

Abstract
Objective
Birth asphyxia (BA) is often associated with seizures that may exacerbate the ensuing hypoxic–ischemic encephalopathy. In rodent models of BA, exposure to hypoxia is used to evoke seizures, that commence already during the insult. This is in stark contrast to clinical BA, in which seizures are typically seen upon recovery. Here, we introduce a term‐equivalent rat model of BA, in which seizures are triggered after exposure to asphyxia.
Methods
Postnatal day 11–12 male rat pups were exposed to steady asphyxia (15 min; air containing 5% O2 + 20% CO2) or to intermittent asphyxia (30 min; three 5 + 5‐min cycles of 9% and 5% O2 at 20% CO2). Cortical activity and electrographic seizures were recorded in freely behaving animals. Simultaneous electrode measurements of intracortical pH, Po2, and local field potentials (LFPs) were made under urethane anesthesia.
Results
Both protocols decreased blood pH to <7.0 and brain pH from 7.3 to 6.7 and led to a fall in base excess by 20 mmol·L–1. Electrographic seizures with convulsions spanning the entire Racine scale were triggered after intermittent but not steady asphyxia. In the presence of 20% CO2, brain Po2 was only transiently affected by 9% ambient O2 but fell below detection level during the steps to 5% O2, and LFP activity was nearly abolished. Post‐asphyxia seizures were strongly suppressed when brain pH recovery was slowed down by 5% CO2.
Significance
The rate of brain pH recovery has a strong influence on post‐asphyxia seizure propensity. The recurring hypoxic episodes during intermittent asphyxia promote neuronal excitability, which leads to seizures only after the suppressing effect of the hypercapnic acidosis is relieved. The present rodent model of BA is to our best knowledge the first one in which, consistent with clinical BA, behavioral and electrographic seizures are triggered after and not during the BA‐mimicking insult.
Keywords: chemoreflex, hypercapnia, hypoxia, neonatal seizures, neuroprotection, perinatal asphyxia
Key Points.
Experimental asphyxia induced severe acidemia and nearly abolished cortical activity
Cortical activity during and after asphyxia was closely linked to changes in brain pH but not Po2
Electrographic seizures closely associated with behavioral convulsions were triggered after but not during intermittent asphyxia
The post‐asphyxia seizures were suppressed when brain pH recovery was slowed down with 5% CO2
1. INTRODUCTION
Birth asphyxia (BA) is one of the leading causes of neonatal mortality, resulting in approximately 1 million neonatal deaths annually. 1 In survivors, BA leads to hypoxic–ischemic encephalopathy (HIE), making them prone to a wide variety of developmental aberrations and lifelong malfunctions of the brain, ranging from minor and major psychiatric and neurological disorders to cerebral palsy. 2 , 3 , 4 The period of recovery after asphyxia is often marked by seizures, some of which are considered to promote HIE‐related trauma. 5
By definition, asphyxia is a combination of systemic hypoxia and hypercapnia, and these two components of asphyxia have distinct—often functionally opposite—actions on the brain. Hypoxia is known to promote neuronal excitability and seizures (Sampath et al., 6 Kawasaki et al., 7 Sun et al., 6 , 8 and references below), whereas an elevation of systemic CO2 produces a fall in brain pH and a consequent decrease in neuronal excitability. 9 , 10 , 11 , 12 Moreover, hypercapnia leads to vasodilation of cerebral arteries and arterioles 13 , 14 and, acting in synergy with neurohormonal factors such as vasopressin, 15 mediates a brain‐sparing increase in cerebral blood flow and oxygenation during the asphyxia‐coupled reduction of O2 availability. 14
Despite the differences in the neurobiological and physiological effects of hypoxia and asphyxia, practically all models on BA employing neonatal rodents are based on exposure of the animals to hypoxia or hypoxia–ischemia. 8 , 16 In such models, seizures are triggered during the exposure to hypoxia. 6 , 8 This is in contrast with observations of human neonates 17 and also large animal models of BA 18 in which seizures are triggered after a period of moderate or severe asphyxia.
To analyze the fundamental differences between hypoxia‐ and (post‐)asphyxia‐induced seizures, we investigated the dependence of seizure generation on changes in brain pH/CO2 and oxygen levels. We used postnatal day (P) 11–12 rats (all males), which, in terms of cortical development, are at a stage that is equivalent to the human term neonate. 19 , 20 Asphyxia was induced by ambient gas mixtures containing 5% or 9% O2 and 20% CO2 (balanced with N2). Two protocols were used in the present study: steady asphyxia (15 min, 5% O2 plus 20% CO2), which mimics an acute complication such as placental abruption or maintained cord compression; and intermittent asphyxia, where the hypoxia is applied in repetitive 5‐min steps at 9% O2 and 5% O2 (at constant 20% CO2) over 30 min to roughly mimic the effects of recurring contractions during prolonged parturition.
In sharp contrast to BA models based on exposure to pure hypoxia, seizures were not observed during the profound brain hypoxia that takes place during asphyxia with 5% O2. However, intense behavioral convulsions, tightly paralleled by electrographic cortical seizures, were triggered during brain pH recovery after the intermittent asphyxia. The seizures were strongly suppressed when the rate of brain pH recovery was slowed down by applying a low level (5%) of CO2 in air. No seizures were seen after steady asphyxia. The striking difference in seizure propensity following the steady and intermittent asphyxia protocols suggests that periodic hypoxic episodes in the asphyxic brain enhance neuronal excitability, 21 , 22 which becomes established as hyperexcitability and seizures once the suppressing effect of the asphyxia‐related hypercapnic acidosis is relieved. Our data as a whole are consistent with the finding that human neonates are typically prone to seizures after but not during parturition.
2. MATERIALS AND METHODS
Methods are described in Appendix S1.
3. RESULTS
3.1. Changes in blood pH, base excess, lactate, and plasma copeptin evoked by experimental asphyxia
To ascertain the translational relevance of the experimental models used in this work, and to compare the hypoxic burden brought about by the steady and intermittent asphyxia protocols, we measured blood gas parameters that are used in the diagnosis of BA. As expected (see Summanen et al., 23 Pospelov et al. 24 ), both types of asphyxia induced a large decrease in blood pH and base excess (BE; Figure 1A,B). Median pH was 6.81 (95% confidence interval [CI] = 6.80–6.87) and 6.93 (95% CI = 6.80–6.95), respectively, as measured immediately at the end of steady and intermittent asphyxia. In the control group at the same time point, median pH was 7.53 (95% CI = 7.51–7.54). The acidemia had a prominent metabolic component, as indicated by a fall in BE of 21.2 (95% CI = 18.9–22.3) mmol·L–1 and 20.6 (95% CI = 19.3–21.2) mmol·L–1 during steady and intermittent protocol, respectively. Blood lactate increased from a baseline of 1.1 (95% CI = 1.0–1.2) mmol·L–1 under control conditions to 10.9 (95% CI = 8.5–14.9] mmol·L–1 and 8.6 (95% CI = 7.0–9.8) mmol·L–1 during steady and intermittent asphyxia, respectively (Figure 1C). In addition, the levels of plasma copeptin, a relevant biomarker of BA, 25 , 26 were highly elevated by the end of both asphyxia protocols (4.41 [95% CI = 1.64–11.15] nM and 6.32 [95% CI = 4.48–7.74] nM for steady and intermittent, respectively vs. 0.22 [95% CI = 0.1–2.5] nM in the control group; Figure 1D).
FIGURE 1.
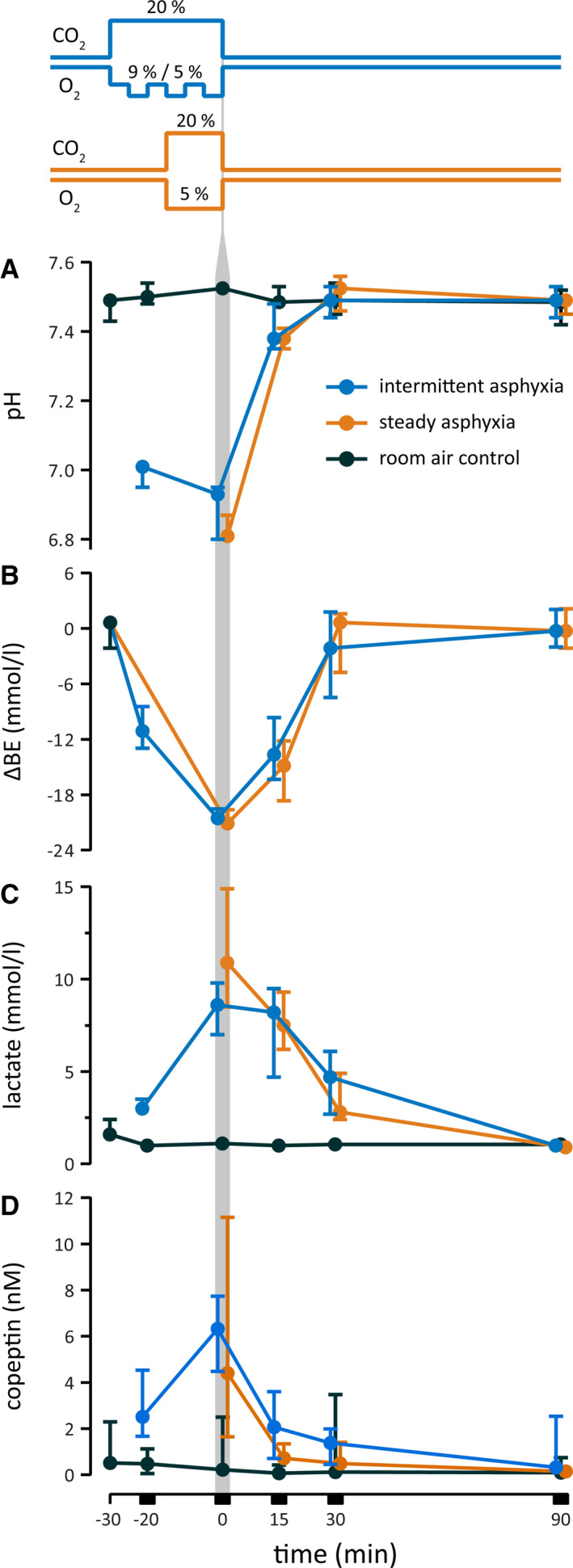
Changes in blood pH (A), base excess (BE; B) lactate (C), and plasma copeptin (D) in postnatal day (P) 11–12 rats exposed to experimental asphyxia. The line graphs on the top illustrate the intermittent (blue) and steady (orange) asphyxia protocols, which start and end in normoxic conditions as indicated by the pre‐ and post‐asphyxia baselines of O2 and CO2. Both protocols decreased pH to <7.0 and BE by 20 mmol·L–1 (ΔBE), and evoked a marked increase in lactate and copeptin. The vertical gray bar marks the end of asphyxia, and, for clarity, the blue symbols have been shifted slightly to the left and the orange symbols slightly to the right. Number of samples per timepoint is 3–5 in the control group and 5–6 in the asphyxia group. The values are median with 95% confidence interval. All data are based on P11–P12 rats in this and subsequent figures
Although the duration of the intermittent asphyxia exposure was twice as long as that of steady asphyxia, the blood gas parameters showed quantitatively similar changes. As described in detail before, 24 rat pups have a remarkable ability to physiologically compensate in response to the 9% O2 challenge. The nearly identical hypoxic burden caused by the two asphyxia protocols is therefore readily explained by the identical total time (15 min) of exposure to 5% O2. This similarity is particularly interesting when comparing the efficacy of steady and intermittent asphyxia to induce post‐asphyxia seizures, as done below.
3.2. Behavioral seizures emerge following intermittent asphyxia, and they are suppressed by graded restoration of normocapnia
We started our experiments on seizure generation in the two asphyxia paradigms using freely moving, non‐instrumented rats. Seizure scoring was based on a modified Racine scale (RS), which did not include non‐convulsive seizures (RS I–II, see Materials and Methods and below). Under control conditions, the pups were for most of the time in apparent sleep. During the exposure to steady or to intermittent asphyxia, the pups initially displayed distress behavior with increased locomotion, but no seizures were observed. Breathing rate decreased gradually and led to apnea and death in three of 17 and two of 17 pups during exposure to steady and intermittent asphyxia, respectively. There was no further mortality.
Notably, convulsive seizures (RS III–V) were never observed after steady asphyxia. The pups displayed some abnormal behavior, including shaking, twitches, and Straub tail, during the first 10 min of recovery. Thereafter, their behavior became indistinguishable from controls. In sharp contrast to this, intense behavioral seizures were seen in seven of 15 pups shortly after intermittent asphyxia, with a median latency of 224 s (range = 178–310 s) from the end of the exposure (Figure 2A and Videos S1, S2). Seizures of increasing severity occurred in succession. They commenced with forelimb clonus (RS III), which was often coupled with rhythmic head‐nodding. This was followed by clonus with loss of righting (RS IV), and the seizures ended after an episode of tonus–clonus with loss of righting (RS V) in all seven seizing animals. The median total duration of seizures was 110 s (range = 66–180 s), and all observed seizures terminated within about 8 min after the end of intermittent asphyxia. This was followed by a period (typically 40–180 s) of total immobility apart from respiratory movements, after which normal behavior gradually resumed. To our knowledge, this is the first description of a rodent model of human full‐term birth asphyxia, in which robust seizures emerge after the termination of the insult, as observed in the neonatal intensive care unit (e.g., Lynch et al. 17 and Discussion).
FIGURE 2.
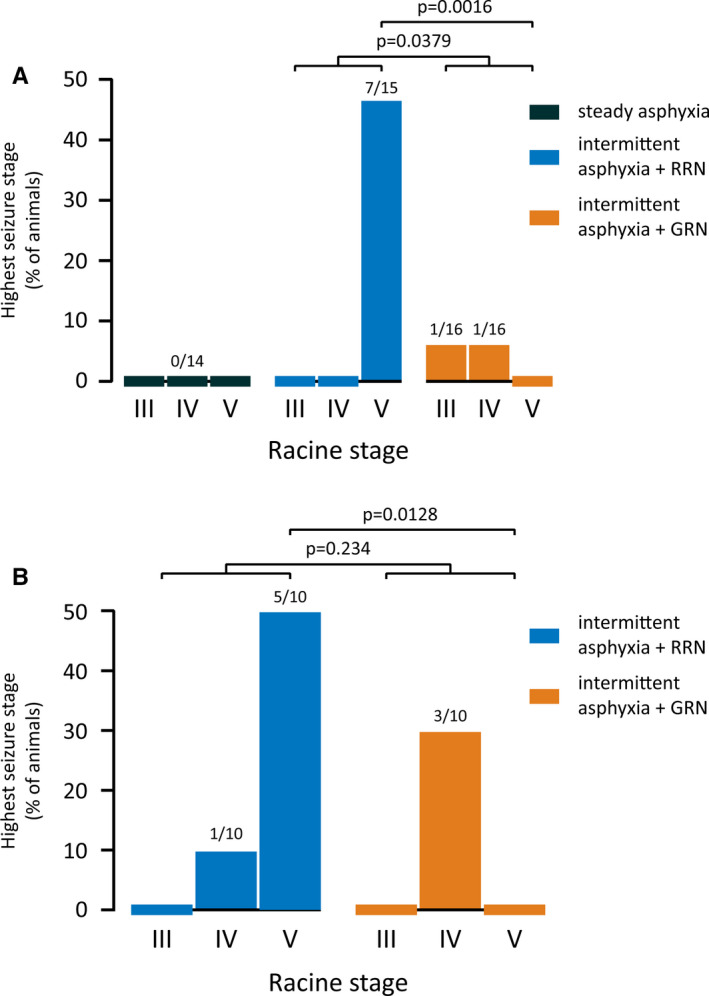
Highest Racine stage of seizures observed after experimental asphyxia. (A) Freely moving, non‐instrumented rats were exposed to steady or intermittent asphyxia followed by rapid (RRN) or graded restoration of normocapnia (GRN). No seizures were seen after steady asphyxia, whereas they were triggered in seven of 15 animals after intermittent asphyxia with RRN. GRN significantly decreased the proportion of animals with RS III–IV seizures and abolished RS V seizures. Sample sizes are given on top of the columns. (B) Similar to A, but the animals had been implanted with epidural cortical electrodes 24 h earlier (see Figure 3 for electrocorticography). GRN abolished RS V seizures also in these experiments
The severe acidosis of the brain that develops during asphyxia shows a prompt recovery to the alkaline direction after the end of the exposure (see Pospelov et al., 24 Bender et al., 27 Remzso et al. 28 ). There is evidence indicating that both the amplitude and the rate of alkaline recovery of brain tissue pH boost neuronal excitability and the triggering of seizures. 29 , 30 Thus, we examined the efficacy of graded restoration of normocapnia (GRN) 31 in suppressing seizures by exposing the rat pups to 5% CO2 in air for 30 min immediately after intermittent asphyxia. Here, rapid restoration of normocapnia (RRN) refers to the bulk of experiments in which the animals were promptly re‐exposed to room air after asphyxia. We found that GRN significantly reduced the occurrence of convulsive seizures compared to RRN (GRN 2/16 vs. RRN 7/15, Barnard test p = .0379; Figure 2A and Video S3). Notably, none of the animals in the GRN group had RS V seizures (p = .0016 compared to RRN). Thus, GRN reduced both seizure incidence and severity.
3.3. Electrocorticographic and behavioral post‐asphyxia seizures in freely moving rats
To examine the effects of intermittent asphyxia on neocortical activity patterns and the relationships between electrographic and behavioral seizures, electrocorticography (ECoG) was recorded at a frontal and a parietal site (Figure 3C). Under control conditions, the ECoG was continuous and, characteristic of this age point, discrete bursts of activity were rarely observed (Figure 3A,B, excerpt a). 32 Immediately following the onset of intermittent asphyxia, the ECoG activity was strongly suppressed. Most of this suppression is caused by the hypercapnic acidosis, as will be demonstrated below (Figures 4 and 5). Nevertheless, during the initial phase of the asphyxia with 9% of O2, about 25% of the ECoG power persisted, whereas the subsequent fall to 5% O2 led to a further decrease with hardly any detectable activity remaining. These effects were constant during the three transitions from 9% to 5% O2 (Figure 3A bottom panel and Figure 3A,B excerpts b and c).
FIGURE 3.
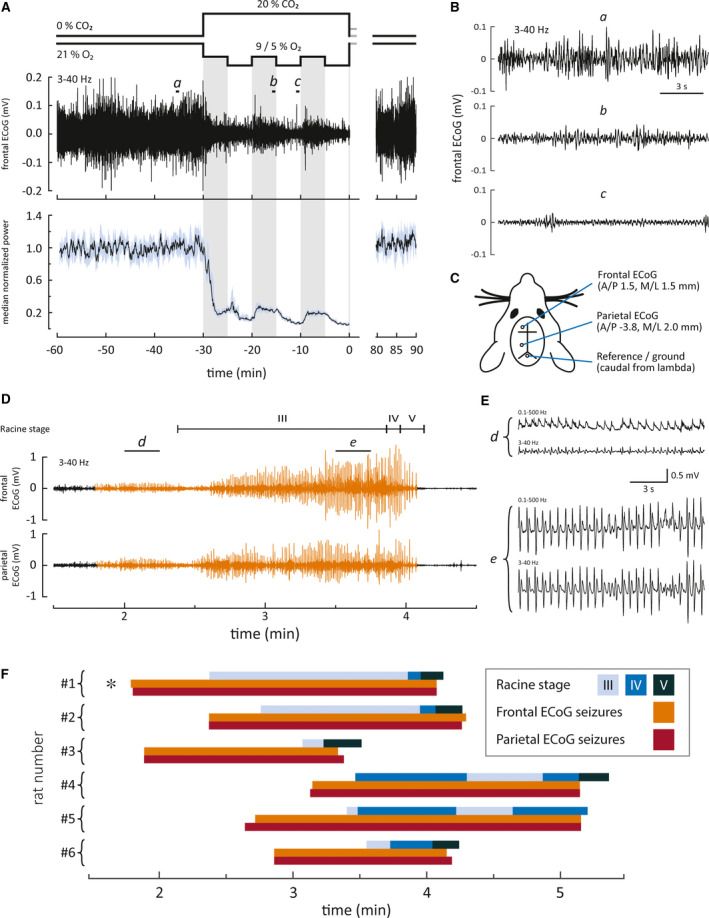
Electrographic and behavioral seizures emerge after but not during intermittent asphyxia. (A) The effect of asphyxia on cortical activity. Top panel features an example of frontal electrocorticography (ECoG) recording and bottom panel displays median power with 95% confidence interval of 18 frontal ECoG recordings. Excerpts a–c marked on the sample ECoG are shown magnified in B. These data demonstrate a profound suppression of cortical activity during asphyxia. (C) Electrode placements (A/P, anterior/posterior; M/L, medial/lateral). (D) An example of a post‐asphyxia recording (asphyxia ends at time 0) showing the temporal relationship between behavioral and electrographic seizures. Panels depict (from top to bottom) Racine stages, frontal ECoG, and parietal ECoG. Electrographic seizures are highlighted in orange. Excerpts d and e from the frontal ECoG displaying early and late stage seizures, respectively, are shown magnified in E. We included two frequency bands in E to show how the seizure activity appears without possible filtering artifacts. (F) Timeline representation of frontal and parietal ECoG seizures and behavioral seizures in the rapid restoration of normocapnia group. Seizures were observed in six of 10 animals. The specimen marked by the asterisk is the same as in D
FIGURE 4.
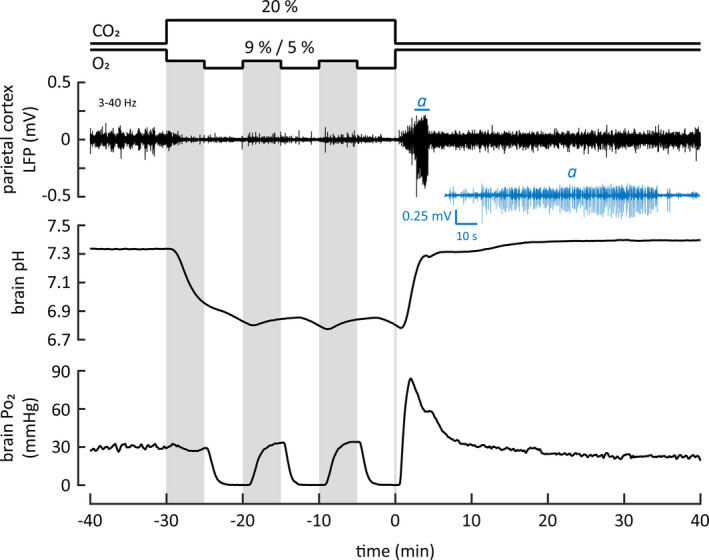
A sample recording with simultaneous monitoring of parietal cortex local field potentials (LFPs), as well as brain pH and Po2. After the intermittent asphyxia, prominent seizure activity was seen in the LFP recording (top panel). Excerpt of the LFP trace (marked with a) including the seizure epoch is shown below in blue. Middle and bottom panels show the pH and Po2 recordings, respectively. To facilitate visual inspection, the three gray vertical columns mark the 9% O2 bouts during the intermittent asphyxia protocol
FIGURE 5.
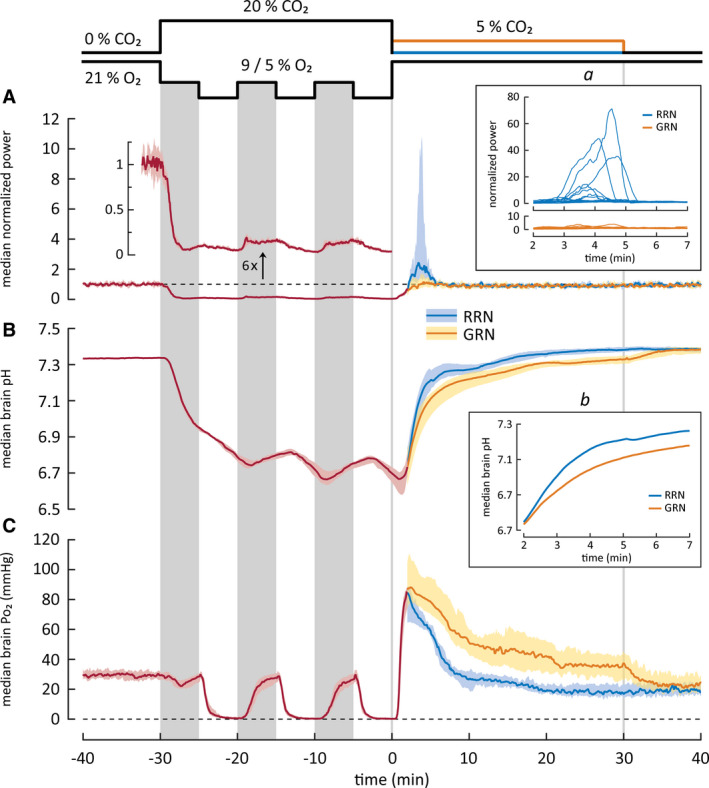
Changes in cortical activity (A) and in brain pH (B) and Po2 (C) in rat pups exposed to intermittent asphyxia, followed by rapid (RRN, n = 17) or graded restoration of normocapnia (GRN, n = 12). All values are median with 95% confidence interval except for insets. The three vertical gray columns on data panels mark the 9% O2 bouts during the intermittent protocol, whereas the two vertical lines indicate the end of asphyxia and the end of GRN. Median data for the RRN and GRN protocols are plotted separately starting at 2 min after the end of asphyxia, as the conditions before this time point are identical in the two protocols. (A) Power of local field potentials (LFPs) in the parietal cortex. The trace with a separate y‐axis is a sixfold magnification of the LFP power and shows the profound suppression of cortical activity during asphyxia (cf. Figure 4). After asphyxia, the activity recovers promptly. Following RRN, but not GRN, a period of hyperexcitability and seizures was seen, and it coincided with the rapid alkaline recovery of brain pH during a time window of 2–7 min after the end of asphyxia. Inset a shows individual LFP power traces during this period. (B) Brain pH decreases by 0.6 pH‐units during the asphyxia and displays only minor modulation (0.12 pH‐units) upon the changes between the 9% and 5% O2 levels. The alkaline recovery of brain pH after RRN is biphasic, and it is slowed down by GRN. The median brain pH during the first recovery phase (2–7 min) is shown magnified in inset b. (C) Brain Po2 falls to apparent zero during the three periods of exposure to 5% O2, but recovers to achieve a normoxic level when the ambient O2 concentration is increased to 9%. After the asphyxia, an overshoot of Po2 is observed. GRN increases the duration but not the amplitude or the rate of the Po2 overshoot
In line with the purely behavioral observations in Figure 2, we never observed electrographic seizure activity during the asphyxia exposure. In contrast, the ECoG recordings showed intense post‐asphyxia seizure activity in six of 10 freely behaving animals, which is in agreement with the seizure incidence in the non‐instrumented animals. The electrographic seizures in the parietal cortex had a median latency of 150 (95% CI = 108–188) s after the end of asphyxia. In all animals, the electrographic seizures appeared practically simultaneously at the recording sites in the parietal and the frontal cortex. The duration of the seizures in the frontal and the parietal cortex was comparable (pooled median duration = 118 [95% CI = 87–137] s), and most of the seizures consisted of spike trains with a 1.2–2.3 Hz discharge frequency (see Figure 3D for an example) with increasing amplitudes toward the end of the seizure epoch. The ECoG bursts in the initial seizure period (Figure 3D,E, excerpt d) were monopolar (without the 3‐Hz high‐pass filtering), and we did not observe slow components (Figure 3D,E, excerpts d and e). The seizure termination was abrupt (delay from high‐amplitude spiking to complete cessation was 0–7 seconds), and it was followed by a strong suppression of the background ECoG. Thereafter, baseline activity gradually recovered.
The ECoG recordings demonstrated a consistent temporal overlap between the convulsive behavioral seizures and the electrographic seizures (Figure 3D,F). The ECoG seizures preceded the convulsive seizures by 19 to 71 s. During this time, transient bouts of abnormal behavior, such as shaking, twitches, and Straub tail, were observed. These aberrant behaviors were reminiscent of non‐convulsive RS I–II seizures, which were excluded from the present analyses (see Materials and Methods). The onset of the convulsive seizures and transition from one seizure stage to another was not marked by any obvious change in the ECoG (Video S4). In four of six cases, RS V seizures, which are considered to originate from the brain stem, 33 , 34 continued for an additional 3–13 s after the cortical seizures had terminated (Figure 3F).
GRN decreased the post‐asphyxia seizure incidence from six of 10 to three of 10 animals as seen in both behavior and ECoG. In agreement with the data on freely moving non‐instrumented animals (Figure 2A), RS V seizures were absent in the instrumented GRN group as well (Barnard test p = 0.0128; Figure 2B). Otherwise, the seizures were qualitatively similar to those observed after RRN. No behavioral or electrographic seizures were observed in the control ECoG group (n = 7).
3.4. Simultaneous recording of neocortical local field potentials, brain pH, and brain oxygen levels during intermittent asphyxia
Although the data presented in Figure 3 show that changes in ambient CO2 and O2 exert robust effects on neuronal excitability (from profound suppression to hyperexcitability during and after intermittent asphyxia, respectively), they provide no information on the contributions of the associated changes in brain pH and brain Po2. Therefore, we made simultaneous recordings of local field potential (LFP) activity and pH as well as Po2 from the parietal cortex in head‐fixed urethane‐anesthetized rats.
The sample recording in Figure 4 shows the quality of the raw data. Exposure to intermittent asphyxia (with the initial 9% O2) caused a fast decline in brain pH and the ongoing LFP activity, and the three shifts in ambient O2 from 9% to 5% produced a further, reversible suppression (cf. Figures 3 and 5). A major decrease in Po2 was caused only by the three exposures to 5% O2. A prompt increase in neuronal excitability took place leading to seizure activity within 2.8 min post‐asphyxia in a manner comparable to what was seen in freely moving animals (Figure 3). Moreover, the seizure pattern was similar, consisting of epileptiform spikes with a crescendo pattern.
3.5. Suppression of LFP activity during intermittent asphyxia is mainly attributable to the fall in brain pH
Simultaneous recordings of LFP, pH, and Po2 were made in a total of 29 animals exposed to intermittent asphyxia and followed by RRN (n = 17) or GRN (n = 12). During the first 5 min of intermittent asphyxia (with 9% O2), the median value of brain pH (RRN and GRN groups combined) decreased rapidly from 7.33 to 6.95, with a further decline to 6.78 during the next 5 min in 5% O2 (Figure 5B). pH recovered only slightly (0.12 units; <20% of the maximum acidosis) in response to the two subsequent 9% O2 bouts, and dropped below 6.7 during the second and third 5% O2 exposures, reaching a minimum of 6.67. In two brain pH recordings with steady asphyxia, the lowest brain pH values were 6.64 and 6.58. They are well within the range of pH minima (6.42–6.79) recorded during the intermittent asphyxia, which further indicates that steady and intermittent asphyxia cause similar hypoxic load.
Median brain Po2 during the pre‐asphyxia baseline was 30.0 mmHg. During the first 5‐min period of intermittent asphyxia with 9% O2, brain Po2 showed initially a small transient fall (nadir at 22 mmHg), but promptly recovered back to the baseline level, indicating a fast re‐establishment of brain normoxia despite the large fall in ambient O2 (Figure 5C). Notably, however, under these conditions, median LFP power was still suppressed to 10% (95% CI = 8.3%–11%) from its baseline level, which is readily explained by the pronounced acidosis. A similar recovery to brain normoxia, associated with a strong suppression of LFP when compared to baseline (to 16%, 95% CI = 13%–19%), was observed during the two subsequent exposures to 9% O2. In contrast to the brain normoxia achieved during the 9% O2 exposures, all three epochs with 5% O2 led to a decrease in brain Po2 to a level where virtually no free oxygen could be detected. Under these conditions, LFP activity was—consistent with the associated acid shift—further suppressed, to 4.1% (95% CI = 3.3%–5.1%) of the baseline power (see magnified power trace in Figure 5A). Thus, all the above data indicate that the LFP power was suppressed by up to 90% despite brain normoxia at 9% ambient O2 because of the simultaneous hypercapnic acidosis.
3.6. Dependence of the post‐asphyxia increase in neuronal excitability and seizures on brain pH changes
After RRN, brain pH recovered in a biphasic manner, apparently reflecting the different elimination rates of CO2 and lactacidosis. 24 During the initial, fast phase starting 2 min and ending 7 min after the asphyxia, brain pH increased from 6.75 to 7.26 (Figure 5B and inset b therein). This was followed by an almost linear and much slower secondary recovery phase (cf. Pospelov et al. 24 ) lasting until 15 min post‐asphyxia. We did not observe an alkaline overshoot of the kind described in P6 rats with steady asphyxia induced by 9% O2 and 20% CO2, 31 indicating that fast brain pH recovery in itself is sufficient in triggering seizures following asphyxia (see also Panaitescu 35 ).
GRN reduced the rate of brain pH recovery after intermittent asphyxia. The fast phase of pH recovery, which in the RRN group took 5 min (see above), was slowed down by a factor of 2.6, to 13 min, by GRN (Figure 5B inset b). The rate of pH recovery during the subsequent slower phase with GRN was closely similar to what was observed after RRN, but it took place at a more acidotic level, with its start set by the slower rate of the preceding fast phase. The full recovery of brain pH following GRN had a third, final phase, which was caused by the change from 5% ambient CO2 to room air, and was completed within 10 min.
Following RRN, there was a prompt increase in neuronal activity. In 12 of 16 animals, this was followed by the emergence of recurring spike bursts (single burst duration < 10 s), which in three cases transformed into prominent seizures (duration = 94–117 s). The power maximum of individual LFP recordings was observed at around 3.5–4.5 min after the asphyxia and showed large variation ranging from 1.3 to 71*baseline power (median = 2.4*; Figure 5A and inset a therein). The low incidence of clear‐cut seizure activity in experiments of the kind illustrated in Figures 4 and 5 is readily explained by the use of urethane anesthesia. 36 Regardless, a period of hyperexcitability, coinciding with the fast phase of pH recovery (2.0–7.0 min after the asphyxia), was consistently associated with RRN (Figure 5A inset a). In contrast to this, in the GRN experiments, the median LFP power returned to the baseline level promptly after the asphyxia with no significant overshoot. Accordingly, the median LFP power 2–7 min after the end of asphyxia was significantly higher in the RRN than in the GRN group (RRN 1.5, 95% CI = 1.1–3.3 vs. GRN 1.1, 95% CI = 0.75–1.5; p = 0.0130, Mann–Whitney U‐test).
The present data indicate that, although seizures are generated only at pH levels relatively close to baseline (>7.0), it is not only the absolute pH but also the rate of change of pH within a defined pH window that is tightly linked to the triggering of seizures. This conclusion is supported by the astonishingly accurate temporal overlap of the major (RRN) and the minor (GRN) increases in LFP power seen during the 2–7‐min window after the asphyxia (Figure 5A inset a).
3.7. Absence of effect of brain O2 on seizure generation after intermittent asphyxia
When the animals in the RRN group were re‐exposed to room air after asphyxia, median brain Po2 recovered rapidly (maximum rate = 140 mmHg/min) with an overshoot to 85 mmHg (cf. 30.0 mmHg during baseline) at 2.0 min after the end of the intermittent asphyxia (Figure 5C). Brain Po2 fell to the baseline level 6.6 min later and showed a transient decline to 16.4 mmHg 26 min after asphyxia. GRN had no effect on the rate (maximum = 146 mmHg/min) or magnitude (peak = 88 mmHg) of the brain Po2 overshoot. However, GRN increased the duration of the overshoot, and median Po2 remained elevated for the whole 30‐min period. Thereafter, the RRN and GRN Po2 curves merged. It is noteworthy that brain Po2 in both the GRN and the RRN group is well above baseline at the time when seizures emerge.
3.8. Rapid and graded restoration of normocapnia have similar effects on rectal temperature and breathing rate
During intermittent asphyxia, mean rectal temperature decreased from 36.6 (95% CI = 36.3–36.8) to 36.0 (95% CI = 35.7–36.3) °C and mean breathing rate from 123 (95% CI = 118–128) to 71 (95% CI = 59–83) min−1 (Figure S1). The rectal temperature recovered to the baseline mean in 18 min after RRN and stabilized to 37.0°C after another 15 min. GRN had no effect on rectal temperature (two‐way repeated measures analysis of variance: Treatment p = .911, Treatment × Time p = .168). The breathing rate recovered promptly with an overshoot to 151 (95% CI = 144–158) min−1 after RRN. GRN appeared to slightly decrease the hyperpnea associated with RRN. However, the effect of GRN on breathing rate was not statistically significant (Treatment p = .196, Treatment × Time p = .624).
4. DISCUSSION
We demonstrate here, for the first time, behavioral convulsions and neocortical electrographic seizures in a rat model that physiologically mimics BA in human full‐term neonates. Consistent with the BA seizures seen in babies, the seizures are observed after, but not during, the asphyxia insult. 17 Our direct and simultaneous measurements of neuronal activity and brain tissue Po2 show beyond any doubt that the post‐asphyxia seizures are not triggered by brain hypoxia. Our intermittent asphyxia model, in which robust seizure activity was observed, satisfies the standard clinical criteria of BA, including a pronounced fall in blood pH from 7.5 to 6.9, associated with a significant metabolic acidosis as indicated by a decrease of 20.6 mM in BE, and an increase in lactate from 1.1 to 8.6 mM. Moreover, the plasma level of the telltale endocrine biomarker of BA, copeptin, 23 , 25 , 26 increased from 0.2 to 6 nM. As a whole, our data demonstrate that the present BA‐seizure model is physiologically valid in that it preserves the organism's innate responses to asphyxia and to the recovery thereof (see also Pospelov et al. 24 ), which have been extensively studied in large‐animal models of BA 14 , 37 and in human neonates. 13 , 26 , 38 This point is of utmost importance because, as discussed below, the changes in the excitability of the brain, including the generation of seizures, are controlled by mechanisms that act not only within the brain, but also at the whole‐organism level.
4.1. Hypercapnia as an endogenous seizure‐suppressing factor during the asphyxia‐linked hypoxia
Asphyxia is, by definition, a combination of systemic hypercapnia and hypoxia. The associated hypercapnic acidosis of the brain leads to suppression of neuronal activity as is presently evident in the ECoG and LFP recordings in Figures 3, 4, 5, 10 , 12 , 39 whereas alkalosis has the opposite effect. 10 , 11 , 39 , 40 A wide variety of neuronal ion channels, which control intrinsic neuronal excitability and synaptic signaling, show a high and functionally synergistic sensitivity to pH. 9 Thus, the suppression of excitability by acidosis during asphyxia is deeply rooted in mammalian evolution (see Ruusuvuori and Kaila 9 ). In line with this, the behavioral and neocortical seizures in our model are not triggered during, but only after the BA‐mimicking insult.
In stark contrast to the above, in both non‐invasive and invasive models of BA and/or HIE in which rat and mouse pups are exposed to pure hypoxia (4%–8% O2 in N2), 6 , 8 , 41 , 42 seizures are triggered already during the hypoxia insult. Strikingly, the highly artificial condition of pure hypoxia leads to a brain alkalosis, 24 , 43 which is known to produce an increase in the excitability as well as seizures especially in the immature brain. 11 , 31 , 40 Here, it is worth re‐emphasizing that peri‐ and neonatal rodents and other mammals are, under non‐experimental conditions, exposed to asphyxia, but not to a condition that could be defined as “birth hypoxia.”
4.2. Post‐asphyxia seizures are not strictly related to the preceding hypoxic load
Our data on blood acid–base parameters (Figure 1) and brain pH indicate that the hypoxic load is nearly identical in the intermittent and steady asphyxia protocols used presently. Thus, the post‐asphyxia seizure generation is not strictly related to the magnitude of the hypoxic load. Whereas the intermittent asphyxia protocol resulted in severe behavioral post‐asphyxia seizures (RS III–V) in about one half of the animals, we never observed seizures following steady asphyxia. A possible scenario that might account for the efficacy of intermittent asphyxia in triggering seizures is that mechanisms that have been shown to promote anoxic/hypoxic LTP in vitro 21 , 44 , 45 are likely to be activated during the 5% O2 bouts of intermittent asphyxia. The enhanced network activity during the periods with 9% O2 (and transient brain normoxia) would then lead to further activity‐dependent potentiation of excitatory connections. Finally, during the recovery from asphyxia, the suppressing effect of the hypercapnic acidosis is quickly removed, unmasking the enhanced excitability, which results in seizure generation. Here, it is worth pointing out that although much work on rodent models has focused on neuronal molecules and signaling mechanisms that are affected as a consequence of seizures, 46 , 47 , 48 little is known about the proximate causes in vivo that render the neonate's brain prone to seizures after BA.
4.3. Post‐asphyxia seizure generation shows a steep dependence on the rate of recovery of brain pH but not oxygen
The convulsive behavioral seizures after intermittent asphyxia were closely paralleled by neocortical seizures as demonstrated by ECoG recordings. Careful reviewing of the video‐ECoG recordings and the movement‐transducer signal showed only minor contamination of the ECoG by movement artifacts even during intense convulsions (see, e.g., Video S4, section 2:20–2:26). The seizures were detected in the frontal and the parietal cortex practically simultaneously, indicating that the post‐asphyxia seizures spread rapidly over large cortical areas (Figure 3). This observation also implies that our two‐site recording is reliable for neocortical seizure detection in the present model. The electrographic seizures initially had a low amplitude (Figures 3 and 4), which increased toward the end of the discharge. The crescendo and the discharge frequency resembled those observed in rat pups during hyperthermia‐induced brain alkalosis. 11 In line with this, both the incidence and the severity of the behavioral as well as the electrographic seizures seen after RRN were strongly reduced by GRN (Figures 3 and 5), which in the present work was achieved by application of 5% CO2 in room air immediately after the asphyxia.
The steep dependence of post‐asphyxia hyperexcitability and seizures on recovery of brain pH (but not O2) was directly demonstrated in simultaneous recordings of cortical pH, Po2, and LFP activity done under light urethane anesthesia. In these experiments, a prominent period of post‐asphyxia hyperexcitability took place, during which some animals (3/16) developed frank seizures (Figure 5). The time window of hyperexcitability and seizures coincided with the fast post‐asphyxia recovery of brain pH and, again, GRN led to a nearly complete abolishment of post‐asphyxia hyperexcitability. Notably, the rate of recovery and overshoot of brain Po2 were similar following GRN and RRN, demonstrating a lack of contribution of the Po2 changes to seizure suppression by GRN. It is obvious from the data in Figures 3 and 5 that a much briefer duration (around 10 min, given the seizure time window of 2–7 min post‐asphyxia) than the preset time of 30 min for the present experiments would have been sufficient for the anticonvulsant action of GRN.
In the fetal sheep model of preterm BA, which has provided large amounts of translationally relevant information, epileptiform discharges emerge within 10–20 min from the end of asphyxia. 49 The magnitude of seizure latency is of similar order as in the present rat model, which is in line with a pH‐dependent mechanism of seizure generation in fetal sheep as well. Importantly, the frequency of the epileptiform discharges in fetal sheep correlates with neuronal loss in the hippocampus and the striatum. 18 , 49 Epileptiform discharges with a brief latency after birth have been reported in human neonates as well, and they are associated with an increased risk of robust electrographic seizures. 50 All these observations suggest that pharmacological interventions done as early as possible to suppress seizures might be highly beneficial for therapeutic outcome.
5. Conclusions
The present work shows that post‐asphyxia changes in brain pH have a pronounced effect on seizure susceptibility. We do not, of course, argue that brain pH changes would be the most important factor in all kinds of post‐asphyxia seizures in human neonates. A number of mechanisms triggered by, for example, energy‐metabolic compromise, free oxygen radicals, and neuroinflammation 51 are likely to contribute, and might be exacerbated by the post‐asphyxia recovery of pH. Notably, the dependence of neuronal excitability on brain pH is particularly steep in the immature brain. 52 Thus, it is reasonable to assume that the large pH changes, which are bound to take place during and following birth asphyxia in human neonates (see Uria‐Avellanal and Robertson 53 ), are likely to have a major influence on the generation and properties of post‐asphyxia seizures. Using an inhaled gas mixture with elevated (5%) CO2 might be difficult in neonatal intensive care units, especially when fast decisions regarding therapeutic interventions have to be made (see Soul et al. 54 ). Therefore, additional strategies targeting post‐asphyxia changes in brain pH should be examined in future work. One such possibility might be to use acetazolamide, which produces a slight hypercapnic acidosis in the brain via inhibition of carbonic anhydrase. 55 Notably, acetazolamide is a safe and highly efficient antiseizure drug, especially in short‐term use. 56
In summary, our work calls into question the translational validity of rodent BA models, which are based on exposure of neonatal animals to pure hypoxia. By far most of the translational impact of preclinical work has come from large‐animal models of BA/HIE, where the experimental protocols typically induce a state of asphyxia, not hypoxia. 14 , 37 , 57 Moreover, large‐animal models have numerous advantages with regard to instrumentation in studies on adaptive and pathophysiological mechanisms associated with BA. On the other hand, standard laboratory rodents offer the important potential for implementing the vast array of well‐established molecular, cellular, and systems‐level techniques that are available for both basic and translational work on the pathophysiology, neurobiology, and behavioral outcome of BA, the associated seizures, and HIE. They also enable efficient screening of potential neuroprotective therapies and antiseizure drugs as done recently by Johne et al. in work on the present model. 58 We are confident that physiologically valid rodent models of the kind described presently will make a significant contribution to research in this highly important field.
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose.
AUTHOR CONTRIBUTIONS
K.K. was responsible for the conception of the study and designed the experiments together with T.A., A.P., J.V. and M.S. T.A., A.P., M.S. and A.A. collected and analysed the data. T.A. prepared all figures. K.K. and T.A. wrote the first draft of the manuscript. They also revised the manuscript according to critical comments from J.V., A.P., S.K. and M.S. All authors read and approved the final manuscript. T.A. and A.P. contributed equally.
Supporting information
Fig S1
Appendix S1
Video S1
Video S2
Video S3
Video S4
ACKNOWLEDGMENTS
We thank Merle Kampura for assistance in the molecular biological analyses, and Maria Partanen, Madara Snepere, and Ann‐Christine Aho for breeding and maintaining the experimental animals. This work was supported by the European Research Council (grant ERC‐2013‐AdG 341116, K.K.), Academy of Finland (grants 294375 and 319237, 2016–2019, K.K.), and Jane and Aatos Erkko Foundation (2017–2020, K.K.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ala‐Kurikka T, Pospelov A, Summanen M, et al. A physiologically validated rat model of term birth asphyxia with seizure generation after, not during, brain hypoxia. Epilepsia.2021;62:908–919. 10.1111/epi.16790
REFERENCES
- 1. Lawn JE, Cousens S, Zupan J. 4 million neonatal deaths: when? Where? Why? Lancet. 2005;365:891–900. [DOI] [PubMed] [Google Scholar]
- 2. Pappas A, Korzeniewski SJ. Long‐term cognitive outcomes of birth asphyxia and the contribution of identified perinatal asphyxia to cerebral palsy. Clin Perinatol. 2016;43:559–72. [DOI] [PubMed] [Google Scholar]
- 3. Rosso IM, Cannon TD, Huttunen T, Huttunen MO, Lönnqvist J, Gasperoni TL. Obstetric risk factors for early‐onset schizophrenia in a Finnish birth cohort. Am J Psychiatry. 2000;157:801–7. [DOI] [PubMed] [Google Scholar]
- 4. Modabbernia A, Mollon J, Boffetta P, Reichenberg A. Impaired gas exchange at birth and risk of intellectual disability and autism: a meta‐analysis. J Autism Dev Disord. 2016;46:1847–59. [DOI] [PubMed] [Google Scholar]
- 5. Glass HC, Grinspan ZM, Shellhaas RA. Outcomes after acute symptomatic seizures in neonates. Semin Fetal Neonatal Med. 2018;23:218–22. [DOI] [PubMed] [Google Scholar]
- 6. Sampath D, White AM, Raol YH. Characterization of neonatal seizures in an animal model of hypoxic‐ischemic encephalopathy. Epilepsia. 2014;55:985–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kawasaki K, Traynelis SF, Dingledine R. Different responses of CA1 and CA3 regions to hypoxia in rat hippocampal slice. J Neurophysiol. 1990;63:385–94. [DOI] [PubMed] [Google Scholar]
- 8. Sun H, Juul HM, Jensen FE. Models of hypoxia and ischemia‐induced seizures. J Neurosci Methods. 2016;260:252–60. [DOI] [PubMed] [Google Scholar]
- 9. Ruusuvuori E, Kaila K. Carbonic anhydrases and brain pH in the control of neuronal excitability. In: Frost S, McKenna R, editors. Carbonic anhydrase: mechanism, regulation, links to disease, and industrial applications. Subcellular Biochemistry, vol. 75. Dordrecht, the Netherlands: Springer; 2014. p. 271–90. [DOI] [PubMed] [Google Scholar]
- 10. Lee J, Taira T, Pihlaja P, Ransom BR, Kaila K. Effects of CO2 on excitatory transmission apparently caused by changes in intracellular pH in the rat hippocampal slice. Brain Res. 1996;706:210–6. [DOI] [PubMed] [Google Scholar]
- 11. Schuchmann S, Schmitz D, Rivera C, Vanhatalo S, Salmen B, Mackie K, et al. Experimental febrile seizures are precipitated by a hyperthermia‐induced respiratory alkalosis. Nat Med. 2006;12:817–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tolner EA, Hochman DW, Hassinen P, Otáhal J, Gaily E, Haglund MM, et al. Five percent CO2 is a potent, fast‐acting inhalation anticonvulsant. Epilepsia. 2011;52:104–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vutskits L. Cerebral blood flow in the neonate. Paediatr Anaesth. 2014;24:22–9. [DOI] [PubMed] [Google Scholar]
- 14. Giussani DA. The fetal brain sparing response to hypoxia: physiological mechanisms. J Physiol. 2016;594:1215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perez R, Espinoza M, Riquelme R, Parer JT, Llanos AJ. Arginine vasopressin mediates cardiovascular responses to hypoxemia in fetal sheep. Am J Physiol Integr Comp Physiol. 1989;256:R1011–8. [DOI] [PubMed] [Google Scholar]
- 16. Vannucci RC, Vannucci SJ. Perinatal hypoxic‐ischemic brain damage: evolution of an animal model. Dev Neurosci. 2005;27:81–6. [DOI] [PubMed] [Google Scholar]
- 17. Lynch NE, Stevenson NJ, Livingstone V, Murphy BP, Rennie JM, Boylan GB. The temporal evolution of electrographic seizure burden in neonatal hypoxic ischemic encephalopathy. Epilepsia. 2012;53:549–57. [DOI] [PubMed] [Google Scholar]
- 18. Bennet L, Dean JM, Wassink G, Gunn AJ. Differential effects of hypothermia on early and late epileptiform events after severe hypoxia in preterm fetal sheep. J Neurophysiol. 2007;97:572–8. [DOI] [PubMed] [Google Scholar]
- 19. Romijn HJ, Hofman MJ, Gransbergen A. At what age is the developing cortex of the rat comparable to that of the full‐term newborn baby? Early Hum Dev. 1991;26:61–7. [DOI] [PubMed] [Google Scholar]
- 20. Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble‐Haeusslein LJ. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. 2013;106–107:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quintana P, Soto D, Poirot O, Zonouzi M, Kellenberger S, Muller D, et al. Acid‐sensing ion channel 1a drives AMPA receptor plasticity following ischemia and acidosis in hippocampal CA1 neurons. J Physiol. 2015;19:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jensen FE, Wang C. Hypoxia‐induced hyperexcitability in vivo and in vitro in the immature hippocampus. Epilepsy Res. 1996;26:131–40. [DOI] [PubMed] [Google Scholar]
- 23. Summanen M, Bäck S, Voipio J, Kaila K. Surge of peripheral arginine vasopressin in a rat model of birth asphyxia. Front Cell Neurosci. 2018;12:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pospelov AS, Puskarjov M, Kaila K, Voipio J. Endogenous brain‐sparing responses in brain pH and PO2 in a rodent model of birth asphyxia. Acta Physiol (Oxf). 2020;229:e13467. [DOI] [PubMed] [Google Scholar]
- 25. Kelen D, Andorka C, Szabó M, Alafuzoff A, Kaila K, Summanen M. Serum copeptin and neuron specific enolase are markers of neonatal distress and long‐term neurodevelopmental outcome. PLoS One. 2017;12:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Evers KS, Wellmann S. Arginine vasopressin and copeptin in perinatology. Front Pediatr. 2016;4:2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bender TM, Johnston JA, Manepalli AN, Mink RB. Association between brain tissue pH and brain injury during asphyxia in piglets. Resuscitation. 2003;59:243–54. [DOI] [PubMed] [Google Scholar]
- 28. Remzso G, Németh J, Varga V, Kovács V, TóthSzuki V, Kaila K, et al. Brain interstitial pH changes in the subacute phase of hypoxic‐ischemic encephalopathy in newborn pigs. PLoS One. 2020;15:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Woodbury DM, Rollins LT, Gardner MD, Hirschi WL, Hogan JR, Rallison ML, et al. Effects of carbon dioxide on brain excitability and electrolytes. Am J Physiol. 1958;192:79–90. [DOI] [PubMed] [Google Scholar]
- 30. Yoshioka H, Nioka S, Miyake H, Zaman A, Sawada T, Chance B. Seizure susceptibility during recovery from hypercapnia in neonatal dogs. Pediatr Neurol. 1996;15:36–40. [DOI] [PubMed] [Google Scholar]
- 31. Helmy MM, Tolner EA, Vanhatalo S, Voipio J, Kaila K. Brain alkalosis causes birth asphyxia seizures, suggesting therapeutic strategy. Ann Neurol. 2011;69:493–500. [DOI] [PubMed] [Google Scholar]
- 32. Cirelli C, Tononi G. Cortical development, electroencephalogram rhythms, and the sleep/wake cycle. Biol Psychiatry. 2015;77:1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pospelov AS, Yukin AY, Blumberg MS, Puskarjov M, Kaila K. Forebrain‐independent generation of hyperthermic convulsions in infant rats. Epilepsia. 2016;57:e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kellaway P, Hrachovy RA. Status epilepticus in newborns: a perspective on neonatal seizures. Adv Neurol. 1983;34:93–9. [PubMed] [Google Scholar]
- 35. Panaitescu A. Oxytocin reduces seizure burden and hippocampal injury in a rat model of perinatal asphyxia. Acta Endocrinol. 2018;14:315–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cain DP, Boon F, Hargreaves EL. Evidence for different neurochemical contributions to long‐term potentiation and to kindling and kindling‐induced potentiation: role of NMDA and urethane‐sensitive mechanisms. Exp Neurol. 1992;116:330–8. [DOI] [PubMed] [Google Scholar]
- 37. Lear CA, Wassink G, Westgate JA, Nijhuis JG, Ugwumadu A, Galinsky R, et al. The peripheral chemoreflex: indefatigable guardian of fetal physiological adaptation to labour. J Physiol. 2018;596:5611–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lear CA, Galinsky R, Wassink G, Yamaguchi K, Davidson JO, Westgate JA, et al. The myths and physiology surrounding intrapartum decelerations: the critical role of the peripheral chemoreflex. J Physiol. 2016;17:4711–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lennox WG. Effect on the electroencephalogram of drugs and conditions which influence seizures. Arch Neurol Psychiatry. 1936;36:1236. [Google Scholar]
- 40. Schuchmann S, Hauck S, Henning S, Grüters‐Kieslich A, Vanhatalo S, Schmitz D, et al. Respiratory alkalosis in children with febrile seizures. Epilepsia. 2011;52:1949–55. [DOI] [PubMed] [Google Scholar]
- 41. Zanelli S, Goodkin HP, Kowalski S, Kapur J. Impact of transient acute hypoxia on the developing mouse EEG. Neurobiol Dis. 2014;68:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zayachkivsky A, Lehmkuhle MJ, Ekstrand JJ, Dudek FE. Ischemic injury suppresses hypoxia‐induced electrographic seizures and the background EEG in a rat model of perinatal hypoxic‐ischemic encephalopathy. J Neurophysiol. 2015;114:2753–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mitsufuji N, Yoshioka H, Tominaga M, Okano S, Nishiki T, Sawada T. Intracellular alkalosis during hypoxia in newborn mouse brain in the presence of systemic acidosis: a phosphorus magnetic resonance spectroscopic study. Brain Dev. 1995;17:256–60. [DOI] [PubMed] [Google Scholar]
- 44. Hsu KS, Huang CC. Characterization of the anoxia‐induced long‐term synaptic potentiation in area CA1 of the rat hippocampus. Br J Pharmacol. 1997;122:671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Di Filippo M, Tozzi A, Costa C, Belcastro V, Tantucci M, Picconi B, et al. Plasticity and repair in the post‐ischemic brain. Neuropharmacology. 2008;55:353–62. [DOI] [PubMed] [Google Scholar]
- 46. Wang Y, Zhan L, Zeng W, Li K, Sun W, Xu ZC, et al. Downregulation of hippocampal GABA after hypoxia‐induced seizures in neonatal rats. Neurochem Res. 2011;36:2409–16. [DOI] [PubMed] [Google Scholar]
- 47. Zhou C, Sun H, Klein PM, Jensen FE. Neonatal seizures alter NMDA glutamate receptor GluN2A and 3A subunit expression and function in hippocampal CA1 neurons. Front Cell Neurosci. 2015;9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lippman‐Bell JJ, Zhou C, Sun H, Feske JS, Jensen FE. Early‐life seizures alter synaptic calcium‐permeable AMPA receptor function and plasticity. Mol Cell Neurosci. 2016;76:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dean JM, Gunn AJ, Wassink G, George S, Bennet L. Endogenous α 2 ‐adrenergic receptor‐mediated neuroprotection after severe hypoxia in preterm fetal sheep. Neuroscience. 2006;142:615–28. [DOI] [PubMed] [Google Scholar]
- 50. Worden LT, Chinappen DM, Stoyell SM, Gold J, Paixao L, Krishnamoorthy K, et al. The probability of seizures during continuous EEG monitoring in high‐risk neonates. Epilepsia. 2019;60:2508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schiering IAM, De Haan TR, Niermeijer JMF, Koelman JH, Majoie CBLM, Reneman L, et al. Correlation between clinical and histologic findings in the human neonatal hippocampus after perinatal asphyxia. J Neuropathol Exp Neurol. 2014;73:324–34. [DOI] [PubMed] [Google Scholar]
- 52. Ruusuvuori E, Kirilkin I, Pandya N, Kaila K. Spontaneous network events driven by depolarizing GABA action in neonatal hippocampal slices are not attributable to deficient mitochondrial energy metabolism. J Neurosci. 2010;30:15638–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Uria‐Avellanal C, Robertson NJ. Na+/H+ exchangers and intracellular pH in perinatal brain injury. Transl Stroke Res. 2014;5:79–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Soul JS, Pressler R, Allen M, Boylan G, Rabe H, Portman R, et al. Recommendations for the design of therapeutic trials for neonatal seizures. Pediatr Res. 2019;85:943–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Swenson ER. Carbonic anhydrase inhibitors and ventilation: a complex interplay of stimulation and suppression. Eur Respir J. 1998;12:1242–7. [DOI] [PubMed] [Google Scholar]
- 56. Reiss WG, Oles KS. Acetazolamide in the treatment of seizures. Ann Pharmacother. 1996;30:514–9. [DOI] [PubMed] [Google Scholar]
- 57. Koehler RC, Yang ZJ, Lee JK, Martin LJ. Perinatal hypoxic‐ischemic brain injury in large animal models: relevance to human neonatal encephalopathy. J Cereb Blood Flow Metab. 2018;38:2092–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Johne M, Römermann K, Hampel P, Gailus B, Theilmann W, Ala‐Kurikka T, et al.Phenobarbital and midazolam suppress neonatal seizures in a noninvasive rat model of birth asphyxia while bumetanide is ineffective. Epilepsia. 2020. doi:10.1111/epi.16778. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Appendix S1
Video S1
Video S2
Video S3
Video S4