Amoeboid-mesenchymal migration plasticity promotes invasion only in complex heterogeneous microenvironments. Sci. Rep 7, 9237. 10.1038/s41598-017-09300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E, Mayor R, 2012. Neural crest migration: interplay between chemorepellents, chemoattractants, contact inhibition, epithelial-mesenchymal transition, and collective cell migration. Wiley Interdiscip. Rev. Dev. Biol 1 (3), 435–445. 10.1002/wdev.28. [DOI] [PubMed] [Google Scholar]
- Tian C, Öhlund D, Rickelt S, Lidström T, Huang Y, Hao L, Zhao RT, Franklin O, Bhatia SN, Tuveson DA, Hynes RO, 2020. Cancer cell-derived matrisome proteins promote metastasis in pancreatic ductal adenocarcinoma. Cancer Res. 80 (7), 1461–1474. 10.1158/0008-5472.CAN-19-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichet M, et al. , 2015. Tumour-derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat. Commun 6, 6993. 10.1038/ncomms7993. England. [DOI] [PubMed] [Google Scholar]
- Tozluoğlu M, et al. , 2013. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat. Cell Biol 15 (7), 751–762. 10.1038/ncb2775. [DOI] [PubMed] [Google Scholar]
- Turner CE, 2000. Paxillin and focal adhesion signalling. Nat. Cell Biol 2 (12), E231–E236. 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- VanderVorst K, et al. , 2019. Wnt/PCP signaling contribution to carcinoma collective cell migration and metastasis. Cancer Res. 79 (8), 1719 LP–1729. 10.1158/0008-5472.CAN-18-2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, et al. , 2007. Coordinated regulation of pathways for enhanced cell motility and chemotaxis is conserved in rat and mouse mammary tumors. Cancer Res. 67 (8), 3505 LP–3511. 10.1158/0008-5472.CAN-06-3714. [DOI] [PubMed] [Google Scholar]
- Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, Chen AC, Sah RL, Taylor SS, Engler AJ, Yang J, 2015. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol 17 (5), 678–688. 10.1038/ncb3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilbaecher KN, Guise TA, McCauley LK, 2011. Cancer to bone: a fatal attraction. Nat. Rev. Cancer 11 (6), 411–425. 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S, Paterson HF, Marshall CJ, 2005. Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat. Cell Biol 7 (3), 255–261. 10.1038/ncb1230. England. [DOI] [PubMed] [Google Scholar]
- Williams KC, et al. , 2019. Invadopodia are chemosensing protrusions that guide cancer cell extravasation to promote brain tropism in metastasis. Oncogene 38 (19), 3598–3615. 10.1038/s41388-018-0667-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, et al. , 2003. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol 160 (2), 267–277. 10.1083/jcb.200209006. United States. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, et al. , 2007. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol 9 (8), 893–904. London: Nature Publishing Group; 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, et al. , 2003. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J. Cell Biol 163 (3), 583–595. 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak MA, et al. , 2004. Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta (BBA)—Mol. Cell Res 1692 (2), 103–119. 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Wu Y, et al. , 2009. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 15 (5), 416–428. 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, et al. , 2015. Roles of the cyclooxygenase 2 matrix metalloproteinase 1 pathway in brain metastasis of breast cancer. J. Biol. Chem 290 (15), 9842–9854. 10.1074/jbc.M114.602185. American Society for Biochemistry and Molecular Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff J, et al. , 2004. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 64 (19), 7022–7029. 10.1158/0008-5472.CAN-04-1449. United States. [DOI] [PubMed] [Google Scholar]
- Wyckoff JB, et al. , 2007. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 67 (6), 2649–2656. 10.1158/0008-5472.CAN-06-1823. United Statefs. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Condeelis J, 2007. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta (BBA)—Mol. Cell Res 1773 (5), 642–652. 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, et al. , 2005. Molecular mechanisms of invadopodium formation. J. Cell Biol 168 (3), 441 LP–452. Available at: http://jcb.rupress.org/content/168/3/441.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, et al. , 2003. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J. Cell Sci 116 (22), 4605 LP–4613. 10.1242/jcs.00792. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R, et al. , 2007. Functional atlas of the integrin adhesome. Nat. Cell Biol 9 (8), 858–867. Nature Publishing Group. Available at: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir E, et al. , 1999. Molecular diversity of cell-matrix adhesions. J. Cell Sci 112 (11), 1655 LP–1669. Available at: http://jcs.biologists.org/content/112/11/1655.abstract. [DOI] [PubMed] [Google Scholar]
- Zervantonakis IK, et al. , 2012. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. U. S. A 109 (34), 13515 LP–13520. 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, et al. , 2018. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. 8 (8), 1006–1025. 10.1158/2159-8290.CD-17-1371. United States. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, et al. , 2015. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527 (7579), 525–530. 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]