

Abstract
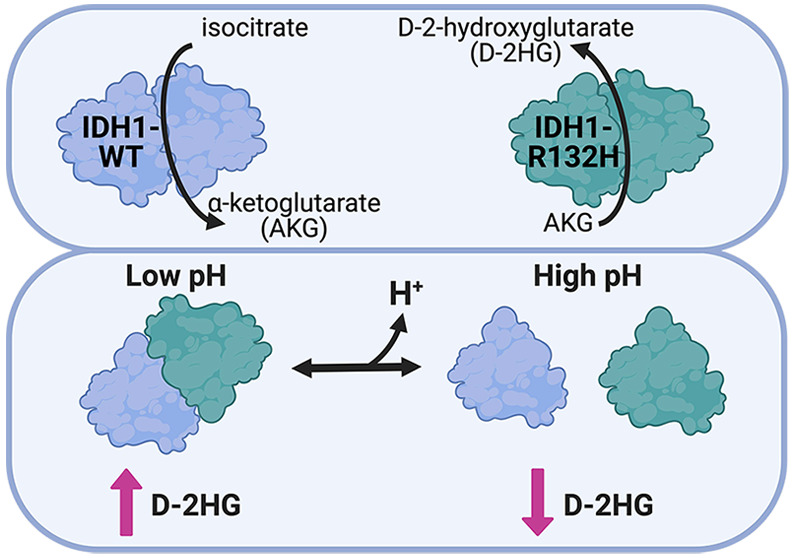
Isocitrate dehydrogenase 1 (IDH1) is a key metabolic enzyme for maintaining cytosolic levels of α-ketoglutarate (AKG) and preserving the redox environment of the cytosol. Wild-type (WT) IDH1 converts isocitrate to AKG; however, mutant IDH1-R132H that is recurrent in human cancers catalyzes the neomorphic production of the oncometabolite d-2-hydroxyglutrate (D-2HG) from AKG. Recent work suggests that production of l-2-hydroxyglutarte in cancer cells can be regulated by environmental changes, including hypoxia and intracellular pH (pHi). However, it is unknown whether and how pHi affects the activity of IDH1-R132H. Here, we show that in cells IDH1-R132H can produce D-2HG in a pH-dependent manner with increased production at lower pHi. We also identify a molecular mechanism by which this pH sensitivity is achieved. We show that pH-dependent production of D-2HG is mediated by pH-dependent heterodimer formation between IDH1-WT and IDH1-R132H. In contrast, neither IDH1-WT nor IDH1-R132H homodimer formation is affected by pH. Our results demonstrate that robust production of D-2HG by IDH1-R132H relies on the coincidence of (1) the ability to form heterodimers with IDH1-WT and (2) low pHi or highly abundant AKG substrate. These data suggest cancer-associated IDH1-R132H may be sensitive to physiological or microenvironmental cues that lower pH, such as hypoxia or metabolic reprogramming. This work reveals new molecular considerations for targeted therapeutics and suggests potential synergistic effects of using catalytic IDH1 inhibitors targeting D-2HG production in combination with drugs targeting the tumor microenvironment.
The metabolic enzyme isocitrate dehydrogenase 1 (IDH1) catalyzes the reversible conversion of isocitrate (ICT) to α-ketoglutarate (AKG) (Figure 1A). While there are two mitochondrial isoforms of isocitrate dehydrogenase (IDH2 and IDH3), IDH1 is the only cytosolic isoform. IDH3 functions in the citric acid cycle and IDH2 maintains the mitochondrial redox state, while IDH1 contributes to cytosolic metabolism by driving NADPH-dependent lipid biosynthesis and maintaining cytosolic pools of AKG.1 Importantly, IDH1 also regulates the redox environment of the cytosol through the generation of NADPH.2
Figure 1.

IDH1 catalysis and structural conformations. (A) Reversible conversion of isocitrate (ICT) to α-ketoglutarate (AKG) catalyzed by WT-IDH1. The neomorphic reaction catalyzed by IDH1 mutants is the irreversible conversion of AKG to d-2-hydroxyglutarate (D-2HG). (B) The active site of IDH1 shows Arg132 (magenta stick) coordinates substrate ICT (green stick) in the enzyme active site. (C) Arg132 also plays a role in dimer formation, coordinating aspartate residues in the quasi-open conformation of the dimer upon NADP+ binding (NADP+-bound). This conformation is an intermediate on the path to the closed, catalytically active conformation (ICT-bound).
Dysregulation or mutation of IDH1 is associated with several diseases, including cancer.3 While the wild-type (WT) enzyme catalyzes the reversible conversion of ICT to AKG, IDH1 mutations at Arg132 confer the ability to catalyze a neomorphic reaction in which AKG is converted to the oncometabolite d-2-hydroxyglutarate (D-2HG) (Figure 1A). This oncometabolite is not produced at high levels in normal tissue and serves as a biomarker for dysplastic or tumorigenic tissue.4 In WT IDH1, Arg132 serves two distinct roles: first in coordinating the substrate isocitrate for oxidation within the enzyme active site5 (Figure 1B) and second to stabilize homodimer formation through an electrostatic interaction with aspartate residues on the adjacent monomer6 (Figure 1C). Because recent work shows that recurrent Arg → His mutations in cancer can confer pH sensitive function to the mutant protein,7 we sought to answer the question of whether D-2HG production by IDH1-R132H is also sensitive to intracellular pH (pHi) dynamics.
This hypothesis that D-2HG production may be sensitive to tumor microenvironment or physiological changes is supported by the disparate outcomes of IDH1-R132H mutation in cancer.8 For example, in glioma the IDH1-R132H mutation is correlated with better prognosis and response,9 while in thyroid cancer10 and AML,11,12 the opposite is true. Indeed, there are several findings that suggest a potential role for hypoxia and low pH in regulating even WT IDH1 function.13 Prior work suggests that WT IDH1 reactions could be sensitive to environment or microenvironment changes, with low buffer pH enhancing the reverse reaction (AKG → ICT)13 and high buffer pH enhancing the forward reaction (ICT → AKG) in vitro.14 These in vitro results with recombinant enzyme have some dependence on the specific buffer systems used, and the most pronounced effects require pH values outside the physiological range (7.0–7.6). However, this work does suggest that IDH1 function may be linked to environmental cues.
Here, we show that IDH1-R132H has pH-dependent production of D-2HG in cells, with increased D-2HG production at lower pH. We confirm that the catalytic activity of recombinant IDH1-R132H is insensitive to pH in vitro, suggesting a more complicated mechanism for pH sensitive production of D-2HG. We reveal that pH-dependent D-2HG production requires heterodimer formation, indicating pH-dependent dimer formation as a potential mechanism. We confirm this mechanism using both native gel electrophoresis assays and ELISA binding assays. Finally, our data provide a missing link in decoupling IDH1 mutation status and D-2HG accumulation to resolve disparate data about (1) whether substrate shuttling is necessary for D-2HG production, (2) reported buffer dependence in prior pH-dependent activity experiments, and (3) varied outcomes for patients with IDH1-R132H mutation.
Results
We recently showed that recurrent Arg → His mutations can confer pH sensitive functions to mutant proteins.7 These results led us to the hypothesis that the increased production of D-2HG observed with IDH1-R132H is also sensitive to pHi. We initially predicted that IDH1-R132H would confer pH-dependent substrate binding (Figure 1B) where at low pH, when the His132 is more likely to be protonated, the WT reaction would be favored (Figure 1A) while increased pH would drive increased production of D-2HG. To test this hypothesis, we stably expressed either IDH1-WT or IDH1-R132H at equal levels in NIH-3T3 cells (Figure 2A).
Figure 2.

IDH1-R132H is required for pH sensitive d-2-hydroxyglutarate production in mammalian cells. (A) Western blot analysis shows equal expression of FLAG-tagged IDH1-WT and IDH1-R132H when stably expressed in NIH-3T3 cells (3T3). Blots shown are duplicate blots with equal loading of 5 μg of total protein in each lane followed by anti-actin and anti-FLAG antibody detection. (B) Intracellular pH (pHi) can be controlled in 3T3 cells using a small molecule inhibitor of NHE1 (EIPA, 1 μM) to decrease pHi and ammonium chloride (NH4Cl, 5 mM) to increase pHi (see Methods for details). (C) 3T3 cells with pHi manipulated as in panel B were assayed for levels of the oncometabolite D-2HG and (D) AKG (see Methods for details). Data in panels B and C are from four biological replicates and shown as box and whisker plots (Tukey display). Data in panel D are from three biological replicates and shown as scatter dot plots (mean ± standard error of the mean). For panels B–D, significance was determined by a two-way analysis of variance with Tukey multiple-comparison correction (*p < 0.05, **p < 0.01, and ***p < 0.001). For panel D, there were no statistically significant differences across the data set.
Importantly, these cell lines also express endogenous IDH1-WT, which in some cases has been shown to be necessary for high D-2HG production with IDH1-R132H,15−17 particularly when the AKG concentration is limiting.16 We can experimentally manipulate pHi in these cell lines over 24 h by incubating with a low concentration of ammonium chloride (NH4Cl, 5 mM) to increase pHi or the plasma membrane Na+–H+ exchanger (NHE1) inhibitor 5-(N-ethyl-N-isopropyl) amiloride (EIPA) to lower pHi (Figure 2B).
D-2HG Production by IDH1-R132H Is Increased at Low pHi
We first measured D-2HG production in cells using a colorimetric assay (see Methods). We found that stable expression of IDH1-WT increased levels of D-2HG over the parental line but showed no dependence on pHi (Figure 2C). This finding is consistent with previous work showing that overexpression of WT IDH1 leads to increased production of D-2HG in cells.18 We also found that stable expression of IDH1-R132H in the background of endogenous IDH1-WT increased D-2HG levels, with the highest level of D-2HG production observed at low pHi (Figure 2C). To confirm that the change in D-2HG production was not the result of altered α-ketoglutarate availability under altered pHi conditions, we also measured levels of AKG using a colorimetric assay. We did not observe significant changes in levels of α-ketoglutarate under any experimental condition tested (Figure 2D). This suggests that the increase in D-2HG observed at lower pHi is not the result of significant changes in the availability of AKG. While we cannot rule out the possibility that the cytosolic pool of AKG is being depleted more rapidly than the mitochondrial pool when IDH1-R132H is overexpressed, equilibrium anapleurotic reactions that replenish both mitochondrial and cytosolic AKG make this possibility unlikely.19 Overall, these data suggest that increased D-2HG production observed at lower pHi is dependent on the expression of IDH1-R132H in these cells.
Our cell data did not fit our initial hypothesis, but they do dovetail with published findings on pH sensitive reduction of AKG by metabolic enzymes. Recent work by Nadtochiy et al. shows that the metabolic enzymes lactate dehydrogenase (LDHA) and malate dehydrogenase (MDH) can produce high levels of L-2HG from AKG specifically at low pHi.20 Subsequent work by Inkelofer et al. confirms this result and suggests that pH sensitive production of L-2HG comes not from titration of LDHA or MDH active site residues, but instead from titration of the substrate metabolite α-ketoglutarate.21 Important to note, however, is the fact that we did not observe pH-dependent production of D-2HG in either IDH1-WT or the parental NIH-3T3 cells (Figure 2C). These results suggest that the pH-dependent production of D-2HG we observed does require the mutant active site residue IDH1-R132H. Therefore, we next tested whether the increase in D-2HG levels observed at lower pHi in cells expressing IDH1-R132H was the result of measurable pH sensitive catalytic activity of that mutant protein.
In Vitro Assays Show the IDH1-WT Homodimer and IDH1-R132H Homodimer Are pH Insensitive
To determine the in vitro activity of IDH1-WT and IDH1-R132H, we expressed and purified the enzymes from Escherichia coli cells (Figure S1). After assembling reaction mixtures with the enzyme, NADP+, and isocitrate, we monitored the production of NADPH as a readout of the forward oxidative reaction and AKG production at pH 7.0 and 7.5 (Figure 3A). We found that IDH1-WT did not show pH-dependent kcat for AKG production [2.2 × 104 ± 1.8 × 103 s–1 at pH 7.0 vs 2.7 × 104 ± 3.6 × 103 s–1 at pH 7.5 (Figure 3D)], nor did IDH1-WT have a pH-dependent Km for ICT [63.0 ± 15.3 μM at pH 7.0 vs 83.0 ± 39.2 μM at pH 7.5 (Figure 3D)]. We note that these values are in the range of the literature, as reported kcat values for the IDH1-WT forward reaction range from 8.6 to 44000 s–1, while the Km for ICT ranges from 6.0 to 110 μM.3,6,16,17,22,23 These findings suggest that the forward oxidative reaction for IDH1-WT is not pH-dependent under these reaction conditions, which is consistent with our data from cells.
Figure 3.

IDH1 homodimer activities in vitro show pH insensitive D-2HG production. See Methods for assembly protocols for in vitro activity assays. (A) Activity of IDH1-WT (micromoles per minute per milligram of protein) for forward oxidative (ICT → AKG) reaction at two buffer pH values (7.0 and 7.5). (B) Activity of IDH1-R132H (micromoles per minute per milligram of protein) for the forward oxidative (ICT → AKG) reaction at two buffer pH values: 7.0 and 7.5. (C) Activity of IDH1-R132H (micromoles per minute per milligram of protein) for the reverse reductive (AKG → D-2HG) reaction at two buffer pH values (7.0 and 7.5). Data in panel A–C are from seven replicates across two protein preparations and shown as scatter plots (mean ± standard error of the mean), with curve fits shown. Significance was determined using Graph Pad Prism Michaelis–Menten curve fitting (see Methods for details) (**p < 0.01) for Vmax. (D) Table of Michaelis–Menten kinetic constants kcat and Km quantified from the data in panels A–C. Significance was determined using Graph Pad Prism Michaelis–Menten curve fitting (see Methods for details) (*p < 0.05, compared to pH 7.5).
In a recent collaborative report, we showed that under ionic buffer conditions, an effect on kcat and Km can be observed with WT IDH1 across a broader pH range (6.2–8.0).14 However, we did not observe this pronounced effect in our buffer system across the narrower physiological pH range tested (pH 7.0–7.5). This discrepancy may suggest that the proposed pH sensitive network at the dimerization interface14 requires larger pH changes to affect IDH1-WT activity. Our observations could also be explained by differences in the effective ionic strength of ionic buffers and zwitterionic buffers when used at similar molar concentrations: while ionic buffers increase ionic strength, zwitterionic buffers have no effect on the ionic strength of buffer solutions.24 We note that the IDH1-WT open conformation is stabilized by a salt-bridge interaction between Arg132 and two or three conserved aspartate residues while the mutant IDH1-R132H dimer does not preserve these close interactions in the apo or NADP+-bound form (Figure 1C).6
We also tested the pH dependence of IDH1-R132H for the forward oxidative reaction by assembling reactions with the enzyme, NADP+, and isocitrate and monitoring the production of NADPH at pH 7.0 and 7.5 (Figure 3B). Here, we found that IDH1-R132H had a pH-independent Km for ICT [27.2 ± 9.0 μM at pH 7.0 vs 23.7 ± 11.0 μM at pH 7.5 (Figure 3D)], but we observed a small but significant difference in kcat for this reaction, with lower production at lower buffer pH [52.0 ± 4.9 s–1 at pH 7.0 vs 79.2 ± 9.1 s–1 at pH 7.5 (Figure 3D)]. As expected, the IDH1-R132H mutant had a lower kcat at both pH values compared to that of WT IDH1 (Figure 3D).
We next tested the pH dependence of IDH1-R132H and IDH1-WT for the reverse, reductive reaction and D-2HG production. After assembling reaction mixtures with the enzyme, NADPH, and AKG, we measured loss of NADPH at pH 7.0 and 7.5. Because this assay monitors NADH consumption, the reaction kinetics reflect the reductive reaction that produces D-2HG as well as the reverse reductive reaction (AKG → ICT). Both IDH1-WT and IDH1-R132H are shown to catalyze the conversion of AKG to ICT, although at very low levels, and mutant IDH1-R132H efficiently catalyzes the production of D-2HG from AKG in vitro. Under our reaction conditions, we were not able to detect any reverse reductive reaction with IDH1-WT. However, the IDH1-R132H mutant catalyzed the reduction of AKG and rapid consumption of NADPH (Figure 3C). This indicates that this assay is an appropriate measure of the reductive generation of D-2HG by the IDH1-R132H enzyme. However, we did not observe a pHi-dependent difference in either kcat or Km with IDH1-R132H (Figure 3D). Taken together, these data suggest that neither IDH1-WT nor IDH1-R132H homodimers exhibit pH sensitive D-2HG production in vitro.
These in vitro data did not clarify a mechanism for the IDH1-R132H-dependent pH sensitive production of D-2HG we observed in cells. However, we noted one significant difference between our cell-based assays (Figure 2) and in vitro kinetic assays (Figure 3). In cells, IDH1-R132H can effectively form a heterodimer complex with endogenous WT IDH1.25 It is possible that while IDH1-WT and IDH1-R132H homodimers we assayed in vitro are pH insensitive for both oxidative and reductive catalysis, the active heterodimer does exhibit pH sensitive catalysis.
Both IDH1-WT and IDH1-R132H Are Required for pH-Dependent Production of D-2HG
To test the hypothesis that pH-dependent WT:R132H heterodimer is driving pH-dependent production of D-2HG in cells, we needed to measure in vitro production of D-2HG under heterodimer reaction conditions. The assays described above monitoring NADPH cannot sufficiently resolve this possibility because IDH1-WT catalytic activity reduces NADP+ while IDH1-R132H catalytic activity oxidizes NADPH, leading to a futile cycle if both enzymes are present and catalytically active. Therefore, we established a protocol to monitor production of AKG and D-2HG by an end point colorimetric assay.
We prepared in vitro reactions with 1:1 WT:R132H ratios with the buffer conditions described above in the presence of ICT and measured levels of AKG and D-2HG by a colorimetric assay (Figure 4A,B). We compared these results to reactions prepared identically with WT or IDH1-R132H alone. We found that both WT homodimer and WT:R132H heterodimer reactions produced significant amounts of AKG from isocitrate, while the R132H:R132H homodimer did not produce detectable amounts of AKG (Figure 4A). These findings are in agreement with our data in cells (Figure 2B) and with previous reports3 suggesting IDH1-R132H is less efficient in converting ICT to AKG compared with IDH1-WT. We did not observe pH-dependent differences in AKG production in our in vitro forward oxidative reactions (Figure 4A). The statistically significant difference in AKG produced with the WT:R132H heterodimer compared to the WT:WT homodimer is likely due to the fact that we used the same amount of total protein in these assays, such that there is exactly half the amount of wild-type protein under the heterodimer condition compared to the WT homodimer condition. If IDH1-R132H is not producing much AKG, as expected, this would account for the reduced amount of AKG produced with the heterodimer.
Figure 4.

Production of D-2HG in vitro is pH sensitive only with WT:R132H heterodimers. Colorimetric analysis of enzymatic reactions prepared with the IDH1-WT homodimer (WT:WT), IDH1-R132H homodimer (R132H:R132H), and heterodimer (WT:R132H) (see Methods for details). Reaction mixtures were prepared at 250 nM total protein (WT:WT, R132H:R132H, or 1:1 WT:R132H). Under these conditions, we measured production of (A) AKG and (B) D-2HG. In all cases, four experimental replicates (each result is the average of two technical replicates) across two protein preparations are shown, with significance determined by two-way analysis of variance with Tukey multiple-comparison correction (*p < 0.05, **p < 0.01, and ***p < 0.001).
We found that only the reaction mixtures containing IDH1-R132H produced significant amounts of D-2HG, and only the WT:R132H heterodimer reactions showed pH-dependent production of D-2HG (Figure 4B), with increased production at lower buffer pH (Figure 4B).
These results suggest that both IDH1-WT and IDH1-R132H are required for pH sensitive production of D-2HG in vitro. These data are also consistent with previous work in cells suggesting that high D-2HG production by IDH1-R132H requires the presence of WT IDH1.26 In the assays described in Figure 4, we could not rule out the possibility that even though the protein was added in 1:1 mixtures, a mixture of the WT:WT homodimer, WT:R132H heterodimer, and R132H:R132H homodimer was being formed and altering our interpretation of these results. To test this hypothesis, we prepared 3:1 and 1:3 mixtures of WT:R132H. In this experiment, maximal heterodimer formation is consistent across these two conditions, allowing us to test for effects of excess IDH1-WT (3:1 mixture) and excess IDH1-R132H (1:3 mixture) on D-2HG formation. We confirmed that we saw the same level of pH-dependent production of D-2HG when we used 3:1 mixtures of WT:R132H or 1:3 mixtures of WT:R132H (Figure S2). This strongly suggests that the heterodimer is the critical catalytic unit for the observed pH-dependent production of D-2HG and that formation of excess WT:WT homodimer or R132H:R132H homodimer in our reactions is not influencing our end point D-2HG assay observations. Taken together, our data suggest that the ability to form the WT:R132H heterodimer is required for pH-dependent production of D-2HG in our in vitro and cell-based assays.
IDH1-WT and IDH1-R132H Heterodimer Formation Is pH-Dependent
To test whether WT:R132H heterodimer formation is pH-dependent, we first measured dimer formation using native gel electrophoresis. We prepared protein at various concentrations in binding buffer at pH 7.0 and 7.8 (see Methods for details). Because we expected the largest difference in binding would be found in the apo (open) conformations, we performed these homodimer and heterodimer binding assays in the absence of substrate. For assays of homodimer formation, we prepared binding assays at pH 7.0 and 7.8 and ran binding reactions on native polyacrylamide gel electrophoresis gels using native gel running buffers prepared at pH 7.0 and 7.8. For assays of homodimer formation, we prepared binding reactions with only IDH1-WT or only IDH1-R132H [0–64 μg of total protein (for details, see Methods)]. For assays of heterodimer formation, we prepared binding assays with constant IDH1-WT (8 μg) and varied IDH1-R132H (0–64 μg of total protein) (for details, see Methods). We found that homodimer formation is pH insensitive for both IDH1-WT (Figure 5A) and IDH1-R132H (Figure 5B). For representative blots, see Figure S3. We observed more dimer formation with the IDH1-WT homodimer compared with the IDH1-R132H homodimer under all conditions. Although reasonable levels of D-2HG production were observed in our in vitro reactions, IDH1-R132H homodimer formation was minimal. These data suggest that either substrate binding stabilizes the homodimer or high levels of IDH1-R132H homodimerization are not required for the neomorphic reaction when abundant AKG substrate is present in the reaction mixture.
Figure 5.
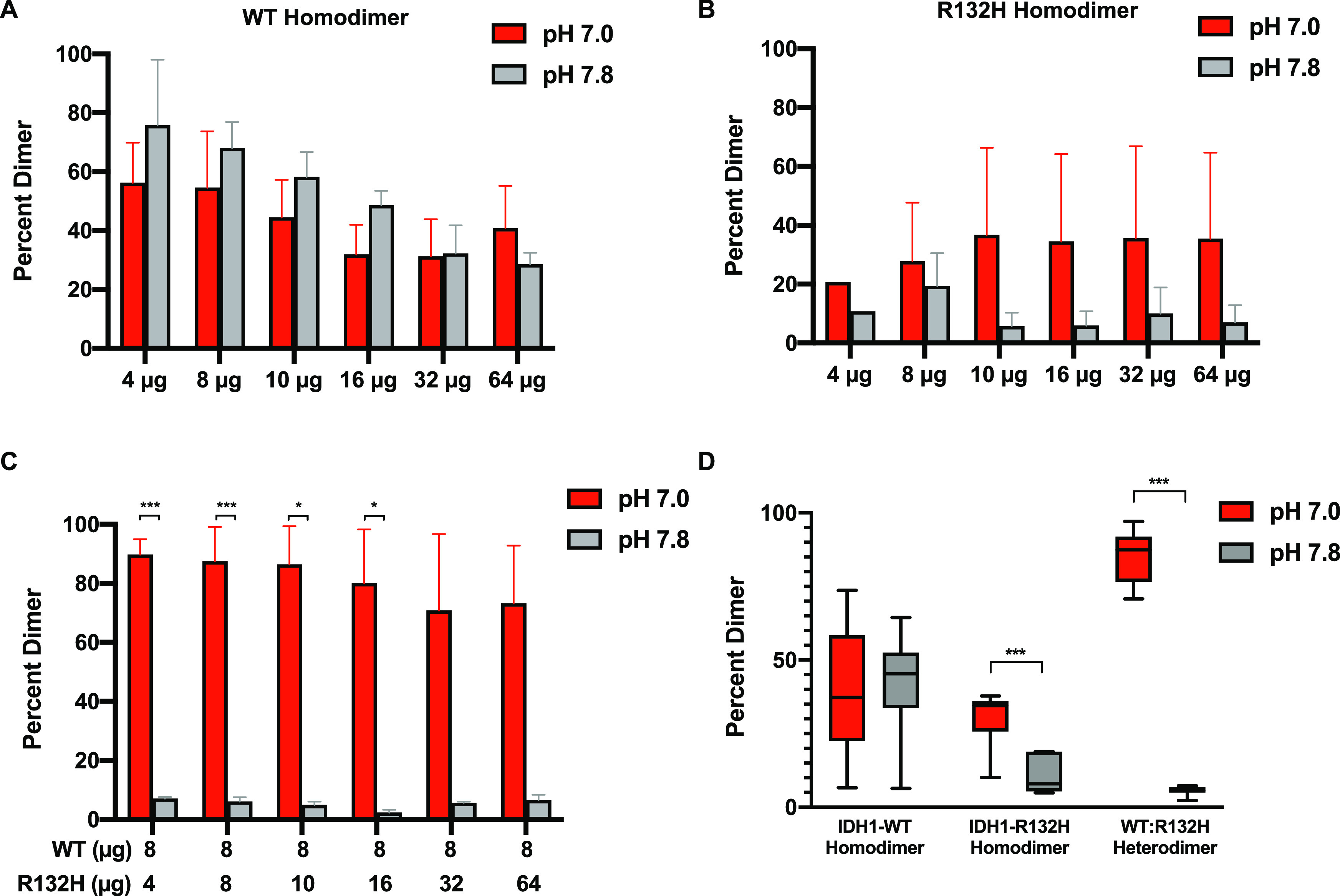
IDH1 heterodimer formation is pH sensitive. IDH1 protein was run on native gels at two buffer pH values, 7.0 and 7.8 (see Methods; Figure S3 for representative gels). Dimer was quantified for (A) IDH1-WT homodimer binding mixtures, (B) IDH1-R132H homodimer binding mixtures, and (C) WT:R132H heterodimer binding mixtures. For panels A–C, quantification of replicate experiments (three experimental replicates, two independent protein preparations). Data shown as column plots (means ± standard deviation). (D) Quantification of replicate experiments as described in panels A–C across all concentrations. Data shown as box and whisker plots; Tukey display. For panels A–D, significance determined by a Student’s t two-tailed test with Holm–Sidak multiple-comparison correction (*p < 0.05, and ***p < 0.001).
It was perhaps surprising to see differences in homodimer formation between WT and R132H, given that most seminal work on mutant IDH1 suggests that the R132H mutant exists in a stable homodimer. However, recent findings suggest that the R132H mutation can be destabilizing to dimer formation under certain buffer and cellular conditions and may contribute to the decrease of the oxidative IDH1 catalytic activity.27 Our results suggest that under the native gel electrophoresis conditions we used, stable dimer formation by IDH1-R132H is attenuated compared with that of IDH1-WT.
With native gel electrophoresis, we found that the level of WT:R132H heterodimer formation is greater at pH 7.0 than at pH 7.8 (Figure 5C, D). In the merged data, we also observed a possible pH sensitive effect for R132H:R132H homodimer formation (Figure 5D), though we note that this effect is weak and that we never observe pH-dependent production of D-2HG by IDH1-R132H alone in vitro (Figures 3 and 4).
One of the concerns with native gel electrophoresis is that separation is dependent on both the size and the native charge of the proteins. We therefore utilized an ELISA binding assay to confirm pH-dependent heterodimerization of WT:R132H observed by native gel electrophoresis. ELISAs have been used previously to measure pH-dependent protein–protein interactions.28 We found pH-independent binding of IDH1-WT to IDH1-WT (Figure 6A, D). We also observed pH-independent binding of IDH1-R132H to IDH1-R132H (Figure 6B,D) and note the calculated Kd was much higher for the R132H:R132H binding interactions [Kd ∼ 2 μM for R132H] under both pH values, compared with 78–96 nM for the WT:WT binding interactions (Figure 6D)]. This result is a confirmation of our native gel assays showing IDH1-R132H does not form homodimers to the same extent as IDH1-WT, regardless of buffer pH. Importantly, we were able to observe distinct pH-dependent binding of IDH1-R132H to IDH1-WT-adsorbed plates (Kd = 59 ± 18 nM at pH 7.0 vs Kd = 329 ± 86 nM at pH 7.5) (Figure 6C, D). These ELISA results support our results obtained from the native gel assay, confirming that the WT:R132H heterodimer is uniquely sensitive to pH. These results support our prediction that the mechanism of pH sensitive D-2HG production is driven by pH-dependent dimerization and that increased WT:R132H heterodimer formation at low pH produces increased D-2HG specifically at low pH.
Figure 6.

IDH1 heterodimer formation is pH sensitive in an ELISA. (A–C) ELISAs for in vitro binding of IDH1 proteins (see Methods for details). (A) WT homodimer assay. IDH1-WT binding to adsorbed IDH1-WT at pH 7.0 and 7.5. (B) R132H homodimer assay. IDH1-R132H binding to adsorbed IDH1-R132H at pH 7.0 and 7.5. (C) Heterodimer assay. IDH1-R132H binding to adsorbed IDH1-WT at pH 7.0 and 7.5. For panels A–C, means ± standard deviation with binding curve fits shown. Data obtained from three replicate assays (each result is the average of two technical replicates) across two protein preparations. (D) Table of binding constants (Kd) calculated from binding curves in panels A–C. Significance was determined using Graph Pad Prism one-site specific nonlinear fitting (see Methods for details) (*p < 0.05, compared to pH 7.5). ⧧ Indicates Kd fits were made, but confidence intervals did not close.
Discussion
Recent work shows that recurrent Arg → His mutations in cancer can confer pH sensitive function to the mutant protein,7 and we asked whether production of D-2HG by IDH1-R132H is sensitive to pHi dynamics. Our data suggest that D-2HG produced by mutant IDH1-R132H is sensitive to pHi dynamics in cells, with increased D-2HG production at lower pHi. Our in vitro results resolve a mechanism by which IDH1 WT:R132H heterodimer formation is pH sensitive, with increased heterodimer formation and D-2HG production at lower pH. This effect is also supported by cell-based assays for D-2HG production as well as colorimetric assays of metabolites from in vitro reactions. Importantly, we show that homodimer formation with both IDH1-WT and IDH1-R132H is pH insensitive in an ELISA, suggesting that the unique interactions between IDH1-WT and IDH1-R132H are important for mediating pH-dependent catalytic activity.
Our data also provide a missing link to resolve conflicting published findings on both wild-type and mutant IDH1 activity. First, our data suggest that substrate shuttling may serve a stronger role in D-2HG production under low-cellular pHi or buffer pH conditions. For example, under high-pH conditions or abundant AKG availability, it is unlikely that substrate shuttling is an important regulatory mechanism as we show IDH1-R132H exists mostly as monomers and forms low levels of the heterodimer with IDH1-WT at high pH. This supports recent data showing that substrate shuttling is not a driver for IDH1 function in cancer cells (which have constitutively increased pHi)27 and recent in vitro results showing a lack of substrate shuttling when assayed at high buffer pH (7.5).29 However, under low-pH conditions or limiting AKG metabolite concentrations, our data suggest that an increased reliance on substrate shuttling may facilitate a high level of D-2HG production due to increased heterodimer formation. Thus, our new data also support previous findings showing mechanistic roles for substrate shuttling in IDH1 catalysis.17
Second, previous work suggests that wild-type IDH1 dimerization relies on a series of electrostatic interactions between conserved aspartate residues (D273, D275, and D279) and Arg132 [Protein Data Bank (PDB) entry 1T0L]30 (Figure 7A). These structural data are supported by our prior collaborative work using computational analyses to identify a pH sensitive network of ionizable residues at the homodimerization interface of WT IDH1.14 Our heterodimer data suggest that maintaining this hydrogen bond network via a positively charged Arg interacting with a negatively charged Asp is important for dimerization. Indeed, the increased heterodimer formation we observe specifically at low pHi, where His132 is more likely to be protonated, supports this hypothesis. Further supporting this idea, those key aspartate residues identified previously are unresolved in R132H:R132H homodimer structures obtained under high-pH (7.5) crystallization conditions (PDB entry 3MAP)6 (Figure 7C) but properly resolved and in the proximity of the His132 residue in crystal structures of R132H:R132H at low-pH (6.8–7.0) crystallization conditions (PDB entry 4KZO)22 (Figure 7C). Unfortunately, we cannot directly compare heterodimer crystal structures, as the only existing heterodimer crystal structure was resolved at high pH (7.5, PDB entry 3MAS)6 and had an unresolved aspartate-rich region. The existing structural data support our findings with native gel and ELISA binding assays showing lower pH stabilizes heterodimer formation and suggest a mechanism by which low pH facilitates coordination of this critical ionizable network (Figure 7D). Future work is needed to explore and confirm this structural hypothesis in greater mechanistic detail.
Figure 7.

Proposed mechanism of pH sensitive heterodimer formation. (A) Crystal structure of the WT:WT homodimer (PDB entry 1T0L).30 Monomer A is colored gray, and monomer B magenta. Regions containing key conserved aspartate residues are colored cyan. Shown as sticks in each monomer: Arg132, Asp273, Asp275, Asp279, with bound NADP+ and isocitrate (ICT) in green stick. (B) Close-up of monomer A (left) and monomer B (right) active sites. Shown as sticks in each monomer are Arg132 (gray in monomer A, magenta in monomer B), Asp273, Asp275, and Asp279 in cyan and bound NADP+ and ICT in green. (C) Crystal structure overlay of an R132H:R132H homodimer at high crystallization pH (gray, PDB entry 3MAP) and at low crystallization pH (magenta, PDB entry 4KZO). Shown as sticks are His132 and key conserved aspartate residues (Asp273, Asp275, and Asp279) that are only resolved in the low-pH structure (magenta). NADP+ and ICT are shown as green stick from the 3MAP structure. (D) Model for pH sensitive heterodimer formation, mediated by the ability of His132 to coordinate key aspartate residues in the heterodimer to promote the quasi-open conformation specifically at low pHi (left).
Finally, our data suggest a potential resolution of conflicting findings regarding the ability of IDH1-R132H mutation to function as a prognostic indicator in cancer. In some cancers, such as glioma, IDH1-R132H is predictive of better patient outcome,8,9 but in other cancers, such as thyroid and acute myeloid leukemia, IDH1-R132H is correlated with poorer prognosis.10,11 Furthermore, therapeutics targeting mutant IDH1 have had mixed results in preclinical studies, with some showing positive outcomes and some showing more aggressive disease.8,31,32 Determining whether and how the cellular environment such as pHi or hypoxia affects IDH1-R132H activity may allow us to decouple D-2HG abundance from IDH1 mutation status in patients.
Methods
Cell Culture
NIH3T3 cells were grown in DMEM with 10% FBS (growth medium). To change the pHi, complete medium was added to cells containing either 1 μM EIPA (Enzo, ALX-550-266-M005) or 5 mM NH4Cl (Sigma-Aldrich, 254134) for 24 h. Cells were then prepared as described in Metabolite Measurement in Mammalian Cells.
Stable Cell Line Generation
NIH3T3 cells were transfected with FLAG-IDH1-WT or FLAG-IDH1-R132H in pCDNA3 vectors using lipofectamine 2000 per the manufacturer’s instructions. Twenty-four hours after transfection, cells were selected with 800 μg/mL G418. After 4 days of selection, low-dilution clonal selection was performed where cells were plated at a density of 0.5 cell/well in 96-well plates in growth medium containing 800 μg/mL Geneticin (G418). Individual surviving clones were screened by Western blotting for expression and clones with equal expression selected for further analysis (see WB in Figure 2).
Intracelluar pH Measurement in Mammalian Cells
For each condition, cells were plated in triplicate at a density of 0.02 × 106 cells per well in a 24-well plate. After treatment with pH control medium (see above), the steady state pHi was measured in cells loaded with the pH sensitive dye 2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein (BCECF) in a NaHCO3-containing buffer and calibrated with nigericin-containing buffers at known pH values, as previously described.7
Western Blot Analyses
For Western blot analyses, cells were washed twice with ice-cold phosphate-buffered saline (PBS) and incubated with 100 μL of ice-cold lysis buffer [50 mM Tris, 150 mM NaCl, 1 mM NaF, 1% Triton X-100, and protease inhibitor cocktail (Roche) (pH 7.5)] for 15 min on ice. Cells were then mechanically disrupted with a cell scraper, and clarified lysate (13000 rpm, 10 min) was used immediately for immunoblotting. For blots, proteins in 5 μg of total cell lysates were separated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). Proteins were then transferred to polyvinylidene difluoride membranes (Immobilon) (100 V, 1 h on ice). Membranes were blocked in 5% fat free milk in TBS-T (0.1% Tween 20 in Tris Buffered Saline (TBS)) for 1 h at room temperature. Primary antibodies were added [1:1000 dilution of mouse anti-flag M2 (Sigma, F1804) or 1:5000 dilution of mouse anti-actin (EMD Millipore, C4: MAB1501R) in 1% milk/TBS-T] and incubated for 1 h at room temperature with shaking. Membranes were washed for 3 × 5 min in TBS-T and then incubated with a 1:10000 dilution of secondary antibodies [anti-mouse HRP (Bio-Rad catalog no. 1706515) in 1% milk/TBS-T] for 1 h at room temperature while being shaken. Membranes were washed for 3 × 5 min in TBS-T while being shaken, developed (Super Signal West Pico, Pierce), and imaged (Alpha Innotech FluorChemQ).
Metabolite Measurement in Mammalian Cells
Cell data were obtained using cells maintained at lower and higher pH values as described above. Levels of AKG and D-2HG were measured using commercial kits (Biovision, D-2HG, catalog no. K213; AKG, catalog no. K677) and per the manufacturer’s instructions. Per kit recommendations, for each biological replicate two technical replicates were used as well as two technical replicates prepared with a spiked-in positive control, and two additional technical replicates without assay enzyme to serve as negative controls.
Recombinant Protein Expression and Purification
IDH1-WT and IDH1-R132H proteins were obtained by in vitro protein expression and purification from E. coli. The protein was expressed in competent E. coli BL21(DE3) cells, with isopropyl β-d-1-thiogalactopyranoside (IPTG) induction (Cf = 0.5 mM) performed overnight at 30 °C. Cells were pelleted at 8000g for 20 min. Bacterial Protein Extraction Reagent (BPER, ThermoFisher catalog no. 78248) was used to lyse the cells, and cells were spun down at 10000g for 20 min. Column chromatography with Ni-NTA resin was used for protein purification. Ni-NTA binding buffer [50 mM Tris and 300 mM NaCl (pH 7.8)] was used as both the binding buffer and the wash buffer, and the protein was eluted using Ni-NTA elution buffer [50 mM Tris, 300 mM NaCl, and 200 mM imidazole (pH 7.8)] and collected in fractions. SDS–PAGE gels were run to check for purity, and elution fractions were pooled for dialysis in a buffer with 20 mM Tris, 20 mM NaCl, 1 mM DTT, and 5% glycerol at pH 7.5 to allow equilibration of imidazole.
A Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) was used to determine the concentration of proteins after dialysis and concentration (Millipore Amicon 10000 molecular weight cutoff, UFC901024D). The purity of concentrated proteins was determined using SDS–PAGE gels. An enzyme activity assay was performed to confirm IDH1-WT and IDH1-R132H activity prior to binding and enzymatic assays.
Homodimer Enzymatic Activity Assays
Reduction and oxidation reactions were performed with IDH1-WT and IDH1-R132H. Each oxidation reaction mixture contained 100 μM NADP+, 400 μM DL-ICT, and 2 mM MgSO4 in 30 mM Tris buffer (pH 7.0 and 7.5). Each reduction reaction mixture contained 400 μM NADPH, 10 mM AKG, and 2 mM MgSO4 in 30 mM Tris (pH 7.0 and 7.5). AKG stock was pH’ed to 7.3 prior to use. Serial dilutions (1:2) of these reaction mixes were performed in buffers prepared identically to that above except without substrate to produce matched buffer conditions with a serial dilution range of substrate concentrations. To initiate reactions, 250 nM enzyme was added to each well using a multipipettor. Controls lacking enzyme and substrate were included for each assay to confirm the specificity of NADPH depletion or production. Activity was determined every 10 s for 20 min by measuring the absorbance at 340 nm using a SpectraMax M3 (or M5) plate reader (Molecular Devices).
Colorimetric Assay Sample Preparation
With Equal Mixtures of WT:R132H (Figure 4)
Each reaction mixture contained 150 mM NaCl, 2 mM MgCl2, and 20 mM Tris-HCl buffer (pH 7.5 and 7.0). Reduction reaction mixtures contained 8 mM ICT, 200 μM NADP+, and 250 nM total protein (IDH1-WT, IDH1-R132H, or 1:1 IDH1-WT:IDH1-R132H). Reaction mixtures run in triplicate were incubated at 24 °C for 1 h; 100 μL reactions were stopped by boiling for 10 min. Samples were then passed through Amicon Ultra filters (Millipore) to remove proteins and processed immediately or flash-frozen and stored at −80 °C. Levels of AKG and D-2HG were measured using commercial kits (Biovision, D-2HG, catalog no. K213; AKG, catalog no. K677) and per manufacturer instructions. Four experimental replicates were analyzed. Per kit recommendations, for each experimental replicate, two technical replicates were used as well as two additional technical replicates prepared with a spiked-in positive control and a background control.
With Unequal Mixtures of WT:R132H (Figure S2)
Each reaction mixture contained 150 mM NaCl, 2 mM MgCl2, and 30 mM Tris-HCl buffer (pH 7.5 and 7.0). Reduction reaction mixtures contained 8 mM ICT, 200 μM NADP+, and 5 μM total protein (for a 3:1 mixture 3.75 μM IDH1-WT and 1.25 μM IDH1-R132H; for a 1:3 mixture 1.25 μM IDH1-WT and 3.75 μM IDH1-R132H). Oxidation reaction mixtures contained 8 mM AKG, 500 μM NADPH, and 5 μM total protein (for a 3:1 mixture 3.75 μM IDH1-WT and 1.25 μM IDH1-R132H; for a 1:3 mixture 1.25 μM IDH1-WT and 3.75 μM IDH1-R132H); 100 μL reactions were run at room temperature for 30 min and then stopped by boiling for 10 min. Levels of D-2HG were measured using commercial kits (Biovision, D-2HG, catalog no. K213) per manufacturer instructions. Five experimental replicates were analyzed. Per kit recommendations, for each experimental replicate, two technical replicates were used as well as two additional technical replicates prepared with a spiked-in positive control and a background control.
Binding Reactions and Native Gels
Native gel assays were performed at pH 7.0 and 7.8 to determine whether IDH1 dimerization occurs and is pH sensitive (see Figure 4 and Figure S3). Recombinant IDH1-WT alone, IDH1-R132H alone, and mixtures of IDH1-WT and IDH1-R132H were run on separate gels; 4 × binding buffers were prepared at these two pH values with 80 mM HEPES and 200 mM NaCl in water. Homodimer gels were loaded with the IDH1 enzyme from 0 to 64 μg, and heterodimer gels contained 8 μg of IDH1-WT in each lane and 0–64 μg of IDH1-R132H, increasing across the lanes of the gel. Binding reaction mixtures were incubated for 20 min at room temperature before being run on Native PAGE gels with BSA as a standard; 5 × native gel running buffers (120 mM Tris and 960 mM glycine) were prepared at pH 7.0 and 7.8. Gels were run for 3 h at 140 V and then stained with Instant Blue dye (Expedeon/Abcam, ab119211) and imaged on a Bio-Rad ChemiDoc gel imager system.
ELISA
Purified protein (either WT or R132H) was bound to high-binding ELISA plates (Corning) at 20 μg/mL in PBS overnight while being shaken at 4 °C. Plates with immobilized protein were blocked with 5% BSA in PBS overnight while being shaken at 4 °C. Plates were washed twice with PBS, and then IDH1-WT or IDH1-R132H were incubated at concentrations from 0 to 2 μM in binding buffer [30 mM Tris, 150 mM NaCl, and 2 mM MgCl2 (pH 7.0 or 7.5)] for 90 min at room temperature while being shaken. Plates were washed with wash buffer (binding buffer with 0.1% Tween 20) for 3 × 5 min and then incubated for 90 min with anti-IDH1 (rabbit, Cell Signaling 3997S, at 1:1000) for detecting WT:WT homodimer) or anti-IDH1-R132H (rat, Origene S TA190113, at 1:1000) for detecting the R132H:R132H homodimer or R132H:WT heterodimer. Plates were washed with wash buffers for 3 × 5 min and then incubated with a 1:2000 dilution of anti-rabbit HRP (1706515, Bio-Rad) for the IDH1 antibody or anti-rat HRP (5204-2504, Bio-Rad) for the IDH1-R132H antibody. In all cases, samples were run in technical replicates and negative controls were used (antibody-supplemented wells with adsorbed protein only, or with no IDH1 adsorbed) and background subtracted from the experimental wells.
Curve Fitting in PRISM
For Michaelis–Menten curve fitting (Figure 3), curves were fit using Michaelis–Menten nonlinear curve fitting in GraphPad PRISM 8.2. No outlier analysis was performed, and curves were fit using each individual replicate Y value as an individual point. R2 for the curve fits ranged from 0.4281 to 0.8101, and none of the curves had ambiguous fits. From these analyses, Km and Vmax (along with standard errors) were obtained. To calculate kcat, we used the kcat nonlinear fit curve in PRISM 8.2, with E(t) constrained to the concentration used in these assays. The resulting kcat ± the standard error of the mean was converted into seconds and then reported in the provided table. Parameters (that can be found in the table in Figure 3D) were compared between pH values using GraphPad PRISM 8.2 nonlinear fit compare mode, with no constraints and kcat, Vmax, and Km individually selected to directly compare parameters across pH values.
For ELISA binding curve fitting (Figure 6), in all cases, curves were fit using one-site specific binding nonlinear curve fitting in GraphPad PRISM 8.2. Parameters (that can be found in the table in Figure 6D) were compared between pH values using GraphPad PRISM 8.2 nonlinear fit compare mode, with no constraints and Kd individually selected to directly compare parameters across pH values. No outlier analysis was performed, and curves were fit using each individual replicate Y value as an individual point. R2 for the curve fits ranged from 0.3015 to 0.8629, and none of the curves had ambiguous fits for Kd. While the software could fit the Kd data for the R132H homodimer, confidence intervals did not close. Thus, we flagged these values in the table as they are near the maximum assay concentrations used, and the R132H homodimer binding curve may not have reached saturation.
Acknowledgments
Biorender was used to prepare the table of contents figure and Figures 1C and 7D.
Glossary
Abbreviations
- pHi
intracellular pH
- DPBS
Dulbecco’s phosphate-buffered saline
- IDH1
isocitrate dehydrogenase 1
- IDH3
isocitrate dehydrogenase 3
- IDH2
isocitrate dehydrogenase 2
- ICT
isocitrate
- ELISA
enzyme-linked immunosorbent assay
- AKG
α-ketoglutarate
- D-2HG
d-2-hydroxyglutarate
- TBS
Tris Buffered Saline
- PBS
Phosphate Buffered Saline
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.1c00059.
Figures S1–S3 (PDF)
Accession Codes
IDH1,O75874
Author Contributions
R.S. and J.F.K. contributed equally to this work. K.A.W. and D.L.B. conceived of the hypothesis, which was developed in collaboration with R.S. J.F.K., R.S., and K.A.W. performed all biochemical experiments and data analysis. All authors contributed to discussions of data and writing manuscript. All authors approved the final version of the manuscript.
This work was supported by National Institutes of Health (NIH) Grant F32 CA177085, a University of Notre Dame startup grant, The Henry Luce Foundation (K.A.W.), and NIH Grant R01 CA197855 (D.L.B.). J.F.K. was supported by a summer research fellowship (RLAC, Harper Cancer Research Institute, University of Notre Dame).
The authors declare no competing financial interest.
Supplementary Material
References
- Bogdanovic E. (2015) IDH1, Lipid Metabolism and Cancer: Shedding New Light on Old Ideas. Biochim. Biophys. Acta, Gen. Subj. 1850 (9), 1781–1785. 10.1016/j.bbagen.2015.04.014. [DOI] [PubMed] [Google Scholar]
- Itsumi M.; Inoue S.; Elia A. J.; Murakami K.; Sasaki M.; Lind E. F.; Brenner D.; Harris I. S.; Chio I. I. C.; Afzal S.; Cairns R. A.; Cescon D. W.; Elford A. R.; Ye J.; Lang P. A.; Li W. Y.; Wakeham A.; Duncan G. S.; Haight J.; You-Ten A.; Snow B.; Yamamoto K.; Ohashi P. S.; Mak T. W. (2015) Idh1 Protects Murine Hepatocytes from Endotoxin-Induced Oxidative Stress by Regulating the Intracellular NADP(+)/NADPH Ratio. Cell Death Differ. 22 (11), 1837–1845. 10.1038/cdd.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L.; White D. W.; Gross S.; Bennett B. D.; Bittinger M. A.; Driggers E. M.; Fantin V. R.; Jang H. G.; Jin S.; Keenan M. C.; Marks K. M.; Prins R. M.; Ward P. S.; Yen K. E.; Liau L. M.; Rabinowitz J. D.; Cantley L. C.; Thompson C. B.; Vander Heiden M. G.; Su S. M. (2009) Cancer-Associated IDH1Mutations Produce 2-Hydroxyglutarate. Nature 462 (7274), 739–744. 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ježek P. (2020) 2-Hydroxyglutarate in Cancer Cells. Antioxid. Redox Signaling 33 (13), 903–926. 10.1089/ars.2019.7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley J. H.; Dean A. M.; Koshland D. E.; Stroud R. M. (1991) Catalytic Mechanism of NADP+-Dependent Isocitrate Dehydrogenase: Implications from the Structures of Magnesium-Isocitrate and NADP+ Complexes. Biochemistry 30 (35), 8671–8678. 10.1021/bi00099a026. [DOI] [PubMed] [Google Scholar]
- Yang B.; Zhong C.; Peng Y.; Lai Z.; Ding J. (2010) Molecular Mechanisms of “off-on Switch” of Activities of Human IDH1 by Tumor-Associated Mutation R132H. Cell Res. 20 (11), 1188–1200. 10.1038/cr.2010.145. [DOI] [PubMed] [Google Scholar]
- White K. A.; Garrido Ruiz D.; Szpiech Z. A.; Strauli N. B.; Hernandez R. D.; Jacobson M. P.; Barber D. L. (2017) Cancer-Associated Arginine-to-Histidine Mutations Confer a Gain in PH Sensing to Mutant Proteins. Sci. Signaling 10, eaam9931. 10.1126/scisignal.aam9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L. E. (2019) Friend or Foe—IDH1Mutations in Glioma 10 Years On. Carcinogenesis 40 (11), 1299–1307. 10.1093/carcin/bgz134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanson M.; Marie Y.; Paris S.; Idbaih A.; Laffaire J.; Ducray F.; El Hallani S.; Boisselier B.; Mokhtari K.; Hoang-Xuan K.; Delattre J.-Y. (2009) Isocitrate Dehydrogenase 1 Codon 132 Mutation Is an Important Prognostic Biomarker in Gliomas. J. Clin. Oncol. 27 (25), 4150–4154. 10.1200/JCO.2009.21.9832. [DOI] [PubMed] [Google Scholar]
- Yoo S.-K.; Lee S.; Kim S.-J.; Jee H.-G.; Kim B.-A.; Cho H.; Song Y. S.; Cho S. W.; Won J.-K.; Shin J.-Y.; Park D. J.; Kim J.-I.; Lee K. E.; Park Y. J.; Seo J.-S. (2016) Comprehensive Analysis of the Transcriptional and Mutational Landscape of Follicular and Papillary Thyroid Cancers. PLoS Genet. 12 (8), e1006239. 10.1371/journal.pgen.1006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnittger S.; Haferlach C.; Ulke M.; Alpermann T.; Kern W.; Haferlach T. (2010) IDH1Mutations Are Detected in 6.6% of 1414 AML Patients and Are Associated with Intermediate Risk Karyotype and Unfavorable Prognosis in Adults Younger than 60 Years and Unmutated NPM1 Status. Blood 116 (25), 5486–5496. 10.1182/blood-2010-02-267955. [DOI] [PubMed] [Google Scholar]
- Upadhyay V. A.; Brunner A. M.; Fathi A. T. (2017) Isocitrate Dehydrogenase (IDH) Inhibition as Treatment of Myeloid Malignancies: Progress and Future Directions. Pharmacol. Ther. 177, 123–128. 10.1016/j.pharmthera.2017.03.003. [DOI] [PubMed] [Google Scholar]
- Wise D. R.; Ward P. S.; Shay J. E. S.; Cross J. R.; Gruber J. J.; Sachdeva U. M.; Platt J. M.; DeMatteo R. G.; Simon M. C.; Thompson C. B. (2011) Hypoxia Promotes Isocitrate Dehydrogenase-Dependent Carboxylation of -Ketoglutarate to Citrate to Support Cell Growth and Viability. Proc. Natl. Acad. Sci. U. S. A. 108 (49), 19611–19616. 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna L. A.; Lesecq Z.; White K. A.; Hoang A.; Scott D. A.; Zagnitko O.; Bobkov A. A.; Barber D. L.; Schiffer J. M.; Isom D. G.; Sohl C. D. (2020) An Acidic Residue Buried in the Dimer Interface of Isocitrate Dehydrogenase 1 (IDH1) Helps Regulate Catalysis and PH Sensitivity. Biochem. J. 477 (16), 2999–3018. 10.1042/BCJ20200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P. S.; Lu C.; Cross J. R.; Abdel-Wahab O.; Levine R. L.; Schwartz G. K.; Thompson C. B. (2013) The Potential for Isocitrate Dehydrogenase Mutations to Produce 2-Hydroxyglutarate Depends on Allele Specificity and Subcellular Compartmentalization. J. Biol. Chem. 288 (6), 3804–3815. 10.1074/jbc.M112.435495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrak B.; Zhao H.; Qi H.; Quinn C.; Gao E.; Boyer J. G.; Concha N.; Brown K.; Duraiswami C.; Wooster R.; Sweitzer S.; Schwartz B. (2011) A Tale of Two Subunits: How the Neomorphic R132H IDH1Mutation Enhances Production of AHG. Biochemistry 50 (21), 4804–4812. 10.1021/bi200499m. [DOI] [PubMed] [Google Scholar]
- Avellaneda Matteo D.; Grunseth A. J.; Gonzalez E. R.; Anselmo S. L.; Kennedy M. A.; Moman P.; Scott D. A.; Hoang A.; Sohl C. D. (2017) Molecular Mechanisms of Isocitrate Dehydrogenase 1 (IDH1) Mutations Identified in Tumors: The Role of Size and Hydrophobicity at Residue 132 on Catalytic Efficiency. J. Biol. Chem. 292 (19), 7971–7983. 10.1074/jbc.M117.776179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer A. M.; Wang B.; Liu H.; Shah H.; Carmona-Fontaine C.; Rustenburg A. S.; Salah S.; Gunner M. R.; Chodera J. D.; Cross J. R.; Thompson C. B. (2017) L-2-Hydroxyglutarate Production Arises from Noncanonical Enzyme Function at Acidic PH. Nat. Chem. Biol. 13, 494. 10.1038/nchembio.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.; Ko B.; Hensley C. T.; Jiang L.; Wasti A. T.; Kim J.; Sudderth J.; Calvaruso M. A.; Lumata L.; Mitsche M.; Rutter J.; Merritt M. E.; DeBerardinis R. J. (2014) Glutamine Oxidation Maintains the TCA Cycle and Cell Survival during Impaired Mitochondrial Pyruvate Transport. Mol. Cell 56 (3), 414–424. 10.1016/j.molcel.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadtochiy S. M.; Schafer X.; Fu D.; Nehrke K.; Munger J.; Brookes P. S. (2016) Acidic PH Is a Metabolic Switch for 2-Hydroxyglutarate Generation and Signaling. J. Biol. Chem. 291 (38), 20188–20197. 10.1074/jbc.M116.738799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer A. M.; Dematteo R. G.; Venneti S.; Finley L. W. S.; Lu C.; Judkins A. R.; Rustenburg A. S.; Grinaway P. B.; Chodera J. D.; Cross J. R.; Thompson C. B. (2015) Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 22 (2), 304–311. 10.1016/j.cmet.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendina A. R.; Pietrak B.; Smallwood A.; Zhao H.; Qi H.; Quinn C.; Adams N. D.; Concha N.; Duraiswami C.; Thrall S. H.; Sweitzer S.; Schwartz B. (2013) Mutant IDH1 Enhances the Production of 2-Hydroxyglutarate Due to Its Kinetic Mechanism. Biochemistry 52 (26), 4563–4577. 10.1021/bi400514k. [DOI] [PubMed] [Google Scholar]
- Brooks E.; Wu X.; Hanel A.; Nguyen S.; Wang J.; Zhang J. H.; Harrison A.; Zhang W. (2014) Identification and Characterization of Small-Molecule Inhibitors of the R132H/R132H Mutant Isocitrate Dehydrogenase 1 Homodimer and R132H/Wild-Type Heterodimer. J. Biomol. Screening 19 (8), 1193–1200. 10.1177/1087057114541148. [DOI] [PubMed] [Google Scholar]
- Stellwagen E.; Prantner J. D.; Stellwagen N. C. (2008) Do Zwitterions Contribute to the Ionic Strength of a Solution?. Anal. Biochem. 373 (2), 407–409. 10.1016/j.ab.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson G. L.; Philip B.; Guthrie M. R.; Cox J. E.; Robinson J. P.; VanBrocklin M. W.; Holmen S. L. (2016) In Vitro Visualization and Characterization of Wild Type and Mutant IDH Homo- and Heterodimers Using Bimolecular Fluorescence Complementation. Cancer Res. Front 2 (2), 311–329. 10.17980/2016.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G.; Reitman Z. J.; Duncan C. G.; Spasojevic I.; Gooden D. M.; Rasheed B. A.; Yang R.; Lopez G. Y.; He Y.; McLendon R. E.; Bigner D. D.; Yan H. (2013) Disruption of Wild-Type IDH1 Suppresses D-2-Hydroxyglutarate Production in IDH1-Mutated Gliomas. Cancer Res. 73 (2), 496–501. 10.1158/0008-5472.CAN-12-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter J. P.; Ward P. S.; Dasgupta T.; Hosios A. M.; Gunawardena J.; Vander Heiden M. G. (2018) Lack of Evidence for Substrate Channeling or Flux between Wildtype and Mutant Isocitrate Dehydrogenase to Produce the Oncometabolite 2-Hydroxyglutarate. J. Biol. Chem. 293 (52), 20051–20061. 10.1074/jbc.RA118.004278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K. A.; Grillo-Hill B. K.; Esquivel M.; Peralta J.; Bui V. N.; Chire I.; Barber D. L. (2018) β-Catenin Is a PH Sensor with Decreased Stability at Higher Intracellular PH. J. Cell Biol. 217 (11), 3965–3976. 10.1083/jcb.201712041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi R.; Subramanian C.; Jackowski S.; Rock C. O. (2012) Cancer-Associated Isocitrate Dehydrogenase Mutations Inactivate NADPH-Dependent Reductive Carboxylation. J. Biol. Chem. 287 (18), 14615–14620. 10.1074/jbc.C112.353946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Zhao J.; Xu Z.; Peng B.; Huang Q.; Arnold E.; Ding J. (2004) Structures of Human Cytosolic NADP-Dependent Isocitrate Dehydrogenase Reveal a Novel Self-Regulatory Mechanism of Activity. J. Biol. Chem. 279 (32), 33946–33957. 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- Popovici-Muller J.; Saunders J. O.; Salituro F. G.; Travins J. M.; Yan S.; Zhao F.; Gross S.; Dang L.; Yen K. E.; Yang H.; Straley K. S.; Jin S.; Kunii K.; Fantin V. R.; Zhang S.; Pan Q.; Shi D.; Biller S. A.; Su S. M. (2012) Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo. ACS Med. Chem. Lett. 3 (10), 850–855. 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X.; Baird D.; Bowen K.; Capka V.; Chen J.; Chenail G.; Cho Y.; Dooley J.; Farsidjani A.; Fortin P.; Kohls D.; Kulathila R.; Lin F.; McKay D.; Rodrigues L.; Sage D.; Touré B. B.; van der Plas S.; Wright K.; Xu M.; Yin H.; Levell J.; Pagliarini R. A. (2017) Allosteric Mutant IDH1 Inhibitors Reveal Mechanisms for IDH1Mutant and Isoform Selectivity. Structure 25 (3), 506–513. 10.1016/j.str.2016.12.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.